
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence(P,C)-cyclometalated complexes derived from naphthyl phosphines: versatile and powerful tools in organometallic chemistry†
Julien
Monot
 ,
Enrico
Marelli
,
Blanca
Martin-Vaca
and
Didier
Bourissou
*
,
Enrico
Marelli
,
Blanca
Martin-Vaca
and
Didier
Bourissou
*
CNRS/Université Paul Sabatier, Laboratoire Hétérochimie Fondamentale et Appliquée (LHFA, UMR 5069), 118 Route de Narbonne, 31062 Cedex 09 Toulouse, France. E-mail: didier.bourissou@univ-tlse3.fr
First published on 2nd May 2023
Abstract
The chemistry of (P,C)-cyclometalated complexes derived from naphthyl phosphines [Np(P,C)M] is presented and analysed in this review. The three main synthetic approaches, namely P-chelation assisted C–H activation, oxidative addition and transmetalation, are described and compared. If a naphthyl framework inherently predisposes a phosphorus atom and transition metal to interact, a rigid metallacycle may induce some strain and distortion, as apparent from the survey of the single-crystal X-ray diffraction structures deposited in the Cambridge Structural Database (77 entries with metals from groups 7 to 11). Generally, the Np(P,C)-cyclometalation imparts high thermal and chemical robustness to the complexes, and a variety of stoichiometric reactions have been reported. In most cases, the metalacyclic structure is retained, but protodecyclometalation and ring-expansion have been sparingly observed. [Np(P,C)M] complexes have also proved to be competent and actually competitive catalysts in several transformations, and they act as key intermediates in some others. In addition, interesting phosphorescence properties have been occasionally pointed out.
 Julien Monot | Julien Monot obtained his PhD degree in 2008 under the supervision of Dr M. Petit and Dr B. Bujoli in Nantes. He then joined Dr E. Lacôte and Pr M. Malacria's group at IPCM, Paris, for a one-year postdoctoral contract supported by the ANR working on the development of NHC-borane chemistry in collaboration with Pr D. Curran from the University of Pittsburgh where he spent two additional years. After a last year as a postdoctoral fellow in the group of Pr V. Gandon at the Université Paris Orsay studying NHC-Ga(III) chemistry, he was recruited as an Assistant Professor at the Université Paul Sabatier in 2012. He is currently working with D. Bourissou and B. Martin-Vaca on metal–ligand cooperative catalysis with group 10 transition metals and the synthesis of original monomers targeting well defined polymers with precise mechanical and chemical properties. |
 Enrico Marelli | Enrico Marelli received his MSc degree at the University of Milan in the group of Prof. C. Gennari, and his PhD degree at the University of St Andrews, working on Ni and Pd catalysis, under the supervision of Professors S. Nolan and R. Goss. He was then awarded a MSCA grant for a postdoc position in the group of Dr D Bourissou, in Toulouse, studying bifunctional complexes of Pd and Au. He currently works as a development chemist in Copenhagen, for H. Lundbeck. |
 Blanca Martin-Vaca | Blanca Martin-Vaca studied chemistry at the University of Oviedo (Spain) where she obtained her PhD degree in 1995 under the supervision of Professor J. Gimeno in organometallic chemistry. She then joined Dr Rudler's group at the University Pierre et Marie Curie (Paris) as a postdoctoral fellow supported by the Spanish M.E.C. (1996–1998), and studied cyclopropanation and other cyclisation reactions with alkoxycarbenes and ylide complexes of tungsten. She was appointed as an Assistant Professor at the University Paul Sabatier (Toulouse) in 1998 and was promoted to a Full Professor in 2008. Her research interests deal with organometallic chemistry, organic catalysis of polymerization and biodegradable polymers. She is interested in non-innocent pincer complexes and their application in metal–ligand cooperative catalysis. She also works on the development of organic catalytic systems for the ring-opening polymerization reactions of lactones and cyclic carbonates (including functionalized ones) allowing the preparation of polyesters and polycarbonates of tuned structures. |
 Didier Bourissou | Didier Bourissou obtained his PhD degree from the Toulouse University with G. Bertrand. He then spent one year as a research associate at the Ecole Polytechnique with F. Mathey and P. Le Floch. Appointed as a Chargé de Recherche CNRS in 1998, he was promoted to a Directeur de Recherche in 2006. From 2006 to 2018, he was an Associate Professor at the Ecole Polytechnique. He has been a Director of the Laboratoire Hétérochimie Fondamentale et Appliquée (LHFA) from 2011 to 2020. He was awarded the Bronze (2005) and Silver (2016) Medals of the CNRS, the Clavel Lespiau Distinction (2006) and Del Duca Grant (2020) from the French Academy of Sciences, the Acros (2009) and Organic Division (2018) Awards from the French Chemical Society in recognition of his work. His research interests concern new bonding situations and reactivity patterns arising from the main group elements, the transition metals and their interplay. He has pioneered ambiphilic ligands in the mid 2000's and developed the concept of σ-acceptor ligands. Part of his research deals with non-innocent pincer complexes and the unusual behavior of the coinage metals, in particular gold. He is also interested in biodegradable polymers (ring-opening polymerization, organic and dual catalysis, and drug delivery systems). |
Introduction
Since the pioneering contributions of Shaw in the early 1970s,1,2 cyclometalated complexes have attracted considerable interest and are now classics in organometallic chemistry. Their interest ranges from fundamental bonding/reactivity aspects to applications in catalysis, materials chemical science, medicine, and others. The easy access, broad structural modularity and versatile coordination properties of phosphines make them very powerful ligands for transition metals. It is thus not surprising that (P,C)-cyclometalated complexes occupy a forefront position.3–6 Here, PCP complexes I with a central benzene ring and two flanking phosphines have become the reference complexes (Chart 1).7 Their pincer structure results in a unique balance of stability/reactivity. The related (P,C)-cyclometalated complexes II featuring a single phosphine sidearm have also been widely investigated. In contrast, the corresponding naphthyl-based (P,C)-cyclometalated complexes III, hereafter denoted as [Np(P,C)M], long remained underdeveloped although they are known for more than 50 years. Significant progress has been achieved over the past 5–10 years however, in particular, in gold(III) chemistry8 where the [Np(P,C)] framework opened new reactivity paths, enabled unprecedented bonding situations to be authenticated and imparted unique catalytic activity.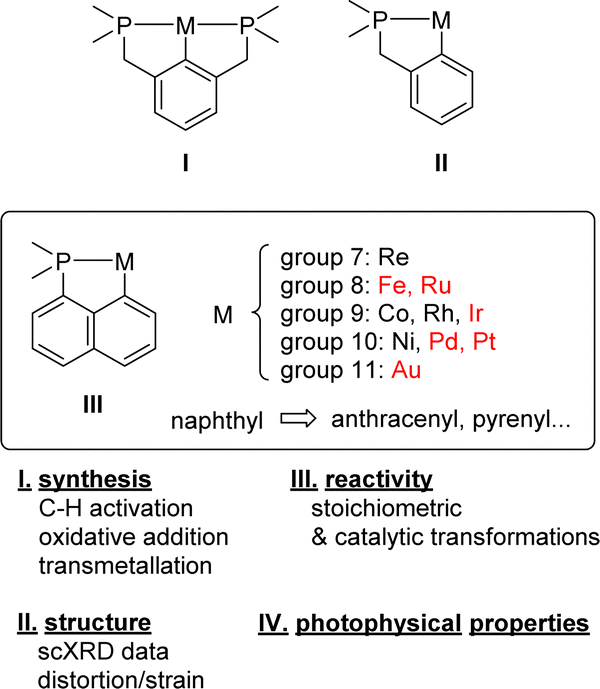 | ||
| Chart 1 (P,C)-cyclometalated complexes I–III, content and organization of this review (metals in red are the most represented ones among [Np(P,C)M] complexes III). | ||
Here, this topic is reviewed. P,C-cyclometalated complexes derived from naphthyl phosphines as well as related systems featuring fused/π-extended systems, such as acenaphthalene, anthracene, phenanthrene and pyrene, are reported. The corresponding two arm stiff PCP complexes are not considered. [Np(P,C)] Complexes have been reported with metals from groups 7 to 11, in line with the preference of a soft phosphine moiety for mid and late transition metals. Emphasis is given to the influence of the rigid naphthyl-based metallacyclic framework on the structure, stability and reactivity of the complexes. Whenever possible, comparison is made with related phenyl-based (P,C)-cyclometalated complexes II.
In the first part, the synthesis of [Np(P,C)M] complexes is presented. The three main synthetic approaches, namely P-chelation assisted C–H activation, oxidative addition and transmetalation, are described and compared. Then, the structure and bonding of [Np(P,C)M] complexes are thoroughly analysed based on crystallographic and computational data. Special attention is given to the distortion/flexibility of a naphthyl spacer and its ability to accommodate a broad range of transition metals, despite its high rigidity. The following section deals with the reactivity of [Np(P,C)M] complexes in stoichiometric reactions and catalytic transformations. The (P,C)-cyclometalated framework is retained in most cases, but not always. Finally, a few photophysical studies performed on [Np(P,C)M] complexes are described.
I. En route to Np(P,C)-cyclometalated complexes
Three main synthetic strategies can be distinguished to access (P,C)-cyclometalated naphthyl complexes, [Np(P,C)M] (Fig. 1). They differ in the nature of pro-ligands and the way the transition metal is introduced. C–H activation, starting from simple 1-naphthyl phosphines, is naturally the most direct and common approach. It is also the oldest one, pioneered by Shaw et al. as early as in 1972. The C–H activation of 1-naphthyl phosphines occurs selectively at the peri position, due to ideal P-chelation (a 5-membered ring with rotation around the P–Cnaphthyl bond as the only degree of freedom). It can be promoted using a basic ligand on the metal or the addition of an exogeneous base/halide scavenger. This route has been used to prepare a broad range of (P,C)-naphthyl complexes with transition metals from groups 7 to 10. Recently, the renewed interest devoted to group 10 and group 11 metals has stimulated studies on alternative pathways to access (P,C)-naphthyl complexes based on the oxidative addition of C–X bonds or on the transmetalation of mercury/tin derivatives. These routes employ prefunctionalized 1-naphthyl phosphines as starting materials, with a halide or a metalloid at the peri position. Naphthalene derivatives with a phosphorus atom bridging the peri positions are also predisposed to give (P,C)-cyclometalated complexes. A few such species have been shown to undergo ring-expansion by the oxidative addition of the strained P–C bond to group 10 metals and gold. These three routes will be successively presented in detail in this section.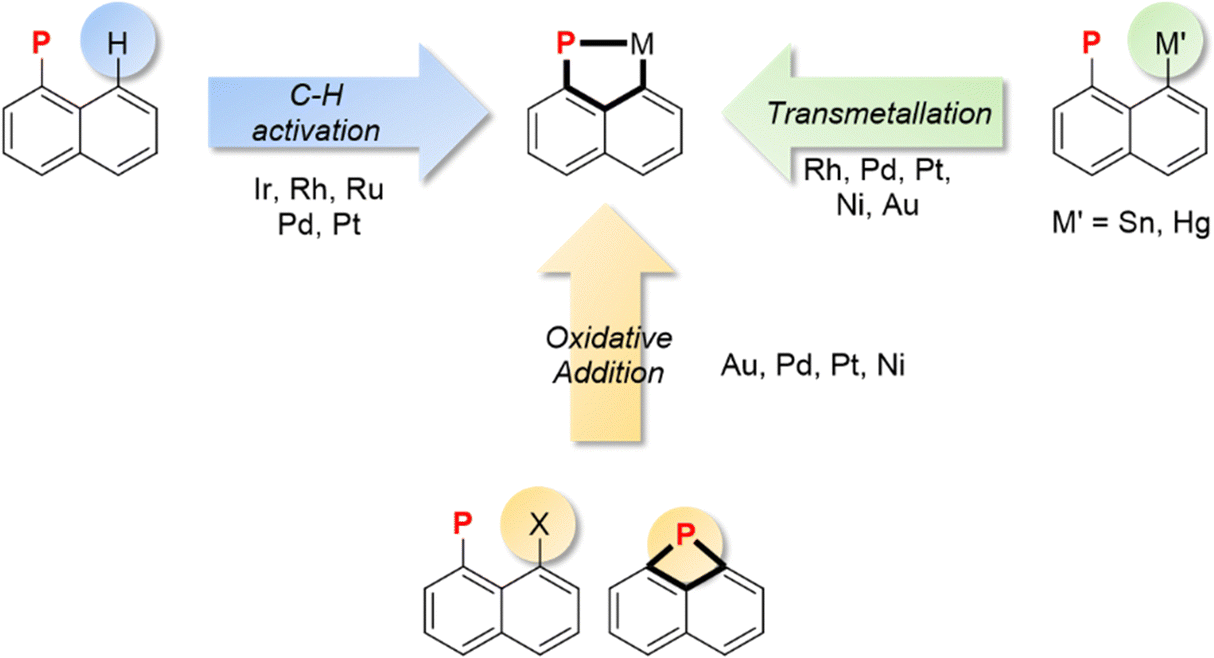 | ||
| Fig. 1 Synthetic routes to access (P,C)-cyclometalated naphthyl complexes [Np(P,C)M]. | ||
1. Via C–H activation
With a unique and simple prefunctionalization on the naphthalene scaffold (to introduce the P atom), the activation of the peri C–H bond of 1-naphthyl phosphines is the most direct and common synthetic route to access [Np(P,C)M] complexes. This strategy was first described by Shaw in 1972 in a comprehensive study targeting [Np(P,C)Ir(III)] complexes.1 Different cyclometalation conditions taking the advantage of P-chelation were reported (Scheme 1).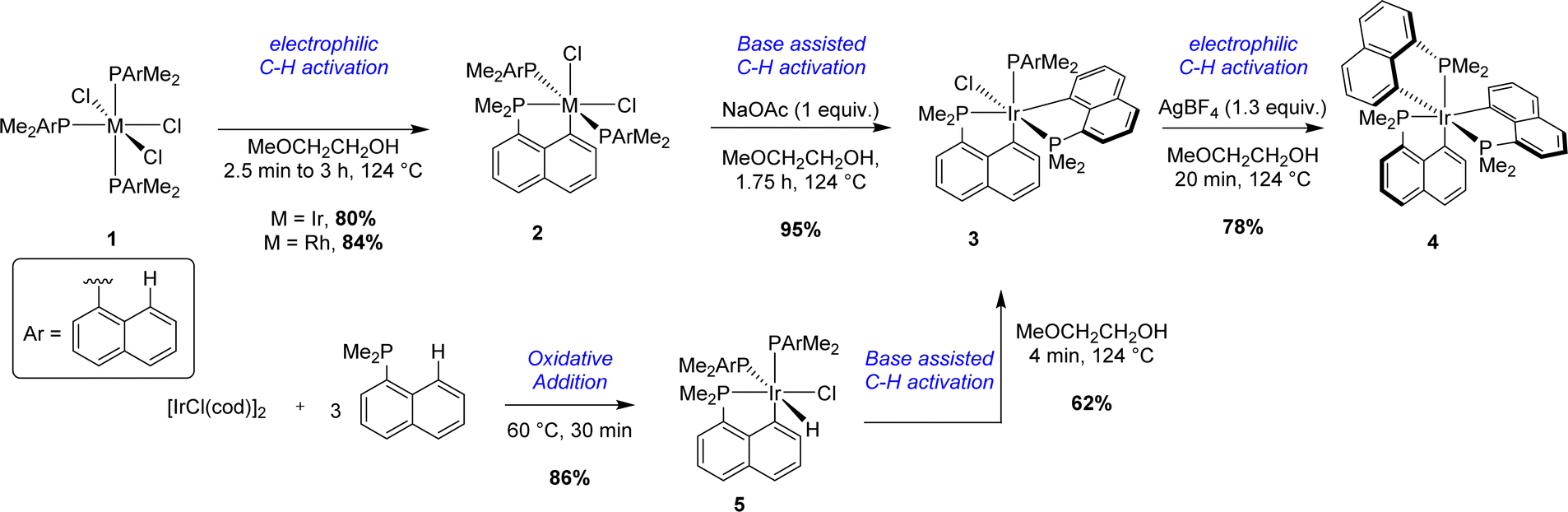 | ||
| Scheme 1 Stepwise cyclometalation of naphthyl phosphines at Ir(III) via electrophilic/base-assisted C–H activations. | ||
Starting from Ir(III) complex 1 bearing three dimethyl 1-naphthyl phosphines coordinated in a meridional fashion, the first cyclometalation reaction was achieved in a good yield (75%) by thermal activation (124 °C) in methyl glycol. A second C–H activation could be efficiently performed by heating mono-cyclometalated iridium complex 2 with an external base, sodium acetate. Interestingly, the coordination mode of the three phosphorus atoms around the iridium center changed from meridional to facial after this second cyclometalation step. In these two steps, an Ir–H species, formed by β-hydride elimination from the alcoholic solvent, was postulated as a key intermediate. This hydride could then act as an internal base, generating H2 as the by-product. No firm evidence for this mechanism was reported however. Concomitantly, the same strategy was tempted to prepare analogous rhodium complexes. The first electrophilic C–H activation worked well (84% yield). However, no further reactivity was observed in the case of [Np(P,C)Rh(III)] complex 2. An alternative pathway was also described to give bis-(P,C)-cyclometalated complex 3 starting from the iridium(I) precursor [IrCl(cod)]2. After a first C–H activation process by oxidative addition to iridium in boiling light petroleum, iridium(III) complex 5 was isolated in a very good yield (86%). 5 evolved quickly under reflux in methyl glycol to give complex 3 with gas evolution, presumably dihydrogen. Finally, the third metalation process was performed by abstraction of the last chloride at iridium with a scavenger (i.e. AgBF4) to give tris-(P,C)-cyclometalated complex 4.
Two years later, Shaw prepared a family of [Np(P,C)Pt(II)] complexes starting from cis-dichloroplatinum(II) precursor 6 (Scheme 2).2 Electrophilic C–H activation was performed by mixing PtCl2 with 6 to yield bridged platinum(II) dimer 7 after prolonged heating in xylenes (139 °C). For monomeric (P,C)-cyclometalated chloroplatinum(II) complex 8, the addition of sodium acetate was mandatory to obtain a good yield (77%). After halide exchange (chloride to iodide), alkyl and dialkylplatinum(II) complexes 10 and 11 were prepared using methyllithium. Pyrolysis at high temperatures (>195 °C) then afforded the corresponding iodo 12 and methyl (P,C)-cyclometalated 13 complexes with methane evolution.
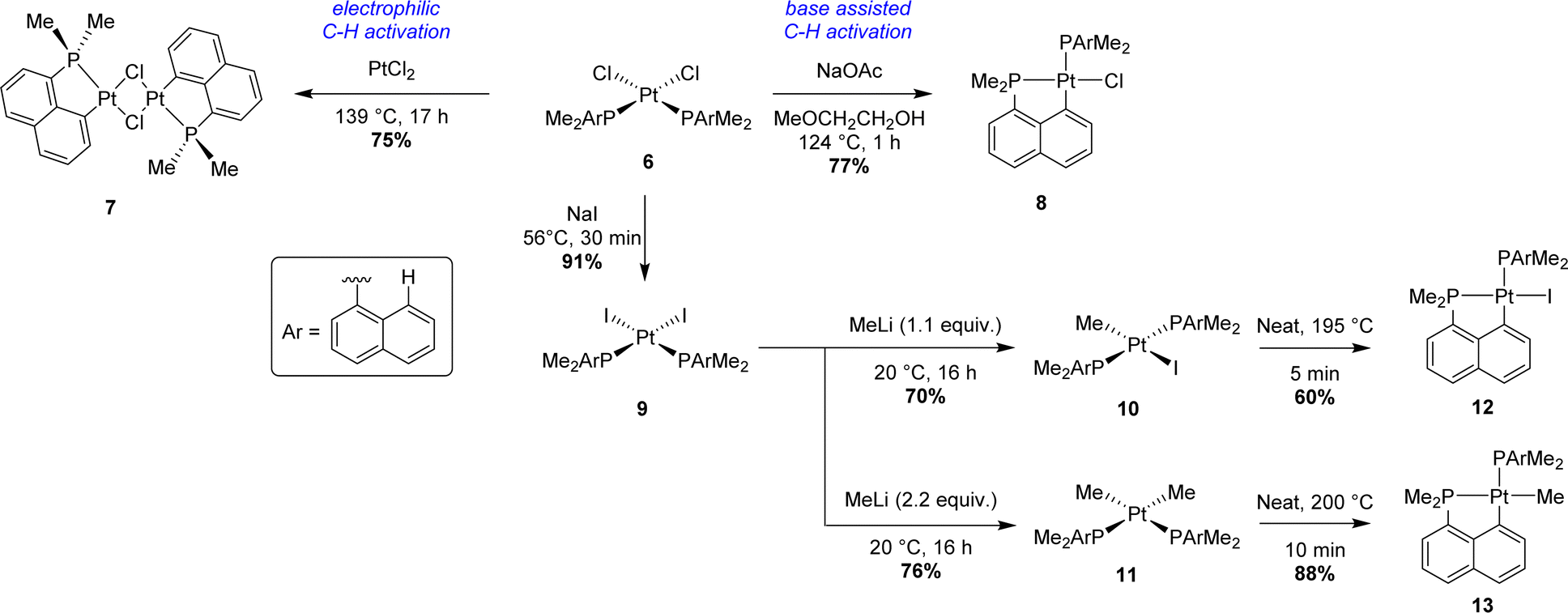 | ||
| Scheme 2 Synthesis of [Np(P,C)Pt(II)] complexes via C–H activation. | ||
In these two seminal reports, Shaw thus depicted four different strategies to cyclometalate 1-naphthyl phosphines, including oxidative addition. The preparations of [Np(P,C)M] complexes via C–H activation reported after 1974 are described in the following sections. Although no detailed mechanistic studies have been carried out on the C–H activation process, these contributions have been organized according to the conditions used to promote cyclometalation:
– Direct (electrophilic) C–H activation by a metal-halide (or metal-pseudohalide) precursor,
– C–H activation in the presence of a mild base (NaOAc, Et3N), bonded to the metal or external, acting in concert with the metal or not,
– C–H activation by a metal-alkyl precursor.
 | ||
| Scheme 3 Synthesis of (P,C)-cyclometalated mono/dinuclear Pt(II) and Pd(II) complexes by electrophilic C–H activation of anthracenyl and pyrenyl mono/diphosphines. | ||
In 2017, Zhu reported the synthesis of two [Np(P,C)Re] complexes starting from Re2(CO)10 (Scheme 4).11 The exact way the peri C–H bond is activated is unknown but the authors invoked P-chelation assistance prior to intramolecular activation.
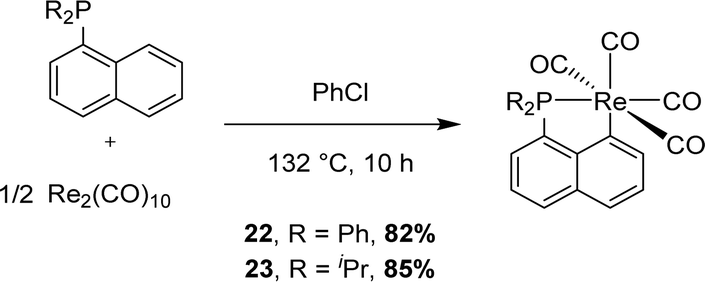 | ||
| Scheme 4 Synthesis of a [Np(P,C)Re(I)] complex via C–H activation. | ||
Following Shaw's pioneering contribution, the groups of Chi and Chou jointly reported in 2010 a stepwise preparation of phosphorescent [Np(Ph2P,C)Ir(III)] complexes (see Section IV).15 First, the bis-N,C-cyclometalated iridium(III) chloride-bridged dimer was splitted by diphenyl 1-naphthyl phosphines upon heating at 100 °C for 24 h (Scheme 5). The initially formed complex 24 with two strong field ligands (carbon and phosphorus) at the trans position to each other could be isomerised into more thermodynamically stable complex 25 (within 48 h at 170 °C). Cyclometalation was then achieved in the presence of sodium acetate (5 eq.) at a high temperature (190 °C). In both cases, a mixture of complexes 26 and 27 was obtained. Based on the absence of thermal interconversion between 26 and 27, the authors proposed a stepwise process: the substitution of chloride by acetate at iridium, followed by C–H bond activation/metalation.
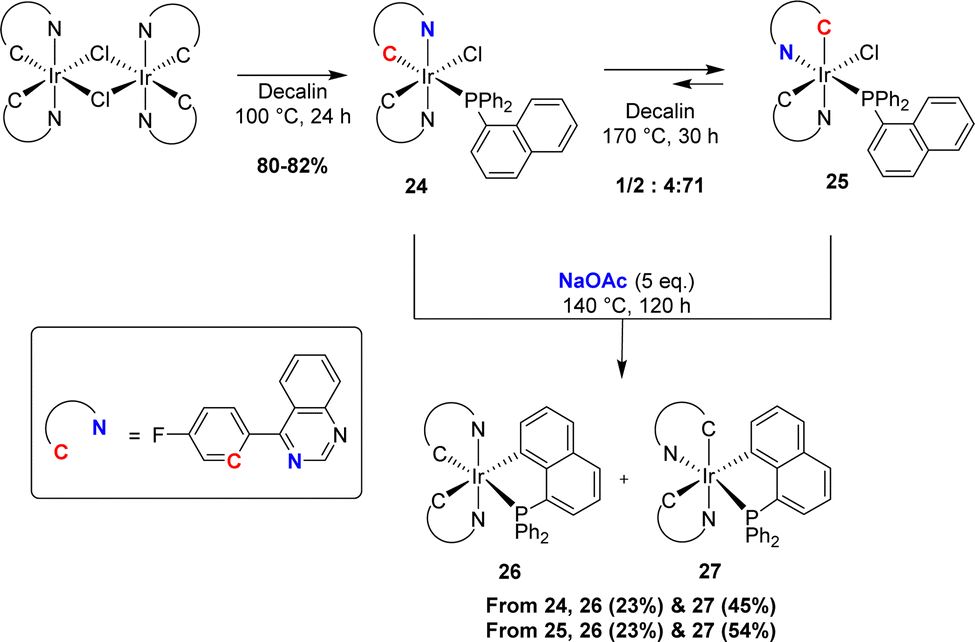 | ||
| Scheme 5 Synthesis of [Np(P,C)Ir(III)] complexes via base-assisted C–H activation. | ||
A year later, the same groups described the preparation of a bis-cyclometalated iridium(III) complex (Scheme 6).16 The electrophilic C–H activation of diphenyl 1-naphthyl phosphines by [IrCl3(tht)3] at 196 °C in decalin first afforded the mono-(P,C)-cyclometalated complex [Np(Ph2P,C)IrCl2(tht)2] (83% isolated yield). Following the addition of diphenyl 1-naphthyl phosphines, the second cyclometalation required the use of sodium acetate (5 equivalents) to give the [Np(Ph2P,C)]2Ir(κ2-OAc) complex.
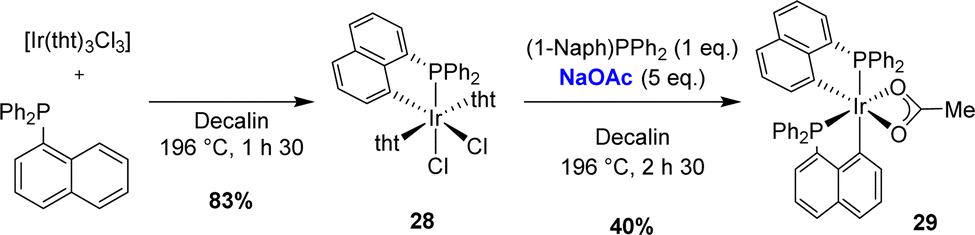 | ||
| Scheme 6 Synthesis of a bis-cyclometalated [Np(P,C)2]Ir(III) complex by subsequent thermal and base-assisted C–H activations. | ||
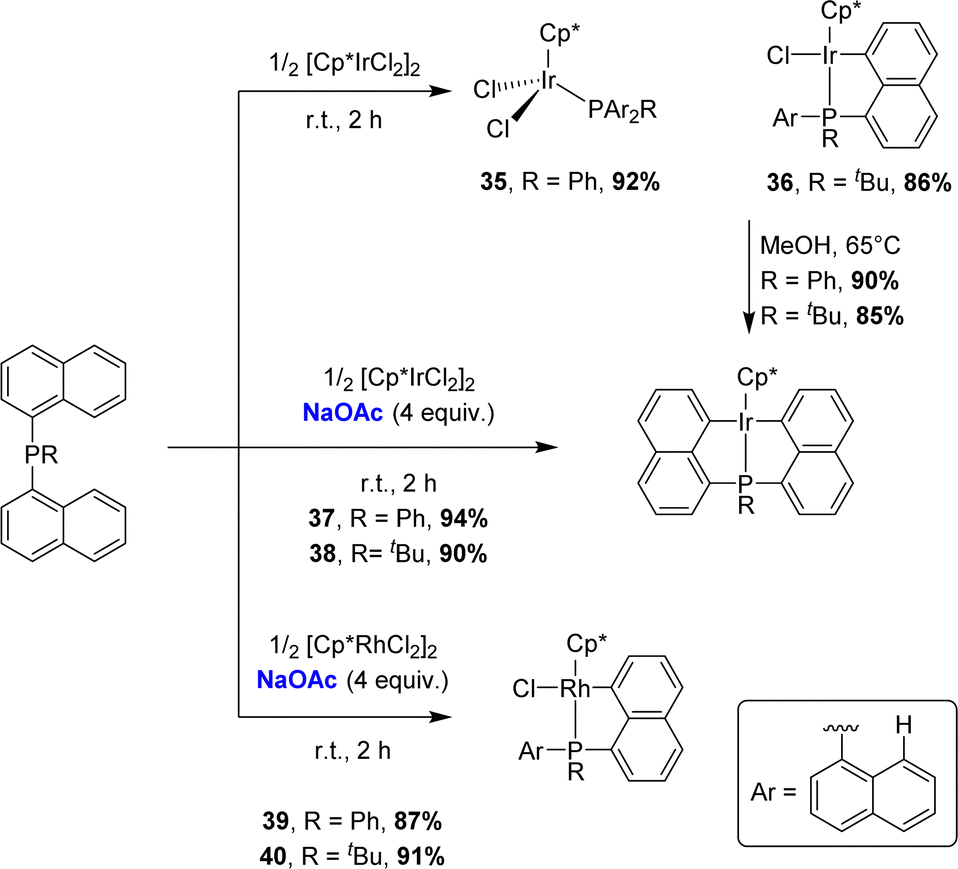
In 2017, the group of Zhu studied in detail the cyclometalation of diphenyl 1-naphthyl phosphines by [Cp*MCl2]2 (M = Ir, Rh) in the presence of sodium acetate (Scheme 7).17 The reactions were performed in methanol at room temperature for 4 h. The cyclometalated complex [Np(Ph2P,C)IrCl(Cp*)] 30 was thereby obtained in a good yield (72%) along with a small amount of the corresponding non-cyclometalated iridium dichloride complex 31. The characterization of complex 31 supports a stepwise P-chelation assisted process for cyclometalation (an independent experiment actually confirmed the conversion of 31 to 30 over time). By comparison, the C–H bond activation seemed to be faster with rhodium, since the rhodium analogue of 31 was not observed during the reaction. In addition, the authors compared the reactivity of Csp2–H and Csp3–H bonds towards cyclometalation using a 1-naphthyl phosphine featuring a methyl group adjacent to phosphorus. Only the Csp2–H bond activation was observed with both iridium and rhodium.
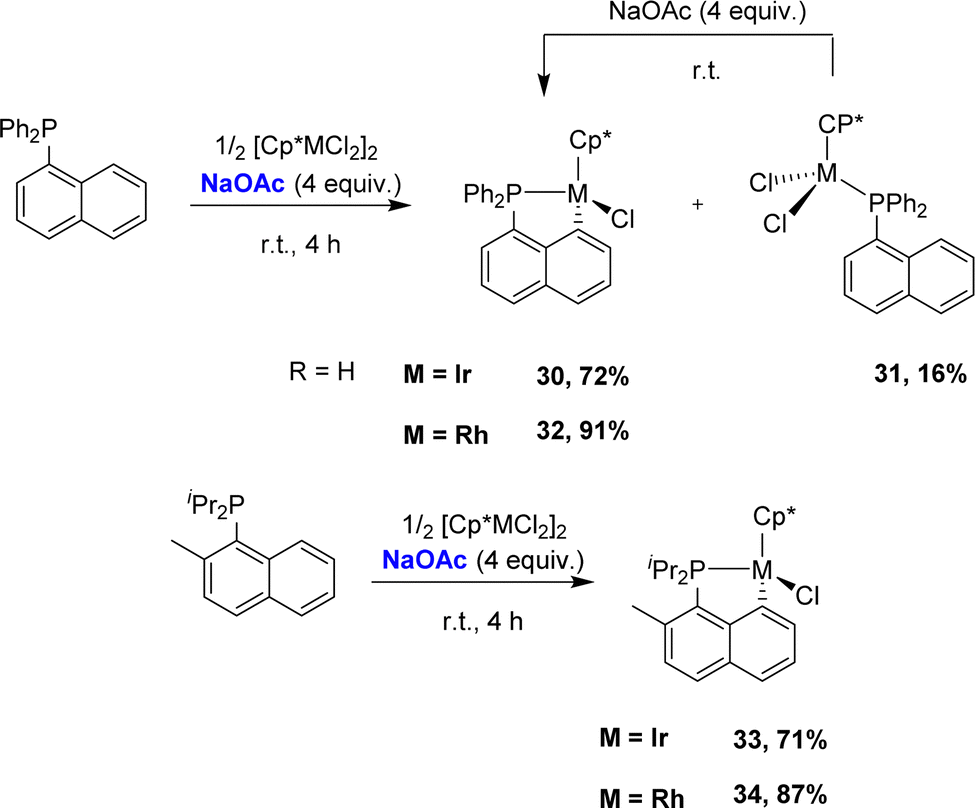 | ||
| Scheme 7 Synthesis of [Np(P,C)Ir(III)] and [Np(P,C)Rh(III)] complexes via base-assisted C–H activation, selectivity for Csp2–H over Csp3–H bonds. | ||
The cyclometalation of bis-1-naphthyl phosphines was then investigated (Scheme 8).18 With [Cp*IrCl2]2, very different outcomes were observed at room temperature depending on the substituent at phosphorus (R = Ph, tBu). Indeed, only P-coordination without cyclometalation was observed for the phenyl phosphine, whereas the tert-butyl phosphine directly afforded the cyclometalated complex [Np(tBu2P,C)IrCl(Cp*)]. Here, C–H activation was probably favoured as it relieved the strain induced by the high steric demand at phosphorus. Upon heating at reflux in methanol, both complexes converted into the corresponding bis-cyclometalated iridium(III) complexes, which were also prepared in high yields without heating using sodium acetate. With [Cp*RhCl2]2, no reaction occurred even upon heating, presumably due to lower electrophilicity of rhodium compared to that of iridium, but the addition of sodium acetate readily promoted cyclometalation, whatever the substituent at phosphorus. Yet, double cyclometalation could not be achieved in this case, even using a stronger base such as tBuOK.
 | ||
| Scheme 8 C–H activation of bis-naphthyl phosphines by [Cp*MCl2]2 (M = Ir, Rh) with/without base assistance. | ||
Zhu also applied the C–H activation route to prepare [Np(P,C)Ru(II)] complexes (Scheme 9).19 Most convenient was to react 1-naphthyl phosphines with [RuCl2(p-cymene)]2 at room temperature in methanol in the presence of sodium acetate (4 eq.). Alternatively, non-cyclometalated phosphine complexes could be first prepared in dichloromethane without a base, and then stirred over silica gel under air to induce cyclometalation. An analogous complex [Np(Ph2P,C)RuCl(C6Me6)] was prepared soon after by Ishii starting from [(η6-C6Me6)RuCl2]2.20 Given that o-tolyl phosphines were shown to undergo Csp3–H cyclometalation under the same conditions, the competition between Csp2–H and Csp3–H activations was assessed by Zhu.19 Using iridium and rhodium, the 1-naphthyl phosphine featuring a methyl group adjacent to phosphorus was found to react with [RuCl2(p-cymene)]2/NaOAc exclusively at the peri Csp2–H site.
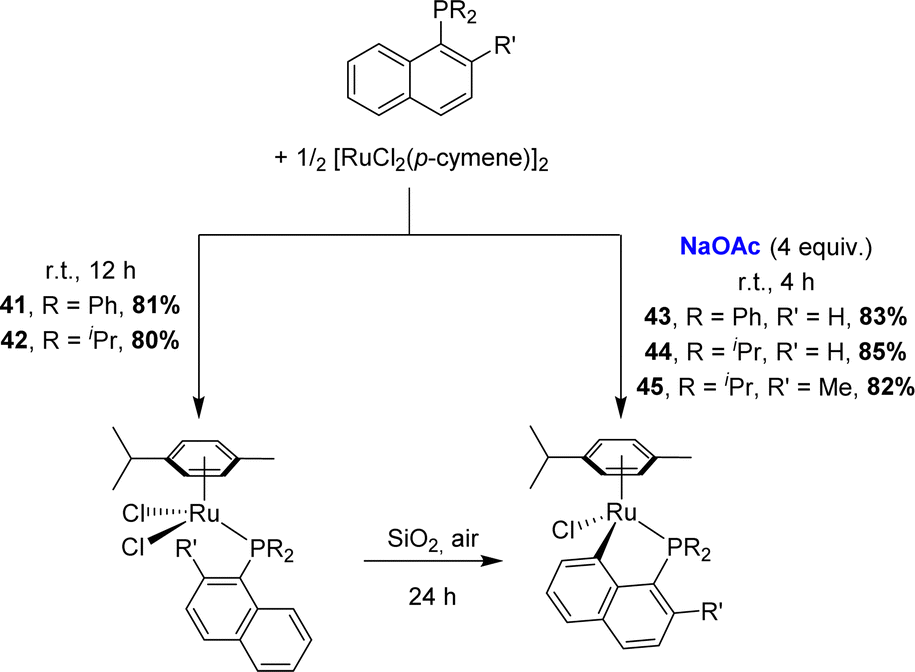 | ||
| Scheme 9 Preparation of [Np(P,C)Ru(II)] complexes via a base or silica gel-assisted C–H activation. | ||
Under the NaOAc-assisted conditions, Grabulosa prepared in 2019 a series of [Np(P,C)Ru(II)] complexes from optically pure P-stereogenic phosphines featuring naphthalene (Chart 2), phenanthrene and pyrene scaffolds for applications in the asymmetric hydrogenation of ketones (see Section III).21 The corresponding (P,C)-cyclometalated complexes were isolated in low yields (4–17%, except 49, 71%) probably due to decomposition during purification by column chromatography. In most cases, they were obtained as diastereoisomeric mixtures (the ruthenium center becomes stereogenic upon cyclometalation). Only in the case of iPr-substituted phosphine, good phosphorus to ruthenium chiral induction was apparently achieved, with the formation of a single diastereoisomer according to 31P NMR spectroscopy.
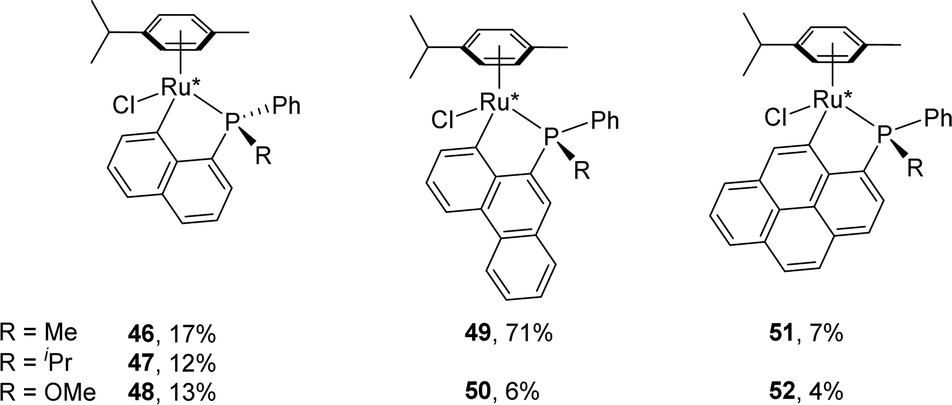 | ||
| Chart 2 (P,C)-cyclometalated Ru(II) complexes derived from optically pure 1-naphthyl, 9-phenanthrenyl and 1-pyrenyl phosphines. | ||
Base-assisted C–H activation has also been applied to prepare (P,C)-cyclometalated naphthyl complexes with group 10 metals. The first examples were reported by Shaw in 1998.22 Starting from the tri-1-naphthyl phosphine, a series of palladacycles were prepared as a result of monocyclometalation. With Pd(OAc)2, a dimeric acetate-bridged complex was obtained (Scheme 10). Subsequent treatment with acetyl-acetone gave the corresponding mononuclear acac complex [Np(Np2P,C)Pd(acac)] which can be alternatively prepared by reacting PNp3 with [Pd(acac)2]. The analogous complex [Np(Np2P,C)Pd(acac)] featuring an hexafluoro acetyl-acetonate co-ligand at palladium was prepared similarly. In 2005, the group of Herrmann applied the same strategy to synthesize palladacycles from the diphenyl 2-methylnaphthyl phosphine.23 Upon treatment with Pd(OAc)2, cyclometalation proceeded readily and efficiently under mild conditions, with complete selectivity for the peri Csp2–H bond.
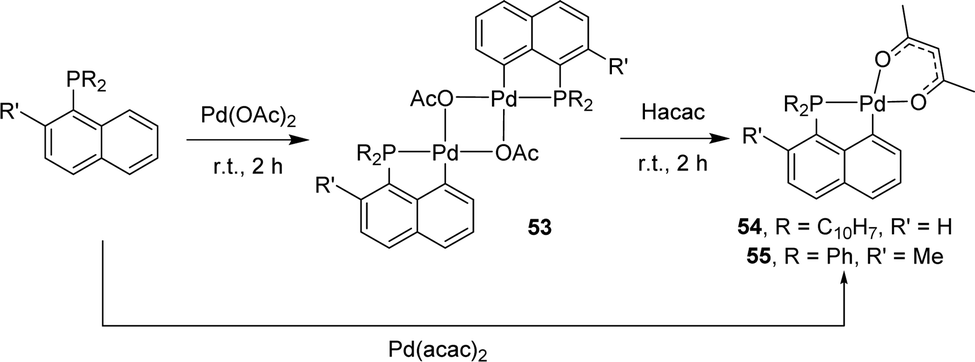 | ||
| Scheme 10 Synthesis of dinuclear OAc-bridged and mononuclear acac [Np(P,C)Pd(II)] complexes by base-assisted C–H activation. | ||
Finally, Vigalok and Vedernikov prepared in 2013 a Pt(II) complex using triethylamine as a base (Scheme 11).24 The naphthyl phosphine featured a methyl group adjacent to phosphorus and again, complete chemoselectivity for Csp2–H over Csp3–H bond activation was observed.
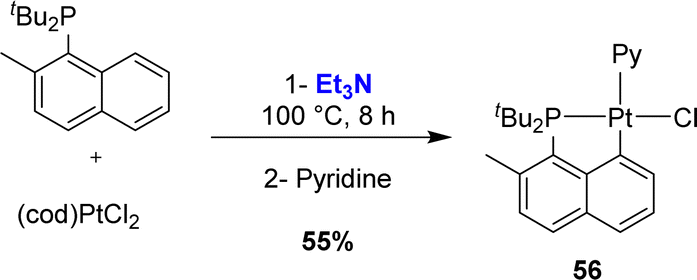 | ||
| Scheme 11 Csp2-selective base-assisted C–H activation using a Pt(II) precursor. | ||
 | ||
| Scheme 12 Synthesis of a [Np(P,C)Pt(II)] complex upon C–H activation by a Pt–Me precursor. | ||
In 2000's, Klein and Beck extended the approach to first-row metals, namely cobalt and iron (Scheme 13).26 The diphenyl 1-naphthyl phosphine was found to react smoothly with trimethylphosphine Fe(II)Me2 and Co(I)Me precursors to give the corresponding (P,C)-cyclometalated complexes. The release of methane acts as a driving force and at the same time imparts irreversibility to cyclometalation.
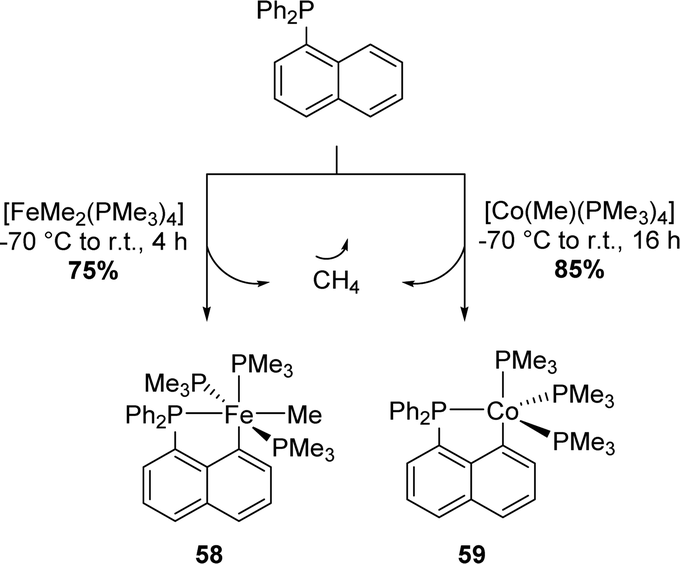 | ||
| Scheme 13 Synthesis of the Np(P,C)-cyclometalated complexes of first row metals by methane elimination. | ||
Metal-silyl precursors may also be used to cyclometalate 1-naphthyl phosphines, as substantiated by Corriu in 1981 (Scheme 14).27 Under UV irradiation, the di-carbonyl silyl iron complex [CpFe(CO)2SiPh2Me] was found to cleanly react with the P-stereogenic phosphine MePhP(Np). The cyclometalated complex [Np(PhMeP,C)FeCp(CO)] was obtained in 85% yield as a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of diastereomers. The silane HSiPh2Me was concomitantly released (90% yield) and no deuterium incorporation was observed when the reaction was performed in deuterated benzene, suggesting that the silyl group indeed participated in the C–H activation process, catching the peri H atom. Of note, the authors attempted to prepare an optically active form of the cyclometalated complex starting from the phosphine in a non-racemic mixture, but sadly, phosphorus epimerization was faster than coordination/cyclometalation under UV irradiation.
:1 mixture of diastereomers. The silane HSiPh2Me was concomitantly released (90% yield) and no deuterium incorporation was observed when the reaction was performed in deuterated benzene, suggesting that the silyl group indeed participated in the C–H activation process, catching the peri H atom. Of note, the authors attempted to prepare an optically active form of the cyclometalated complex starting from the phosphine in a non-racemic mixture, but sadly, phosphorus epimerization was faster than coordination/cyclometalation under UV irradiation.
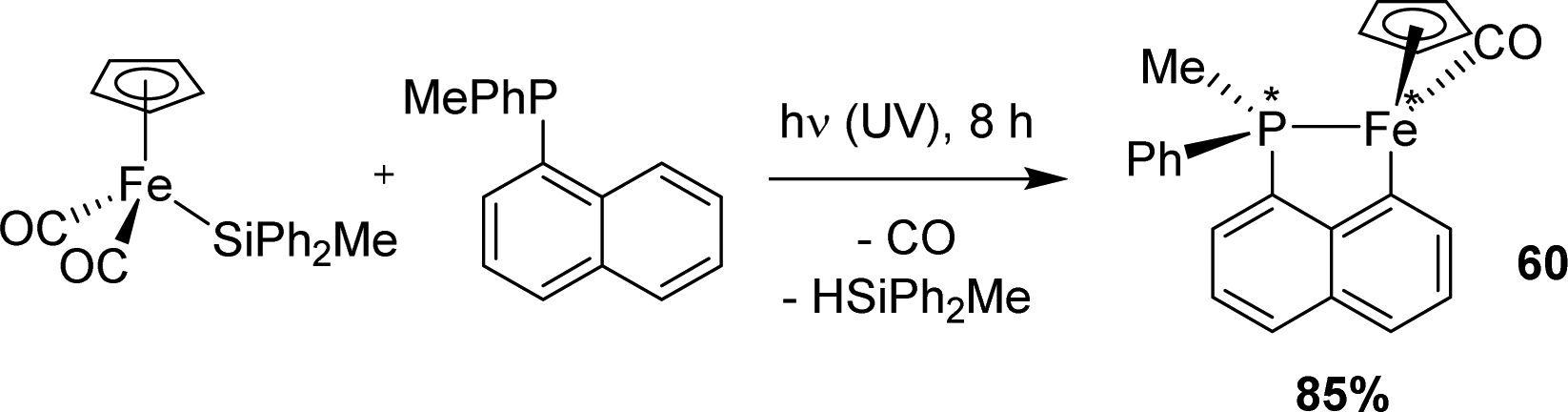 | ||
| Scheme 14 Synthesis of a [Np(P,C)Fe(II)] complex by silane elimination under UV irradiation. | ||
 | ||
| Scheme 15 Synthesis of a dinuclear hydroxy-bridged [Np(P,C)Pd(II)] complex by O2-promoted C–H activation. | ||
2. Via oxidative addition
In 2002, the group of Mizuta reported an original P(III) peri-bridged naphthalene derivative (Scheme 16). This highly strained structure is prone to ring-expansion, as substantiated by the preparation of a [Np(P,C)Pt(II)] complex.29,30 The phosphorus lone pair was first masked with a W(CO)5 fragment. Upon treatment with [Pt(PPh3)4], one of the P–C bonds oxidatively added to platinum and one of the PPh3 co-ligands at platinum were then displaced by CO. Related [Np(P,C)Pd(II)] complexes were then synthesized by the same group using a similar strategy, using [Pd(PPh3)4] as the palladium(0) precursor.31 The phosphorus atom of the strained four-membered phosphacycle was preliminarily oxidized with hydrogen peroxide or elemental sulfur to give the corresponding P(V) pro-ligands. Oxidative addition of the strained P–C bond proceeded easily at room temperature to give the desired phosphapalladacycles in high yields (81–86%). Only one triphenylphosphine co-ligand remained coordinated to the palladium center. The complexes adopt dimeric structures with the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) E moiety (E = O, S) in a bridging mode. These results show the possibility to obtain (P,C)-cyclometalated complexes by the insertion of low valent metals into the strained four-membered ring of phosphorus peri-bridged naphthalenes, but the generality and practical utility of this approach are limited by the access to the starting P compounds.
E moiety (E = O, S) in a bridging mode. These results show the possibility to obtain (P,C)-cyclometalated complexes by the insertion of low valent metals into the strained four-membered ring of phosphorus peri-bridged naphthalenes, but the generality and practical utility of this approach are limited by the access to the starting P compounds.
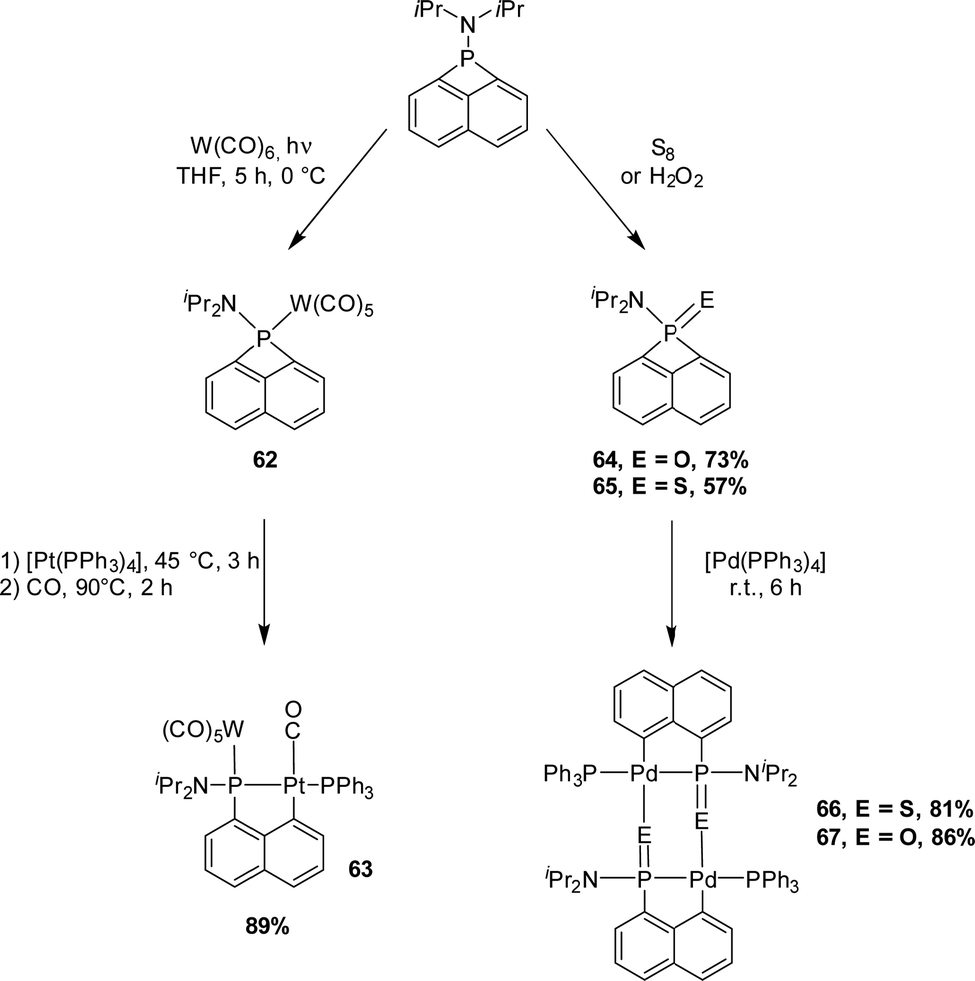 | ||
| Scheme 16 Phosphorus peri-bridged naphthalene derivatives and their ring expansion into (P,C)-cyclometalated naphthyl complexes by oxidative addition to Pt and Pd. | ||
Naphthyl phosphines with halogen at the peri position are readily available from 1,8-dihalogenonaphthalenes.32 They are predisposed to undergo P-chelation assisted oxidative addition, as first demonstrated by our group in 2014 with gold.33 In the context of a lively debate about the feasibility of oxidative addition at gold, we described the straightforward synthesis of (P,C)-cyclometalated gold(III) complexes through the oxidative addition of a Csp2–I bond to a single gold(I) center under mild conditions (Scheme 17).33
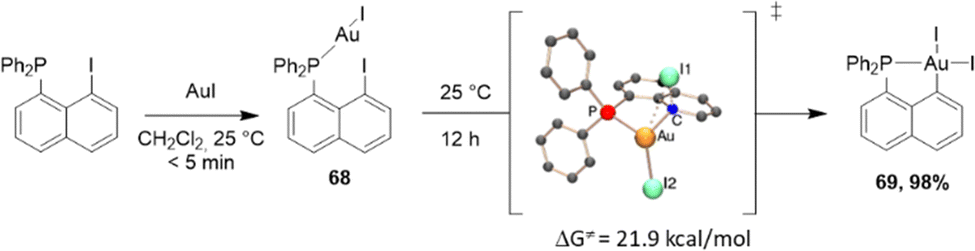 | ||
| Scheme 17 Synthesis of a [Np(P,C)Au(III)] complex by the P-chelation assisted oxidative addition of a peri-iodo naphthyl phosphine to Au. | ||
P-chelation assistance was clearly substantiated by the spectroscopic characterization of the instantaneously formed gold(I) complex, followed by the progressive build-up of the [Np(P,C)Au(III)I2] complex. Kinetic and computational studies shed light on the intramolecular and unimolecular characters of the process involving a three-center transition state. With the corresponding peri-bromo phosphine, oxidative addition could also be successfully achieved upon increasing the reaction temperature to 130 °C (instead of 25 °C for the peri-iodo phosphine, in line with the relative strengths of C–Br and C–I bonds). However, no reaction was observed with the corresponding peri-chloro phosphine, even after several hours at 130 °C in xylene. Calculations confirmed the higher activation barrier for the oxidative addition of the C–Cl bond (ΔG‡ = 39.7 kcal mol−1, compared to 32.1 kcal mol−1 for C–Br and 21.9 kcal mol−1 for C–I). Furthermore, in this case, the formation of the gold(III) complex is uphill in energy (+0.3 kcal mol−1) in contrast to the oxidative additions of the C–I and C–Br bonds (−9.1 and −7.2 kcal mol−1, respectively). Analogous [Np(P,C)AuI2] was subsequently prepared using the same approach using iPr and Cy substituents at phosphorus.34,35
We also studied the behaviour of copper in such a process and reacted peri-halo naphthyl phosphines with various Cu(I) complexes (Scheme 18).36,37 P-chelation was found to efficiently assist the oxidative addition of the C–I bond to copper, but the reaction did not stop there. The ensuing copper(III) species spontaneously underwent P–C reductive elimination to yield new peri-bridged phosphonio naphthalene. DFT calculations confirmed the feasibility and easiness of the C–I oxidative addition and P–C reductive elimination (activation barriers of 9.5 and 7.6 kcal mol−1 in DCM, respectively). The formation of the peri-bridged phosphonio naphthalene compound is exergonic by 12.3 kcal mol−1.
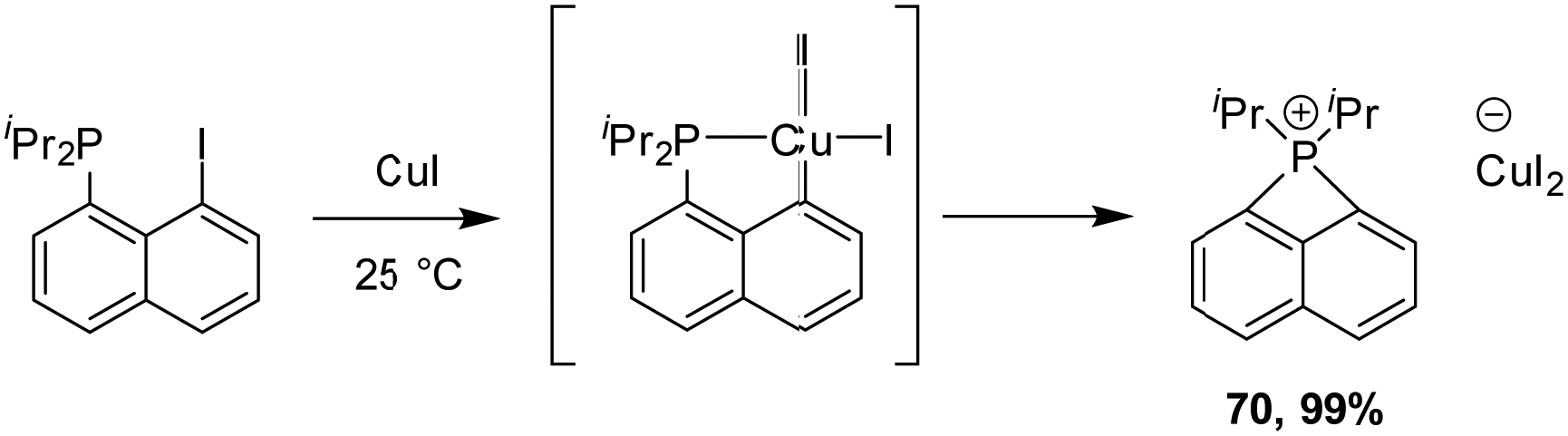 | ||
| Scheme 18 Formation of peri-bridged naphthyl phosphonium by P-chelation assisted C–I oxidative addition to Cu, followed by P–C reductive elimination. | ||
The generality of the reaction was then examined. Replacing the iPr substituents at phosphorus either by a diamino (iPr)NCH2CH2N(iPr) moiety or Ph groups led to similar results. The halogen atom at the peri position was then varied, demonstrating that P-chelation enables copper to also activate C–Br and C–Cl bonds. Finally, efficient catalytic conditions were established to obtain peri-bridged naphthyl-phosphonium using only 1–5 mol% of copper(I) starting from peri iodo- or bromo-naphthyl phosphines (Scheme 19).38
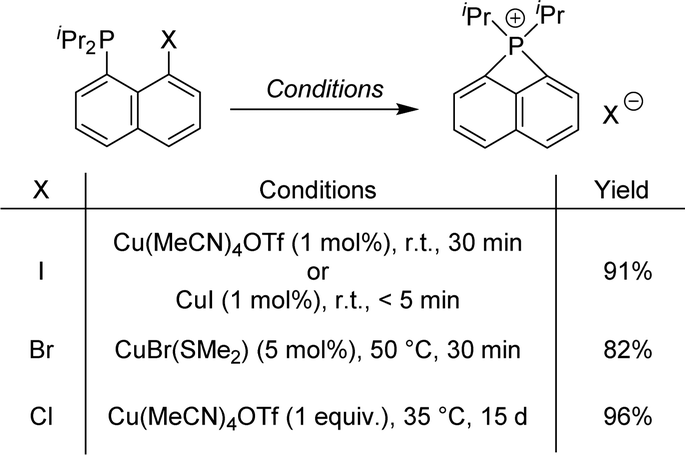 | ||
| Scheme 19 Copper-catalyzed/mediated formation of peri-bridged naphthyl phosphoniums. | ||
The ring strain of peri-bridged phosphonio naphthalenes is apparent from the acute CperiPCperi (77.34°) and PCperiCbridge (86.99° and 86.57°) bond angles. Given the rarity of intermolecular (not chelation-assisted) oxidative additions to gold, we wondered about the possibility to insert gold into the P–C bond and to access thereby [Np(P,C)Au(III)] complexes. This proved indeed feasible, although it required relatively harsh conditions (Scheme 20).
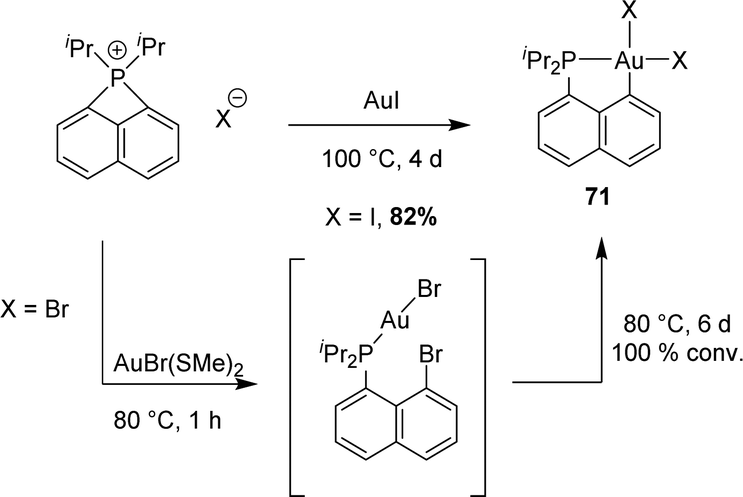 | ||
| Scheme 20 Reactions of the peri-bridged naphthyl phosphonium with the corresponding gold(I) precursor. | ||
In 2021, Beckmann extended the P-chelation strategy and prepared [AcNp(Ph2P,C)Ni(II)] complexes (AcNp refers to acenaphthyl) via the oxidative addition of C–X bonds (X = F, Cl, Br, and I) peri to phosphorus (Scheme 21).39 Once again, halide was observed to have a major impact on the rate of the C–X bond activation. Indeed, the oxidative addition of the C–I bond proceeded at –78 °C and that of the C–Br/C–Cl bonds at room temperature, while heating at 50 °C was required for the C–F bond. Highly air-sensitive dinuclear complexes were obtained. Addition of THF or MeCN subsequently gave rise to mononuclear complexes for X = Cl, Br, and I but not for X = F.
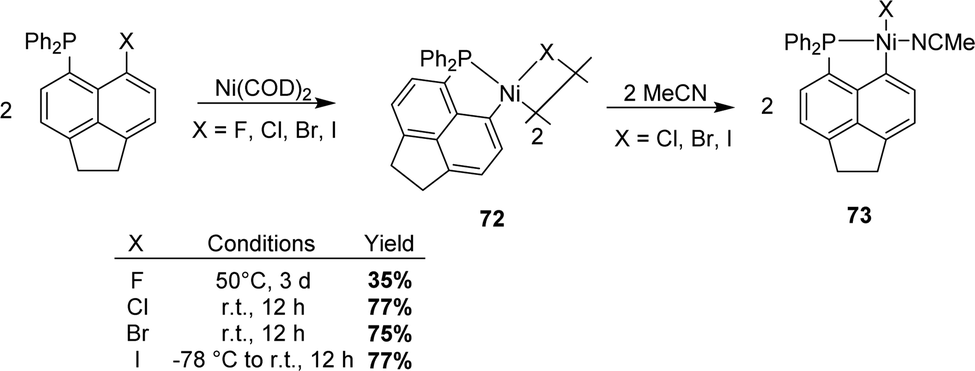 | ||
| Scheme 21 Synthesis of [AcNp(P,C)Ni(II)] complexes by the P-chelation assisted oxidative addition of peri-halo naphthyl phosphines to Ni. | ||
3. Via transmetalation
Recently, Beckmann has developed a complementary transmetalation route (Scheme 22). Accordingly, the [AcNp(Ph2P,C)] group was transferred from mercury or tin to groups 9–11 transition metals (Rh, Ir, Ni, Pd, and Au). Transmetalation from gallium to copper was also observed once, leading to a Cu cluster.40 The mercury(II)/tin(IV) derivatives are readily prepared from the corresponding peri-bromo acenaphthyl phosphine. They are non-reductive and bench stable.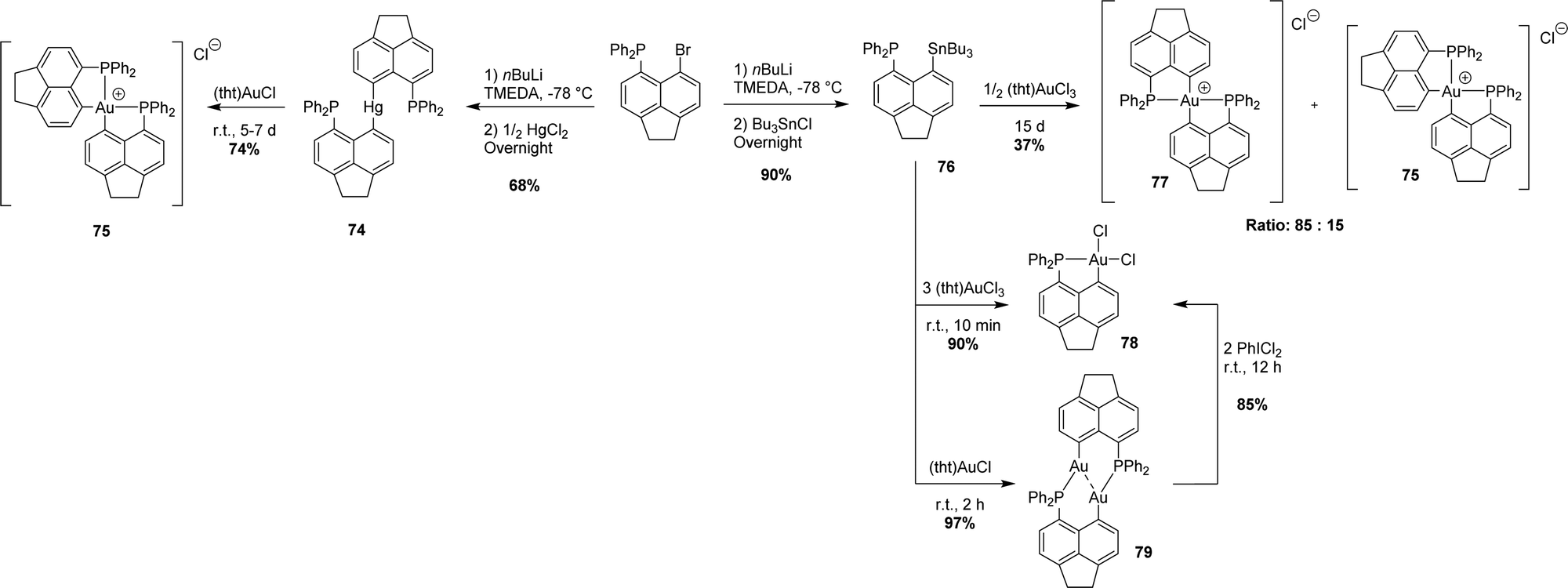 | ||
| Scheme 22 Synthesis of [AcNp(P,C)Au(III)] complexes via mercury and tin to gold transmetalation. | ||
A transmetalation/redox process was first reported.41 The [AcNp(Ph2P,C)]2Hg(II) derivative slowly reacted with (tht)AuCl (tht = tetrahydrothiophene) to give the cationic cis-[AcNp(Ph2P,C)]2Au+ complex along with mercury(0). Note that the same gold(III) complex was obtained later on in the mixture with its trans isomer (cis/trans ratio = 15:85) upon the reaction of the [AcNp(Ph2P,C)SnnBu3] derivative with half an equivalent of (tht)AuCl3.42 The factors controlling the cis/trans stereochemistry of gold(III) complexes remain unknown. When reacted with an excess of (tht)AuCl3, the tin derivative afforded the neutral [AcNp(Ph2P,C)AuCl2] complex (90% yield). This mononuclear gold(III) species could also be prepared in two steps, reacting the tin derivative with one equivalent of the gold(I) precursor (tht)AuCl, and oxidizing the ensuing (P,C)-bridged gold(I) dinuclear complex with PhICl2.
Besides gold(III) complexes, the transmetalation/redox strategy was applied to the preparation of bis-(P,C)-cyclometalated rhodium(III) and platinum(IV) complexes by reacting [AcNp(Ph2P,C)]2Hg(II) with [Rh(CO)2Cl]2 and [PtCl2(CH3CN)2], respectively (Scheme 23).43
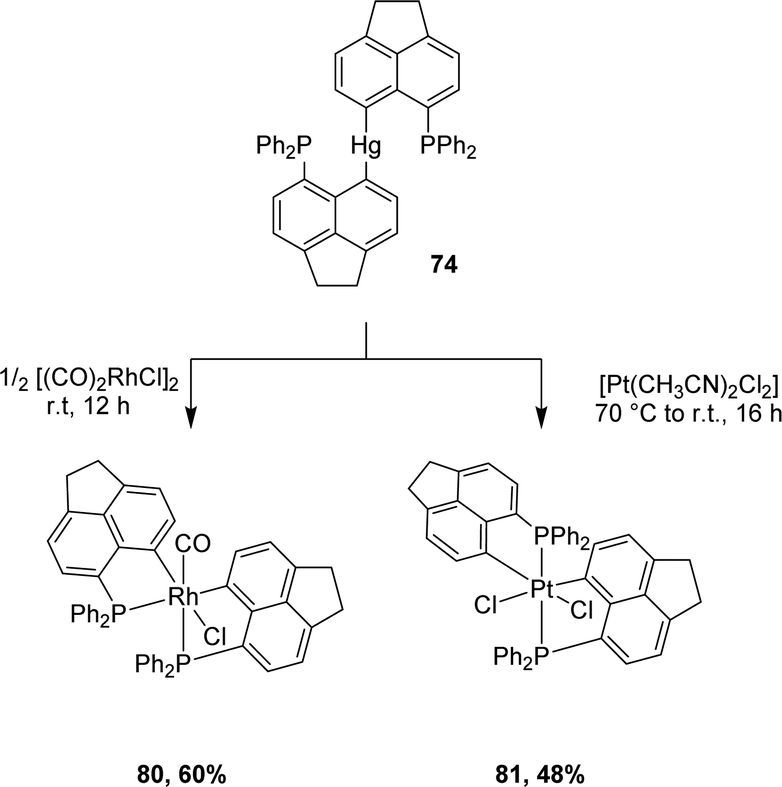 | ||
| Scheme 23 Synthesis of [AcNp(P,C)Rh(III)] and [AcNp(P,C)Pt(IV)] complexes via mercury to rhodium/platinum transmetalation. | ||
The transmetalation approach also works with palladium, as substantiated by the reaction of [AcNp(Ph2P,C)SnnBu3] with [PdCl2(CH3CN)2] (Scheme 24).43 This led to a (P,C)-cyclometalated chloro-bridged dinuclear palladium(II) complex [AcNp(Ph2P,C)PdCl]2 analogous to the nickel complex obtained previously by the P-chelation assisted oxidative addition route.
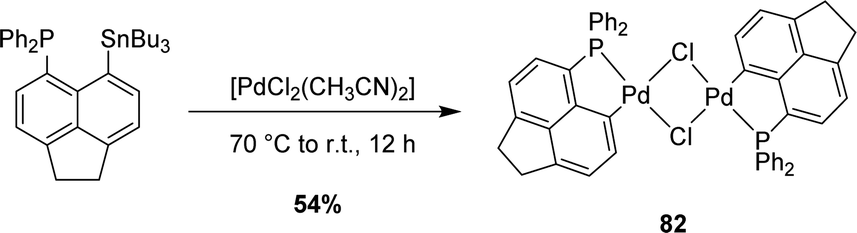 | ||
| Scheme 24 Synthesis of a [AcNp(P,C)Pd(II)] complex via tin to palladium transmetalation. | ||
II. Structure and bonding analyses
A CCDC database search reveals 77 X-ray structures of (P,C)-naphthyl cyclometalated complexes. The key geometric features were collected (see the ESI,† Table S1) so as to analyse the geometric constraints and distortions. These data have been sorted according to the metal: from group 7 (2 Re complexes) to group 8 (6 Fe and 8 Ru complexes), group 9 (2 Co, 1 Rh and 6 Ir complexes), group 10 (4 Ni, 13 Pd and 13 Pt complexes), and group 11 (22 Au complexes).First, looking at the bond distances, the P–M and M–Cperi distances were compared with the sum of the corresponding covalent radii,44 referring to the r parameters (Charts 3a and b). None of the structurally characterized complex shows a r(P–M) factor of above 1.00. About 20% of the structures, mainly Au complexes, possess a r(P–M) factor of 0.95–1.00. All the other complexes fall in the 0.90 ± 0.05 range. The (P,C)-naphthyl backbone is thus very suited and very well accommodate all metals. It does not induce significant constraint on the P–M and C–M bond lengths. This feature is even more apparent from the r(M–Cperi) values that marginally deviate from 1.00 (87% fall in the 1.00 ± 0.05 range).
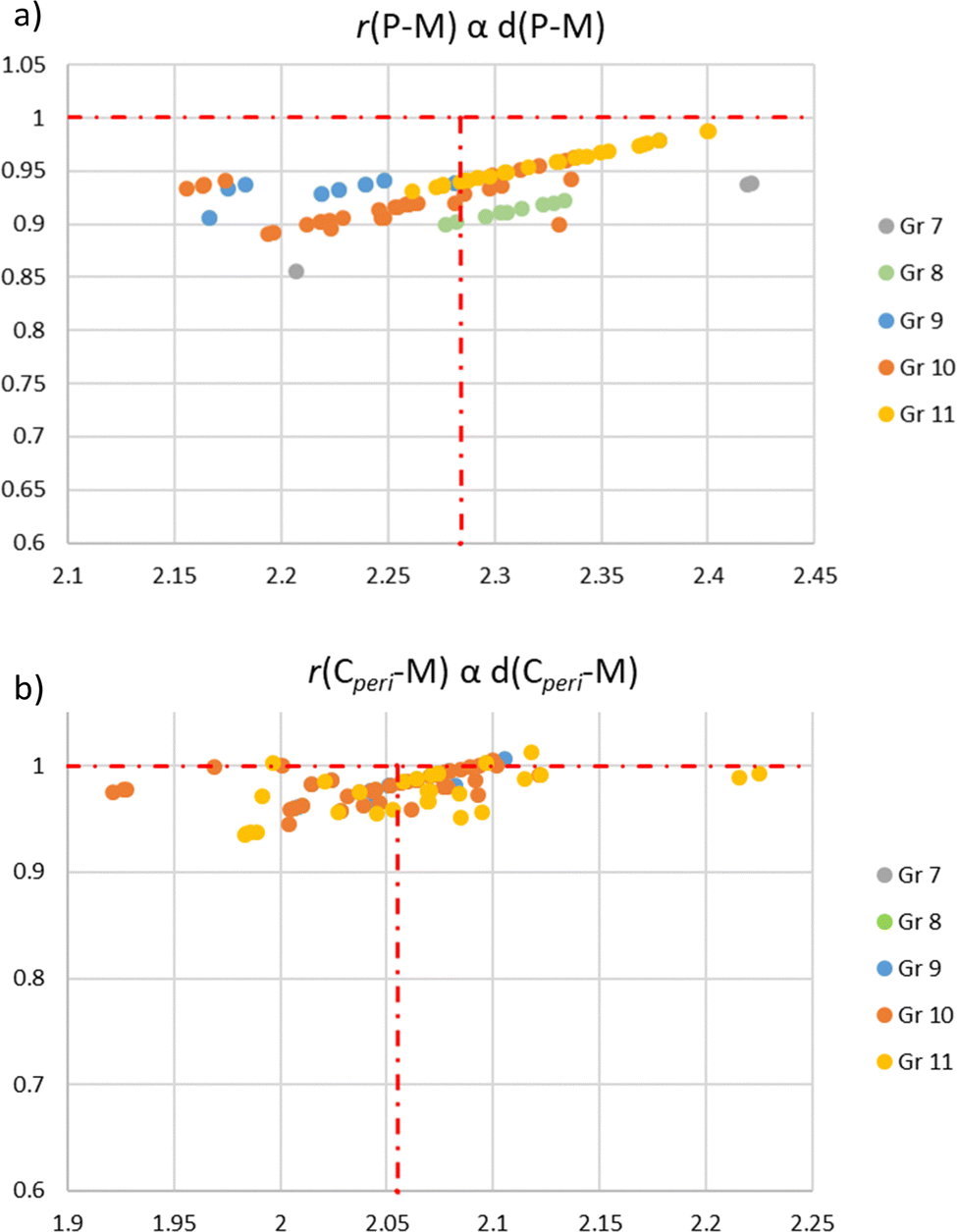 | ||
| Chart 3 (a) Plot of the r(P–M) factor [r(P–M) = d(P–M)/(rcov(P) + rcov(M))] against the P–M bond distance for Gr 7 to 11 transition metals, and the average P–M bond distance (2.284 Å) is indicated by a vertical red dotted line. (b) Plot of the r(Cperi–M) factor [r(Cperi–M) = d(Cperi–M)/(rcov(C) + rcov(M))] against the Cperi–M bond distance for Gr 7 to 11 transition metals, and the average Cperi–M bond distance (2.055 Å) is indicated by a vertical red dotted line. | ||
For bond angles, the bite angle (P–M–Cperi) varies very little, regardless of the transition metal and complex, with 88% of the registered structures displaying a value in between 82 and 85° (Chart 4a). Some in-plane distortions of the naphthalene scaffold are however observed, as apparent from the Cbridge–Cperi–M and P–Cperi–Cbridge bay angles (Charts 4b and c). While the Cbridge–Cperi–M bay angle does not deviate much from the ideal 120° value (by 2° at most), the P–Cperi–Cbridge bay angle is systematically found below 120°, with 85% of the structures falling in the range of 110–115°. The ability of the (P,C)-naphthyl framework to accommodate different metals thus seems to mainly arise from the P atom and the Cperi–P bond.
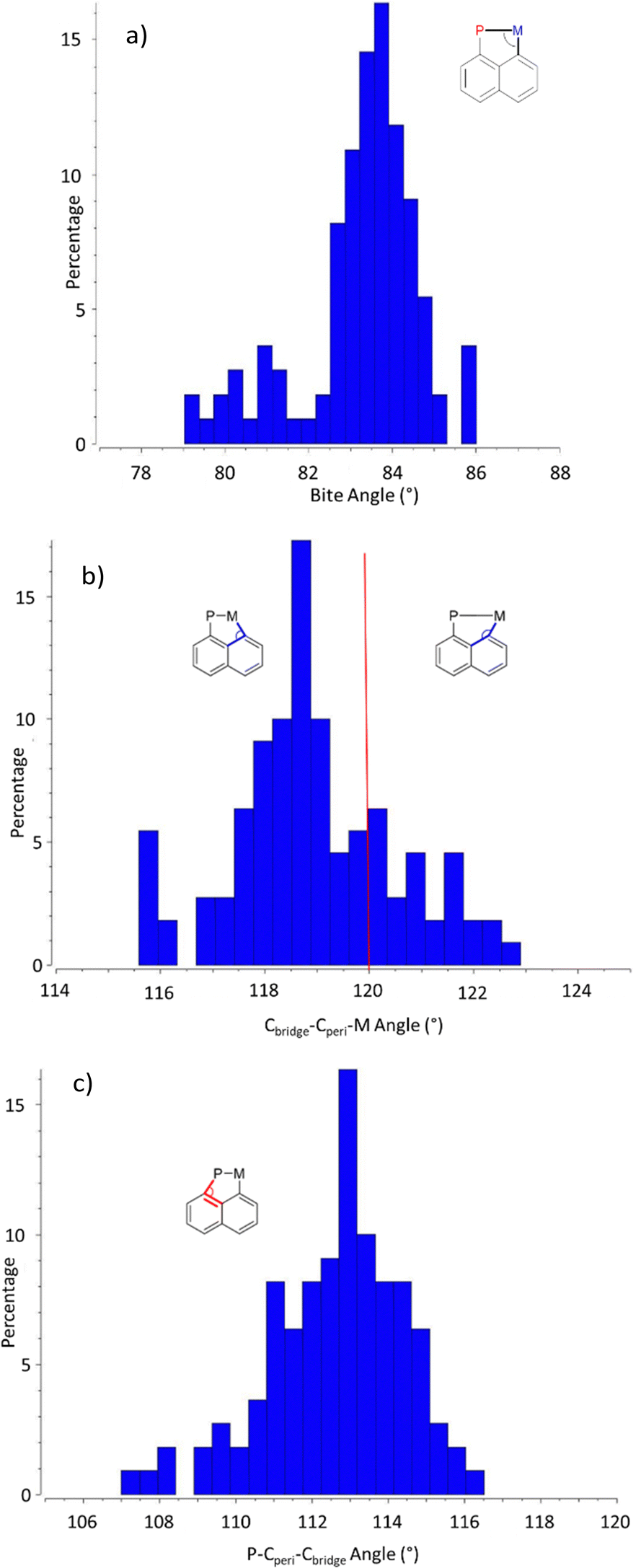 | ||
| Chart 4 Histograms showing the distribution (in percentage) of the bite angle (a), the Cbridge–Cperi–M bond (b) and the P–Cperi–Cbridge bond angle (c) among crystallographically characterized [Np(P,C)M] complexes. | ||
Out-of-plane distortions were also examined through the deviations of the P and M atoms at peri positions from the naphthalene plane (tilt angles α/α′, Charts 5a and b). In general, only minor deviations are observed. No correlation was found between the variations of α,α′ and r(P–M) (Charts S1 and S2, ESI†). The largest deviations concern the P atom (with out-of-plane distortions α up to 10° and 94% of the structures in the 4–10° range) while the metal usually remains close to the naphthalene plane (71% of the structures display α′ values between 1 and 5°). It is to note that even if the deviations are small, the P and M atoms are systematically tilted in opposite directions (up and down the naphthalene plane), probably to minimize steric repulsions between the substituents at phosphorus and the co-ligands at M.
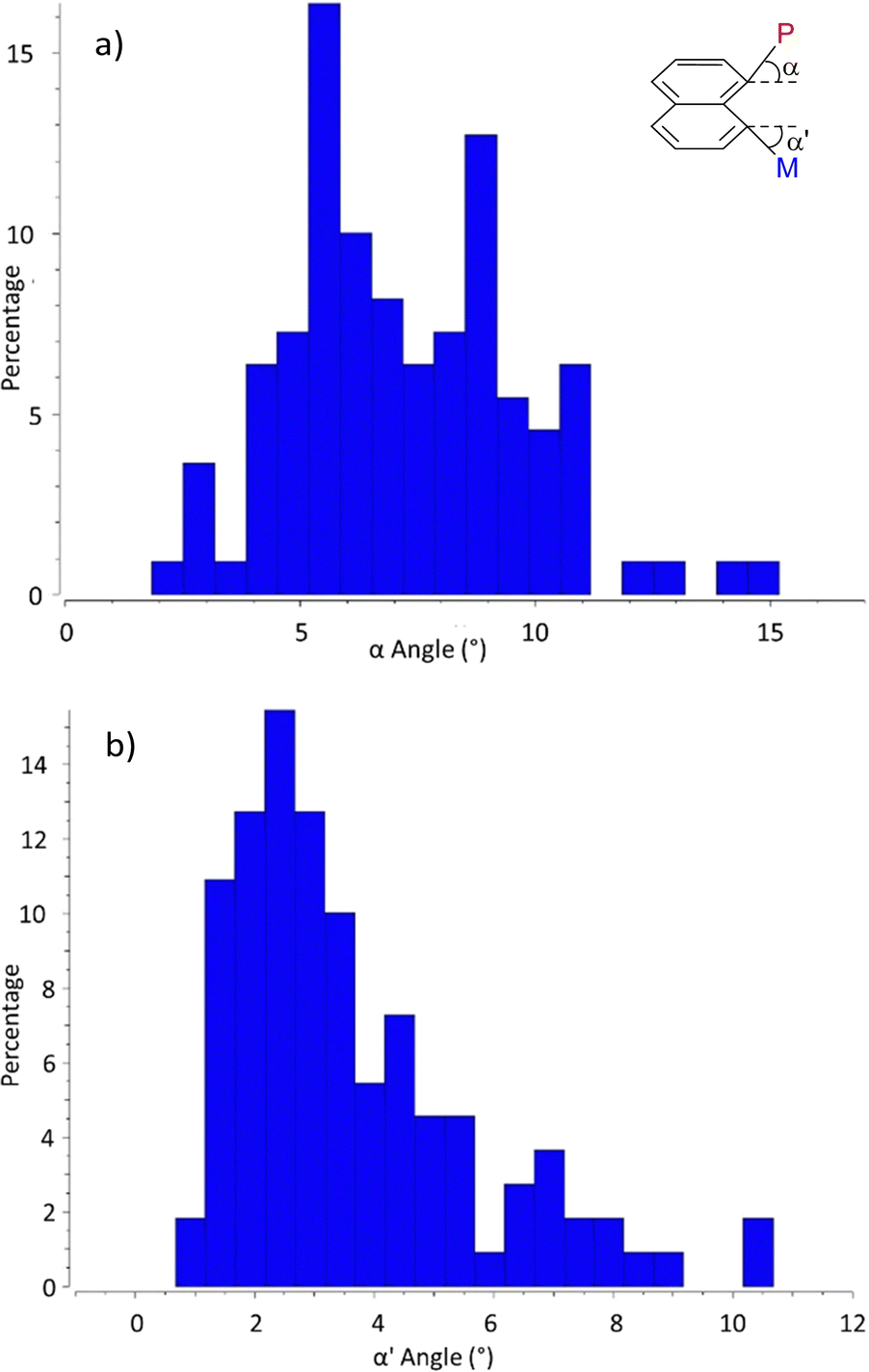 | ||
| Chart 5 Out-of-plane deviations of the P and M atoms at peri positions from the naphthalene plane illustrated by the distribution of the tilt angles α (a) and α′ (b). | ||
The bonding situation within [Np(P,C)M] complexes has been occasionally assessed by computational means. Mebs, Hupf and Beckmann performed atom-in-molecules, electron localizability indicators and non-covalent interaction analyses on rhodium, nickel, palladium, platinum and gold complexes.39,42,43 In all cases, the P–M and C–M bonds were found to be of polar covalent nature. The covalent and ionic contributions are slightly lower in the P–M bonds than in the C–M bonds.
III. Applications of [Np(P,C)M] cyclometalated complexes: stoichiometric reactivity and catalytic transformations
In this section, the stoichiometric reactivity and catalytic application of cyclometalated naphthyl (P,C) complexes will be presented. It is organized per metal, starting from gold(III), then palladium(II), platinum(II/IV), iron(II) and ruthenium(II). In most cases, the metalacyclic structure is retained and these reactions are reported first. Finally, miscellaneous examples involving the cleavage of the M–C bond are discussed.1. [Np(P,C)Au(III)] complexes
In the previous section, we presented the easy access to cyclometalated (P,C)-Au(III) complexes via chelate-assisted oxidative addition of the C–X bond to gold(I). The rigid nature of the (P,C) naphthyl ligand and its strong coordination to gold gives highly stable derivatives and allows us to explore the reactivity of gold(III) complexes.Most of the elementary steps (migratory insertion, β-H elimination, etc.) known with isoelectronic metals (nickel(II), palladium(II) and platinum(II)) were not described with gold(III) complexes a decade ago. In this section, we present our work consisting of meticulous experimental and theoretical studies aiming to demonstrate the ability of gold(III) to efficiently undergo these elementary transformations.
For this purpose, we took advantage of the selective modulation of cyclometalated (P,C)-AuI2 giving easy access to 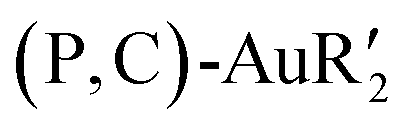 complexes by the reaction with the corresponding Grignard reagent (Scheme 25).34,45,46 In the presence of one equivalent of an organometallic reactant, the organic fragment sits trans to the phosphorus as apparent from NMR (JC–P ≥ 100 Hz for trans isomer compared to 5.7 Hz for methyl at the cis position in Np(P,C)-AuMe2) and in line with trans effects.
complexes by the reaction with the corresponding Grignard reagent (Scheme 25).34,45,46 In the presence of one equivalent of an organometallic reactant, the organic fragment sits trans to the phosphorus as apparent from NMR (JC–P ≥ 100 Hz for trans isomer compared to 5.7 Hz for methyl at the cis position in Np(P,C)-AuMe2) and in line with trans effects.
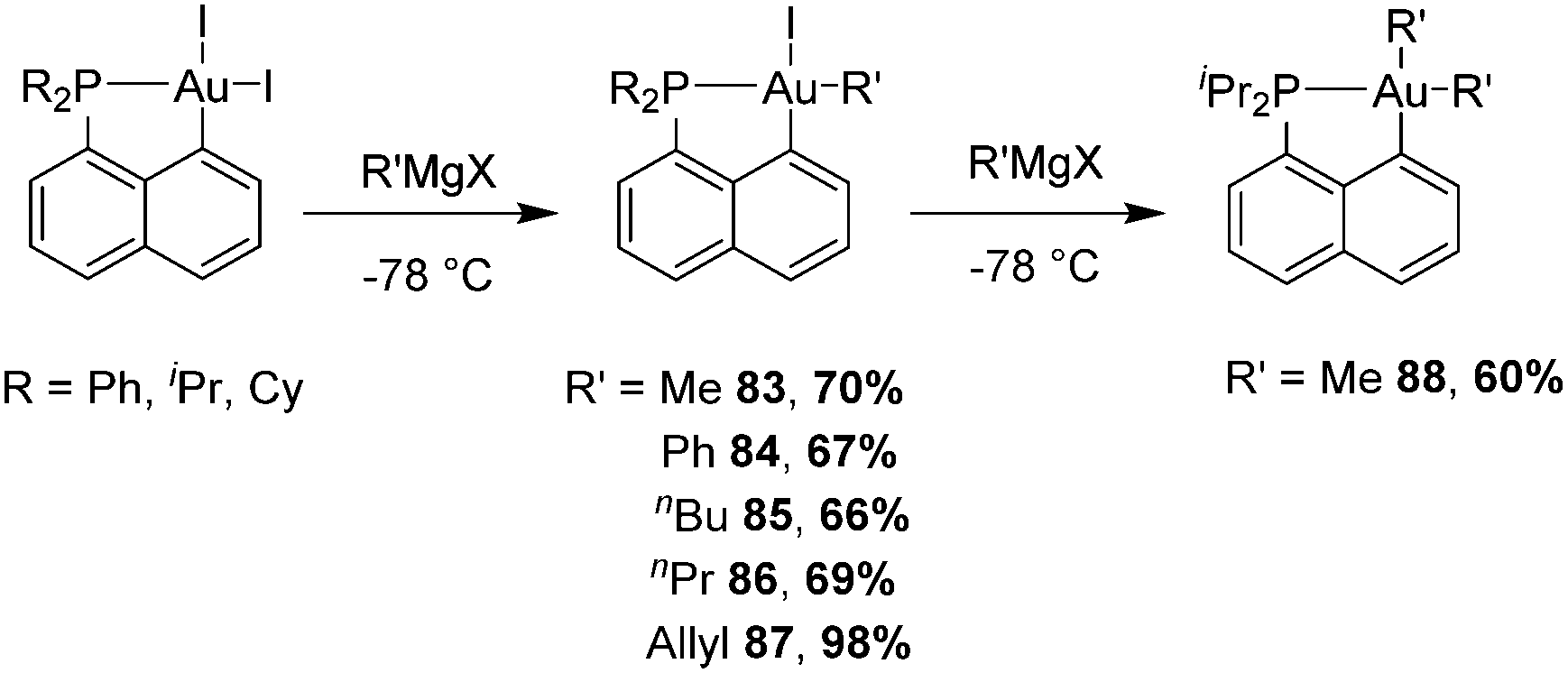 | ||
| Scheme 25 Synthesis of mono- and dialkyl [Np(P,C)Au(III)] complexes using Grignard reagents. | ||
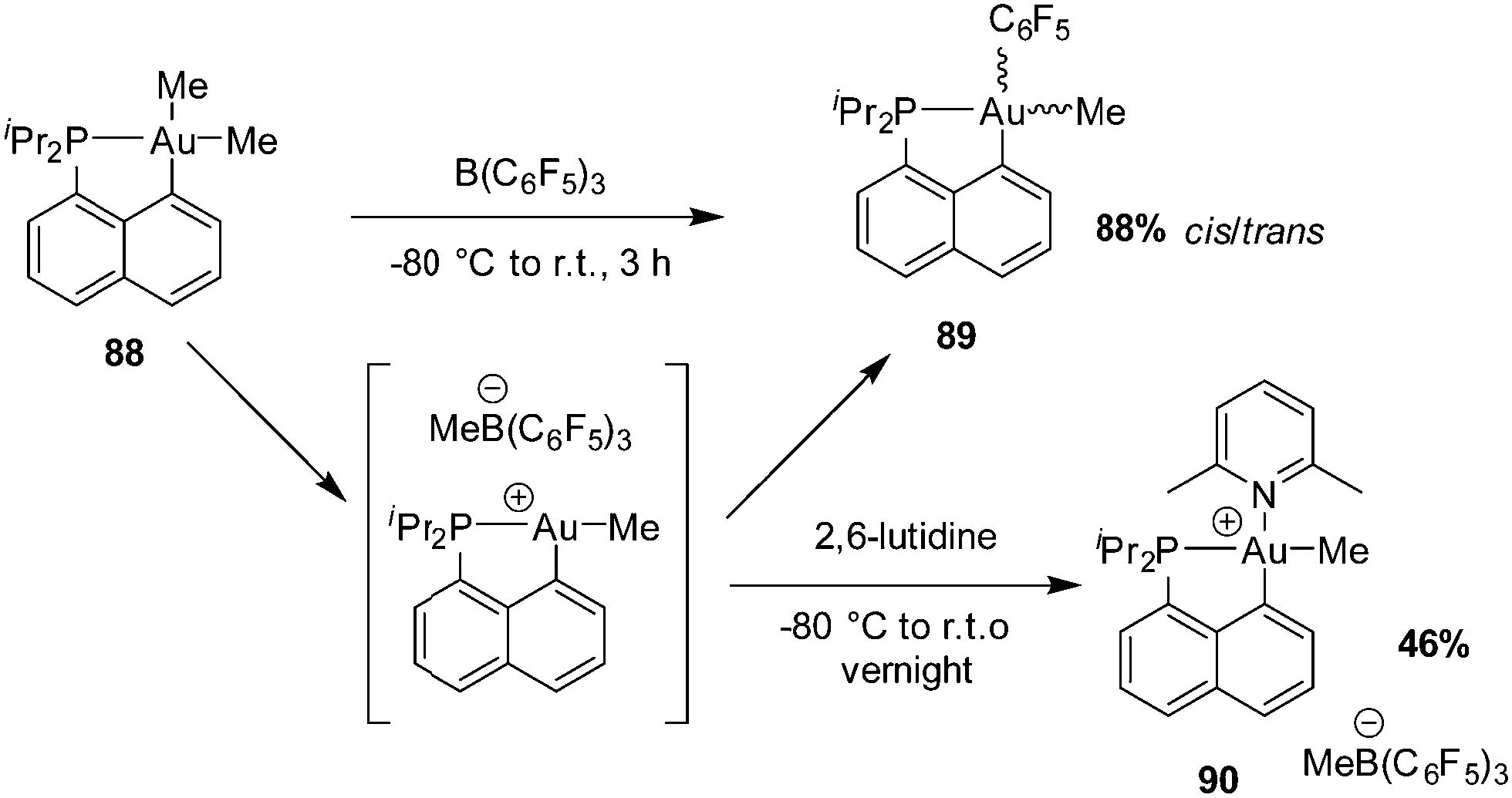 | ||
| Scheme 26 Generation and rearrangement/trapping of the 3-coordinate cationic Au(III) complex [Np(P,C)AuMe]+[MeB(C6F5)3]−. | ||
The reaction of [Np(P,C)AuMe2] with B(C6F5)3 was then carried out in the presence of alkenes with the aim to achieve migratory insertion. Using ethylene or styrene, no sign of alkene incorporation was observed, but with norbornene (nbe), a strained and more reactive alkene, double insertion into the Au(III)–Me bond occurred (Scheme 27). The ensuing gold(III) complex was isolated and fully characterized (HRMS, NMR spectroscopy, and XRD) after trapping with tetrabutylammonium chloride (or pyridine). Syn additions across the CC double bond of nbe attested unequivocally to the inner-sphere migratory addition process (as opposed to the usual outer-sphere addition anti to gold).
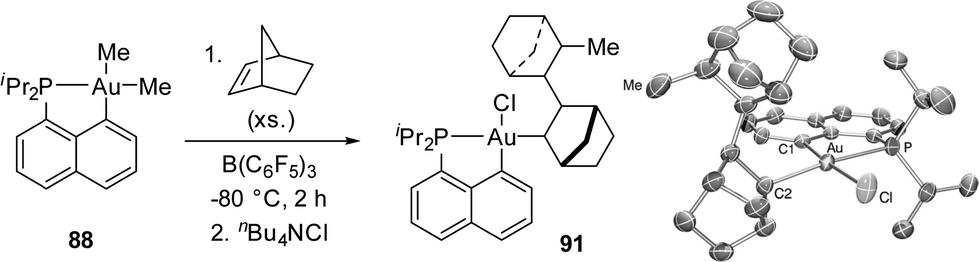 | ||
| Scheme 27 Double insertion of norbornene into an Au(III)–Me bond. | ||
The mechanism of this reaction was thoroughly investigated by computational and experimental means (Scheme 28).34,47 Accordingly, it classically involves coordination–insertion of alkene and because of the electronic dissymmetry of the cyclometalated (P,C) ligand, the first insertion was found slightly more demanding energetically than the second one (activation barriers of 18.7 and 15.7 kcal mol−1, respectively), in line with the observation of double insertion. Low-temperature monitoring by NMR spectroscopy enabled detecting two key intermediates: the first π-alkene complex and the double insertion product before trapping with chloride. The latter species represents the first example of an agostic complex with gold. Coordination of the γ C–H bond of the bis-norbornyl moiety to gold was apparent from the reduced 1JCH coupling (97 Hz) as well as NBO and AIM analyses.48
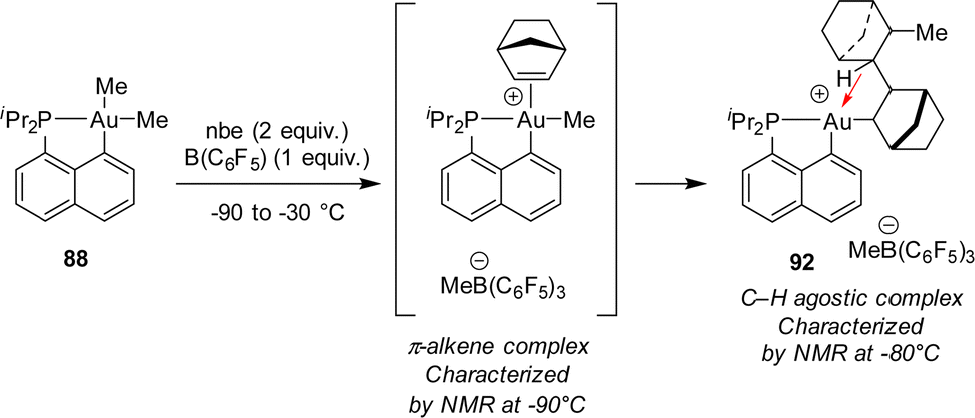 | ||
| Scheme 28 π-Alkene and C–H agostic complexes authenticated upon low-temperature NMR monitoring of the double nbe insertion at Au(III). | ||
We then envisioned to extend the migratory insertion of alkenes to gold(III) aryl complexes, with the idea to access and characterize π-arene gold(III) complexes (Scheme 29).49,50 Such species were proposed as the initial stage of the C–H activation of arenes at gold(III). The [Np(P,C)Au(Ph)I] complex was reacted with AgSbF6 in the presence of norbornene. Here, mono-insertion took place rapidly and cleanly to give a π-arene gold(III) complex. According to NMR spectroscopy, XRD analysis (of the p-methoxy derivative) and DFT calculations, the arene moiety is η2-coordinated to gold. The reaction profile of nbe insertion into the Au(III)–Ph bond was analysed by DFT calculations. Coordination–insertion was found to be facile (with an even lower activation barrier than that for the Au(III)–Me complex, 10.7 vs. 18.7 kcal mol−1). Then, cis/trans isomerization proceeds readily to give the π-arene gold(III) complex obtained. Of note, the auration of π-coordinated arene could be triggered easily by adding a base such as the proton sponge. This demonstrates the relevance of such π-arene complexes to the electrophilic metalation of arenes at gold(III).
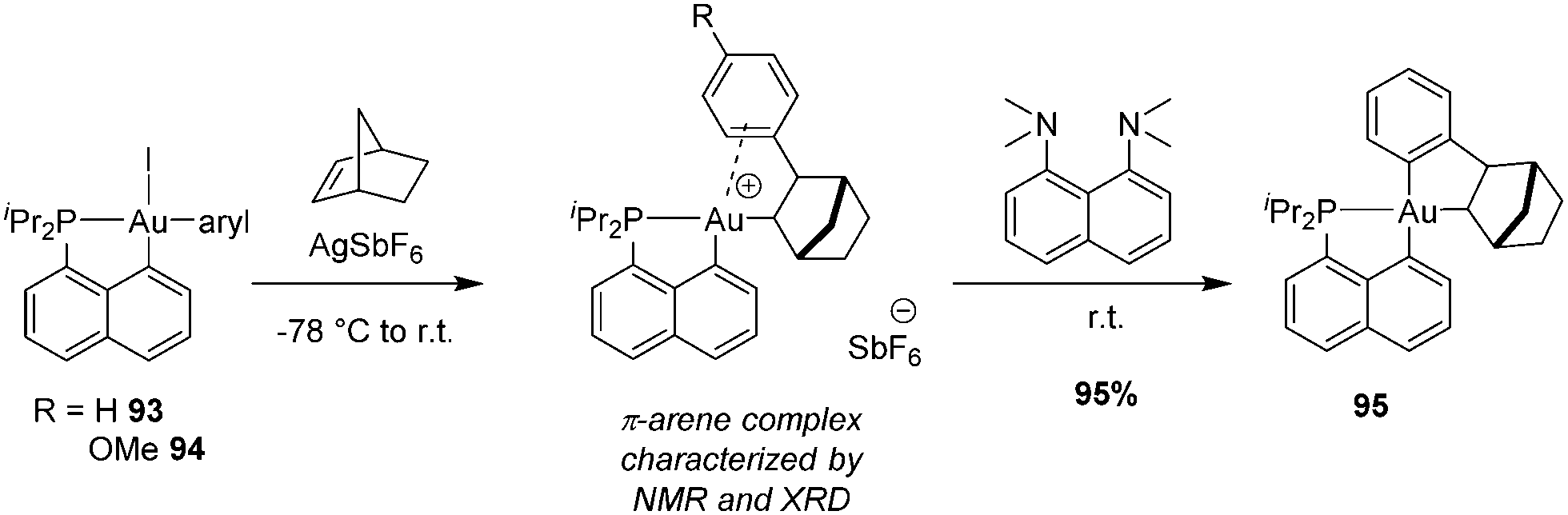 | ||
| Scheme 29 Insertion of norbornene into Au(III)–aryl bonds and the base-assisted cyclometalation of the ensuing π-arene Au(III) complex. | ||
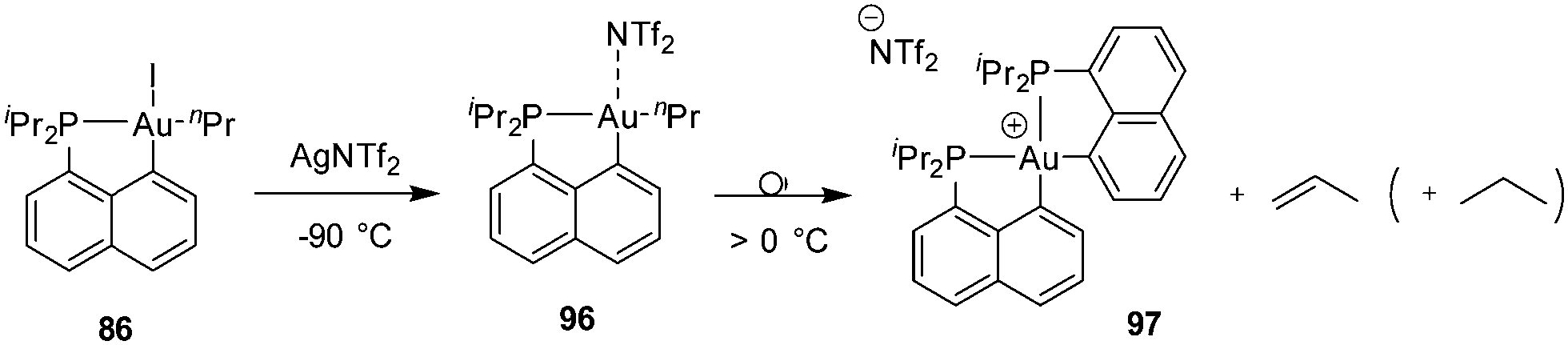 | ||
| Scheme 30 β-Hydride elimination from the cationic [Np(P,C)Au(III)] n-propyl complex. | ||
A similar process was observed from the corresponding n-butyl gold(III) complex, with the release of cis and trans 2-butenes, as well as butane (Scheme 31). The formation of internal alkenes indicates that the initially generated 1-butene rapidly re-inserts into the Au(III)–H bond resulting in a chain walking process. Computational studies have estimated the activation barriers for such β-H eliminations to 8–14 kcal mol−1 (compared to <2 kcal mol−1 for the olefin re-insertion into the Au(III)–H bond). In addition, reacting the corresponding [Np(P,C)Au(III)] methyl complex with ethylene (7 bars) at 50 °C afforded a mixture of propene and butenes. Here, the slow insertion of ethylene into the Au(III)–Me bond is followed by rapid β-H elimination.
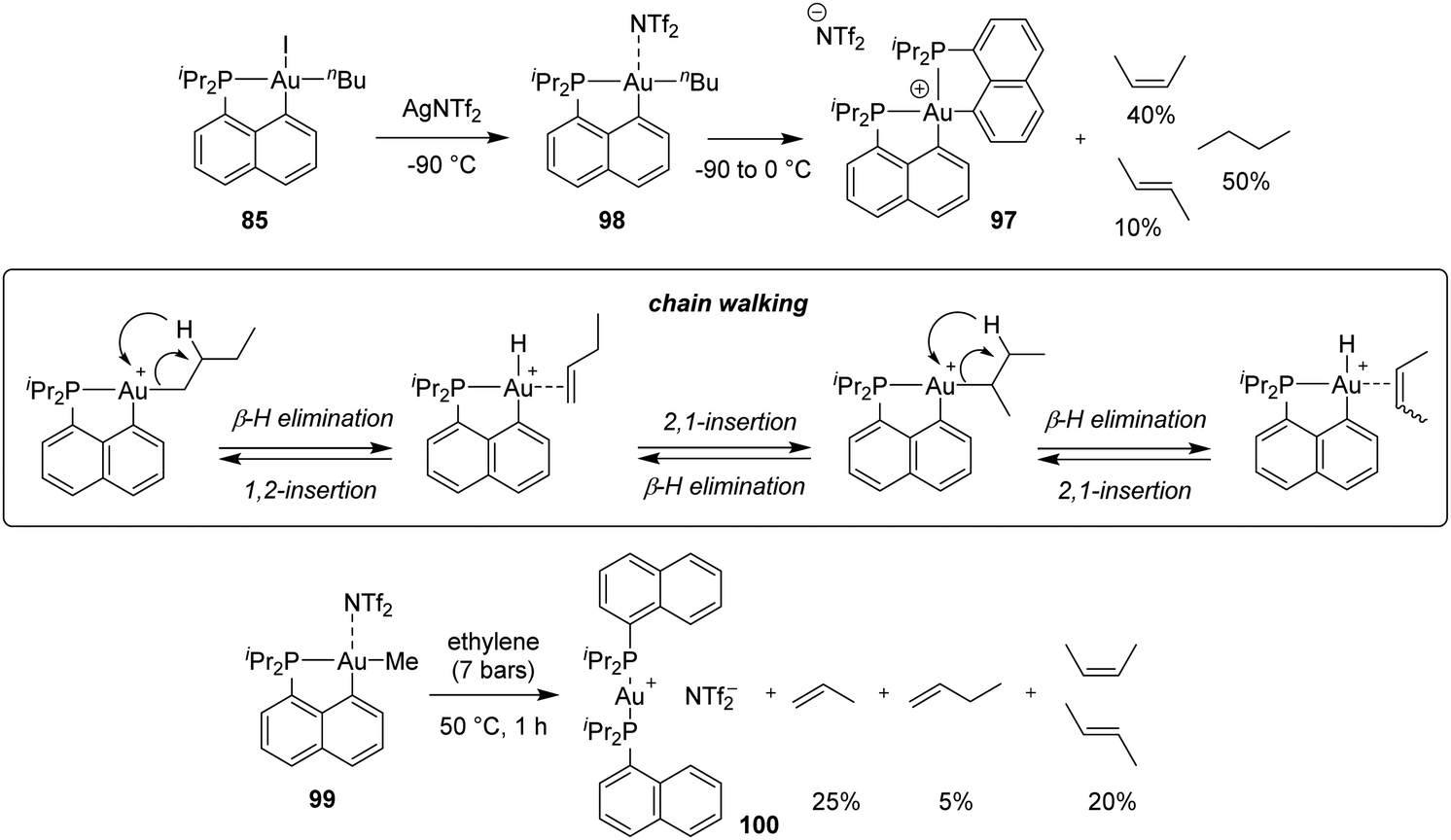 | ||
| Scheme 31 Reactions of the cationic [Np(P,C)Au(III)] n-butyl and methyl complexes involving β-H elimination. | ||
A reaction sequence involving coordination–insertion and β-hydride elimination was also observed by reacting the [Np(P,C)Au(III)Ph] complex with ethylene (Scheme 32).49 In this case, styrene re-insertion is very facile, much more than its dissociation. As a result, the reaction gives a gold(III) styryl complex stabilized by π-arene coordination.
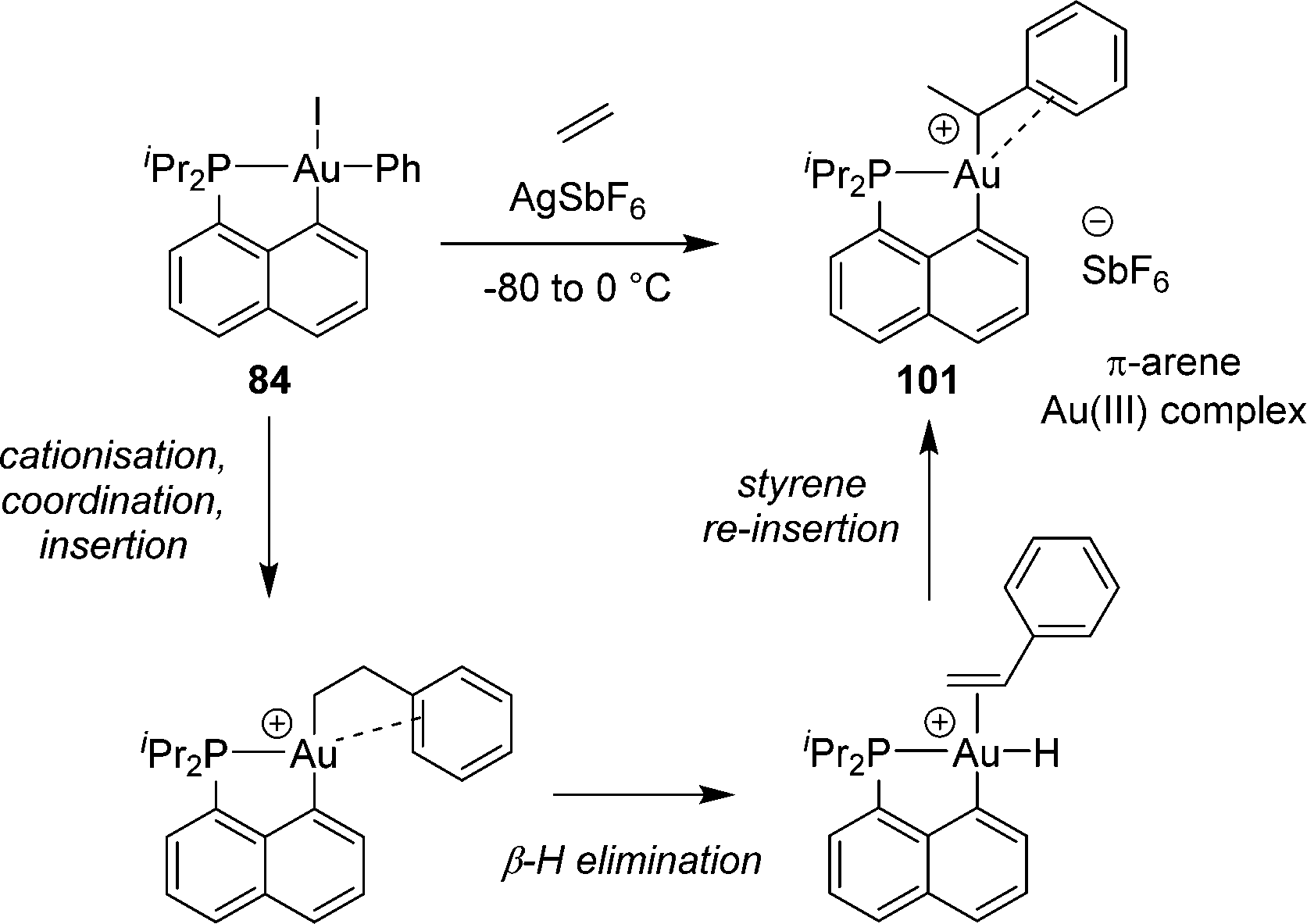 | ||
| Scheme 32 Formation of a [Np(P,C)Au(III)] π-arene complex by a coordination–insertion/β-hydride elimination/isomerisation sequence. | ||
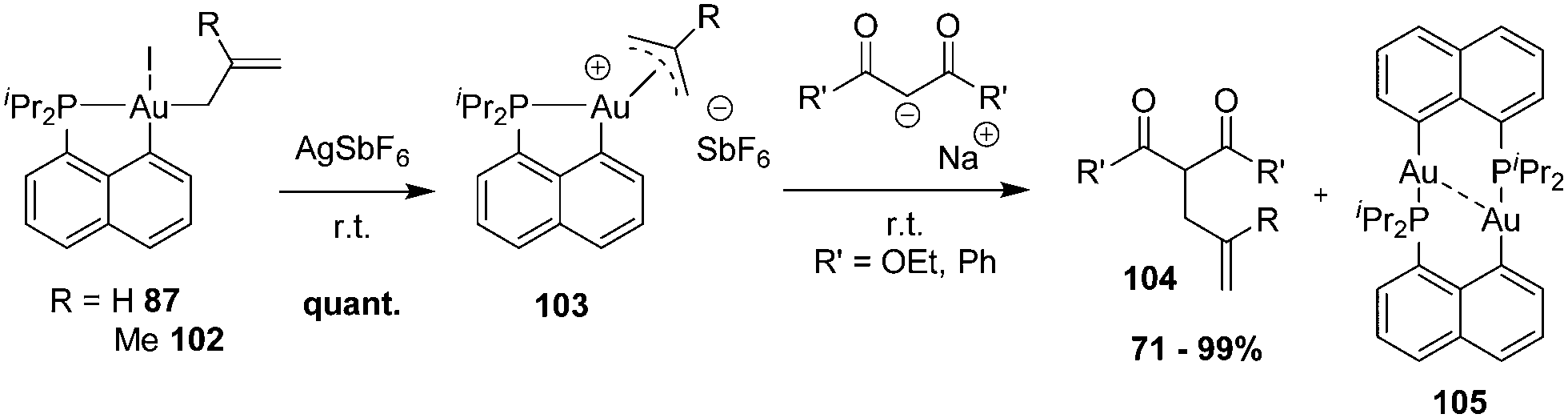 | ||
| Scheme 33 Synthesis of cationic [Np(P,C)Au(III)] π-allyl complexes and their C–C coupling with β-dicarbonyl carbanions. | ||
A meticulous mechanistic study combining in situ NMR monitoring at low temperatures and DFT calculations revealed a complex and rich picture for this apparently simple transformation (Scheme 34).35 If the C-allylation product is readily obtained whatever the π-allyl substitution (R = H, Me) and β-dicarbonyl nucleophile (R′ = OEt, Ph), nucleophilic attack can occur initially at either the terminal or the central positions of π-allyl, as well as at the metal itself. Reactive intermediates resulting from these 3 competitive paths were actually all authenticated spectroscopically and/or crystallographically. For palladium(II) π-allyl complexes, the regioselectivity of the nucleophilic addition is under orbital rather than charge control. The close inspection of the molecular orbital diagram for [Np(P,C)Au(III)] π-allyl complexes showed the presence of three vacant orbitals with major contributions of gold and the terminal/central positions of π-allyl within a small energy gap. This explains why nucleophilic attacks to the 3 sites are intrinsically in competition.
 | ||
| Scheme 34 Three reactive sites for the addition of β-dicarbonyl nucleophiles to [Np(P,C)Au(III)] π-allyl complexes and the ensuing products authenticated experimentally. | ||
Of note, π-allyl gold(III) complexes were amenable to catalytic allylation. Using a P^N hemilabile ligand, we could cross-couple allyl acetates and alcohols with indoles with complete selectivity for the branched C3-allylated products.52
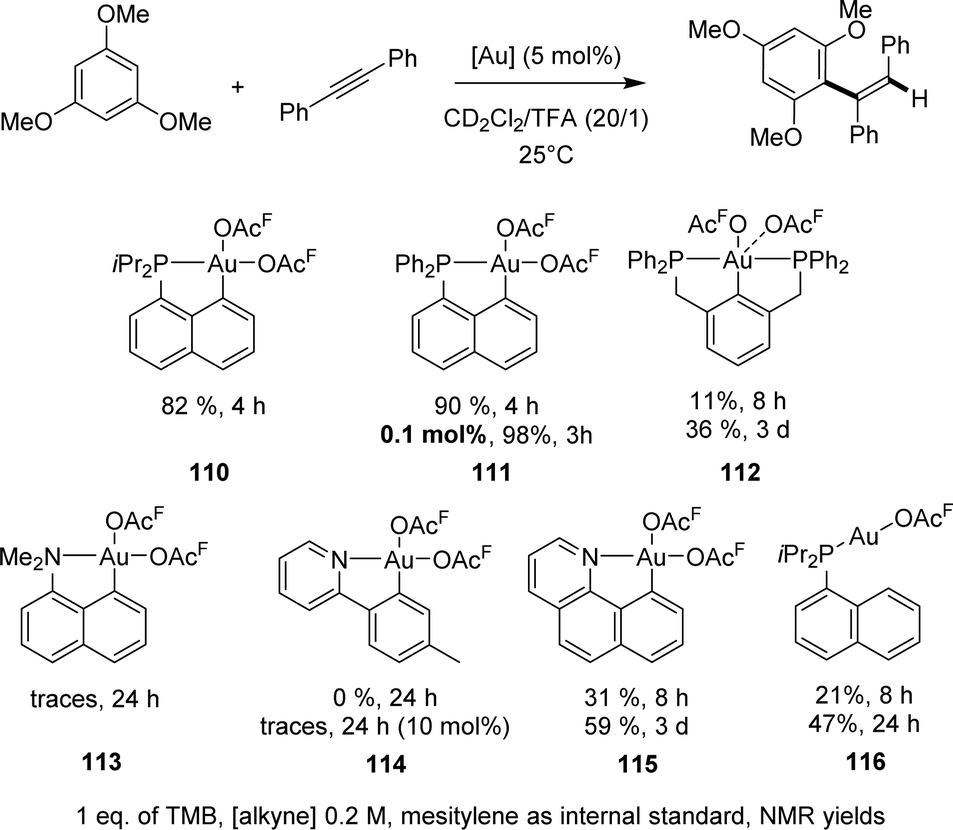 | ||
| Scheme 35 Comparison of the catalytic activities of a range of gold complexes in the hydroarylation of diphenylacetylene with 1,3,6-trimethoxybenzene. | ||
[Np(P,C)Au(III)] complexes showed high activity with a rather broad scope of substrates (Chart 6). The reaction works efficiently with internal alkynes featuring aryl, alkyl, ester and keto substituents, as well as terminal alkynes. Other electron-rich arenes than TMB can also be used, including dialkoxy-benzenes, durene and mesitylene. In all cases, hydroarylation is fully regioselective, and it is generally fully stereoselective, affording the anti-addition product.
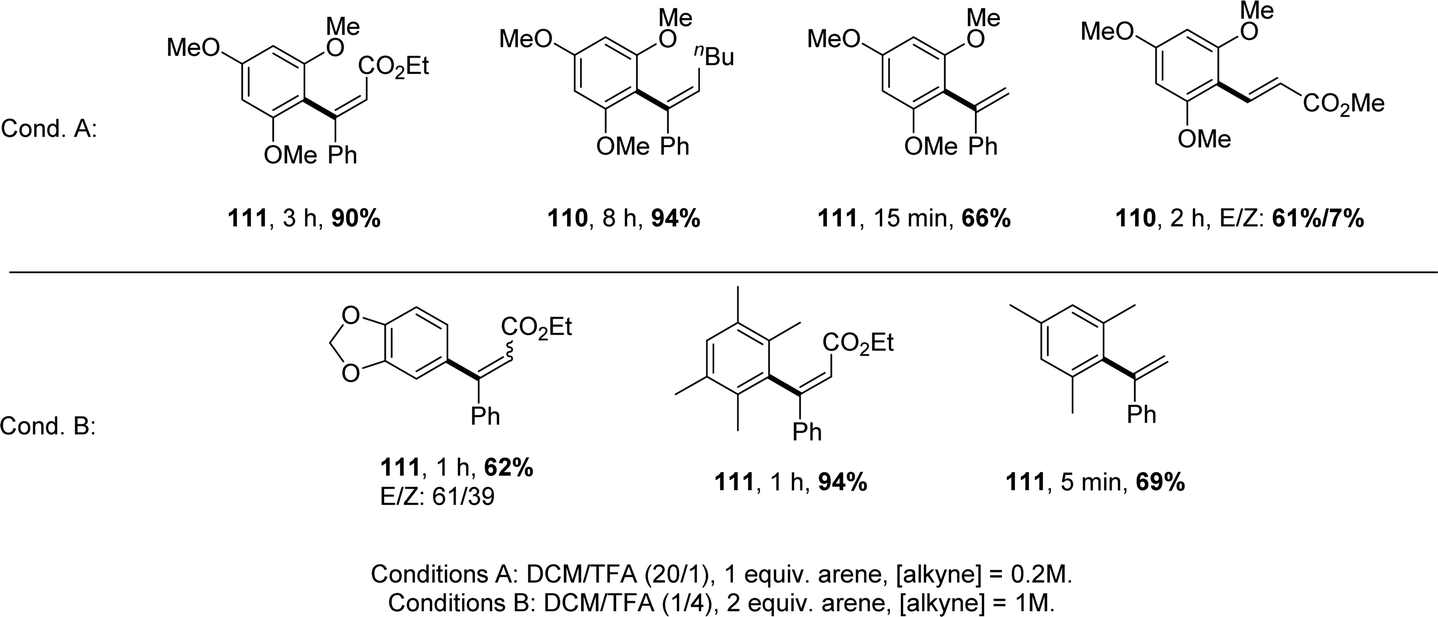 | ||
| Chart 6 Selected examples of intermolecular hydroarylation of alkynes catalysed by [Np(P,C)Au(III)] complexes. | ||
A priori, two paths are conceivable to account for such Au(III)-catalyzed arene/alkyne coupling (Scheme 36). One involves the π-coordination of the alkyne to gold yielding 119, the outer-sphere nucleophilic addition of the arene and finally protodeauration. The other one starts by the C–H activation of the arene affording 117, followed by the migratory insertion of the alkyne and finally protodeauration. The outer-sphere path was initially inferred indirectly, based on the observed formation of the anti-addition products. A thorough mechanistic study was then performed,56 to track reactive intermediates and better understand the factors at play. Accordingly, using B(C6F5)3 as the co-catalyst instead of TFA as the co-solvent enabled us to characterize by low temperature NMR spectroscopy the cationic complex [Np(P,C)Au(OAcF)]+, the active species easily shifting from κ2 to κ1-OAcF coordination, and its TMB adduct [Np(P,C)Au(OAcF)(TMB)]+, an off-cycle resting state and actually the first Wheland-type σ-arene complex of Au(III) to be authenticated. The key π-alkyne complex was also generated and characterized in the gas phase by mass spectrometry using ion-molecule collision techniques. The energy profiles for the coupling of TMB/DPA were also analysed computationally and the outer-sphere path was found to be much less demanding energetically than the inner-sphere mechanism.
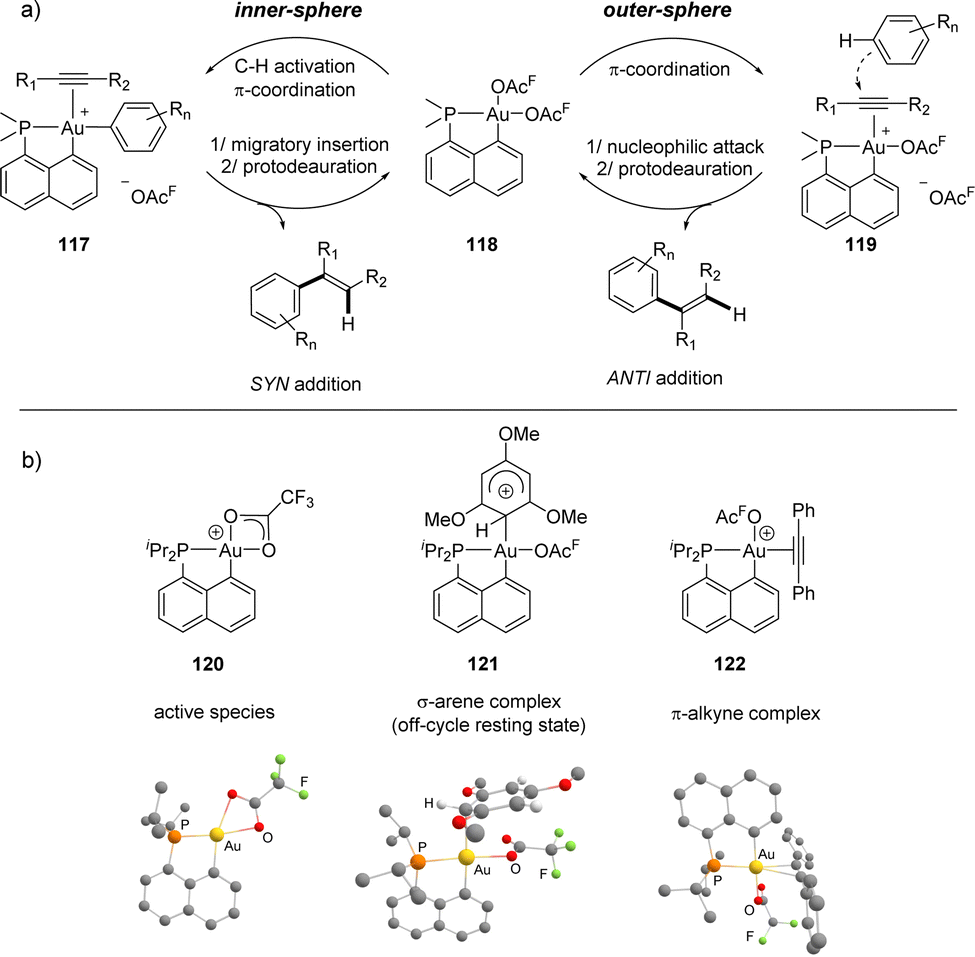 | ||
| Scheme 36 (a) Schematic representation of the inner- and outer-sphere mechanisms conceivable for the hydroarylation of alkynes catalyzed by [Np(P,C)Au(III)] complexes. (b) Related intermediates authenticated experimentally and optimized geometry computed at the SMD(CH2Cl2)-B97D/SDD+f(Au)/6-31G** (other atoms) level of theory. | ||
S)Ph2 side-arm is coordinated to gold, and the other remains pendant. Variable-temperature 31P NMR indicated a dynamic situation, with the rapid exchange of the two P(S)Ph2 moieties at room temperature. The 13C NMR signal for the carbene center was found at δ 18.2 ppm, very much shielded compared to those of electrophilic cationic Au(I) carbene complexes (δ from 200 to 321 ppm), indicating a completely different bonding situation and an electron-rich carbene center. Consistently, DFT calculations revealed a high energy HOMO centered at the carbene center. The nucleophilic, Schrock-type character of the carbene center was actually substantiated experimentally by Wittig-type reactions with phenylisothiocyanate and CS2.
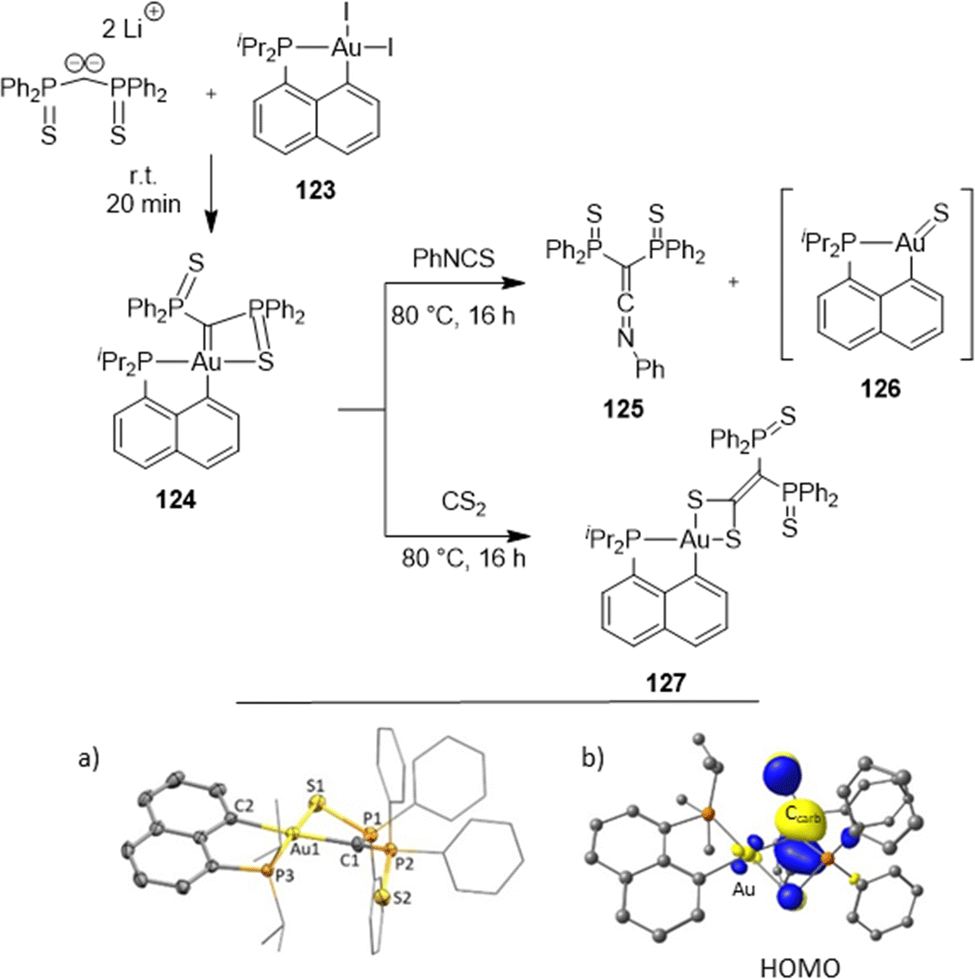 | ||
| Scheme 37 Synthesis and reactivity of a nucleophilic [Np(P,C)Au(III)] carbene complex. X-ray structure (a) and its HOMO (b). | ||
2. [Np(P,C)Pd(II)] complexes
In 1998, Shaw demonstrated the high catalytic activity and robustness of [Np(P,C)Pd] acetate and acetylacetonate complexes 128 and 129 in the Heck reaction (Scheme 38).22 The coupling of iodobenzene and styrene could be achieved at extremely low catalytic loadings (down to 0.0001 mol%) under forcing conditions (up to 13 days at 95 °C) to reach TON as high as 1120000. Of note, related cyclometalated complexes derived from benzyl phosphines led to significant lower TON (900 at best). A few years later, Herrmann extended this study to bromo- and chloro-arenes, working at 130 °C and 0.1 mol% catalyst loading.23 Under these conditions, the Heck coupling products could be obtained in high yields from bromo-arenes (with TON up to 1,000) but not from chloro-arenes (29% at best).
 | ||
| Scheme 38 Catalytic applications of [Np(P,C)Pd] complexes 128 and 129. | ||
With these more challenging substrates, the [Bn(P,C)Pd] complexes perform best, with TON values up to 329000 achieved from bromo-arenes.
Conversely, Hou obtained good results with the rigid (P,C)-naphthyl ligand in the intermolecular [4+2] cycloaddition of bicyclic alkenes with terminal ynones affording polysubstituted furans.58 An exhaustive catalyst screening was conducted, from simple commercially available Pd compounds to (P,C)-cyclometalated complexes derived from binaphthyl, biphenyl, benzyl and naphthyl phosphines. Good yields (>60%) were only obtained when using [Np(P,C)Pd] complexes.
Recently, Yamamoto applied 9-(diphenylphosphino)anthracene-based phosphapalladacycles as catalysts for the conjugate addition of arylboronic acids to electron-deficient alkenes (Scheme 39).59In situ tests using Pd(OAc)2 and phosphines showed the higher performance of the anthracenyl phosphine (95% yield in coupling product from phenylboronic acid and PhCHCHC(O)Ph) over the corresponding naphthyl phosphine (Ph2PNp, 53%) and triphenylphosphine (<5%). Using the preformed [Ant(P,C)Pd] complex with P(OEt)3 as the co-ligand, the catalytic loading could be decreased to 0.2–0.5 mol% keeping high reactivity over a large substrate scope.
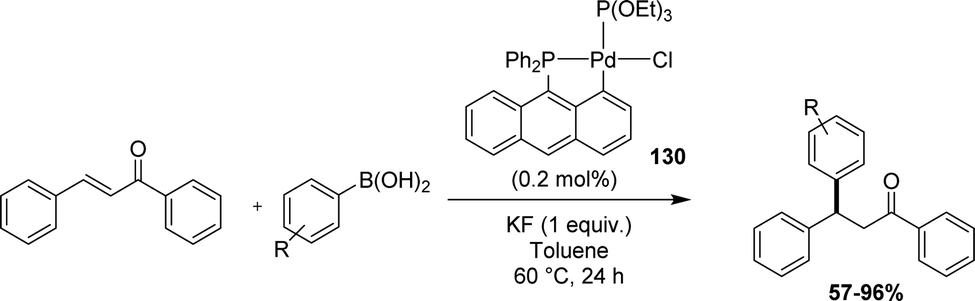 | ||
| Scheme 39 Catalytic application of an [An(P,C)Pd] complex in the conjugate addition of arylboronic acids to electron-deficient alkenes. | ||
3. [Np(P,C)Pt(II/IV)] complexes
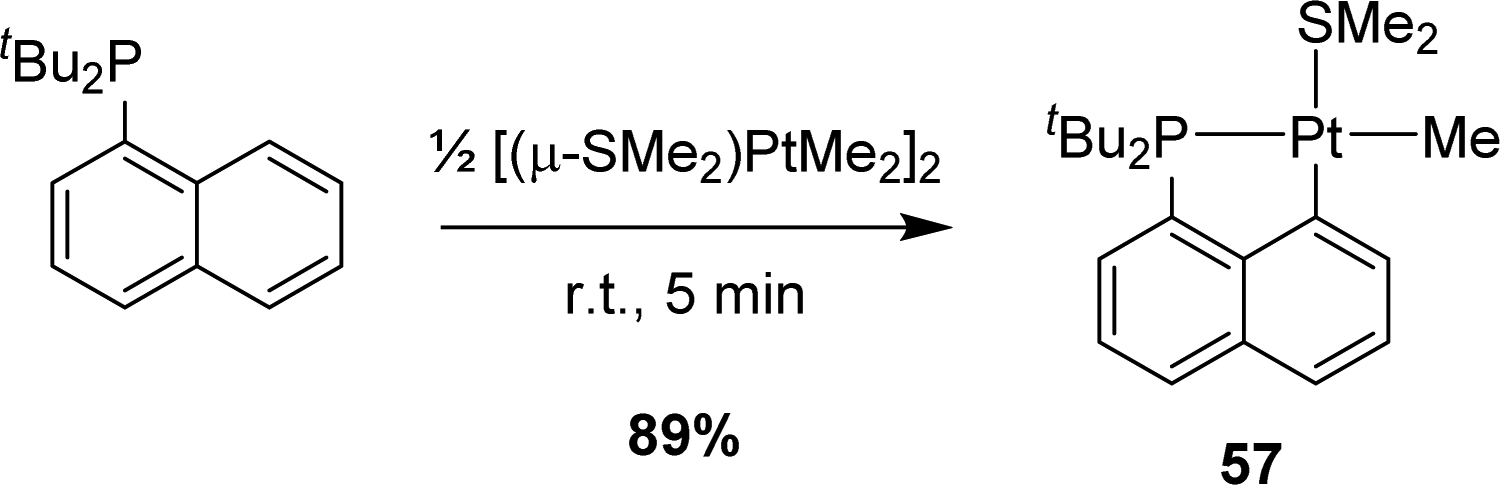
In 2011, Goldberg reported the C–H activation of benzene upon thermolysis (100 °C) of a cyclometalated [Np(P,C)Pt(II)] methyl complex in C6D6 (Scheme 40).25 Methane was identified as the co-product (with CH3D as the major isotopomer) and the addition of an excess of SMe2 was found to inhibit the C–H activation. The authors thus proposed a mechanism involving the displacement of SMe2 by benzene-D6 at platinum, followed by the oxidative addition of a C–D bond/reductive elimination of CH3D or a σ-bond metathesis process.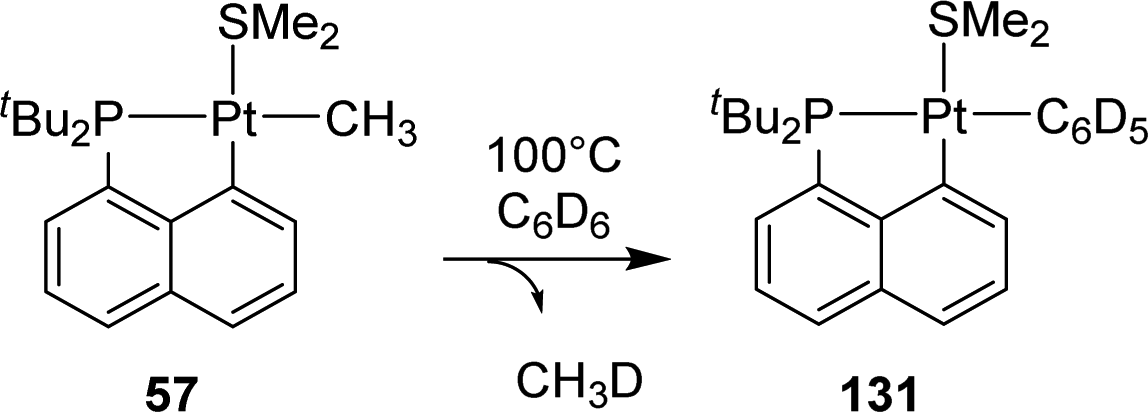 | ||
| Scheme 40 C–H bond activation of benzene by a [Np(P,C)Pt(II)] methyl complex. | ||
In the context of the electrophilic fluorination of arenes with high valent transition metals, Vigalok and Verdernikov investigated the oxidation of [Np(P,C)Pt(II)] aryl complexes with XeF2 (Scheme 41).24 The ensuing platinum(IV) complexes showed moderate stability and evolved at 20–60 °C via an 1,3-aryl migration from the platinum center to the β-carbon of the naphthyl fragment to yield (P,C,C)Pt(II) pincer complexes. Based on DFT calculations, the authors proposed a stepwise mechanism for this unprecedented Csp2–Csp2 coupling. Following pyridine dissociation and C–C coupling, the aryl group would migrate from the α-position to the β-position of the naphthyl ring to generate a naphthylidene complex. Finally, deprotonation of the naphthyl ring would restore the aromatic naphthalene system.
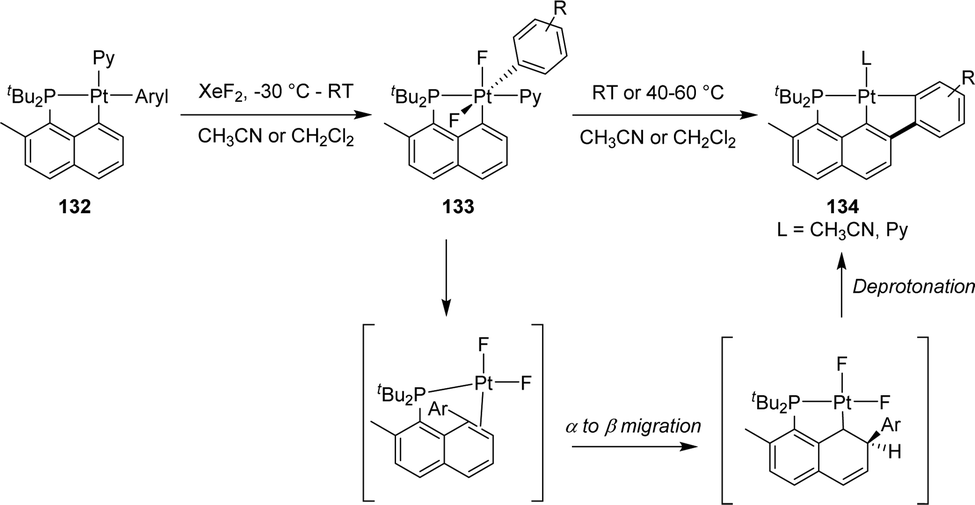 | ||
| Scheme 41 Electrophilic fluorination of [Np(P,C)Pt(II)] aryl complexes leading to (P,C,C)Pt(II) pincer complexes. | ||
4. [Np(P,C)Fe(II)] complexes
The [Np(P,C)Fe(II)] methyl complex was found by Beck and Sun to be unstable thermally (Scheme 42).60 The iron center is in octahedral geometry with the Me group at the cis position to the carbon atom of the (P,C)-cyclometalated ligand. Upon gentle heating (40 °C, 4 h), reductive elimination took place to give the corresponding C-methylated naphthyl diphenylphosphine. Nonetheless, some stoichiometric reactions with the retention of the cyclometalated Np(P,C) framework were also reported. With carbon monoxide, the ligand exchange of one trimethylphosphine co-ligand proceeded smoothly at room temperature, while C–H activation occurred with terminal alkynes to give the corresponding acetylide complexes with release of methane.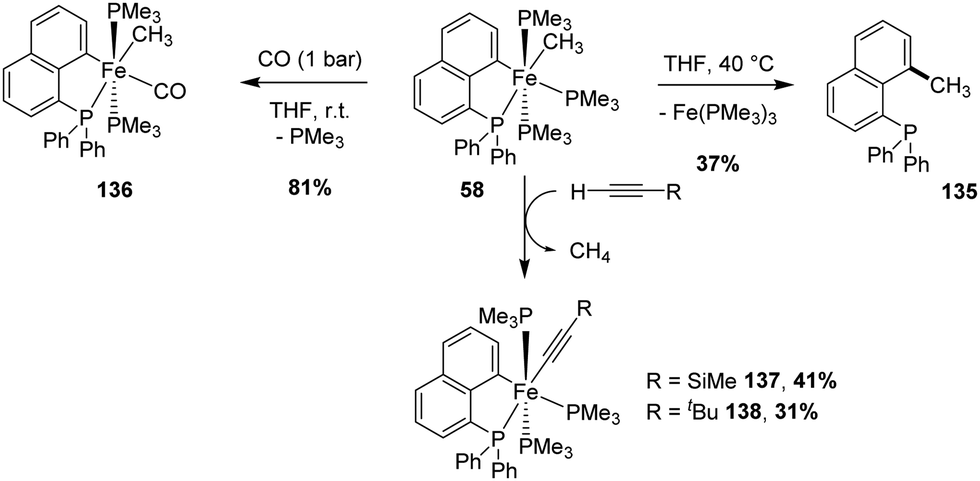 | ||
| Scheme 42 Stoichiometric reactions of a [Np(P,C)Fe(II)] methyl complex. | ||
5. [Np(P,C)Ru(II)] complexes
[Np(P,C)Ru(II)] complexes were applied to the catalytic reduction of ketones by transfer hydrogenation with isopropanol. In 2007, Zhu compared the catalytic activities of various p-cymene phosphine complexes under similar conditions: 0.5 mol% catalyst loading, 82 °C (boiling iPrOH), 48 h, KOH (20 eq.) (Scheme 43).19 Whatever the substituent at phosphorus (Ph or iPr), the [Np(P,C)Ru(II)] complex showed lower activity (70–90% yields for the reduction of benzophenone) compared to the non-cyclometalated complex (>98% yield) and the benzyl cyclometalated complex (96% yield). However, a significant increase in yields was observed upon introducing a methyl group at the o-position of phosphorus of the [Np(iPr2P,C)Ru] complex (from 70 to 98%).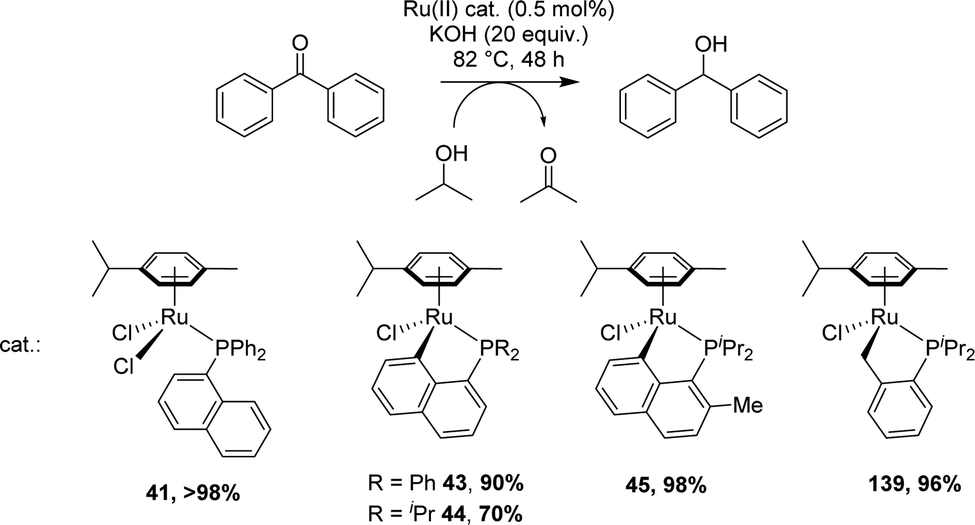 | ||
| Scheme 43 Reduction of ketones catalyzed by p-cymene phosphine Ru(II) complexes. | ||
Two years later, Grabulosa envisioned to develop an asymmetric version by chirality transfer from a P-stereogenic center to the ruthenium center (Scheme 44).21 Starting from homochiral naphthyl phosphines, the cyclometalation step proceeded with disappointingly low diastereoselectivity (d.r. = 1:2.4 for R = Me and d.r. = 1:1.6 for R = MeO). Only the iPr-substituted phosphine induced full control of the ruthenium chiral center, but unfortunately, the cyclometalated complex was formed in a very low yield (15%). Evaluation of these [Np(P,C)Ru(II)] complexes in asymmetric transfer hydrogenation of acetophenone reveals very low, if any, enantioselectivity (ee < 5%). Nevertheless, it is interesting to note that cyclometalation of the naphthyl phosphinite ligand (R = OMe) resulted in a noticeable increase of the catalytic activity (from 45% yield in reduction product after 3 h with the non-cyclometalated complex, to 99% yield with the cyclometalated complex).
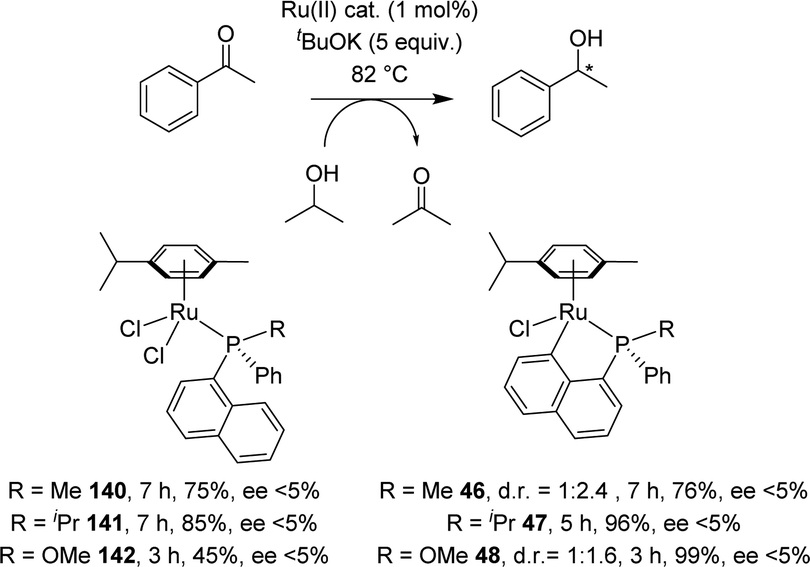 | ||
| Scheme 44 Attempts for Ru-catalyzed asymmetric hydrogenation of acetophenone with P-chiral naphthyl phosphine complexes. | ||
6. Miscellaneous examples involving M–C cleavage
[Np(P,C)M] metalacycles are very robust and in all the previously described reactions and catalyses, they remain intact throughout the transformations. However, M–C bond cleavage was found to occur in a few cases, presented hereafter.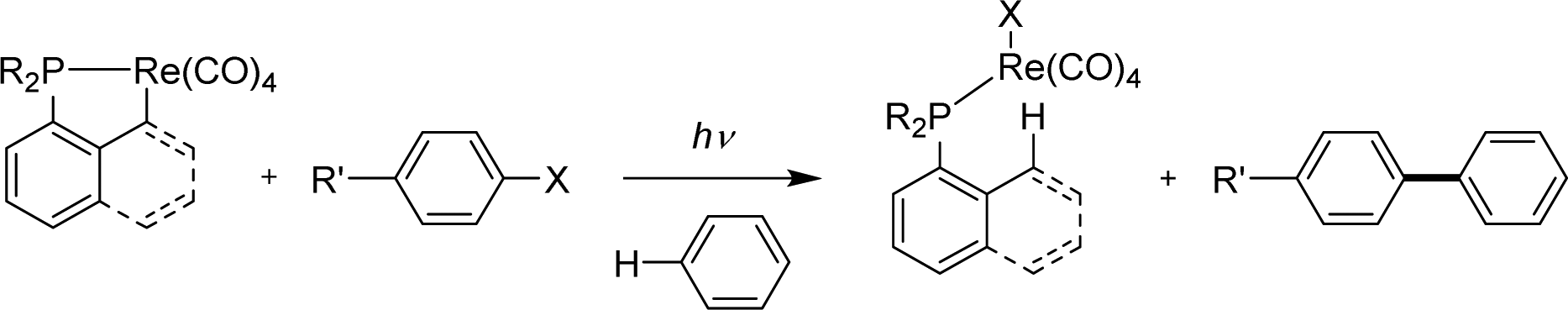 | ||
| Scheme 45 Aryl halides/benzene coupling upon the photochemical reaction of cyclometalated rhenium complexes. | ||
Taking advantage of the facile cyclometalation of naphthyl phosphines at rhenium in the presence of a base, the cross-coupling of 4-bromoanisole and benzene (used as the solvent) was then possible under catalytic conditions, in the presence of 1 equivalent of sodium hydroxide (Scheme 46). Of note, the ancillary Np(P,C) ligand was found to outperform its more flexible Csp3 variant (59 vs. 26% yield) and replacing the Ph groups at phosphorus for iPr led to an increase in stability of the rhenium catalyst (no free ligand observed with iPr) and thus a better yield in the coupling product (68%).
 | ||
| Scheme 46 Re-catalyzed arylation of benzene with aryl halides. | ||
 | ||
| Scheme 47 Ru-catalyzed C5-selective alkylation of naphthyl phosphines involving [Np(P,C)Ru(II)] complexes. | ||
 | ||
| Scheme 48 Rh-catalyzed C–H arylation of 1-naphthyl diphenylphosphines involving [Np(P,C)Rh] complexes. | ||
 | ||
| Scheme 49 Rh-catalyzed C–H arylation of 1-pyrenyl diphenylphosphines involving [Np(P,C)Rh] complexes. | ||
 | ||
| Scheme 50 Reaction of a [Np(P,C)Ru(II)] complex with internal alkynes leading to phosphanelium salts. | ||
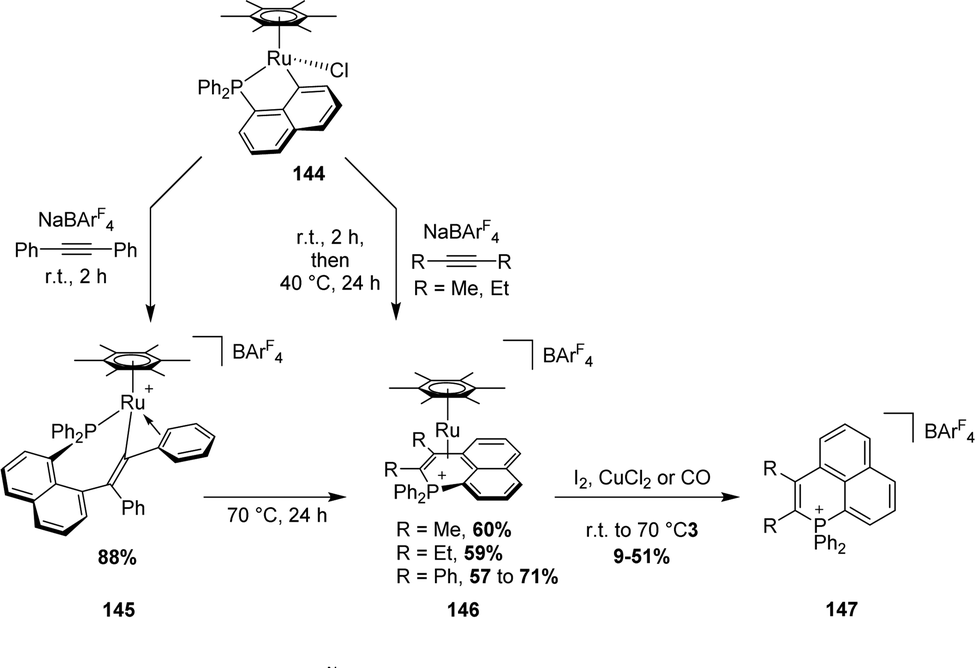
Recently, Ishii synthesized the analogous alkenyl rhodium(III) complex coordinated with pentamethylcyclopentadienyl (Cp*) by the 1,2-insertion of diphenylacetylene in the presence of NaBArF4 (Scheme 51).63 An addition of 1.0 equivalent of di-p-tolylacetylene to this complex results in an equilibrium between the two corresponding alkenylrhodium complexes with a 43:57 ratio. The authors proposed that an alkyne exchange occurs via β-carbon elimination, for which the activation barrier was estimated computationally to be accessible (15.6 kcal mol−1). This represents one of the rare examples of β-carbon elimination from an alkenyl complex (i.e. the reverse process of alkyne insertion).
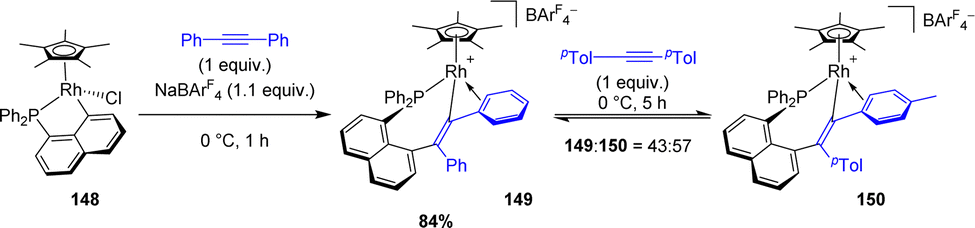 | ||
| Scheme 51 Synthesis of complex 149 by alkyne insertion in the Cperi–Ru bond and the exchange reaction of the alkenyl moiety using di-p-tolylacetylene. | ||
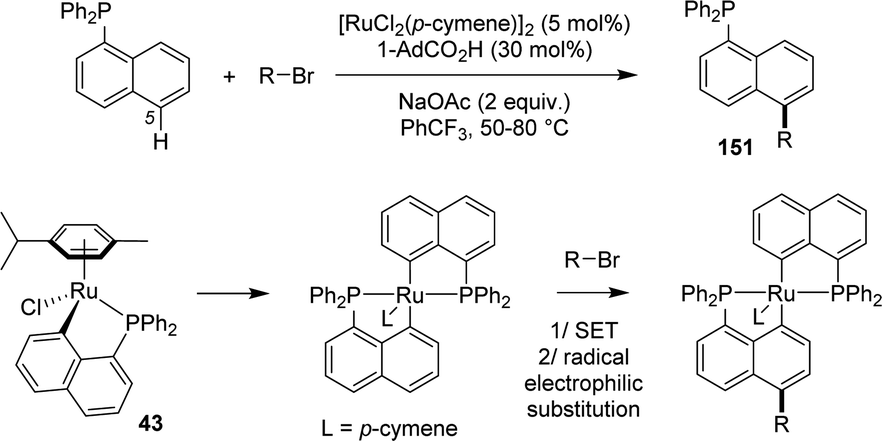
Recently, Zeng reported an original Ru-catalyzed alkylation of naphthyl phosphines occurring selectively on the remote C5 position (Scheme 47).64 The transformation proceeds under relative mild conditions (50–80 °C) with bromo-alkanes as alkylating reagents. It takes advantage of the facile and reversible formation of [Np(P,C)Ru] complexes by phosphine-directed cyclometalation/protodemetalation. Bromo alkanes are proposed to oxidize the ruthenium center via a single-electron transfer (SET), the alkylation would then occur by radical electrophilic substitution on the naphthyl ring. The high C5-regioselectivity (C5/others >20/1) is tentatively assigned to the inductive effect of the Np(P,C)Ru metallacycle.
In 2018, Yu and Che described jointly the Rh-catalyzed peri arylation of naphthyl phosphines (Scheme 48).65 Using 2.5 mol% of [Rh(cod)Cl]2 and 3 equivalents of tBuOLi as a base, naphthyl phosphines could be efficiently coupled with bromo-arenes (within 24 h at 110 °C) as well as chloro-arenes (within 24 h at 150 °C) in good to high yields. The reaction is fully selective for peri arylation and works with bromo/chloro hetero-arenes as well. Mechanistic studies by mass spectrometry suggest a Rh(I)/Rh(III) catalytic cycle. The bis-phosphine complex [Np(P,C)Rh(PPh2Np)] was proposed to be the active species.
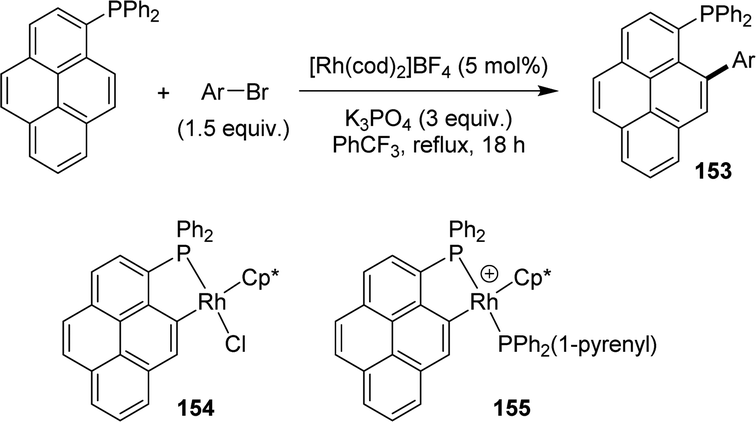
Very recently, Hierso and Roger extended this methodology to polycyclic aromatic hydrocarbons, in particular 1-pyrenyl diphenylphosphine (Scheme 49).66 The reaction works well with a large scope of bromo-arenes substituted by electron-donating and electron-withdrawing groups, as well as bromo-heteroarenes. It is fully selective for the arylation of the position peri to phosphorus, and is also applied to phenanthryl and fluoranthenyl phosphines. The [Rh(I)(cod)2]BF4 and [Rh(Cp*)Cl2]2 complexes can be used and gave similar results. (P,C)-cyclometalated Rh(III) complexes were prepared and their activities were evaluated. The neutral species gave poor results and was assumed to be a deactivated out-of-cycle resting state, while the di-phosphine cationic complex showed high catalytic activity.
IV. Luminescence properties and optoelectronic applications of [Np(P,C)M] complexes
1. [Np(P,C)Pt(II)] complexes
In 2009, Yip reported an interesting study on (P,C)-cyclometalated platinum complexes derived from pyrenes.10 The fluorescence properties of pyrene derivatives are well-known and widely used, while phosphorescence is usually very weak. Phosphorus substitution and subsequent cyclometalation of the peri C–H bond were envisioned as a mean to introduce Pt on the pyrene ring and benefit from the internal heavy-atom effect to enhance phosphorescence. Accordingly, the mono and dimetallic platinum complexes 18 and 20 showed phosphorescence at λem 611–627 nm in oxygen-free acetonitrile solution at room temperature, with quantum yields of 0.44–1.5% and lifetimes of 31.3–63.7 μs (Fig. 2). The heavy-atom effect of Pt was supported by the very weak phosphorescence of the related Pd dinuclear complex under the same conditions (λem 650 nm, quantum yields < 0.001%). Cyclometalation is beneficial, as apparent from comparison with the corresponding metalated platinum(II) complex lacking (P,C)-cyclometalation.67 The related complexes with dangling (non-cyclometalated) Pt centers showed very weak phosphorescence if any.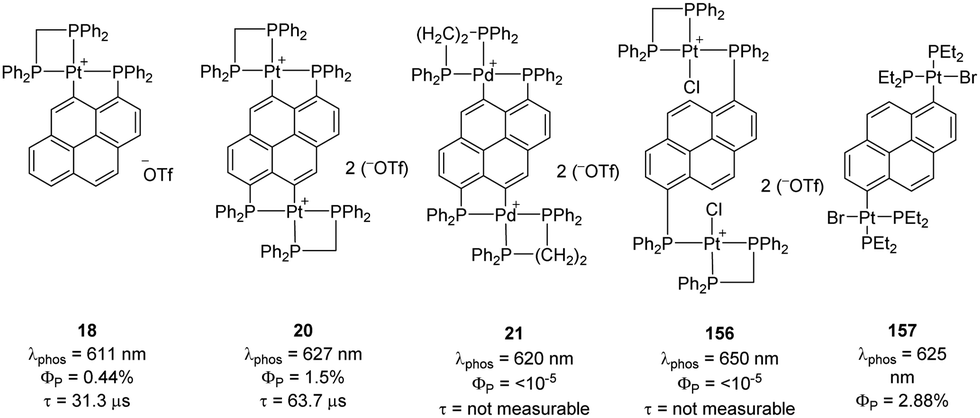 | ||
| Fig. 2 Phosphorescence properties of (P,C)-cyclometalated and non-cyclometalated pyrene derivatives. | ||
Note that the same authors performed a similar photophysical study on analogous anthracene-derived complexes two years before.9 The bis-cyclometalated complexes were found to emit at λem ∼ 460–510 nm in the dichloromethane solution at room temperature, with quantum yields of 1–6% and short half-lifetimes <2 ns (Fig. 3). Only weak fluorescence was observed in this case, with no apparent enhancement of phosphorescence due to the heavy-atom effect. Comparison of the syn and anti-isomers revealed little changes, while the corresponding mono-cyclometalated complex showed weaker fluorescence (0.16% quantum yield) with a too-short lifetime to be measured.
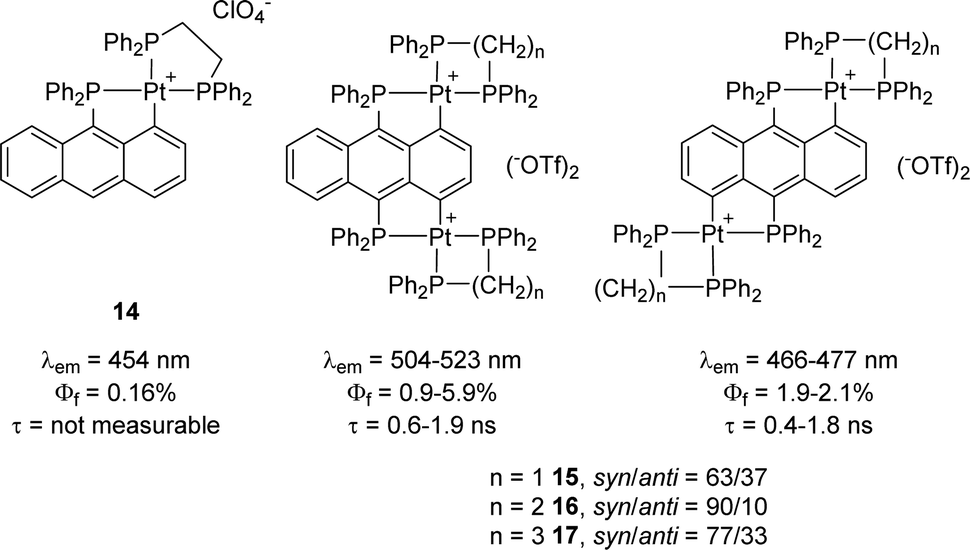 | ||
| Fig. 3 Fluorescence properties of (P,C)-cyclometalated Pt complexes derived from anthracene (similar data were observed with dppm and dppp instead of dppe as ancillary diphosphines). | ||
2. [Np(P,C)Ir(III)] complexes
Cyclometalated iridium(III) complexes such as [(phenyl-pyridyl)3Ir] are strong phosphorescent emitters of interest for optical devices such as organic light-emitting diodes (OLEDs). Varying the ligands at iridium offers a simple means to modulate and eventually improve the stability and photophysical properties of the complexes. In this regard, Chi and Chou jointly studied in 2010 heteroleptic complexes combining two C^N and one P^C cyclometalated ligands (Fig. 4).15 Attempts to prepare analogous complexes from benzyldiphenylphosphine failed, preventing the authors to assess the influence of the naphthyl ring on the luminescence properties. Whatsoever, complexes 26 and 27 proved robust (as long as the N^C ligand is equipped with a fluorine atom) and phosphorescent in oxygen-free dichloromethane solution at room temperature with λem of 530–650 nm and quantum yields from 1.6 to 100%. Significantly higher quantum yields were systematically observed for the complexes in which the two nitrogen atoms occupy cis positions. The impact of the N^C cyclometalated ligand at iridium was thoroughly examined. Thanks to time-dependent DFT calculations, the emission transitions were shown to involve MLCT (metal-to-ligand charge transfer) and LLCT (ligand-to-ligand charge transfer) or MLCT and ILCT (intra-ligand charge transfer), depending on the N^C ligand. In particular, the (P,C)-naphthyl ligand was proposed to act as the CT acceptor when iridium is ligated by the phenyl-pyridine ligand. Of note, the proof of the feasibility of incorporating such complexes in devices was substantiated. Orange-red emitting OLEDs with decent quantum efficiencies (up to 12.5% at 500 cd m−2 brightness) were prepared.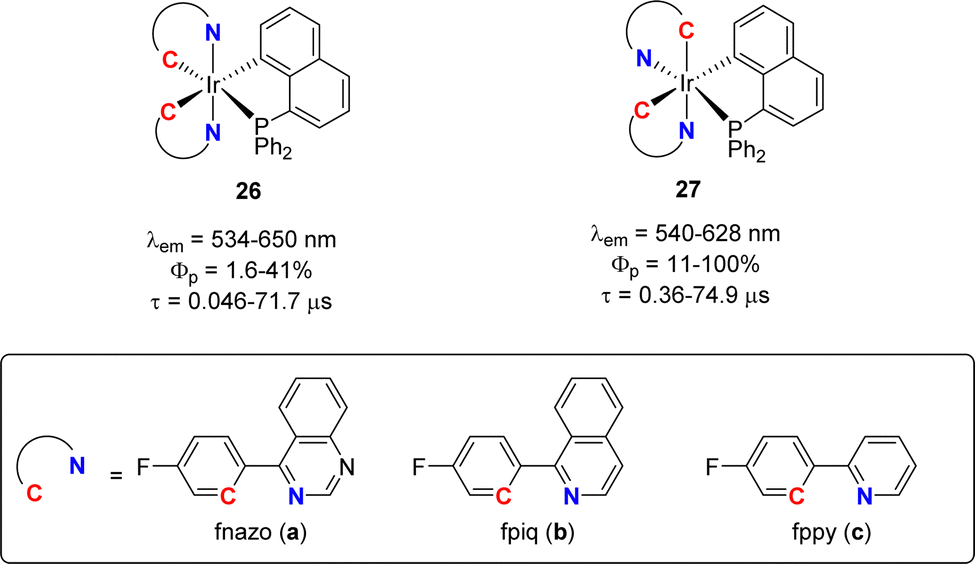 | ||
| Fig. 4 Phosphorescence properties of heteroleptic tris-cyclometalated Ir(III) complexes. | ||
In a following study, the same authors further extended the structural diversity of iridium(III) emitters.16 Complexes with one N^N azolate ligand and two P^C-cyclometalated ligands derived from diphenyl 1-naphthyl phosphine (or its isoquinoline analog) were shown to also display phosphorescence (with λem of 530–640 nm and quantum yields from 11 to 100%) (Fig. 5).
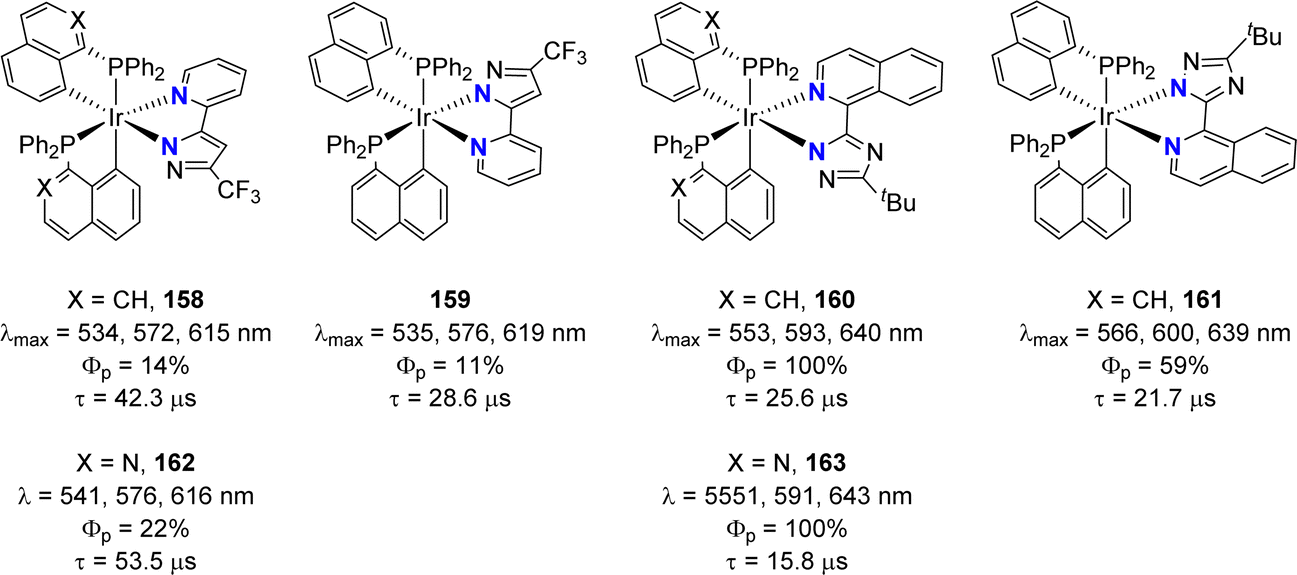 | ||
| Fig. 5 Structural modulation of the phosphorescent heteroleptic cyclometalated Ir(III) complexes: naphthalene and isoquinoline-derived phosphines and N^N azolate ligands. | ||
Conclusions
As shown in this review, (P,C)-cyclometalated complexes derived from naphthyl phosphines have been discovered early on but long remained neglected. They have however attracted growing interest recently and been shown to display rich chemistry and open interesting perspectives. The chemistry of the [Np(P,C)] complexes is now much more developed than that of the related [Np(N,C)] complexes derived from the well-known 1,8-dimethylaminonaphthyl ligand DAN.68–71The historical P-chelation assisted C–H activation route pioneered by Shaw 50 years ago remains the most straightforward and general route to prepare [Np(P,C)M] complexes. This shows complete selectivity for the Csp2–H bond at the peri position to phosphorus, even when a methyl group is present next to phosphorus. peri-Halo naphthyl phosphines are readily available. Their P-chelation assisted oxidative addition has become a valuable alternative route to C–H activation. First introduced to demonstrate the feasibility of Csp2–X oxidative addition to a single gold(I) center, it has then been extended to nickel. A third complementary route applied to groups 7–11 metals relies on transmetalation from stable mercury or tin peri-metalated naphthyl phosphines.
From a structural viewpoint, the [Np(P,C)] framework is well suited and nicely accommodates metals from groups 7 to 11, as apparent from the P–M and Cperi–M bond lengths, which only slightly deviate from the respective sums of the covalent radii. The naphthyl moiety imparts rigidity, but retains some flexibility, by bending the Cperi–P bond toward the metal and out of the naphthalene plane.
Reactivity studies on [Np(P,C)M] complexes long remained sparse and isolated, but the robustness and synthetic potential of palladium, platinum and ruthenium complexes were recognized early on in several transformations (the Heck reaction, the conjugate addition of aryl boronic acids to electron-deficient alkenes, the transfer hydrogenation of ketones, etc.). Np(P,C)-cyclometalation turned to be particularly fruitful in gold(III) chemistry, where it has opened new reactivity paths (migratory insertion, β-H elimination, and nucleophilic addition to π-allyl) and has enabled many new bonding situations to be authenticated (C–H agostic, π and σ-arene, π-allyl, and Schrock-type nucleophilic carbene complexes), resulting ultimately in unique catalytic activity in the intermolecular hydroarylation of alkynes. [Np(P,C)M] complexes have also been shown recently to be key intermediates in the Ru-catalyzed alkylation of the remote C5 position of naphthyl phosphines and Rh-catalyzed arylation of the peri position of naphthyl/pyrenyl phosphines.
Very little is known yet about the photophysical properties of [Np(P,C)M] complexes. Interesting phosphorescence properties have been occasionally pointed out but further studies are certainly needed to better understand the very influence of Np(P,C)-cyclometalation and develop its use in opto-electronic systems.
It is likely that the chemistry of [Np(P,C)M] complexes will keep developing and new facets will be discovered in near future. Although not all transition metals may form stable [Np(P,C)M] complexes, there is certainly room to expand the actual diversity. First-raw metals are certainly worth to consider and explore thoroughly. There is no manganese complex known to date, only one tetranuclear copper(I) cluster and very few complexes with iron, cobalt and nickel. So far, only alkyl and aryl groups have been introduced at phosphorus. The substitution pattern may be varied further using heteroaryl, alkoxy, and amino groups to significantly decrease/increase the electron-density at phosphorus. Other P-containing groups than phosphines may also be incorporated. The rich chemistry displayed by [Np(P,C)Au(III)] complexes will certainly stimulate research towards highly reactive complexes, basic transformations and catalytic applications with other metals. Being readily accessible, broadly modulable and very robust, the [Np(P,C)M] platform is very attractive and holds much potential. To date, applications have focused essentially on catalysis, but materials science and medicinal chemistry are certainly worth to be considered too. Indeed, [Np(P,C)M] complexes may well complement and eventually outperform the commonly used rigid (N,C)-cyclometalated complexes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Support from the Centre National de la Recherche Scientifique, the Université de Toulouse and the Agence Nationale de la Recherche is gratefully acknowledged. E. M. thanks the European Commission for a MCIF (Au-MLC – 841877).References
- J. M. Duff and B. L. Shaw, J. Chem. Soc., Dalton Trans., 1972, 2219–2225 RSC.
- J. M. Duff, B. E. Mann, B. L. Shaw and B. Turtle, J. Chem. Soc., Dalton Trans., 1974, 139–145 RSC.
- M. E. van der Boom and D. Milstein, Chem. Rev., 2003, 103, 1759–1792 CrossRef CAS PubMed.
- M. Albrecht, Chem. Rev., 2010, 110, 576–623 CrossRef CAS PubMed.
- G. C. Dickmu and I. P. Smoliakova, Coord. Chem. Rev., 2020, 409, 213203 CrossRef CAS.
- R. Cerón-Camacho, M. A. Roque-Ramires, A. D. Ryabov and R. Le Lagadec, Molecules, 2021, 26, 1563 CrossRef PubMed.
- M. Albrecht and G. Van Koten, Angew. Chem., Int. Ed., 2001, 40, 3750–3781 CrossRef CAS PubMed.
- L. Rocchigiani and M. Bochmann, Chem. Rev., 2021, 121, 8364–8451 CrossRef CAS PubMed.
- J. Hu, R. Lin, J. H. K. Yip, K. Y. Wong, D. L. Ma and J. J. Vittal, Organometallics, 2007, 26, 6533–6543 CrossRef CAS.
- J. Hu, J. H. K. Yip, D. L. Ma, K. Y. Wong and W. H. Chung, Organometallics, 2009, 28, 51–59 CrossRef CAS.
- R. Sun, T. Wang, S. Zhang, X. Chu and B. Zhu, RSC Adv., 2017, 7, 17063–17070 RSC.
- D. H. Ess, W. A. Goddard and R. A. Periana, Organometallics, 2010, 29, 6459–6472 CrossRef CAS.
- D. L. Davies, S. A. Macgregor and C. L. McMullin, Chem. Rev., 2017, 117, 8649–8709 CrossRef CAS PubMed.
- K. M. Altus and J. A. Love, Commun. Chem., 2021, 4, 173 CrossRef PubMed.
- B. S. Du, C. H. Lin, Y. Chi, J. Y. Hung, M. W. Chung, T. Y. Lin, G. H. Lee, K. T. Wong, P. T. Chou, W. Y. Hung and H. C. Chiu, Inorg. Chem., 2010, 49, 8713–8723 CrossRef CAS PubMed.
- C. H. Lin, Y. Chi, M. W. Chung, Y. J. Chen, K. W. Wang, G. H. Lee, P. T. Chou, W. Y. Hung and H. C. Chiu, Dalton Trans., 2011, 40, 1132–1143 RSC.
- R. Sun, S. Zhang, X. Chu and B. Zhu, Organometallics, 2017, 36, 1133–1141 CrossRef CAS.
- S. Zhang, X. Chu, T. Li, Z. Wang and B. Zhu, ACS Omega, 2018, 3, 4522–4533 CrossRef CAS PubMed.
- R. Sun, X. Chu, S. Zhang, T. Li, Z. Wang and B. Zhu, Eur. J. Inorg. Chem., 2017, 3174–3183 CrossRef CAS.
- T. Kuwabara, T. Kato, K. Takano, S. Kodama, Y. Manabe, N. Tsuchida, K. Takano, Y. Minami, T. Hiyama and Y. Ishii, Chem. Commun., 2018, 54, 5357–5360 RSC.
- A. Grabulosa, J. Granell and M. Font-Bardia, J. Organomet. Chem., 2019, 896, 51–58 CrossRef CAS.
- L. Shaw, Chem. Commun., 1998, 1361–1362 RSC.
- G. D. Frey, C. P. Reisinger, E. Herdtweck and W. A. Herrmann, J. Organomet. Chem., 2005, 690, 3193–3201 CrossRef CAS.
- I. S. Dubinsky-Davidchik, I. Goldberg, A. Vigalok and A. N. Vedernikov, Chem. Commun., 2013, 49, 3446–3448 RSC.
- K. A. Grice, W. Kaminsky and K. I. Goldberg, Inorg. Chim. Acta, 2011, 369, 76–81 CrossRef CAS.
- H. F. Klein, R. Beck, U. Flörke and H. J. Haupt, Eur. J. Inorg. Chem., 2003, 1380–1387 CrossRef CAS.
- G. Cerveau, G. Chauviere, E. Colomer and R. J. P. Corriu, J. Organomet. Chem., 1981, 210, 343–351 CrossRef CAS.
- M. L. Scheuermann, D. W. Boyce, K. A. Grice, W. Kaminsky, S. Stoll, W. B. Tolman, O. Swang and K. I. Goldberg, Angew. Chem., Int. Ed., 2014, 53, 6492–6495 CrossRef CAS PubMed.
- T. Mizuta, T. Nakazono and K. Miyoshi, Angew. Chem., Int. Ed., 2002, 41, 3897–3898 CrossRef CAS PubMed.
- T. Mizuta, Y. Iwakuni, T. Nakazono, K. Kubo and K. Miyoshi, J. Organomet. Chem., 2007, 692, 184–193 CrossRef CAS.
- T. Mizuta, N. Tanaka, Y. Iwakuni, K. Kubo and K. Miyoshi, Organometallics, 2009, 28, 2808–2817 CrossRef CAS.
- S. Bontemps, M. Devillard, S. Mallet-Ladeira, G. Bouhadir, K. Miqueu and D. Bourissou, Inorg. Chem., 2013, 52, 4714–4720 CrossRef CAS PubMed.
- J. Guenther, S. Mallet-Ladeira, L. Estevez, K. Miqueu, A. Amgoune and D. Bourissou, J. Am. Chem. Soc., 2014, 136, 1778–1781 CrossRef CAS PubMed.
- F. Rekhroukh, R. Brousses, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2015, 54, 1266–1269 CrossRef CAS PubMed.
- J. Rodriguez, M. S. M. Holmsen, Y. García-Rodeja, E. D. Sosa Carrizo, P. Lavedan, S. Mallet-Ladeira, K. Miqueu and D. Bourissou, J. Am. Chem. Soc., 2021, 143, 11568–11581 CrossRef CAS PubMed.
- C. Blons, M. Duval, D. Delcroix, H. Olivier-Bourbigou, S. Mallet-Ladeira, E. D. S. Carrizo, K. Miqueu, A. Amgoune and D. Bourissou, Chem. – Eur. J., 2018, 24, 11922–11925 CrossRef CAS PubMed.
- Silver was found to behave similarly to copper in this process, affording peri-bridged phosphonium salts via [Np(P,C)Ag(III)] intermediates: G. Szaloki, K. Miqueu and D. Bourissou, Mendeleev Commun., 2022, 32, 78–79 CrossRef CAS.
- M. Duval, C. Blons, S. Mallet-Ladeira, D. Delcroix, L. Magna, H. Olivier-Bourbigou, E. D. Sosa Carrizo, K. Miqueu, A. Amgoune, G. Szaloki and D. Bourissou, Dalton Trans., 2020, 49, 13100–13109 RSC.
- S. Furan, M. Vogt, K. Winkels, E. Lork, S. Mebs, E. Hupf and J. Beckmann, Organometallics, 2021, 40, 1284–1295 CrossRef CAS.
- S. Furan, M. Molkenthin, K. Winkels, E. Lork, S. Mebs, E. Hupf and J. Beckmann, Organometallics, 2021, 40, 3785–3796 CrossRef CAS.
- E. Hupf, E. Lork, S. Mebs and J. Beckmann, Inorg. Chem., 2015, 54, 1847–1859 CrossRef CAS PubMed.
- T. G. Do, E. Hupf, E. Lork, J. F. Kögel, F. Mohr, A. Brown, R. Toyoda, R. Sakamoto, H. Nishihara, S. Mebs and J. Beckmann, Eur. J. Inorg. Chem., 2019, 647–659 CrossRef CAS.
- S. Furan, E. Lork, S. Mebs, E. Hupf and J. Beckmann, Z. Anorg. Allg. Chem., 2020, 646, 856–865 CrossRef CAS.
- B. Cordero, V. Gomez, A. E. Platero-Prats, M. Reves, J. Echeverria, E. Cremades, F. Barragan and S. Alvarez, Dalton Trans., 2008, 2832–2838 RSC.
- F. Rekhroukh, L. Estevez, S. Mallet-Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, J. Am. Chem. Soc., 2016, 138, 11920–11929 CrossRef CAS PubMed.
- J. Rodriguez, G. Szaloki, E. D. Sosa Carrizo, N. Saffon-Merceron, K. Miqueu and D. Bourissou, Angew. Chem., Int. Ed., 2020, 59, 1511–1515 CrossRef CAS PubMed.
- F. Rekhroukh, L. Estevez, C. Bijani, K. Miqueu, A. Amgoune and D. Bourissou, Organometallics, 2016, 35, 995–1001 CrossRef CAS.
- F. Rekhroukh, L. Estévez, C. Bijani, K. Miqueu, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2016, 55, 3414–3418 CrossRef CAS PubMed.
- F. Rekhroukh, C. Blons, L. Estevez, S. Mallet-Ladeira, K. Miqueu, A. Amgoune and D. Bourissou, Chem. Sci., 2017, 8, 4539–4545 RSC.
- C. Blons, A. Amgoune and D. Bourissou, Dalton Trans., 2018, 47, 10388–10393 RSC.
- M. S. M. Holmsen, A. Nova, S. Øien-Ødegaard, R. H. Heyn and M. Tilset, Angew. Chem., Int. Ed., 2020, 59, 1516–1520 CrossRef CAS PubMed.
- J. Rodriguez, D. Vesseur, A. Tabey, S. Mallet-Ladeira, K. Miqueu and D. Bourissou, ACS Catal., 2022, 993–1003 CrossRef CAS.
- C. Blons, S. Mallet-Ladeira, A. Amgoune and D. Bourissou, Angew. Chem., Int. Ed., 2018, 57, 11732–11736 CrossRef CAS PubMed.
- C. Jia, D. Piao, J. Oyamada, W. Lu, T. Kitamura and Y. Fujiwara, Science, 2000, 287, 1992–1995 CrossRef CAS PubMed.
- C. Jia, W. Lu, J. Oyamada, T. Kitamura, K. Matsuda, M. Irie and Y. Fujiwara, J. Am. Chem. Soc., 2000, 122, 7252–7263 CrossRef CAS.
- M. S. M. Holmsen, C. Blons, A. Amgoune, M. Regnacq, D. Lesage, E. D. Sosa Carrizo, P. Lavedan, Y. Gimbert, K. Miqueu and D. Bourissou, J. Am. Chem. Soc., 2022, 144, 22722–22733 CrossRef CAS PubMed.
- A. Pujol, M. Lafage, F. Rekhroukh, N. Saffon-Merceron, A. Amgoune, D. Bourissou, N. Nebra, M. Fustier-Boutignon and N. Mézailles, Angew. Chem., Int. Ed., 2017, 56, 12264–12267 CrossRef CAS PubMed.
- G. C. Ge, D. L. Mo, C. H. Ding, L. X. Dai and X. L. Hou, Org. Lett., 2012, 14, 5756–5759 CrossRef CAS PubMed.
- M. Shimizu and T. Yamamoto, Tetrahedron Lett., 2020, 61, 152257 CrossRef CAS.
- R. Beck, T. Zheng, H. Sun, X. Li, U. Flörke and H. F. Klein, J. Organomet. Chem., 2008, 693, 3471–3478 CrossRef CAS.
- K. L. Lu, H. H. Lee, C. M. Wang and Y. S. Wen, Organometallics, 1994, 13, 593–599 CrossRef CAS.
- T. Kato, T. Kuwabara, Y. Minami, T. Hiyama and Y. Ishii, Bull. Chem. Soc. Jpn., 2019, 92, 1131–1141 CrossRef CAS.
- T. Iwamoto, K. Shibuya, T. Takakuwa, T. Kuwabara and Y. Ishii, Organometallics, 2022, 41, 182–186 CrossRef CAS.
- Y. Fu, C. H. Chen, M. G. Huang, J. Y. Tao, X. Peng, H. B. Xu, Y. J. Liu and M. H. Zeng, ACS Catal., 2022, 12, 5036–5047 CrossRef CAS.
- X. Luo, J. Yuan, C. D. Yue, Z. Y. Zhang, J. Chen, G. A. Yu and C. M. Che, Org. Lett., 2018, 20, 1810–1814 CrossRef CAS PubMed.
- C. Sire, H. Cattey, A. Tsivery, J. C. Hierso and J. Roger, Adv. Synth. Catal., 2022, 364, 440–452 CrossRef CAS.
- W. Y. Heng, J. Hu and J. H. K. Yip, Organometallics, 2007, 26, 6760–6768 CrossRef CAS.
- C. Arlen, M. Pfeffer, O. Bars and D. Grandjean, J. Chem. Soc., Dalton Trans., 1983, 1535–1544 RSC.
- E. Wehman, G. van Koten, M. Knotter, H. Spelten, D. Heijdenrijk, A. N. S. Mak and C. H. Stam, J. Organomet. Chem., 1987, 325, 293–309 CrossRef CAS.
- E. Wehman, G. van Koten, J. T. B. H. Jastzebski, H. Ossor and M. Pfeffer, J. Chem. Soc., Dalton Trans., 1988, 2975–2981 RSC.
- W. J. J. Smeets, A. L. Spek, J. A. M. van Beek and G. van Koten, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1992, 48, 745–747 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cs00564f |
| This journal is © The Royal Society of Chemistry 2023 |