
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Brightness of fluorescent organic nanomaterials
Anila Hoskere
Ashoka
,
Ilya O.
Aparin
,
Andreas
Reisch
and
Andrey S.
Klymchenko
 *
*
Laboratoire de Bioimagerie et Pathologies, UMR 7021 CNRS, Université de Strasbourg, 74 route du Rhin, 67401, Illkirch, France. E-mail: andrey.klymchenko@unistra.fr
First published on 20th June 2023
Abstract
Brightness is a fundamental property of fluorescent nanomaterials reflecting their capacity to absorb and emit light. In sensing materials, brightness is crucial for high-sensitivity (bio)molecular detection, while in optical bioimaging it ensures high spatial and temporal resolution. Fluorescent organic nanoparticles (NPs) are particularly attractive because of their superior brightness compared to organic dyes. With the ever-growing diversity of organic nanomaterials, it is important to establish universal principles for measuring and estimating their brightness. This tutorial review provides definitions of brightness and describes the major approaches to its analysis based on ensemble and single-particle techniques. We present the current chemical approaches to fight Aggregation-Caused Quenching (ACQ) of fluorophores, which is a major challenge in the design of bright organic nanomaterials. The main classes of fluorescent organic NPs are described, including conjugated polymer NPs, aggregation-induced emission NPs, and NPs based on neutral and ionic dyes. Their brightness and other properties are systematically compared. Some brightest examples of bulk solid-state emissive organic materials are also mentioned. Finally, we analyse the importance of brightness and other particle properties in biological applications, such as bioimaging and biosensing. This tutorial will provide guidelines for chemists on the design of fluorescent organic NPs with improved performance and help them to estimate and compare the brightness of new nanomaterials with literature reports. Moreover, it will help biologists to select appropriate materials for sensing and imaging applications.
 Anila Hoskere Ashoka | Anila Hoskere Ashoka received his PhD degree in Organic Chemistry from the National Chemical Laboratory (India) in 2018. Then he carried out post-doctoral research with Dr. Andrey Klymchenko at the Laboratory of Bioimaging and Pathologies, University of Strasbourg. Currently, he is working as a development scientist at Beckman Coulter Life Sciences, India. |
 Ilya O. Aparin | Ilya Aparin graduated from the Chemistry Department of M. V. Lomonosov Moscow State University with a master's degree in organic chemistry (2012) and defended his PhD in bioorganic chemistry on fluorescent DNA probes. In 2019 he had been awarded with Marie Curie postdoctoral fellowship and joined Klymchenko's team at the University of Strasbourg. Over the past 10 years, his research projects have been dedicated to fluorescent bioconjugates for analytical application. Currently, Ilya holds the position of a senior scientist at “DNA script” startup at Paris. |
 Andreas Reisch | Andreas Reisch studied chemistry in Germany and France and obtained his PhD in 2009 for research on biomimetic polymers and surfaces at the Institut Charles Sadron, Strasbourg. As a postdoc in the group of Joseph B. Schlenoff at the Florida State University, he worked on polymeric bulk biomaterials. He is currently associate professor at the Faculty of Pharmacy of the University of Strasbourg. His research focuses on functional polymeric materials, notably fluorescent nanoparticles, and how to use polymer chemistry and kinetics of assembly to control their properties and interactions with biological systems. |
 Andrey S. Klymchenko | Andrey Klymchenko was born in Kherson, Ukraine. He received his PhD degree in 2003 from Kyiv Taras Shevchenko University. After postdoctoral work at the University of Strasbourg and the Catholic University of Leuven, he joined CNRS at the University of Strasbourg in 2006. He got promoted to CNRS Research Director in 2014, and he was an ERC Consolidator fellow in 2015–2020. He is a leader of “Nanochemistry and Bioimaging” group and co-founder of BrightSens Diagnostics SAS. His research interests include functional fluorescent molecules and nanomaterials for biosensing, imaging and theranostics. |
Key learning points(1) Definition of brightness and methods for its estimation for fluorescent nanomaterials at the ensemble and single-particle level.(2) Approaches to prevent aggregation-caused quenching in order to design bright organic nanomaterials. (3) Key classes of bright organic nanoparticles with systematic comparison of their brightness. (4) Insights on the choice of fluorescent NPs for biosensing and bioimaging applications. (5) Current challenges and perspectives in bright organic nanomaterials. |
1. Introduction
Even though the fluorescence imaging field has been dominated since quite some time by fluorescent dyes1 and fluorescent proteins,2 they present a fundamental limitation in terms of fluorescence brightness. In chemistry, the fluorescence brightness is defined as a product of molar extinction coefficient and fluorescence quantum yield, which for molecular emitters is physically limited to ∼300![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1.1,3 Brightness is a key property that defines the number of photons that can be collected for a given time period from the fluorescent probe. It determines the detection sensitivity in biosensing as well as spatial and temporal resolution in bioimaging.1 The limitations of classical fluorescent molecular and biomolecular probes in terms of brightness have stimulated the development of nanoscale materials (nanoparticles, NPs), because their molar extinction coefficient can be 10–1000-fold higher than that of molecular dyes.4,5 Among them, particularly attractive in terms of flexibility, rich surface chemistry and biocompatibility are fluorescent organic NPs,6 such as conjugated polymer NPs,7–9 aggregation-induced emission (AIE) NPs,10–12 dye-loaded polymeric NPs,5,13etc. Fluorescent organic NPs can be defined as fluorescent nanoscale materials composed mainly (if not exclusively) of organic components, in contrast to other types of luminescent NPs, such as quantum dots (QDs),14 dye-loaded silica NPs,15 metal nanoclusters,16 metal–organic framework NPs,17 carbon dots,18etc. There are several excellent reviews focused on the synthesis and applications of fluorescent organic nanomaterials.5–8,10,11,13,19 These reviews highlight the importance of critical characteristics of fluorescent organic NPs, such as size, aspect ratio, optical properties, surface functionalization, stealth properties, colloidal stability, multimodality, biodegradability, biocompatibility, toxicity and biodistribution (Fig. 1). In this tutorial review, we will focus on the fluorescence brightness of organic NPs, which has not yet been systematically addressed in the current literature. With the large variety of developed fluorescent organic NPs, the lack of a common comparative approach in terms of brightness makes it difficult to choose the proper tool for sensing and imaging applications. Moreover, brightness determination is a complex issue, which often leads to misinterpretation and incorrect comparison between fluorescent NPs of different nature. This tutorial review will provide a clear methodology for theoretical and experimental estimation of brightness of organic NPs. It will also describe basic concepts for designing bright nanomaterials, in particular, how to prevent the fundamental problem of aggregation-caused quenching (ACQ) of dyes in organic nanomaterials.5,10 Then, a systematic comparison will be given for key classes of organic NPs in terms of brightness and structural characteristics, such as size and intrinsic organization of emitters within the nanomaterial. We will also discuss other characteristics of nanomaterials, affecting the performance of fluorescence biosensing and bioimaging: (i) absorption and emission wavelength, which should be shifted to the red in order to achieve deeper tissue penetration and lower photodamage; (ii) photostability should be high in order to collect a maximum of photons; (iii) ON/OFF switching (blinking), which should be suppressed for tracking application or used for super-resolution microscopy; (iv) compatibility with two-photon excitation, which is particularly suitable for tissue imaging. Finally, we will provide insight on how brightness, other optical properties and size of NPs define their biological sensing applications. Overall, we provide a tutorial for chemists, physicists and biologists, to facilitate the design of new NPs, estimate their brightness and compare it with existing NPs, and further choose the right fluorescent tool for a given biological application.
000 M−1 cm−1.1,3 Brightness is a key property that defines the number of photons that can be collected for a given time period from the fluorescent probe. It determines the detection sensitivity in biosensing as well as spatial and temporal resolution in bioimaging.1 The limitations of classical fluorescent molecular and biomolecular probes in terms of brightness have stimulated the development of nanoscale materials (nanoparticles, NPs), because their molar extinction coefficient can be 10–1000-fold higher than that of molecular dyes.4,5 Among them, particularly attractive in terms of flexibility, rich surface chemistry and biocompatibility are fluorescent organic NPs,6 such as conjugated polymer NPs,7–9 aggregation-induced emission (AIE) NPs,10–12 dye-loaded polymeric NPs,5,13etc. Fluorescent organic NPs can be defined as fluorescent nanoscale materials composed mainly (if not exclusively) of organic components, in contrast to other types of luminescent NPs, such as quantum dots (QDs),14 dye-loaded silica NPs,15 metal nanoclusters,16 metal–organic framework NPs,17 carbon dots,18etc. There are several excellent reviews focused on the synthesis and applications of fluorescent organic nanomaterials.5–8,10,11,13,19 These reviews highlight the importance of critical characteristics of fluorescent organic NPs, such as size, aspect ratio, optical properties, surface functionalization, stealth properties, colloidal stability, multimodality, biodegradability, biocompatibility, toxicity and biodistribution (Fig. 1). In this tutorial review, we will focus on the fluorescence brightness of organic NPs, which has not yet been systematically addressed in the current literature. With the large variety of developed fluorescent organic NPs, the lack of a common comparative approach in terms of brightness makes it difficult to choose the proper tool for sensing and imaging applications. Moreover, brightness determination is a complex issue, which often leads to misinterpretation and incorrect comparison between fluorescent NPs of different nature. This tutorial review will provide a clear methodology for theoretical and experimental estimation of brightness of organic NPs. It will also describe basic concepts for designing bright nanomaterials, in particular, how to prevent the fundamental problem of aggregation-caused quenching (ACQ) of dyes in organic nanomaterials.5,10 Then, a systematic comparison will be given for key classes of organic NPs in terms of brightness and structural characteristics, such as size and intrinsic organization of emitters within the nanomaterial. We will also discuss other characteristics of nanomaterials, affecting the performance of fluorescence biosensing and bioimaging: (i) absorption and emission wavelength, which should be shifted to the red in order to achieve deeper tissue penetration and lower photodamage; (ii) photostability should be high in order to collect a maximum of photons; (iii) ON/OFF switching (blinking), which should be suppressed for tracking application or used for super-resolution microscopy; (iv) compatibility with two-photon excitation, which is particularly suitable for tissue imaging. Finally, we will provide insight on how brightness, other optical properties and size of NPs define their biological sensing applications. Overall, we provide a tutorial for chemists, physicists and biologists, to facilitate the design of new NPs, estimate their brightness and compare it with existing NPs, and further choose the right fluorescent tool for a given biological application.
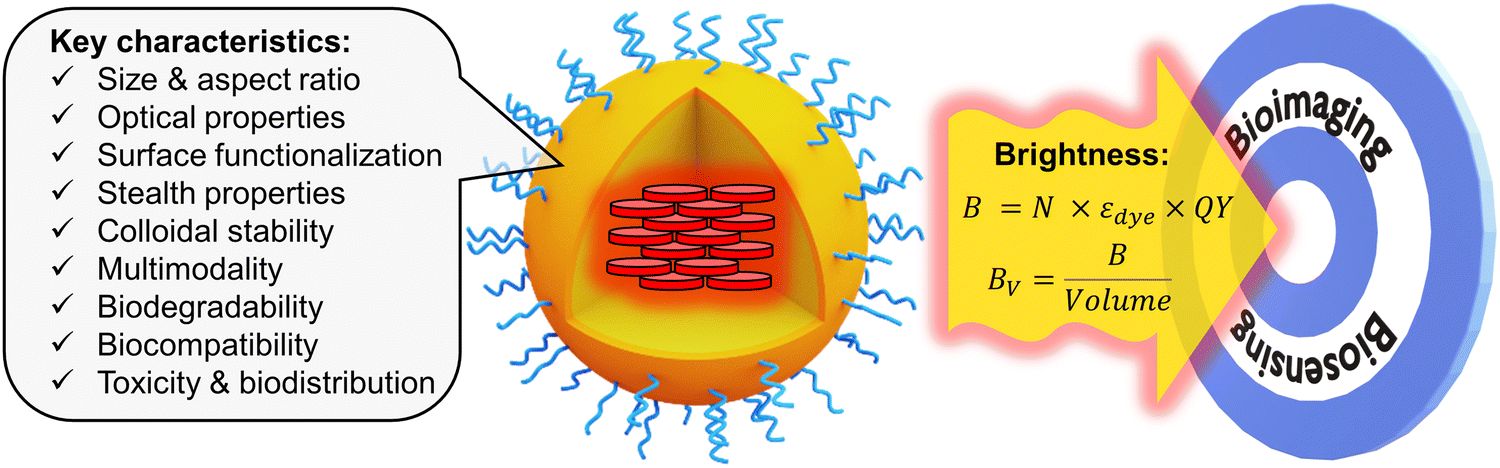 | ||
| Fig. 1 Fluorescent organic NPs, their key characteristics with focus on brightness. | ||
2 Brightness of fluorescent organic nanoparticles
2.1 Definitions of brightness
On a fundamental level, fluorescence brightness refers to the number of photons emitted by a probe per unit time upon irradiation with a given irradiance (excitation power density), where the irradiance corresponds to the power per surface. For practical reasons, the brightness is expressed in various ways depending on the context and experimental method used.20,21 In the chemical sciences, it is expressed based on molar extinction coefficient (ε) and quantum yield (QY) of an emitter:| Brightness = ε × QY | (1) |
The quantum yield is expressed as the following:
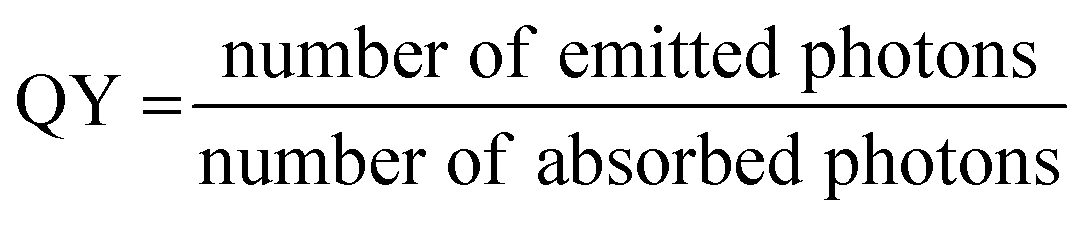 | (2) |
| σfluo = σ × QY | (3) |
| σ = 3.82 × 10−21 × ε | (4) |
| Brightness (B) = N × εdye × QY | (5) |
Here, the brightness is an ensemble value, which reflects the mean brightness of a large population of NPs, typically also averaged over relatively long measurement times (in the range of tens of seconds to minutes for most spectrofluorometers). On the other hand, brightness can be directly estimated in single-particle measurements22 either with respect to a reference emitter (relative brightness) or by precisely estimating the excitation and emission photon flux (also called light power density or irradiance) from single particles (absolute brightness).23 In the latter case the single particle brightness can, for example, be defined as:24
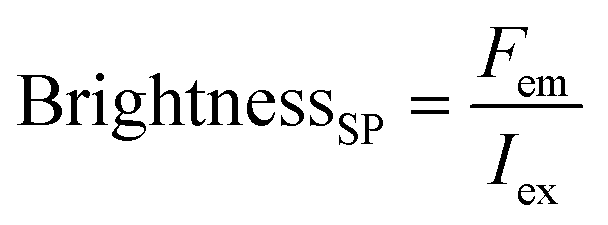 | (6) |
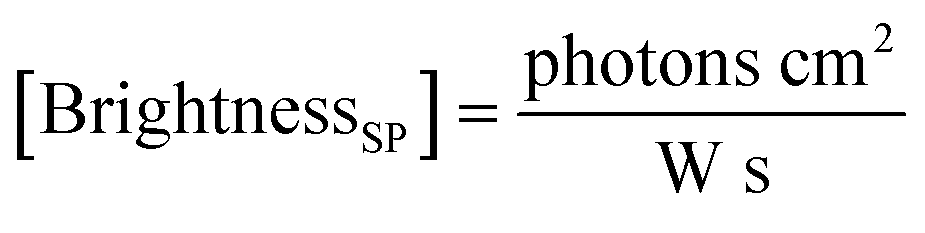 | (7) |
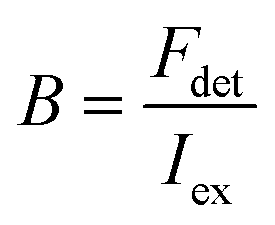 | (8) |
In the case of organic NPs, the particle size has a direct influence on the brightness. For a given loading of the NPs with dyes, the number of dyes per NP, is proportional to the particle volume, which means to the power of three of the diameter. If there is no major influence of the size on the QY, this then also means that the brightness is proportional to volume. This behaviour is expected for all types of NPs, where the dye is encapsulated inside the NPs, for example in dye-loaded polymeric NPs, AIE NPs or conjugated polymer NPs. One should note that variation of NP size in this case does not have a direct effect on the absorption and fluorescence spectra of NPs. This is different from some inorganic NPs like QDs, nanodiamonds, carbon dots, etc.21
In order to compare the inherent brightness of fluorescent nanomaterials of different sizes, it is therefore interesting to define a brightness per volume (BV):
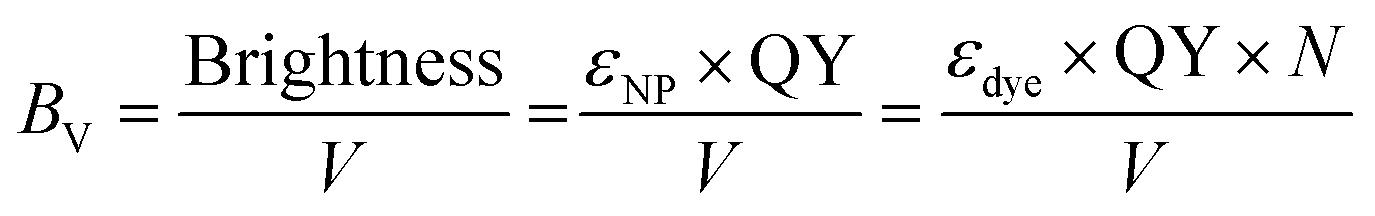 | (9) |
In case of dye-based NPs, N can be expressed as follows:
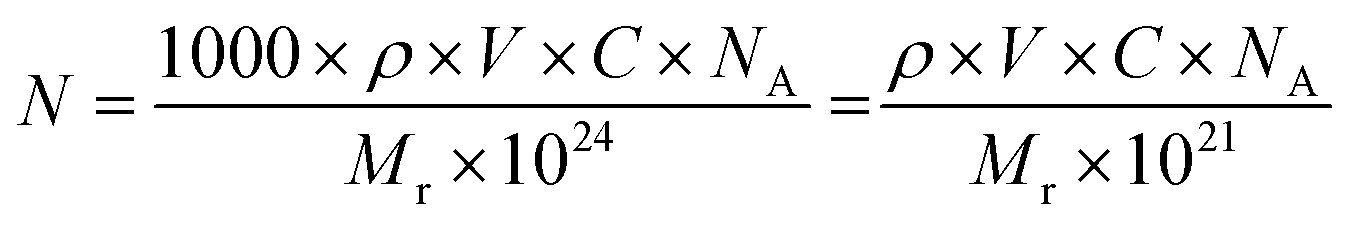 | (10) |
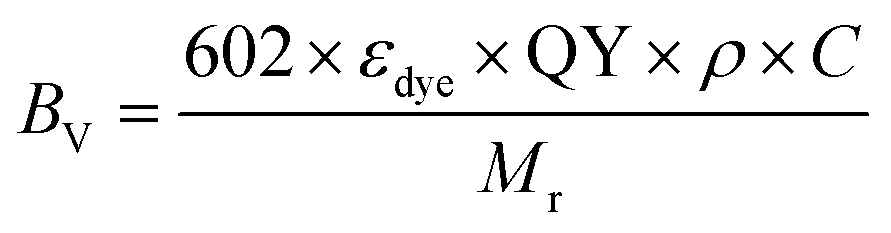 | (11) |
Another point to be considered is the appearance of inner filter effects that can occur at very high local dye concentrations inside NPs even at the level of a single particle. According to the Beer–Lambert law the absorbance is expressed as follows:
| A = εdye × l × c | (12) |
Furthermore, the influence of the particle size on the QY should be considered. On the one hand, larger size decreases surface to volume ratio, which could decrease the fraction of species quenched by interaction with aqueous medium. On the other hand, larger NPs may favour quenching processes linked to energy transfer effects occurring inside the NPs. Studies in our group on dye-loaded polymeric NPs showed no clear influence of the particle size on the QY.25 However, systems that allow to study precisely this effect without varying other parameters are rare.
It should be noted that most of the definitions of brightness given above could be applied to other types of nanomaterials, such as quantum dots, carbon dots, or metal–organic framework NPs. However, these materials cannot be always regarded to contain several individual emitters, and, therefore, their molar absorption coefficient is estimated for the whole particle.
Finally, the particle size is important, because most of biological applications require bright NPs of the smallest possible size (see below). Therefore, both particle brightness and BV will be further used in the review to analyse brightness of most representative examples of NPs reported in the literature.
2.2. Methods for measuring brightness
The methods to determine brightness experimentally can be distinguished depending on whether they are based on ensemble or single-particle measurements. While the ensemble methods are relatively simple to implement, they rely on several assumptions. Single-particle methods, on the other hand, are more demanding to realize, but they give access to the actual particle brightness and its distribution, in conditions similar to those in which these probes are used in optical microscopy experiments.| εNP = N × εdye | (13) |
Determining the number N of dyes (or emitters) per NP is less straightforward.26 In principle, N can be determined if the concentration of the dyes in the NPs and the particle size are known:
| N = [dye] × VNP | (14) |
A variety of approaches available to determine the quantum yields of fluorescent NPs have been reviewed in detail previously.30 Relative methods remain the most used and, a priori, the easiest. In this case, the fluorescence intensity integrated over the whole emission range INP and absorbance ANP at the excitation wavelength of the NPs are compared to those of a reference dye, IR and AR, respectively, measured under the same spectrofluorometer settings.
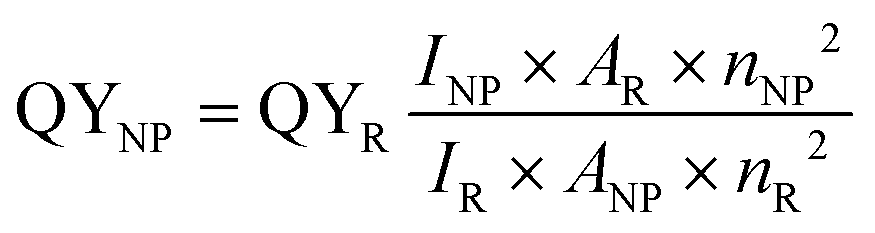 | (15) |
An alternative is the determination of absolute QYs, which can, for example, be obtained using an integrating sphere.30 Integrating spheres collect all photons absorbed, scattered and emitted by the sample and can nowadays be implemented in standard spectrofluorometers. However, a precise calibration of the set-up is required to achieve a high precision.
A general limitation of these approaches is that they typically do not allow obtaining any information on the distribution of particle brightness over the individual nanoparticles.
 | ||
| Fig. 2 Single-particle brightness analysis. (A) Simplified analysis workflow that includes: detection of local maxima, defining and measuring the intensity at regions of interest around maxima (raw signal: Iraw) and around central regions (background: Ibkg). The signal is then obtained by subtraction of the background from the raw signal. (B,C) Example of single-particle imaging by wide-field microscopy (B) and intensity analysis (C) for 20 nm DNA-modified dye-loaded polymeric NPs (organic NPs) vs. reference particles QD-605. In case of QD-605, the source irradiance was increased 65-fold to achieve comparable intensity with organic NPs. Other experimental settings were identical. Excitation wavelength was 550 nm. Panels B and C reproduced with permission from ref. 36. Copyright American Chemical Society. | ||
Next, the image analysis is performed, where the signal of the individual particles (or luminescent spots) is first measured and the background is subtracted (Fig. 2). Simple ImageJ algorithms allow automatic localisation of the brightest spots in an image. Then, a circular region of interest (ROI) with fixed area is defined around the determined localizations and the mean (or total) intensity in the ROI is measured.24 Ideally, the local background signal can be subtracted by measuring the intensity in a band around the ROI (Fig. 2). Further, from this data, the mean intensity and the intensity histograms can be generated by analysing hundreds of NPs of interest and reference NPs. An example of such analysis is provided for DNA-modified dye-loaded polymeric NPs in Fig. 2.36
Generally, the relative particle brightness depends on the reference and to some extent the instrumental settings. Therefore, attempts have been made to introduce procedures to measure absolute brightness of nanomaterials. In particular, a brightness parameter B defined by eqn (8) requires determining the number of detected photons Fdet, established based on the microscope and camera parameters.23,37 It is possible to go one step further and determine the single-particle brightness as defined by eqn (6),24 which is, in principle, independent of the microscope settings and allows comparison of nanomaterials, despite using different setups. However, this approach requires measuring precisely the excitation irradiance, which is done by dividing the irradiation power at the sample position by the illuminated surface area for a given objective. Moreover, it requires converting the recorded fluorescence signal into the emitted photon flux. In this case, the number of detected photons per second Fdet is determined for a given spot according to:
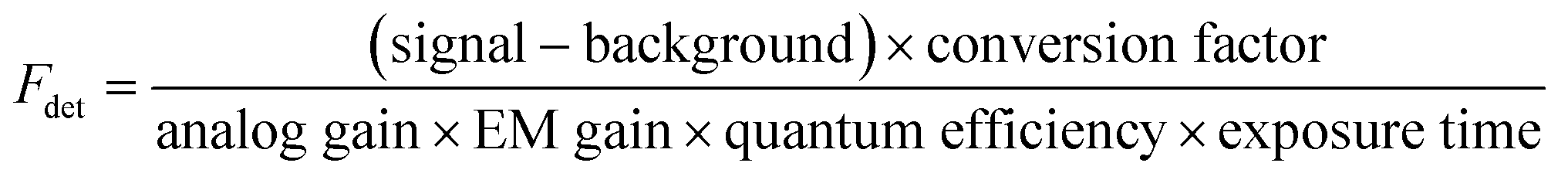 | (16) |
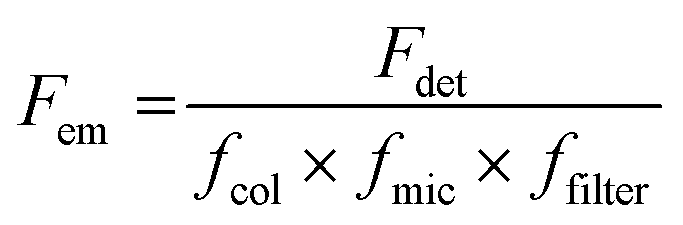 | (17) |
A method apart to measure single-particle brightness is based on FCS.38 However, it is rarely used, because it requires a dedicated FCS setup, and, moreover, the excitation power used in the focal spot is very high and the particle size should be much smaller than the focal spot.
Generally, the brightness measured at the single-particle level correlates with that measured for a particle ensemble in solution. For example, we showed that single-particle brightness of dye-loaded NPs vs. reference NPs, e.g. QDs, could be predicted based on the estimations of their extinction coefficient and quantum yield.24,35,36 However, the estimations never gave perfect match between the two methods. The primary reason for this is that the excitation power density (irradiance) in the single-particle microscopy technique (∼1 W cm−2) is ∼1000-fold higher than that used in the fluorometer (∼1 mW cm−2). This high irradiance can produce saturation effects inside NPs as well as photobleaching, which will contribute to the decrease in the actual brightness. The second reason is the difference in the detection optics, which generally requires dichroic mirrors and filters in the case of fluorescence microscope, which should be taken into account when the NPs of interest are compared with the reference NPs. Therefore, for new fluorescent nanomaterials it is always better to estimate both ensemble and single-particle brightness, which would facilitate in the future comparison between different nanomaterials.
3. Brightness and aggregation-caused quenching: basic design concepts of fluorescent NPs
To achieve high brightness in fluorescent nanomaterials, one needs to assemble a large number of dyes, having high fluorescence quantum yield and extinction coefficient, in the small volume of a nanoparticle. However, at high local concentration within nanomaterials, organic dyes, which are usually flat aromatic structures, tend to form non-emissive pi-stacked aggregates with face-to-face assembly. This process, called aggregation-caused quenching (ACQ), is a major challenge in preparation of bright organic nanomaterials. According to the exciton theory, adapted by Kasha to organic fluorophores,39,40 dye assemblies could be classified into H- and J-aggregates. These aggregates can be described by the Coulombic intermolecular coupling (Jc), which, in case of parallel dye dipoles, are given by the following equation:40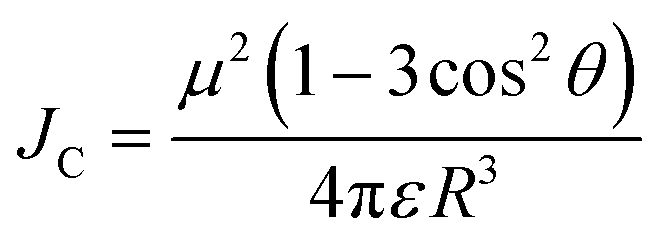 | (18) |
 | ||
| Fig. 3 Principles of dye aggregation based on Kasha's exciton theory (A) and the most established methods to prevent ACQ in fluorescent organic nanomaterials (B). | ||
Importantly, the emission of J-aggregate is allowed, whereas that of the H-aggregate is forbidden (Fig. 3). H-Aggregation is the most common cause of ACQ in nanomaterials based on dyes. Moreover, H-aggregation leads to hypochromism, which is a reduction of the molar extinction coefficient of dyes within the aggregate, that additionally decreases the brightness of the nanomaterials. Other mechanisms of ACQ should be considered, which include excimer formation, excited state intermolecular charge and electron transfer20 within the aggregated dyes as well as inner filter effect within large dye ensembles (see above). J-Aggregates of dyes are attractive for building emissive bulk materials,41 however their tendency to form 1D nanostructures makes it difficult to assemble fluorescent NPs and their Stokes shift is too small for conventional bioimaging. Therefore, to prevent ACQ in fluorescent nanomaterials, one should go beyond H- and J-aggregation, which implies control of dye–dye spacing and orientation (Fig. 3).
We can identify the following major approaches to prevent ACQ in organic nanomaterials. First, one should mention the use of conjugated polymers.42,43 In this case, the fluorophores are made of conjugated units, which are aligned along the conjugated polymer chain. The spacing between fluorophore units is controlled by the rigid pi-conjugated bonds. Moreover, bulky side groups on these fluorophore units prevent pi-stacking and thus ensure formation of emissive particles. The other approaches to prevent ACQ concern specially designed fluorescent molecules, i.e. dyes assembled into nanomaterials without covalent pi-conjugation between them. One of the most popular approaches is the use of aggregation-induced emission (AIE) dyes, proposed by Tang and co-workers in 2001.44 In this case, the fluorophore is twisted along its conjugated structure, which generates a propeller-like topology. AIE dyes (AIEgens) are poorly emissive in solution, whereas they light up in the aggregated state because their intramolecular rotation is restricted, while their propeller topology prevents formation of H-aggregates (Fig. 3). The AIE concept gave rise to a great variety of highly emissive (nano)materials for a variety of applications, especially in bioimaging.10,45 Alternatively, conventional fluorophores could be redesigned in order to prevent ACQ. The primary approach is to introduce bulky side groups into organic fluorophores. In contrast to AIE, the flat conjugated structure of the dye is maintained (or disturbed to a minor extent), whereas the side groups create a steric hindrance against pi-stacking and H-aggregation.46–48 However, this method needs multistep organic synthesis and it cannot be simply transposed to different conventional dyes. Finally, a promising approach to prevent ACQ is based on bulky hydrophobic counterions as insulators of charged organic fluorophores (Fig. 3).5,33,49 The large diameter of these counterions (around 1 nm) ensures good spacing between fluorophores and thus prevents their H-aggregation. The approach is particularly suitable for cationic cyanines and rhodamines, which are known for their outstanding brightness and photostability. Importantly, this approach can work in both pure dye salts50–53 and in dye-loaded polymeric nanoparticles.5,33 Below, we will discuss all these approaches for different classes of organic nanomaterials and analyse their optical properties with focus on the achieved brightness. The structural and spectroscopic data of nanomaterials, discussed in the review, are presented in Table 1.
| Composition | NPs diameter (nm) | Dye content (wt%) | λ abs (nm) | λ em (nm) | QY (%) | B (M−1 cm−1) | B V (M−1 m−1 nm−3) | Singe NP brightness | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| a B and BV are brightness and per-volume brightness of NPs estimated based on eqn (5) and (9) or (11), respectively. For rhodamine 6G we assume the volume of the molecule of 1 nm3. Single NP brightness is experimental value obtained from single-particle microscopy measurements. The brightness of QD-685, QD-605 and QD-705 was estimated for the following excitation wavelengths: 488. b 532. c 532 (c) nm, respectively. d Data from ref. 82. | |||||||||
| Molecular organic dyes | |||||||||
| Rhodamine 6G | 530 | 552 | 90 | 1.0 × 105 | 1.0 × 105 | 31 | |||
| Quantum dots | |||||||||
| QD-585 | 585 | 60 | 1.8 × 105b |
||||||
| QD-605 | 605 | 52 | 3.0 × 105c |
54 | |||||
| QD-705 | 705 | 49 | 1.0 × 106c |
54 | |||||
| Conjugated polymer NPs | |||||||||
| MEH-PPV | 15 | 100 | 485 | 590 | 1 | 1.2 × 106 | 651 | 55 | |
| PFBT | 15 | 100 | 450 | 545 | 7 | 5.1 × 105 | 2900 | 55 | |
| PFBT | 10 | 100 | 450 | 545 | 30 | 3 × 106 | 5700 | 30 × QD-565 | 56 |
| PFPV | 15 | 100 | 445 | 510 | 8 | 1.2 × 107 | 6520 | 55 | |
| PFBT-DBSOC6 10-COOH | 23 | 50 | 455 | 712 | 15 | 1.1 × 107 | 1760 | 2 × QD-705 | 57 |
| PF-TC6FQ-BODIPY | 26 | 100 | 495 | 723 | 33 | 1.7 × 107 | 1954 | 3 × QD-705 | 58 and 59 |
| Pttc-TFQ-BODIPY | 28 | 100 | 490 | 724 | 51 | 5.1 × 107 | 4480 | 7 × QD-705 | 60 |
| PF-TC6FQ | 21 | 100 | 493 | 652 | 47 | 2.4 × 107 | 5070 | 8 × QD-655 | 61 |
| PFDBT5–PFBT | 15 | 80 | 450 | 650 | 56 | 1.6 × 107 | 9500 | 15 × QD-655 | 42 |
| PFGBDP/PFDHTBT–BDP720 | 31 | 100 | 528 | 721 | 42 | 1.8 × 108 | 11300 |
83 × QD-705 | 62 |
| Aggregation-induced emission NPs | |||||||||
| TPETPAFN | 33 | — | 510 | 670 | 25 | 1.1 × 107 | 560 | 10 × QD-655 | 63 |
| BTPEBT | 30 | 33 | 422 | 547 | 63 | 3.5 × 107 | 1980 | 64 | |
| BTPEBT-V2 | 29 | 40 | 418 | 547 | 62 | 2.8 × 107 | 2870 | 65 | |
| BTPEBD | 32 | 33 | 436 | 574 | 90 | 5.3 × 107 | 3100 | 66 | |
| PTZ–BT–TPA | 100 | 33 | 483 | 656 | 39 | 2.4 × 109 | 4610 | 67 | |
| DTPA–TBZ | 50 | 33 | 652 | 929 | 11 | 4.0 × 107 | 614 | 68 | |
| NPs based on conventional dyes | |||||||||
| Nile red/PS | 100 | 0.70 | 570 | 635 | 23 | 7 × 107 | 134 | 69 | |
| DiD/PLGA | 66 | 0.72 | 650 | 667 | 21 | 1.3 × 107 | 84 | 70 | |
| NPs based on dyes bearing bulky side groups | |||||||||
| Mes-BODIPY/PS | 16 | 3.5 | 526 | 540 | 77 | 5.1 × 106 | 2300 | 71 | |
| BODIPY/PS-PEG | 60 | 1.3 | 529 | 544 | 35 | 1.4 × 108 | 470 | 72 | |
| BDP4–PEG(1000)–PMAO | 14 | 18 | 532 | 560 | 60 | 2.5 × 106 | 1740 | 5 × QD-585 | 73 |
| PDI-Cl/co-polymerized | 40 | 2.4 | 520 | 550 | 50 | 1.0 × 107 | 310 | 74 | |
| LR/PLGA | 38 | 5.0 | 575 | 605 | 50 | 7.5 × 106 | 261 | 18 × QD-585 | 75 |
| NPs based on charged dyes with bulky counterions | |||||||||
| R12/F5-TPB | 14 | 100 | 560 | 580 | 32 | 2.7 × 107 | 18700 |
76 | |
| R12/F12-TPB | 19 | 100 | 560 | 580 | 60 | 6.8 × 107 | 19100 |
45 × QD-585 | 76 |
| R18/F5-TPB/PLGA | 40 | 5 | 560 | 580 | 20 | 1.8 × 107 | 600 | 6 × QD-605 | 33 |
| R18/F5-TPB/PMMA-MA | 15 | 5 | 560 | 580 | 60 | 3.0 × 106 | 1600 | 10 × QD-585 | 77 |
| R18/F5-TPB/PMMA-MA | 34 | 23 | 560 | 580 | 31 | 8.9 × 107 | 4330 | 100 × QD-585 | 24 |
| R18/F5-TPB/PCL | 33 | 23 | 560 | 580 | 34 | 8.9 × 107 | 4730 | 24 | |
| R18/F5-TPB/PMMA-N3 | 40 | 23 | 560 | 580 | 46 | 1.9 × 108 | 5790 | 100 × QD-605 | 35 |
| R18/F5-TPB/PEMA-N3 | 20 | 33 | 560 | 580 | 52 | 3.9 × 107 | 9480 | 87 × Q-605 | 35 |
| BlueCy/TPB/PMMA-MA | 40 | 12 | 425 | 475 | 17.3 | 2.3 × 107 | 687 | 70 × QD-525 | 78 |
| R6G-C18/F12-TPB/PMMA-N3 | 44 | 250 mM | 530 | 570 | 23 | 1.1 × 108 | 2490 | 79 | |
| Cy5/F12TPB/PEMA-N3 | 16 | 23 | 652 | 682 | 42 | 9.7 × 106 | 4530 | 22 × QD-705 | 54 |
| PhSP18/F5-TPB/PMMA-MA | 40 | 29 | 507 | 675 | 40 | 1.2 × 107 | 3840 | 50 × QD-605 | 80 |
| DiI/TPB/lipid droplets | 87 | 8 | 553 | 575 | 14 | 2.5 × 108 | 725 | 81 | |
| Cyanostar | 16 | — | 560 | 580 | 30 | 1.5 × 107 | 5000d | 20 × FS | 82 |
4. Classes of fluorescent organic nanoparticles
4.1 Conjugated polymer nanoparticles
NPs prepared from conjugated polymers represent an established class of nanomaterials with exceptional photophysical properties. Owing to their large molar extinction coefficients, tunable emission, high fluorescence quantum yield, and photostability, conjugated polymer nanoparticles (CPNs) or polymer dots (Pdots, particle size <30 nm) are versatile tools for various applications.43,83,84 Because of their biocompatibility and inherent optical properties, CPNs are widely used in single photon,85–93 multiphoton,94,95 photoacoustic96,97 and super-resolution24 imaging applications. CPNs have been used to develop various ultrasensitive biosensors98–100 and they were successfully used for cell labelling,56,101 flow cytometry102 and theranostics applications.103–105 We recommend the readers several excellent reviews focused on design and applications of CPNs and Pdots.83,84,99,106–108 Below we analyse the brightness of typical examples of CPNs.In the seminal work, McNeill and co-workers prepared CPNs based on PFPV, polyfluorene benzothiadiazole (PFBT) and MEH-PPV, absorbing and emitting in the visible (Fig. 4).55 It was shown that the small size of NPs was crucial to achieve high QY values, therefore, the focus was made on small CPNs of 15 nm diameter. MEH-PPV emitting in the red region showed the weakest QY values (1%), but high extinction coefficient of particles of 1.2 × 108 M−1 cm−1. As a result, the brightness of these NPs according to eqn (5) was 1.2 × 106 M−1 cm−1 and per-volume brightness (BV) according to eqn (9) of 650 M−1 cm−1 nm−3 (Table 1). PFPV and PFBT operating in the green and orange region, respectively, showed higher QY around 7 and 8%, respectively. Their corresponding brightness reached values of 1.2 × 107 and 5.1 × 106 M−1 cm−1, with BV of 6500 and 2900 M−1 cm−1 nm−3. Later on, PFBT NPs were further improved by encapsulation into the block copolymer PS–PEG–COOH (Fig. 4).56 PFBT dots had small size of 10 nm and high extinction coefficient (1 × 107 M−1 cm−1) and QY of 30%. The achieved brightness was 3× 106 M−1 cm−1, while BV reached 5700 M−1 cm−1 nm−3 (Table 1). According to single-particle measurements by fluorescence microscopy, these NPs were 30-fold brighter than QD-565 (excitation at 488 nm), which positioned semiconductor polymer NPs as ultrabright nanomaterials.
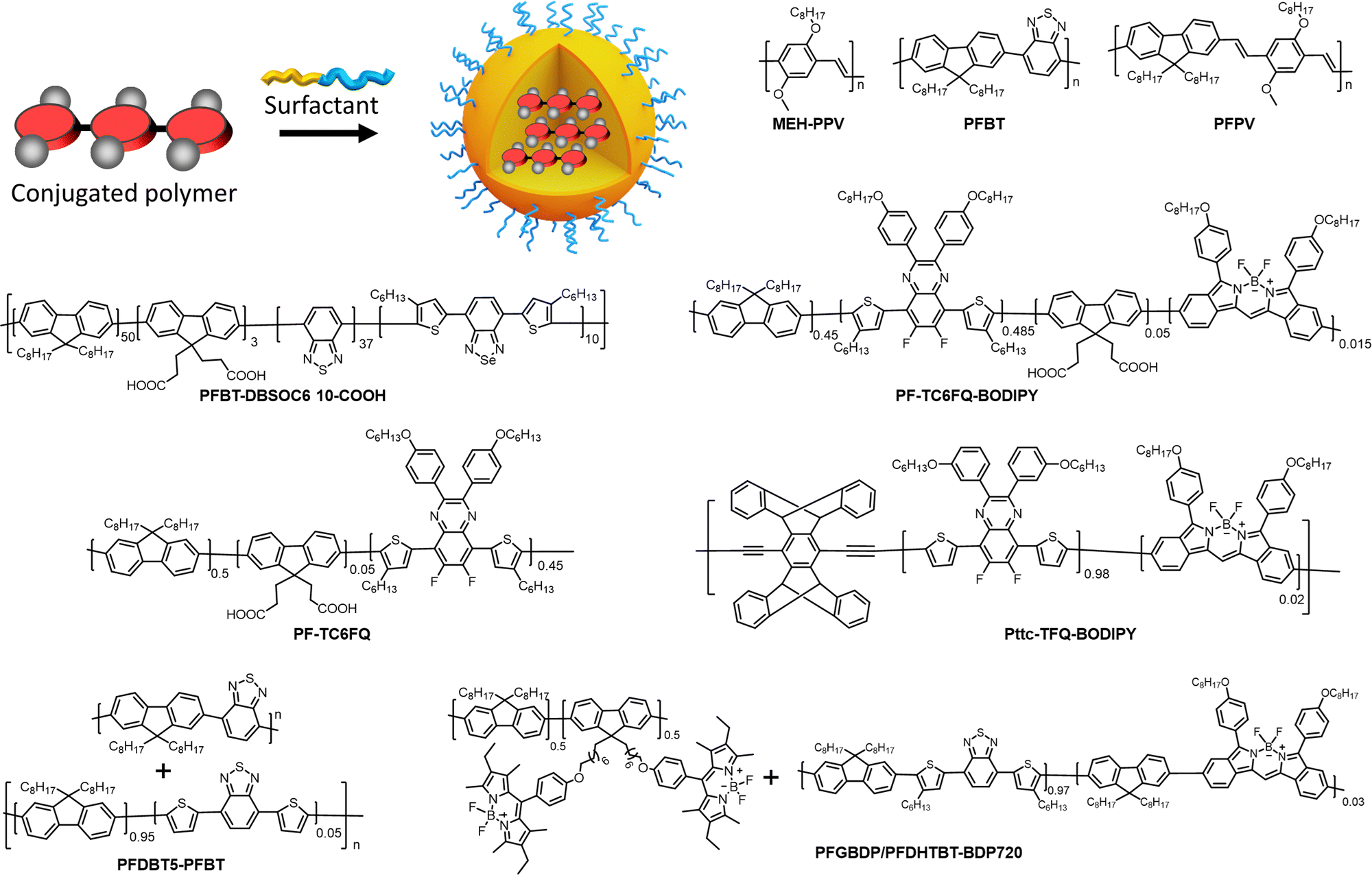 | ||
| Fig. 4 Fluorescent NPs based on conjugated polymers: preparation and their chemical structures. | ||
With efforts from various research groups, good quantum yields and brightness were achieved for CPNs that absorb and emit in the visible region.109,110 However, CPNs that emit in the far-red to NIR region suffer from poor quantum yield due to the ACQ of large pi-conjugated polymer units. Probes with far-red to NIR emission are very useful for bioimaging because of low light-scattering and auto-fluorescence of live tissues in the NIR region, allowing imaging at higher penetration depth.111 Therefore, there is a significant interest in developing bright CPNs that emit in far-red to NIR window. Chen et al. developed dithienylbenzoselenadiazole (DBS) based NIR emitting donor–acceptor (D–A) type Pdots (PFBT-DBSOC6 10-COOH) (Fig. 4).57 DBS-based Pdots exhibited large extinction coefficient (7.4 × 107 M−1 cm−1). The long-alkyl chains present on thiophene units reduced the close packing of DBS monomers in the Pdots, which in turn helped to maintain reasonably high quantum yields (15%). The resulting 29 nm DBS based Pdots had a brightness of 1.1 × 107 M−1 cm−1 and BV of 1758 M−1 cm−1 nm−3 (Table 1). These Pdots were 2 times brighter than Qdot 705 (excitation at 488 nm).
A promising direction in designing of NIR CPNs is to incorporate a powerful fluorophore BODIPY directly into the conjugation backbone, as exemplified by PF-TC6FQ-BODIPY (Fig. 4). The BODIPY unit serves as energy acceptor used at low molar ratio with respect to energy donor quinoxaline units, which allows to shift emission to the red and decrease self-quenching phenomenon for the donor units. The obtained 26 nm CPNs showed far-red to NIR emission (723 nm) with good QY (33%), with brightness of 1.7 × 107 M−1 cm−1 and BV of 1950−1 cm−1 nm−3 (Table 1).58,59 In order to further address the ACQ in NIR fluorescent Pdots, Chen and co-workers developed BODIPY-containing donor–acceptor conjugated polymers bearing AIE-active tetraphenylethene (TPE) unit and a bulky pentiptycene (pttc) unit (Pttc-TFQ-BODIPY). A control polymer containing polyfluorene donors conjugated to BODIPY based acceptor was used for comparison.60 The resulting 28 nm sized Pdots showed absorption in the visible region (490 nm) with remarkably high extinction coefficients (1 × 108 M−1 cm−1) and emission in the NIR window (724 nm). Modification in the polymer backbone with bulky substituents significantly increased the QY of the resulting Pdots. Pdots derived from bulky Pttc-based polymer and tetraphenyl ethylene (TPE)-based polymer exhibited high QY of 51% and 37%, respectively, compared to those from the control polymer (7%). The BV value of Pttc-TFQ-BODIPY Pdots was 4481 M−1 cm−1 nm−3 (Table 1), whereas single-molecule microscopy suggested that they were 5 times brighter than QD 705 when excited at 473 nm. The same group reported quinoxaline based semiconducting polymer dots PF-TC6FQ (Fig. 4).61 These 21 nm sized NPs showed far-red emission and high QY of 46% and BV of 4961 M−1 cm−1 nm−3, whereas they were 8 times brighter than Qdot 655 (excitation at 488 nm).
Chiu and co-workers developed semiconducting polymer blend dots (PBdots) for in vivo tumor targeting. D–A type PBdots, PFDBT5–PFBT were prepared by PFBT polymer donor and deep red-emitting PFDBT5 polymer as acceptor (Fig. 4).42 The resulting 15 nm PBdots showed absorption (450 nm) and emission (650 nm) in the visible to far-red region. PBdots had large molar extinction coefficient (ε = 3.7 × 107 M−1 cm−1) and excellent quantum yield (QY = 56%), which is so far one of the highest QY reported for CPNs. These PBdots exhibited an average BV of 9500 M−1 cm−1 nm−3. Single-particle imaging suggested that PBdots were 15 times brighter than Qdot 655 (excitation at 488 nm). Later on, Wu and Chiu groups reported ultrabright, narrow band, NIR emissive blend Pdots, PFGBDP/PFDHTBT-BDP720.62 These Pdots were prepared from D1/D2-A type conjugated polymers. D1-Polymer was constructed by grafting green emitting BODIPY to a polyfluorene (PFO) backbone and D2-A was constructed by introducing BODIPY720 acceptor into PFDHTBT conjugated polymer (Fig. 4). The resulting 31 nm Pdots showed absorption in the visible region (528 nm) and NIR emission (721 nm). These Pdots exhibited extremely high extinction coefficient of 4.2 × 108 M−1 cm−1. Quantum yield of such Pdots was 40%, so that brightness and BV reached very high values: 1.8 × 108 M−1 cm−1 and 11308 M−1 cm−1 nm−3, respectively (Table 1), the latter being the highest per-volume brightness reported for CPNs. Single-particle brightness measurements showed that Pdots PFGBDP/PFDHTBT-BDP720 were 83 times brighter than Qdot 705 when excited at 532 nm.
Overall, analysis of the brightness of CPNs from various literature reports suggest that along with fine tuning of the particle size, careful molecular engineering of the polymers is particularly important to achieve high brightness. On the one hand, optimum conjugation is necessary to obtain higher extinction coefficients. On the other hand, bulky non-planar substituents on the polymer chain are necessary to improve the quantum yield by preventing ACQ. Donor–acceptor type conjugated polymers with an optimized acceptor ratio is so far the most successful design to obtain bright longer wavelength emissive CPNs. An emerging direction is reaching NIR-II region with CPNs, which can further boost tissue penetration depth,105,112 however, brightness of these systems still needs improvement. An important tendency is that for similar polymers, CPNs of smaller size showed higher QY values and thus higher per-volume brightness (Table 1).55 Larger sizes probably increase the chances of the fluorescence quenching by a fraction of the polymer present in the dark state within NPs. Therefore, design of bright CPNs operating in red to NIR regions will require both optimal small size and careful molecular engineering, which includes strong conjugation, bulky side groups, and eventually the use of donor–acceptor configuration.
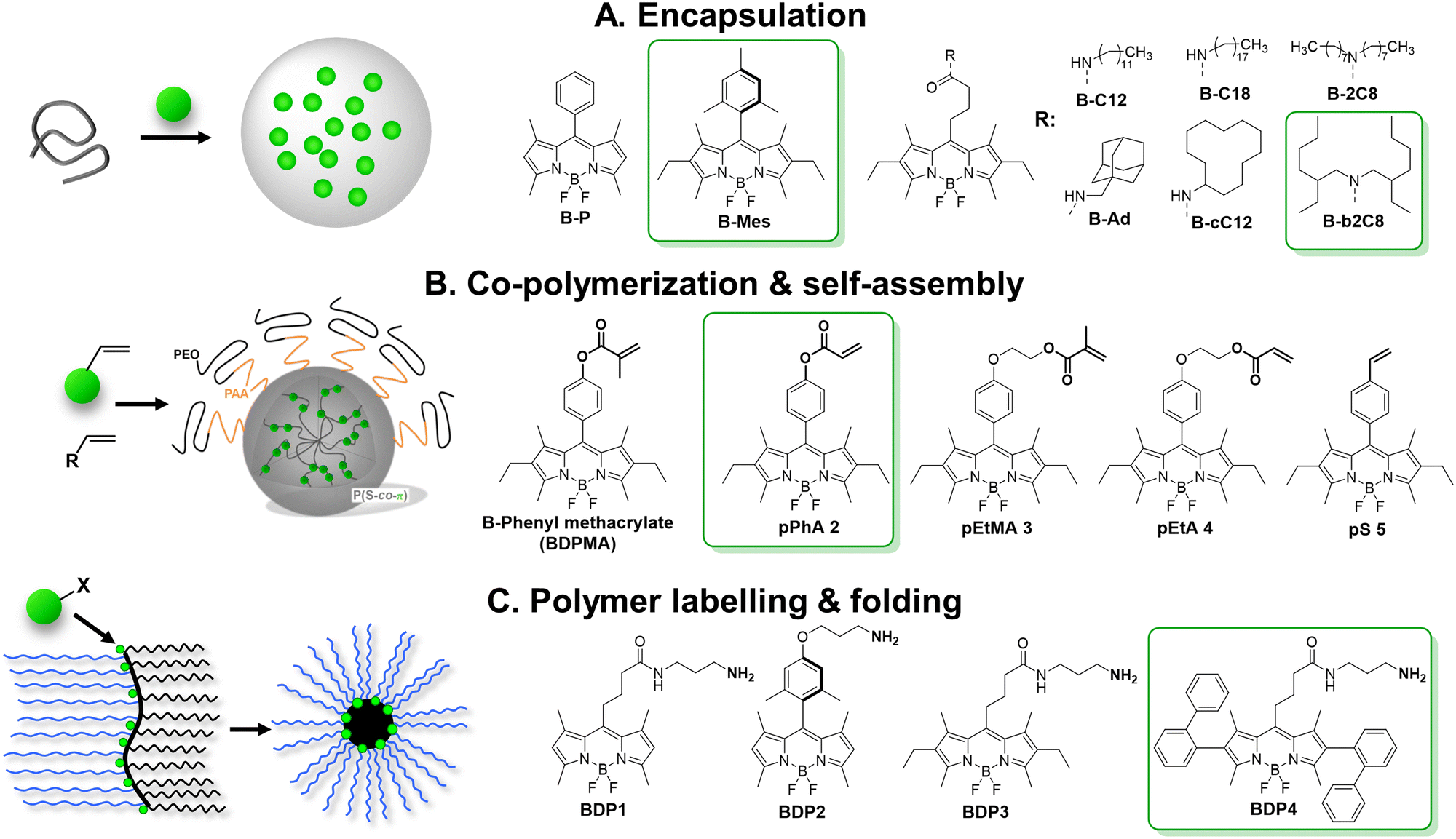
4.2. Aggregation-induced emission nanoparticles
The AIE concept is particularly suitable to prevent ACQ in nanomaterials, because the dyes are specifically designed to be highly emissive in the solid state. Since the first report on AIE materials by Tang et al.,44 tremendous research efforts were done to develop bright fluorescent (nano)materials with varied optical properties and broad range of applications, such as organic light emitting diodes (OLEDs),113,114 sensing,115,116 single photon,117 multiphoton,65,118 NIR-II bioimaging,68,119,120 image-guided surgery67,121 and phototherapy.122–125 There are several excellent reviews on the mechanism and applications of this remarkable phenomenon.126–129 In this tutorial review, we only highlight the strategies focused on improving the brightness of AIE NPs of different colour.In an early study, Tang and Liu groups reported red emitting NPs (AIE dots) based on dicyano-substituted stilbene bearing tetraphenylethylene (TPE) and triphenylamino (TPA) units (TPETPAFN, Fig. 5), which were coated with the lipid–PEG conjugate DSPE–PEG and functionalised with a cell penetrating peptide.63 The obtained NPs displayed an average size of 33 nm, quantum yield of 25%63 and extinction coefficient of 4.2 × 107 M−1. Having brightness of 1.1 × 107 M−1 cm−1 (BV = 560 cm−1 nm−3, Table 1), these AIE dots were 10 times brighter than QD 655 in single-particle measurements (488 nm excitation). The same teams also developed AIE dots based on 4,7-bis[4-(1,2,2-triphenylvinyl)phenyl]benzo-2,1,3-thiadiazole (BTPEBT, Fig. 5) encapsulated in a DSPE–PEG2000 shell.64 The resulting 30 nm sized AIE-dots showed absorption with large molar extinction coefficient (5.9 × 107 M−1 cm−1) and green emission high quantum yield (QY = 63%). Their brightness reached 3.5 × 107 M−1 cm−1 and BV was 1980 M−1 cm−1 nm−3. AIE dots based on BTPEBT coated with DSPE–PEG-coating (BTPEBT-V2, Table 1) were also developed for two-photon imaging application65 with slightly smaller particle size (29 nm) and improved BV (2870 M−1 cm−1 nm−3).
 | ||
| Fig. 5 Scheme of preparation of AIE NPs and chemical structure of AIEgens. | ||
Liu and co-workers further developed AIE dots based on BTPEBD (4,7-bis[4-(1,2,2-triphenylvinyl)phenyl]benzoxadiazole),66 where benzothiadiazole of BTPEBT was replaced with a benzoxadiazole unit (Fig. 5). The orange emissive BTPEBD-AIE nanoparticles had an average size of 32 nm and exceptionally high QY of 90%. BTPEBD AIE dots showed high brightness 5.3 × 107 M−1 cm−1 and their BV reached up to 3100 M−1 cm−1 nm−3 (Table 1).
Tang and co-workers recently reported the AIEgen PTZ–BT–TPA presenting both planar and twisted units based on phenathiazine (PTZ), benzothiadiazole (BT) and triphenylamine (TPA).67 The resulting PTZ–BT–TPA dye had high extinction coefficient (6.24 × 104 M−1 cm−1) and AIE properties. The NPs prepared from PTZ–BT–TPA encapsulated in DSPE–PEG2000 had an average diameter of 100 nm and QY of 39%. PTZ–BT–TPA particles had very high brightness of 2.4 × 109 M−1 cm−1 and their BV reached as high as 4600 M−1 cm−1 nm−3 (Table 1), which is one of the highest values reported for AIE NPs to date. Meng and co-workers reported D–A–D type AIE dots that emit in the NIR-II window (900–1700 nm), attractive due to higher penetration depth for in vivo imaging applications.68 The dye was designed by coupling triphenylamine (TPA) donor units on either side of the thiadiazolobenzotriazole (TBZ) acceptor core (Fig. 5). The 50 nm AIE dots prepared from DTPA–TBZ displayed long-wavelength absorption (652 nm) and NIR-II emission (929 nm). DTPA–TBZ AIE dots had a quantum yield of 11% and brightness of 4.0 × 107 M−1 cm−1. Even though their BV was lower (614 M−1 cm−1 nm−3) compared to other AIE systems, it is significant considering emission in the NIR-II window.
Overall, the AIE concept enables preparation of bright NPs with high quantum yields and high dye concentration, because the NPs core is generally composed of pure dye. Nevertheless, their brightness is currently limited by molar extinction coefficients of AIEgens, which are smaller compared to conventional dyes. On the one hand, the twisted molecular structures of AIEgens help them to achieve good quantum yield by reducing ACQ, on the other hand, their twisted non-planar architecture decreases the pi-conjugation and thus the molar extinction coefficients. Thus, it will be important to find a balance between the conjugation and non-planar structure to improve both the quantum yield and extinction coefficient of AIE nanomaterials. Recent report by Tang co-workers on PTZ–BT–TPA AIEgen67 showed a path forward to achieve this balance. Moreover, the future efforts will be devoted to the development of bright AIEgens in NIR-I and NIR-II windows.
4.3. Nanomaterials derived from conventional organic dyes
The opportunity to assemble nanomaterials from typical fluorescent dyes looks particularly attractive. Indeed, a great variety of fluorescent dyes with desired optical properties, e.g. BODIPYs, perylene diimides (PDIs), rhodamines, cyanines, etc., are available and chemically assessable. Moreover, they exhibit high molar extinction coefficients and fluorescence QYs, which makes them bright single-molecule emitters with BV values reaching 105 M−1 cm−1 nm−3 for rhodamine 6G (Table 1). However, highly-emissive nanomaterials cannot be obtained straight from conventional fluorescent dyes, because of the above-mentioned phenomenon of ACQ. Indeed, at high loading within a particle matrix (above 1 wt%) most of these dyes undergo clusterization and strong self-quenching. Moreover, non-covalently dye-loaded NPs tend to release significant amounts of encapsulated dyes leading to pronounced fluorescence background. Therefore, preparation of bright NPs from conventional dyes requires addressing simultaneously the problems of ACQ and dye leakage. Conceptually, methods to prevent ACQ in solid state are different for neutral and charged dyes, therefore hereafter they are presented separately.To prevent ACQ in neutral dyes, the most common approach is to introduce bulky side groups.46–48 In contrast to AIE approach, it exploits “classical” flat dyes, and their pi-stacking is prevented by introduced out-of-plane bulky groups.46,47,130,131 Importantly, these bulky side groups also decrease the tendency of dyes to crystallize and increase their hydrophobicity, both favouring preparation of NPs. This method was effectively applied in case two dye families: BODIPYs and PDIs, which are presented below.
BODIPY are bright organic dyes typically with high QY close to 1.0 and extinction coefficient above 70000 cm−1 M−1. However, their flat chromophore core favours ACQ in solid state. In some early works, it was shown that sterically hindered bulky substituents introduced to BODIPY core prevented ACQ and yielded materials emissive in the solid-state.132,133 The classical example of sterically hindered dye is Mes-BODIPY, which was encapsulated into cross-linked polystyrene NPs (16 nm) by swelling in dichloromethane followed by evaporation.71 At 76 dyes loaded per particle, the QY remained high (77%) and achieved brightness of NPs was 5.1 × 106 M−1 cm−1 with BV of 2300 M−1 cm−1 nm−3. We compared Mes-BODIPY with a series of hydrophobic BODIPY derivatives (Fig. 6A) after their encapsulation into PLGA nanoparticles, prepared by nanoprecipitation.75 BODIPY with meso-alkyl substituents were systematically less resistant to ACQ (QY = 10–20% vs. 50% for Meso-BODIPY at 50 mM loading), probably due to their planar structure. On the other hand, their most hydrophobic analogues were stable against dye leakage from NPs in aqueous media with foetal bovine serum or being internalized into live cell, probably because they were better encapsulated inside the hydrophobic core of NPs.
 | ||
| Fig. 6 Schemes of preparation of fluorescent NPs based on BODIPY derivatives. Reproduced with permission from ref. 72 and 73. Copyright American Chemical Society. | ||
A fruitful strategy to control the dye ACQ and encapsulation is to covalently graft dyes to the polymer backbone. Clavier with co-workers synthesized a reactive analogue of sterically hindered BODIPY and co-polymerized them in miniemulsion to yield NPs (Fig. 6B).72 The synthesis relied on polymerization-induced self-assembly (PISA) – folding of growing chains of amphiphilic PEGylated polyacrylate–polystyrene block copolymer forming core–shell nanoparticles. All five BODIPY monomers demonstrated excellent inclusion rate (from 0.92 to 0.98) targeting 3 dyes per a chain (15–20 kDa, ∼8 wt% dye loading). Up to 5000 BODIPY acrylates per particle were loaded for 60–90 nm size of NPs, while reserving relatively high QY values (35%). By theoretical calculation they were 200–2000 brighter NPs than quantum dots (1.4 × 108 M−1 cm−1) with BV of 470 M−1 cm−1 nm−3. Later on, the same group showed that brightness could be further increased by inclusion of higher dye content, but QY gradually decreased presumably due to ACQ.134
We followed a quite different strategy, where sterically hindered BODIPY dyes were covalently attached to an amphiphilic polymer capable to form single-polymer NPs of small size (Fig. 6C).73 In this case, poly(maleic anhydride-alt-1-octadecene) (PMAO, 30 kDa) was modified in two steps with BODIPY dye and PEG groups. In water this polymer folded into small single-chain NPs, bearing the dye in the hydrophobic core and PEG groups on the surface. Among tested BODIPYs, BDP4 bearing two diphenyl substituents was the most resistant to ACQ: no hypochromism or absorbance band broadening or significant H-aggregate band appeared upon increasing dye content from 2 to 50 mol%, compared to other dyes (BDP1–3). BDP4–PEG(1000)–PMAO NPs showed QY of 60% at 50 mol% dye content (i.e. 18 wt%) and high 2.5 × 106 M−1 cm−1 brightness for monomolecular nanoparticles of 14 nm in diameter, with BV of 1740 M−1 cm−1 nm−3 (Table 1).73 Single-particle measurements showed that these NPs were 5-fold brighter than QD-585 (at 532 nm excitation).
Polyaromatic imides are a unique class of organic emitters, characterized by high brightness and phenomenal photostability. On the other hand, their poor solubility and tendency to aggregation due to strong pi–pi stacking makes them particularly prone to ACQ. For example, perylendiimide (PDI) forms non-emissive (H-aggregate) and emissive red-shifted (J-aggregate or excimer) species in solid state depending on substituents in the bay and imide sides (Fig. 7), as summarized by Würthner and co-workers in a recent review.135 However, the vast majority of the examples deals with bulk materials without formulation of NPs. Wong and co-authors designed PDI insulated with branched aromatic substituents at imide positions.46 Compound bPDI4 bearing bulkiest groups (Fig. 7), exhibited QY of 29% and BV of 8700 M−1 cm−1 nm−3 in solid state, whereas by inclusion into PMMA thick film at a relatively high (120 mM) concentration, QY raised up to 70%, but the BV value decreased twice (3700 M−1 cm−1 nm−3). Würthner and co-workers reported PBI9d with highly sterically hindered substituent, which gave QY of 17% and BV of 8300 M−1 cm−1 nm−3.136 Even more efficient is shielding of PDI dye with bulky substituents at the bay region. For example, PDI(Ph) with four ortho-phenyl-phenoxy groups exhibited highest QY in crystals (59%) with dye extinction coefficient of 42300 M−1 cm−1 and BV of ∼18000 M−1 cm−1 nm−3 (calculated with parameters of X-ray crystal structure).137 Even higher brightness could be achieved with N,N′-dicyclohexylperylene diimide sterically fully enwrapped with two 2,4,6-tris(4-tert-butylphenyl)phenoxy groups at bay positions (PBI3c, Fig. 7).47 Quantum yield of PBI-3c in monocrystals reaches 84% along with BV of almost 30000 M−1 cm−1 nm−3. However, the possibility to transform these ultra-bright bulk materials into bright fluorescent NPs has not been demonstrated do date.
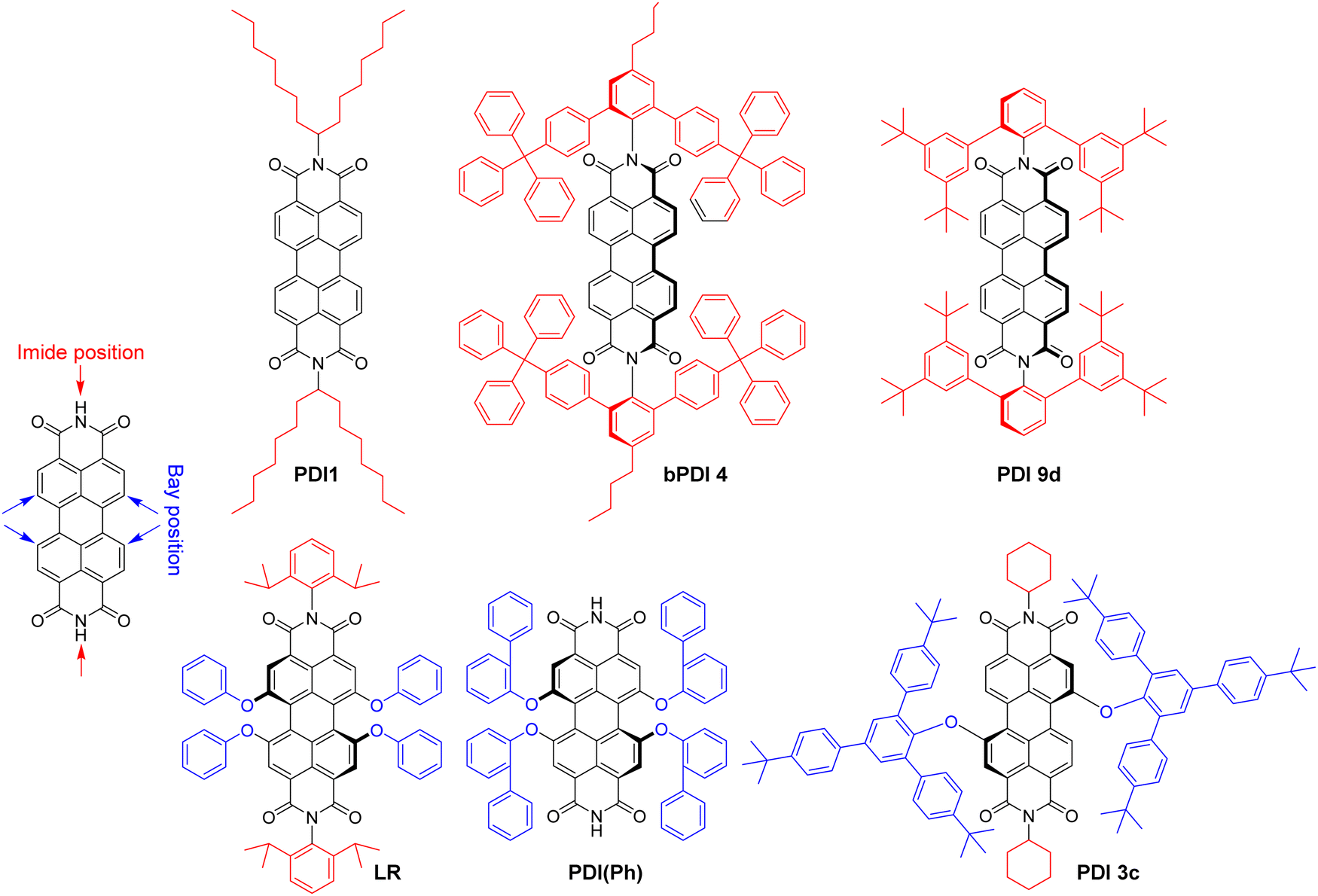 | ||
| Fig. 7 Molecular structure of perylene diimides (PDI) with bulky substituents. | ||
The examples of incorporation of PDI dyes into nanoparticles are very rare, and limited to derivatives with much smaller side groups. In an early study, Li and co-workers incorporated PDI derivative bearing chlorines at the bay regions (PDI-Cl) into polymer chains by co-polymerization.74 40 nm cross-linked polymer NPs were obtained with PDI-Cl dye loading of 2.4 wt% and QY value of 50%. The achieved brightness and BV values were 1.0 × 107 M−1 cm−1 and 310 M−1 cm−1 nm−3, respectively. Single-particle measurements revealed brightness equivalent to 50–220 PDI dye monomers. We studied the role of bulky substituent in the bay and imide regions for the formulation of fluorescent polymeric nanoparticles based on biodegradable polymer PLGA. We selected PDI-1 bearing branched alkyl groups in the imide part and Lumogen Red (LR, Fig. 7), substituted with bulky groups at the bay region, and encapsulated them into PLGA NPs by co-precipitation.75 PDI-1 showed a tendency to aggregation that increased from 0.02 to 1 wt% dye loading, observed as a broadening in the absorbance spectra and a rise in the red shifted excimer band in the emission spectra, whereas QY decreased from 67 to 31%. In contrast, LR showed better resistance to ACQ, displaying exclusively green emission of the molecular form up to 5 wt% loading and decrease in QY from 97% (0.02 wt%) to 47% (5 wt%). At 5 wt% LR loading, 38 nm NPs showed a brightness of 7.5 × 106 M−1 cm−1 with BV of 261 M−1 cm−1 cm−3, whereas in single-particle microscopy experiments they were 18-fold brighter than quantum dots emitting in the same range (QD-585 at 532 nm excitation).
Overall, neutral fluorescent dyes such as BODIPYs and PDIs, are bright mono-molecular emitters but tend to strong ACQ in solid state. This could be partially resolved by introduction of bulky and branched substituents, leading to a brighter fluorescence at high local dye concentration. Therefore, PDI derivatives reach record-breaking brightness per volume values in form of bulk solid materials. However, owing to their large flat pi-conjugated structure, they tend to crystallize, which makes it challenging to prepare them in form of NPs, either as pure dye NPs or encapsulated into polymer or lipid NPs. Nonetheless, appropriate functionalization of bulky and/or reactive groups allows fabrication of NPs of relatively high brightness. Further efforts should be directed to design of neutral dyes that keep the balance between the bulkiness of side groups and capacity to be encapsulated into organic NPs, while preserving their high brightness and other spectral characteristics in NPs.
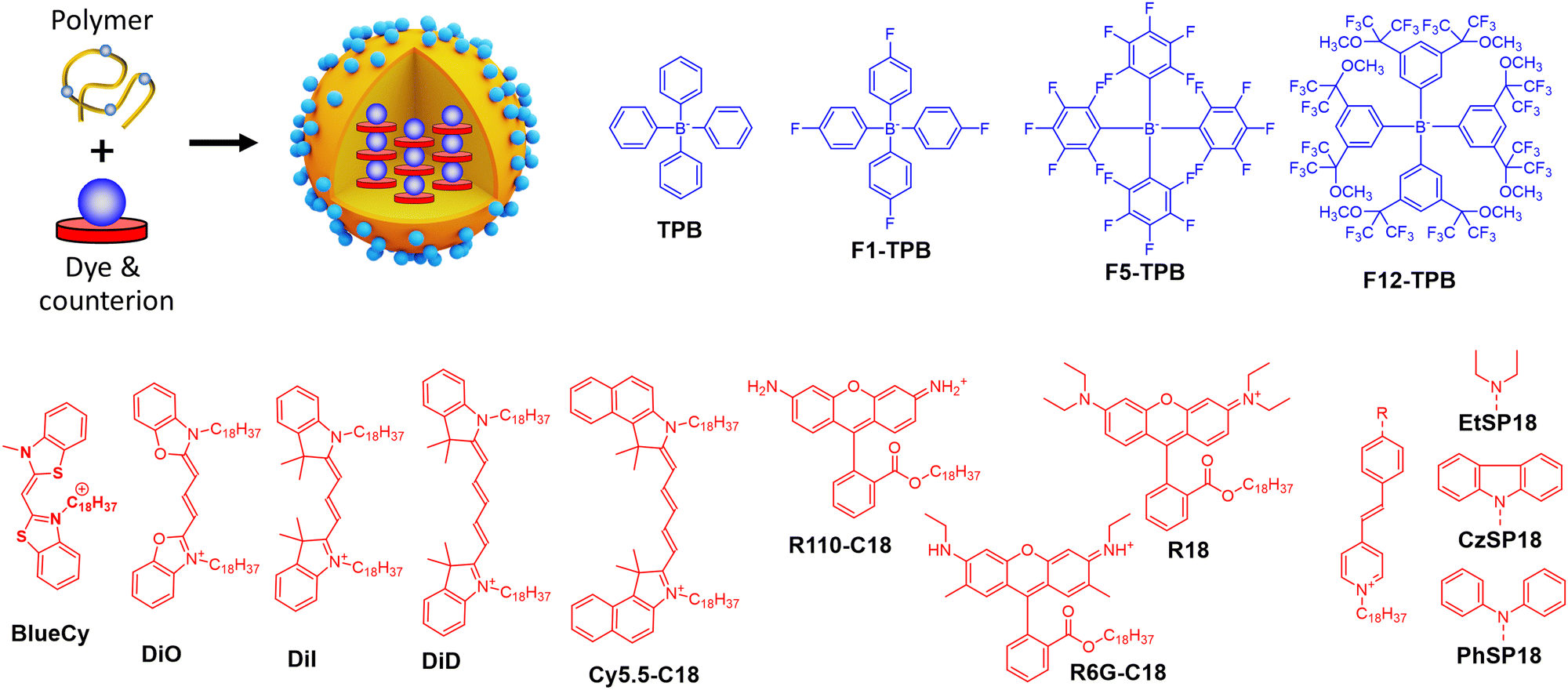 | ||
| Fig. 8 Dye-loaded polymeric NPs and examples of bulky counterions paired with cationic dyes for the NPs preparation. | ||
Insulation of positively-charged fluorophores could be efficiently performed with organic counterions. Earlier works of Yao et al.51 and Warner and co-workers52 demonstrated that bulky hydrophobic anions decrease ACQ in pure ion pairs in solid state form. Previously, we described preparation of fluorescent organic NPs from pure dye salts of alkyl esters of rhodamine B with different tetraphenylborate counterions (TPB, F1-TPB, F5-TPB, F12-TPB, Fig. 8) by nanoprecipitation.76 The increase in the alkyl ester chain length as well as higher fluorination level and bulkiness of the tetraphenylborate counterions significantly improved fluorescence quantum yield and stability of the obtained NPs. Thus, O-dodecyl rhodamine B (R12) with F5-TPB and F12-TPB NPs gave 14 and 19 nm NPs, respectively, with quantum yields of 40 and 60% respectively. Theoretical calculation based on eqn (5) gave brightness (B) values of 2.7 × 107 M−1 cm−1 and 6.8 × 107 M−1 cm−1 for R12/F5-TPB and R12/F12-TPB NPs, respectively, while eqn (11) gave impressive BV values: 18700 and 19100 M−1 cm−1 nm−3, respectively (Table 1). According to FCS measurements, the experimental single-particle brightness of R12/F12-TPB NPs reached 2.4 × 107 M−1 cm−1, which was equivalent to 540 single rhodamines or 45 QDs-585. However, they were not sufficiently stable in cells undergoing dye release, even though NPs with more fluorinated counterions were more stable.
To formulate stable dye-loaded NPs, we proposed to pair cationic dyes with bulky hydrophobic counterions and encapsulate them into the matrix of polymeric NPs by nanoprecipitation. In our first report, octadecyl rhodamine B (R18) paired with F5-TPB was encapsulated at 5 wt% (vs. polymer) into 40 nm PLGA NPs (Fig. 8). Owing to a QY of 21%, their estimated brightness reached 1.8 × 107 M−1 cm−1 with relatively low BV values of 600 M−1 cm−1 nm−3 (Table 1).33 Single-particle microscopy suggested that the single-particle brightness corresponded to 6 QDs-585 at 532 nm excitation and revealed neatly complete ON/OFF particle blinking, caused by fast dye–dye energy transfer.33 The same dye was then encapsulated at 5 wt% into much smaller (15 nm) NPs built of sulfonated PMMA derivative, yielding NPs with improved BV of 1600 M−1 cm−1 nm−3,77 due to a high fluorescence quantum yield (60%) in PMMA-based matrix. They were 10-fold brighter than QD-585 at 532 nm excitation. Further studies showed that more hydrophobic PMMA matrix ensured systematically higher quantum yields compared to PLGA, which could be explained by more even distribution of the dye without clustering inside the particle core. In this study, for 30 wt% loading vs. polymer (23 wt% of total NP mass) the brightness values reached 8.9 × 107 M−1 cm−1 for 34 nm PMMA-MA NPs, which means the BV values of 4730 M−1 cm−1 nm−3 (Table 1).24 According to single-particle microscopy, PMMA-MA NPs reached brightness equivalent to 100 QDs-585 (at 532 nm excitation), while their blinking was largely supressed compared to PLGA NPs. Moreover, 40 nm PMMA-based NPs loaded with 30 wt% of R18/F5-TPB vs. polymer (23 wt% of total NP mass) were decorated with DNA, yielding DNA nanoprobes with 46% QY, corresponding to a brightness of 1.9 × 108 M−1 cm−1 and BV of 5790 M−1 cm−1 nm−3 (Table 1).35 Single-particle measurements showed that these NPs were equivalent to 100 QD-605 excited at 488 nm. Next, replacing the PMMA matrix with the more hydrophobic PEMA enabled encapsulation of 50 wt% (33 wt% of total NP mass) of the dye into a smaller NPs’ core of 20 nm functionalized with DNA.36 These NPs displayed even higher QY of 52% than PMMA-MA NPs and a brightness of 3.8 × 107 M−1 cm−1, whereas the BV value reached 9480 M−1 cm−1 nm−3 (Table 1). Single-molecule microscopy showed that these 20 nm NPs were 87-fold brighter than QDs-605 measured at 550 nm excitation and they showed more stable emission (lower NP blinking) than their PMMA-based analogues. Remarkably, high fluorophore content in polymeric NPs enables highly efficient FRET to few acceptor fluorophores on the surface of NPs, leading also to increase in the overall QY.35,36
Thereafter, the concept of bulky tetraphenylborate counterion gained versatility towards different classes of fluorophores. Other hydrophobic rhodamine and cyanine dyes demonstrated compatibility with tetraphenylborates for encapsulation into a polymer matrix in order to broaden spectral operating range of fluorescent NPs (Fig. 8 and Table 1). For example, BlueCy/F5-TPB emitting in blue spectral range at 12 wt% in 40 nm PMMA-MA NPs enhanced drastically QY up to 17% compared to 0.1–0.3% in solution.78 Brightness of the NPs was ∼70-fold higher than for QD-525 at 470 nm excitation, with brightness of 2.3 × 107 M−1 cm−1 and BV of 690 M−1 cm−1 nm−3 (Table 1). Octadecyl-substituted analogue of rhodamine 6G was mostly efficient with F12-TPB counterion in 44 nm PMMA NPs at 250 mM (vs. polymer) loading with QY of 23%, brightness of 1.1 × 108 M−1 cm−1 and BV of 2490 M−1 cm−1 nm−3.79 F12-TPB was also found to be particularly efficient to prevent ACQ of cyanine dyes in NPs based on PLGA138 and PMMA54 polymers. For example, cyanine 5 dye DiD loaded at 30 wt% (23% of total NP mass) into 16 nm PEMA-based NPs showed QY of 42%, which corresponded to a particle brightness of 9.7 × 106 M−1 cm−1 and BV of 4530 M−1 cm−1 nm−3.54 Single-particle measurements showed that these small NPs were 22-fold brighter than QD-705 at 640 nm excitation. Bulky counterions can also enhance QY of cationic AIE dyes.139 Previously, we studied a styryl pyridinium dye family: EtSP18, CzSP18, and PhSP18 (Fig. 8), which presented or not AIE properties.80 Among them EtSP18, which was not emissive in solution or solid state, lighted up only in the presence of bulky hydrophobic counterions. We named this phenomenon “ionic AIE”, due to the key role of bulky counterions that light up non-emissive dyes in the solid state. Inside polymeric NPs these ion pairs exhibited efficient fluorescence: PhSP18/F5-TPB encapsulated in 40 nm PMMA NPs at 40 wt% dye loading (29% of total NP mass) showed 40% QY and fluorescence brightness of 1.2 × 108 M−1 cm−1 with BV value of 3840 M−1 cm−1 nm−3, which was equivalent to 50 QD-605 at 488 nm excitation in the single-particle microscopy measurements.80
Ionic dye insulation with bulky counterions was also found applicable to lipid nanoemulsions. Dioctadecyl Cy3 dye – DiI with TPB counterion was loaded into labrafac nanoemulsion up to 8 wt%.81 The increase in the dye loading from 0.1 to 8 wt% vs. oil core, decreased quantum yield by only 3.5-fold to 14%, which led to 87 nm nanoemulsion droplets encapsulating ≈12000 dyes per droplet. This value corresponded to a brightness of 2.5 × 108 M−1 cm−1, close to that obtained experimentally (8.0 × 107 M−1 cm−1) by single-particle microscopy with reference particles (FluoSpheres) of known brightness. However, the BV value of these lipid NPs (725 M−1 cm−1 nm−3) was significantly lower compared to the polymer NPs, which can be explained by both lower QY and lower dye loading.
Overall, ionic dye insulation with bulky hydrophobic counterions appears as a universal concept to prevent ACQ in charged dyes of different nature. Counterions play here multiple roles: (i) preventing dyes from H-aggregation, related to pi-stacking; (ii) enhance dye encapsulation into polymeric and lipid NPs because of high hydrophobicity of the obtained ion pair; and (iii) providing highly rigid surrounding for a dye.
Recently, the teams of Laursen and Flood presented an analogous concept called SMILES (i.e. small-molecule ionic isolation lattices), which used a supramolecular counterion complex built of the small inorganic anions (BF4−, PF6− and ClO4−) with cyanostar – a planar conjugated macrocycle (Fig. 9A).141 Two cyanostars form a cage complex with one anion such as ClO4−, PF6−, BF4−. At the same time, this planar counterion complex alternates with dyes in stacked columns in crystal structure with mean dye–dye distance around 15 Å. This splitting distance prevents to some extent ACQ through the H-aggregation. This approach was successfully applied to a variety of cationic dyes, including triangulenium dyes (Fig. 9B), rhodamines and cyanines in form of bulk materials, such as films and crystals. The highest BV values were obtained with rhodamine 3B perchlorate and cyanine 3 hexafluorophosphate dyes 9700 M−1 cm−1 nm−3 (QY 29%) and 11800 M−1 cm−1 nm−3 (QY 25%), respectively. Later work showed that using energy transfer within mixed rhodamine R3B – cyanine DIOC2-based FRET SMILES, even higher quantum yields can be achieved (65%), reaching an impressive BV value of 32200 M−1 cm−1 nm−3 in bulk materials.140 The same groups used the SMILES concept with rhodamine derivative R12 in order to obtain small NPs of 16 nm size stabilized with DSPE–PEG lipid.82 The obtained NPs contained 400 dyes exhibiting QY of 30%, which corresponded to brightness of 1.5 × 107 M−1 cm−1 and BV of 5000 M−1 cm−1 nm−3 (Table 1). In the single-molecule microscopy, they were 20-fold brighter than the reference NPs 40 nm FluoSpheres®.
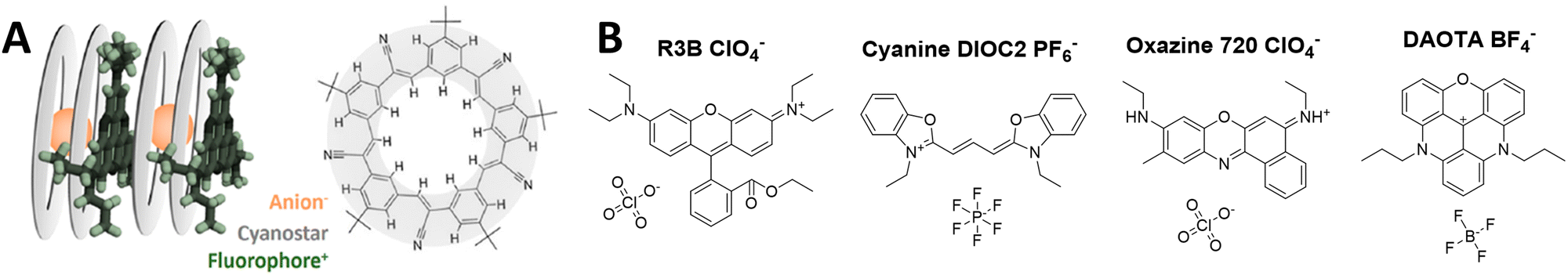 | ||
| Fig. 9 (A) Visual representation of SMILES concept and cyanostar microcyte and (B) fluorescent dyes insulated with anion-2-cyanostar complex. Panel A reproduced with permission from ref. 140. Copyright American Chemical Society. | ||
5. Brightness and biological applications
Owing to their unique optical properties, multifunctional surface chemistry and nanometric size, fluorescent NPs cover an immense range of biological applications.4 On the one hand, fluorescent organic NPs are advanced optical probes in biological media, cells and live animals, allowing high-contrast, high-resolution,5,22,142 and multimodal (e.g. photoacoustic) biological/biomedical imaging13,106 as well as theranostics applications involving phototherapy.8,12,124,129 On the other hand, they are powerful platforms for building sensors for small molecules and biomolecules36,83,103 as well as temperature and mechanical forces.19 Here, we will focus on some examples of biosensing and bioimaging applications where brightness of fluorescent organic NPs plays a particularly important role.5.1. Bioimaging applications
Brightness is essential to achieve high spatial and temporal resolution in fluorescence imaging. Single-particle tracking is probably the most demanding in terms of high brightness.142 It was originally developed for semiconductor QDs, for example, in tracking membrane receptors, which allowed tracking molecular diffusion with higher resolution and longer time compared to organic dye Cy3.143 Organic nanoparticles present attractive alternative to QDs for continuous single-particle tracking, because they can be much brighter and they generally do not blink (with some exceptions).21,144 McNeill and co-workers showed that high brightness of conjugated polymer NPs enabled fast tracking (50 Hz rate) with precision down to 1 nm and applied that to study complex diffusion behaviour in fixed cells (Fig. 10A).145 In a recent study using arginine modified CPNs, it was shown that particle entry inside the cells by endocytosis can be tracked at the single-particle level.146 Thus, Blanchard-Desce and co-workers showed that NPs assembled from push–pull dyes can be tracked with high precision inside live cells.147 Using single-particle tracking, we studied diffusion of dye-loaded polymeric NPs inside the cytosol as a function of their size. This study revealed critical size of NPs around 23 nm, below which NPs can diffuse freely in the cytosol (Fig. 10B).25 Further studies, in fixed and permeabilized cells suggested that small size (<20 nm) was also crucial for penetration of DNA-functionalized NPs inside the cells and detection of mRNA targets by fluorescence in situ hybridization (FISH), in line with earlier data on ultrasmall QDs.54 As the particle brightness decays as power of three of its diameter, a compromise should be made between small NPs size and the number of encapsulated dyes per particle. Based on the previous studies, the size around 10–20 nm could be considered as a good compromise. On the other hand, single-particle tracking in small animals requires much higher brightness, because of strong background and light scattering from the tissues. In the early studies, to track single NPs inside zebrafish embryo, we designed lipid NPs containing ∼10000 cyanine dyes with bulky counterion (brightness was 2.5 × 108 M−1 cm−1).81 One should note that in addition to high brightness, in vivo tracking requires compatibility with near-infrared imaging modalities, such as efficient two-photon absorption cross-section and/or near-infrared absorption/emission operating range. Thus, using polymeric matrix, we prepared ultrabright NPs of 74 nm dimeter by DLS and BV of 4280 M−1 cm−1 nm−3, which according to two-photon FCS were 150-fold brighter than commercial Nile Red-loaded FluoSpheres. They enabled single-particle tracking directly in brain of live mice by two-photon microscopy and detection of NPs crossing the brain blood barrier.148
 | ||
| Fig. 10 Application of bright organic NPs for bioimaging. (A) Tracking 15 nm PFBT Pdots in macrophage-like J774 cells. Left: Transmission image of a fixed cell. The colour marks indicate the locations of NPs: particle bound to the membrane (blue), outside the cell (green), and in the cell interior (red). Right: The trajectories for the three particles. Reproduced with permission from ref. 145. Copyright American Chemical Society (B) epi-fluorescence and phase-contrast images of HeLa cell microinjected with 32 and 17 nm dye-loaded PMMA-based NPs. Injection points are indicated by arrows. Scale bars, 10 μm. Insets show distributions of particle sizes obtained by TEM. NPs were loaded with 10 wt% of R18/F5-TPB and coated with Tween 80. Reproduced with permission from ref. 25. Copyright John Wiley and Sons. (C) Tracking transplanted neurons in vivo. D16 hESC-derived neurons were labelled with 30 nm TPETPAFN AIE-NPs for 24 h prior to transplantation into mouse brain striatum. Brain tissues were collected 24 h, 2 weeks, and 1 month post-transplantation. Scale bar: 100 μm, enlarged panel scale bar: 50 μm. Reproduced with permission from ref. 149. Copyright Elsevier. (D) Principle of cell barcoding by 40 nm dye-loaded polymeric NPs of three different colours: blue, green and red loaded with DiO/F12-TPB, DiI/F12-TPB and DiD/F12-TPB, respectively. (E) Tracking multiple RGB barcoded cell populations. The large micrograph shows a confocal image six cell types (HeLa, KB, 293T, U87, RBL, and CHO) mixed and co-cultured for 24 h. Each cell type was labelled with an RGB barcode (orange, cyan, green, red, magenta, and blue, respectively), also shown separately in the smaller images. Images are superpositions of the three NP channels with identical settings and of the membrane channel in grey. Scale bar is 100 μm. (F) Tracking RGB barcoded cancer cells in zebrafish embryo: six batches of D2A1 cells were labelled with fluorescent NPs generating RGB barcodes (green, red, blue, yellow, magenta, and cyan) and imaged 3 h post-injection. (9 D–F) – Reproduced with permission from ref. 138. Copyright John Wiley and Sons. (G) Top: Standard TIRF image of immobilized 40 nm dye-loaded NPs (PLGA, 5 wt% R18/F5-TPB); bottom: the same field after applying a super-localization procedure, showing capacity to resolve two particles (scale bar, 200 nm). Reproduced with permission from ref. 33. Copyright Springer Nature. (H) Dual-color superresolution (SOFI) imaging of subcellular structures labelled with small (10 and 13 nm) photoblinking Pdots. Left: Wide field imaging of clathrin coated pits labeled with PFO (green) Pdots and microtubule labelled with PFTBT5 (red) Pdots. Top right: Magnified region show in white box in left panel. Bottom right: SOFI image generated by analysing 500 frames of raw data from the wide-field image. Reproduced with permission from ref. 144. Copyright American Chemical Society. (I) STED imaging of the microtubule structures labeled using the AIE NPs (14–16 nm): confocal (left) and super-resolution STED (right) images of the microtubules. Reproduced with permission from ref. 150. Copyright John Wiley and Sons. | ||
Cell tracking with NPs is another important application, especially given the growing interest in cell-based therapies. High brightness of NPs is crucial here, because cells can endocytose limited number of NPs and their tracking in vivo would require the strongest possible signal. Liu and co-workers designed 32–33 nm AIE dots functionalized with cell penetrating peptides in order to label cells. Due to their high brightness, stability and cell internalization, AIEs dots operating in green (BTPEBT) and red (TPETPAFN) regions allowed simultaneous discrimination of different populations of cancer cells both in culture medium and in animal organs.63 The red 30 nm AIE Dots (TPETPAFN) bearing TAT peptide were successfully applied for long-term labelling of neurons and their tracking in mouse brain striatum in various time points post-transplantation.149 These bright AIE dots allowed tracking neuronal grafts for up to 1 month (Fig. 10C). Using dye-loaded polymeric NP of different colour, which can be efficiently endocytosed by cells, we made long-term barcoding of cells and further tracking in vitro and in vivo on zebrafish (Fig. 10D–F).138 High brightness of NPs was important for the long-term tracking (over up to two weeks), because after each cell division the number of NPs per cell (initially ∼10000) was divided by two. However, it still remains a challenge to extend the tracking time and achieve their tracking in small animals at sufficient depth.
High brightness of NPs is of particular interest for super-resolution imaging, because localization precision is intrinsically connected with the number of collected photons of the particle.142 However, this application is particularly challenging because NPs should be sufficiently small and present suitable optical properties. In particular, for single-molecule localization microscopy, it requires ON–OFF switching behaviour. Previously, we showed a phenomenon of collective blinking of >100 dyes within a dye-loaded polymeric particle, due to ultrafast dye-communication (Fig. 10G).33 Single-molecule localization microscopy (SMLM) imaging of single NPs revealed spots having width at half-maximum of 35 ± 7 nm, corresponding to their diameter, and possibility to resolve them at interparticle distances below the diffraction limit. Later on, Pdots were described based on PFO and PFTBT5, presenting small size (10 and 13 nm) and fluorescence blinking.144 They enabled two-colour SMLM (SOFI) imaging subcellular structures in cells with resolution down to 181 nm (Fig. 10H).144 Moreover, Wu, Sun and co-workers described photo-crosslinkable AIE NPs functionalized with streptavidin.150 The obtained NPs of three different colours, small size (14–16 nm) and high fluorescence quantum yields (24–39%) enabled STED-based super-resolution imaging of tubulin with resolution reaching 95 nm (Fig. 10I).
One should note that in this tutorial review, we focused only on a few examples of bioimaging applications of organic NPs, where brightness is particularly critical. One should mention other important applications, such as targeted imaging of tumours using NIR-I and NIR-II spectral regions151,152 as well as combination of bioimaging with photodynamic therapy,112,153 where CPNs and AIE NPs were particularly successful.
5.2. Biosensing applications
NP brightness is also crucial for biological detection. The brightness defines the number of NPs that can be detected above the background and, therefore, the sensitivity of the assay. Indeed, NPs with 1000-fold higher brightness can be detected at 1000-fold lower concentration, typically from nM to pM range,35 compared to molecular probes, detectable in the μM–nM range. However, to work as probes for analytes, the sensing mechanism should be implemented that couples a molecular recognition event with the fluorescence response of the particle. In this respect, one of the most universal approaches is Förster resonance energy transfer (FRET), because it is highly sensitive to distances at the molecular scale around Förster radius of ∼5 nm (Fig. 11A).154 However, the general problem of fluorescent NPs is their limited FRET efficiency to a single acceptor, because their size is generally larger than the Förster radius. For example, in the case of QDs, multiple acceptors per particle are needed to achieve efficient FRET in biosensors.155 In this respect, conjugated polymer NPs are of particular interest for preparation of bright nanoprobes with efficient FRET for amplified biological sensing.103 Indeed, they exhibit high brightness and outstanding capacity to transfer energy due to fast excitation energy migration.83 A more recent example of bright organic NPs capable to undergo efficient FRET are dye-loaded NPs that use bulky hydrophobic counterions.156 At high dye loading the counterion not only prevent ACQ, but also ensures proper dye–dye spacing for ultrafast energy migration towards the FRET acceptor (Fig. 11A). As a result ∼10000 dyes could efficiently transfer energy to a single dye acceptor, which generated giant antenna effect (signal amplification factor) of ∼1000, allowing detection of single molecules at ambient light-like conditions.156
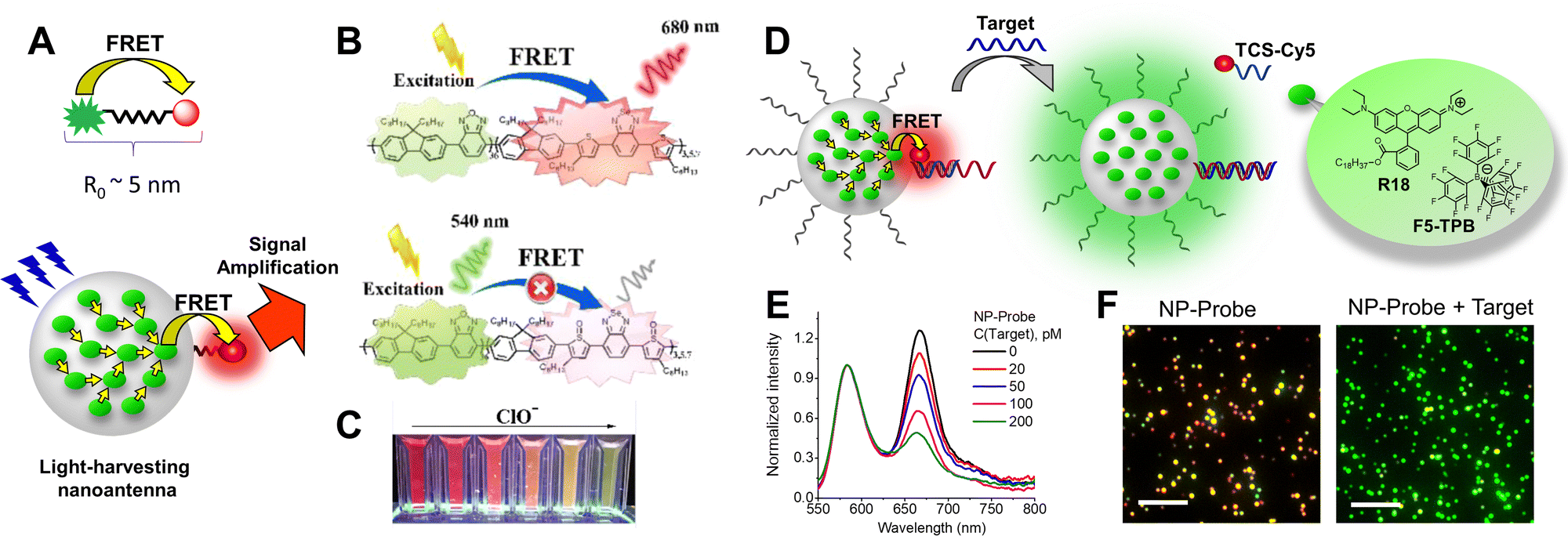 | ||
| Fig. 11 Applications of bright NPs for biosensing. (A) Top: FRET based molecular probe showing that the applications are limited to distances around the Förster radius. Bottom: FRET based nanoprobe where large number of donors are coupled due to excitation energy transfer (small yellow arrows) in order to ensure efficient transfer to a single acceptor. (B) Schematic Illustration of Pdot from donor–acceptor conjugated polymer. (C) Photographs of PFOBT36SeTBT5 Pdots with the addition of ClO− taken under normal laboratory lighting and illumination with a UV light at 365 nm. (B and C) – Reproduced with permission from ref. 158. Copyright American Chemical Society. (D) DNA-functionalized dye-loaded polymeric nanoparticle (NP-Probe) for FRET-based detection of nucleic acids by stand displacement principle. R18 and its bulky counterion F5-TPB are also shown. (E) Fluorescence spectral response of NP-probe to the growing concentrations of the nucleic acid target. (F) Colour response of surface-immobilized NP-probe to the nucleic acid target (100 pM) at the single-particle level. (D–F) – Reproduced with permission from ref. 35. Copyright American Chemical Society. | ||
The early examples of amplified sensing using light-harvesting and effective FRET in organic NPs were reported by Chiu and co-workers for conjugated polymer NPs (Fig. 11B).157 They developed CPNs of ∼23 nm size composed of donor and acceptor conjugated polymer units as probes for reactive oxygen and nitrogen species (ROS and RONS).157 The obtained nanoprobes exhibited strong acceptor emission in the intact form, but under action of ROS hypochlorite, the acceptor was bleached, which led to the enhancement of the donor emission (Fig. 11B and C). High brightness and operation in the far red region enabled ratiometric detection in hypochlorite with good limit of detection in solution (0.5 μM) as well as in cells and small animals.157 In another study, CPNs were functionalized with near-infrared dyes as FRET acceptors sensitive to ROS and nitrogen species. The obtained 78 nm nanoprobe generated a ratiometric response with a limit of detection for RONS at 10 nM and allowed their detection in cells and imaging inflammation in mice.158 More recently, FRET concept was applied for AIE NPs, using tetraphenylethylene derivative as a FRET donor and a hypochlorite-sensitive acceptor, allowing limit of detection of 105 nM and application to live cells.159 Based on bright dye-loaded polymeric NPs as light-harvesting nanoantenna undergoing efficient FRET to minimal amount of phosphorescent Pt–porphyrin, we developed a nanoprobe for amplified oxygen sensing with minimized cytotoxicity.78 Among the most challenging targets for amplified detection are nucleic acids, because they are present in biological samples at concentrations far below nanomolar. To this end, we grafted DNA to our light-harvesting nanoantenna based on 40 nm dye-loaded polymer NPs (Fig. 11D). Efficient energy transfer to few acceptors hybridized at the NPs surface enabled amplified detection of nucleic acids in solution and on surfaces (Fig. 11E and F) with the limit of detection of 0.5 and 10 pM, respectively.35 Further improved nanoprobe of 16 nm size with higher dye loading and per-volume brightness (9480 M−1 cm−1 nm−3) enabled detection of single nucleic acid copies that could switch emission of the whole particle after hybridization on its surface.36 One should note that in this case the limit of detection around 1 pM was defined by the kinetics of hybridization and not by the particle brightness.36 The concept was successfully applied for detection of microRNA cancer markers from cell extracts,160 showing compatibility with simple detection by a RGB camera of a smartphone.79 The performance of this system was dramatically enhanced by the recently found phenomenon of efficient long-distance FRET between dye-loaded NPs.161 In a recent report by Tian and co-workers, the light-harvesting nanoantenna concept was successfully realized for conjugated polymer NPs, resulting in the nanoprobes with a limit of detection of 1.7 pM for microRNA, which were further applied for RNA sensing at the single cell level.109
6. Conclusions and future perspectives
The field of fluorescent organic nanomaterials developed rapidly over the last decade. The high interest in these materials is explained by rich chemistry that enables tuning of their structural and optical characteristics. The holy grail property of these NPs is their fluorescence brightness, which can be 1000-fold higher than that of organic dyes, which opens new possibilities for biological and biomedical applications. In this tutorial review, we provide the first systematic analysis of brightness for a large variety of fluorescent organic nanomaterials. To this end, we discussed the definitions of brightness, which is essentially a product of extinction coefficient and fluorescence quantum yield of the nanoparticle. Moreover, we highlighted the importance to analyse brightness per volume, which allows comparison of brightness of NPs independently of their size. We described methods used for measuring and evaluating brightness of NPs in bulk (ensemble) and at the single-particle level. One should note that both types of measurements should be performed: while the first one provides a robust value directly linked to optical properties of NPs in solution, the latter shows the single-particle performance of NPs under the microscope. The discrepancy between these two methods frequently originates from >1000-fold higher excitation power (irradiance), used in the single-particle measurements. Then, we present current concepts to fight ACQ: aggregation-induced emission, the use of bulky side groups for neutral dyes and large hydrophobic counterions for ionic dyes. Moreover, the use of conjugated polymer NPs is a special approach, where the design combines conjugation of fluorophores through rigid pi-conjugated bonds and the use of bulky side groups.The core of this review is dedicated to the presentation of the major classes of organic NPs and the systematic analysis of their properties. From this analysis it is clear that conjugated polymer NPs are among the brightest nanomaterials reported to date, however, they require a rather complex molecular design in combination with energy transfer. The future challenges here will be to further improve their extinction coefficient and develop a robust methodology to obtain small particle size, which is a key to achieve high fluorescence quantum yield. The AIE NPs showed relatively good brightness, however the current challenge in the field is to further increase the extinction coefficient of these dyes. A fruitful approach in this sense would be to combine flat, well-conjugated motifs with propeller-shaped AIE units. Neutral dyes with bulky side groups enable preparation of ultrabright macroscopic materials, but preparation of stable and bright NPs based on these fluorophores remains a challenge. Therefore, formulation of NPs and/or integration of these dyes into the polymeric matrix should be further improved. Finally, ionic dyes in combination with bulky hydrophobic counterions enabled preparation of ultrabright nanomaterials of different colour and size, having brightness per volume close to those of conjugated polymer NPs. This approach is particularly efficient because it uses cyanine and rhodamine derivatives, which are among the brightest dyes reported do date. However, bulky counterions present high molecular weight, which remains a limitation for further improvement of the per-volume brightness. Moreover, for all organic nanomaterials, the general challenge is to enhance their brightness in the NIR spectral regions, where all of them present relatively small fluorescence quantum yield. Overall, organic NPs show total brightness from 107 till 109 M−1 cm−1, which is ≥100 times higher than that of typical organic dyes. On the other hand, their per-volume brightness for the best examples ranges from 5000 to 12000 M−1 cm−1 nm−3, which is still significantly lower compared to organic dyes in molecular form (e.g. BV for rhodamine 6G is 105 M−1 cm−1 nm−3). We expect that with proper molecular design and nano-formulation, these values can be further improved, reaching those reported for bulk materials. However, one should also note that the brightness of nanomaterials of a given size has a physical limit because of the Beer–Lambert law. Indeed, once the particle is capable to absorb close to 100% of photons, further enhancement of the extinction coefficient would be counterproductive. Moreover, in these cases the problem of re-absorption of emitted photons could occur, which would even decrease the overall brightness of the nanomaterial.
In bioimaging applications, brightness is particularly important for single-particle tracking, both at the cellular and small animal level. For intracellular applications, a compromise should be made with the particle size, which should be kept close to that of proteins. Brightness is also crucial for super-resolution imaging, where the number of photons improves both spatial and temporal resolution of the methods. The future developments here will focus on obtaining smallest possible NPs presenting highest possible brightness with or without blinking. Biosensing is a key application of fluorescent NPs, where the brightness affects directly the sensitivity of the method. Here, high brightness should be combined with proper functionalization of NPs, using for instance nucleic acids and proteins. Moreover, a variety of colours should be achieved for these NPs in order to access multiplexing capabilities. We expect that significant efforts will be dedicated in the coming years to bright fluorescent organic NPs as probes for biomolecular sensing based on FRET or affinity (binding) principles. Overall, it is clear that the future of organic nanomaterials is bright, but a lot more research is needed to make them indispensable tools in the biological and biomedical laboratories.
Conflicts of interest
A. R. and A. S. K. are co-inventors of a filed patent applications on fluorescent polymeric nanoparticles and cofounders of BrightSens Diagnostics SAS. Other co-authors have no conflict of interest to disclose.Acknowledgements
This work was supported by the European Research Council ERC Consolidator Grant BrightSens 648525, ERC Proof of Concept Grant AmpliFISH 899928, and Marie Curie postdoctoral research grant DNANanoProbes 846571 by the European Union. A. S. K. would like to dedicate his contribution to this work to Ukrainian people fighting with courage for their freedom and for peace in the whole continent.References
- J. B. Grimm and L. D. Lavis, Nat. Methods, 2022, 19, 149–158 CrossRef CAS PubMed.
- B. N. G. Giepmans, S. R. Adams, M. H. Ellisman and R. Y. Tsien, Science, 2006, 312, 217–224 CrossRef CAS PubMed.
- L. D. Lavis and R. T. Raines, ACS Chem. Biol., 2008, 3, 142–155 CrossRef CAS PubMed.
- W. R. Algar, M. Massey, K. Rees, R. Higgins, K. D. Krause, G. H. Darwish, W. J. Peveler, Z. Xiao, H.-Y. Tsai, R. Gupta, K. Lix, M. V. Tran and H. Kim, Chem. Rev., 2021, 121, 9243–9358 CrossRef CAS PubMed.
- A. Reisch and A. S. Klymchenko, Small, 2016, 12, 1968–1992 CrossRef CAS PubMed.
- H. S. Peng and D. T. Chiu, Chem. Soc. Rev., 2015, 44, 4699–4722 RSC.
- C. F. Wu and D. T. Chiu, Angew. Chem., Int. Ed., 2013, 52, 3086–3109 CrossRef CAS PubMed.
- J. C. Li and K. Y. Pu, Chem. Soc. Rev., 2019, 48, 38–71 RSC.
- L. H. Feng, C. L. Zhu, H. X. Yuan, L. B. Liu, F. T. Lv and S. Wang, Chem. Soc. Rev., 2013, 42, 6620–6633 RSC.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- Y. N. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
- G. X. Feng and B. Liu, Acc. Chem. Res., 2018, 51, 1404–1414 CrossRef CAS PubMed.
- S. Y. Ong, C. Zhang, X. Dong and S. Q. Yao, Angew. Chem., Int. Ed., 2021, 60, 17797–17809 CrossRef CAS PubMed.
- K. D. Wegner and N. Hildebrandt, Chem. Soc. Rev., 2015, 44, 4792–4834 RSC.
- M. Montalti, L. Prodi, E. Rampazzo and N. Zaccheroni, Chem. Soc. Rev., 2014, 43, 4243–4268 RSC.
- C. Zhang, C. Li, Y. Liu, J. Zhang, C. Bao, S. Liang, Q. Wang, Y. Yang, H. Fu, K. Wang and D. Cui, Adv. Funct. Mater., 2015, 25, 1314–1325 CrossRef CAS.
- J. Della Rocca, D. M. Liu and W. B. Lin, Acc. Chem. Res., 2011, 44, 957–968 CrossRef CAS PubMed.
- J. J. Liu, R. Li and B. Yang, ACS Cent. Sci., 2020, 6, 2179–2195 CrossRef CAS PubMed.
- D. Yan, Chem. – Eur. J., 2015, 21, 4880–4896 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 3rd edn, 2006 Search PubMed.
- U. Resch-Genger, M. Grabolle, S. Cavaliere-Jaricot, R. Nitschke and T. Nann, Nat. Methods, 2008, 5, 763–775 CrossRef CAS PubMed.
- H. Shen, L. J. Tauzin, R. Baiyasi, W. X. Wang, N. Moringo, B. Shuang and C. F. Landes, Chem. Rev., 2017, 117, 7331–7376 CrossRef CAS PubMed.
- Y. Tian, J. Halle, M. Wojdyr, D. Sahoo and I. G. Scheblykin, Methods Appl. Fluoresc., 2014, 2, 035003 CrossRef CAS PubMed.
- A. Reisch, K. Trofymchuk, A. Runser, G. Fleith, M. Rawiso and A. S. Klymchenko, ACS Appl. Mater. Interfaces, 2017, 9, 43030–43042 CrossRef CAS PubMed.
- A. Reisch, D. Heimburger, P. Ernst, A. Runser, P. Didier, D. Dujardin and A. S. Klymchenko, Adv. Funct. Mater., 2018, 28, 1805157 CrossRef.
- E.-M. Laux, T. Behnke, K. Hoffmann and U. Resch-Genger, Anal. Methods, 2012, 4, 1759–1768 RSC.
- B. K. Wilson and R. K. Prud'homme, J. Colloid Interface Sci., 2021, 604, 208–220 CrossRef CAS PubMed.
- D. Wöll, RSC Adv., 2014, 4, 2447–2465 RSC.
- A. S. Klymchenko, E. Roger, N. Anton, H. Anton, I. Shulov, J. Vermot, Y. Mely and T. F. Vandamme, RSC Adv., 2012, 2, 11876–11886 RSC.
- C. Würth, D. Geißler, T. Behnke, M. Kaiser and U. Resch-Genger, Anal. Bioanal. Chem., 2015, 407, 59–78 CrossRef PubMed.
- C. Würth, M. Grabolle, J. Pauli, M. Spieles and U. Resch-Genger, Anal. Chem., 2011, 83, 3431–3439 CrossRef PubMed.
- S. Hatami, C. Würth, M. Kaiser, S. Leubner, S. Gabriel, L. Bahrig, V. Lesnyak, J. Pauli, N. Gaponik, A. Eychmüller and U. Resch-Genger, Nanoscale, 2015, 7, 133–143 RSC.
- A. Reisch, P. Didier, L. Richert, S. Oncul, Y. Arntz, Y. Mely and A. S. Klymchenko, Nat. Commun., 2014, 5, 4089 CrossRef CAS PubMed.
- T. Yudhistira, E. C. Da Silva, A. Combes, M. Lehmann, A. Reisch and A. S. Klymchenko, Small Methods, 2023, e2201452, DOI:10.1002/smtd.202201452.
- N. Melnychuk and A. S. Klymchenko, J. Am. Chem. Soc., 2018, 140, 10856–10865 CrossRef CAS PubMed.
- N. Melnychuk, S. Egloff, A. Runser, A. Reisch and A. S. Klymchenko, Angew. Chem., Int. Ed., 2020, 59, 6811–6818 CrossRef CAS PubMed.
- Y. X. Tian, A. Merdasa, M. Peter, M. Abdellah, K. B. Zheng, C. S. Ponseca, T. Pullerits, A. Yartsev, V. Sundstrom and I. G. Scheblykin, Nano Lett., 2015, 15, 1603–1608 CrossRef CAS PubMed.
- G. Abbandonato, K. Hoffmann and U. Resch-Genger, Nanoscale, 2018, 10, 7147–7154 RSC.
- M. Kasha, H. R. Rawls and M. A. El-Bayoumi, Pure Appl. Chem., 1965, 11, 371–392 CAS.
- N. J. Hestand and F. C. Spano, Chem. Rev., 2018, 118, 7069–7163 CrossRef CAS PubMed.
- F. Würthner, T. E. Kaiser and C. R. Saha-Möller, Angew. Chem., Int. Ed., 2011, 50, 3376–3410 CrossRef PubMed.
- C. Wu, S. J. Hansen, Q. Hou, J. Yu, M. Zeigler, Y. Jin, D. R. Burnham, J. D. McNeill, J. M. Olson and D. T. Chiu, Angew. Chem., Int. Ed., 2011, 50, 3430–3434 CrossRef CAS PubMed.
- C. Wu and D. T. Chiu, Angew. Chem., Int. Ed., 2013, 52, 3086–3109 CrossRef CAS PubMed.
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740–1741, 10.1039/B105159H.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
- B. Zhang, H. Soleimaninejad, D. J. Jones, J. M. White, K. P. Ghiggino, T. A. Smith and W. W. H. Wong, Chem. Mater., 2017, 29, 8395–8403 CrossRef CAS.
- D. Schmidt, M. Stolte, J. Süβ, A. Liess, V. Stepanenko and F. Würthner, Angew. Chem., Int. Ed., 2019, 58, 13385–13389 CrossRef CAS PubMed.
- F. Wurthner, C. R. Saha-Moller, B. Fimmel, S. Ogi, P. Leowanawat and D. Schmidt, Chem. Rev., 2016, 116, 962–1052 CrossRef PubMed.
- B. Andreiuk, A. Reisch, E. Bernhardt and A. S. Klymchenko, Chem. – Asian J., 2019, 14, 836–846 CrossRef CAS PubMed.
- Z. M. Ou, H. Yao and K. Kimura, Bull. Chem. Soc. Jpn., 2007, 80, 295–302 CrossRef CAS.
- H. Yao and K. Ashiba, RSC Adv., 2011, 1, 834–838 RSC.
- D. K. Bwambok, B. El-Zahab, S. K. Challa, M. Li, L. Chandler, G. A. Baker and I. M. Warner, ACS Nano, 2009, 3, 3854–3860 CrossRef CAS PubMed.
- S. Das, D. Bwambok, B. El-Zahab, J. Monk, S. L. De Rooy, S. Challa, M. Li, F. R. Hung, G. A. Baker and I. M. Warner, Langmuir, 2010, 26, 12867–12876 CrossRef CAS PubMed.
- S. Egloff, N. Melnychuk, E. Cruz Da Silva, A. Reisch, S. Martin and A. S. Klymchenko, ACS Nano, 2022, 16, 1381–1394 CrossRef CAS PubMed.
- C. Wu, B. Bull, C. Szymanski, K. Christensen and J. McNeill, ACS Nano, 2008, 2, 2415–2423 CrossRef CAS PubMed.
- C. Wu, T. Schneider, M. Zeigler, J. Yu, P. G. Schiro, D. R. Burnham, J. D. McNeill and D. T. Chiu, J. Am. Chem. Soc., 2010, 132, 15410–15417 CrossRef CAS PubMed.
- C.-P. Chen, Y.-C. Huang, S.-Y. Liou, P.-J. Wu, S.-Y. Kuo and Y.-H. Chan, ACS Appl. Mater. Interfaces, 2014, 6, 21585–21595 CrossRef CAS PubMed.
- C.-S. Ke, C.-C. Fang, J.-Y. Yan, P.-J. Tseng, J. R. Pyle, C.-P. Chen, S.-Y. Lin, J. Chen, X. Zhang and Y.-H. Chan, ACS Nano, 2017, 11, 3166–3177 CrossRef CAS PubMed.
- Z. Zhang, Y. Yuan, Z. Liu, H. Chen, D. Chen, X. Fang, J. Zheng, W. Qin and C. Wu, ACS Appl. Mater. Interfaces, 2018, 10, 26928–26935 CrossRef CAS PubMed.
- W.-K. Tsai, C.-I. Wang, C.-H. Liao, C.-N. Yao, T.-J. Kuo, M.-H. Liu, C.-P. Hsu, S.-Y. Lin, C.-Y. Wu, J. R. Pyle, J. Chen and Y.-H. Chan, Chem. Sci., 2019, 10, 198–207 RSC.
- H.-Y. Liu, P.-J. Wu, S.-Y. Kuo, C.-P. Chen, E.-H. Chang, C.-Y. Wu and Y.-H. Chan, J. Am. Chem. Soc., 2015, 137, 10420–10429 CrossRef CAS PubMed.
- L. Chen, D. Chen, Y. Jiang, J. Zhang, J. Yu, C. C. DuFort, S. R. Hingorani, X. Zhang, C. Wu and D. T. Chiu, Angew. Chem., Int. Ed., 2019, 58, 7008–7012 CrossRef CAS PubMed.
- K. Li, Z. Zhu, P. Cai, R. Liu, N. Tomczak, D. Ding, J. Liu, W. Qin, Z. Zhao, Y. Hu, X. Chen, B. Z. Tang and B. Liu, Chem. Mater., 2013, 25, 4181–4187 CrossRef CAS.
- G. Feng, C. Y. Tay, Q. X. Chui, R. Liu, N. Tomczak, J. Liu, B. Z. Tang, D. T. Leong and B. Liu, Biomaterials, 2014, 35, 8669–8677 CrossRef CAS PubMed.
- D. Ding, C. C. Goh, G. Feng, Z. Zhao, J. Liu, R. Liu, N. Tomczak, J. Geng, B. Z. Tang, L. G. Ng and B. Liu, Adv. Mater., 2013, 25, 6083–6088 CrossRef CAS PubMed.
- J. Liu, M. Evrard, X. Cai, G. Feng, N. Tomczak, L. G. Ng and B. Liu, J. Mater. Chem. B, 2018, 6, 2630–2636 RSC.
- J. Qi, X. Duan, W. Liu, Y. Li, Y. Cai, J. W. Y. Lam, R. T. K. Kwok, D. Ding and B. Z. Tang, Biomaterials, 2020, 248, 120036 CrossRef CAS PubMed.
- Y. Xu, C. Li, R. Xu, N. Zhang, Z. Wang, X. Jing, Z. Yang, D. Dang, P. Zhang and L. Meng, Chem. Sci., 2020, 11, 8157–8166 RSC.
- T. Behnke, C. Würth, E.-M. Laux, K. Hoffmann and U. Resch-Genger, Dyes Pigm., 2012, 94, 247–257 CrossRef CAS.
- A. Wagh, S. Y. Qian and B. Law, Bioconjugate Chem., 2012, 23, 981–992 CrossRef CAS PubMed.
- R. Méallet-Renault, A. Hérault, J.-J. Vachon, R. B. Pansu, S. Amigoni-Gerbier and C. Larpent, Photochem. Photobiol. Sci., 2006, 5, 300–310 CrossRef PubMed.
- C. Grazon, J. Rieger, R. Méallet-Renault, B. Charleux and G. Clavier, Macromolecules, 2013, 46, 5167–5176 CrossRef CAS.
- M. Collot, J. Schild, K. T. Fam, R. Bouchaala and A. S. Klymchenko, ACS Nano, 2020, 14, 13924–13937 CrossRef CAS PubMed.
- Z. Tian, A. D. Shaller and A. D. Q. Li, Chem. Commun., 2009, 180–182, 10.1039/B815507K,.
- K. Trofymchuk, J. Valanciunaite, B. Andreiuk, A. Reisch, M. Collot and A. S. Klymchenko, J. Mater. Chem. B, 2019, 7, 5199–5210 RSC.
- I. Shulov, S. Oncul, A. Reisch, Y. Arntz, M. Collot, Y. Mely and A. S. Klymchenko, Nanoscale, 2015, 7, 18198–18210 RSC.
- A. Reisch, A. Runser, Y. Arntz, Y. Mély and A. S. Klymchenko, ACS Nano, 2015, 9, 5104–5116 CrossRef CAS PubMed.
- P. Ashokkumar, N. Adarsh and A. S. Klymchenko, Small, 2020, 16, 2002494 CrossRef CAS PubMed.
- C. Severi, N. Melnychuk and A. S. Klymchenko, Biosens. Bioelectron., 2020, 168, 112515 CrossRef CAS PubMed.
- I. O. Aparin, N. Melnychuk and A. S. Klymchenko, Adv. Opt. Mater., 2020, 8, 2000027 CrossRef CAS.
- V. N. Kilin, H. Anton, N. Anton, E. Steed, J. Vermot, T. F. Vandamme, Y. Mely and A. S. Klymchenko, Biomaterials, 2014, 35, 4950–4957 CrossRef CAS PubMed.
- J. Chen, S. M. A. Fateminia, L. Kacenauskaite, N. Baerentsen, S. Grønfeldt Stenspil, J. Bredehoeft, K. L. Martinez, A. H. Flood and B. W. Laursen, Angew. Chem., Int. Ed., 2021, 60, 9450–9458 CrossRef CAS PubMed.
- Y. Jiang and J. McNeill, Chem. Rev., 2017, 117, 838–859 CrossRef CAS PubMed.
- J. Yu, Y. Rong, C.-T. Kuo, X.-H. Zhou and D. T. Chiu, Anal. Chem., 2017, 89, 42–56 CrossRef CAS PubMed.
- S. Kim, C.-K. Lim, J. Na, Y.-D. Lee, K. Kim, K. Choi, J. F. Leary and I. C. Kwon, Chem. Commun., 2010, 46, 1617–1619 RSC.
- J. Liu, J. Geng and B. Liu, Chem. Commun., 2013, 49, 1491–1493 RSC.
- M. Peters, N. Zaquen, L. D’Olieslaeger, H. Bové, D. Vanderzande, N. Hellings, T. Junkers and A. Ethirajan, Biomacromolecules, 2016, 17, 2562–2571 CrossRef CAS PubMed.
- C. Yang, H. Liu, Y. Zhang, Z. Xu, X. Wang, B. Cao and M. Wang, Biomacromolecules, 2016, 17, 1673–1683 CrossRef CAS PubMed.
- P. Koralli, A. D. Nega, L. E. Vagiaki, A. Pavlou, M. G. Siskos, A. Dimitrakopoulou-Strauss, V. G. Gregoriou and C. L. Chochos, Mater. Chem. Front., 2020, 4, 2357–2369 RSC.
- Q. Miao, C. Xie, X. Zhen, Y. Lyu, H. Duan, X. Liu, J. V. Jokerst and K. Pu, Nat. Biotechnol., 2017, 35, 1102–1110 CrossRef CAS PubMed.
- T. Repenko, A. Rix, S. Ludwanowski, D. Go, F. Kiessling, W. Lederle and A. J. C. Kuehne, Nat. Commun., 2017, 8, 470 CrossRef PubMed.
- D. Ding, J. Liu, G. Feng, K. Li, Y. Hu and B. Liu, Small, 2013, 9, 3093–3102 CrossRef CAS PubMed.
- G. Feng, K. Li, J. Liu, D. Ding and B. Liu, Small, 2014, 10, 1212–1219 CrossRef CAS PubMed.
- P. Liu, S. Li, Y. Jin, L. Qian, N. Gao, S. Q. Yao, F. Huang, Q.-H. Xu and Y. Cao, ACS Appl. Mater. Interfaces, 2015, 7, 6754–6763 CrossRef CAS PubMed.
- C. Wu, C. Szymanski, Z. Cain and J. McNeill, J. Am. Chem. Soc., 2007, 129, 12904–12905 CrossRef CAS PubMed.
- T. F. Abelha, P. R. Neumann, J. Holthof, C. A. Dreiss, C. Alexander, M. Green and L. A. Dailey, J. Mater. Chem. B, 2019, 7, 5115–5124 RSC.
- D. Gao, P. Zhang, Y. Liu, Z. Sheng, H. Chen and Z. Yuan, Nanoscale, 2018, 10, 19742–19748 RSC.
- C. Wu, B. Bull, K. Christensen and J. McNeill, Angew. Chem., Int. Ed., 2009, 48, 2741–2745 CrossRef CAS PubMed.
- Z. Tian, J. Yu, C. Wu, C. Szymanski and J. McNeill, Nanoscale, 2010, 2, 1999–2011 RSC.
- K. Sun, Y. Yang, H. Zhou, S. Yin, W. Qin, J. Yu, D. T. Chiu, Z. Yuan, X. Zhang and C. Wu, ACS Nano, 2018, 12, 5176–5184 CrossRef CAS PubMed.
- M. Fan, Y. Zhou, Y. Guo, J. Song and X. Duan, Anal. Methods, 2017, 9, 3255–3259 RSC.
- X. Wang, Y.-Z. Hu, A. Chen, Y. Wu, R. Aggeler, Q. Low, H. C. Kang and K. R. Gee, Chem. Commun., 2016, 52, 4022–4024 RSC.
- W. Wu, G. C. Bazan and B. Liu, Chem, 2017, 2, 760–790 CAS.
- T. Yang, L. Liu, Y. Deng, Z. Guo, G. Zhang, Z. Ge, H. Ke and H. Chen, Adv. Mater., 2017, 29, 1700487 CrossRef PubMed.
- J. Li and K. Pu, Chem. Soc. Rev., 2019, 48, 38–71 RSC.
- H.-S. Peng and D. T. Chiu, Chem. Soc. Rev., 2015, 44, 4699–4722 RSC.
- Y. Yuan, W. Hou, W. Qin and C. Wu, Biomater. Sci., 2021, 9, 328–346 RSC.
- Y. Wang, L. Feng and S. Wang, Adv. Funct. Mater., 2019, 29, 1806818 CrossRef.
- F. Xiao, X. Fang, H. Li, H. Xue, Z. Wei, W. Zhang, Y. Zhu, L. Lin, Y. Zhao, C. Wu and L. Tian, Angew. Chem., Int. Ed., 2022, 61, e202115812 CAS.
- L. R. MacFarlane, H. Shaikh, J. D. Garcia-Hernandez, M. Vespa, T. Fukui and I. Manners, Nat. Rev. Mater., 2021, 6, 7–26 CrossRef CAS.
- A. H. Ashoka, S.-H. Kong, B. Seeliger, B. Andreiuk, R. V. Soares, M. Barberio, M. Diana and A. S. Klymchenko, Biomaterials, 2020, 261, 120306 CrossRef CAS PubMed.
- C. Xie, W. Zhou, Z. Zeng, Q. Fan and K. Pu, Chem. Sci., 2020, 11, 10553–10570 RSC.
- H. Zhang, B. Zhang, Y. Zhang, Z. Xu, H. Wu, P.-A. Yin, Z. Wang, Z. Zhao, D. Ma and B. Z. Tang, Adv. Funct. Mater., 2020, 30, 2002323 CrossRef CAS.
- J. Huang, H. Nie, J. Zeng, Z. Zhuang, S. Gan, Y. Cai, J. Guo, S.-J. Su, Z. Zhao and B. Z. Tang, Angew. Chem., Int. Ed., 2017, 56, 12971–12976 CrossRef CAS PubMed.
- Z. Song, R. T. K. Kwok, D. Ding, H. Nie, J. W. Y. Lam, B. Liu and B. Z. Tang, Chem. Commun., 2016, 52, 10076–10079 RSC.
- J. Chen, X. Chen, Q. Huang, W. Li, Q. Yu, L. Zhu, T. Zhu, S. Liu and Z. Chi, ACS Appl. Mater. Interfaces, 2019, 11, 32689–32696 CrossRef CAS PubMed.
- F. Hu, X. Cai, P. N. Manghnani, Kenry, W. Wu and B. Liu, Chem. Sci., 2018, 9, 2756–2761 RSC.
- A. Nicol, W. Qin, R. T. K. Kwok, J. M. Burkhartsmeyer, Z. Zhu, H. Su, W. Luo, J. W. Y. Lam, J. Qian, K. S. Wong and B. Z. Tang, Chem. Sci., 2017, 8, 4634–4643 RSC.
- J. Qi, C. Sun, A. Zebibula, H. Zhang, R. T. K. Kwok, X. Zhao, W. Xi, J. W. Y. Lam, J. Qian and B. Z. Tang, Adv. Mater., 2018, 30, 1706856 CrossRef PubMed.
- P. Alam, W. He, N. L. C. Leung, C. Ma, R. T. K. Kwok, J. W. Y. Lam, H. H. Y. Sung, I. D. Williams, K. S. Wong and B. Z. Tang, Adv. Funct. Mater., 2020, 30, 1909268 CrossRef CAS.
- G. Deng, X. Peng, Z. Sun, W. Zheng, J. Yu, L. Du, H. Chen, P. Gong, P. Zhang, L. Cai and B. Z. Tang, ACS Nano, 2020, 14, 11452–11462 CrossRef CAS PubMed.
- X. Cai, A. Bandla, C. K. Chuan, G. Magarajah, L.-D. Liao, D. B. L. Teh, B. K. Kennedy, N. V. Thakor and B. Liu, Mater. Horiz., 2019, 6, 311–317 RSC.
- J. Huang, B. He, Z. Zhang, Y. Li, M. Kang, Y. Wang, K. Li, D. Wang and B. Z. Tang, Adv. Mater., 2020, 2003382 CrossRef CAS PubMed.
- Z. Zhang, W. Xu, M. Kang, H. Wen, H. Guo, P. Zhang, L. Xi, K. Li, L. Wang, D. Wang and B. Z. Tang, Adv. Mater., 2020, 2003210 CrossRef CAS PubMed.
- W. Wu, D. Mao, S. Xu, M. Panahandeh-Fard, Y. Duan, F. Hu, D. Kong and B. Liu, Adv. Funct. Mater., 2019, 29, 1901791 CrossRef CAS.
- J. Li, J. Wang, H. Li, N. Song, D. Wang and B. Z. Tang, Chem. Soc. Rev., 2020, 49, 1144–1172 RSC.
- X. Cai and B. Liu, Angew. Chem., Int. Ed., 2020, 59, 9868–9886 CrossRef CAS PubMed.
- S. Xu, Y. Duan and B. Liu, Adv. Mater., 2020, 32, 1903530 CrossRef CAS PubMed.
- R. Zhang, Y. Duan and B. Liu, Nanoscale, 2019, 11, 19241–19250 RSC.
- M.-J. Lin, Á. J. Jiménez, C. Burschka and F. Würthner, Chem. Commun., 2012, 48, 12050–12052 RSC.
- V. Caponetti, J. W. Trzcinski, A. Cantelli, R. Tavano, E. Papini, F. Mancin and M. Montalti, Front. Chem., 2019, 7, 168 CrossRef CAS PubMed.
- D. Zhang, Y. Wen, Y. Xiao, G. Yu, Y. Liu and X. Qian, Chem. Commun., 2008, 4777–4779, 10.1039/B808681H.
- T. Ozdemir, S. Atilgan, I. Kutuk, L. T. Yildirim, A. Tulek, M. Bayindir and E. U. Akkaya, Org. Lett., 2009, 11, 2105–2107 CrossRef CAS PubMed.
- C. Grazon, J. Rieger, B. Charleux, G. Clavier and R. Méallet-Renault, J. Phys. Chem. C, 2014, 118, 13945–13952 CrossRef CAS.
- F. Würthner, C. R. Saha-Möller, B. Fimmel, S. Ogi, P. Leowanawat and D. Schmidt, Chem. Rev., 2016, 116, 962–1052 CrossRef PubMed.
- M. Mahl, K. Shoyama, A.-M. Krause, D. Schmidt and F. Würthner, Angew. Chem., Int. Ed., 2020, 59, 13401–13405 CrossRef CAS PubMed.
- M. Stolte, T. Schembri, J. Süβ, D. Schmidt, A.-M. Krause, M. O. Vysotsky and F. Würthner, Chem. Mater., 2020, 32, 6222–6236 CrossRef CAS.
- B. Andreiuk, A. Reisch, M. Lindecker, G. Follain, N. Peyriéras, J. G. Goetz and A. S. Klymchenko, Small, 2017, 13, 1701582 CrossRef PubMed.
- N. Adarsh and A. S. Klymchenko, Nanoscale, 2019, 11, 13977–13987 RSC.
- L. Kacenauskaite, S. G. Stenspil, A. H. Olsson, A. H. Flood and B. W. Laursen, J. Am. Chem. Soc., 2022, 144, 19981–19989 CrossRef CAS PubMed.
- C. R. Benson, L. Kacenauskaite, K. L. VanDenburgh, W. Zhao, B. Qiao, T. Sadhukhan, M. Pink, J. Chen, S. Borgi, C.-H. Chen, B. J. Davis, Y. C. Simon, K. Raghavachari, B. W. Laursen and A. H. Flood, Chem, 2020, 6, 1978–1997 CAS.
- D. Y. Jin, P. Xi, B. M. Wang, L. Zhang, J. Enderlein and A. M. van Oijen, Nat. Methods, 2018, 15, 415–423 CrossRef CAS PubMed.
- M. Dahan, S. Lévi, C. Luccardini, P. Rostaing, B. Riveau and A. Triller, Science, 2003, 302, 442–445 CrossRef CAS PubMed.
- X. Chen, Z. Liu, R. Li, C. Shan, Z. Zeng, B. Xue, W. Yuan, C. Mo, P. Xi, C. Wu and Y. Sun, ACS Nano, 2017, 11, 8084–8091 CrossRef CAS PubMed.
- J. B. Yu, C. F. Wu, S. P. Sahu, L. P. Fernando, C. Szymanski and J. McNeill, J. Am. Chem. Soc., 2009, 131, 18410–18414 CrossRef CAS PubMed.
- Y. Luo, Y. P. Han, X. J. Hu, M. Yin, C. F. Wu, Q. Li, N. Chen and Y. Zhao, Cell Proliferation, 2019, 52, e12556 CrossRef PubMed.
- E. Genin, Z. Gao, J. A. Varela, J. Daniel, T. Bsaibess, I. Gosse, L. Groc, L. Cognet and M. Blanchard-Desce, Adv. Mater., 2014, 26(2258–2261), 2257 CrossRef PubMed.
- I. Khalin, D. Heimburger, N. Melnychuk, M. Collot, B. Groschup, F. Hellal, A. Reisch, N. Plesnila and A. S. Klymchenko, ACS Nano, 2020, 14, 9755–9770 CrossRef CAS PubMed.
- S. E. Jang, L. Qiu, X. Cai, J. W. L. Lee, W. Zhang, E. K. Tan, B. Liu and L. Zeng, Biomaterials, 2021, 271, 120747 CrossRef CAS PubMed.
- X. Fang, X. Chen, R. Li, Z. Liu, H. Chen, Z. Sun, B. Ju, Y. Liu, S. X.-A. Zhang, D. Ding, Y. Sun and C. Wu, Small, 2017, 13, 1702128 CrossRef PubMed.
- Z. Zhang, X. Fang, Z. Liu, H. Liu, D. Chen, S. He, J. Zheng, B. Yang, W. Qin, X. Zhang and C. Wu, Angew. Chem., Int. Ed., 2020, 59, 3691–3698 CrossRef CAS PubMed.
- J. Lin, X. Zeng, Y. Xiao, L. Tang, J. Nong, Y. Liu, H. Zhou, B. Ding, F. Xu, H. Tong, Z. Deng and X. Hong, Chem. Sci., 2019, 10, 1219–1226 RSC.
- X. Gu, X. Zhang, H. Ma, S. Jia, P. Zhang, Y. Zhao, Q. Liu, J. Wang, X. Zheng, J. W. Y. Lam, D. Ding and B. Z. Tang, Adv. Mater., 2018, 30, e1801065 CrossRef PubMed.
- N. Hildebrandt, C. M. Spillmann, W. R. Algar, T. Pons, M. H. Stewart, E. Oh, K. Susumu, S. A. Díaz, J. B. Delehanty and I. L. Medintz, Chem. Rev., 2017, 117, 536–711 CrossRef CAS PubMed.
- I. L. Medintz, A. R. Clapp, H. Mattoussi, E. R. Goldman, B. Fisher and J. M. Mauro, Nat. Mater., 2003, 2, 630–638 CrossRef CAS PubMed.
- K. Trofymchuk, A. Reisch, P. Didier, F. Fras, P. Gilliot, Y. Mely and A. S. Klymchenko, Nat. Photonics, 2017, 11, 657–663 CrossRef CAS PubMed.
- L. Wu, I. C. Wu, C. C. DuFort, M. A. Carlson, X. Wu, L. Chen, C. T. Kuo, Y. L. Qin, J. B. Yu, S. R. Hingorani and D. T. Chiu, J. Am. Chem. Soc., 2017, 139, 6911–6918 CrossRef CAS PubMed.
- K. Pu, A. J. Shuhendler and J. Rao, Angew. Chem., Int. Ed., 2013, 52, 10325–10329 CrossRef CAS PubMed.
- K. Nie, B. Dong, H. Shi, L. Chao, M. Long, H. Xu, Z. Liu and B. Liang, J. Lumin., 2020, 220, 117018 CrossRef CAS.
- S. Egloff, N. Melnychuk, A. Reisch, S. Martin and A. S. Klymchenko, Biosens. Bioelectron., 2021, 179, 113084 CrossRef CAS PubMed.
- D. S. Biswas, P. Gaki, E. C. Da Silva, A. Combes, A. Reisch, P. Didier and A. S. Klymchenko, Adv. Mater., 2023 DOI:10.1002/adma.202301402.
| This journal is © The Royal Society of Chemistry 2023 |