
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnnatural helical peptidic foldamers as protein segment mimics
Peng
Sang
*a and
Jianfeng
Cai
 *b
*b
aTianjian Laboratory of Advanced Biomedical Sciences, School of Pharmaceutical Sciences, Zhengzhou University, Zhengzhou 450001, China. E-mail: pengsang@zzu.edu.cn
bDepartment of Chemistry, University of South Florida, Tampa, FL 33620, USA. E-mail: jianfengcai@usf.edu
First published on 4th July 2023
Abstract
Unnatural helical peptidic foldamers have attracted considerable attention owing to their unique folding behaviours, diverse artificial protein binding mechanisms, and promising applications in chemical, biological, medical, and material fields. Unlike the conventional α-helix consisting of molecular entities of native α-amino acids, unnatural helical peptidic foldamers are generally comprised of well-defined backbone conformers with unique and unnatural structural parameters. Their folded structures usually arise from unnatural amino acids such as N-substituted glycine, N-substituted-β-alanine, β-amino acid, urea, thiourea, α-aminoxy acid, α-aminoisobutyric acid, aza-amino acid, aromatic amide, γ-amino acid, as well as sulfono-γ-AA amino acid. They can exhibit intriguing and predictable three-dimensional helical structures, generally featuring superior resistance to proteolytic degradation, enhanced bioavailability, and improved chemodiversity, and are promising in mimicking helical segments of various proteins. Although it is impossible to include every piece of research work, we attempt to highlight the research progress in the past 10 years in exploring unnatural peptidic foldamers as protein helical segment mimics, by giving some representative examples and discussing the current challenges and future perspectives. We expect that this review will help elucidate the principles of structural design and applications of existing unnatural helical peptidic foldamers in protein segment mimicry, thereby attracting more researchers to explore and generate novel unnatural peptidic foldamers with unique structural and functional properties, leading to more unprecedented and practical applications.
 Peng Sang | Peng Sang received his PhD (2014, supervisors: Prof. Yuhong Zhang and Prof. Jianwei Zou) from Zhejiang University. He worked as a postdoctoral researcher in Prof. X. Peter Zhang's group at the University of South Florida during 2014–2015, and in the lab of Prof. Jianfeng Cai during 2015–2022. He then joined the School of pharmaceutical sciences at Zhengzhou University in 2022 and was promoted to full professor in 2023. His current research interests mainly focus on medicinal chemistry, including the discovery of peptidomimetic lead compounds related to tumors and metabolic diseases. |
 Jianfeng Cai | Jianfeng Cai is currently a USF Preeminent professor in the Department of Chemistry, at the University of South Florida. He received his BS and MS from Nanjing University in 1997 and 2000, respectively. After obtaining his PhD from the Department of Chemistry, Washington University in St. Louis in 2006, he did his postdoctoral research at Yale University Chemistry Department from 2007 to 2009. He joined the USF Chemistry Department in 2009 as an assistant professor and was promoted to associate professor in 2015 and full professor in 2018. His current research interest includes designing and synthesizing peptidomimetics that mimic the structure and function of proteins/peptides, and developing antimicrobial, anti-cancer, and anti-Alzheimer's disease agents. |
Key learning points(1) The basic concept and importance of unnatural helical peptidic foldamers as protein segment mimics.(2) The main approaches to constructing unnatural helical peptidic foldamers. (3) Representative examples of protein segment mimicry based on diverse unnatural helical peptidic foldamers. (4) Application of unnatural helical peptidic foldamers for supramolecular self-assembly. (5) The challenges and opportunities that remain for developing unnatural peptidic foldamers as protein helical segment mimics. |
1. Introduction
Natural proteins exhibit enormous structures and functions, which have inspired the design of functional peptides that play a growing important role in chemical, biological, medical, and materials sciences owing to their canonical folding structures (e.g. α-helix, β-sheet). They have found wide applications in the mimicry of host-defense peptides,1–3 disease-related protein–protein interactions (PPIs),4–6 catalysis,7–9 molecular recognition,10–12 and supramolecular design.13 However, conventional helical peptides often suffer from the loss of secondary structure, susceptibility to proteolytic degradation, and difficulty in penetrating intact cells, significantly limiting their applications.14–16 In 1996, the groups of Seebach and Gellman independently reported the secondary helical structure of β-peptides17,18 (Fig. 1), and Gellman coined the term “foldamer” in 1998.19 Since then, several classes of helical foldamers have been developed.20–28 The unnatural helical peptidic foldamers can mimic the structure and function of natural α-helices at various levels and overcome the limitations of functional conventional helical peptides in biomedical applications.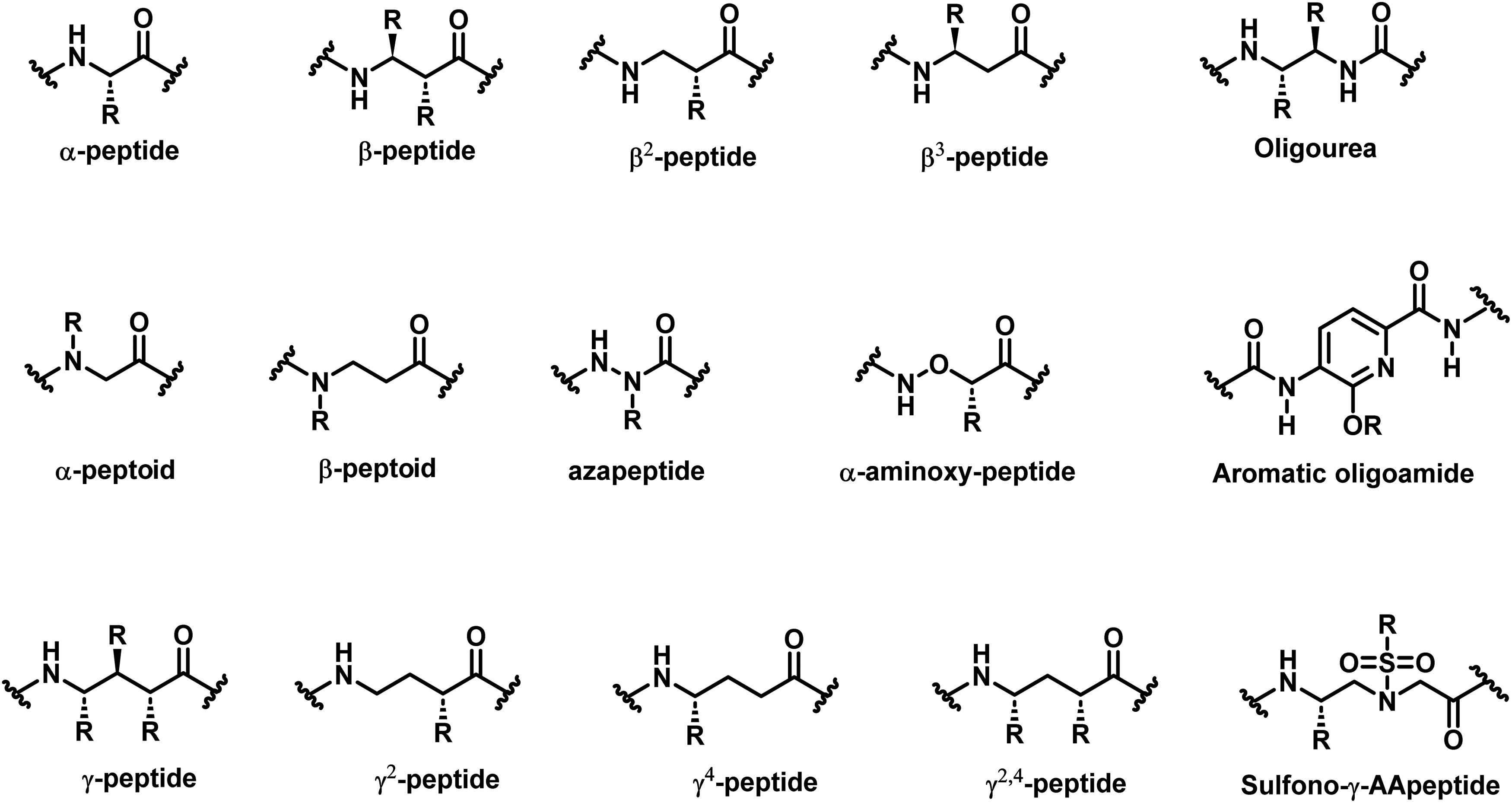 | ||
| Fig. 1 Structure of the α-peptide residue and examples of unnatural helical peptidic foldamer residues. | ||
As alternatives to the conventional α-helix, in recent years more and more unnatural helical peptidic foldamers (Fig. 1) have come into sight, such as β-peptides,20 peptoids,22,23 oligoureas,24 α-aminoxy-peptides,29 azapeptides,25 aromatic oligoamides,27 γ-peptides,30 sulfono-γ-AApeptides14 and so forth. Unlike conventional α-helical peptides, unnatural helical peptidic foldamers are generally composed of unnatural amino acids and possess well-defined folding propensity with discrete structural parameters. The unnatural backbones and modular synthesis endow helical peptidic foldamers with unique advantages, including superior resistance to proteolytic degradation, enhanced bioavailability, improved chemodiversity and versatile design in their mimicry of helical segments of proteins.
Although unnatural helical peptidic foldamers fold into predictable, well-defined and robust helices with helical parameters that differ from those of the conventional α-helix, in most cases, the formation of their helical structures is still largely dependent on intramolecular hydrogen bonding as in the case of conventional helical peptides.14,17,20,27 Of course, for a specific unnatural helical peptidic foldamer system, salt bridges, cation–π interaction, π–π interaction and conformation rigidification, and so forth may also play a dominant role individually or in combination.
Based on the understanding of the intrinsic forces underlying the formation of unnatural helical peptidic foldamers, one can recognize that the design or discovery of the most basic units that assemble into helical foldamers, i.e., the identification of unnatural amino acid building blocks, is crucial. Such unnatural amino acids that researchers have developed so far include N-substituted glycine, N-substituted-β-alanine, β-amino acid, urea, α-aminoxy acid, α-aminoisobutyric acid, aza-amino acid, aromatic amide, γ-amino acid, sulfono-γ-AA amino acid, and others (Fig. 1). It is these building blocks that are built into different forms to obtain the desired unnatural helical peptidic foldamers bearing various functions.
In this review, we tend to provide a brief and educational overview of the progress in the past 10 years in exploring helical peptidic foldamers based on non-natural molecular backbones as the mimics of protein helical segments by highlighting some achievements in this field, with a focus on the principles of their structural design, general characteristics of their intrinsic folding properties, their applications in protein segment mimicry, as well as the current challenges and future perspectives. Due to the limited space, it is impossible to include all of the research endeavours. Also in this review, we only focus on the foldamers forming helical structures. The classes of foldamers which are developed to mimic/recognize hot spots involved in PPIs but which themselves do not adopt well-defined helical conformations will not be discussed in detail here.31–33 Helical foldamers bearing either homogeneous or heterogeneous backbones will be underlined; however, the hybrid structures with insertion of the minimal number of unnatural residues (less than 3) into canonical peptide sequences have limited influence on the entire structure of the natural α-helix, and they will be highlighted with just a few examples in this review.
We hope that this review will help to guide general readers to understand the principles of structural design and applications of existing unnatural helical peptidic foldamers for protein segment mimicry, thereby attracting more researchers to explore and generate new classes of unnatural peptidic foldamers with unique structural and properties for unprecedented and practical applications.
2. The crystal structures of unnatural helical peptidic foldamers
Different from the conventional helical peptides or their functionalized analogs, helical foldamers built on non-natural frameworks have unique structural features; understanding their folding principle is of scientific interest. Since structure ultimately governs function, it is crucial to determine the structures of helical foldamers to elucidate the structure–function relationship, based on which the new helical foldamers can be designed and/or evolved with predictable function through structural tailoring or functional group modification. Understanding the intrinsic mechanisms for the formation, stabilization and variation of the helical structure of foldamers will accelerate molecular engineering or structural design endeavors in building unnatural helical peptidic foldamers. This section will elaborate on the investigations, information gathered, and insights gained by some research groups on the representative crystal structures of unnatural helical peptidic foldamers, including their secondary, tertiary, and even quaternary structures.2.1 Homogeneous structures
Homogeneous structures represent a large body of important helical foldamers, which are composed of the same type of unnatural amino acid residues. To date, there are a number of types of homogeneous helical peptidic foldamers reported with well-defined single crystal structures, such as β-peptides, peptoids, oligoureas, α-aminoxy-peptides, aromatic oligoamides, γ-peptides and sulfono-γ-AApeptides.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯H–N hydrogen bonds between backbone amide groups; these hydrogen bonds occur in 14-atom rings. The 12-helix here, on the other hand, is a helical foldamer containing i, i + 3 CO⋯H–N hydrogen bonds that occur in 12-atom rings, whereas the M/N-helix represents a helical foldamer containing both M- and N-atom pseudo-hydrogen-bonded rings. All the following types of helical structures are named based on this convention, and we will not explain them individually. β-Peptides containing no constraining residues also form helical structures (Fig. 2, left-handed helical β-peptide 5).
O⋯H–N hydrogen bonds between backbone amide groups; these hydrogen bonds occur in 14-atom rings. The 12-helix here, on the other hand, is a helical foldamer containing i, i + 3 CO⋯H–N hydrogen bonds that occur in 12-atom rings, whereas the M/N-helix represents a helical foldamer containing both M- and N-atom pseudo-hydrogen-bonded rings. All the following types of helical structures are named based on this convention, and we will not explain them individually. β-Peptides containing no constraining residues also form helical structures (Fig. 2, left-handed helical β-peptide 5).
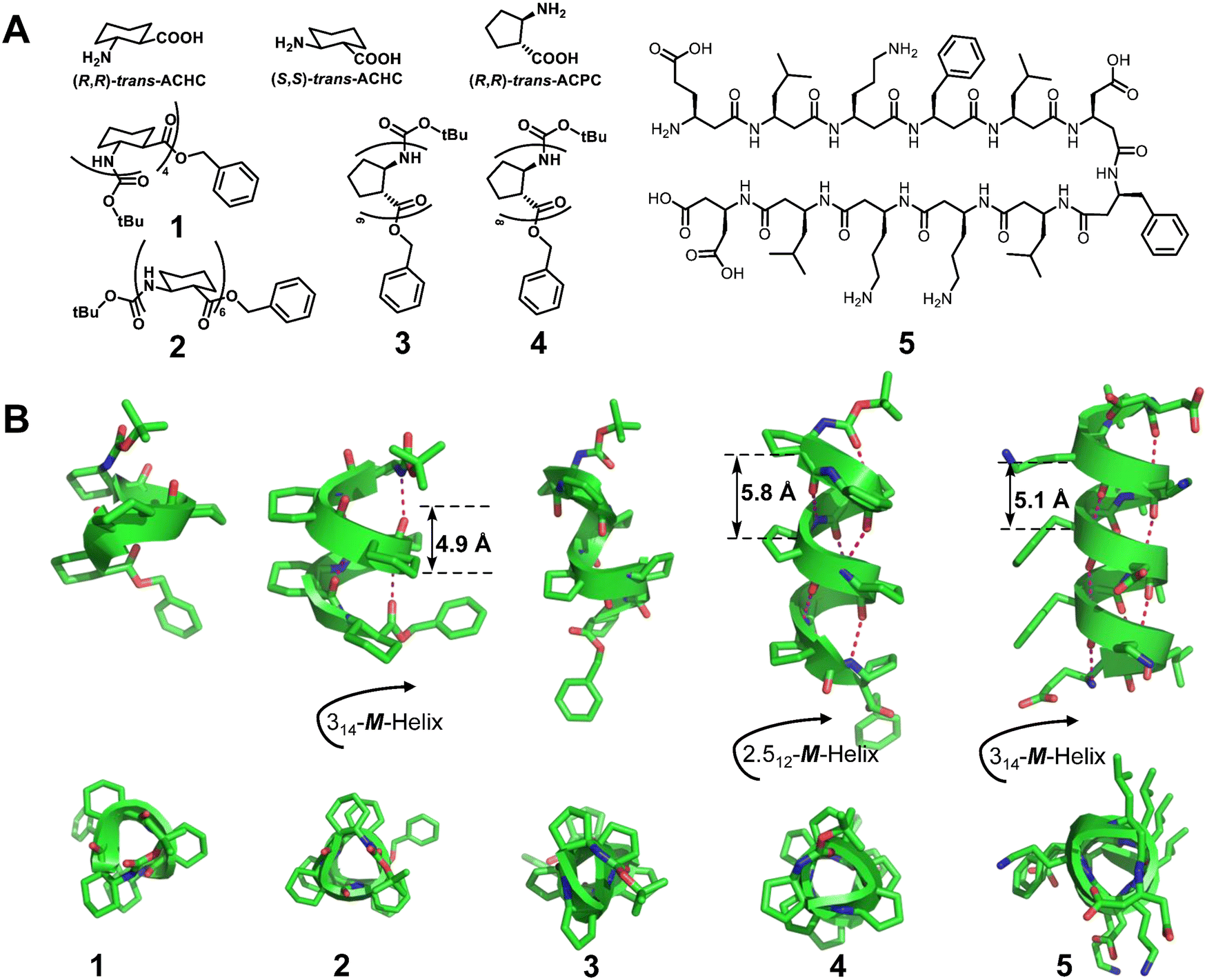 | ||
| Fig. 2 (A) Chemical structures of trans-ACHC, trans-ACPC and β-peptides 1, 2, 3, 4, and 5; (B) crystal structures of β-peptides 1 (CCDC 1207811), 2 (CCDC 1247871), 3 (CCDC 1291846), 4 (CCDC 1291854) and 5 (CCDC 633286). The magenta dashed lines indicate intramolecular hydrogen bonds. Some hydrogen atoms are omitted for clarity. | ||
Barron et al.42 reported a homogeneous (R)-N-(1-cyclohexylethyl) glycine pentamer (6) formed by peptoid oligomers with α-chiral aliphatic side chains, which has a left-handed helix with cis-amide bonds (Fig. 3). This is the first X-ray crystal structure of peptoids. Then, the Blackwell group22 and the Taillefumier group43 found a series of homogeneous right-handed helical peptoids with α-chiral aromatic (S)-N-(1-naphthylethyl) side chains or nonaromatic α-chiral (S)-N-(1-tert-butylethyl) side chains, respectively (Fig. 3, 7 and 8). These results also suggest that the helical sense of the peptoid was dictated by the stereochemistry of the α-chiral side chain, with R or S stereocenters producing a left- or right-handed helix, respectively.
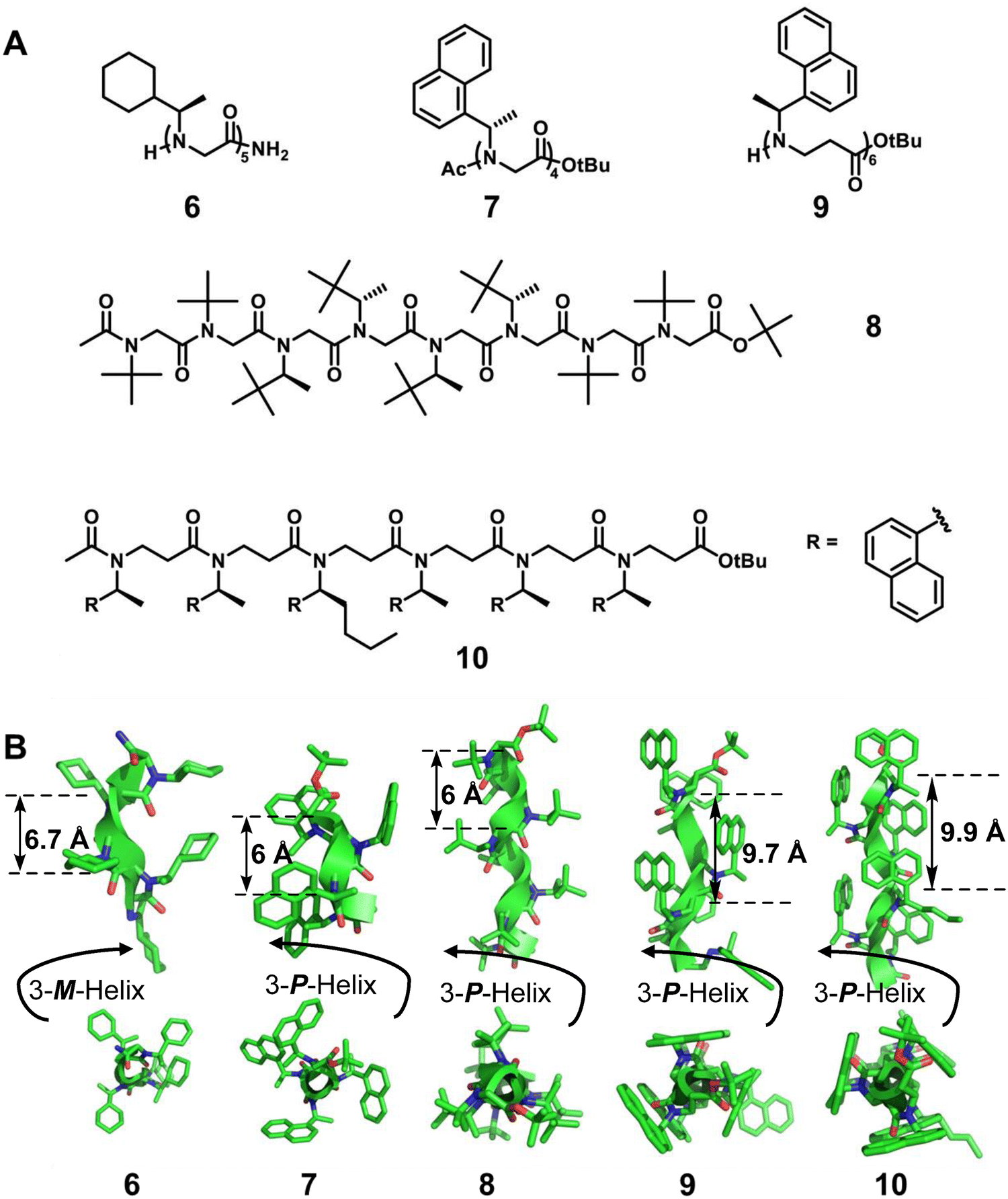 | ||
| Fig. 3 (A) Chemical structures of representative peptoids 6, 7, 8, 9 and 10; (B) crystal structures of peptoids 6 (CCDC 224613), 7 (CCDC 787852), 8 (CCDC 1561295), 9 (CCDC 1054163) and 10 (CCDC 1857129). | ||
The additional methylene group in the backbone of β-peptoids may potentially give rise to altered amide rotamer equilibria compared to α-peptoids. Despite the challenges related to the lack of hydrogen bonding combined with the increased flexibility of the backbone, β-peptoids containing N-(S)-1-(1-naphthyl)ethyl side chains can fold into unique triangular prism-shaped helices. This is reflected in a series of single crystal studies of the Olsen group (Fig. 3, right-handed helical β-peptoid 9).23 By the introduction of amino groups onto robustly folded β-peptoid helices, they also reported the first example of the X-ray crystal structure of a linear β-peptoid containing more than one type of side chain (Fig. 3, right-handed helical β-peptoid 10).41
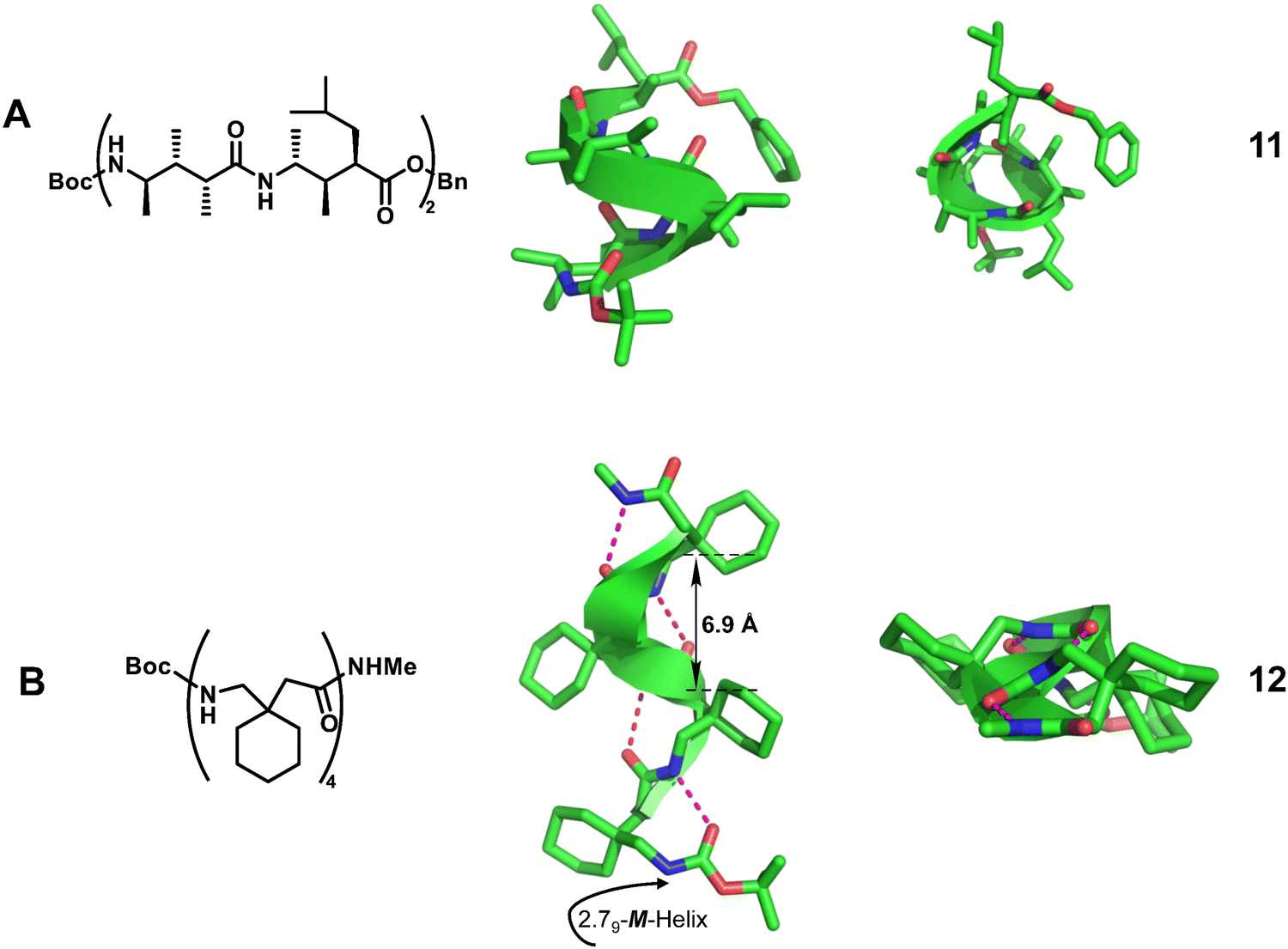 | ||
| Fig. 4 Representative helical γ-peptides. (A) Chemical and crystal structure of 11 (CCDC 159803); (B) chemical and crystal structure of 12 (CCDC 266652). The magenta dashed lines indicate intramolecular hydrogen bonds. Some hydrogen atoms are omitted for clarity. | ||
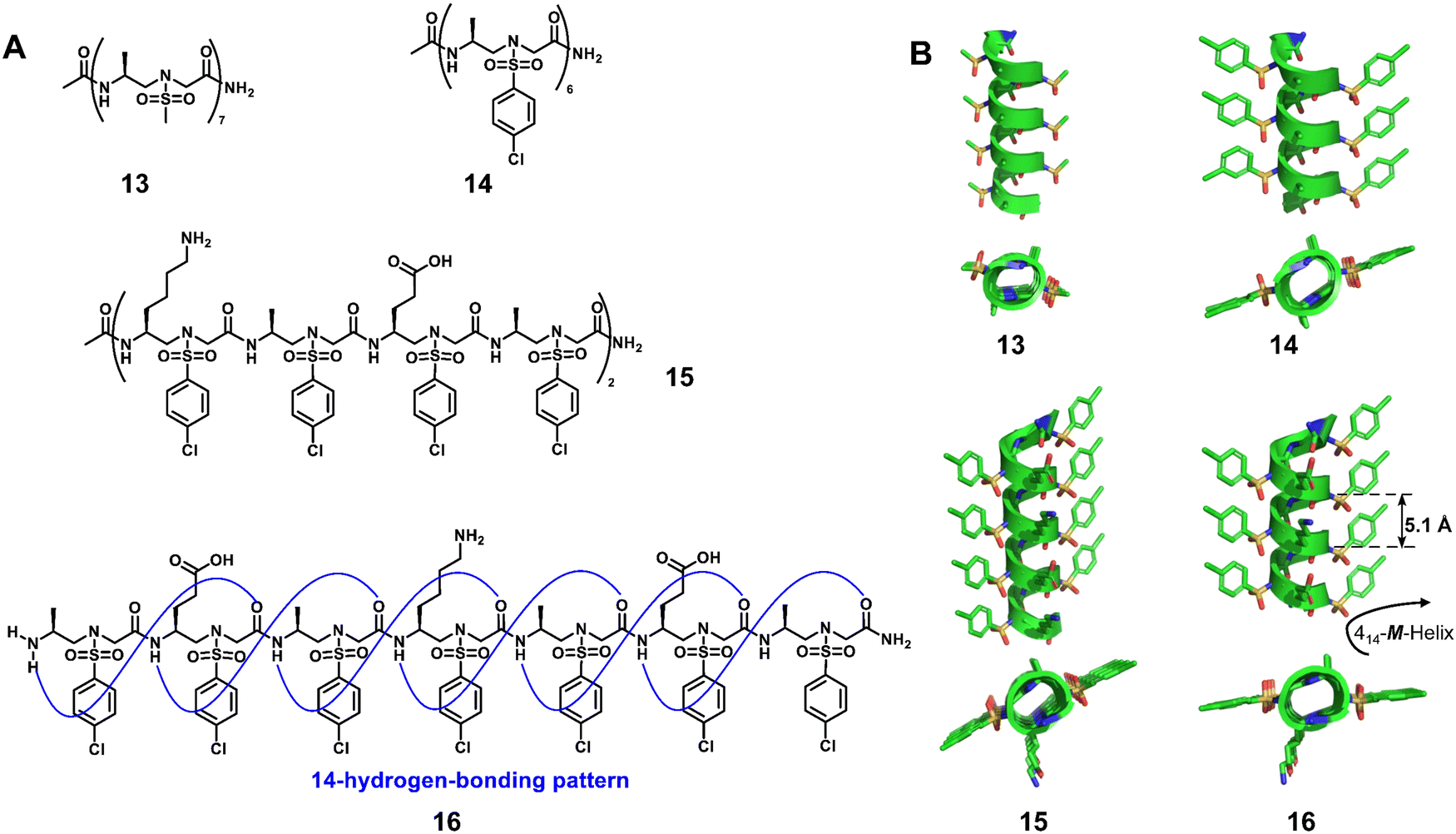 | ||
| Fig. 5 Helical L-sulfono-γ-AApeptides. (A) Chemical structures of 13, 14, 15 and 16; (B) crystal structures of 13 (CCDC 1841094), 14 (CCDC 1841091), 15 (CCDC 1841092) and 16 (CCDC 1841093). | ||
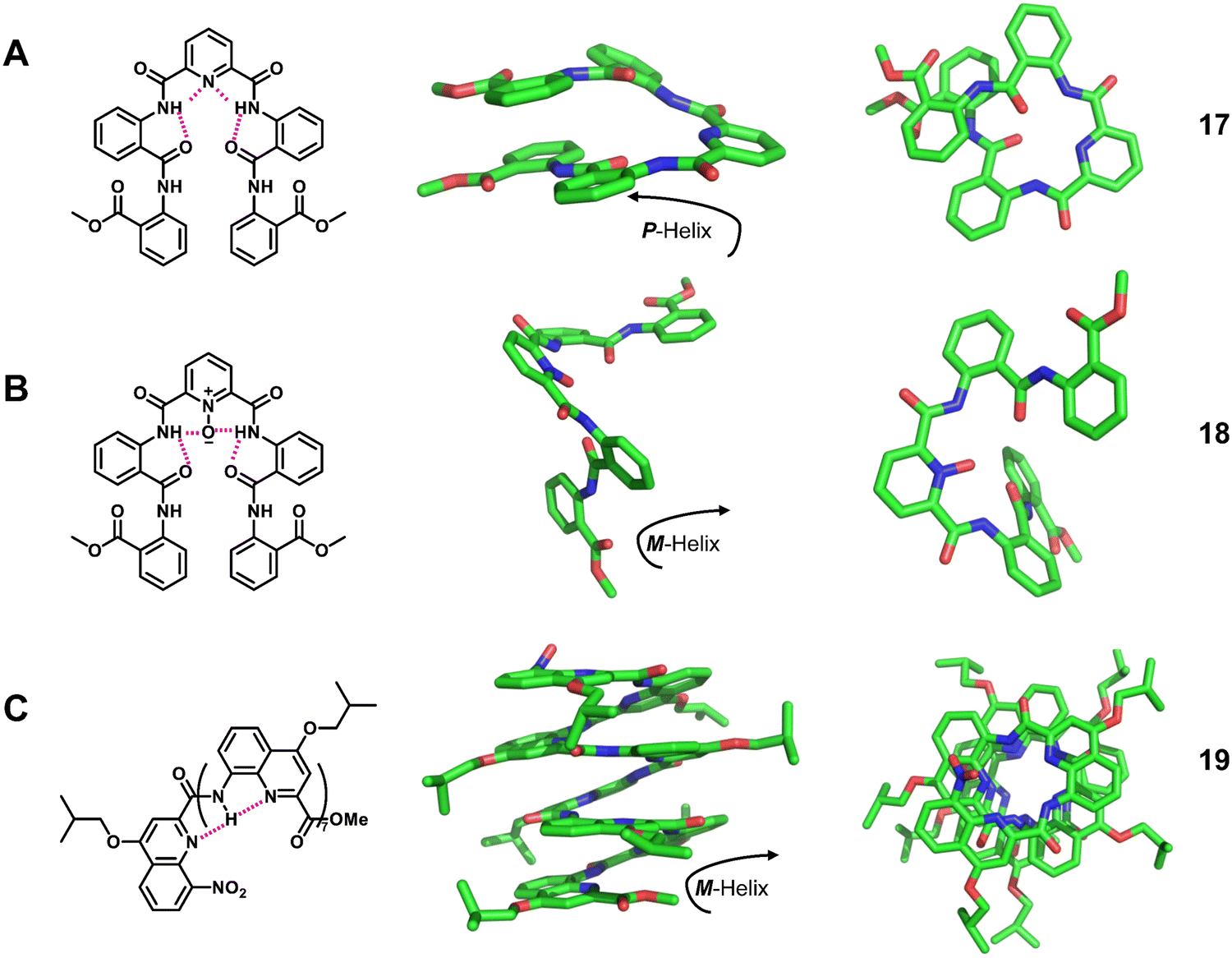 | ||
| Fig. 6 Representative helical aromatic oligoamides. Chemical and crystal structure of right-handed foldamer 17 (CCDC 1233815), left-handed foldamer 18 (CCDC 1233817) and left-handed foldamer 19 (CCDC 208068). The magenta dashed lines indicate intramolecular hydrogen bonds. Some hydrogen atoms are omitted for clarity. | ||
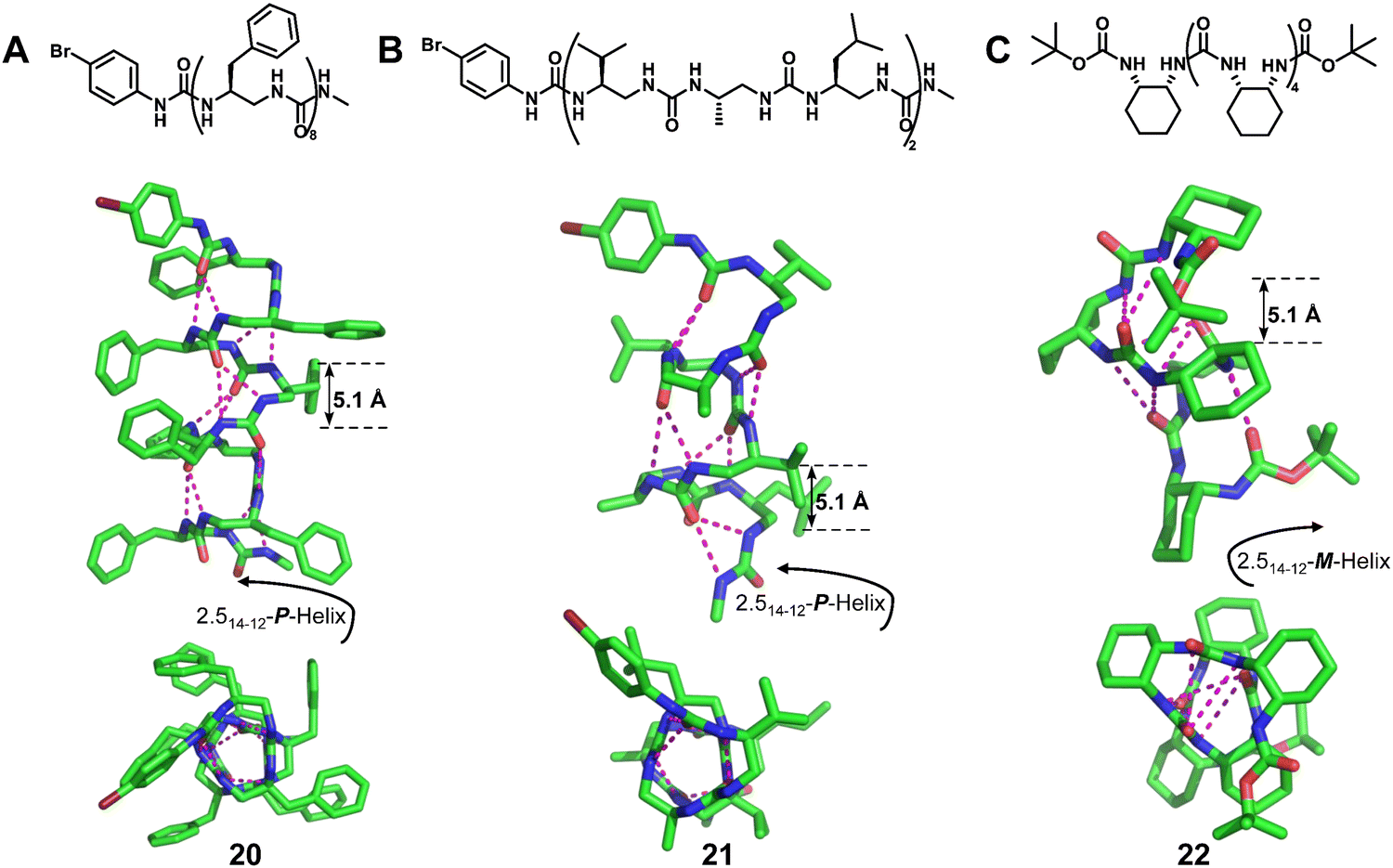 | ||
| Fig. 7 Representative examples of helical N,N′-linked oligoureas. Chemical and crystal structure of right-handed foldamer 20 (CCDC 750017), right-handed foldamer 21 (CCDC 750016) and left-handed foldamer 22 (CCDC 1477894). The magenta dashed lines indicate intramolecular hydrogen bonds. Some hydrogen atoms are omitted for clarity. | ||
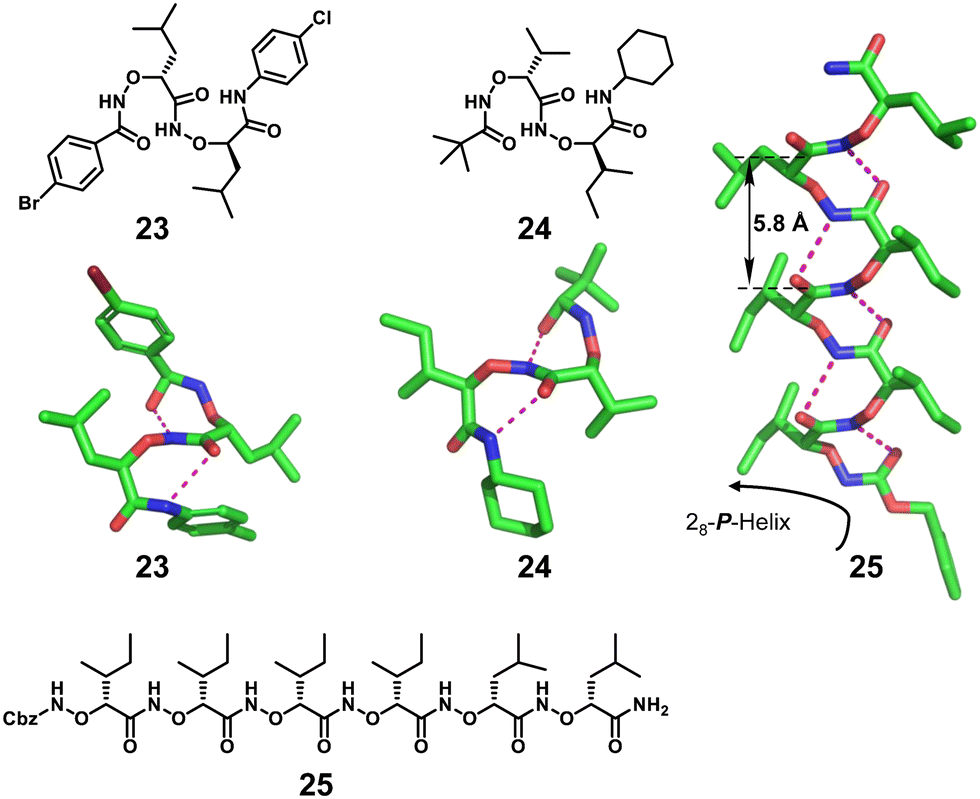 | ||
| Fig. 8 Chemical and crystal structures of helical right-handed α-aminoxy-peptides 23, 24 and 25 (CCDC 1420774). The magenta dashed lines indicate intramolecular hydrogen bonds. Some hydrogen atoms are omitted for clarity. | ||
2.2 Heterogeneous structures
Introducing α-amino acids (or different kinds of unnatural amino acids) into the backbone of unnatural helical peptidic foldamers is the most convenient way to increase structural and chemical diversity. There are several possible combinations for the α-amino acids and unnatural residues in a heterogeneous helical structure, including different ratios between the α-amino acids and the unnatural building blocks (or two different types of unnatural residues).![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1 Hybrid structure.
1
:1 Hybrid structure.
1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 hybridization is one of the most common structural types to form hybrid helical peptidic foldamers. This important proportional form has led to the construction of well-defined artificial structures that exhibit a variety of helical configurations. The substitution pattern of residues on unnatural amino acids has a profound effect on the type of helical structure and the stability of helical conformation. Typical examples of the 1:1 hybrid crystal structure include α/β peptides,74 α/γ peptides,75–78 α/sulfono-γ-AA peptides,79–82 β/γ peptides,83 urea/isosteric amide peptides,84 urea/isosteric carbamate peptides,84,85etc. Gellman and co-workers reported a large set of α/β peptide crystal structures with 1:1 residue hybrid alternation (Fig. 9).74 The nature of these helical structures changes with the cyclic constraint embedded in the β residues and other structural parameters. The chain length is another important factor in the helix formation of these foldamers; longer ones prefer the 14/15-helix over the 11-helix.
:1 hybridization is one of the most common structural types to form hybrid helical peptidic foldamers. This important proportional form has led to the construction of well-defined artificial structures that exhibit a variety of helical configurations. The substitution pattern of residues on unnatural amino acids has a profound effect on the type of helical structure and the stability of helical conformation. Typical examples of the 1:1 hybrid crystal structure include α/β peptides,74 α/γ peptides,75–78 α/sulfono-γ-AA peptides,79–82 β/γ peptides,83 urea/isosteric amide peptides,84 urea/isosteric carbamate peptides,84,85etc. Gellman and co-workers reported a large set of α/β peptide crystal structures with 1:1 residue hybrid alternation (Fig. 9).74 The nature of these helical structures changes with the cyclic constraint embedded in the β residues and other structural parameters. The chain length is another important factor in the helix formation of these foldamers; longer ones prefer the 14/15-helix over the 11-helix.
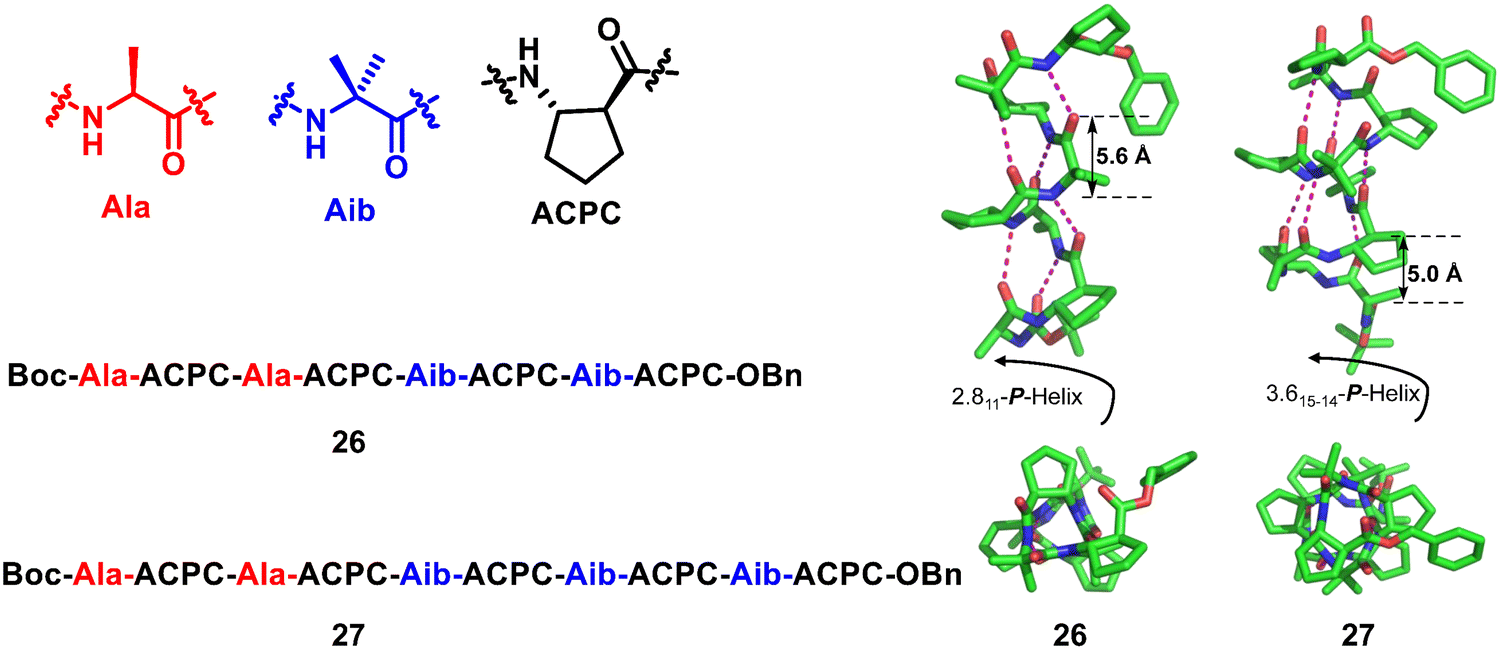 | ||
| Fig. 9 Chemical and crystal structures of representative helical 1:1 hybrid α/β peptides 26 (CCDC 640724) and 27 (CCDC 640726). The magenta dashed lines indicate intramolecular hydrogen bonds. Some hydrogen atoms are omitted for clarity. | ||
In a series of single crystal structures obtained by the Gopi group,75,78 the Balaram group86 and the Gellman group,76,77 the helical configurations of the 1:1 hybrid α/γ peptide foldamers include the 12-helix (right-handed foldamers 28 and 29)75,78,86 and the 12/10-helix (right-handed foldamer 30)76,77 (Fig. 10). For the formation of these two helical configurations, geometrically constrained γ-amino acids play an important role. We have also recently reported the formation of robust pinwheel-shaped 13-helix structures in the 1:1 α/sulfono-γ-AA peptides where the bulky sulfonamide groups and intramolecular hydrogen bonding induce helical conformation of the backbone (Fig. 11).79–82
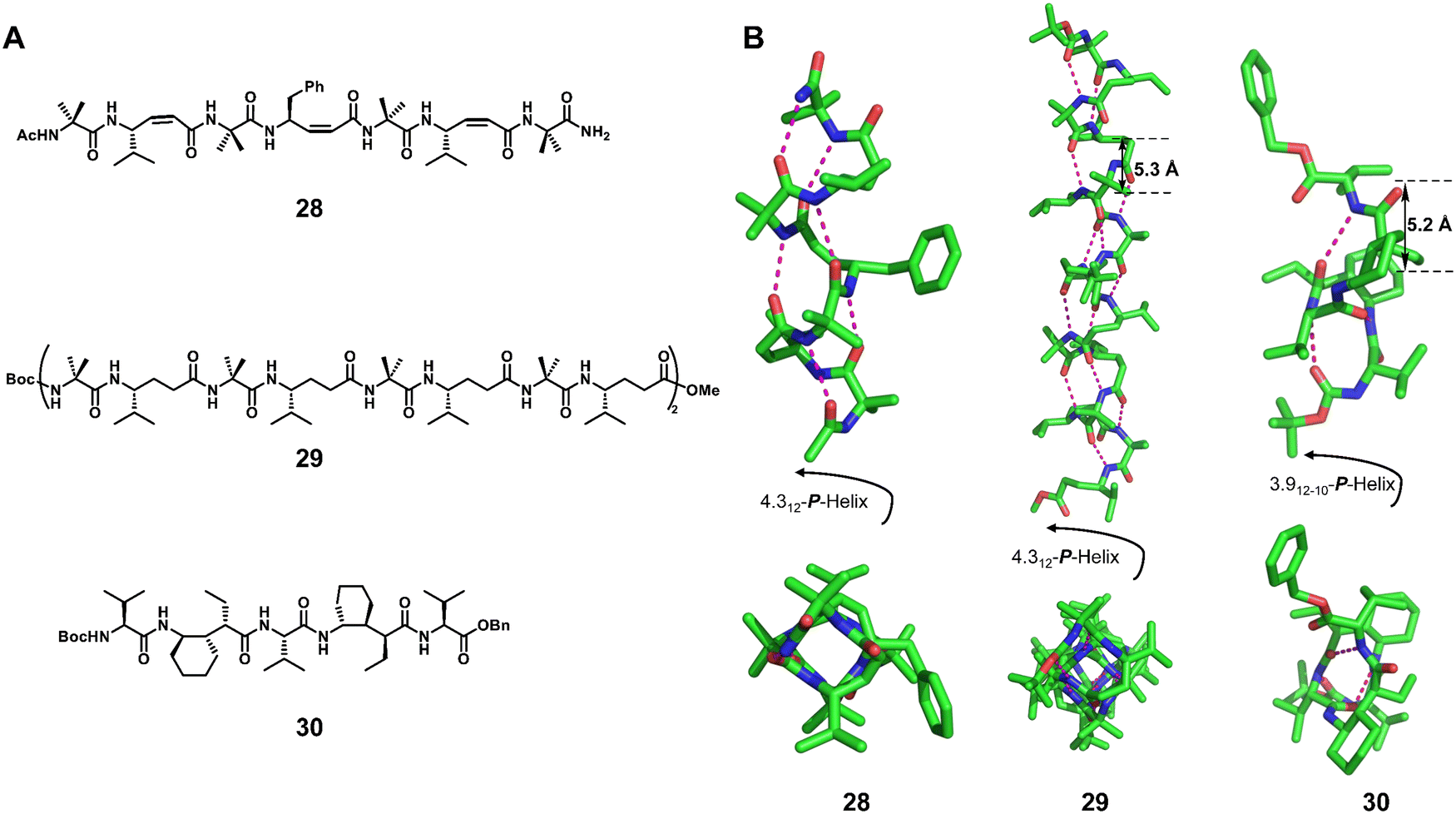 | ||
| Fig. 10 Chemical (A) and crystal structures (B) of representative helical 1:1 hybrid α/γ peptides 28 (CCDC 1437322), 29 (CCDC 881178) and 30 (CCDC 1056030). The magenta dashed lines indicate intramolecular hydrogen bonds. Some hydrogen atoms are omitted for clarity. | ||
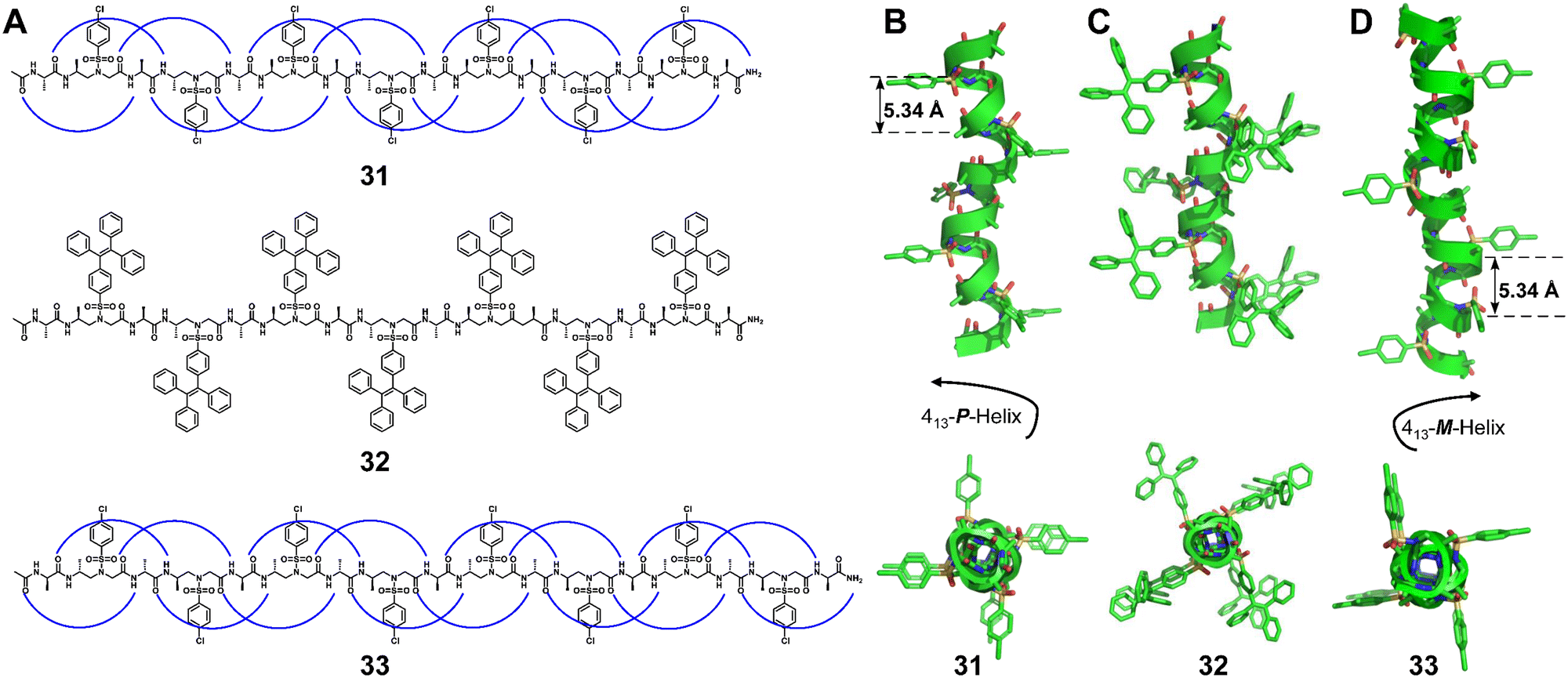 | ||
| Fig. 11 Chemical and crystal structures of representative 1:1 L-α/L-sulfono-γ-AApeptides 31 (A and B, CCDC 1541641) and 32 (A and C, CCDC 1946119), and 1:1 D-α/D-sulfono-γ-AApeptide 33 (A and D, CCDC 1976024). | ||
The helical handedness of the 1:1 α/sulfono-γ-AA peptide foldamers is controlled by the chirality of their chiral side chains and irrelevant to achiral sulfonyl side chains. For example, 1:1 L-α/L-sulfono-γ-AA hybrid foldamers exhibit a right-handed 13-helix pattern (Fig. 11),79–82 while 1:1 D-α/D-sulfono-γ-AA hybrid foldamers form a left-handed 13-helix (Fig. 11).82 Notably, the racemate of these two types of foldamer forms a heterochiral coiled-coil-like dimer meshing together in a manner akin to two gears of opposite sense.82 More intriguingly, dimerization of the 1:1 L-α/L-sulfono-γ-AA hybrid foldamer through a covalent bond at a deliberately selected position formed a stable peptidomimetic zipper, with a unique tertiary structure.80 The 3D self-assembly is driven by intra/intermolecular hydrogen bonding and C–Cl⋯Cl–C halogen bonding.
Besides 1:1 α/unnatural amino acid hybrid helical foldamers, 1:1 β/γ peptides (Fig. 12, right-handed foldamer 34), urea/isosteric amide peptides (Fig. 12, right-handed foldamer 35) and urea/isosteric carbamate peptides (Fig. 12, right-handed foldamer 36) also demonstrate interesting helical structures. Interestingly, a βγ-dipeptidic segment in the β/γ peptides has the same number of atoms between the N- and C-termini as an α-tripeptide, and both helices contain 13-atom ring H-bonds.83 This suggests that the β/γ-peptide may be a promising unnatural foldamer for functional mimicry of natural α-helices. For 1:1 alternate urea/isosteric amide peptides and urea/isosteric carbamate peptides, the folding is dominated by urea units whose prominent helical-folding character counterbalances the slightly lower or limited helix forming ability of isosteric amide and isosteric carbamate units, respectively.84
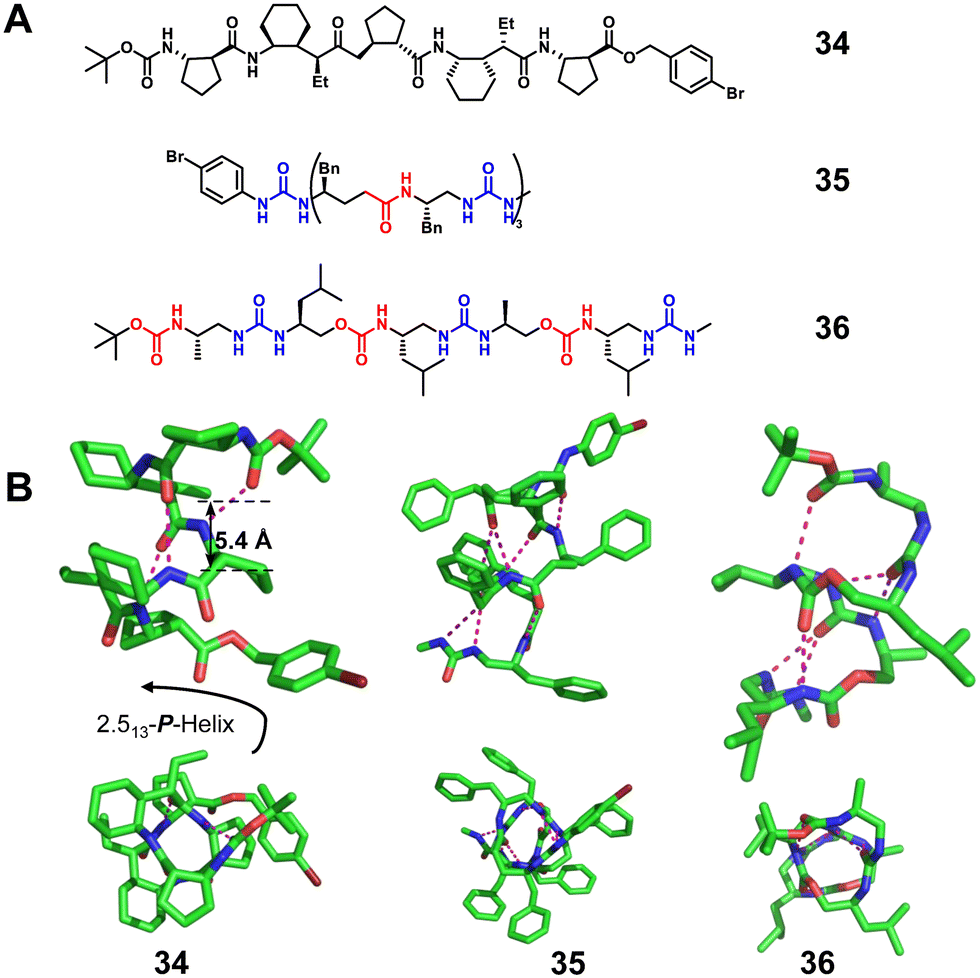 | ||
| Fig. 12 Representative examples of 1:1 β/γ peptide 34 (CCDC 812441), 1:1 urea/isosteric amide peptide 35 (CCDC 913814) and 1:1 urea/isosteric carbamate peptide 36 (CCDC 913811). The magenta dashed lines indicate intramolecular hydrogen bonds. Some hydrogen atoms are omitted for clarity. | ||
:1 hybrid structure.
Mixing α amino acids with other unnatural residue types such as β or γ-amino acids in a 2:1 pattern was also expected to significantly enlarge the diversity of unnatural helical structures.87–89 The Gellman group reported several crystal structures of 2:1 α/β-peptides88 (Fig. 13A, right-handed foldamer 37) and α/γ-peptides87 (Fig. 13B, left-handed foldamer 38). The crystal structures of 2:1 α/β-peptides are fully helical, and all helices display the i, i + 3 CO⋯H–N hydrogen bonding pattern.88 Similarly, each of the 2:1 α/γ-peptides displays a helical conformation with the maximum number of the i, i + 3 CO⋯H–N hydrogen bonds (12-atom hydrogen-bonded rings) formed in each case.87 This internal hydrogen-bonding pattern is comparable to that of the 10-helix observed for some regular foldamers consisting of only α amino acids.
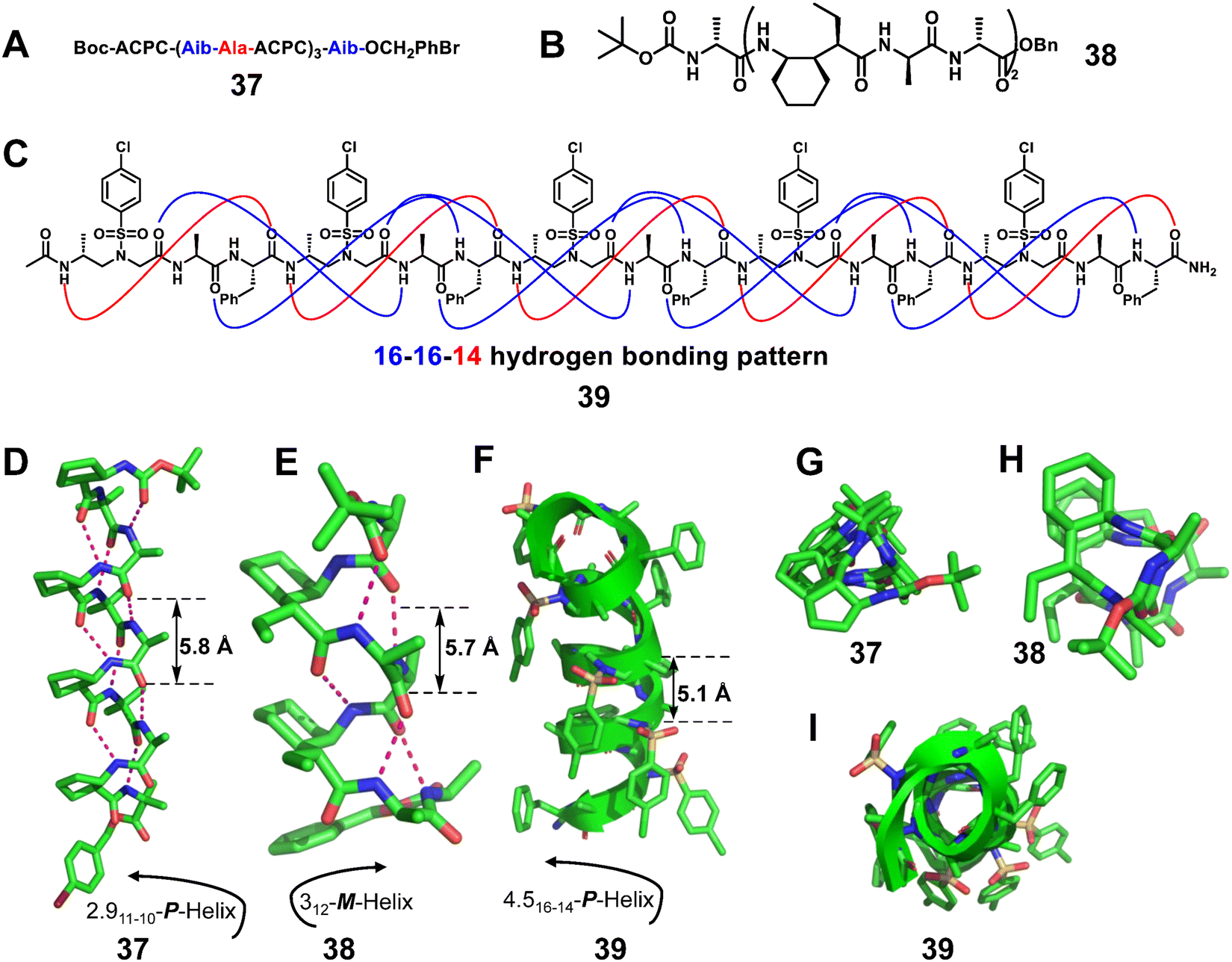 | ||
| Fig. 13 Representative examples of 2:1 α/β-peptide 37 (A, D and G, CCDC 685821), 2:1 α/γ-peptide 38 (B, E and H, CCDC 867043) and 2:1 L-α/D-sulfono-γ-AApeptide 39 (C, F and I, CCDC 1564298). The magenta dashed lines indicate intramolecular hydrogen bonds. Some hydrogen atoms are omitted for clarity. | ||
Our group reported the X-ray crystal structures of heterogeneous helical foldamers consisting of a 2:1 pattern of L-α/D-sulfono-γ-AA amino acids, and elucidated their folded conformation at the atomic level (Fig. 13C, 39).89 All crystals were shown to adopt well-defined right-handed helical conformations with the pattern of 16/14 helix.
:2 hybrid structure.
The Gellman group87,88,90 reported the crystal structures of representative 1:2 α/unnatural amino acid hybrid helical foldamers. They explored this type of helical secondary structure in α/β-peptide foldamers (Fig. 14A, right-handed foldamer 40) and α/γ-peptide foldamers (Fig. 14B, right-handed foldamer 41). The two groups of crystal structures here provide similar helical secondary structures. The i, i + 3 CO⋯H–NH-bonded helices were formed by backbones that contain either a 1:2 repeating pattern of α- and β-amino acid residues or a 1:2 repeating pattern of α- and γ-amino acid residues. These new structures should be valuable additions to unnatural helical foldamer scaffolds that can be used to create specifically functionalized surfaces by rational design.
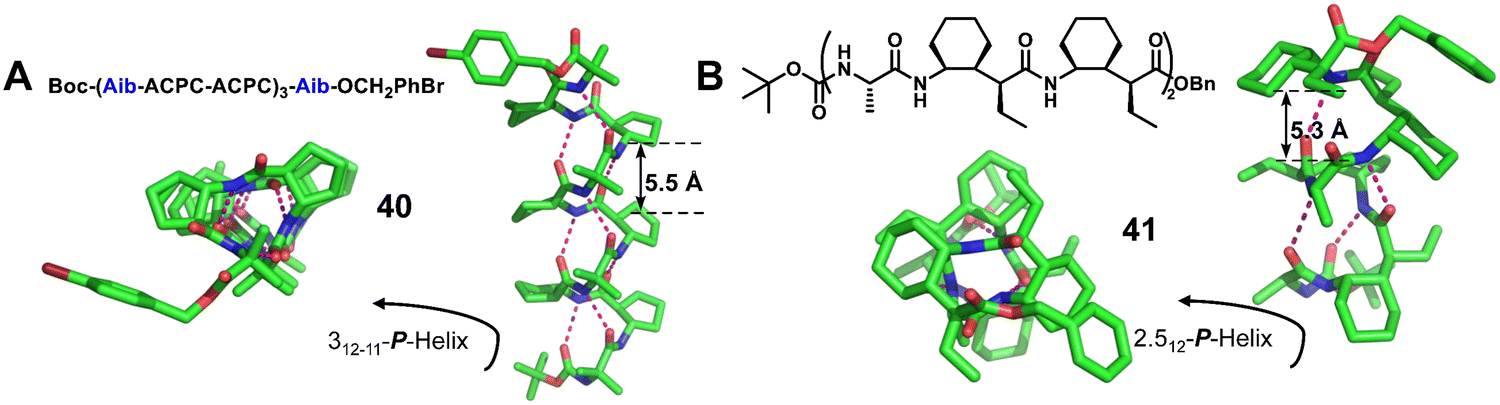 | ||
| Fig. 14 Representative examples of 1:2 α/β-peptide 40 (A, CCDC 685819) and 1:2 α/γ-peptide 41 (B, CCDC 867046). The magenta dashed lines indicate intramolecular hydrogen bonds. Some hydrogen atoms are omitted for clarity. | ||
3. Applications of unnatural helical peptidic foldamers
Due to their relatively canonical folding behaviours, functionalized conventional helical peptides have extensive applications in biology, catalysis, molecular recognition, supramolecular interaction, etc. Unnatural helical peptidic foldamers, which are structurally different from the conventional α-helix, have also gained increasing attention and show promise in conventional protein helical segment mimicry. Specifically, owing to their enhanced resistance to proteolytic degradation, better bioavailability and improved chemodiversity, they enjoy significant merits for biological applications like host-defense peptide mimicry and modulation of disease-related protein–protein interactions. Although many structures are lipophilic and crystallized from organic solvents, circular dichroism and stability studies revealed that their helical conformations did not change in protic solvents. On the basis of the well-established helical parameters and folded conformations, the design of functional unnatural helical peptidic foldamers opens a new avenue for the development of protein segment mimics. Below, we will just highlight a few typical examples developed in the past 10 years.3.1 Biological applications
3.1.1.1 β-Peptides. Palecek et al. reported that homogeneous β-peptides based on aurein 1.2, which is a helical host defense peptide, exhibited antifungal activity (Fig. 15A, 42).95 They next designed α/β-peptide analogues with different α/β-amino acid organizations by replacing α-amino acid residues with β-amino acid residues to generate ααβ, αααβ and ααβαααβ repeating units.94 The helical α/β-peptides showed high selectivity against Candida albicans over human red blood cells. Park et al. reported that the enantiomeric glycosylated cationic block co-β-peptides inhibit biofilm formation by Staphylococcus aureus (Fig. 15B, 43).99 The copolymer induces helical structure formation and subsequent antibacterial activity through the interaction of cationic β-amino acid residues with anionic bacterial membranes. Even in vivo, these copolymers were able to eradicate S. aureus biofilms effectively. As can be seen from these examples, the incorporation of β-amino acids enhances the antifungal activity and selectivity of the natural helical antimicrobial peptide.
 | ||
| Fig. 15 Representative lead compounds of antibacterial β-peptides 42 (A) and 43 (B). | ||
3.1.1.2 Peptoids. The peptoids also show resistance to proteolysis and display low cytotoxicity, which are attractive properties for biological applications as antimicrobial molecules, and thus have continued to develop in recent years. Differences in side chains and backbones between peptoids and natural peptides should affect their mode of action and activity. Seo et al. introduced several aromatic side chains for the hydrophobization of peptoids to improve selectivity against bacteria (Fig. 16, 44).104 However, higher hydrophobization of peptoids does not always equate with potent activity, but depends on the type of bacteria, as reported by Jenssen et al.105 Faure and co-workers reported the introduction of the cationic 1,2,3-triazolium moiety to develop antimicrobial peptoids, which showed potent activities and selectivity against Enterococcus faecalis and S. aureus strains (Fig. 16, 45).106 In addition, longer nonamer peptoids show different mechanisms of action, such as pore formation or membrane penetration, compared to 6-mer peptoids.106 Peptide/peptoid hybrids were also useful in developing AMPs with various activities and selectivity, which are embodied in α-peptide/α-peptoid hybrids (Fig. 16, 46) and Lys-based α-peptide/β-peptoid hybrids (Fig. 16, 47).96,107
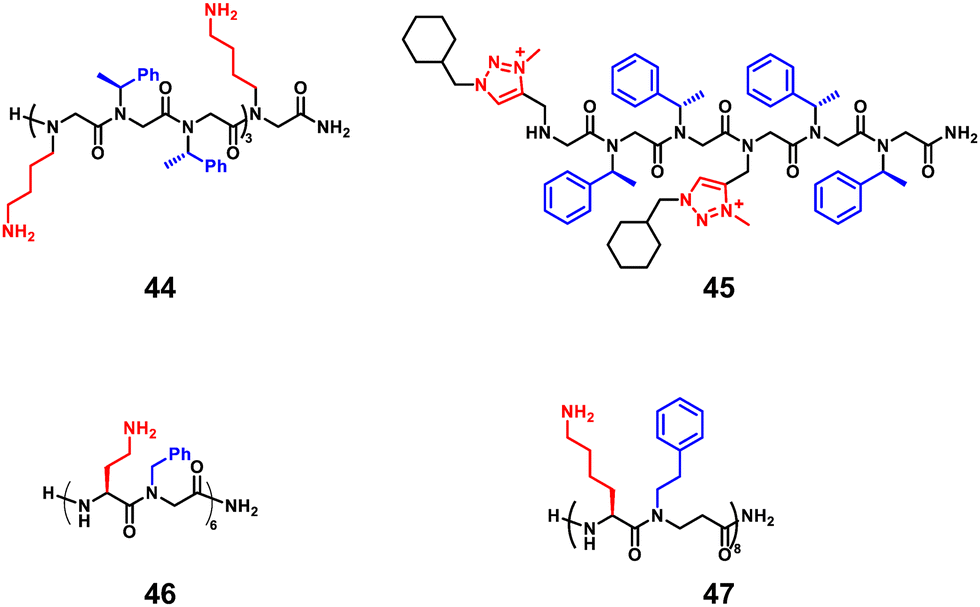 | ||
| Fig. 16 Representative lead compounds of antibacterial peptoids. | ||
3.1.1.3 γ-Peptides. The bactericidal mechanism of γ-peptides is the same as that of traditional AMPs, which is achieved by disrupting bacterial membranes. Maillard et al. developed unique foldamers composed of constrained heterocyclic γ-amino acids built around a thiazole ring, named ATC (4-amino (methyl)-1,3-thiazole-5-carboxylic acid).92 Amphipathic ATC-based helical foldamers showed strong antimicrobial activity against Gram-positive and Gram-negative bacteria without significant hemolytic activity (Fig. 17A, 48). In addition, Gopi and colleagues reported α/γ-hybrid antimicrobial peptides containing α-Lys and γ-Leu residues, which showed potent antimicrobial activity by inhibiting bacterial growth, causing cell morphological changes and destroying bacterial membranes (Fig. 17B, 49).108
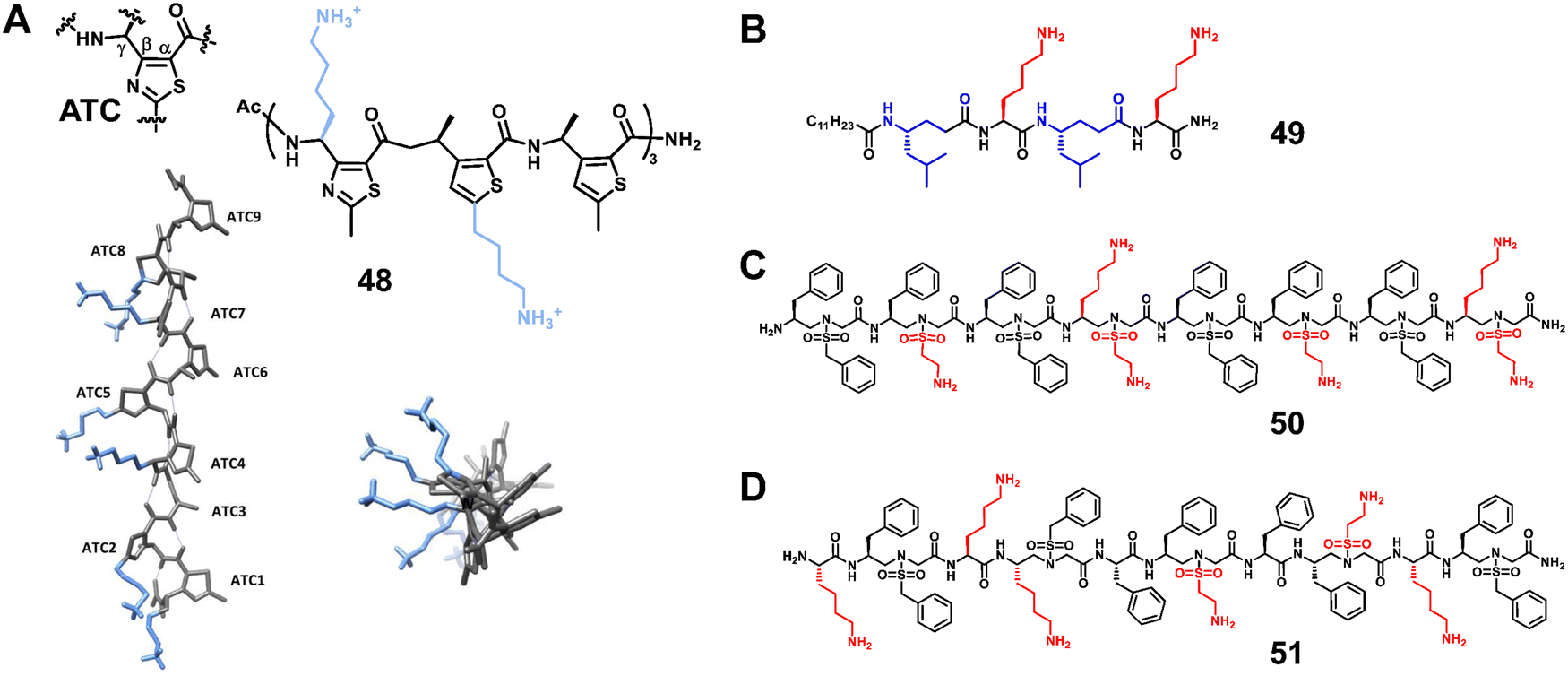 | ||
| Fig. 17 Representative lead compounds of antibacterial γ-peptides 48 (A) and 49 (B) and sulfono-γ-AApeptides 50 (C) and 51 (D). Reproduced with permission from ref. 92. Copyright 2020, American Chemical Society. | ||
3.1.1.4 Sulfono-γ-AApeptides. Our group designed and synthesized a series of sulfono-γ-AApeptides, and investigated their application in host-defense peptide mimicry.101,103 The lead homogeneous sulfono-γ-AApeptides showed broad-spectrum and potent activity against a range of multi-drug-resistant Gram-positive and Gram-negative bacteria (Fig. 17C, 50).103 The heterogeneous helical 1
:1 α/sulfono-γ-AApeptides also showed potent activity against Gram-positive and Gram-negative bacteria (Fig. 17D, 51).101 These sulfono-γ-AApeptides are highly resistant to enzymatic degradation relative to their natural peptide analogues.
3.1.1.5 Oligoureas. Meanwhile, as another family of potent antimicrobial foldamers, oligoureas have been further developed. The membrane lytic activity of natural AMPs is usually highly dependent on their secondary structures, whereas oligoureas not only show higher biostability but also maintain their tendency to fold in water, which allows them to be used as AMP mimics. Guichard et al. reported an amphiphilic cationic helical oligourea AMP mimetic that was found to be active against bacterial forms of Bacillus anthracis encountered in vivo and to exert partial protection in cutaneous and inhalational models of infection with B. anthracis (Fig. 18A, 52).109 Very recently, Kichler et al. reported new types of helical oligoureas derived from cell-penetrating foldamers by altering the distribution of histidine and arginine-type residues on the surface of the oligourea helix (Fig. 18B, 53).97 In addition, they also prepared and tested novel amphiphilic block cofolders consisting of oligourea and peptide stretches, in which polar and charged residues are located in the peptide stretches and more hydrophobic residues are located in the oligourea stretches. Some of them were found to show potent antibacterial activity even in the presence of 50% serum and also have promising antifungal properties.
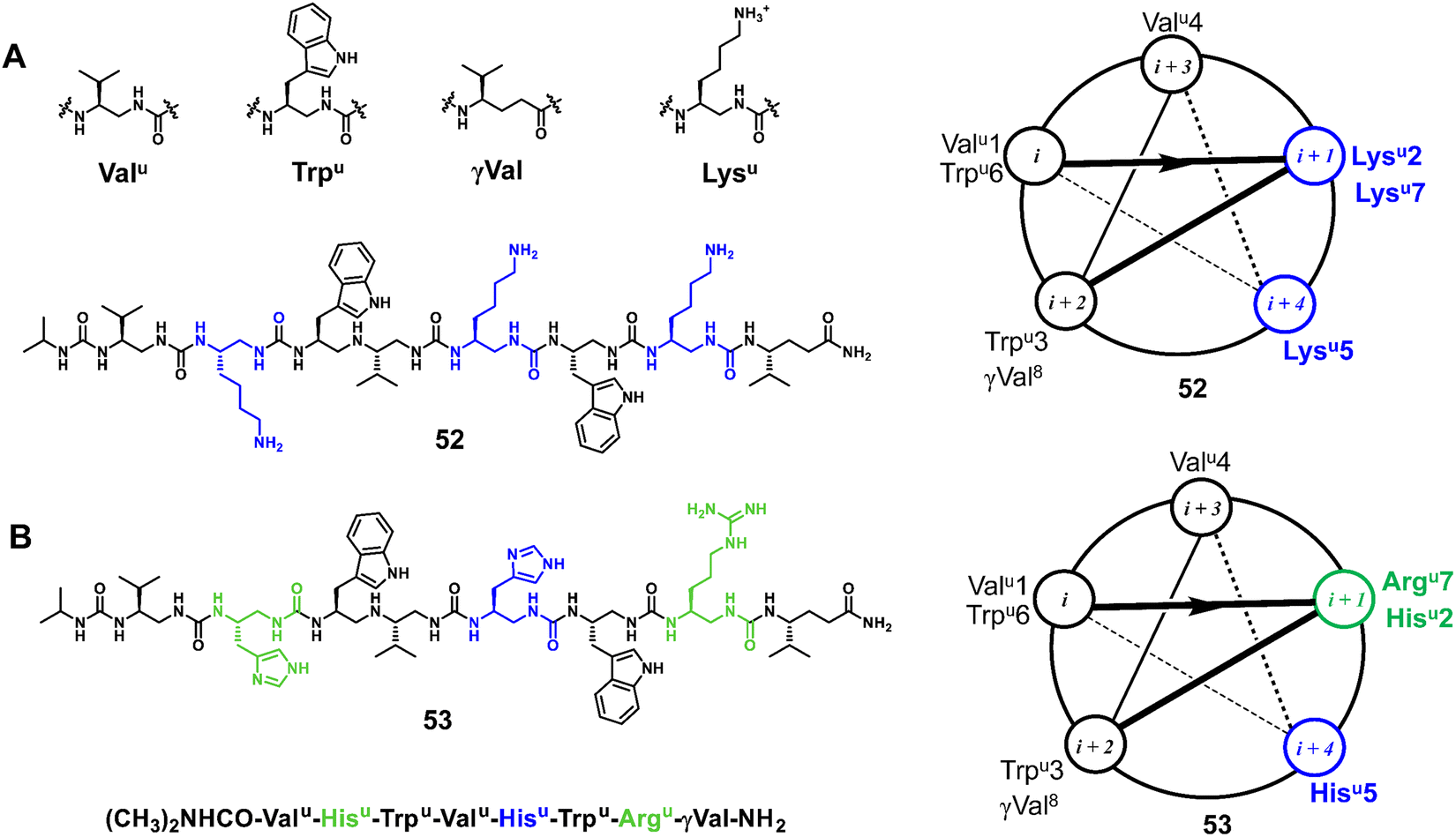 | ||
| Fig. 18 Representative lead compounds of antibacterial oligoureas 52 (A) and 53 (B). | ||
3.1.2.1 p53–MDM2/MDMX. p53 is a tumor suppressor that regulates cell division and prevents tumor formation. MDM2 and MDMX are oncogenic proteins, which share considerable structural homology, and their binding to the N-terminus of p53 causes the inhibition of p53's transcriptional activity and promotes its degradation.114 The interaction between p53 and MDM2/MDMX plays a crucial role in the homeostasis of mammalian cells, and overexpression of MDM2/MDMX reduces the expression level of p53, leading to the exacerbation of cancers (Fig. 19A).115–120 After decades of development, the interaction of p53–MDM2/MDMX has been well studied, which provides a good platform to verify the effectiveness of helical unnatural mimics.
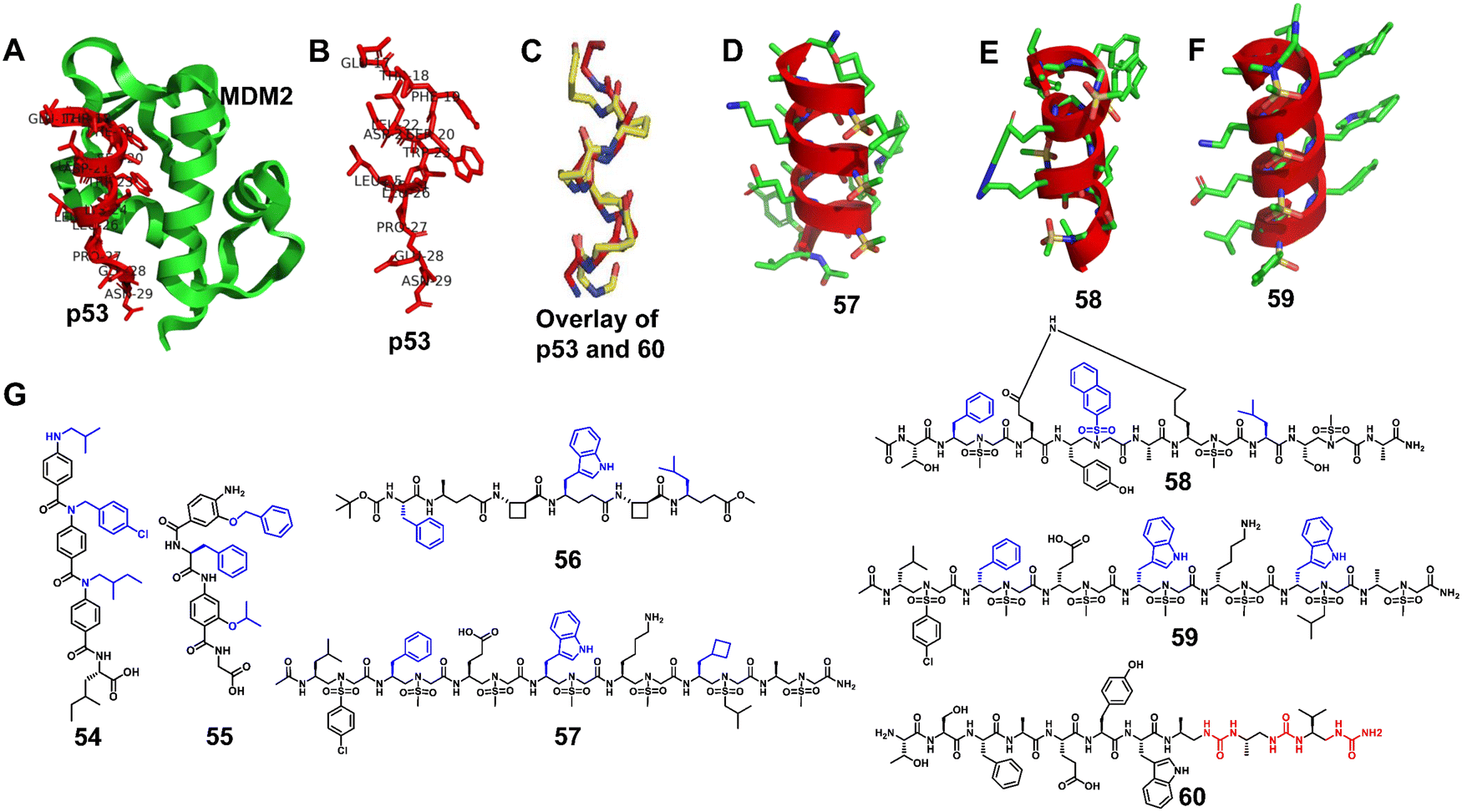 | ||
| Fig. 19 (A) Interaction of p53 with the crystal structure of MDM2 (PDB: 1YCR); (B) p53; (C) overlay of p53 and 60; (D–F) cartoon structures of 57–59; (G) chemical structures of lead compounds 54-60. | ||
Wilson et al. synthesized and tested a library of 73 trimeric aromatic foldamers (Fig. 19B, 54) in a high-content cell screening setting (U2OS osteosarcoma cells assessing for cell number, caspase-3 level, autophagy, and actin filament arrangements as the end points).121 The design was biased toward p53/MDM2 protein–protein interaction, and screening afforded foldamers with μM activity in disrupting this interaction. They next combined α-amino acids with an oligoamide residue to generate hybrid polyaromatic amino acid foldamers (Fig. 19B, 55).122 The introduction of chirality enhances the ability of the designed molecule to recognize the MDM2 protein, and the central amino acid enables more in-depth probing of the conformational space. Notably, the hybrid foldamer retained proteolytic stability. Wilson and Aitken also reported that hybrid α/β/γ-peptides composed of α- and γ-amino acids and trans-2-aminocyclobutanoic acid (tACBC) form 12- or 13-membered helices (Fig. 19B, 56).123 The designated α/β/γ-peptides bound MDM2 to inhibit the p53/MDM2 interaction and showed high tolerance against digestive enzymes. Our group developed three new classes of sulfono-γ-AApeptide (left-handed homogeneous L-sulfono-γ-AApeptides,124 right-handed 1:1 α/L-sulfono-γ-AApeptides125 and right-handed homogeneous D-sulfono-γ-AApeptides53) inhibitors to mimic the helical domain of p53 and inhibit the p53–MDM2/MDMX interaction (Fig. 19, 57–59). The resulting lead homogeneous L-sulfono-γ-AApeptide is one of the most potent unnatural peptidomimetic inhibitors against this interaction reported so far. The 1:1 α/L-sulfono-γ-AApeptides could bind tightly to MDM2/MDMX and block the p53–MDM2/MDMX interaction. The lead D-sulfono-γ-AApeptide can also bind well to MDM2 and competitively block the p53–MDM2 interaction at the binding site. Very recently, Guichard and co-workers designed the urea peptide TSFAEYWuAuLuAu. They introduced Wu at the seventh position to improve protease resistance and introduced AuLuAu to induce helix formation (Fig. 19, 60).126 This helical urea-type peptide binds to MDM2 with high affinity.
3.1.2.2 BCL-2 family. Another important type of PPI involves the BH3 helical segment of the BCL-2 family. The BCL-2 family has attracted great interest in the oncology community, especially the MCL-1 and BCL-xL proteins. They are regulated by the apoptosis regulators Bak and Bad in response to cellular stress, and overexpression of the BCL-2 family allows oncogenic cells to escape cell death.127,128
Structure-guided rational design of α/β-peptide foldamers was used by Fairlie et al. to optimize binding through the synergistic modification of three residues, leading to double-digit nM binders to MCL-1, albeit at the cost of affinity for Bcl-xL (Fig. 20A–C, 61).129 Gellman et al. developed hybrid helical α/β peptides as binders of the BH3-cleft from BCL-xL (Fig. 20A–C, 62).130 The systematic study was performed on BCL-xL and MCL-1, starting from an 18-mer Bim BH3 domain. The best lead compound led to affinity in the 50 nM range for BCL-xL and 170 nM range for MCL-1. It was possible to increase affinity by increasing the peptide length to a 26-mer, and the best compound was shown to have a similar single-digit nanomolar affinity for four members of the BCL-2 family (BCL-2, BCL-xL, BCL-w, and MCL-1), making this unnatural peptidic foldamer a tighter binder than the corresponding α-peptide. In addition, the hybrid α/β peptide proved to be more resistant to proteolysis relative to the Bim BH3 α-peptide. Gellman et al. further investigated the isomeric β3-to-β2-amino acid substitution within a helical α/β-peptide. The results showed that β2 residues can be helix-destabilizing but can also increase the peptide's affinity for a protein binding partner (Fig. 20A–C, 63).131
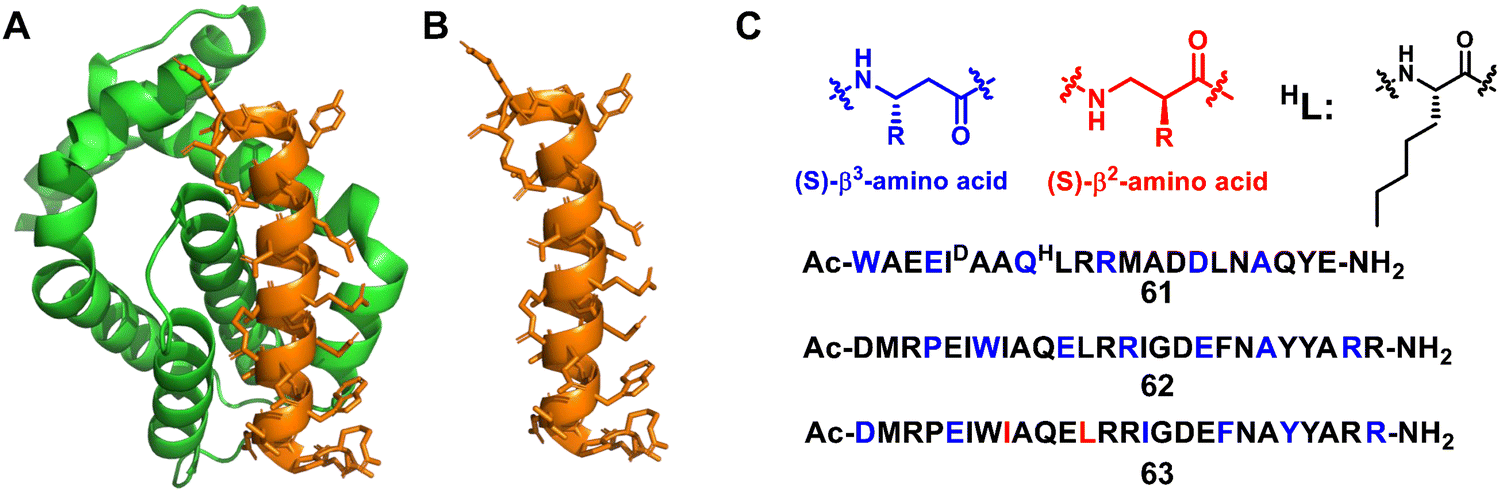 | ||
| Fig. 20 (A) The structure of Bcl-xL from the Bim/Bcl-xL complex (PDB: 3FDL); (B) the Bim BH3 α-helix; (C) sequences of lead compounds 61–63; β3 and β2 residues are highlighted in blue and red, respectively. | ||
3.1.2.3 GLP-1 mimic. GLP-1 (glucagon-like peptide 1)132–134 is a physiological peptide secreted from L-cells in the small intestine, which binds to the GLP-1 receptor (GLP-1R)135–138 on pancreatic β-cells and stimulates insulin secretion. However, the half-life of GLP-1 is very short, and it will be rapidly degraded by proteases.139–143 Unnatural helical peptidic foldamers can improve the stability of GLP-1, which is essential for the development of diabetes drugs.
Starting from the 7–37 N-terminus peptide, a hybrid foldamer was developed by Gellman et al. to introduce β-amino acids at selected positions (26, 30, and 34) but it led to a large drop in activity in a cAMP production assay (Fig. 21, 64).144 However, the replacement of the β-amino acids with their cyclic analogues introduced a more rigid conformation, leading to activity in the same range as that of GLP-1 and avoiding rapid proteolytic degradation. Further research showed that replacement of α-amino acids with β-amino acids generally resulted in a modest decrease in the stimulation of cAMP production and β-arrest in recruitment, but for some replacement sets cAMP production is more affected than β-arrest in recruitment.145 The central portion of GLP-1 appears to be critical for achieving bias toward β-arrest in recruitment. The same group also investigated several new α/β hybrid foldamers of GLP-1 with each containing five α to β replacements in the C-terminal half of the peptide, which helps to establish the scope of the replacement of α-amino acids with β-amino acids (Fig. 21, 65).146
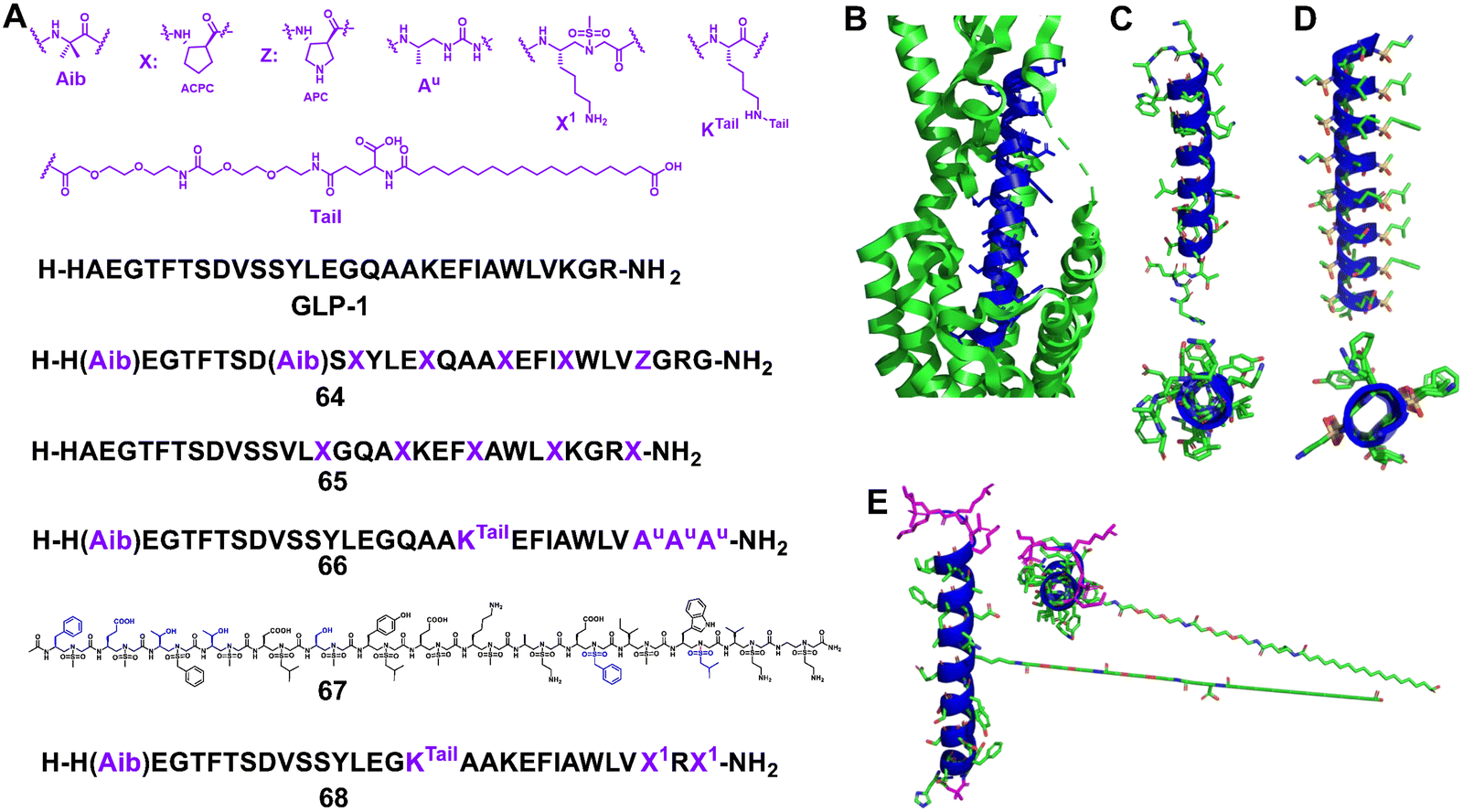 | ||
| Fig. 21 (A) Sequences of GLP-1 and lead compounds 64–68; (B) GLP-1 binds to GLP-1R (Protein Data Bank: 5VAI). GLP-1 (7–36) is shown in blue and GLP-1R is represented as a green cartoon; (C) the helical domain of GLP-1; (D) cartoon structure of 67; (E) cartoon structure of 68. | ||
In 2019, Goudreau and Guichard et al. reported that peptide–oligourea hybrid foldamers could be used to mimic GLP-1 (Fig. 21, 66).147 In their report, the urea-peptide, in which residues 14–21 of GLP-1 are replaced with YuEuAuAuAuAu, exhibited high degradation stability in mouse plasma while maintaining its blood glucose inhibitory effect in mice. They have also shown that hybrid GLP-1 with a single ureido residue replacement at position 2 exhibits antidiabetic properties and longer duration of action via selective enhancement of cAMP signalling and altered GLP-1R trafficking.148
Our group reported the use of helical sulfono-γ-AApeptides with entire unnatural backbones to mimic the structure and function of GLP-1 (Fig. 21D, 67).149 Our findings show that these new helical foldamers can mimic the residues on multiple faces of the GLP-1 α-helical domain and achieve nM potencies. Among them, the lead foldamer showed potent GLP-1R agonistic activity in both cell-based experiments and oral glucose tolerance tests. Notably, the degradation of the lead sulfono-γ-AApeptide was not observed in the enzymatic stability and human serum stability experiments, thus circumventing the poor stability of regular GLP-1 peptides, which augments their biological potential. This work also represents the first example of foldameric peptidomimetics based on an entire unnatural backbone for GLP-1 mimicking.
However, as there are still conformational differences between the GLP-1 α-helix and homogeneous sulfono-γ-AApeptides, the activity of homogeneous sulfono-γ-AApeptide based GLP-1 foldamers was observed with at least 300-fold loss compared to natural GLP-1, hindering their future clinical development as GLP-1R agonists. Very recently, our group developed a series of novel α/sulfono-γ-AApeptide hybrids to modify and improve the pharmaceutical properties of GLP-1, by replacing certain amino acid residues with unnatural sulfono-γ-AA building blocks (Fig. 21E, 68).150 After investigation of the structure–activity relationship of these GLP-1 analogues, the optimized peptidomimetic hybrids were proved to be potent GLP-1R agonists with even higher potency than GLP-1, and with remarkably improved stability (t1/2 > 14 days in serum vs. t1/2 < 24 h for GLP-1). Moreover, after long PEG and fatty acid chains were conjugated to the α/sulfono-γ-AApeptides’ main backbone, the yielded peptide hybrids demonstrated much-prolonged bioactivity and favourable PK in vivo.
3.1.2.4 PTH1 mimic. The parathyroid hormone receptor 1 (PTHR1) is a member of the B-family of G protein-coupled receptors (GPCR); these receptors are activated by long polypeptide hormones. Parathyroid hormone 1 (PTH1) is an 84 amino acid long natural agonist of PTHR1, and an N-terminal fragment of PTH1, PTH (1–34), is used clinically to treat osteoporosis.151–154 However, conventional peptides in the 20–40-mer length range are rapidly degraded by proteases, which limits their biomedical utility.
Gellman et al. modified the 1–34 fragment of PTH1, which is known to adopt a helical conformation upon receptor binding, with up to 7 positions substituted with β-amino acids (Fig. 22, 69).151 Importantly, the modified peptides retained full agonism. Notably, the EC50 values were comparable to that of the parent 1–34 PTH1 sequence. In addition, two modified peptide analogues were studied in vivo to assess their effect on calcium levels. One of the two exhibited similar intrinsic activity at the receptor but with a much longer duration of action. This property may be caused by β-amino acid substitutions, resulting in increased proteolytic stability.
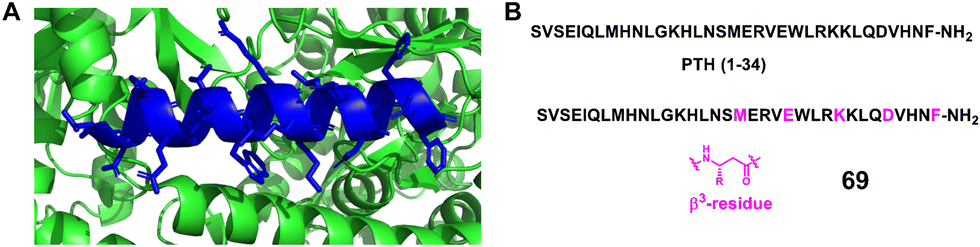 | ||
| Fig. 22 (A) The crystal structure of PTH (15–34) bound to the extracellular domain of PTHR1 (PDB: 3C4M). The extracellular domain is represented as a green cartoon, and the PTH (15–34) ligand is shown in blue; (B) sequences of PTH (1–34) and mimics 69. | ||
They further used the PTHR1-ligand system to explore the impact of the broadly distributed replacement of α-amino acid residues with β-amino acid residues on susceptibility to proteolysis and agonist activity.152 This work has led to the discovery of new PTHR1 agonists, manifesting potent agonist activity in cellular assays and displaying remarkable resistance to proteolysis, and remaining active after extended exposure to simulated gastric fluid.
3.1.2.5 ASF1. Anti-silencing function 1 (ASF1)155–158 is a conserved H3/H4 histone chaperone involved in histone dynamics during replication, transcription, and DNA repair. It is overexpressed in proliferating tissues including many tumors, and hence has emerged as a promising therapeutic target. Depletion of ASF1 inhibits the growth of various cancer cell lines and enhances the sensitization of cells to chemotherapeutic agents. Very recently, Guichard, Ochsenbein and coworkers reported a foldamer/peptide hybrid inhibitor of ASF1 by using oligoureas as α-helix mimics (Fig. 23).159 Based on the C-terminal sequence of H3, an ASF1-inhibiting urea peptide with four urea residues inserted in the centre of the helix was designed to form the interaction interface between ASF1 and H3. The urea-peptide hybrid showed high stability against proteolysis under stringent conditions compared to the cognate α-helical peptide and demonstrated its binding to ASF1 at low μM values. In addition, the X-ray co-crystal structure of the foldamer and ASF1 was also obtained and analyzed. These results are expected to make a significant contribution to the future development of ASF-targeted drug discovery.
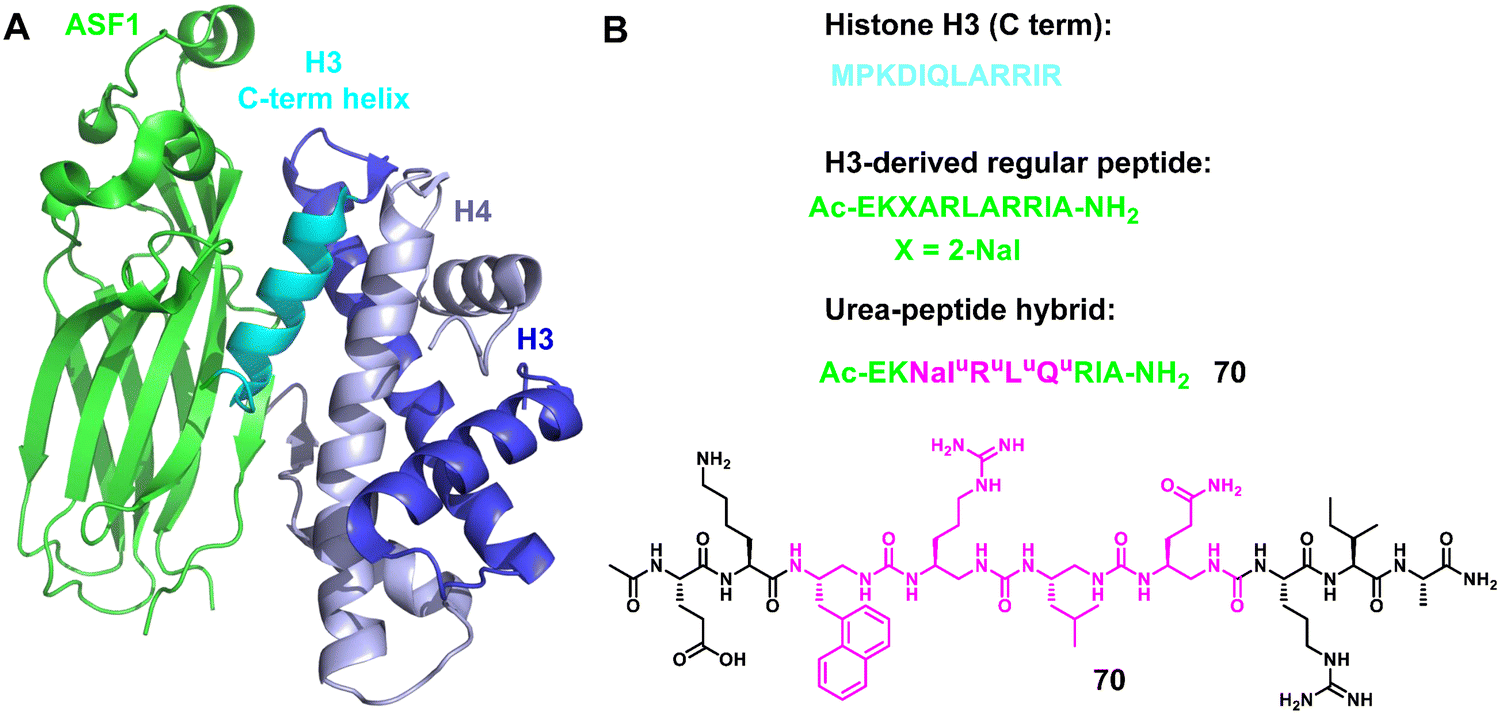 | ||
| Fig. 23 (A) Structure of the ternary complex hASF1A (1–175)-H3-H4 (PDB: 2io5); (B) sequences of regular H3 C-term helix and helical oligourea α-helix mimics 70. | ||
3.1.2.6 VEGF mimic. The vascular endothelial growth factor (VEGF)160,161 is an essential factor in pathophysiology angiogenesis. In cancer cells, VEGF is overexpressed and promotes angiogenesis, causing an exacerbation of cancer.162 Disruption of VEGF/VEGF receptor interaction in cancer patients inhibits the development of new and pre-existing tumor blood vessels. Consequently, VEGF has become an important therapeutic target for handling solid tumors.
About a decade ago, Gellman's group began to develop α/β-peptide inhibitors of VEGF signaling.163 Their initial work to develop α/β-peptides that bind to the receptor-recognition sites on the VEGF homodimer was based on disulfide-crosslinked 19-mer peptides discovered by Fairbrother through phage display (Fig. 24A and B).163 They found that resistance to proteolysis could be enhanced by replacing α-amino acids with β-amino acids, but these substitutions resulted in decreased affinity for VEGF. They then designed new α/β-peptides based on the three-helix bundle Z-domain scaffold developed by phage display targeting the VEGF monomer (Fig. 24C).164 The designed two-helix α/β-peptides exhibited high affinity toward VEGF, which was similar to that of the parent α-peptide. The two-helix α/β-peptides showed anti-proliferation activity against HUVEC cells, indicating that they antagonize VEGF/VEGF receptor signaling. They further investigated how a two-helix “mini-Z-domain” could be modified to contain β and other nonproteinogenic residues by iterative incorporation of unnatural residues while retaining the target binding epitope.165 The resulting α/β-peptides are less prone to proteolysis than their parent α-peptide, and some of these α/β-peptides are comparable to the full-length Z-domain in terms of affinity for receptor-recognition surfaces on the VEGF homodimer.
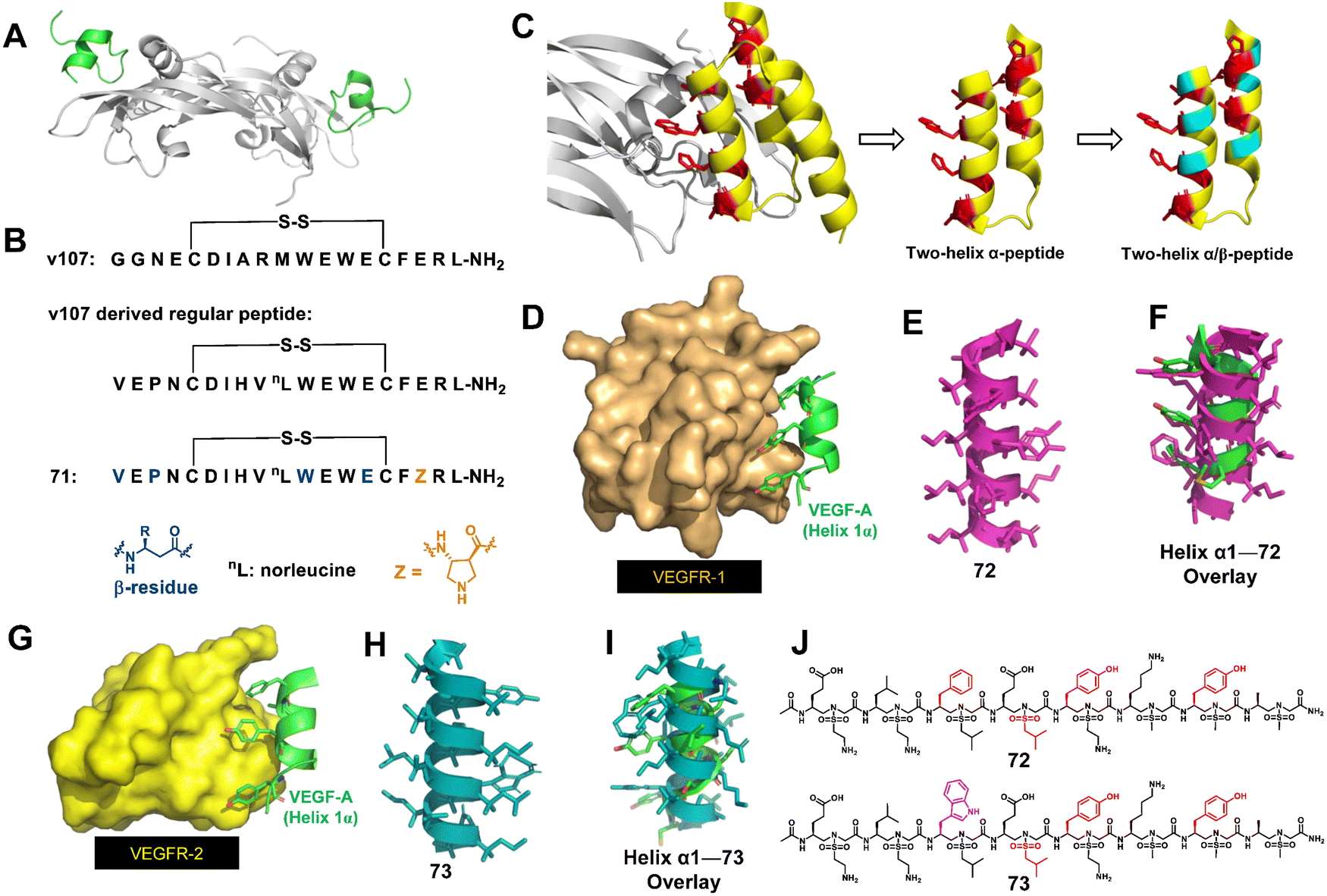 | ||
| Fig. 24 (A) High-resolution structure of the VEGF9–108 homodimer (gray) bound to peptide v107 (v107 is green; PDB: 1KAT); (B) sequences of the regular peptide and helical α-helix mimics 71. (C) Strategy for the design of α/β-peptide mimics of Z-VEGF (shown in yellow and red). The red residues indicate selected residues that contact VEGF8–109 (shown in gray) in the cocrystal structure. Sites targeted for non-natural amino acid substitutions are shown in teal. The figure is based on PDB: 3S1K. (D) Binding interaction of key residues on helix-α1 of VEGF-A (green) with VEGFR-1 (bronze), PDB: 1flt; (E) sulfono-γ-AApeptide mimic 72; (F) superimposition of 72 (magenta) with critical residues of helix-α1 (green); (G) binding interaction of key residues on helix-α1 of VEGF-A (green) with VEGFR-2 (yellow), PDB: 3V2A; (H) sulfono-γ-AApeptide mimic 73; (I) overlay of 73 (teal) with critical residues of helix-α1 (green); (J) chemical structures of sulfono-γ-AApeptides 72 and 73. | ||
Recently, our group developed helical homogeneous sulfono-γ-AApeptide foldamers to mimic the critical helix binding segment on VEGF-A (helix α1) to inhibit the interaction of VEGF-A with VEGFR-1 and VEGFR-2 (Fig. 24D–I).166 Cell-based angiogenesis assays demonstrated that these unnatural foldamers could be either pro- or anti-angiogenic and upregulate or downregulate angiogenesis, and thus the angiogenesis can be effectively switched and regulated. The distinct angiogenesis signaling is due to the specific binding of our helical mimetics to VEGFR-1 or VEGFR-2, respectively. Targeting VEGFR-1 specifically (72) is expected to release more VEGF from VEGFR-1 binding and shift the dial for VEGFR-2 interactions, leading to the amplification of angiogenesis. Conversely, specific binding to VEGFR-2 (73) inhibits the VEGF-A/VEGFR-2 PPI, thereby blocking the angiogenic signaling pathway. Therefore, these two unnatural helical peptides 72 and 73 with only one residue difference can be used to intervene in disease conditions caused by angiogenesis imbalance. Another highlight of this work was that 72 and 73 were very stable and did not show any significant degradation, whereas the peptide bearing native amino acid residues was completely degraded.
3.1.2.7 β-Catenin/BCL9. Wnt/β-catenin signalling is crucial in both normal embryonic development and throughout the life cycle of an organism.167–170 Furthermore, aberrant Wnt signalling has been implicated in a variety of diseases, especially cancer and fibrosis. As a central mediator of signal transduction, β-catenin controls the expression of several critical genes that regulate the cell cycle and apoptosis. The transcriptional activation of the Wnt/β-catenin signalling pathway depends on the formation of the β-catenin supercomplex involving BCL9 or BCL9-like (B9L), as well as the T cell factor (Tcf)/lymphoid enhancer-binding factor (Lef) family of transcriptional factors. The crystal structure of the β-catenin/BCL9/TCF-4 ternary complex revealed that the helical segment of BCL9 (351–374) interacts with the binding groove in β-catenin.171 The key residues of BCL9 (R359, L363, L366, I369 and L373) are located on the same side of the BCL9 helix and form hydrophilic and hydrophobic contacts with the binding surface of β-catenin (Fig. 25B). Therefore, directly targeting the β-catenin/BCL9 PPI is a promising strategy to block the Wnt pathway.
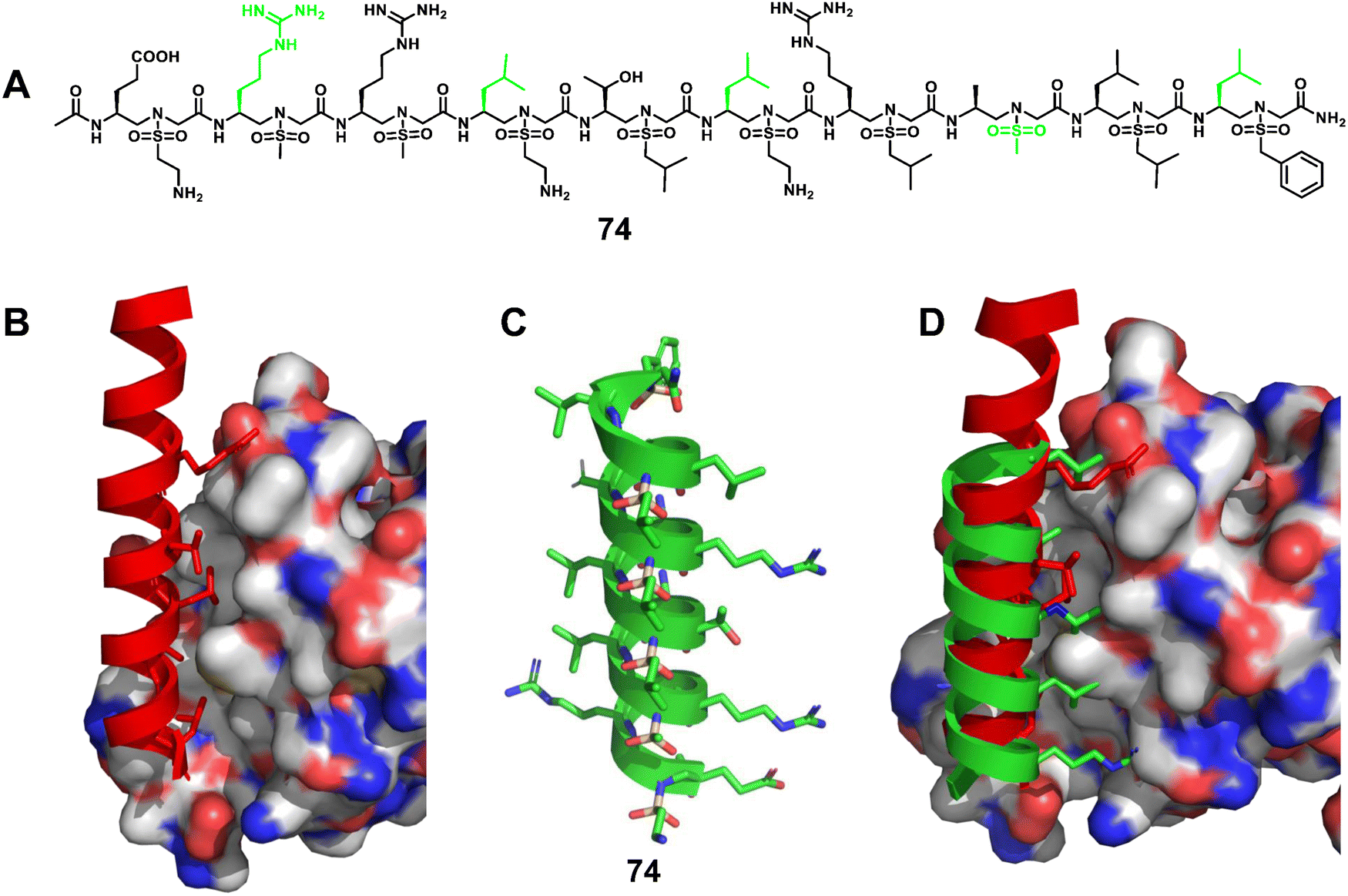 | ||
| Fig. 25 (A) Chemical structure of sulfono-γ-AApeptide 74 investigated for the disruption of the β-catenin/BCL9 interaction; (B) cartoon representation of the residues of BCL9 (red) critical for binding to β-catenin, shown as sticks, PDB: 2GL7; (C) proposed cartoon structure of 74; (D) overlay of 74 with the critical residues of BCL9 on the binding surface of β-catenin (PDB: 2GL7). | ||
Elucidation of the mechanism of interaction between β-catenin and BCL9 basded on their co-crystal complex has facilitated the discovery of inhibitors targeting the β-catenin/BCL9 PPI, but there are still only a few reports. Based on the obtained single crystals, we designed and synthesized a series of homogeneous sulfono-γ-AApeptides to mimic key residues of the BCL9 helix (Fig. 25A–D).172 The competition assay revealed that sulfono-γ-AApeptides can selectively disrupt the β-catenin/BCL9 PPI with higher potency. More interestingly, these sulfono-γ-AApeptides could enter SW480 cancer cells, bind to β-catenin and disrupt the β-catenin/BCL9 PPI, and showed superior cellular activity than regular BCL9. In addition, enzymatic stability studies showed the remarkable stability of the helical sulfono-γ-AApeptides without degradation for 24 h in the presence of pronase. BCL9 as a reference was completely degraded by pronase, leaving no intact peptides. This work not only represents a successful example of a fully unnatural helical peptidic foldamer that mimics α-helices and disrupts PPIs, but is also an excellent example of a potent, selective, and cell-permeable unnatural foldamer that disrupts the β-catenin/BCL9 PPI.
3.1.2.8 Glucagon mimic. Glucagon173–175 is an insulin counter-regulatory hormone secreted by the α-cells of the pancreas that raises blood glucose levels by stimulating gluconeogenesis and glycogenolysis, thus circumventing a hypoglycaemic state. Glucagon exerts its action by binding to its receptor, glucagon receptor (GCGR), one of class B G protein coupled receptors (GPCRs), expressed in a variety of tissues including liver and brain. Glucagon is considered an effective therapy for treating severe hypoglycaemia; however, its plasma half-life is only a couple of minutes, which limits its wide clinical applications.176–179 More recently, our group reported a series of α/sulfono-γ-AApeptide hybrids to modify and modulate the properties of glucagon (Fig. 26A).180 These compounds were rationally designed to introduce unnatural sulfono-γ-AApeptide residues and long-chain fatty acids into native glucagon. The best lead compound showed enhanced stability toward proteolytic degradation and prolonged in vivo activity (Fig. 26B). This work is the first example of glucagon stabilization using sulfono-γ-AA amino acid residues.
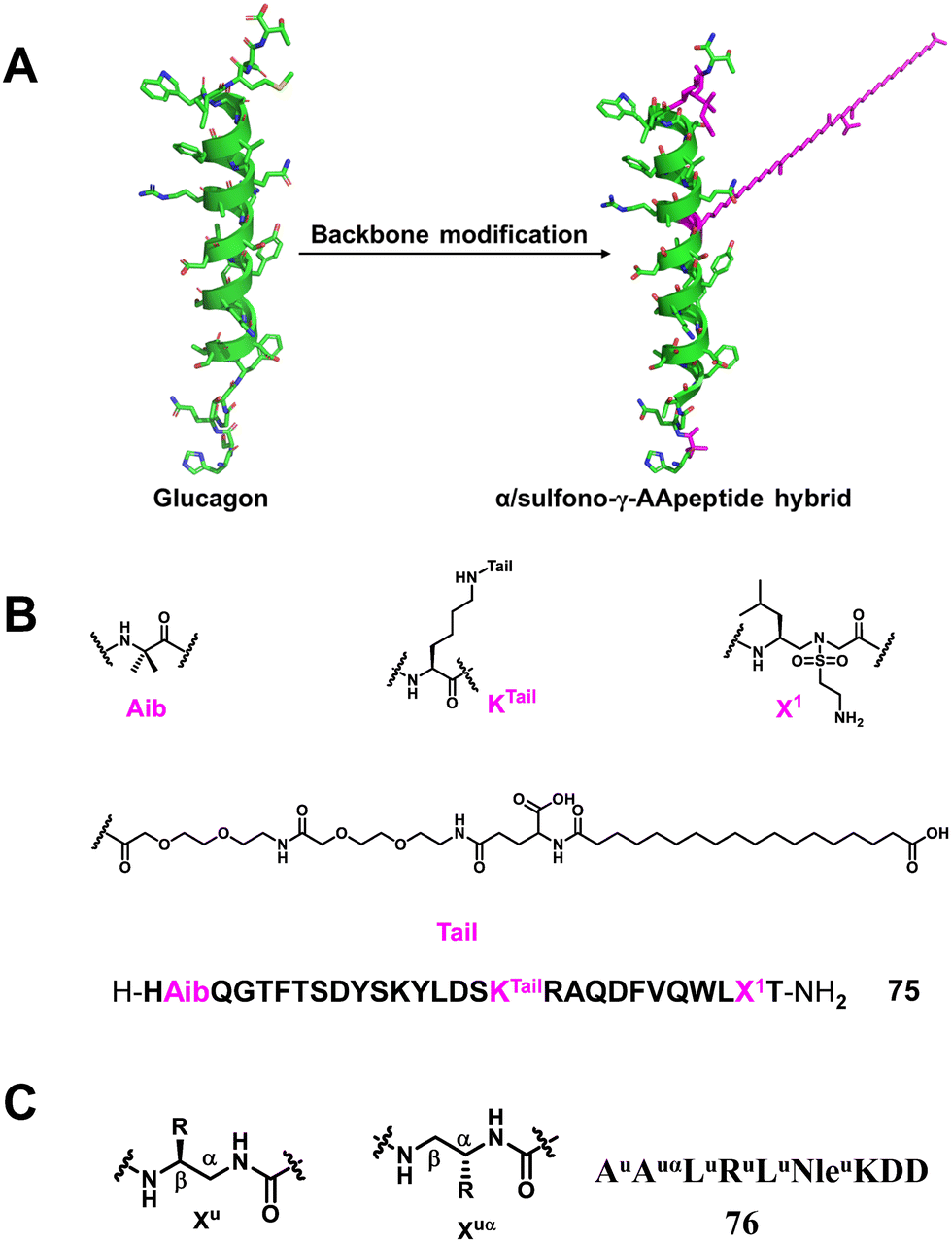 | ||
| Fig. 26 (A) Strategy for the design of α/sulfono-γ-AApeptide mimics; (B) sequence of the lead α/sulfono-γ-AApeptide hybrid glucagon analogue 75; (C) the lead peptide–oligourea hybrid 76 derived from SRC peptides and binding to hVDR LBD. | ||
3.1.2.9 SRC/VDR. The vitamin D receptor (VDR), a nuclear hormone receptor (NHR), is implicated in the regulation of many biological functions such as bone homeostasis, cell growth, and immunity. For VDR to transcribe 1,25-dihydroxyvitamin D3 [1,25-(OH)2D3] as a ligand, molecular binding of the VDR ligand-binding domain (LBD) to coactivators such as steroid receptor coactivators (SRC1, SRC2, SRC3) is required.181 Therefore, inhibiting NHR–SRC interaction is a potential therapeutic strategy to treat cancer associated with elevated expression of SRC. A series of peptide–oligourea hybrids based on the structure of SRC1-2 and SRC2-3 tridecapeptides consisting of a central consensus LXXLL motif were designed by Guichard and co-workers to inhibit VDR, and coactivator interactions were also developed.126 What they designed was a hexa-urea (AuAuαLuRuLuNleuKDD, 76) peptide in which residues 1–10 of SRC2–3 (ENALLRYLLDKDD) were replaced, and the lead peptide–oligourea hybrid showed 10-times stronger binding affinity to VDR compared with SRC2–3 (Fig. 26C). This strategy can generate peptide analogs that are significantly resistant to proteolytic degradation, a characteristic often desired when developing peptide therapeutics.
3.1.2.10 Virus entry. Unnatural helical foldamers have been used to mimic natural virus entry peptides to block infectivity. During human immunodeficiency virus-1 (HIV-1) infection, the envelope glycoprotein subunit gp41 folds into a stable six-helix bundle structure (6HB) formed by the interaction of the C-terminal heptad repeat (CHR) segment with the N-terminal heptad repeat (NHR) segment, promoting viral and cell membrane fusion.182,183 Therefore, CHR mimics may inhibit the formation of bundles and inhibit the fusion process of HIV. Based on the previous work of their group, Gellman and co-workers synthesized α/β-peptides containing cyclic β-amino acid residues based on the C-terminal heptad-repeat (CHR) segment of HIV protein gp41 (Fig. 27A–D).184 α/β-Peptides with the ααβαααβ pattern were designed to stabilize the helix structure through the formation of a salt bridge. The α/β-peptides showed higher affinity against the NHR segment than the parent α-peptide (Fig. 27A, 77 and 78). Moreover, the introduction of β-amino acids into the sequence led to >1000-fold improvement in the half-life of the peptides against digestive enzymes (Fig. 27, 78). Thus, these unnatural α/β-peptide foldamers increased the affinity toward target proteins and improved peptide stability via the incorporation of a dense ionic side-chain array, demonstrating the potential development of therapeutic reagents.
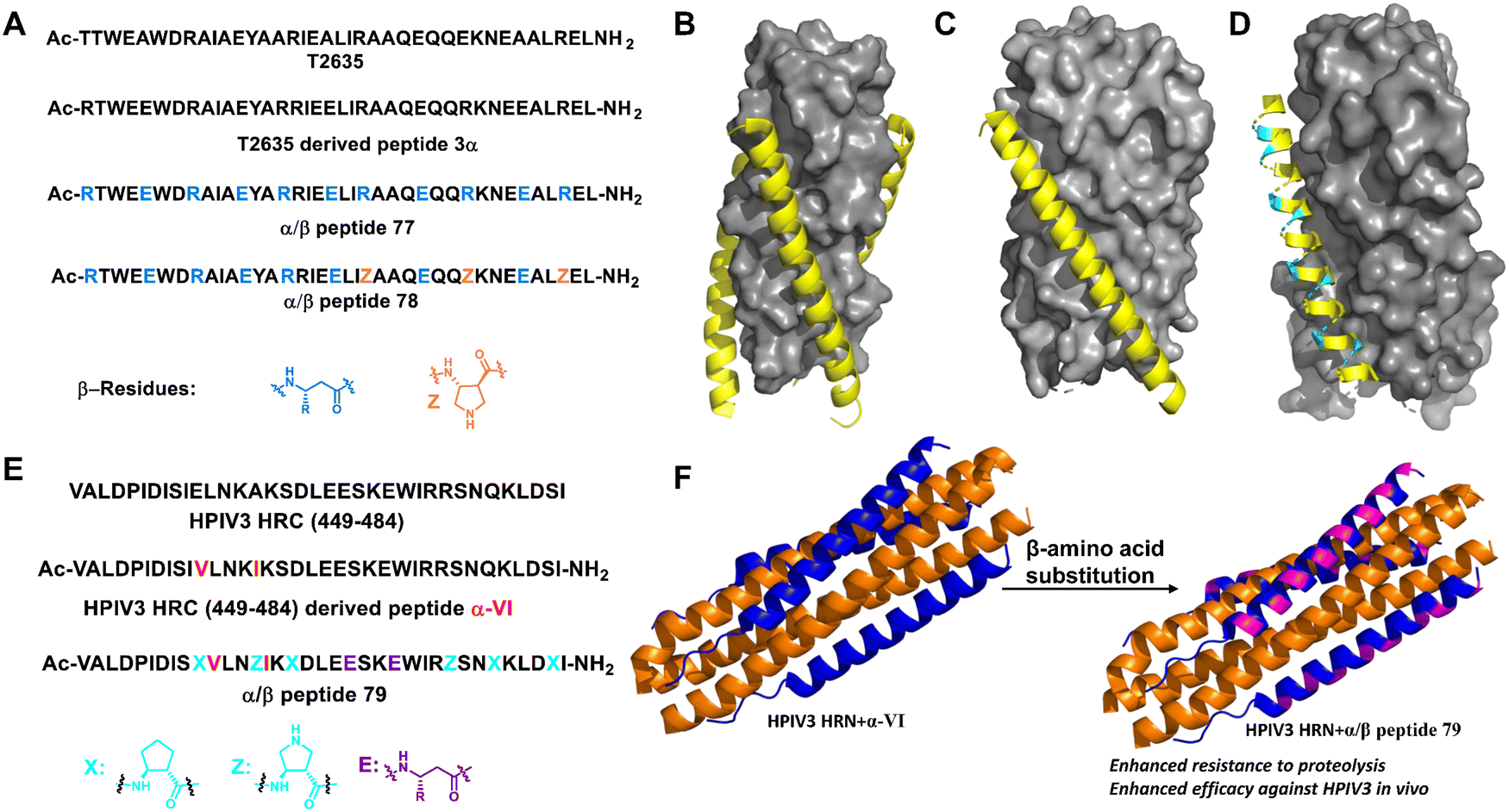 | ||
| Fig. 27 (A) Sequences of T-2635, 3α and α/β-peptides (77 and 78); (B) structure of T-2635 + N36 (PDB: 3F4Y). The gray surface represents N36 and the yellow ribbon represents T-2635; (C) structure of gp41-5 + 3α (PDB: 4DZU); (D) structure of gp41-5 + 77 (PDB: 4DZV). For parts C and D, the gray surface represents gp41-5, the yellow ribbon represents α residues, and the cyan ribbon represents β3 residues; (E) sequences of HPIV3 HRC peptide (449–484), α-VI and α/β-peptide 79; (F) six-helix bundle formed by HPIV3 HRN (139–189) (orange) and α-VI (blue) (PDB: 6NYX); (G) six-helix bundle formed by HPIV3 HRN (139–189) (orange) and α/β peptide (blue, β residues are represented in magenta) (PDB: 6VJO). | ||
Human parainfluenza virus (HPIV) infection is initiated by viral glycoprotein-mediated fusion between viral and host cell membranes. Once activated in the vicinity of the target cell, the viral fusion protein (F) undergoes a series of conformational changes, first extending the trimer subunit to allow hydrophobic domains to be inserted into the target cell membrane, and then refolding the trimer into a stable post-fusion state to promote the fusion of the virus and host cell membrane. Lipopeptides from the C-terminal heptapeptide repeat (HRC) segment of HPIV3 F inhibit infection by interfering with the structural transitions of the trimeric F assembly.185,186 However, the in vivo stability of this natural antiviral peptide is not high. Gellman and Moscona et al. modified the HRC peptide backbone by partially replacing α-amino acid residues with β-amino acid residues (Fig. 27E and F). The best α/β-lipopeptides exhibited improved persistence in vivo and higher anti-HPIV3 antiviral activity in animals relative to native α-lipopeptides.187
3.1.2.11 Amyloid protein aggregation. Abnormal protein aggregation leads to the formation of amyloid fibrils. This phenomenon has been linked to the development of Type 2 diabetes (T2D), Alzheimer's disease (AD), Parkinson's disease (PD), and so on.188–193 Unnatural helical peptidic foldamers can be successfully used to modulate the aggregation of important amyloid proteins such as islet amyloid polypeptide (IAPP), amyloid β (Aβ), and α-synuclein, thereby facilitating the development of anti-amyloid therapeutic agents.194–201
Miranker et al. reported a carboxylate functionalized oligoquinolineamide-based pentameric helical foldamer 80 for targeting the N-terminal helical domain of IAPP, which can well inhibit the aggregation of IAPP under lipid-catalyzed conditions (Fig. 28).194,195 10 equivalents of 80 completely suppressed IAPP aggregation (t50 > 16 h). They next developed a more potent IAPP aggregation antagonist 81 based on oligoquinolineamide which was functionalized with ethyl and carboxylate groups in an alternate manner (Fig. 28).196,197 It showed significant IAPP aggregation inhibition at sub-stoichiometric concentrations under lipid-catalyzed conditions.
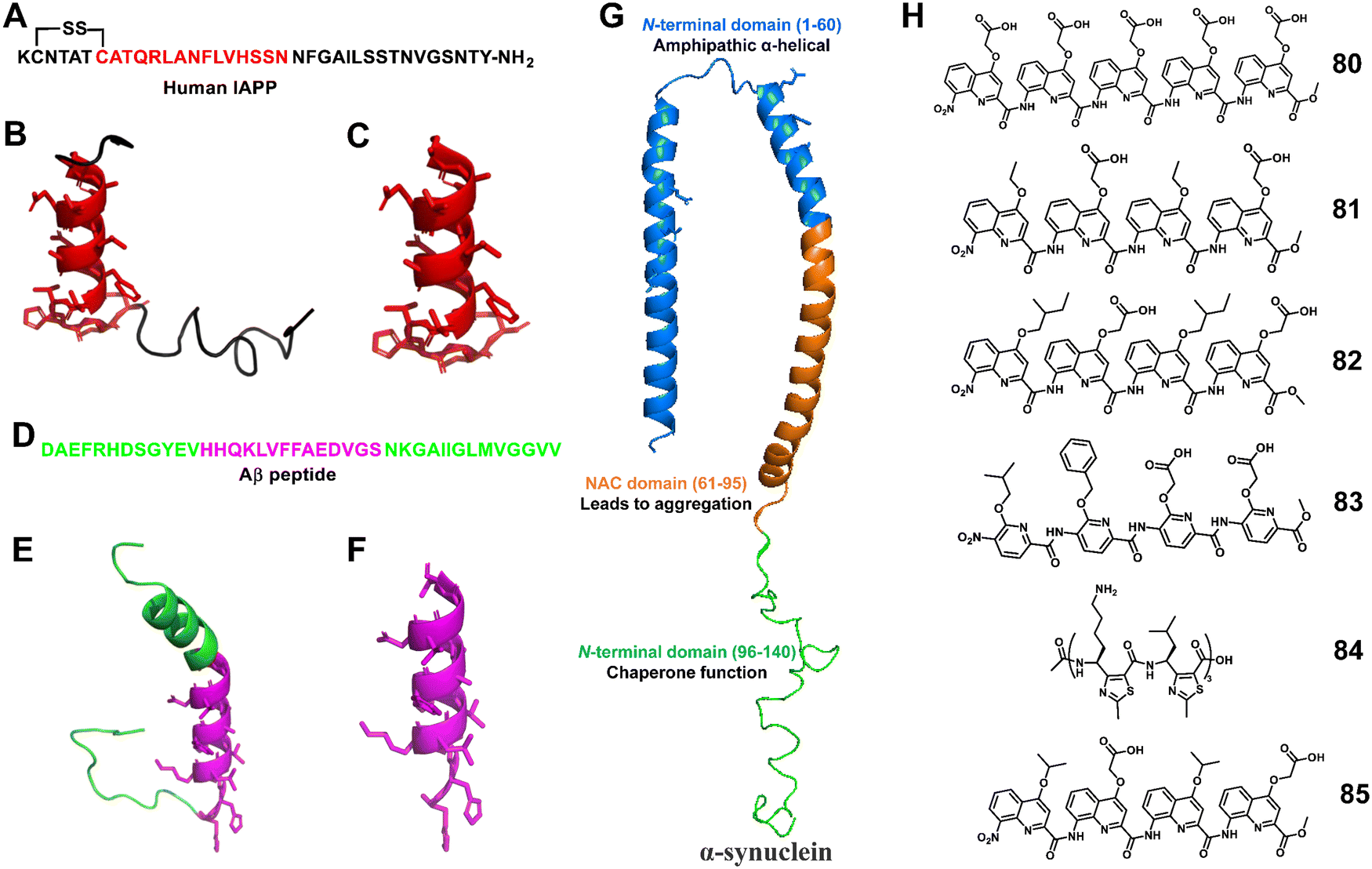 | ||
| Fig. 28 (A) Human IAPP primary sequence; (B) structure of IAPP (PDB: 5MGQ); (C) IAPP N-terminal helix subsegment; (D) sequence of the Aβ (1–40) peptide; (E) structure of the Aβ peptide (PDB:1BA4); (F) the positively charged subdomain of Aβ with side chains; (G) structure of α-synuclein (PDB: 1XQ8); (H) lead compounds 80–85. | ||
The Hamilton group employed an unnatural helical foldamer to modulate Aβ42 self-assembly. The di-anionic tetraquinolineamide-based foldamer 82 completely inhibited the Aβ42 aggregation at an equivalent concentration.198 They screened another library of unnatural helical foldamers which were anionic oligopyridylamide based compounds for modulation of Aβ42 aggregation by targeting cationic and hydrophobic segments.19983, a tetrapyridylamide based compound containing two carboxylic acid groups, completely suppressed Aβ42 aggregation including the oligomerization process at an equivalent concentration (Fig. 28). This compound also constrained Aβ42 in the α-helical state and prevented the formation of the β-sheet structure.
Moreover, Kaffy, Maillard and Ongeri demonstrated that the homogeneous thiazole-based γ-peptides adopted the 9-helix structure and interacted with amyloid-β peptides and islet amyloid polypeptide (IAPP).200 The cationic oligomer 84 showed the ability to delay the early oligomerization event of both amyloid-β peptide (Aβ1–42) and IAPP at 1/1 and 10/1 ratios, respectively. Their efforts revealed that the homogeneous thiazole-based γ-peptides could be applicable for developing PPI inhibitors by mimicking the helical structures.
Kumar et al. recently reported that oligoquinolineamide-based helical compounds modulated α-synuclein aggregation (Fig. 28).201 They screened a library of oligoquinolineamides containing carboxylic acid and different hydrophobic groups as side chains for inhibitors of α-synuclein aggregation. They identified 85 as a potent inhibitor that modulated α-synuclein aggregation by interacting with the protein's N-terminus.
3.1.2.12 Other PPIs. The Huc group developed unique aromatic oligoamide-based foldamers that adopt single-helix structures and mimic the negatively charged phosphate moieties of B-DNA.202 These mimics alter the activity of DNA-interacting enzymes used as targets for cancer therapy and they are cytotoxic only in the presence of a transfection agent. Subsequently, these phosphorylated quinoline foldamers were conjugated as a payload to trastuzumab for selective delivery to breast cancer cells.203 These phosphorylated foldamers are also expected to be used in the development of PPI inhibitors.
3.2 Catalytic applications
Enzymes, mainly proteins, are powerful catalysts capable of selectively catalyzing highly complex biochemical reactions under mild reaction conditions.204–206 However, their inherent fragility, combined with their low thermal stability, low tolerance to solvent conditions, poor substrate diversity and high production costs, severely affects their attractiveness. Therefore, there is a great need for the development of enzyme mimics. De novo design and synthesis of highly stable non-native enzymes capable of catalyzing a wide range of chemical reactions under variable conditions is a great challenge. Unnatural helical peptidic foldamers are well-stabilized oligomers with a strong tendency to adopt specific helical conformations and represent a unique platform for catalyst design utilizing the principles of enzyme function.207,208 Here, we summarize the last decade of efforts to develop unnatural helical peptidic foldamers that catalyze reactions. To date, the number of studies in this area has been modest, but the accomplishments so far suggest great potential for further development.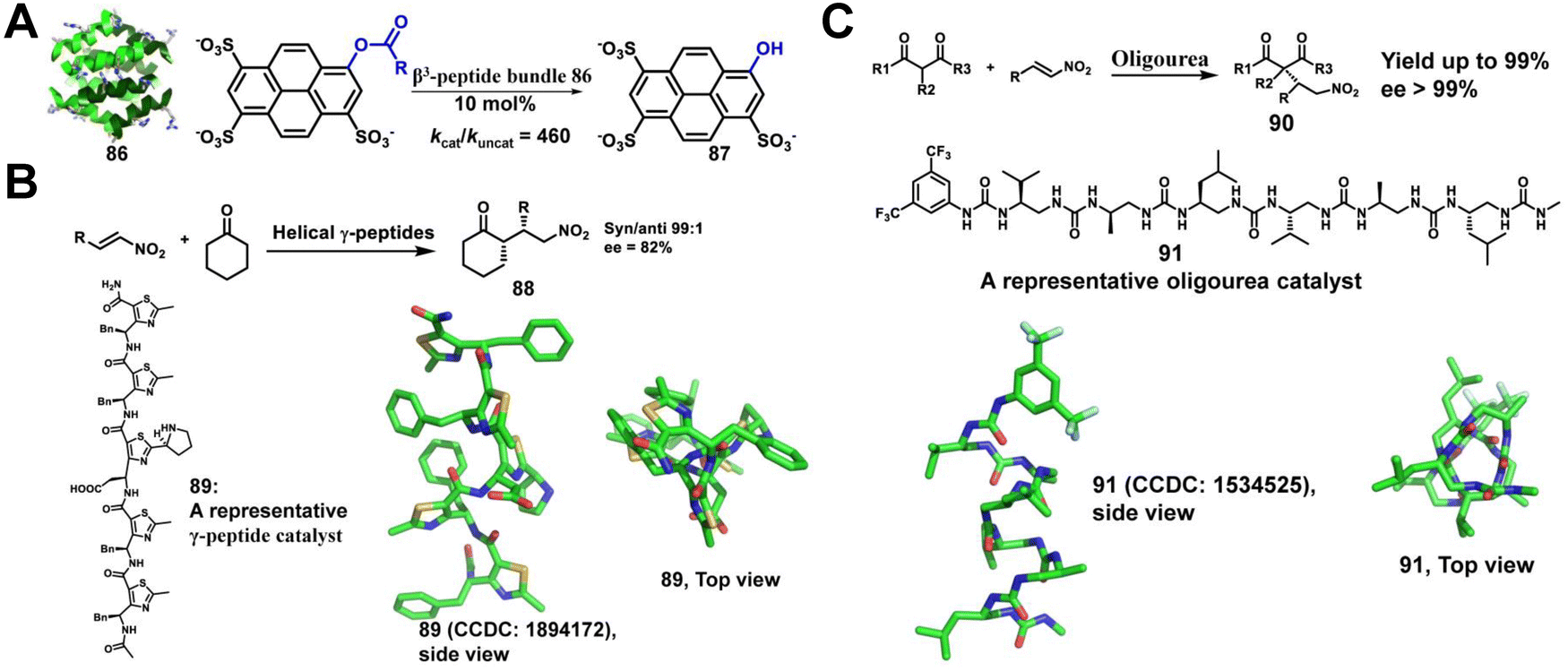 | ||
| Fig. 29 (A) Structure of the octameric Zwit-EYYK bundle 86 (CCDC 804687) and the hydrolysis of 8-acetoxypyrene-1,3,6-trisulfonate by the β3-peptide catalyst; (B) helical γ-peptides containing thiazole-based subunits catalyze the stereospecific nitro-Michael addition reaction of cyclohexanone and β-trans-nitrostyrene; (C) the enantioselective conjugate addition of dimethylmalonate to nitroalkenes catalyzed by helical oligourea. | ||
Maillard and co-workers have reported that helical γ-peptides containing thiazole-based subunits catalyze the stereospecific nitro-Michael addition reaction of cyclohexanone and β-trans-nitrostyrene (Fig. 29B).210 In this system, the two reactive groups required for bifunctional catalysis, a pyrrolidine and a carboxylic acid, are located within a single γ residue. However, the authors demonstrated a modest increase in yield and stereoselectivity as the foldamer embedded with γ residues grew longer, highlighting that the helical secondary structure is likely to contribute to the reaction outcome.
Palomo, Guichard, and colleagues extended the concept of unnatural foldamer catalysis to aliphatic N,N′-linked oligoureas (Fig. 29C).211 The urea-based foldamers form a helix with ∼2.5 residues per turn, which is stabilized by bifurcated H-bonds between each backbone carbonyl and both N–H groups of the urea group at position i +2. They envisioned that helical oligo(thio)urea foldamers could be engineered to catalyze enantioselective C–C bond formation. The catalytic system they designed consists of an oligourea hexamer as the H-bonding chiral component and a tertiary amine as a base component. This system facilitates the enantioselective addition of malonates to nitro-olefins at very low chiral catalyst loading.
The catalytic applications of unnatural helical foldamers in Gellman's group have focused on bifunctional catalysis of aldol reactions by α/β-peptide helices bearing amine diads.212–214 The helical backbone has a repeating motif of one α-amino acid residue and two β-amino acid residues (αββ). They found that a diad comprising two secondary amines supports bifunctional catalysis of crossed aldol reactions, in which formaldehyde is the electrophilic reagent (Fig. 30A).212 Replacement of one secondary amine with a primary amine generated a catalyst for the macrocyclization of aldol condensation, but catalysis of crossed aldol reactions with formaldehyde was lost (Fig. 30B).213 Very recently, they reported an unexpected finding that a diad with two primary amine groups is required for bifunctional catalysis of crossed aldol reactions with aryl aldehydes as electrophiles (Fig. 30C).214 The effect of amine identity on reaction selectivity might be impossible to identify without the use of a tunable unnatural helical foldamer scaffold, which demonstrated that the unnatural helical peptidic foldamer catalyst displays activity distinct from that of smaller and more conventional catalysts.
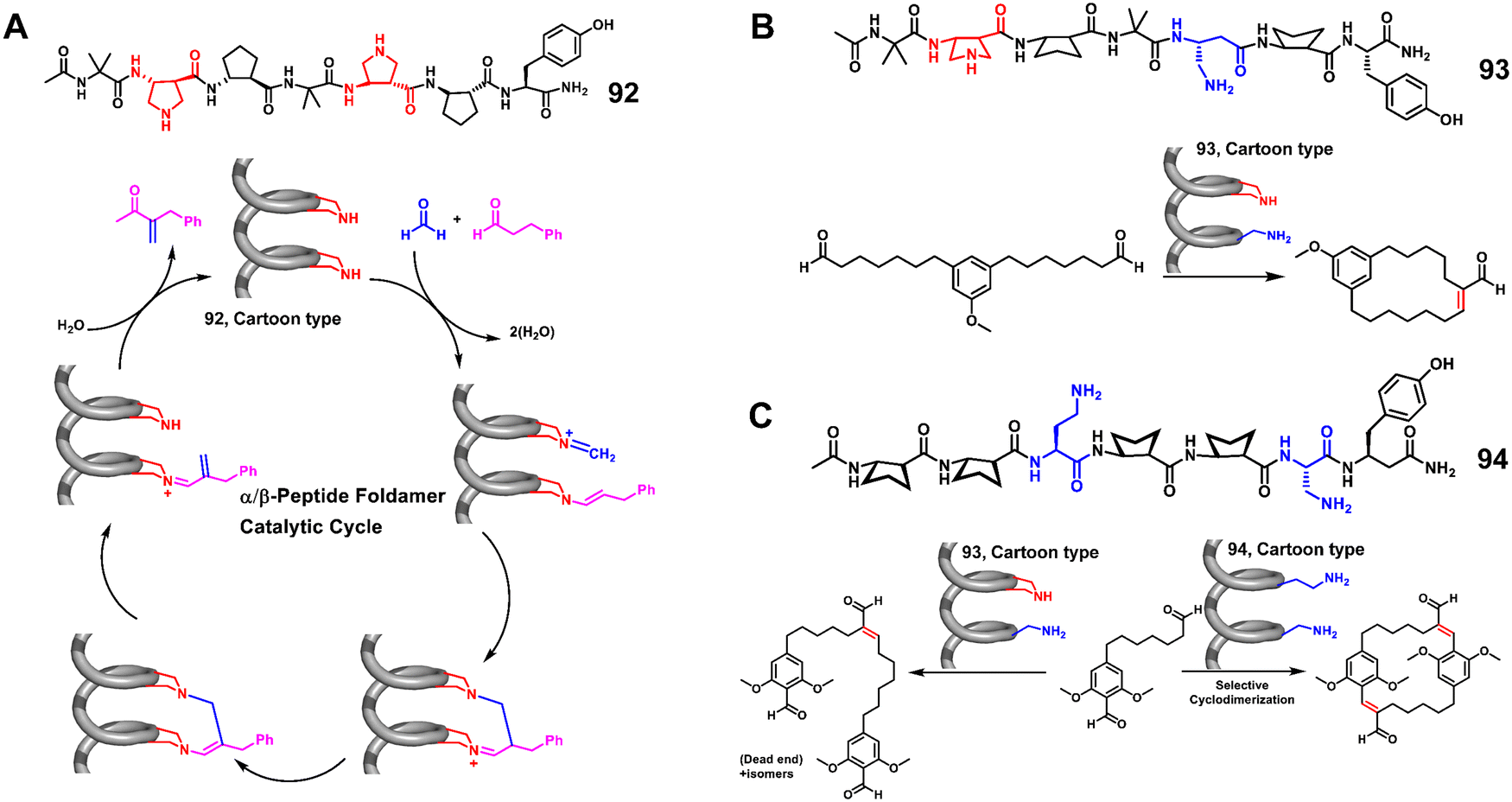 | ||
| Fig. 30 (A) The α/β peptide foldamer catalyzed crossed aldol reaction; (B) the α/β peptide foldamer catalyzed macrocyclization; (C) the α/β peptide foldamer catalyzed cyclodimerization of dialdehyde. | ||
In native proteins, a functional heterodimeric chorismate mutase (CM) catalytic site is formed at the interface of four α-helix bundles. Cleavage of the N-terminal helix spanning the dimer to generate single-helix and triple-helix fragments eliminates enzyme function. However, linking the leucine zipper dimerization domain to two polypeptides results in the spontaneous assembly of the two fragments, returning a cleavage enzyme with wild-type-like activity. Hilvert et al. developed a functional CM through the non-covalent association of helical α/β peptide foldamers with the inactive triple helical domain of the enzyme, in which the single helical domain was replaced by an α/β-peptide surrogate (Fig. 31).215 Linking the leucine-zipper dimerization domains to the folded body ensures their association with the triple helix fragment. Although the unnatural α/β-peptide foldamers alone were unable to catalyze the conversion of chorismate, enzymatic-like activity was observed in the hybrid assemblies.
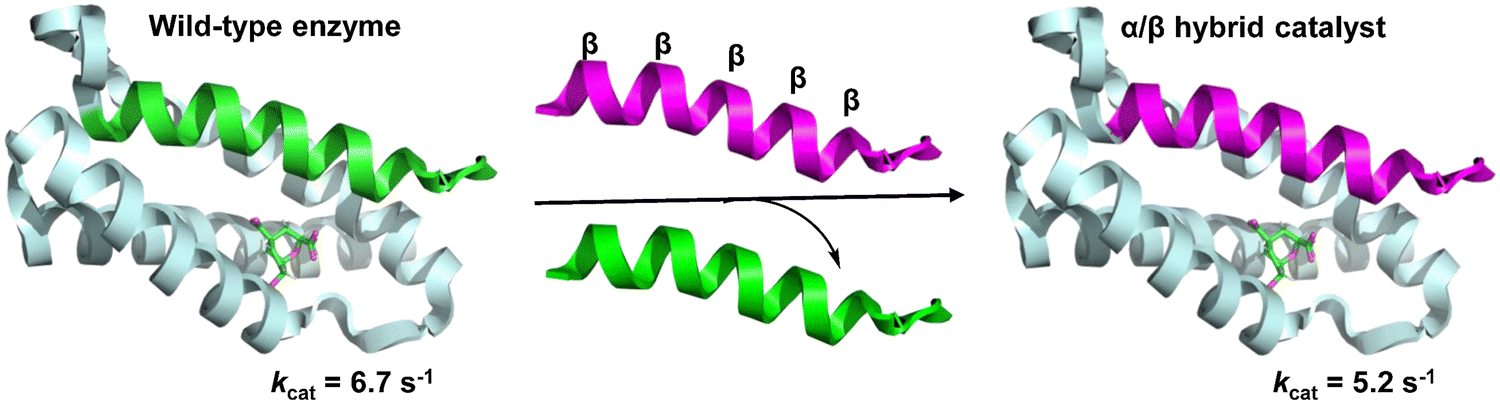 | ||
| Fig. 31 A functional heterodimeric chorismate mutase (CM) through the non-covalent association of an α/β-peptide foldamer with an inactive three-helix domain of the enzyme (PDB: 2gtv). | ||
3.3 Mimics of zinc finger
Zinc fingers16,216,217 are involved in specific double-stranded DNA recognition and are the most common class of DNA-binding proteins throughout biology, and their folding properties have been extensively studied. Folding and metal bonding are closely related. The folding of these motifs is driven by and completely dependent on binding to Zn2+ ions. Zinc finger domains are often arranged in tandem as part of larger DNA-binding proteins, resulting in refined DNA sequence-specific recognition by docking into the major groove of the double helix. The mimicry of helical regions of their structures using unnatural helical peptidic foldamers can improve their folding stability.218–220In 2017, Horne et al. created mimics of zinc finger 3 based on the human transcription factor Sp1 (Sp1–3), in which β3 residues or N-Me-α residues with side chains of substituted α residues, respectively, are incorporated into the helix and hairpin area (Fig. 32A–C).221 The resulting hetero-backbone oligomers displayed native-like metal coordination and higher thermodynamic folding stability than the native backbone, as well as an NMR structure indistinguishable from the prototypic domain. An unexpected observation in this work was that the modification of the metal-binding steering resulted in the variant being unable to fold or bind the metal.
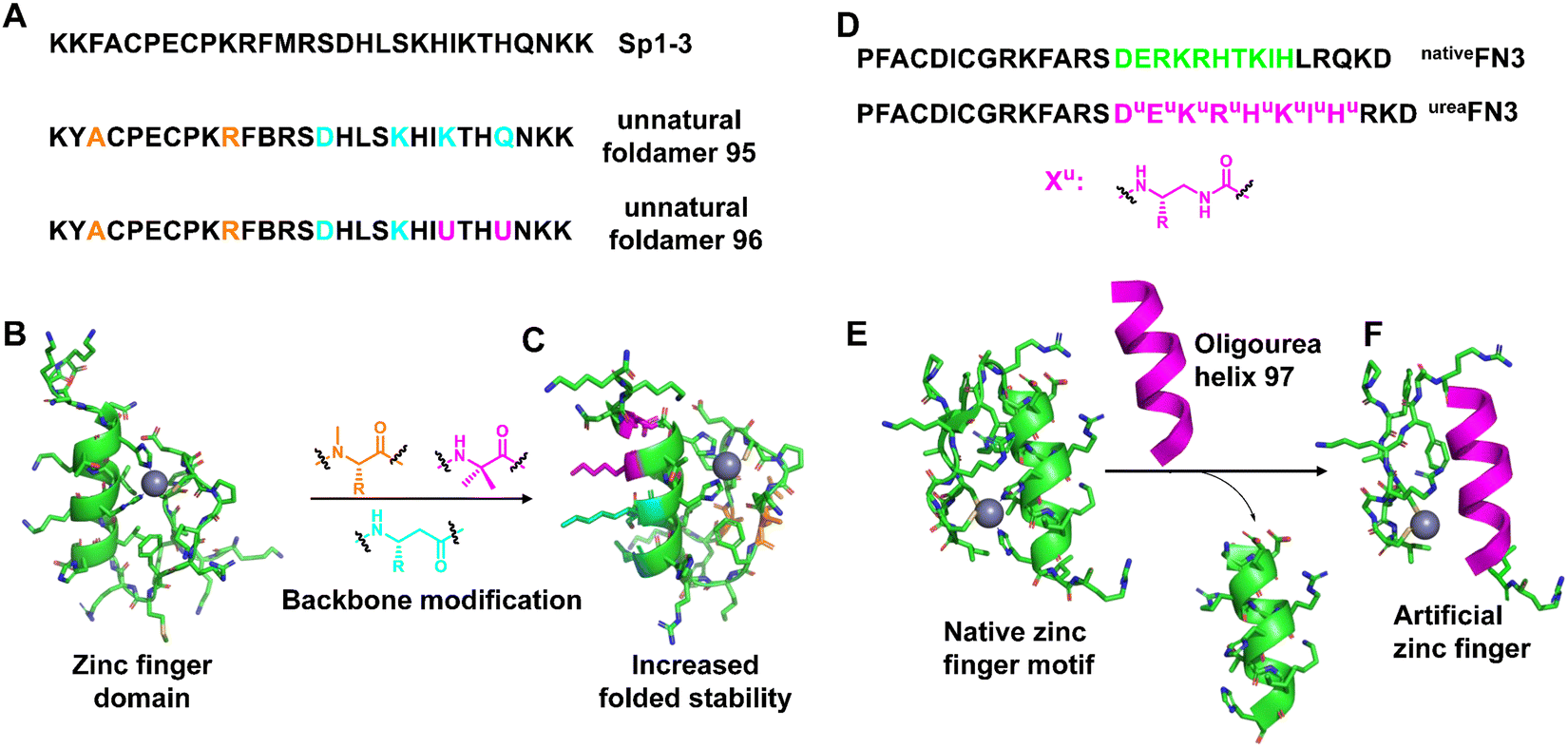 | ||
| Fig. 32 (A) Sequences of Sp1–3 and unnatural foldamers (95 and 96); (B) NMR structure of Sp1–3 (PDB: 1SP1); (C) the folded NMR structure of the unnatural foldamer (PDB: 5US3); (D) sequence of designed composite zinc finger ureaFN3 and the native zinc finger nativeFN3 derived from Zif268; (E) the structure of native FN3 (PDB: 1AAY); (F) view of a representative conformation of oligourea 97 highlighting coordination to the zinc metal ion. | ||
Cys2His2 zinc fingers are repetitive protein motifs involving specific double-stranded DNA recognition of about 30 residues consisting of two anti-parallel β-sheet strands connected by a turn to an α-helix package. This folded prototype is stabilized by a single zinc ion coordinated by a pair of cysteine residues located in the β-sheet and a pair of histidine residues located at the C-terminus of the α-helix in a tetrahedral geometry. Based on their seminal groundwork, Guichard et al. replaced the 10-residue-long original α-helical fragment in the Cys2His2 zinc finger 3 of the transcription factor Egr1 with an oligourea sequence with two appropriately spaced imidazole side chains (Fig. 32D–F).222 The results show that the ability of the peptide/oligourea hybrid to coordinate with zinc ions is not affected by the introduction of the unnatural foldamer insert. In addition to native-like folds and metal-binding sites, chimeric zinc finger domains are also capable of binding double-stranded DNA, underscoring the efficient structural mimicry of the protein fragment.
3.4 Supramolecular self-assembly
Unnatural helical peptidic foldamers play an important role in the self-assembly of higher-order structures. Self-assembly of unnatural helical peptidic foldamers has been considered one of the most attractive and active research areas in supramolecular chemistry since 1998.19 In the past ten years alone, there have been more than 100 related research papers and reviews, mainly including discrete self-assembly, extended self-assembly and self-assembly into solid three-dimensional structures.223–230 For each type of self-assembly, here we will give some representative examples (within the last decade) to illustrate the application of unnatural helical peptidic foldamers in supramolecular self-assembly.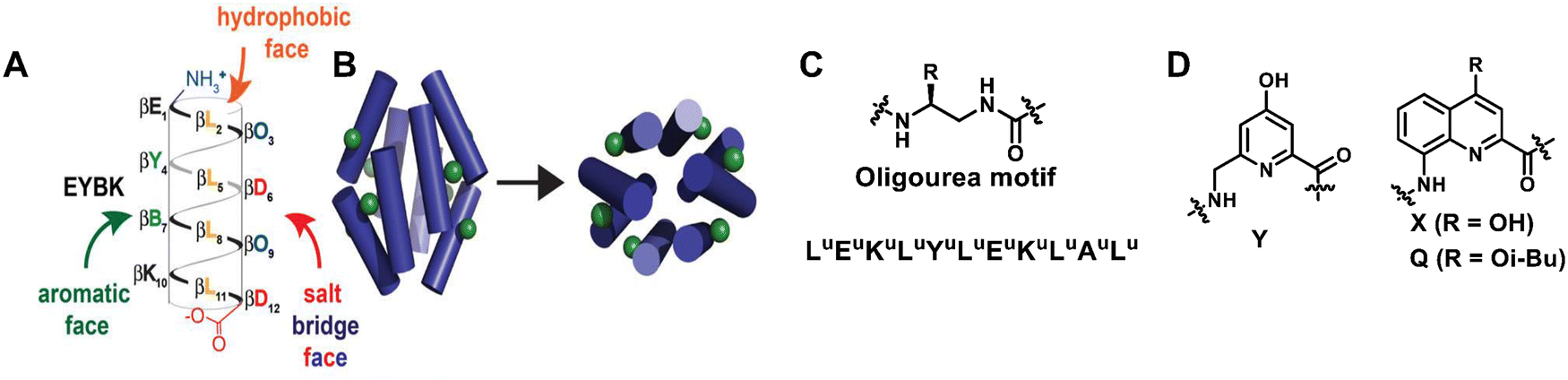 | ||
| Fig. 33 (A) Sequence of β-boronopeptide EYBK. B represents 4-borono-β3-homophenylalanine. (B) Cylinders show the predicted location of phenylboronic acid (PBA) side chains (green) on the surface of an octameric bundle; Reproduced with permission from ref. 231. Copyright 2013, American Chemical Society. (C) sequence of amphiphilic oligourea helix H1; (D) chemical structures of 6-aminomethyl-4-hydroxypicolinic acid (Y) and 8-amino-2-quinolinecarboxylic acid derivatives (X and Q). | ||
Building on previous work on oligoureas, the more recent work by Guichard et al. has moved towards artificial glycolipid binding proteins.232 For the first time, they achieved adaptive binding of alkyl glycosides by unnatural helical bundles in water. They previously used X-ray crystallography to show that a central nonpolar cavity formed within an H1 oligourea 6-helix bundle (empty in the apo form) can accommodate simple alkanols and that this binding does not affect the original structure of the bundle. By using larger and more complex guest molecules that could not fit into this pre-existing cavity, they showed that the previously described oligourea 6-helix bundle could be rearranged into a closely related but distinct bundle to accommodate the new guest molecules (Fig. 33C). This study showed that water-soluble amphiphilic oligourea helical foldamers hold promise as monomers for further development of unnatural protein-like assemblies with a diversity of shapes and functions.
The use of aromatic oligoamides with well-placed lipophilicity and hydrogen-bonded side chains can also give single-helical aromatic foldamers the necessary possibility to form bundles in nonpolar solvents.233 Ac-YQXQQYQXQQ-OMe and O2N-QXQQYQQQ-OME are two types of aromatic oligoamides (Fig. 33D). In both sequences, the residues (Y and X) display hydroxyl groups on the surface of the helix and are therefore able to form interchain hydrogen bonds with the exposed oxygen atoms of backbone amide groups. Due to the additional Y residues in the longer sequence Ac-YQXQQYQXQQ-OMe, the arrangement of Y and X residues results in two pairs of hydroxyl groups in two different columnar arrangements along a helical plane. This in turn allows for the formation of perfectly parallel C3 symmetric trimers in the crystal and solution, where each folded molecule forms a total of eight hydrogen bonds with adjacent helices. Conversely, the lack of a regular double-columnar arrangement of hydrogen bond donors in the shortened sequence O2N-QXQQYQXQQ-OMe results in the formation of a major six-fold hydrogen bond dimer bundle in the solution. These results indicate the richness and uniqueness of abiotic tertiary structure behaviour.
Gong et al. synthesized short tetrapeptides with two aromatic γ-amino acid residues at both ends and either the βα or αβ central sequence (α = Gly and β = β-Ala).234 Self-pairing chains are formed, resulting in linear hydrogen-bonded duplexes. The two sequences showed different self-assembly, as shown in Fig. 34. Achiral γβαγ tetrapeptides form extended β-turns, and the extended helices employed by their γαβγ sequence isomers dimerize into double helices, stabilized by intermolecular hydrogen bonds and π-stacking. On the other hand, the introduction of chiral amino acids (in place of Gly or -Ala) was shown to improve duplex stability, even in polar solvents, and provide major helical significance while reducing the helical inversion process. Double helices can also be formed by short homooligomers of achiral γ-amino acids. In the case of α,β-unsaturated-γ-peptides, X-ray diffraction demonstrates that the self-assembled β-double helices were stabilized only by intermolecular hydrogen bonds (Fig. 35).235
 | ||
| Fig. 34 Distinct folding and assembling behaviour of γβαγ and γαβγ foldamers. Reproduced with permission from ref. 234. Copyright 2019, American Chemical Society. | ||
 | ||
| Fig. 35 (A) Crystal structure of 98 (CCDC 1812184); (B) top view of 98; (C) chemical structure of 98. Interstrand H-bonds observed in the crystal structure of 98 are highlighted. | ||
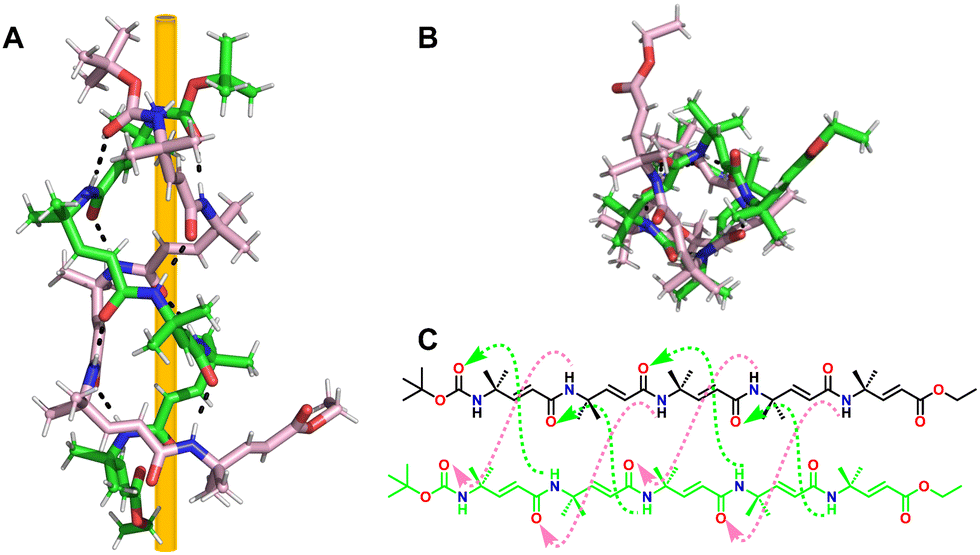
Our group has established a hydrogen bond-driven three-dimensional (3D) assembly of a peptidomimetic zipper using α/sulfono-γ-AApeptide zippers, which are directionally assembled through two layers of hydrogen bond connectors to interact in a de novo lattice arrangement.80 The high-resolution crystal structure of the dimer reveals interesting 3D self-assembly driven by intramolecular/intermolecular hydrogen bonds and C–Cl⋯Cl–C halogen bonds, resulting in a novel porous supramolecular polymer structure with enhanced stability and excellent gas adsorption capability characteristics. Our finding paves a new way for the supramolecular assembly of synthetic tertiary peptides or other building units into novel architectures with enhanced stability and discrete functions (Fig. 36). Subsequently, we reported the intriguing 3D supramolecular self-assembly of a L-sulfono-γ-AApeptide/α-peptide hybrid foldamer driven by orthogonal C–X⋯π and C–X⋯X–C halogen bonding, hydrogen bonding and intermolecular π–π interactions (Fig. 37).81 Our findings demonstrate that halogen interactions in the α/L-sulfono-γ-AA peptidic foldamer can be precisely designed and predicted, thereby permitting the future structure-based design of self-assembled architectures with new functions.
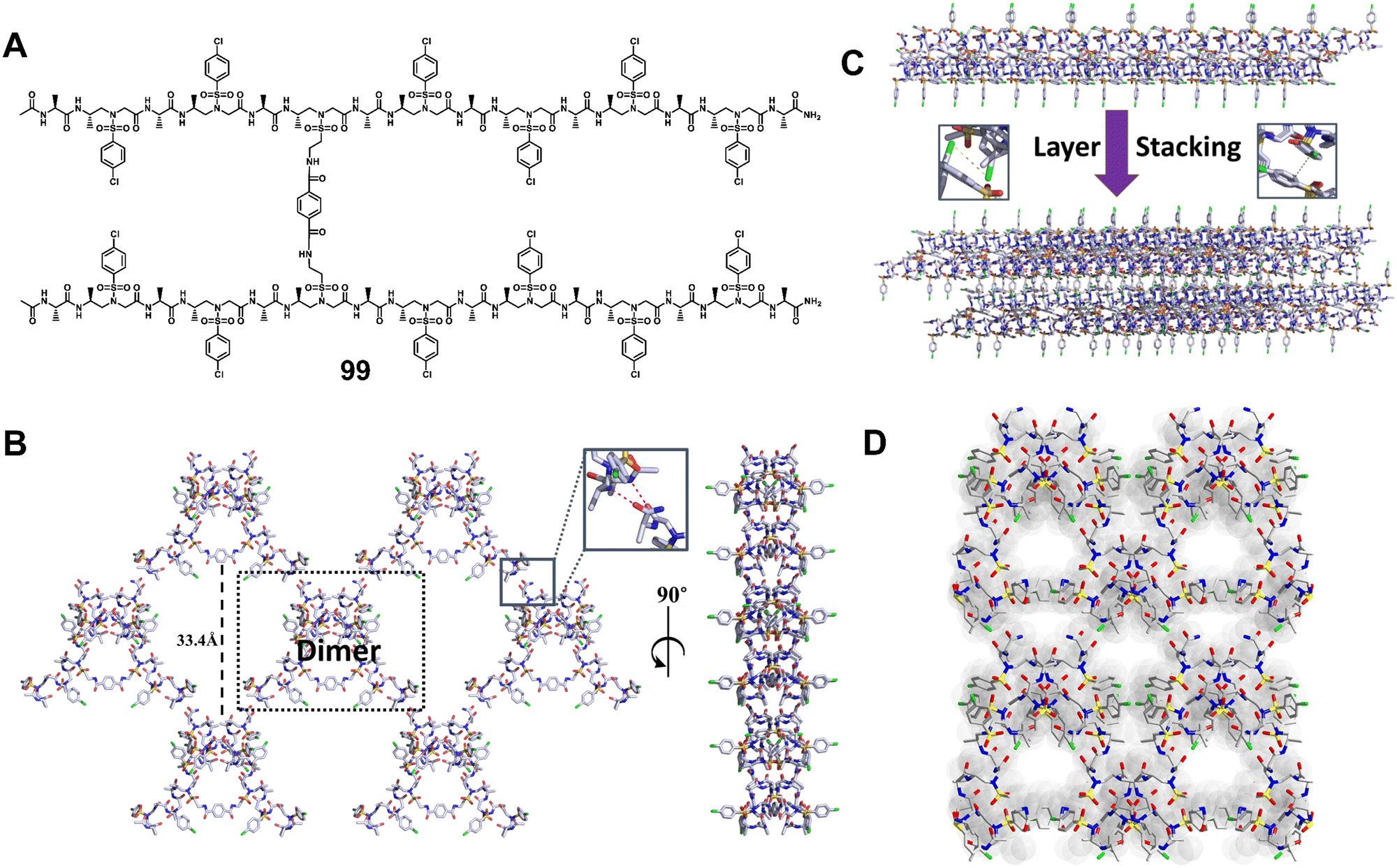 | ||
| Fig. 36 (A) Chemical structure of dimer 99. (B) 2D supramolecular network of dimer 99 (shown in the dashed black box) formed through 2D self-assembly in terms of intra/intermolecular hydrogen bond interactions (magenta dashed line in the inset). (C) Adjacent 2D supramolecular networks in the 3D network were packed laterally and held by the C–Cl⋯Cl–C halogen bond and hydrophobic interactions. Inset: Representation of the C–Cl⋯Cl–C halogen bond (magenta dashed line). (D) Three-dimensional supramolecular network of dimer 99 formed through 3D self-assembly in terms of intra/intermolecular hydrogen bonding and interhelical C–Cl⋯Cl–C halogen bonding interactions. Reproduced with permission from ref. 80. Copyright 2018, American Chemical Society. | ||
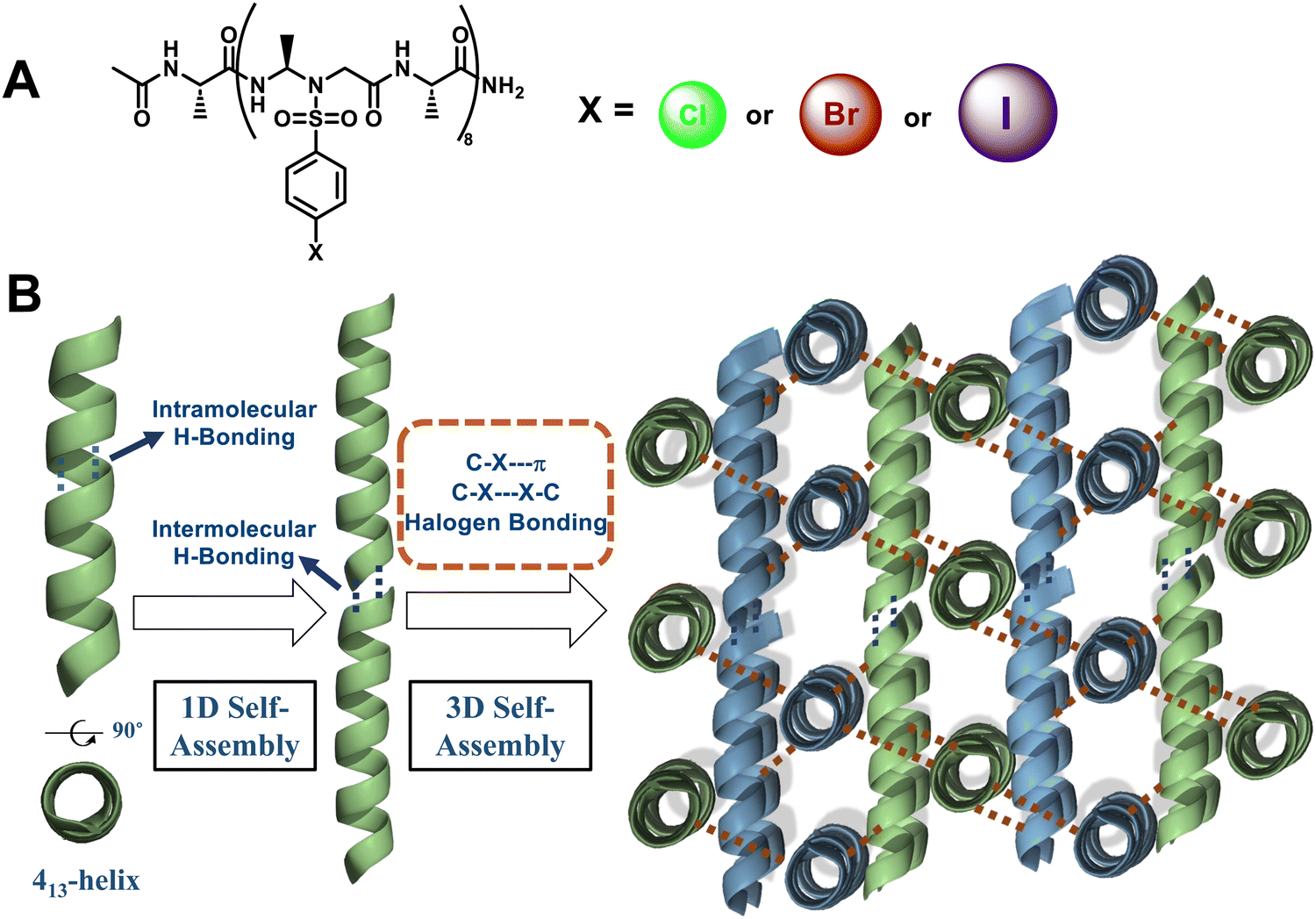 | ||
| Fig. 37 (A) Chemical structures of the 1:1 L-sulfono-γ-AApeptide/α-peptide hybrid foldamer; (B) concept of the self-assembly process from the halogenated sulfono-γ-AApeptide foldamer to 3D supramolecular assembly. | ||
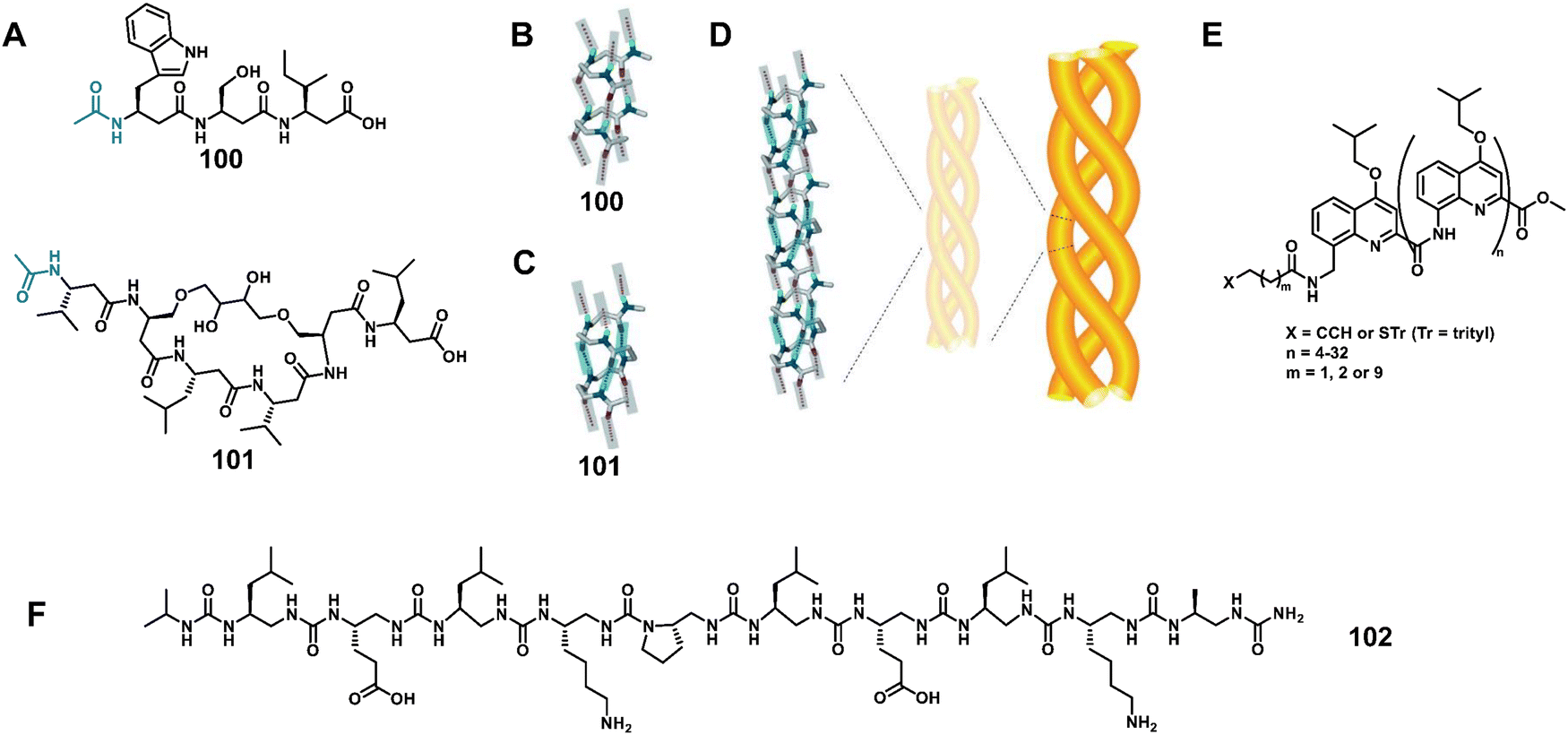 | ||
| Fig. 38 (A) Representative structures of β3-tripeptide 100 and β3-hexapeptide 101; (B and C) crystal structures of 100 and 101; (D) supposed “self-twining” process at the basis of the higher-order hierarchical self-assembly from nanorods to individual fibrils, and then to fibers; Reproduced with permission from ref. 236. Copyright 2013, John Wiley and Sons. (E) chemical formulae of helical oligoquinoline foldamers employed in the study; (F) chemical structure of 102. | ||
Huc and Bassani reported the investigation of vertical and horizontal charge transport over long distances in helical oligo-quinolinecarboxamide foldamers organized as single monolayers on Au or SiO2 (Fig. 38E).246 From the experimental and theoretical results, it emerges that the combination of through-space and through-bond charge transport channels is an essential component in designing efficient 1D semiconducting materials. This study showed that helical foldamer architectures may provide a route for achieving charge transport over long distances by combining multiple charge transport pathways.
Guichard et al. have demonstrated that an amphiphilic oligourea foldamer helix B1 is capable of self-assembly into a variety of supramolecular tubular structures (Fig. 38F).247 The self-assembly process, including the speed and final structure of the assembly, can be readily tuned through the use of alcohol additives, with both alcohol concentration and polarity affecting the assembly process. Interestingly, the alcohol effect reported here, that fibrillation of the oligourea foldamer helix is maximized at alcohol concentrations (e.g. IPA) between 20 and 30%, is similar to the behaviour of many known natural proteins that are predisposed to fibrillation.
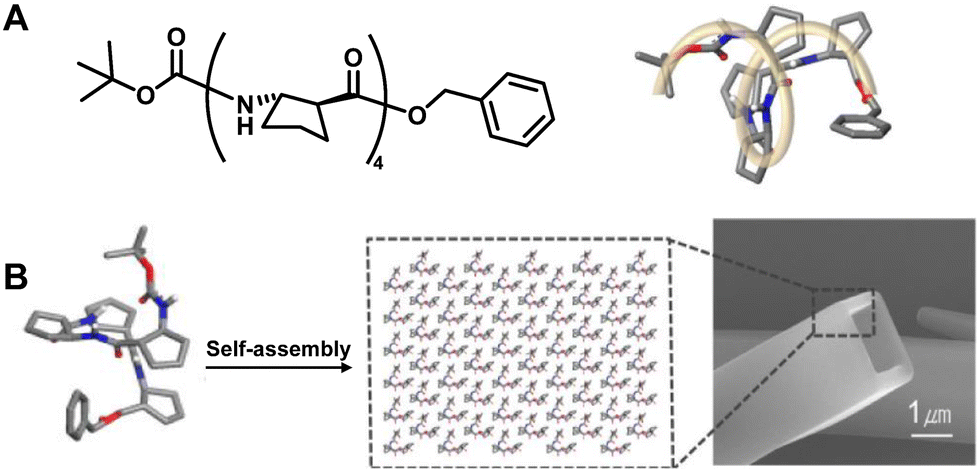 | ||
| Fig. 39 (A) Chemical and crystal structure of trans-(S,S)-ACPC4103; (B) schematic representation of the self-assembly process for the formation of tubular morphology. Reproduced with permission from ref. 251. Copyright 2012, American Chemical Society. | ||
Lee's group has also extensively studied the aqueous self-assembly in solid shapes of surfactant-containing solutions of 1:1 alternating α/β hybrid foldamers.255–259 In 2015, they demonstrated that 11-helix foldamer 104 self-assembles to repeatedly form a new set of foldectures with unique triangular double-pyramid morphology (Fig. 40).256 The 3-fold symmetric shape observed from the self-assembly of 104 is different from the 4-fold symmetric shape resulting from the 12-helix foldamer, implying that the foldamer secondary structure constrains the symmetry elements in foldecture shape and that new and useful foldecture shapes may be available from other peptide secondary structures.
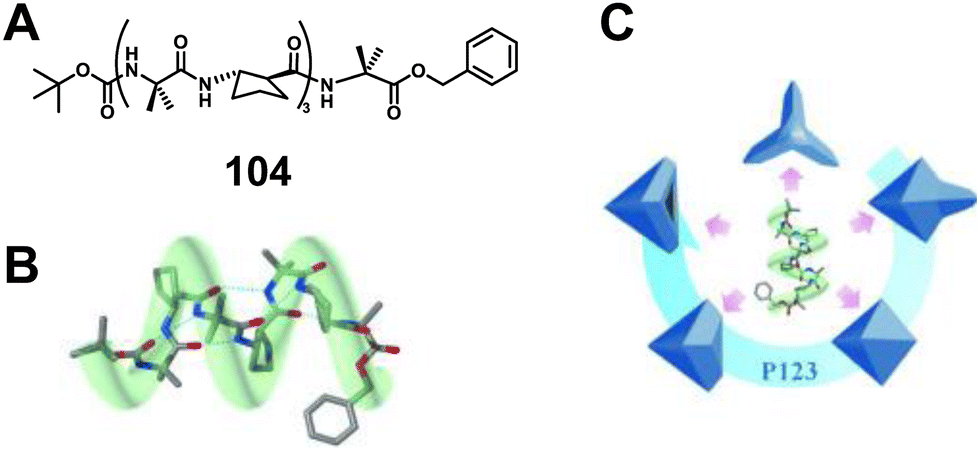 | ||
| Fig. 40 (A) Chemical structure of 104; (B) 11-helical molecular model of α/β-heptamer 104; (C) illustrations of various microstructures having 3-fold symmetry derived from the self-assembly of α/β-heptamer 104. Reproduced with permission from ref. 256. Copyright 2015, John Wiley and Sons. | ||
4. Challenges and perspectives
Benefiting from the unique helical structure and inherent folding stability, unnatural helical peptidic foldamers have many advantages that are generally challenging to achieve in natural α helices, such as stable and predictable structures, significantly improved resistance to proteolytic degradation, enhanced chemical diversity and ability to mimic and improve the function of biomolecules. However, due to the enormous structures and functions of natural molecules and the relatively preliminary understanding of unnatural helical folds, there is still much room for improvement in different aspects, including in-depth mechanistic insights, designing functional foldamers with new frameworks, regulation of properties/activities using chemical modification, prediction of structure and function through quantitative simulation models, etc. Below we discuss some of the challenges and prospects in this area.4.1 Mechanistic insights for molecular recognition
Over the past two decades, although great progress has been made in understanding the conformational properties and corresponding folding-driven mechanisms of helical foldamer frameworks, limited mechanistic understanding of their specific applications, e.g., protein binding and recognition, has restricted their future design and application. In other words, probing the structural information of helical foldamers in the presence of binding partners could be imperative for optimization and development. For instance, upon binding to the protein targets, whether there are just interactions between side chains of the helical foldamer, and/or conformational changes in the partial or entire helical scaffold, is uncertain. It is not surprising that in the presence and absence of binding partners the folding conformation of helical foldamers could be largely different, which is very common for canonical peptides. It is expected that biophysical characterization of the helical foldamer/protein complex, such as X-ray crystallography, cryo-electron microscopy imaging, 2D-NMR, and others, will be very crucial to reveal the mysteries. Particularly, the recent advance in cryo-EM technology would greatly accelerate the elucidation of the structure of helical foldamers in the context of their binding partners. The findings could serve as new foundation to guide future development and new design.4.2 Design of unnatural helical foldamers with new backbones
Although there is significant progress in the development of unnatural helical peptidic foldamers, their application as the helical domain of proteins is still limited, possibly owing to their structural difference from regular α-helices. As proteins display an enormous set of structures and functions, it is imperative to explore helical foldamers with new backbones and new functions. Fortunately, the current design principles of unnatural helical peptidic foldamers can be extended to develop new classes of helical foldamers built on novel molecular scaffolds, based on either homogeneous or heterogeneous backbones. In addition to intramolecular hydrogen bonding which is crucial for the folding propensity of most helical foldamer types, the chemical nature of molecular entities could also be of utmost importance in endowing the intrinsic folding propensity for some classes. For instance, sulfonamido moieties are found to be critical for the formation of well-defined helical structures in sulfono-γ-AApeptides. Therefore, one could adopt a bottom-up strategy to build foldamers with new backbones, by constructing building blocks containing unique functional groups aiding in the formation of certain 3D conformation, and then conjoining them together. Another plausible way could be blending the current known molecular entities of helical foldamers to create foldamers with new heterogeneous backbones. For example, 1:1 or 1:2 ratios of β-amino acids and sulfono-γ-AA amino acids are very likely to produce a new class of unnatural helical foldamers, as 1:1 α/sulfono-γ-AApeptide hybrids are known to form robust helical structures in solution and solid state.
4.3 Regulation of properties/activities using chemical modification
There have been many ways to tune the properties/activities and stability of the regular α helices, such as the inclusion of unnatural amino acids, side-chain cross-linking, glycosylation, long fatty acid conjugation, etc. Although unnatural helical foldamers have high resistance to enzymatic degradation, there is a great need to modulate/tune the properties/activities of the foldamers to increase their potential for applications. For instance, on top of currently known folding mechanisms, non-covalent interactions of side chains and the introduction of di-sulfide bonds can enhance the stability and consolidate the folded conformation of the helical foldamers. Another example is regarding cell permeability. Although the molecular backbone affects cell permeability, the side chains play a more important role. Our recent discoveries show that helical sulfono-γ-AApeptides are much more cell-permeable than canonical helical peptides bearing similar side functional groups. However, upon side-chain stapling, the macrocyclized sulfono-γ-AApeptides exhibit even higher helicity, enhanced cell permeability, and much more potent biological activity. Some of these chemical approaches have been successfully employed in the development of helical foldamers that modulate protein–protein interactions of p53–MDM2/MDMX, β-catenin/BCL9, GLP-1/GLP-1R, VEGF/VEGFR, etc. Last but not least, chemical modifications could be applied to create artificial proteins arising from multiple helical foldameric sequences. The introduction of certain side chains (such as charged or hydrophobic groups) onto individual sequences at certain positions could promote the self-assembly of sequences to form quaternary protein-mimicking structures. Covalent linkers can also be introduced between monomeric sequences to create non-natural proteins bearing tertiary structures. Similarly, some reactive ligands or metal ions can be introduced onto these tertiary and quaternary structures to build artificial enzymes.4.4 Theoretical simulation and prediction
Theoretical insights into unnatural helical foldamers are still limited and require a significant advancement. Most of the current computational simulation programs are built for proteins bearing canonical amino acids, and the Alphafold program can even predict the structures of a vast majority of known proteins. However, there is no general program built for unnatural helical peptidic foldamers as their backbones and frameworks are different from each other. As such, it is difficult to theoretically predict the property/activity of unnatural helical peptidic foldamers. Although the existing crystal structures have successfully explained different types of unnatural folding patterns and guided the discovery and design of multiple novel functional unnatural helical foldamers, the current mechanistic understanding and theoretical calculations are still in their infancy. At the current stage, different models may be developed to study the mechanism of folding/action of various unnatural foldamers, and then their outcomes could be compared with each other. In addition, one should be optimistic due to the recent advances in artificial intelligence (AI). With more and more crystal structures obtained for unnatural helical foldamers, through machine learning, AI may be able to formulate a reasonable rule to predict the folding conformation of many classes of helical foldamers.5. Conclusions
In summary, this review highlights the structure and function of helical unnatural peptidic foldamers and their progress over the past decade as protein domain mimics. These unnatural helical peptidic foldamers generally contain only unnatural amino acids, or hybridized residues comprising of unnatural amino acids and natural α-amino acids in certain patterns. Despite the great progress in the study of unnatural helical peptidic foldamers in the past two decades, the development of novel unnatural helical peptidic foldamers, especially those capable of mimicking the long α-helix, is still limited. Considering that the single crystal structure of unnatural helical peptidic foldamers may not be consistent with the inferred structures based on 2D NMR, CD studies, and molecular dynamics simulations, it is very necessary to further study the single crystal structures of more or novel unnatural helical peptidic foldamers. From the existing crystal structures of unnatural helical peptidic foldamers, the formation of helical structures depends on intramolecular planar amide bonds, substitution patterns of functional groups in the main chain, and conformational constraints, which seems to illustrate the origin of the unique folding patterns of unnatural foldamers. The exact interaction forces are attributed to the backbone or side-chain groups with through-space electron delocalization, arising from hydrogen bonding, halogen bonding, ionic bonding and other noncovalent interactions, which all help rigidifying the conformation of the helical foldamers, thus favouring the formation of stable unnatural helical peptidic foldamers.Meanwhile, the known crystal structures could also help guide the discovery and design of new unnatural helical peptidic foldamers. With the reports of various unnatural helical peptidic foldamers in recent years, their applications in natural protein mimicry, including host-defense peptide mimicry, modulation of disease-related protein–protein interactions, catalysis, molecular recognition and supramolecular design, have gradually been recognized. An increasing number of methods, such as nuclear magnetic resonance (NMR) spectroscopy, cocrystal analysis, and cryo-EM analysis, have been introduced to uncover the essence of general characteristics of unnatural helical peptidic foldamers.
While early research mainly focused more on the fundamental study of secondary structures and tertiary structures, the thorough and comprehensive investigation of unnatural helical peptidic foldamers as protein segment mimics for biological applications is still in its early stage. It is of great significance to further explore their mechanism in functional applications utilizing explicit single-crystal structures and molecular packings. Of course, it is imperative to obtain the structural information of the complexes formed by helical foldamers and their binding partners, which could guide their new design and optimization. Moreover, although the mimicry of proteins by unnatural helical peptidic foldamers remains difficult to fine-tune, recent progress does imply the feasibility and great potential to tune their properties/activity, stability, and cell permeability through molecular and technological efforts, which also sheds new light on the folding mechanism in turn.
Currently, unnatural helical peptidic foldamers have exhibited promising applications in the fields of chemistry, biology, medicine, and materials. Compared with native helical peptides, their increased resistance to proteolytic degradation, enhanced bioavailability, and improved chemical diversity give them unique advantages, which also make them promising for drug discovery and delivery, practical functional material development and other biological applications. Future exploration of their emerging applications is highly desired.
With the growing interest in this area, we can expect rapid development in both fundamental and practical aspects. More attention from the diversified scientific community will be beneficial to reveal more insights into unnatural helical peptidic foldamers that mimic helical segments of active proteins. This helps to uncover the underlying mechanisms that guide the rational design of unnatural helical peptidic foldamers towards efficient and/or high activity and pave the way for the development of emerging applications. We also believe that the development of unnatural helical peptidic foldamers will facilitate in-depth understanding of other related fields such as bioorthogonal chemistry and nano-pharmaceutical formulations. We look forward to witnessing rapid growth and new achievements in this active and fascinating field in the future.
Conflicts of interest
There are no conflicts to declare.Note added after first publication
This article replaces the version published on 4th July 2023, which contained errors in Fig. 1.Acknowledgements
The work was supported by NIH R01AI152416 and NIH R01 AI149852.References
- A. R. P. Silva, M. S. Guimarães, J. Rabelo, L. H. Belén, C. J. Perecin, J. G. Farías, J. H. P. M. Santos and C. O. Rangel-Yagui, J. Mater. Chem. B, 2022, 10, 3587–3600 RSC.
- O. N. Silva, W. F. Porto, S. M. Ribeiro, I. Batista and O. L. Franco, Drug Discovery Today, 2018, 23, 1666–1671 CrossRef CAS PubMed.
- V. Basso, D. Q. Tran, A. J. Ouellette and M. E. Selsted, J. Fungi, 2020, 6, 241 CrossRef CAS.
- P.-Y. Yang, H. Zou, E. Chao, L. Sherwood, V. Nunez, M. Keeney, E. Ghartey-Tagoe, Z. Ding, H. Quirino, X. Luo, G. Welzel, G. Chen, P. Singh, A. K. Woods, P. G. Schultz and W. Shen, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 4140–4145 CrossRef CAS PubMed.
- P. M. Levine, A. T. Balana, E. Sturchler, C. Koole, H. Noda, B. Zarzycka, E. J. Daley, T. T. Truong, V. Katritch, T. J. Gardella, D. Wootten, P. M. Sexton, P. McDonald and M. R. Pratt, J. Am. Chem. Soc., 2019, 141, 14210–14219 CrossRef CAS PubMed.
- E. S. Leshchiner, A. Parkhitko, G. H. Bird, J. Luccarelli, J. A. Bellairs, S. Escudero, K. Opoku-Nsiah, M. Godes, N. Perrimon and L. D. Walensky, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 1761–1766 CrossRef CAS PubMed.
- O. Zozulia, M. A. Dolan and I. V. Korendovych, Chem. Soc. Rev., 2018, 47, 3621–3639 RSC.
- Y. Zheng, Y. Cong, E. W. Schmidt and S. K. Nair, Acc. Chem. Res., 2022, 55, 1313–1323 CrossRef CAS PubMed.
- K. Kudo, Catalytic Asymmetric Synthesis, 2022, pp. 157–198 DOI:10.1002/9781119736424.ch5.
- T. Li, X.-M. Lu, M.-R. Zhang, K. Hu and Z. Li, Bioact. Mater., 2022, 11, 268–282 CrossRef CAS PubMed.
- P. B. Crowley, Acc. Chem. Res., 2022, 55, 2019–2032 CrossRef CAS PubMed.
- K. Jung, N. Corrigan, E. H. H. Wong and C. Boyer, Adv. Mater., 2022, 34, 2105063 CrossRef CAS PubMed.
- N. J. Sinha, M. G. Langenstein, D. J. Pochan, C. J. Kloxin and J. G. Saven, Chem. Rev., 2021, 121, 13915–13935 CrossRef CAS PubMed.
- P. Sang, Y. Shi, B. Huang, S. Xue, T. Odom and J. Cai, Acc. Chem. Res., 2020, 53, 2425–2442 CrossRef CAS PubMed.
- Y. Shi, P. Teng, P. Sang, F. She, L. Wei and J. Cai, Acc. Chem. Res., 2016, 49, 428–441 CrossRef CAS PubMed.
- W. S. Horne and T. N. Grossmann, Nat. Chem., 2020, 12, 331–337 CrossRef CAS.
- D. H. Appella, L. A. Christianson, I. L. Karle, D. R. Powell and S. H. Gellman, J. Am. Chem. Soc., 1996, 118, 13071–13072 CrossRef CAS.
- D. Seebach, M. Overhand, F. N. M. Kühnle, B. Martinoni, L. Oberer, U. Hommel and H. Widmer, Helv. Chim. Acta, 1996, 79, 913–941 CrossRef CAS.
- S. H. Gellman, Acc. Chem. Res., 1998, 31, 173–180 CrossRef CAS.
- R. P. Cheng, S. H. Gellman and W. F. DeGrado, Chem. Rev., 2001, 101, 3219–3232 CrossRef CAS PubMed.
- Ł. Berlicki, L. Pilsl, E. Wéber, I. M. Mándity, C. Cabrele, T. A. Martinek, F. Fülöp and O. Reiser, Angew. Chem., Int. Ed., 2012, 51, 2208–2212 CrossRef PubMed.
- J. R. Stringer, J. A. Crapster, I. A. Guzei and H. E. Blackwell, J. Am. Chem. Soc., 2011, 133, 15559–15567 CrossRef CAS.
- J. S. Laursen, P. Harris, P. Fristrup and C. A. Olsen, Nat. Commun., 2015, 6, 7013 CrossRef CAS PubMed.
- L. Fischer, P. Claudon, N. Pendem, E. Miclet, C. Didierjean, E. Ennifar and G. Guichard, Angew. Chem., Int. Ed., 2010, 49, 1067–1070 CrossRef CAS PubMed.
- C. Proulx, D. Sabatino, R. Hopewell, J. Spiegel, Y. G. Ramos and W. D. Lubell, Future Med. Chem., 2011, 3, 1139–1164 CrossRef CAS PubMed.
- P. Wilhelm, B. Lewandowski, N. Trapp and H. Wennemers, J. Am. Chem. Soc., 2014, 136, 15829–15832 CrossRef CAS PubMed.
- D.-W. Zhang, X. Zhao, J.-L. Hou and Z.-T. Li, Chem. Rev., 2012, 112, 5271–5316 CrossRef CAS PubMed.
- J. Garric, J.-M. Léger and I. Huc, Angew. Chem., Int. Ed., 2005, 44, 1954–1958 CrossRef CAS.
- J. E. Jones, V. Diemer, C. Adam, J. Raftery, R. E. Ruscoe, J. T. Sengel, M. I. Wallace, A. Bader, S. L. Cockroft, J. Clayden and S. J. Webb, J. Am. Chem. Soc., 2016, 138, 688–695 CrossRef CAS PubMed.
- B. Legrand and L. T. Maillard, ChemPlusChem, 2021, 86, 629–645 CrossRef CAS PubMed.
- B. B. Lao, I. Grishagin, H. Mesallati, T. F. Brewer, B. Z. Olenyuk and P. S. Arora, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 7531–7536 CrossRef CAS PubMed.
- L. Palanikumar, L. Karpauskaite, M. Al-Sayegh, I. Chehade, M. Alam, S. Hassan, D. Maity, L. Ali, M. Kalmouni, Y. Hunashal, J. Ahmed, T. Houhou, S. Karapetyan, Z. Falls, R. Samudrala, R. Pasricha, G. Esposito, A. J. Afzal, A. D. Hamilton, S. Kumar and M. Magzoub, Nat. Commun., 2021, 12, 3962 CrossRef CAS PubMed.
- D. Maity, S. Kumar, F. Curreli, A. K. Debnath and A. D. Hamilton, Chem. – Eur. J., 2019, 25, 7265–7269 CrossRef CAS PubMed.
- D. H. Appella, L. A. Christianson, D. A. Klein, M. R. Richards, D. R. Powell and S. H. Gellman, J. Am. Chem. Soc., 1999, 121, 7574–7581 CrossRef CAS.
- D. H. Appella, J. J. Barchi, S. R. Durell and S. H. Gellman, J. Am. Chem. Soc., 1999, 121, 2309–2310 CrossRef CAS.
- D. H. Appella, L. A. Christianson, I. L. Karle, D. R. Powell and S. H. Gellman, J. Am. Chem. Soc., 1999, 121, 6206–6212 CrossRef CAS.
- M.-r Lee, T. L. Raguse, M. Schinnerl, W. C. Pomerantz, X. Wang, P. Wipf and S. H. Gellman, Org. Lett., 2007, 9, 1801–1804 CrossRef CAS PubMed.
- E. Vaz, W. C. Pomerantz, M. Geyer, S. H. Gellman and L. Brunsveld, ChemBioChem, 2008, 9, 2254–2259 CrossRef CAS PubMed.
- D. H. Appella, L. A. Christianson, D. A. Klein, D. R. Powell, X. Huang, J. J. Barchi and S. H. Gellman, Nature, 1997, 387, 381–384 CrossRef CAS PubMed.
- H.-S. Lee, F. A. Syud, X. Wang and S. H. Gellman, J. Am. Chem. Soc., 2001, 123, 7721–7722 CrossRef CAS PubMed.
- I. Wellhöfer, K. Frydenvang, S. Kotesova, A. M. Christiansen, J. S. Laursen and C. A. Olsen, J. Org. Chem., 2019, 84, 3762–3779 CrossRef PubMed.
- C. W. Wu, K. Kirshenbaum, T. J. Sanborn, J. A. Patch, K. Huang, K. A. Dill, R. N. Zuckermann and A. E. Barron, J. Am. Chem. Soc., 2003, 125, 13525–13530 CrossRef CAS.
- O. Roy, G. Dumonteil, S. Faure, L. Jouffret, A. Kriznik and C. Taillefumier, J. Am. Chem. Soc., 2017, 139, 13533–13540 CrossRef CAS.
- J. S. Laursen, J. Engel-Andreasen and C. A. Olsen, Acc. Chem. Res., 2015, 48, 2696–2704 CrossRef CAS.
- T. Hintermann, K. Gademann, B. Jaun and D. Seebach, Helv. Chim. Acta, 1998, 81, 983–1002 CrossRef CAS.
- S. Hanessian, X. Luo, R. Schaum and S. Michnick, J. Am. Chem. Soc., 1998, 120, 8569–8570 CrossRef CAS.
- D. Seebach, M. Brenner, M. Rueping and B. Jaun, Chem. – Eur. J., 2002, 8, 573–584 CrossRef CAS.
- D. Seebach, M. Brenner, M. Rueping, B. Schweizer and B. Jaun, Chem. Commun., 2001, 207–208, 10.1039/B008377L.
- P. G. Vasudev, N. Shamala, K. Ananda and P. Balaram, Angew. Chem., Int. Ed., 2005, 44, 4972–4975 CrossRef CAS PubMed.
- P. Sang, Y. Shi, L. Wei and J. Cai, RSC Chem. Biol., 2022, 3, 805–814 RSC.
- F. She, P. Teng, A. Peguero-Tejada, M. Wang, N. Ma, T. Odom, M. Zhou, E. Gjonaj, L. Wojtas, A. van der Vaart and J. Cai, Angew. Chem., Int. Ed., 2018, 57, 9916–9920 CrossRef CAS PubMed.
- H. Wu, Q. Qiao, Y. Hu, P. Teng, W. Gao, X. Zuo, L. Wojtas, R. W. Larsen, S. Ma and J. Cai, Chem. – Eur. J., 2015, 21, 2501–2507 CrossRef CAS PubMed.
- P. Sang, Y. Shi, P. Higbee, M. Wang, S. Abdulkadir, J. Lu, G. Daughdrill, J. Chen and J. Cai, J. Org. Chem., 2020, 85, 10552–10560 CrossRef CAS PubMed.
- Y. Hamuro, S. J. Geib and A. D. Hamilton, Angew. Chem., Int. Ed. Engl., 1994, 33, 446–448 CrossRef.
- Y. Hamuro, S. J. Geib and A. D. Hamilton, J. Am. Chem. Soc., 1996, 118, 7529–7541 CrossRef CAS.
- R. Gopalakrishnan, A. I. Frolov, L. Knerr, W. J. Drury, III and E. Valeur, J. Med. Chem., 2016, 59, 9599–9621 CrossRef CAS.
- M. Zwillinger, P. S. Reddy, B. Wicher, P. K. Mandal, M. Csékei, L. Fischer, A. Kotschy and I. Huc, Chem. – Eur. J., 2020, 26, 17366–17370 CrossRef CAS.
- I. Saraogi and A. D. Hamilton, Chem. Soc. Rev., 2009, 38, 1726–1743 RSC.
- J. M. Rodriguez and A. D. Hamilton, Angew. Chem., Int. Ed., 2007, 46, 8614–8617 CrossRef CAS PubMed.
- H. Jiang, J.-M. Léger and I. Huc, J. Am. Chem. Soc., 2003, 125, 3448–3449 CrossRef CAS PubMed.
- V. Semetey, D. Rognan, C. Hemmerlin, R. Graff, J.-P. Briand, M. Marraud and G. Guichard, Angew. Chem., Int. Ed., 2002, 41, 1893–1895 CrossRef CAS PubMed.
- J. Fremaux, L. Fischer, T. Arbogast, B. Kauffmann and G. Guichard, Angew. Chem., Int. Ed., 2011, 50, 11382–11385 CrossRef CAS PubMed.
- B. Legrand, C. André, E. Wenger, C. Didierjean, M. C. Averlant-Petit, J. Martinez, M. Calmes and M. Amblard, Angew. Chem., Int. Ed., 2012, 51, 11267–11270 CrossRef CAS PubMed.
- R. Wechsel, J. Raftery, D. Cavagnat, G. Guichard and J. Clayden, Angew. Chem., Int. Ed., 2016, 55, 9657–9661 CrossRef CAS PubMed.
- G. W. Collie, K. Pulka-Ziach and G. Guichard, Chem. Sci., 2016, 7, 3377–3383 RSC.
- P. Milbeo, M. Simon, C. Didierjean, E. Wenger, E. Aubert, J. Martinez, M. Amblard, M. Calmès and B. Legrand, Chem. Commun., 2020, 56, 7921–7924 RSC.
- Y. R. Nelli, S. Antunes, A. Salaün, E. Thinon, S. Massip, B. Kauffmann, C. Douat and G. Guichard, Chem. – Eur. J., 2015, 21, 2870–2880 CrossRef CAS PubMed.
- N. Pendem, C. Douat, P. Claudon, M. Laguerre, S. Castano, B. Desbat, D. Cavagnat, E. Ennifar, B. Kauffmann and G. Guichard, J. Am. Chem. Soc., 2013, 135, 4884–4892 CrossRef CAS PubMed.
- J. Fremaux, B. Kauffmann and G. Guichard, J. Org. Chem., 2014, 79, 5494–5502 CrossRef CAS PubMed.
- K. Burgess, H. Shin and D. S. Linthicum, Angew. Chem., Int. Ed. Engl., 1995, 34, 907–909 CrossRef CAS.
- D. Yang, F.-F. Ng, Z.-J. Li, Y.-D. Wu, K. W. K. Chan and D.-P. Wang, J. Am. Chem. Soc., 1996, 118, 9794–9795 CrossRef CAS.
- X. Li, Y.-D. Wu and D. Yang, Acc. Chem. Res., 2008, 41, 1428–1438 CrossRef CAS PubMed.
- D. Diedrich, A. J. R. Moita, A. Rüther, B. Frieg, G. J. Reiss, A. Hoeppner, T. Kurz, H. Gohlke, S. Lüdeke, M. U. Kassack and F. K. Hansen, Chem. – Eur. J., 2016, 22, 17600–17611 CrossRef CAS PubMed.
- S. H. Choi, I. A. Guzei, L. C. Spencer and S. H. Gellman, J. Am. Chem. Soc., 2008, 130, 6544–6550 CrossRef CAS PubMed.
- M. Ganesh Kumar, V. J. Thombare, M. M. Katariya, K. Veeresh, K. M. P. Raja and H. N. Gopi, Angew. Chem., Int. Ed., 2016, 55, 7847–7851 CrossRef CAS PubMed.
- M. W. Giuliano, S. J. Maynard, A. M. Almeida, L. Guo, I. A. Guzei, L. C. Spencer and S. H. Gellman, J. Am. Chem. Soc., 2014, 136, 15046–15053 CrossRef CAS PubMed.
- B. F. Fisher, L. Guo, B. S. Dolinar, I. A. Guzei and S. H. Gellman, J. Am. Chem. Soc., 2015, 137, 6484–6487 CrossRef CAS PubMed.
- S. V. Jadhav, R. Misra and H. N. Gopi, Chem. – Eur. J., 2014, 20, 16523–16528 CrossRef CAS PubMed.
- Y. Shi, G. Yin, Z. Yan, P. Sang, M. Wang, R. Brzozowski, P. Eswara, L. Wojtas, Y. Zheng, X. Li and J. Cai, J. Am. Chem. Soc., 2019, 141, 12697–12706 CrossRef CAS PubMed.
- P. Teng, Z. Niu, F. She, M. Zhou, P. Sang, G. M. Gray, G. Verma, L. Wojtas, A. van der Vaart, S. Ma and J. Cai, J. Am. Chem. Soc., 2018, 140, 5661–5665 CrossRef CAS PubMed.
- P. Teng, G. M. Gray, M. Zheng, S. Singh, X. Li, L. Wojtas, A. van der Vaart and J. Cai, Angew. Chem., Int. Ed., 2019, 58, 7778–7782 CrossRef CAS PubMed.
- P. Teng, M. Zheng, D. C. Cerrato, Y. Shi, M. Zhou, S. Xue, W. Jiang, L. Wojtas, L.-J. Ming, Y. Hu and J. Cai, Commun. Chem., 2021, 4, 58 CrossRef CAS PubMed.
- L. Guo, A. M. Almeida, W. Zhang, A. G. Reidenbach, S. H. Choi, I. A. Guzei and S. H. Gellman, J. Am. Chem. Soc., 2010, 132, 7868–7869 CrossRef CAS PubMed.
- N. Pendem, Y. R. Nelli, C. Douat, L. Fischer, M. Laguerre, E. Ennifar, B. Kauffmann and G. Guichard, Angew. Chem., Int. Ed., 2013, 52, 4147–4151 CrossRef CAS PubMed.
- Y. R. Nelli, L. Fischer, G. W. Collie, B. Kauffmann and G. Guichard, Pept. Sci., 2013, 100, 687–697 CrossRef CAS PubMed.
- K. Basuroy, B. Dinesh, N. Shamala and P. Balaram, Angew. Chem., Int. Ed., 2012, 51, 8736–8739 CrossRef CAS PubMed.
- L. Guo, W. Zhang, I. A. Guzei, L. C. Spencer and S. H. Gellman, Tetrahedron, 2012, 68, 4413–4417 CrossRef CAS.
- S. H. Choi, I. A. Guzei, L. C. Spencer and S. H. Gellman, J. Am. Chem. Soc., 2009, 131, 2917–2924 CrossRef CAS PubMed.
- P. Teng, N. Ma, D. C. Cerrato, F. She, T. Odom, X. Wang, L.-J. Ming, A. van der Vaart, L. Wojtas, H. Xu and J. Cai, J. Am. Chem. Soc., 2017, 139, 7363–7369 CrossRef CAS PubMed.
- M. A. Schmitt, S. H. Choi, I. A. Guzei and S. H. Gellman, J. Am. Chem. Soc., 2006, 128, 4538–4539 CrossRef CAS PubMed.
- H. Yokoo, M. Hirano, T. Misawa and Y. Demizu, ChemMedChem, 2021, 16, 1226–1233 CrossRef CAS PubMed.
- C. Bonnel, B. Legrand, M. Simon, M. Clavié, A. Masnou, E. Jumas-Bilak, Y. K. Kang, P. Licznar-Fajardo, L. T. Maillard and N. Masurier, J. Med. Chem., 2020, 63, 9168–9180 CrossRef CAS PubMed.
- E. Teyssières, J.-P. Corre, S. Antunes, C. Rougeot, C. Dugave, G. Jouvion, P. Claudon, G. Mikaty, C. Douat, P. L. Goossens and G. Guichard, J. Med. Chem., 2016, 59, 8221–8232 CrossRef PubMed.
- M.-R. Lee, N. Raman, S. H. Gellman, D. M. Lynn and S. P. Palecek, ACS Chem. Biol., 2017, 12, 2975–2980 CrossRef CAS PubMed.
- M.-R. Lee, N. Raman, S. H. Gellman, D. M. Lynn and S. P. Palecek, ACS Chem. Biol., 2014, 9, 1613–1621 CrossRef CAS PubMed.
- N. Frederiksen, P. R. Hansen, F. Björkling and H. Franzyk, Molecules, 2019, 24, 4429 CrossRef CAS PubMed.
- L. Tallet, E. Frisch, M. Bornerie, C. Medemblik, B. Frisch, P. Lavalle, G. Guichard, C. Douat and A. Kichler, Molecules, 2022, 27, 1749 CrossRef CAS PubMed.
- B. Mojsoska, R. N. Zuckermann and H. Jenssen, Antimicrob. Agents Chemother., 2015, 59, 4112–4120 CrossRef CAS PubMed.
- K. Zhang, Y. Du, Z. Si, Y. Liu, M. E. Turvey, C. Raju, D. Keogh, L. Ruan, S. L. Jothy, S. Reghu, K. Marimuthu, P. P. De, O. T. Ng, J. R. Mediavilla, B. N. Kreiswirth, Y. R. Chi, J. Ren, K. C. Tam, X.-W. Liu, H. Duan, Y. Zhu, Y. Mu, P. T. Hammond, G. C. Bazan, K. Pethe and M. B. Chan-Park, Nat. Commun., 2019, 10, 4792 CrossRef PubMed.
- N. Mookherjee, M. A. Anderson, H. P. Haagsman and D. J. Davidson, Nat. Rev. Drug Discovery, 2020, 19, 311–332 CrossRef CAS PubMed.
- F. She, A. Nimmagadda, P. Teng, M. Su, X. Zuo and J. Cai, Biomacromolecules, 2016, 17, 1854–1859 CrossRef CAS PubMed.
- R. Mourtada, H. D. Herce, D. J. Yin, J. A. Moroco, T. E. Wales, J. R. Engen and L. D. Walensky, Nat. Biotechnol., 2019, 37, 1186–1197 CrossRef CAS PubMed.
- Y. Li, H. Wu, P. Teng, G. Bai, X. Lin, X. Zuo, C. Cao and J. Cai, J. Med. Chem., 2015, 58, 4802–4811 CrossRef CAS PubMed.
- J. Lee, D. Kang, J. Choi, W. Huang, M. Wadman, A. E. Barron and J. Seo, Bioorg. Med. Chem. Lett., 2018, 28, 170–173 CrossRef CAS PubMed.
- B. Mojsoska, R. N. Zuckermann and H. Jenssen, Antimicrob. Agents Chemother., 2015, 59, 4112–4120 CrossRef CAS PubMed.
- R. Shyam, N. Charbonnel, A. Job, C. Blavignac, C. Forestier, C. Taillefumier and S. Faure, ChemMedChem, 2018, 13, 1513–1516 CrossRef CAS PubMed.
- N. Molchanova, H. Wang, P. R. Hansen, N. Høiby, H. M. Nielsen and H. Franzyk, Front. Microbiol., 2019, 10, 275 CrossRef PubMed.
- S. N. Benke, H. V. Thulasiram and H. N. Gopi, ChemMedChem, 2017, 12, 1610–1615 CrossRef CAS PubMed.
- S. Antunes, J.-P. Corre, G. Mikaty, C. Douat, P. L. Goossens and G. Guichard, Bioorg. Med. Chem., 2017, 25, 4245–4252 CrossRef CAS PubMed.
- T. A. Edwards and A. J. Wilson, Amino Acids, 2011, 41, 743–754 CrossRef CAS PubMed.
- V. Azzarito, K. Long, N. S. Murphy and A. J. Wilson, Nat. Chem., 2013, 5, 161–173 CrossRef CAS PubMed.
- M. Pasco, C. Dolain and G. Guichard, in Comprehensive Supramolecular Chemistry II, ed. J. L. Atwood, Elsevier, Oxford, 2017, pp. 89–125 DOI:10.1016/B978-0-12-409547-2.12565-X.
- K. Tsuchiya, T. Kurohara, K. Fukuhara, T. Misawa and Y. Demizu, Processes, 2022, 10, 924 CrossRef CAS.
- C. J. Brown, S. Lain, C. S. Verma, A. R. Fersht and D. P. Lane, Nat. Rev. Cancer, 2009, 9, 862–873 CrossRef CAS PubMed.
- C. Zhan, L. Zhao, X. Wei, X. Wu, X. Chen, W. Yuan, W.-Y. Lu, M. Pazgier and W. Lu, J. Med. Chem., 2012, 55, 6237–6241 CrossRef CAS PubMed.
- C. Li, M. Pazgier, M. Liu, W.-Y. Lu and W. Lu, Angew. Chem., Int. Ed., 2009, 48, 8712–8715 CrossRef CAS PubMed.
- M. Liu, C. Li, M. Pazgier, C. Li, Y. Mao, Y. Lv, B. Gu, G. Wei, W. Yuan, C. Zhan, W.-Y. Lu and W. Lu, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 14321–14326 CrossRef CAS PubMed.
- M. Liu, M. Pazgier, C. Li, W. Yuan, C. Li and W. Lu, Angew. Chem., Int. Ed., 2010, 49, 3649–3652 CrossRef CAS PubMed.
- S. Chen, X. Li, W. Yuan, Y. Zou, Z. Guo, Y. Chai and W. Lu, RSC Adv., 2017, 7, 9989–9997 RSC.
- M. Pazgier, M. Liu, G. Zou, W. Yuan, C. Li, C. Li, J. Li, J. Monbo, D. Zella, S. G. Tarasov and W. Lu, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 4665–4670 CrossRef CAS PubMed.
- A. Barnard, K. Long, H. L. Martin, J. A. Miles, T. A. Edwards, D. C. Tomlinson, A. Macdonald and A. J. Wilson, Angew. Chem., Int. Ed., 2015, 54, 2960–2965 CrossRef CAS PubMed.
- V. Azzarito, J. A. Miles, J. Fisher, T. A. Edwards, S. L. Warriner and A. J. Wilson, Chem. Sci., 2015, 6, 2434–2443 RSC.
- C. M. Grison, J. A. Miles, S. Robin, A. J. Wilson and D. J. Aitken, Angew. Chem., Int. Ed., 2016, 55, 11096–11100 CrossRef CAS PubMed.
- P. Sang, Y. Shi, J. Lu, L. Chen, L. Yang, W. Borcherds, S. Abdulkadir, Q. Li, G. Daughdrill, J. Chen and J. Cai, J. Med. Chem., 2020, 63, 975–986 CrossRef CAS PubMed.
- Y. Shi, P. Sang, J. Lu, P. Higbee, L. Chen, L. Yang, T. Odom, G. Daughdrill, J. Chen and J. Cai, J. Med. Chem., 2020, 63, 13187–13196 CrossRef CAS PubMed.
- L. Cussol, L. Mauran-Ambrosino, J. Buratto, A. Y. Belorusova, M. Neuville, J. Osz, S. Fribourg, J. Fremaux, C. Dolain, S. R. Goudreau, N. Rochel and G. Guichard, Angew. Chem., Int. Ed., 2021, 60, 2296–2303 CrossRef CAS PubMed.
- J. M. Adams and S. Cory, Science, 1998, 281, 1322–1326 CrossRef CAS PubMed.
- A. Letai, M. C. Bassik, L. D. Walensky, M. D. Sorcinelli, S. Weiler and S. J. Korsmeyer, Cancer Cell, 2002, 2, 183–192 CrossRef CAS PubMed.
- B. J. Smith, E. F. Lee, J. W. Checco, M. Evangelista, S. H. Gellman and W. D. Fairlie, ChemBioChem, 2013, 14, 1564–1572 CrossRef CAS PubMed.
- M. D. Boersma, H. S. Haase, K. J. Peterson-Kaufman, E. F. Lee, O. B. Clarke, P. M. Colman, B. J. Smith, W. S. Horne, W. D. Fairlie and S. H. Gellman, J. Am. Chem. Soc., 2012, 134, 315–323 CrossRef CAS PubMed.
- G. A. Eddinger and S. H. Gellman, Angew. Chem., Int. Ed., 2018, 57, 13829–13832 CrossRef CAS PubMed.
- B. Manandhar and J.-M. Ahn, J. Med. Chem., 2015, 58, 1020–1037 CrossRef CAS PubMed.
- Daniel J. Drucker, Cell Metab., 2016, 24, 15–30 CrossRef CAS PubMed.
- D. J. Drucker, Cell Metab., 2018, 27, 740–756 CrossRef CAS PubMed.
- S. Runge, H. Thøgersen, K. Madsen, J. Lau and R. Rudolph, J. Biol. Chem., 2008, 283, 11340–11347 CrossRef CAS PubMed.
- C. R. Underwood, P. Garibay, L. B. Knudsen, S. Hastrup, G. H. Peters, R. Rudolph and S. Reedtz-Runge, J. Biol. Chem., 2010, 285, 723–730 CrossRef CAS PubMed.
- A. Jazayeri, M. Rappas, A. J. H. Brown, J. Kean, J. C. Errey, N. J. Robertson, C. Fiez-Vandal, S. P. Andrews, M. Congreve, A. Bortolato, J. S. Mason, A. H. Baig, I. Teobald, A. S. Doré, M. Weir, R. M. Cooke and F. H. Marshall, Nature, 2017, 546, 254–258 CrossRef CAS PubMed.
- Y. Zhang, B. Sun, D. Feng, H. Hu, M. Chu, Q. Qu, J. T. Tarrasch, S. Li, T. Sun Kobilka, B. K. Kobilka and G. Skiniotis, Nature, 2017, 546, 248–253 CrossRef CAS PubMed.
- K. Hupe-Sodmann, G. P. McGregor, R. Bridenbaugh, R. Göke, B. Göke, H. Thole, B. Zimmermann and K. Voigt, Regul. Pept., 1995, 58, 149–156 CrossRef CAS PubMed.
- E. G. Siegel, G. Scharf, B. Gallwitz, R. Mentlein, C. Morys-Wortmann, U. R. Fölsch and W. E. Schmidt, Eur. J. Clin. Invest., 1999, 29, 610–614 CrossRef CAS PubMed.
- E. G. Siegel, B. Gallwitz, G. Scharf, R. Mentlein, C. Morys-Wortmann, U. R. Fölsch, J. Schrezenmeir, K. Drescher and W. E. Schmidt, Regul. Pept., 1999, 79, 93–102 CrossRef CAS PubMed.
- Q. Xiao, J. Giguere, M. Parisien, W. Jeng, S. A. St-Pierre, P. L. Brubaker and M. B. Wheeler, Biochemistry, 2001, 40, 2860–2869 CrossRef CAS PubMed.
- L. Jessen, B. A. Aulinger, J. L. Hassel, K. J. Roy, E. P. Smith, T. M. Greer, S. C. Woods, R. J. Seeley and D. A. D'Alessio, Endocrinology, 2012, 153, 5735–5745 CrossRef CAS PubMed.
- L. M. Johnson, S. Barrick, M. V. Hager, A. McFedries, E. A. Homan, M. E. Rabaglia, M. P. Keller, A. D. Attie, A. Saghatelian, A. Bisello and S. H. Gellman, J. Am. Chem. Soc., 2014, 136, 12848–12851 CrossRef CAS PubMed.
- M. V. Hager, L. M. Johnson, D. Wootten, P. M. Sexton and S. H. Gellman, J. Am. Chem. Soc., 2016, 138, 14970–14979 CrossRef CAS PubMed.
- B. P. Cary, M. V. Hager and S. H. Gellman, ChemBioChem, 2019, 20, 2834–2840 CrossRef CAS PubMed.
- J. Fremaux, C. Venin, L. Mauran, R. H. Zimmer, G. Guichard and S. R. Goudreau, Nat. Commun., 2019, 10, 924 CrossRef PubMed.
- J. Fremaux, C. Venin, L. Mauran, R. Zimmer, F. Koensgen, D. Rognan, S. Bitsi, M. A. Lucey, B. Jones, A. Tomas, G. Guichard and S. R. Goudreau, Chem. Sci., 2019, 10, 9872–9879 RSC.
- P. Sang, Z. Zhou, Y. Shi, C. Lee, Z. Amso, D. Huang, T. Odom, V. T. B. Nguyen-Tran, W. Shen and J. Cai, Sci. Adv., 2020, 6, eaaz4988 CrossRef CAS PubMed.
- Y. Shi, C. Lee, P. Sang, Z. Amso, D. Huang, W. Zhong, M. Gu, L. Wei, V. T. B. Nguyen-Tran, J. Zhang, W. Shen and J. Cai, Acta Pharm. Sin. B, 2023, 13, 1648–1659 CrossRef CAS PubMed.
- R. W. Cheloha, A. Maeda, T. Dean, T. J. Gardella and S. H. Gellman, Nat. Biotechnol., 2014, 32, 653–655 CrossRef CAS PubMed.
- R. W. Cheloha, B. Chen, N. N. Kumar, T. Watanabe, R. G. Thorne, L. Li, T. J. Gardella and S. H. Gellman, J. Med. Chem., 2017, 60, 8816–8833 CrossRef CAS PubMed.
- J.-P. Vilardaga, G. Romero, P. A. Friedman and T. J. Gardella, Cell. Mol. Life Sci., 2011, 68, 1–13 CrossRef CAS PubMed.
- A. A. Pioszak and H. E. Xu, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 5034–5039 CrossRef CAS PubMed.
- F. Abascal, A. Corpet, Z. A. Gurard-Levin, D. Juan, F. Ochsenbein, D. Rico, A. Valencia and G. Almouzni, Mol. Biol. Evol., 2013, 30, 1853–1866 CrossRef CAS PubMed.
- A. Corpet, L. De Koning, J. Toedling, A. Savignoni, F. Berger, C. Lemaître, R. J. O'Sullivan, J. Karlseder, E. Barillot, B. Asselain, X. Sastre-Garau and G. Almouzni, EMBO J., 2011, 30, 480–493 CrossRef CAS PubMed.
- X. Liang, X. Yuan, J. Yu, Y. Wu, K. Li, C. Sun, S. Li, L. Shen, F. Kong, J. Jia, M. Björkholm and D. Xu, EBioMedicine, 2017, 21, 104–116 CrossRef PubMed.
- Y. Wu, X. Li, J. Yu, M. Björkholm and D. Xu, Cell Death Dis., 2019, 10, 76 CrossRef PubMed.
- J. Mbianda, M. Bakail, C. André, G. Moal, M. E. Perrin, G. Pinna, R. Guerois, F. Becher, P. Legrand, S. Traoré, C. Douat, G. Guichard and F. Ochsenbein, Sci. Adv., 2021, 7, eabd9153 CrossRef CAS PubMed.
- D. Hanahan and Robert A. Weinberg, Cell, 2011, 144, 646–674 CrossRef CAS PubMed.
- M. Klagsbrun and M. A. Moses, Chem. Biol., 1999, 6, R217–R224 CrossRef CAS PubMed.
- W. Risau, Nature, 1997, 386, 671–674 CrossRef CAS PubMed.
- H. S. Haase, K. J. Peterson-Kaufman, S. K. Lan Levengood, J. W. Checco, W. L. Murphy and S. H. Gellman, J. Am. Chem. Soc., 2012, 134, 7652–7655 CrossRef CAS PubMed.
- J. W. Checco, D. F. Kreitler, N. C. Thomas, D. G. Belair, N. J. Rettko, W. L. Murphy, K. T. Forest and S. H. Gellman, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 4552–4557 CrossRef CAS PubMed.
- J. W. Checco and S. H. Gellman, ChemBioChem, 2017, 18, 291–299 CrossRef CAS PubMed.
- S. Abdulkadir, C. Li, W. Jiang, X. Zhao, P. Sang, L. Wei, Y. Hu, Q. Li and J. Cai, J. Am. Chem. Soc., 2022, 144, 270–281 CrossRef CAS PubMed.
- R. Nusse and H. Clevers, Cell, 2017, 169, 985–999 CrossRef CAS PubMed.
- J. N. Anastas and R. T. Moon, Nat. Rev. Cancer, 2013, 13, 11–26 CrossRef CAS PubMed.
- T. N. Grossmann, J. T.-H. Yeh, B. R. Bowman, Q. Chu, R. E. Moellering and G. L. Verdine, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 17942–17947 CrossRef CAS PubMed.
- L. Dietrich, B. Rathmer, K. Ewan, T. Bange, S. Heinrichs, T. C. Dale, D. Schade and T. N. Grossmann, Cell Chem. Biol., 2017, 24, 958–968 CrossRef CAS PubMed.
- J. Sampietro, C. L. Dahlberg, U. S. Cho, T. R. Hinds, D. Kimelman and W. Xu, Mol. Cell, 2006, 24, 293–300 CrossRef CAS PubMed.
- P. Sang, M. Zhang, Y. Shi, C. Li, S. Abdulkadir, Q. Li, H. Ji and J. Cai, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 10757–10762 CrossRef CAS PubMed.
- M. J. Pereira, K. Thombare, A. Sarsenbayeva, P. G. Kamble, K. Almby, M. Lundqvist and J. W. Eriksson, Mol. Cell. Endocrinol., 2020, 503, 110696 CrossRef CAS PubMed.
- L. Wu, Y. Zhai, J. Lu, Q. Wang and F. Sun, Protein Expression Purif., 2013, 89, 232–240 CrossRef CAS PubMed.
- A. Evers, T. Haack, M. Lorenz, M. Bossart, R. Elvert, B. Henkel, S. Stengelin, M. Kurz, M. Glien, A. Dudda, K. Lorenz, D. Kadereit and M. Wagner, J. Med. Chem., 2017, 60, 4293–4303 CrossRef CAS PubMed.
- M. K. Åm, I. Dirnena-Fusini, A. L. Fougner, S. M. Carlsen and S. C. Christiansen, Sci. Rep., 2020, 10, 13735 CrossRef PubMed.
- V. T. Thieu, B. D. Mitchell, O. J. Varnado and B. M. Frier, Diabetes, Obes. Metab., 2020, 22, 469–479 CrossRef PubMed.
- C. Worth, D. Yau, M. Salomon Estebanez, E. O’Shea, K. Cosgrove, M. Dunne and I. Banerjee, Clin. Endocrinol., 2020, 92, 387–395 CrossRef PubMed.
- I. Banerjee, M. Salomon-Estebanez, P. Shah, J. Nicholson, K. E. Cosgrove and M. J. Dunne, Diabetic Med., 2019, 36, 9–21 CrossRef CAS PubMed.
- P. Sang, H. Zeng, C. Lee, Y. Shi, M. Wang, C. Pan, L. Wei, C. Huang, M. Wu, W. Shen, X. Li and J. Cai, J. Med. Chem., 2021, 64, 13893–13901 CrossRef CAS PubMed.
- G. D. Roodman and J. J. Windle, J. Clin. Invest., 2005, 115, 200–208 CrossRef CAS PubMed.
- L. M. Johnson, W. S. Horne and S. H. Gellman, J. Am. Chem. Soc., 2011, 133, 10038–10041 CrossRef CAS PubMed.
- W. S. Horne, L. M. Johnson, T. J. Ketas, P. J. Klasse, M. Lu, J. P. Moore and S. H. Gellman, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 14751–14756 CrossRef CAS PubMed.
- L. M. Johnson, D. E. Mortenson, H. G. Yun, W. S. Horne, T. J. Ketas, M. Lu, J. P. Moore and S. H. Gellman, J. Am. Chem. Soc., 2012, 134, 7317–7320 CrossRef CAS PubMed.
- A. L. Greninger, K. Rybkina, M. J. Lin, J. Drew-Bear, T. C. Marcink, R. C. Shean, N. Makhsous, M. Boeckh, O. Harder, F. Bovier, S. R. Burstein, S. Niewiesk, B. K. Rima, M. Porotto and A. Moscona, J. Clin. Invest., 2021, 131, e150506 CrossRef CAS PubMed.
- L. C. Rijsbergen, K. S. Schmitz, L. Begeman, J. Drew-Bear, L. Gommers, M. M. Lamers, A. L. Greninger, B. L. Haagmans, M. Porotto, R. L. D. Swart, A. Moscona and R. D. D. Vries, mBio, 2022, 13, e03831 CrossRef CAS PubMed.
- V. K. Outlaw, R. W. Cheloha, E. M. Jurgens, F. T. Bovier, Y. Zhu, D. F. Kreitler, O. Harder, S. Niewiesk, M. Porotto, S. H. Gellman and A. Moscona, J. Am. Chem. Soc., 2021, 143, 5958–5966 CrossRef CAS PubMed.
- J. D. Sipe, M. D. Benson, J. N. Buxbaum, S.-I. Ikeda, G. Merlini, M. J. M. Saraiva and P. Westermark, Amyloid, 2016, 23, 209–213 CrossRef CAS PubMed.
- F. Emamzadeh, J. Res. Med. Sci., 2016, 21, 29 CrossRef PubMed.
- J. Collinge, Nature, 2016, 539, 217–226 CrossRef PubMed.
- G. Hoffner and P. Djian, Mol. Neurobiol., 2015, 52, 1297–1314 CrossRef CAS PubMed.
- A. Mukherjee, D. Morales-Scheihing, P. C. Butler and C. Soto, Trends Mol. Med., 2015, 21, 439–449 CrossRef CAS PubMed.
- A. K. Mankad, I. Sesay and K. B. Shah, Curr. Probl. Cancer, 2017, 41, 144–156 CrossRef PubMed.
- S. Kumar, Mark A. Brown, A. Nath and Andrew D. Miranker, Chem. Biol., 2014, 21, 775–781 CrossRef CAS PubMed.
- S. Kumar and A. D. Miranker, Chem. Commun., 2013, 49, 4749–4751 RSC.
- S. Kumar, M. Birol, D. E. Schlamadinger, S. P. Wojcik, E. Rhoades and A. D. Miranker, Nat. Commun., 2016, 7, 11412 CrossRef CAS PubMed.
- M. Birol, S. Kumar, E. Rhoades and A. D. Miranker, Nat. Commun., 2018, 9, 1312 CrossRef PubMed.
- S. Kumar, A. Henning-Knechtel, I. Chehade, M. Magzoub and A. D. Hamilton, J. Am. Chem. Soc., 2017, 139, 17098–17108 CrossRef CAS PubMed.
- S. Kumar, A. Henning-Knechtel, M. Magzoub and A. D. Hamilton, J. Am. Chem. Soc., 2018, 140, 6562–6574 CrossRef CAS PubMed.
- J. Kaffy, C. Berardet, L. Mathieu, B. Legrand, M. Taverna, F. Halgand, G. Van Der Rest, L. T. Maillard and S. Ongeri, Chem. – Eur. J., 2020, 26, 14612–14622 CrossRef CAS PubMed.
- J. Ahmed, T. C. Fitch, C. M. Donnelly, J. A. Joseph, T. D. Ball, M. M. Bassil, A. Son, C. Zhang, A. Ledreux, S. Horowitz, Y. Qin, D. Paredes and S. Kumar, Nat. Commun., 2022, 13, 2273 CrossRef CAS PubMed.
- K. Ziach, C. Chollet, V. Parissi, P. Prabhakaran, M. Marchivie, V. Corvaglia, P. P. Bose, K. Laxmi-Reddy, F. Godde, J.-M. Schmitter, S. Chaignepain, P. Pourquier and I. Huc, Nat. Chem., 2018, 10, 511–518 CrossRef CAS PubMed.
- V. Corvaglia, I. Ait Mohamed Amar, V. Garambois, S. Letast, A. Garcin, C. Gongora, M. Del Rio, C. Denevault-Sabourin, N. Joubert, I. Huc and P. Pourquier, Pharmaceuticals, 2021, 14, 624 CrossRef CAS PubMed.
- B. M. Nestl, S. C. Hammer, B. A. Nebel and B. Hauer, Angew. Chem., Int. Ed., 2014, 53, 3070–3095 CrossRef CAS PubMed.
- I. Schomburg, A. Chang, S. Placzek, C. Söhngen, M. Rother, M. Lang, C. Munaretto, S. Ulas, M. Stelzer, A. Grote, M. Scheer and D. Schomburg, Nucleic Acids Res., 2012, 41, D764–D772 CrossRef PubMed.
- B. Legrand, J. Aguesseau-Kondrotas, M. Simon and L. Maillard, Catalysts, 2020, 10, 700 CrossRef CAS.
- Z. C. Girvin and S. H. Gellman, J. Am. Chem. Soc., 2020, 142, 17211–17223 CrossRef CAS PubMed.
- G. Maayan, M. D. Ward and K. Kirshenbaum, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 13679–13684 CrossRef CAS PubMed.
- P. S. P. Wang, J. B. Nguyen and A. Schepartz, J. Am. Chem. Soc., 2014, 136, 6810–6813 CrossRef CAS PubMed.
- J. Aguesseau-Kondrotas, M. Simon, B. Legrand, J.-L. Bantigniès, Y. K. Kang, D. Dumitrescu, A. Van der Lee, J.-M. Campagne, R. M. de Figueiredo and L. T. Maillard, Chem. – Eur. J., 2019, 25, 7396–7401 CrossRef CAS PubMed.
- D. Bécart, V. Diemer, A. Salaün, M. Oiarbide, Y. R. Nelli, B. Kauffmann, L. Fischer, C. Palomo and G. Guichard, J. Am. Chem. Soc., 2017, 139, 12524–12532 CrossRef PubMed.
- Z. C. Girvin and S. H. Gellman, J. Am. Chem. Soc., 2018, 140, 12476–12483 CrossRef CAS PubMed.
- Z. C. Girvin, M. K. Andrews, X. Liu and S. H. Gellman, Science, 2019, 366, 1528–1531 CrossRef CAS PubMed.
- M. K. Andrews, X. Liu and S. H. Gellman, J. Am. Chem. Soc., 2022, 144, 2225–2232 CrossRef CAS PubMed.
- C. Mayer, M. M. Müller, S. H. Gellman and D. Hilvert, Angew. Chem., Int. Ed., 2014, 53, 6978–6981 CrossRef CAS PubMed.
- S. R. Rao, S. L. Schettler and W. S. Horne, ChemPlusChem, 2021, 86, 137–145 CrossRef CAS PubMed.
- K. Ożga and Ł. Berlicki, ACS Bio Med Chem Au, 2022, 2, 316–327 CrossRef PubMed.
- C. A. Gersbach, T. Gaj and C. F. Barbas, III, Acc. Chem. Res., 2014, 47, 2309–2318 CrossRef CAS PubMed.
- K. L. George and W. S. Horne, Acc. Chem. Res., 2018, 51, 1220–1228 CrossRef CAS PubMed.
- S. H. Yoo, B. Li, C. Dolain, M. Pasco and G. Guichard, in Methods in Enzymology, ed. E. J. Petersson, Academic Press, 2021, vol. 656, pp. 59–92 Search PubMed.
- K. L. George and W. S. Horne, J. Am. Chem. Soc., 2017, 139, 7931–7938 CrossRef CAS PubMed.
- C. M. Lombardo, V. Kumar M. V., C. Douat, F. Rosu, J.-L. Mergny, G. F. Salgado and G. Guichard, J. Am. Chem. Soc., 2019, 141, 2516–2525 CrossRef CAS PubMed.
- S. Rinaldi, Molecules, 2020, 25, 3276 CrossRef CAS PubMed.
- E. Yashima, N. Ousaka, D. Taura, K. Shimomura, T. Ikai and K. Maeda, Chem. Rev., 2016, 116, 13752–13990 CrossRef CAS PubMed.
- X. Yan, P. Weng, D. Shi and Y.-B. Jiang, Chem. Commun., 2021, 57, 12562–12574 RSC.
- Y. Ferrand and I. Huc, Acc. Chem. Res., 2018, 51, 970–977 CrossRef CAS PubMed.
- P. S. P. Wang and A. Schepartz, Chem. Commun., 2016, 52, 7420–7432 RSC.
- K. Kulkarni, N. Habila, M. P. Del Borgo and M.-I. Aguilar, Front. Chem., 2019, 7, 70 CrossRef CAS PubMed.
- V. Koehler, A. Roy, I. Huc and Y. Ferrand, Acc. Chem. Res., 2022, 55, 1074–1085 CrossRef CAS PubMed.
- M. Szefczyk, Nanoscale, 2021, 13, 11325–11333 RSC.
- M. S. Melicher, J. Chu, A. S. Walker, S. J. Miller, R. H. G. Baxter and A. Schepartz, Org. Lett., 2013, 15, 5048–5051 CrossRef CAS PubMed.
- S. H. Yoo, J. Buratto, A. Roy, E. Morvan, M. Pasco, K. Pulka-Ziach, C. M. Lombardo, F. Rosu, V. Gabelica, C. D. Mackereth, G. W. Collie and G. Guichard, J. Am. Chem. Soc., 2022, 144, 15988–15998 CrossRef CAS PubMed.
- S. De, B. Chi, T. Granier, T. Qi, V. Maurizot and I. Huc, Nat. Chem., 2018, 10, 51–57 CrossRef CAS PubMed.
- Y. Zhang, Y. Zhong, A. L. Connor, D. P. Miller, R. Cao, J. Shen, B. Song, E. S. Baker, Q. Tang, S. V. S. R. K. Pulavarti, R. Liu, Q. Wang, Z.-L. Lu, T. Szyperski, H. Zeng, X. Li, R. D. Smith, E. Zurek, J. Zhu and B. Gong, J. Am. Chem. Soc., 2019, 141, 14239–14248 CrossRef CAS PubMed.
- R. Misra, S. Dey, R. M. Reja and H. N. Gopi, Angew. Chem., Int. Ed., 2018, 57, 1057–1061 CrossRef CAS PubMed.
- M. P. Del Borgo, A. I. Mechler, D. Traore, C. Forsyth, J. A. Wilce, M. C. J. Wilce, M.-I. Aguilar and P. Perlmutter, Angew. Chem., Int. Ed., 2013, 52, 8266–8270 CrossRef CAS PubMed.
- R. S. Seoudi, A. Dowd, M. D. Borgo, K. Kulkarni, P. Perlmutter, M.-I. Aguilar and A. Mechler, Pure Appl. Chem., 2015, 87, 1021–1028 CrossRef CAS.
- S. Motamed, M. P. Del Borgo, K. Kulkarni, N. Habila, K. Zhou, P. Perlmutter, J. S. Forsythe and M. I. Aguilar, Soft Matter, 2016, 12, 2243–2246 RSC.
- K. Luder, K. Kulkarni, H. W. Lee, R. E. Widdop, M. P. Del Borgo and M.-I. Aguilar, Chem. Commun., 2016, 52, 4549–4552 RSC.
- K. Kulkarni, S. Motamed, N. Habila, P. Perlmutter, J. S. Forsythe, M.-I. Aguilar and M. P. Del Borgo, Chem. Commun., 2016, 52, 5844–5847 RSC.
- R. S. Seoudi, M. G. Hinds, D. J. D. Wilson, C. G. Adda, M. Del Borgo, M.-I. Aguilar, P. Perlmutter and A. Mechler, Nanotechnology, 2016, 27, 135606 CrossRef PubMed.
- M. P. D. Borgo, K. Kulkarni, M. A. Tonta, J. L. Ratcliffe, R. Seoudi, A. I. Mechler, P. Perlmutter, H. C. Parkington and M.-I. Aguilar, APL Bioeng., 2018, 2, 026104 CrossRef PubMed.
- A. J. Christofferson, Z. S. Al-Garawi, N. Todorova, J. Turner, M. P. Del Borgo, L. C. Serpell, M.-I. Aguilar and I. Yarovsky, ACS Nano, 2018, 12, 9101–9109 CrossRef CAS PubMed.
- K. Kulkarni, J. Hung, A. J. Fulcher, A. H. P. Chan, A. Hong, J. S. Forsythe, M.-I. Aguilar, S. G. Wise and M. P. Del Borgo, ACS Biomater. Sci. Eng., 2018, 4, 3843–3847 CrossRef CAS PubMed.
- S. Motamed, M. P. Del Borgo, K. Zhou, K. Kulkarni, P. J. Crack, T. D. Merson, M.-I. Aguilar, D. I. Finkelstein and J. S. Forsythe, Front. Bioeng. Biotechnol., 2019, 7, 315 CrossRef PubMed.
- A. Méndez-Ardoy, N. Markandeya, X. Li, Y.-T. Tsai, G. Pecastaings, T. Buffeteau, V. Maurizot, L. Muccioli, F. Castet, I. Huc and D. M. Bassani, Chem. Sci., 2017, 8, 7251–7257 RSC.
- S. H. Yoo, G. W. Collie, L. Mauran and G. Guichard, ChemPlusChem, 2020, 85, 2243–2250 CrossRef CAS PubMed.
- S. Kwon, A. Jeon, S. H. Yoo, I. S. Chung and H.-S. Lee, Angew. Chem., Int. Ed., 2010, 49, 8232–8236 CrossRef CAS PubMed.
- S. Kwon, H. S. Shin, J. Gong, J.-H. Eom, A. Jeon, S. H. Yoo, I. S. Chung, S. J. Cho and H.-S. Lee, J. Am. Chem. Soc., 2011, 133, 17618–17621 CrossRef CAS PubMed.
- S. Kwon, K. Kang, A. Jeon, J. H. Park, I. S. Choi and H.-S. Lee, Tetrahedron, 2012, 68, 4368–4373 CrossRef CAS.
- J. Kim, S. Kwon, S. H. Kim, C.-K. Lee, J.-H. Lee, S. J. Cho, H.-S. Lee and H. Ihee, J. Am. Chem. Soc., 2012, 134, 20573–20576 CrossRef CAS PubMed.
- S. H. Yoo, T. Eom, S. Kwon, J. Gong, J. Kim, S. J. Cho, R. W. Driver, Y. Lee, H. Kim and H.-S. Lee, J. Am. Chem. Soc., 2015, 137, 2159–2162 CrossRef CAS PubMed.
- S. Kwon, B. J. Kim, H.-K. Lim, K. Kang, S. H. Yoo, J. Gong, E. Yoon, J. Lee, I. S. Choi, H. Kim and H.-S. Lee, Nat. Commun., 2015, 6, 8747 CrossRef CAS PubMed.
- J. Gong, T. Eom, W. Lee, A. Roy, S. Kwon, H. Kim and H.-S. Lee, ChemPlusChem, 2019, 84, 481–487 CrossRef CAS PubMed.
- J.-H. Eom, J. Gong, R. Jeong, R. W. Driver and H.-S. Lee, Solid State Sci., 2015, 48, 39–43 CrossRef CAS.
- J.-H. Eom, J. Gong, S. Kwon, A. Jeon, R. Jeong, R. W. Driver and H.-S. Lee, Angew. Chem., Int. Ed., 2015, 54, 13204–13207 CrossRef CAS PubMed.
- J.-H. Eom, R. Jeong, J. Gong, R. W. Driver and H.-S. Lee, Bull. Korean Chem. Soc., 2015, 36, 2583–2584 CrossRef CAS.
- J. Gong, J.-H. Eom, R. Jeong, R. W. Driver and H.-S. Lee, Solid State Sci., 2017, 70, 1–5 CrossRef CAS.
- D. Lim, H. Kim, J. Gong, J.-H. Eom, E. Yoon, R. W. Driver, M.-H. Baik and H.-S. Lee, Chem. – Eur. J., 2019, 25, 2226–2233 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |