
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe challenge of peptide nucleic acid synthesis
K. P.
Nandhini
ab,
Danah Al
Shaer
 ab,
Fernando
Albericio
*bc and
Beatriz G.
de la Torre
*a
ab,
Fernando
Albericio
*bc and
Beatriz G.
de la Torre
*a
aKwaZulu-Natal Research Innovation and Sequencing Platform (KRISP), School of Laboratory Medicine and Medical Sciences, College of Health Sciences, University of KwaZulu-Natal, Durban 4041, South Africa. E-mail: garciadelatorreb@ukzn.ac.za
bPeptide Science Laboratory, School of Chemistry and Physics, University of KwaZulu-Natal, Westville, Durban 4000, South Africa. E-mail: albericio@ukzn.ac.za
cCIBER-BBN, Networking Centre on Bioengineering, Biomaterials and Nanomedicine, and Department of Organic Chemistry, University of Barcelona, Marti i Franques 1-11, 08028 Barcelona, Spain
First published on 3rd April 2023
Abstract
Peptide nucleic acids (PNAs) are an important class of DNA/RNA mimics that can hybridize complementary chains of nucleic acids with high affinity and specificity. Because of this property and their metabolic stability, PNAs have broad potential applications in different fields. Consisting of a neutral polyamide backbone, PNAs are prepared following the method used for peptide synthesis. In this regard, they are prepared by the sequential coupling of the protected monomers on a solid support using a similar approach to solid-phase peptide synthesis (SPPS). However, PNA synthesis is a little more challenging due to issues of the difficulty on the preparation of monomers and their solubility. Furthermore, the PNA elongation is jeopardized by intra/inter chain aggregation and side reactions. These hurdles can be overcome using different protecting group strategies on the PNA monomer, which also dictate the approach followed to prepare the oligomers. Herein, the main synthetic strategies driven by the protecting group scheme are discussed. However, there is still ample scope for further enhancement of the overall process.
 K. P. Nandhini | Nandhini KP (Nandhini Kalavathi Palanisamy) was born in Coimbatore (India). She obtained her Bachelor of Technology (BTech) degree at Sathyabama University in Tamil Nadu (India) and her Master of Science (MS) at the University of Skövde (Sweden). During that time, she was an Erasmus student at the University of Copenhagen (Denmark) under the supervision of Prof. Paul Robert Hansen. After that, she moved to the University of Kwazulu-Natal (South Africa) for carrying out the PhD under the supervisions of Professors Beatriz G. de la Torre and Fernando Albericio. Her PhD research involves both classic organic chemistry techniques and solid-phase strategies (SPS) for the synthesis of Peptides and Peptide Nucleic Acids (PNA). |
 Danah Al Shaer | Danah Alshaer is from Jordan, she was born in Kuwait. She obtained her BSc and MSc in chemistry from the University of Jordan. Her Master project was focused on the synthesis of new sequestering agents for some lanthanides. She was Lecturer in the University of Hail (Saudi Arabia) (2009–2017). Then, she joined the groups of Professors Fernando Albericio and Beatriz G de la Torre at the University of KwaZulu-Natal (South Africa) for the PhD. Her research was focused on the solid-phase synthesis and physico-chemical characterization of peptide-based HOPO containing ligands for Fe(III) chelation, as potential bacterio-static agents. She currently works as a medicinal chemist, drug discovery scientist I, in Evotec Ltd. (Abingdon-United Kingdom). |
 Fernando Albericio | Fernando Albericio's career has taken him through Europe, the USA, Latin America, and Asia to Africa (presently Research Professor at the University of KwaZulu-Natal (South Africa)). His research covers practically all aspects of peptide synthesis, as well as the synthesis of small molecules with therapeutic activities, especially against tuberculosis, infectious diseases, and cancer. His group is also involved in developing new strategies for drug delivery. He is involved in the third mission of the University: the transference of knowledge and technology to society. He was co-founder and general director of the Barcelona Science Park and is co-promotor of BioDurban. |
 Beatriz G. de la Torre | Beatriz G. de la Torre obtained her PhD from the University of Barcelona (Spain). After a dilated career in Spain, she is presently a research Professor at KRISP, College of Health Sciences, University of KwaZulu-Natal (Durban, South Africa). She has been working extensively on glyco, nucleo-, and lipopeptides. Her scientific interests are focused more on the discovery of new antimicrobial peptides, including those for fighting tuberculosis, peptide-based vaccines, and peptide-based drug-delivery systems. Lastly, she is also deeply involved in developing GREEN Solid-Phase Peptide Synthesis Strategies. |
1. Introduction
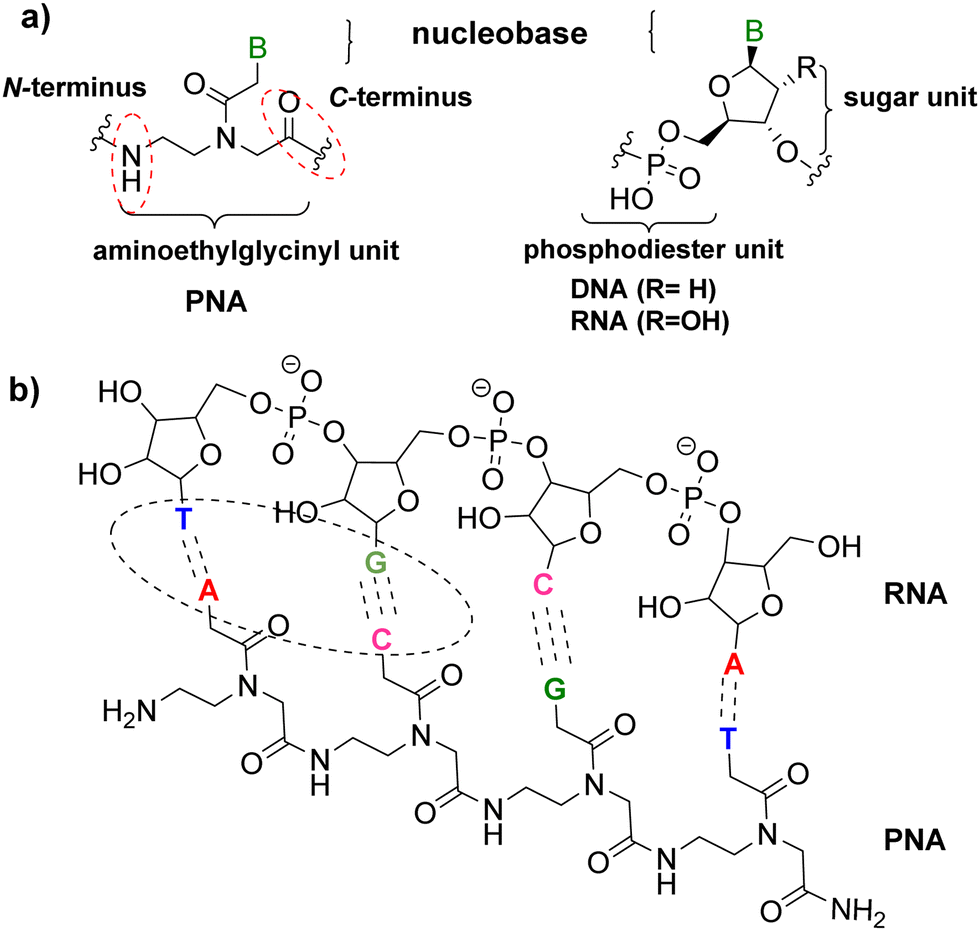
In the last two decades, interest in oligonucleotides as therapeutic drugs has grown due to their demonstrated capacity to modulate the expression of certain genes and provide treatments for serious genetic disorders. Between 2016 and 2021, the United States Food and Drug Administration (FDA) approved eleven oligonucleotide drugs with a range of applications, some of which act as short interfering nucleotides and others as gene antisense nucleotides.1 However, oligonucleotides in natural forms have weak binding affinity and membrane permeability, and they are also readily digestible, thus leading to low bioavailability. Hence, chemical modifications of the nucleobases, phosphodiester backbones, and sugar moieties of antisense oligonucleotides were necessary to improve target affinity, specificity and cellular uptake, decrease toxicity, and reduce manufacturing costs.2 To enhance the stability of oligonucleotides against degradation inside cells, several strategies have been exploited for the sugar–phosphate backbone, such as phosphorothioation,3,4 phosphoramidation5,6 and phosphoromethylation7 of the phosphate, 2′-fluorination or methylation of the ribose ring,8 and replacement of the whole backbone with a peptidyl-like one to render a peptide nucleic acid (PNA).PNAs were chemically developed at the Buchardt laboratory in 19919,10 as an N-(2-aminoethyl)glycine (AEG) unit that replaces the sugar–phosphodiester backbone of natural oligonucleotides, on which the nucleobases are connected. PNAs were hypothesized to form the genetic molecules in the very early forms of life on earth,11,12 a notion also supported by the presence of the aminoethyl glycine molecule in cyanobacteria.13 The distances between the nucleobases linked to the AEG backbone are approximately the same as the natural phosphodiester–sugar backbone of DNA/RNA, thus allowing hybridization of their complementary chain in a sequence-specific fashion.14–16 Therefore, PNAs are DNA/RNA analogues that are capable of forming hydrogen bonds with DNA/RNA nucleobases, obeying the Watson–Crick or Hoogsteen bonding rules (Fig. 1).14,15,17,18 Furthermore, being the PNA backbone non-ionic, unlike DNA/RNA, prevents electrostatic repulsions and the duplex PNA–DNA/RNA shows high thermal stability and is not affected by low ionic strength media. Moreover, PNAs are not susceptible to hydrolytic (enzymatic) cleavage as are not easily recognized by proteases or other enzymes.18,19
 | ||
| Fig. 1 (a) General structure of PNA vs. DNA or RNA monomers. (b) PNA–RNA duplex via Watson–Crick base pairing. | ||
PNAs show a greater capacity to inhibit reverse transcription than phosphorothioate oligonucleotides.20 Therefore, PNAs attracted attention as antisense and anti-gene agents for drug therapy applications,21,22 and more recently in gene editing. Beyond these applications, they have also been developed as biomolecular tools, molecular probes, and biosensors.23,24
The possibility of broadening PNA applications through the development of PNA conjugates with other biomolecules such as peptides and DNA25,26 has fuelled the development of various synthetic strategies. Furthermore, the possibility of modifying the classic PNA peptide backbone (the aminoethylglycyl) at the α-, β-, γ-positions, or the use of cyclic backbones, allows tuning the PNA conformations and hybridization capability.27,28 The modification on the γ-position is one of the most attractive to be exploited in many applications.18,22,29–31 The γ-substituted chiral PNA has shown several advantages compared with AEG–PNA such as minimal self-aggregation, good solubility, and stronger PNA–DNA duplex formation. It has been demonstrated that γ-PNA adopt a well-defined helical conformation left or right-handed depending on the chirality on the γ-stereocenter. The right-handed helix is afforded by the L-enantiomer which hybridizes to DNA/RNA with high affinity and sequence selectivity while the left-handed-helix shows less affinity than the achiral PNA.32 This is the reason why the enantiomeric purity of the γ-monomers is crucial for the synthesis of the oligomers.33
Although PNAs are closer to DNA/RNA complexes from a structural perspective and in terms of biological application, their synthesis takes advantage of the solid-phase synthesis (SPS) methodology developed for peptides.34–37 This review seeks to provide a synthetic overview of the chemical construction of PNA monomers, which involves the preparation of the AEG backbone and the nucleobase acetic acid, and it gives special attention to the protecting scheme used during the preparation of the monomers as the scheme will determine the strategy to be followed for PNA SPS. This review also aims to shed light on possible areas to explore in order to improve current synthetic strategies.
2. Synthesis of PNA at a glance (SPS)
As mentioned above, PNAs are synthesized using the SPS approach, which was initially developed for the synthesis of peptides. In this regard, the strategy is basically the same as that used for peptides. The C-terminal monomer is anchored to a linker through its carboxylic group on a solid support, while the primary amino function of the backbone and the exocyclic amino of the base are conveniently masked through temporary and permanent protecting groups, respectively. The temporary protecting group of the amino function is then removed, followed by the incorporation of the next monomer through an amide bond. At the end of PNA chain elongation, the permanent protecting group is removed from the bases and PNA is cleaved from the resin concomitantly (Scheme 1). | ||
| Scheme 1 General PNA SPS, showing the temporary (PG1) and permanent (PG2) protecting groups on the backbone and nucleobase, respectively, during synthesis. | ||
The synthesis of PNA was first achieved more than 30 years ago.9 However, as its preparation is not as straightforward as that of peptides, a large number of improvements by the Danish group that initiated the PNA synthesis and by other groups have since followed. In this regard, there are several challenges to overcome before achieving an optimum strategy that makes these molecules appealing from both a chemical and biological perspective. Some of the hurdles to overcome are that the nucleobases of PNA are more difficult to protect than most amino acid side-chains and PNA monomers show poor solubility,38 which is translated into inefficient coupling and the tendency to self-aggregate on resin. These factors limit PNA SPS to relatively short sequences.27,39 Thus, the choice of the protection for the backbone amino group and base (PG1, PG2 in Scheme 1) is crucial.
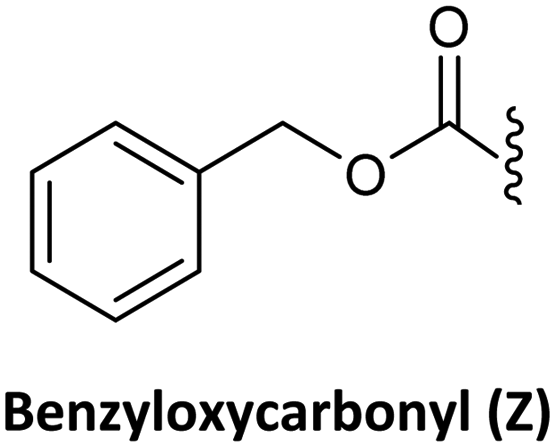
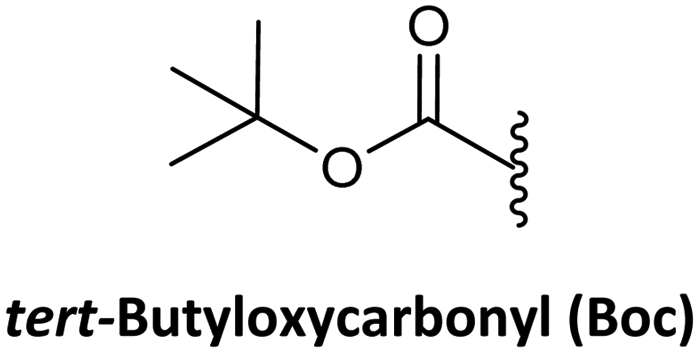
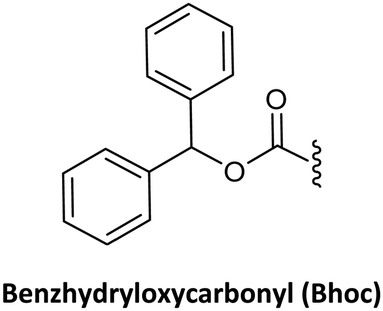
As in solid-phase peptide synthesis (SPPS), the first strategy developed by Buchardt's group was based on tert-butyloxycarbonyl (Boc) for the temporary protection of the amino function and benzyloxycarbonyl (Z) for the permanent protection of the exocyclic amino of the nucleobases, while p-methylbenzhydrylamine resin (MBHA) was used for linking the C-terminal monomer to the resin. An acid-labile amino protecting group such as Boc on the AEG backbone is compatible with Z on the nucleobase acetic acid. However, in this strategy, the strong conditions required to cleave PNA from the resin which involves trifluoroacetic acid (TFA)/hydrogen fluoride (HF) or trifluoromethanesulphonic acid (TFMSA), are quite harsh and not available in all research laboratories. Furthermore, the use of HF is banned in many countries. With the idea of using a milder scheme, Coull and co-workers developed the so-called 9-fluorenylmethyloxycarbonyl (Fmoc)/benzhydryloxycarbonyl (Bhoc) strategy.40 More recently, Franzyk and co-workers demonstrated the efficient synthesis and assembly of PNA following the Fmoc/Boc strategy.41
The base-labile protecting group Fmoc, which has mild removal conditions (20% piperidine in DMF), was introduced on the AEG backbone. However, the repetitive use of basic conditions to remove the temporary N-terminal protecting group has two major drawbacks. The first is the cyclization of AEG moieties of the backbone where the free amino group attacks the α-carboxyl group (Fig. 2A). This intramolecular reaction is favoured because of the formation of a stable six-membered lactam (ketopiperazine), leading to the loss of the monomer and, therefore, to deletion sequences. When preparing C-terminal carboxylic acid PNAs, this secondary reaction occurs dramatically upon attachment of the first PNA monomer to the functionalized resin.42 The initial attachment of a glycine residue for instance, as a spacer to the cleavable linker, for instance, helps prevent resin cleavage during the first deprotection because the six-member ring formation cannot take place. Given that this side reaction occurs mainly under basic conditions, the use of the right protecting group can also help prevent ketopiperazine formation. The second drawback is the transacylation of the nucleobase moiety from the α-amine of glycine to the free amine group of the ethylene diamine moiety (Fig. 2B). In this case, the secondary amine released can be acylated by the next monomer, thus leading to branched PNA. As in ketopiperazine formation, the driving force of the reaction is that it goes through a five-member ring intermediate.
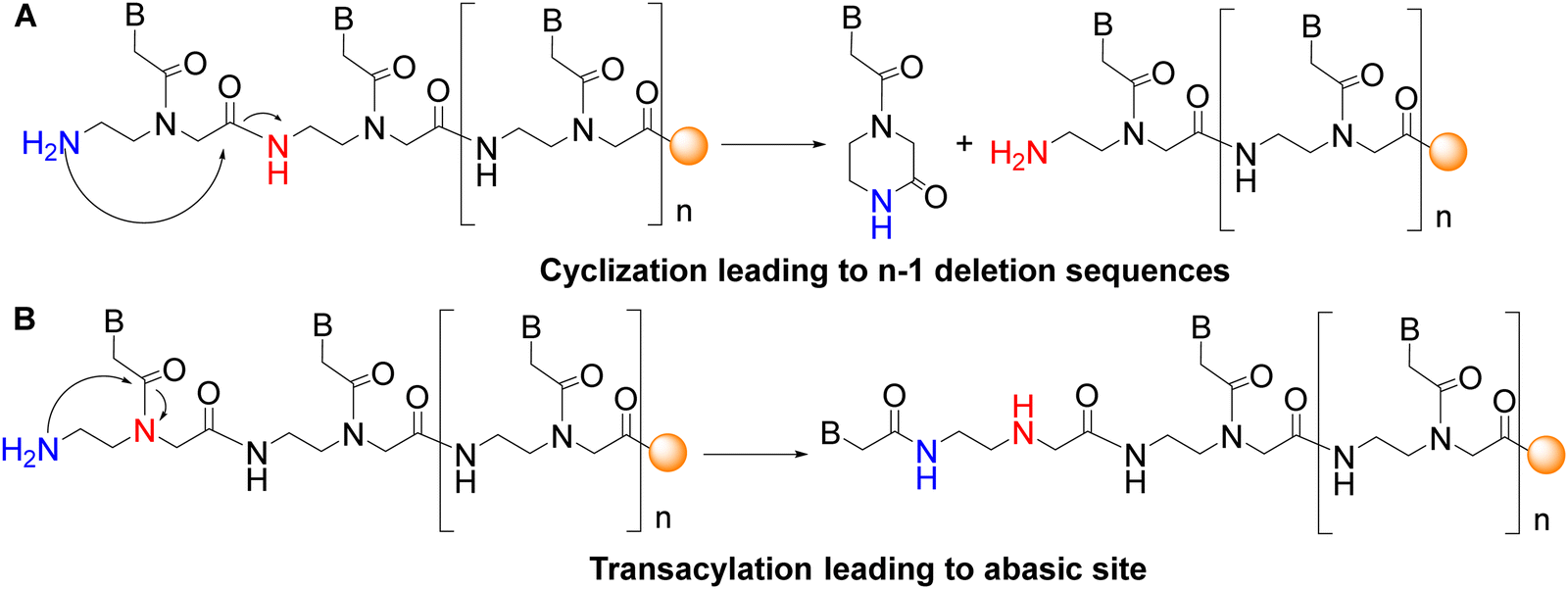 | ||
| Fig. 2 Major side reactions when using the Fmoc strategy.39 | ||
Although PNA oligomers can be synthesized using standard Boc/Z15,17,43 and Fmoc/Bhoc42 and the corresponding monomers are commercially available, the drawbacks outlined earlier emphasize the need to explore other strategies or enhance current ones. The alternative approaches using different combinations of protecting groups on the PNA monomer are discussed below.
A major problem during the elongation of the PNA sequence is the great tendency to have chain aggregation and therefore the low loading resins have been preferred. Furthermore, the use of more hydrophilic resins as polyethyleneglycol (PEG)-based resins, such as Tenta-Gel44 or ChemMatrix45 have been also used to reduce the aggregation.
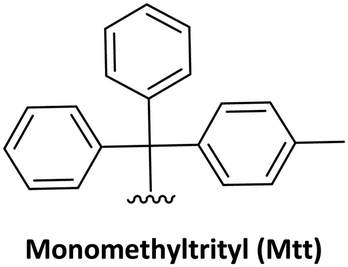
At the end of the synthesis, the cleavage and final deprotection of the permanent protecting groups is also a key for the choice of the strategy to follow. The standard solid supports (resin and linker) used for Boc/Z and Fmoc/Bhoc methodology require strong acid treatment, HF or TFMSA, and TFA. As the AEG-based PNAs does not have sensitive moieties as the peptides do, simple scavengers as H2O are very frequently used.39 However, when the target molecule is a PNA–peptide hybrid the proper scavengers should be included in the cleavage solution following the recommendations for peptides.46 Finally, these harsh cleaving conditions are incompatible with the synthesis of other types of modified PNAs like PNA–DNA chimeras. In that cases, the best supports are the ones commonly used in solid phase oligonucleotide synthesis which are cleavable by ammonia treatment. Thus, the nucleobases have to be protected with protecting groups labile to these conditions and the temporary amino protecting group preferable removed in mild acidic conditions as is the case of Mmt.25,47,48 But, in a more recent study Inagaki et al.49 demonstrated the compatibility of Fmoc as temporary amino protecting group with Bz protection for the nucleobases due to a significant slower kinetics of the Bz removal compared to Fmoc in presence of piperidine.
3. Elongation of the PNA oligomer
3.1 Solid-phase strategy
The synthesis of PNA should be considered in two steps. The first is the preparation of the protected monomers, which is preferably carried out in solution. The second step involves the elongation of the PNA chain, which is carried out on a solid support. For successful PNA synthesis, as pointed out before, the choice of the amino and base protecting groups is crucial. These groups can be orthogonal or not, thus the deprotection scheme should be carefully designed. Awareness of the possible side reactions and the drawbacks associated with each protection scheme can help identify the most appropriate option.The concept of orthogonality in SPPS was first demonstrated in 1985 by Barany and Albericio, using three distinct kinds of protecting groups.50 In an orthogonal protection scheme, each group is removed independently by a different chemical mechanism and can be removed in any order. The main advantage of an orthogonal protection scheme is that the removal conditions can be optimized to ensure the full deprotection of the chemical function without altering the rest. The Fmoc/Bhoc scheme is a good example of orthogonality because these groups are removed by base and acid, respectively. Orthogonality is desirable but not paramount for efficient synthesis.51
The concept of compatibility applied to a protection scheme is when two protecting groups are removed from the same molecule using the same kind of chemical reagent but at different concentrations, and then the removal of the first one does not damage the integrity of the second. However, the order of removal cannot be inverted as in the case of orthogonality.51
Table 1 shows the protecting groups explored in PNA SPS, classified on the basis of the chemical mechanism involved in the removal, indistinctly of whether the function to be removed is temporary [amino function (N-terminal)] or permanent (nucleobase).
| Entry | Name and structure | Lability | PG1/PG2 | Removal protocol | Ref. |
|---|---|---|---|---|---|
| a Used as protecting group for N3-T to prevent alkylated under Mitsunobu conditions. b Used as protecting group for O4-T to prevent aggregation during PNA synthesis. | |||||
| 1 |
|
Acid- and reduction-labile | PG2 | (1) HF (2.5% thioanisole) for 1 h @ 0 °C | 42,43 and 52–56 |
| (2) 0.5 ml anisole for 2–12 h, then cooled in N2 bath, followed by HF @ 0 °C for 30–45 min | |||||
| (3) 36% solution of dry HBr in AcOH for 15 min to 1 h @ RT | |||||
| (4) Refluxing anhydrous TFA for 30 min | |||||
| (5) Refluxing 18.5 mol% thioanisole in TFA for 3 h @ 25 °C | |||||
| (6) Catalytic hydrogenation | |||||
| (7) 1 M BBr3 in DCM for 1 h @ −10 °C and for 2 h @ 25 °C | |||||
| 2 |
|
Acid-labile | PG1 and PG2 | (1) 50% TFA in DCM (1 × 2 min and 1 × 30 min) | 43,57 and 58 |
(2) TFA/m-cresol (95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5) for 4 min :5) for 4 min |
|||||
| (3) TFA/DCM (1:1) for 1 h |
|||||
| 3 |
|
Acid-labile | PG2 | (1) 50% TFA in DCM (1 × 2 min and 1 × 30 min) | 58–61 |
| 4 |
|
Acid-labile | PG1 | (1) 1% TFA in DCM (5 min) or 10 min in neat hexafluoroisopropanol (HFIP) | 62 |
| 5 |

|
Acid-labile | PG1 and PG2 | (1) 3% trichloroacetic acid in methylene chloride (3 min) | 25,63 and 64 |
| (2) 1% TFA in DCM (5–10 min) with HFIP | |||||
| 6 |

|
Acid-labile | PG1 | (1) Successive 30 s treatments with TFA/MeOH/DCM (1:2:97) |
65 |
| 7 |
|
Acid-labile | PG2 | (1) TFA/m-cresol (4:1) for 90 min |
40 and 66 |
| 8 |

|
Base-labile | PG1 | (1) 20% piperidine in H2O @ 0 °C for 30–45 min | 42,57 and 67 |
| (2) 20% piperidine in DMF (1 × 2 min, 1 × 8 min) | |||||
| 9 |
|
Base-labile | PG1 | (1) 2% hydrazine in DMF | 26,68 and 69 |
| (2) 20% NH2OH·HCl/imidazole (1/0.75 equiv.) in NMP/DMF (5/1) | |||||
| 10 |
|
Base-labile | PG1 | (1) 0.8 M 4-methoxybenzenethiol and 0.4 M DIEA in DMF @ 40 °C for 10 min | 56 and 70–74 |
| (2) 3.89 mol% Al in crystals of HgCl2; under THF–H2O overnight @ RT | |||||
| (3) 69.7 mol% Zn powder in AcOH for 14 h @ RT | |||||
| (4) 50% aq. H3PO2 for 2 h with substrate under refluxing THF | |||||
| (5) 2–3 equiv. thiophenol in 3 equiv. K2CO3 for 3–5 h @ RT | |||||
| (6) 5 equiv. of NaBH4 in EtOH for 1–3 h @ RT | |||||
| (7) 20 equiv. PS-thiophenoxide resin in THF–EtOH (1:1) containing substrate; shaking for 2 h @ RT |
|||||
| 11 |

|
Base-labile | PG2 | (1) Ammonia (32%) or methylamine (40%) @ 60 °C for 20 h | 25,63,75 and 76 |
| (2) 28% ammonia @ RT for 5 h | |||||
| (3) Conc. aq. ammonia solution @ 50 °C for 6 h | |||||
| 12 |

|
Base-labile | PG2 | (1) Ammonia (32%) or methylamine (40%) @ 60 °C for 20 h | 25,63,75 and 76 |
| (2) 28% ammonia @ RT for 5 h | |||||
| (3) Conc. aq. ammonia solution @ 50 °C for 6 h | |||||
| 13 |
|
Base-labile | PG2 | (1) Ammonia (32%) or methylamine (40%) @ 60 °C for 20 h | 25,63,75 and 76 |
| (2) 28% ammonia @ RT for 5 h | |||||
| (3) Conc. aq. ammonia solution @ 50 °C for 6 h | |||||
| 14 |
|
Base-labile | PG2 | (1) Ammonia (32%) or methylamine (40%) @ 60 °C for 20 h | 25,63,75 and 76 |
| (2) 28% ammonia @ RT for 5 h | |||||
| (3) Conc. aq. ammonia solution @ 50 °C for 6 h | |||||
| 15 |
|
Base-labile | PG2 | (1) Ammonia (32%) or methylamine (40%) @ 60 °C for 20 h | 25,63,75 and 76 |
| (2) 28% ammonia @ RT for 5 h | |||||
| (3) Conc. aq. ammonia solution @ 50 °C for 6 h | |||||
| 16 |

|
Reduction-labile | PG1 | (1) Dithiothreitol (DTT) (0.5 M) in AcOH (1 × 2 min, 1 × 8 min) | 77 |
| 17 |

|
Photo-labile | PG1 | (1) Photolysis at wavelengths >300 nm; additives: N2H4, NH2OH·HCl, or semicarbazide·HCl (several hours) | 56 and 78 |
| 18 |

|
Photo-labile | PG1 | (1) Photolysis at wavelengths >300 nm | 79 |
| 19 |
|
Reduction-labile | PG1 | (1) 1 M THF solution of trimethyl phosphine and tributyl phosphine @ 23 °C for 5 min | 66 |
| 20 |

|
Reduction-labile | PG1 | (1) 1 M trimethyl phosphine in THF/H2O (95:5) for 5 min |
80 |
| 21 |

|
Reduction-labile | PG1 | (1) 3 M SnCl2 in 20 mM HCl in DMF @ 60 °C for 15 min | |
| (2) Catalytic hydrogenation | 61 | ||||
| 22 |
|
Acid-labile | PG2 | (1) 0.6 M AlCl3 in anisole @ RT for 30 min, followed by alkaline aqueous workup | 65 |
| 23 |
|
Acid-labile | PG2 | (1) Removed in cleavage conditions: | 81 |
| TFA/TIS/m-cresol/H2O 85:5:5:5 |
|||||
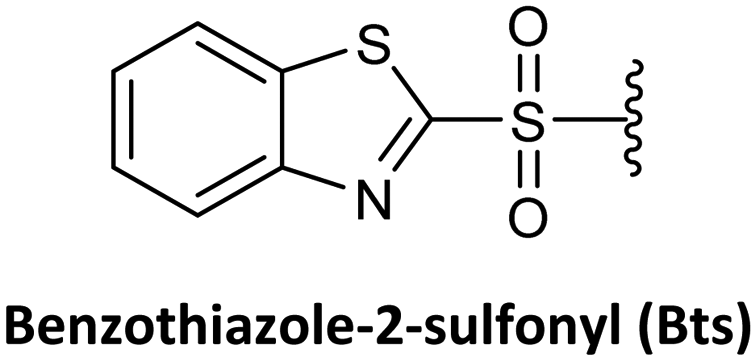
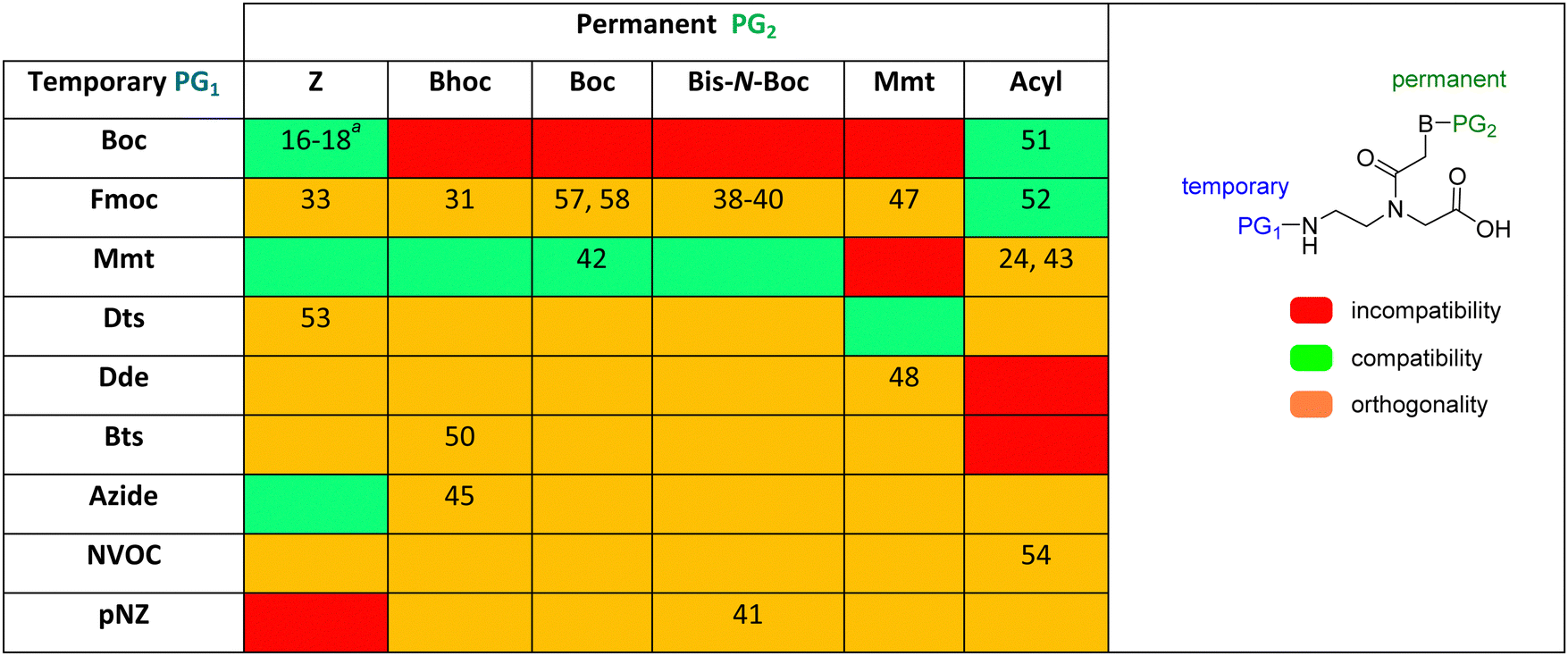
As mentioned above, Boc/Z and Fmoc/Bhoc are the two protection schemes most widely used for PNA synthesis; however, their drawbacks have led to the exploration of alternatives. The Fmoc group has also been studied with other permanent protecting groups for nucleobases, such as Z, Mmt, Acyl, Boc, and bis-N-Boc42,57–60,67,76,82–84 Alternatively, other temporary protecting groups, such as Mmt, Dts, Dde, Bts, azide, Nvoc, and pNZ, for the amino-terminal of the AEG backbone have been tested.
The Mmt/Boc strategy was established because of the convenience of using only TFA to remove both protecting groups. This is an example of protecting group compatibility. Mmt and Boc are compatible because the diluted TFA solution used to remove the former leaves the latter unaltered.62 The combination of Mmt for the backbone amino group and acyl protecting for the exocyclic amino group of the bases allowed the synthesis of PNA–DNA conjugates in SPS, thereby minimizing the risk of depurination.25,63
The use of Dts as a temporary protecting group at the N-terminal of the PNA monomers showed the same performance for the PNA synthesis compared to the use of Boc-monomers. However, using thiolytic reagents under mild acidic conditions to remove the Dts moiety and a shorter time than the usually described for the Boc strategy for the neutralization steps helped to minimize the typical side reactions explained above.77
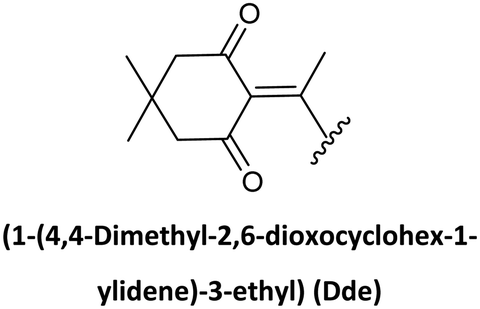
Along with preventing side reactions during PNA SPS, the need to prepare peptide–PNA conjugates to enhance, for instance, cellular uptake has driven the development of new protecting groups for PNA synthesis. In this regard, Bradley's lab developed the use of Dde as a temporary protecting group. In the search for N-terminal protecting groups for PNA monomers that are compatible/orthogonal with the standard Fmoc-amino acids and their side chain protection, they first explored the use of the Alloc group, which is removed under neutral conditions using Pd(0) catalysis. However, this protecting group did not render satisfactory results for chain elongation. Therefore, the Dde group emerged as the alternative since it is stable under Fmoc removal conditions. Nevertheless, the most used conditions to remove Dde, namely hydrazine solution, cause partial deprotection of the Fmoc group, hence Dde removal was tested using hydroxylamine, in which the Fmoc group showed full stability. Thus, the synthesis of Dde/Mmt protected PNA monomers was successful as well as their use for PNA oligomers and PNA/peptide conjugates.26,68,69
Lee et al.74 reported a novel type of cyclic PNA monomer where the Bts group was used as both protecting and self-activating group for the effective preparation of PNA oligomers. The amino group of the backbone carrying the Bts group cyclizes through the carboxyl group, giving a piperazinone, in which the carbonyl group is activated and prone to attack by nucleophiles such as the amine of PNAs (Scheme 2). The Bts monomers were synthesized in combination with the Bhoc protecting group for the exocyclic amino group of the bases. The removal of Bts in each cycle of the oligomerization was performed by means of thiols in the presence of base. The application of these monomers in the synthesis of a 15-mer PNA demonstrated their high performance. Although Bts cyclic monomers could be ideal for the large-scale SPS of PNA oligomers, the use of malodorous thiols is a huge limitation for their general implementation.
 | ||
| Scheme 2 PNA synthesis using Bts-protected/self-activated monomers. | ||
NVOC has also been used as protecting group for the amino function of the PNA backbone in combination with anisoyl protection for adenine and cytosine, and isobutyryl for guanine.78 The advantage of NVOC lies in its photolytic cleavage by irradiation at >300 nm, thus circumventing the need for commonly used deprotection reagents such as TFA or piperidine. The smooth deprotection conditions make these monomers suitable for photolithographic methods for the synthesis of PNA microarrays and PNA–DNA chimeras.85 Another photolabile group, namely NPPOC, has also been developed for the same application.79
Another strategy developed involves the use of the azide group to mask the N-terminal amine of the AEG backbone.66 The azide group was made to react with three phosphines: Me3P, Bu3P, and Ph3P.
Only the two alkyl phosphines rendered the iminophosphorane derivative rapidly. However, the hydrolysis to amine was slow in both cases. This problem was circumvented using the pre-activated N-hydroxybenzotriazole ester of the incoming monomer, although it was successful only in the case of the Me3P derivative. Along the same line, the protecting group Azoc was developed.80 In this case, full removal of the protecting group was achieved with a solution of Me3P in 5 min. Azoc is compatible with Fmoc, Alloc, and other protecting groups used in peptide synthesis. The Boc group was chosen to protect the exocyclic amino group of the bases for azide and Boc for Azoc.
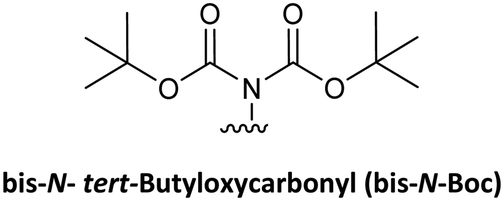
Although protecting groups such as Mmt, Dde, and Azide were successfully used to synthesize PNA monomers and oligomers and they avoided many of the disadvantages of protecting groups such as Fmoc and Dts and Bts, their scale-up was problematic due to the need for expensive starting material. With these considerations in mind, Huang et al.61 explored the use of pNZ as protecting group for the AEG backbone. This protecting group had already demonstrated effectiveness in peptide synthesis and can be produced cost-effectively from the non-expensive pNZ chloride at gram scale. The pNZ group is removed by nitro-reducing methods, the use of SnCl2 under acidic catalysis being described as preferable. Thus, this protecting group is orthogonal with Fmoc, Boc, and Alloc, which are all widely used in peptide chemistry. Nevertheless, because of the higher acidic lability of the Bhoc group, which is frequently used as protecting group of the bases in PNA monomers, the authors of this work preferred the combination with bis-N-Boc protection. Previous reports have compared bis-N-Boc protection with that afforded by Bhoc and Boc.59,60 The bis-N-Boc group offered higher stability than Bhoc and the same performance as Boc. However, as bis-N-Boc reduces one hydrogen-bond donor in the monomer, it is easier to solubilize in organic solvents.
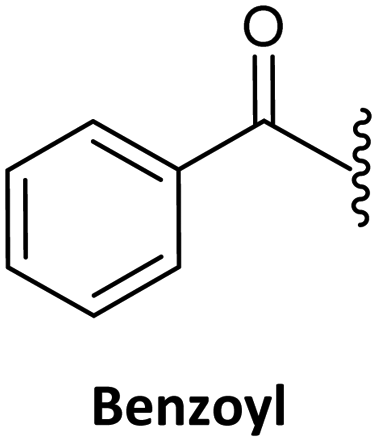
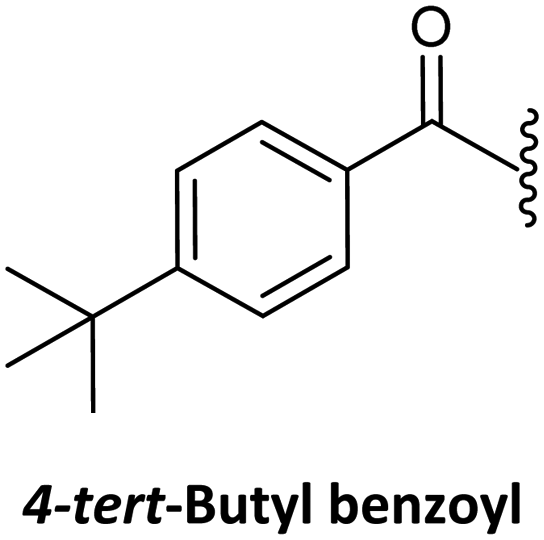
Finally, regarding the protection of the exocyclic amino group of the nucleobases, attention has also been given to acyl groups such as anisoyl, benzoyl, isobutanoyl, t-butyl benzoyl, and isobutyryl, which were already established in oligonucleotide synthesis.76
Table 2 shows a summary of protecting groups explored for PNA synthesis. Their combination as protecting groups for the amino backbone and exocyclic bases are highlighted in different colours on the basis of incompatibility, compatibility, and orthogonality. As can be appreciated from the table, there is still room to explore several combinations of protecting groups to improve PNA synthesis.
| a Numbers correspond to references. |
|---|
|
PNA chain elongation is achieved mainly monomer by monomer. However, from a structural perspective, given that the repetitive units consist of three moieties, namely glycine, ethylenediamine, and acyl-nucleobase, a subunit approach has also been used for oligomer elongation. This sub-monomeric approach can comprise two- or three-component assemblies.86,87
The first synthetic attempt at the two-component strategy was made by Seitz's group86 using the HYCRON linker (hydroxycrotyl-oligoethylene glycol-n-alkanoyl), an allylic anchor that contains a mini-chain of polyethylene glycol (PEG) to facilitate cleavage of the oligomer from the resin, a process achieved with Pd(0) catalysis.88 This linker offers a complete orthogonal protection scheme since both acid- and base-labile protecting groups can be used as temporary N-protection. The sub-monomeric strategy requires an extra temporary protecting group for the secondary amine of the AEG moiety. In the mentioned work, the N-terminal was protected with Boc, while Fmoc was used to protect the secondary amine of the backbone. Once the first component is attached to the resin as a preformed linker, which contains its own linker and the first AEG backbone moiety, the Fmoc group is removed, and the nucleobase acetic acid (second component) with the base protected with Z is introduced. At this point, the protected monomer can be obtained from the resin by Pd(0)-catalyzed cleavage, or Boc/Z monomers are incorporated until the oligomer has been completed (Scheme 3). Although only the first monomer was built on solid phase, the authors of that study found that the yield of the resulting PNA oligomer was markedly increased. The convenience of obtaining the protected oligomer lies in the possibility of introducing modifications to the C-terminus of the oligomer.
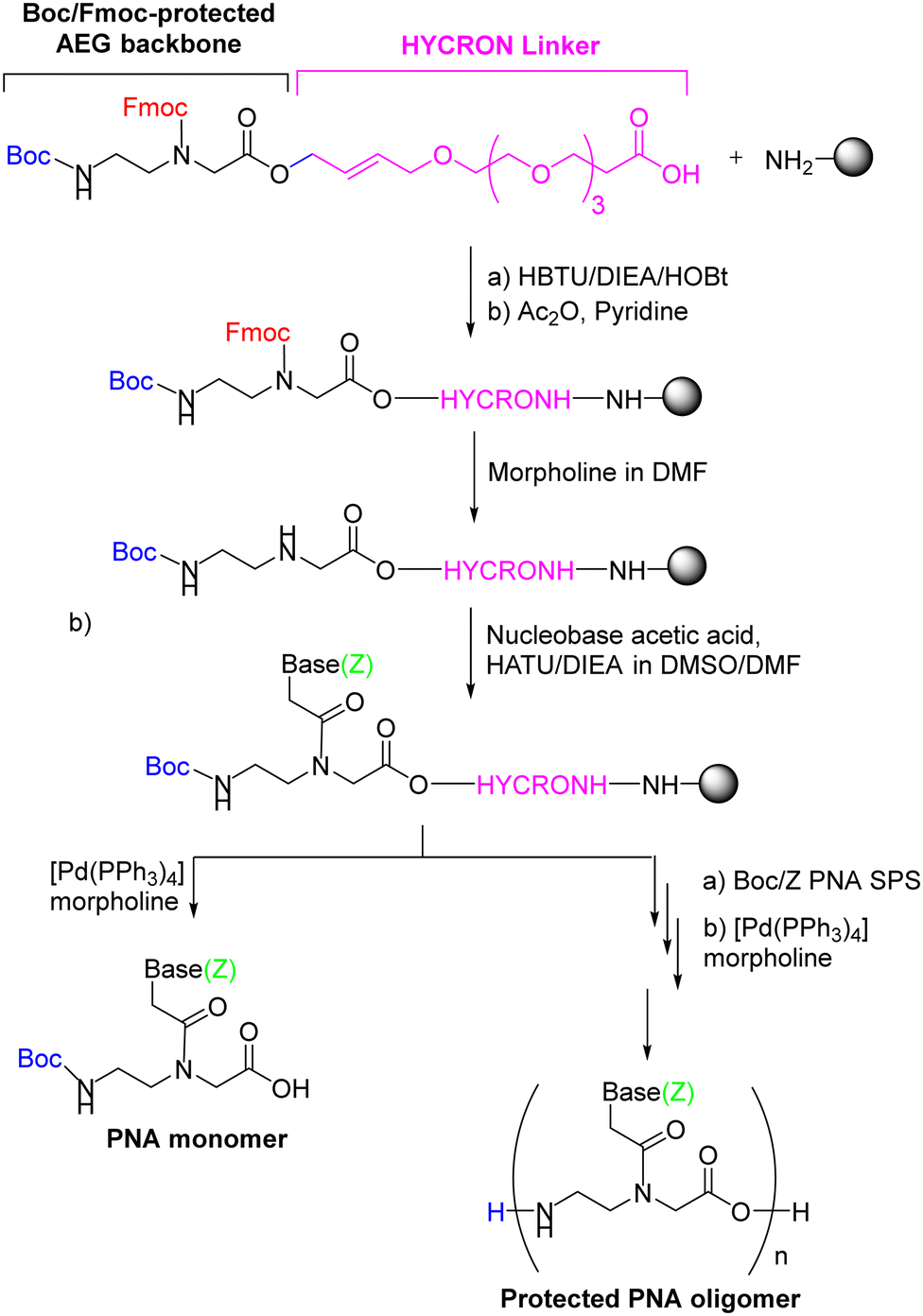 | ||
| Scheme 3 The first PNA monomer-HYCRON conjugate built from scratch to obtain a high-yield end product (oligomer). | ||
Following the two-subunit monomeric approach, Condom, Patino, and co-workers89 elongated first the PNA backbone using Boc as temporary protecting group and a variety of orthogonal protecting groups for the secondary amine, each one for each kind of nucleobase. Once the backbone had been elongated, the selective removal of each protecting group, followed by the incorporation of the corresponding nucleobase acetic acid derivative, led to the target sequence (Scheme 4).
 | ||
| Scheme 4 Sub-monomer PNA SPS through two subunit approach: secondary amine protected Boc-PNA backbone and nucleobase acetic acid derivatives. | ||
A further step forward in the field was the strategy to construct the PNA monomer entirely in solid phase from three components.
Richter and Zuckermann87 were the pioneers in the three-component strategy, which sought to circumvent the solubility issues associated with the protected PNA monomers. Using bromoacetic acid, Moz-ethylenediamine (Moz = 4-methoxybenzyloxycarbonyl), and 1-carboxymethyl-thymine as subunits, they were able to synthesize successfully an 8-mer thymine oligomer.
Later, in the in the work developed by Viirre and Hudson,65,90 the synthesis was started by incorporating Fmoc–Gly–OH (or another amino acid) onto Wang resin, which allows the detachment of the products by treatment with a high concentration of TFA. Then, after removal of the Fmoc group, the amino terminus was reprotected in the form of o-nitrobenzenesulphonamide (o-Ns), with a double function: masking the amine nucleophilicity at the same time that activating it for participating in a Fukuyama–Mitsunobu reaction. Thereby the formation of the AEG backbone bearing the primary amino group protected with dimethoxytrityl (DMT) was facilitated (Scheme 5). In the next step, the secondary amine was deprotected by thiolysis of the o-NS group and made reacting with the nucleobase derivatized as acid fluoride and protected with the p-methoxybenzyl (PMB) group. At this point, DMT can be removed using a low concentration of TFA, and the cycle from the Fmoc-amino acid coupling can be reinitiated and repeated to obtain the oligomer (Scheme 5, route A), or the amino group could be protected again with Fmoc followed by cleavage from the resin using a high concentration of TFA obtaining the protected PNA monomers for further use in PNA SPS (Scheme 5, route B).
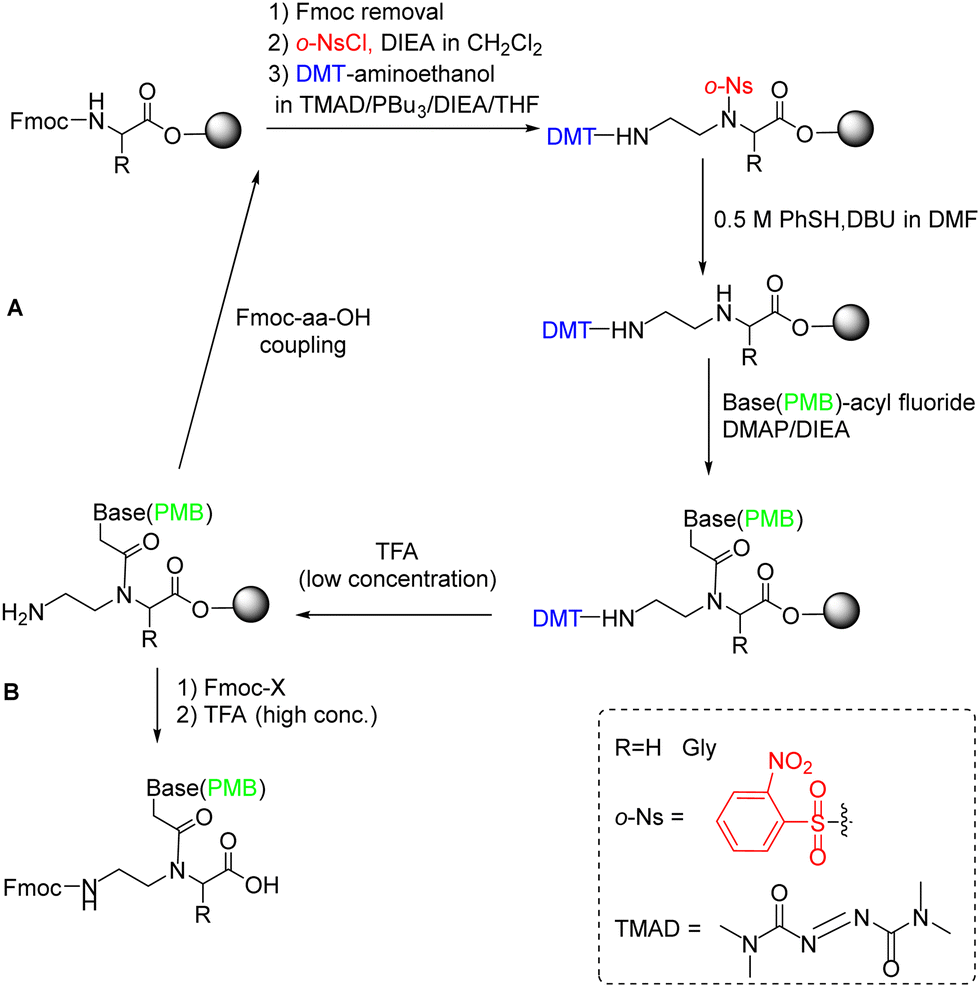 | ||
| Scheme 5 Sub-monomer PNA SPS using the Fukuyama–Mitsunobu reaction. (A) PNA oligomer synthesis and (B) Protected PNA monomer synthesis. | ||
The advantage of the three-component approach is that it allows the introduction of other amino acid residues instead of Gly in a straightforward manner. Such modifications introduce variability into the oligomer as chirality, charges that improve solubility, and functionalities to allow conjugations without modifying the binding to nucleic acids—all properties that are highly desirable in many PNA oligomers.
3.2 Solution-phase strategy
Although solid phase is the mode of choice for PNA synthesis, as occurs with peptides, several interesting strategies have been developed for the preparation of these molecules in solution. The group of Condom, Patino, and co-workers91 proposed the following three strategies for PNA synthesis in solution following an N → C chain elongation (the solid-phase mode is C → N): (i) the carboxylic acid of one protected monomer is reacted with the primary amino group of an AEG ester. Once this AEG ester has been incorporated, the nucleobase acetic acid derivative is attached in, and the ester hydrolysed. Then, the next AEG ester is incorporated and the cycle repeated until the full-length oligomer is built (Scheme 6A); (ii) based on a fully protected polyamide backbone (discussed above), where the whole backbone is built first, with all the secondary amines protected with different compatible amino protecting groups that are sequentially removed for the incorporation on each one the corresponding nucleobase acetic acid derivatives to finally give the desired PNA (Scheme 6B); (iii) shorter fragments, di- or trimers, are prepared by one of the previously mentioned strategies and the condensation takes place after hydrolysis of the ester of the N-fragment and the removal of the protecting group from the C-fragment (Scheme 6C). | ||
| Scheme 6 PNA synthesis in solution. (A) N → C chain elongation; (B) fully protected PNA backbone, followed by removal of protecting group sequentially for nucleobase acetic acid addition and (C) condensation between PNA di- or trimers after the removal of protecting groups from N- and C-fragments. | ||
These solution strategies are recommended only for the synthesis of PNAs with short sequences. The second strategy was successfully applied independently by the groups of Condom and van Boom for the preparation of cyclic PNAs consisting of two to six monomer units.92–96
Recently, Periyalagan and Hong described a variation of the third strategy for the preparation of trimers, where the carboxylic group to react is activated in the form of pentafluorophenyl ester and the incoming monomer has both the primary amine and the carboxylic group free. The trimers were used for the synthesis of PNAs in solution and solid phase, demonstrating great performance for solution synthesis up to 12-mer.97
Finally, Chiba and co-workers,98 who have extensively developed tag-assisted liquid phase peptide synthesis (LPPS),99 used this methodology to prepare PNAs. This LPPS approach is based on SPPS, where the solid support is substituted by a hydrophobic soluble linker, making the growing PNA soluble in solvents such as THF. After the incorporation of each monomer, PNA is collected by precipitation by adding a polar solvent such as acetonitrile (ACN).
4. Synthesis of PNA monomers
Although the preparation of the monomers in solid phase, like the examples shown in Schemes 3 and 4, can be appealing from the perspective of efficiency as it circumvents a tiresome workup and chromatographic purification after each step, most monomers have been prepared in solution. Most syntheses of PNA monomers reported in the literature start with the separate preparation of the AEG backbone with protected amino and carboxylic acid ends (Scheme 7A) and the protected nucleobase carboxylic acid derivative (Scheme 7B). The protected nucleobases are then introduced through an acylation reaction of the α-amine of the glycine moiety in the backbone (Scheme 7C). Finally, the protection of the carboxylic group of the glycine moiety is removed (Scheme 7D). To select the temporary and permanent protecting groups (PG1 and PG2), two points should be taken into consideration. The first is that the route through which the AEG backbone is prepared should be suitable for an efficient amino temporary protecting group PG1. The second point is that the protecting group of the C-terminus of the monomer (PG3) should be removable under conditions that do not affect the amino/base protecting groups PG1 or PG2, respectively.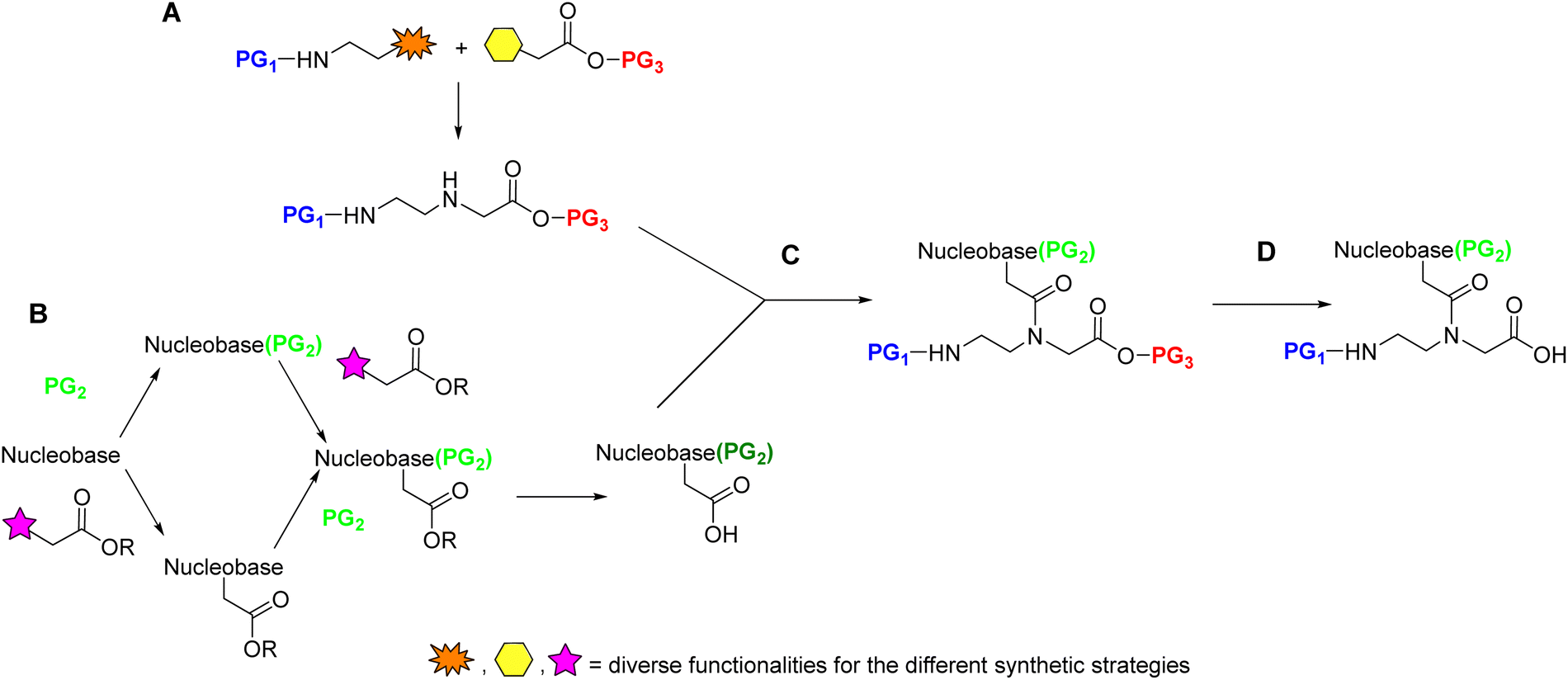 | ||
| Scheme 7 Schematic representation of the subunit synthesis of PNA monomers. | ||
4.1 Synthesis of the AEG backbone
The AEG backbone can be synthesized by distinct pathways:39 (i) alkylation;75 (ii) the Fukuyama–Mitsunobu100 reaction; and (iii) reductive amination,101 which are discussed below. | ||
| Scheme 8 Protected PNA backbone obtained by alkylation reaction. | ||
The obtention of the dialkylated amine as a by-product, along with unreacted amine, has been reported when using PG–EDA in the presence of base as starting material. Instead, a high excess of the unprotected EDA (9-fold excess) can substitute the use of a base and limit the likelihood of over-alkylation, affording the free N-terminal backbone with high yield.26,42,106 However, the overall yield was dramatically reduced after synthesis completion when attempting to achieve the desired PNA backbone with N-terminal protecting groups such as Fmoc, Dde, and Mmt.26,42,58,107
Advances made in PNA backbone synthesis have brought about the development of protocols with distinct protecting group schemes and synthetic methodologies. However, initially little attention was paid to the Fmoc strategy for PNA synthesis due to both costly materials and challenging synthetic routes rendering low yields. Although later on, the Fmoc strategy was probably the main applied approach, there are not many reports in the literature describing their synthesis.42,59,60
Thomson and co-workers42 efficiently synthesized the backbone Fmoc–AEG–OtBu starting from A, and PG3 was tBu (Scheme 8). For the obtention of the final monomer, Z was used as protecting group for the nucleobases. However, although the chain elongation to synthesize PNA oligomers could be run under smooth conditions, the final deprotection required treatment with HF. Based on this approach, Feagin and co-workers107 attempted the same strategy but substituting t-butyl ester by a benzyl ester, which has a significant advantage over the methyl/ethyl ester in that it can be removed by catalytic hydrogenation avoiding the basic treatment. Nevertheless, the reaction led to ketopiperazine and benzyl alcohol (Scheme 9). Thus, the less bulky nature of the benzyl ester favoured the cyclization, as was previously proven when using methyl ester analogues instead of t-butyl ester. In a second attempt, a “three-step process” was described to obtain Fmoc-EDA. First, EDA was mono-protected using Boc anhydride, followed by Fmoc protection on the other primary amine of EDA. Finally, Boc was removed under acidic conditions to yield the Fmoc–EDA as TFA salt. However, the following step of alkylation with benzyl bromoacetate failed because of the instability of the free base on the intermediate. The free amine group is basic enough to promote the cleavage of the Fmoc group under the reaction conditions. Finally, the third attempt was successful. Here, the Boc–AEG–OBn backbone was first synthesized, and the Boc protecting group was swapped for Fmoc (Scheme 9).
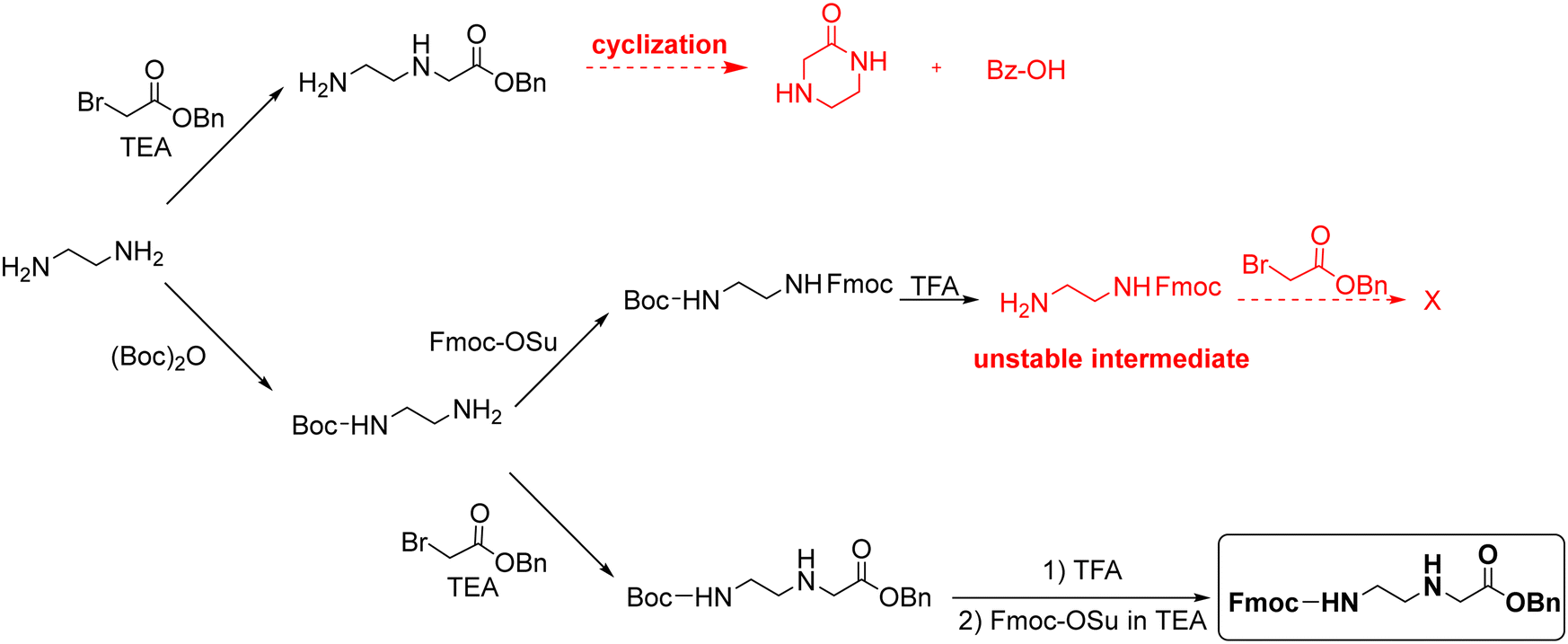 | ||
| Scheme 9 Synthesis of Fmoc–AEG–OBn backbone. | ||
The previously reported synthesis was considered a good strategy due to effortless scalability using low-cost starting materials, in spite of giving only a 32% overall yield. More recently, a new approach has been reported by the Franzyk lab in which EDA was alkylated with chloroacetic acid to obtain the unprotected PNA backbone. The AEG conversion into p-toluenesulfonic acid (PTSA) salt gave stability to the compound and allowed prolonged storage. Finally, the unprotected AEG was esterified and protected with Fmoc.108
Given some advantages of using Mmt as PG1 because of the compatibility of Boc protection for the nucleobases, the synthesis of the Mtt-AEG-OMe backbone was successfully achieved with a good overall yield. The described process consisted of first protecting EDA by reaction with 4-methyl trityl chloride, followed by its alkylation with methyl bromoacetate in the presence of TEA.62
Malamgari et al. adopted the Fukuyama N-alkylation reaction for AEG synthesis. Unlike the aforementioned approach where the EDA was alkylated by haloacetate derivatives, here the α-amino group of the Gly ester was alkylated by N-Boc-bromoethylamine.109 Thus, the glycine ester was first N-protected by o-nosyl chloride, followed by the alkylation reaction with the protected amine alkyl halide in the presence of DBU to obtain the fully protected AEG backbone. The o-nosyl protection was essential to prevent an over-alkylated side-product. Later, the o-nosyl group was selectively removed by thiolysis to afford the backbone in excellent yield (Scheme 10).
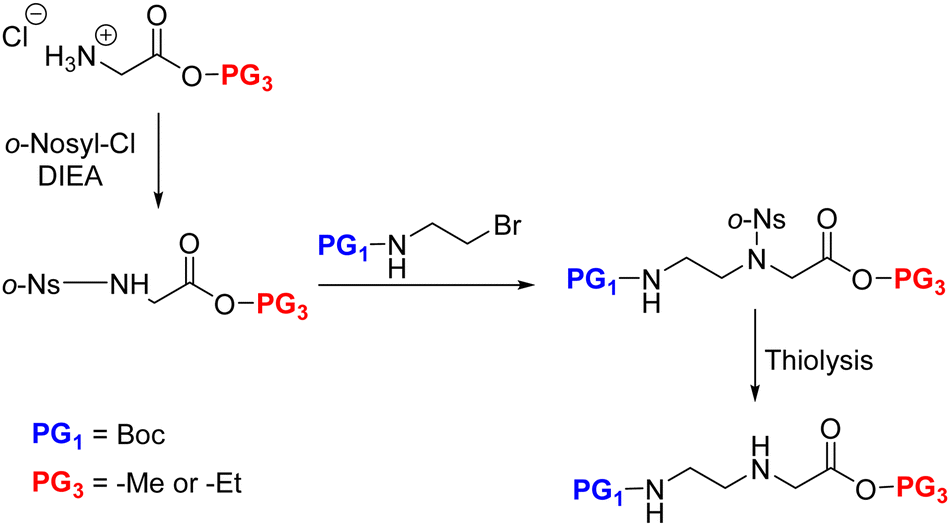 | ||
| Scheme 10 Synthesis of the fully protected AEG backbone via N-alkylation of amino acid (glycine). | ||
This approach has further merit as it can be easily adopted for the synthesis of chiral PNA monomers based on different amino acids of interest.
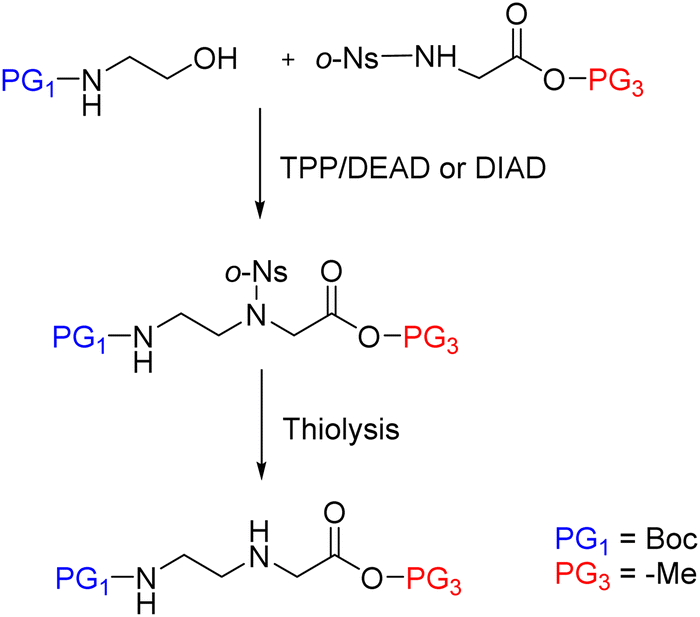 | ||
| Scheme 11 PNA backbone synthesis by Fukuyama–Mitsunobu reaction. | ||
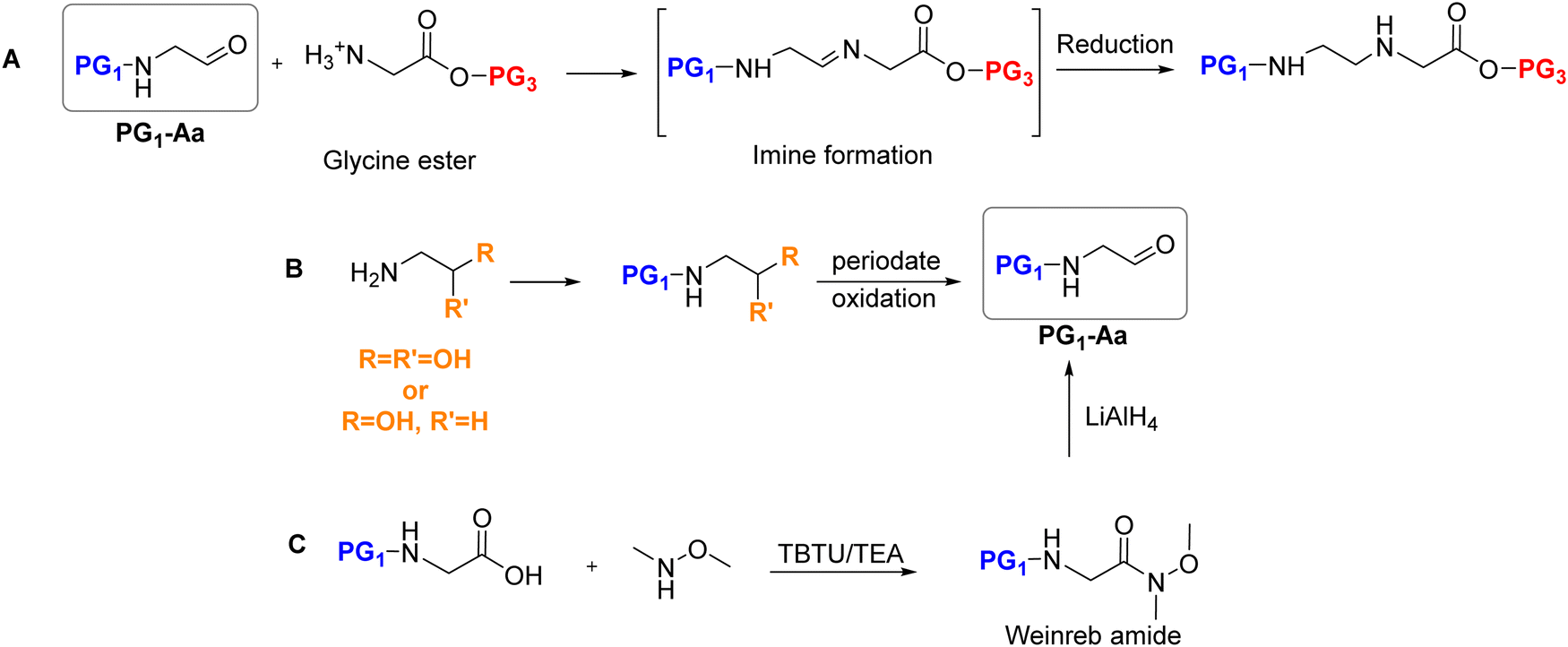 | ||
| Scheme 12 (A) Synthesis of the fully protected AEG backbone by reductive amination; (B) and (C) synthesis of the key aldehyde intermediate. | ||
The second critical step in this process is the reduction of the imine intermediates to form the final protected backbone. The final yields of PG1-AEG obtained in various studies depended on the reducing reagent used, the N-protecting group, and the ester. The trend observed is that reduction using 10% Pd/C under H2 gave acceptable yields. However, more potent reducing agents like NaBH4 led to lower yields and are therefore not suitable reagents for this purpose. In the case of using NaCNBH3 as a reductant, the moderate yields obtained were improved when the reaction was performed in the presence of AcOH.125 Although, it has not been reported for AEG backbone, Volpi et al.126 performed the reductive amination by transfer hydrogenation promoted by an Ir(III) catalyst127 in the presence of a mixture of formic acid and DIEA. The results were successful, reacting the (PG1-Aa) and D-Lys(Boc)-OH or DArg(Pbf)-OH. In this work the carboxylic acid did not require protection since it was used in a two-subunit monomeric approach.
4.2 Synthesis of protected nucleobase acetic acid derivatives
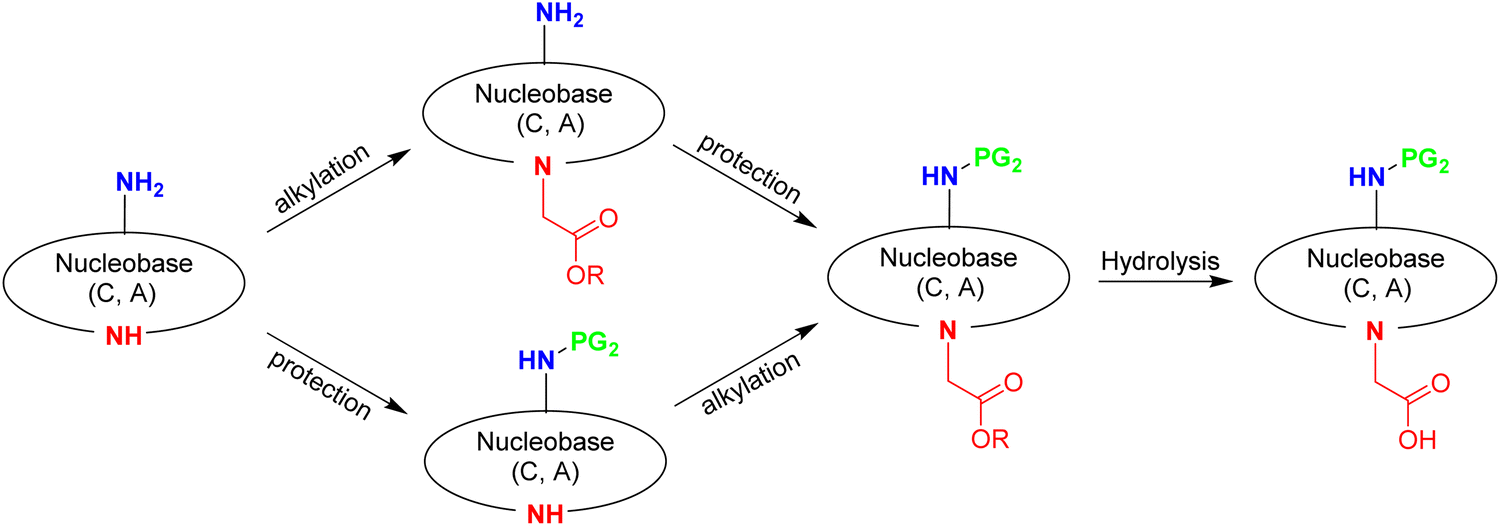
In this section, the four DNA canonical bases (T, C, A, and G) will be discussed along with the pseudo-complementary nucleobases (pcNB) 2-thiouracil (SU) and 2,6-diaminopurine (D).128 However, many other modified bases have been used as part of PNA monomers.129,130Prior to assembly onto the AEG backbone, the nucleobases have to undergo two modifications. One is common to all of them, namely the need to carry a methyl carboxylic acid, which will act as a linker between the nucleobase and the AEG backbone. This group is placed on the endocyclic N1 in the case of thymine and cytosine and on N9 in the case of adenine and guanine. The other modification is the protection of the exocyclic amine group of cytosine (N4), adenine (N6), and guanine (N2). while thymine does not require protection (Fig. 3). However, the protocols described for the protection of the exocyclic amino group are not common for the three nucleobases. The following discussion about the synthesis of the building blocks to be attached to the backbone is divided into three sections: (i) derivatization of thymine; (ii) derivatization of cytosine and adenine; and (iii) derivatization of guanine.
 | ||
| Fig. 3 Protected exocyclic amino group on the nucleobase acetic acids. | ||
 | ||
| Scheme 13 (A) Two basic routes for the synthesis of thyminyl acetic acid; (B) synthesis of O4-allyl-protected thyminyl acetic acid and (C) synthesis of N3-Boc-protected thyminyl acetic acid. | ||
Although thymine does not require further protection, owing to on-resin aggregation during the synthesis of poly-T PNAs, the use of ‘protected thymine’ was found not only to solve this issue but also to improve coupling efficiency. One of the alternatives to protect thymine adopted the notion from O4-allyl protection of thymidine applied successfully in oligonucleotide synthesis134 to prepare O4-allyl-protected Taa for PNA monomer synthesis.81O4-allyl-protection is achieved by activating the O4-carbonyl oxygen of Taa using 2-mesitylenesulfonyl chloride, followed by treatment with allyl alcohol to give the desired product in high yield (Scheme 13B).81 The second alternative consists of introducing a Boc protecting group on N3-position. In this synthesis, the benzyl ester of the bromoacetic acid was used for the alkylation. Thus after the introduction of the Boc group, the product was subjected to catalytic hydrogenation to give the desired product in quantitative yield (Scheme 13C).83
 | ||
| Scheme 14 Two basic routes followed in the synthesis of protected exocyclic amino cytosinyl and adeninyl acetic acid. | ||
The most relevant synthesis described in the literature for Caa and Aaa derivatives can be found in Tables 4 and 5.
It is important to highlight that adenine is more complex than cytosine because it has more reactive sites. For instance, carbamate formation can occur at both N1 and N6 although the N1 carbamate is hydrolyzed during the work-up. When N6 of adenine is protected, the alkylation can take place at N7 and N9, but the product can be purified by crystallization.42
The initial reports of synthetic procedures were devoted to the use of Z as protecting group (Table 3, #1; Table 4, #1). The laboratory of Nielsen-Buchardt carried out the protection and then the alkylation for Caa17 and then the opposite, alkylation–protection for Aaa.15 In a later study, the same group found that the use of benzyl chloroformate reagent gave some problems for Aaa, but the more sophisticated Rappoport's reagent (BzlOCOImEt+ BF4−) rendered better results.43 However, Thomson et al.,42 using a protection-alkylation route, found that Rappoport's reagent, which in their opinion was tedious to prepare, gave a mixture of difficult purification. Thus, in this case, benzyl chloroformate was preferred.
| # | PG2 | Reagent | Ester | Routea | Yield% | Ref. |
|---|---|---|---|---|---|---|
| a P = protection, A = alkylation. b Yields correspond to ref. 43. | ||||||
| 1 | Z | Benzyl chloroformate, pyridine | Methyl | P + A | 56/40b | 16 and 43 |
| Benzyl chloroformate, DMAP | t-Butyl | P + A | 43/59 | 42 | ||
| 2 | t-Butyl benzoyl | t-Butyl benzoyl chloride, TEA | Methyl | P + A | 61/36 | 63 |
| Ethyl | 46/63 | 76 | ||||
| 3 | Benzoyl | Benzoyl chloride, pyridine | Benzyl | A + P | 79/56 | 135 |
| Methyl | P + A | 62/45 | 47 | |||
| 4 | Mmt | Mmt-Cl, N-ethylmorpholine (NEM) | Methyl | P + A | 42/66 | 67 |
| 45/66 | 26 | |||||
| 5 | An | Anisoyl chloride, pyridine | Methyl | P + A | 93/48 | 76 and 78 |
| 81/82 | ||||||
| 6 | Bhoc | Benzhydrol, CDI | Benzyl | A + P | 91/71 | 66 |
| 7 | Boc | t-Butanol, CDI | Benzyl | A + P | 92/65 | 80 |
| 8 | Cl-Bhoc | 4,4′-Dichlorobenzhydrol, CDI | Benzyl | A + P | 92/76 | 80 |
| 9 | bis-N-Boc | (Boc)2O, TEA, DMAP (cat.) | Ethyl methyl | A + P | —/83 | 59 |
| (a) (Boc)2O, DMAP | P + A | 97/69–78 | 58, 60 and 61 | |||
| (b) NaHCO3 | ||||||
| # | PG2 | Reagent | Ester | Routea | Yield % | Ref. |
|---|---|---|---|---|---|---|
| a P = protection, A = alkylation. b Yields correspond to ref. 43. | ||||||
| 1 | Z | PhCH2OCOImEt+ BF4− (Rapoport's reagent) | Ethyl | A + P | 73/63b | 16 and 43 |
| Benzyl chloroformate, NaH | t-Butyl | P + A | 44/53 | 42 | ||
| 2 | Benzoyl | Benzoyl chloride, pyridine | Methyl | A + P | 75/46 | 47 |
| Ethyl | 44/35 | 75 | ||||
| 3 | An | Anisoyl chloride, TEA, or pyridine | Methyl | P + A | 82/37 | 63 |
| Methyl | 74/37 | 76 | ||||
| t-Butyl | 74/54 | |||||
| Ethyl | A + P | 64/70 | ||||
| 4 | Mmt | Mmt-Cl, pyridine -NEM, or NaOH | Methyl | P + A | 77/52 | 67 |
| Ethyl | A + P | 70/78 | 26 | |||
| 5 | Bhoc | Benzhydrol, CDI | Benzyl | A + P | 72/76 | 66 |
| Ethyl | 68/69 | 31 | ||||
| 6 | Boc | t-Butanol, CDI | Benzyl | A + P | 84/47 | 80 |
| 7 | Cl-Bhoc | 4,4′-Dichlorobenzhydrol, CDI | Benzyl | A + P | 84/73 | 80 |
| 8 | bis-N-Boc | (Boc)2O, TEA, DMAP (cat.) | Ethyl | A + P | —/61 | 59 |
| (a) (Boc)2O, DMAP | Benzyl | P + A | 95/70 | 58 and 60 | ||
| (b) NaHCO3 | Methyl | 55 (overall) | 61 | |||
Benzoyl47,75,135 (Table 3, #3; Table 4, #2) and anisoyl (Table 3, #5; Table 4, #3)25,63 were proposed as acyl protecting group for both cytosine and adenine. Nevertheless, for the synthesis of Caa, the t-butylbenzoyl group has also been described. The introduction of this protecting group aimed to avoid the precipitation of the final monomer during its incorporation into a PNA sequence, as observed in the case of being protected by the benzoyl and anisoyl groups (Table 3, #2).63,76
The use of TFA labile protecting groups such as Mmt (Table 3, #4; Table 4, #4), Boc (Table 3, #7; Table 4, #6), Bhoc (Table 3, #6; Table 4, #5), Cl-Bhoc (Table 3, #8; Table 4, #7), and Bis-N-Boc (Table 3, #9; Table 4, #8) had been gradually implemented due to: (i) the search of protecting groups friendlier to be removed; (ii) improved solubility of the final monomer; and (iii) compatibility with protecting groups of the amino of the AEG backbone removable in mild conditions. Mmt has been used to protect the nucleobases in combination with Fmoc67 and Dde26 as AEG protecting groups. In these two studies, the Caa derivative was synthesized similarly (protection–alkylation), but Aaa was prepared using both protection–alkylation67 and alkylation–protection strategies.26 Bhoc to protect nucleobases is the most widely used approach when Fmoc is the protecting group at the N-terminal of the AEG backbone since these monomers are commercially available. However, few studies describe the synthesis of these monomers. The Bhoc protecting group is preferred over Boc because it confers greater solubility to the final monomer, and it shows higher lability to TFA (1% of TFA).66 Like Boc, Bhoc can be removed at the same time as the PNA chain is cleaved from the solid support. Given its high acid lability, Bhoc was not a good candidate as nucleobase protector when the synthesis of protected PNA fragments for convergent strategy was the target. To increase acid stability, Cl-Bhoc was studied. However, it was still found to be too labile. In this case, the best alternative was found to be Boc.80
The bis-N-Boc group (Table 3, #9; Table 4, #8) was first described for adenine protection136 and later as an intermediate in the synthetic route of Boc-protected purines.137 The implementation of this protecting group in the synthesis of PNA monomers, independently carried out by Wojciechowski and Hudson59 and Porcheddu et al.,60 was driven by the high cost associated with the preparation of the Bhoc derivative. Furthermore, the introduction of Bhoc requires CDI or triphosgene, and Bhoc is not stable to catalytic hydrogenation (H2/Pd) or even mild acidolysis.59 The bis-N-Boc group has a double advantage because, on the one hand, it improves the solubility of the final monomer and, on the other hand, the absence of H, at position 6 avoids the interchain aggregation, thereby facilitating elongation of the PNA chain, similar to the role of Pro in peptide chemistry.138 While Wojciechowski and Hudson carried out first the alkylation and then the protection, Porcheddu et al. did the opposite. In both cases, the bis-N-Boc group was introduced using readily affordable reagents such as Boc2O on a large scale without significant purification of intermediates. Although performing the alkylation first prevents Boc introduction in other positions of the nucleobase, the strategy of Porcheddu et al. is more convenient in terms of yields and purity of the final products and has been adopted by other groups.58,61 In this strategy, the extra Boc introduced in position 1 in the case of cytosine and 9 in case of adenine, is easily removed by a short treatment with a NaHCO3 solution (Scheme 15).
 | ||
| Scheme 15 The synthesis of bis-N-Boc cytosine and adenine. | ||
2A-6Cl changes the reactivity of the molecule and renders alkylation at position 9 as the main product, along with alkylation at position 7. In the first route described by the Nielsen-Buchardt laboratory, 2A-6Cl was directly alkylated using bromoacetic acid, followed by the Cl substitution by a benzyloxy protecting group. The amino group at position 2 was not protected because of its low reactivity (Scheme 16). The overall yield obtained was 23%. However, this derivative presented two further problems; the benzyloxy group was not stable to the TFA treatment used to remove the Boc group, which was protecting the backbone in the final monomer, and the free amino at position 2 was partially acetylated during the capping step in the chain elongation process.43
 | ||
| Scheme 16 Synthesis of O6 protected Gaa from 2A-6Cl: N9 alkylation and 6-Cl replacement by 6-O-PG2. | ||
Later, Thompson et al.42 developed an alternative strategy to circumvent some of the problems previously mentioned. First, 2A-6Cl was alkylated with allyl bromide, giving a ratio of 3:1 between regioisomers at positions 9 and 7, which were easily separated. The Cl derivative was then converted into the guanidine one by refluxing with HCl, and the amino at position 2 was protected with the Z group. Finally, the allyl group that was masking the carboxylic acid, was submitted to ozonolysis followed by oxidation leading to Gaa (Scheme 17). All steps except the allylation, which requires purification, were performed with relatively good yields. The target product was obtained with an overall yield of 40%.
 | ||
| Scheme 17 Synthesis of N2 protected Gaa from 2A-6Cl: N9 allylation; conversion of 6-Cl to the O derivative; N2 protection; oxidation of the N9 alkene to N9 acid. | ||
Although the synthetic route described before gives an acceptable overall yield, it was not generally implemented by other research groups. Instead, much attention was given to the procedure starting from 2A-6Cl, in which it is first alkylated, followed by the 2-amino protection, and, finally, hydrolysis of the ester with the concomitant replacement of the 6-Cl by 6-OC (Scheme 18). This methodology has been used to synthesize Gaa protected with Mmt,26,67 Bhoc,66 Cl-Bhoc, Boc80 and iso-butyryl (iBu),139 achieving moderate yields for the first two mentioned protecting groups (30–40%), but a good yield for the rest (around 70%).
 | ||
| Scheme 18 Synthesis of N2 protected Gaa from 2A-6Cl: N9 alkylation; N2 protection; conversion of the 6-Cl to O derivative. | ||
By a similar route, a Gaa derivative with an extra protection at the O6 position was obtained. After the alkylation with the benzyl bromoacetate, 6-Cl was replaced by 6-OBzl. This reaction took place with concomitant removal of the benzyl ester, the free acid was again esterified in form of ethyl ester, and position 2 was protected in the form of iBu75 or as bis-N-Boc.59 Although these routes involved more steps, the overall yield was approximately 40% in both cases. The advantage of these derivatives was their solubility and minimization of the interchain aggregation caused by hydrogen bonds during the PNA chain elongation.
Porcheddu et al.60 attempted to directly protect guanine in a similar way as they did with adenine using Boc2O. In addition to bis-N-Boc in the amino at position 2, the Boc group also introduced at positions 1 and 9. However, in contrast to what happened with Aaa, in this case, the extra Boc groups could not be removed. But, when 2A-6Cl was used as starting material, the introduction of Boc at position 1 did not take place and the one at position 9 could be easily removed, as it was in the case of C and A (Scheme 15). After that, alkylation, followed by the substitution of the 6Cl by O, was carried out. That was done in two steps, the first by reaction with trimethylamine to give the ammonium salt and finally hydrolysis with NaOH. The overall yield was 50%. Similarly, Huan et al.61 followed the same synthetic scheme using the 6-OBzl guanine instead of 2A-6Cl as starting material. The benzyloxy moiety present in the final PNA monomer favours its solubility in organic solvents.
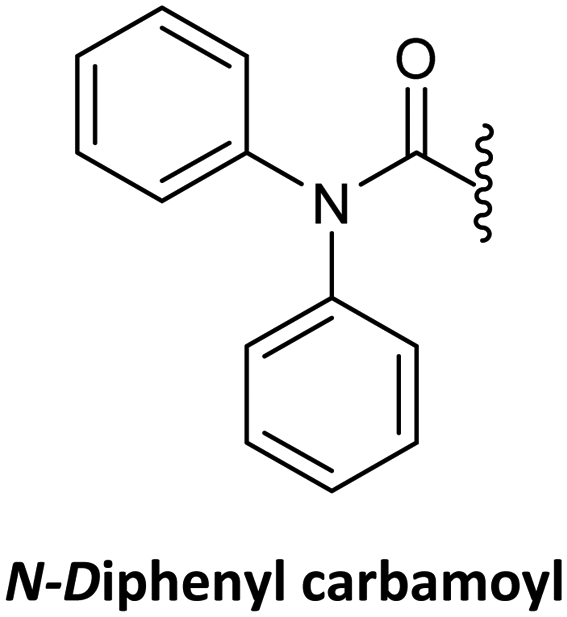
As mentioned earlier, the direct alkylation of the guanine is not a convenient starting point, but the synthesis of Gaa starting by acylation of the amine group at position 2 of guanine is possible. However, modification of position 2 alone is not enough to achieve regioselective alkylation thus, this step requires a further purification.63 Alternatively, the incorporation of the diphenylcarbamoiyl derivative at position 6 was shown to improve alkylation regioselectivity in the case of glycosidation.140 In the case of alkylation, this substitution also enhanced the regioselectivity of the alkylation at position 9,25,76 but this improvement was found to be dependent on the alkylating reagent.76
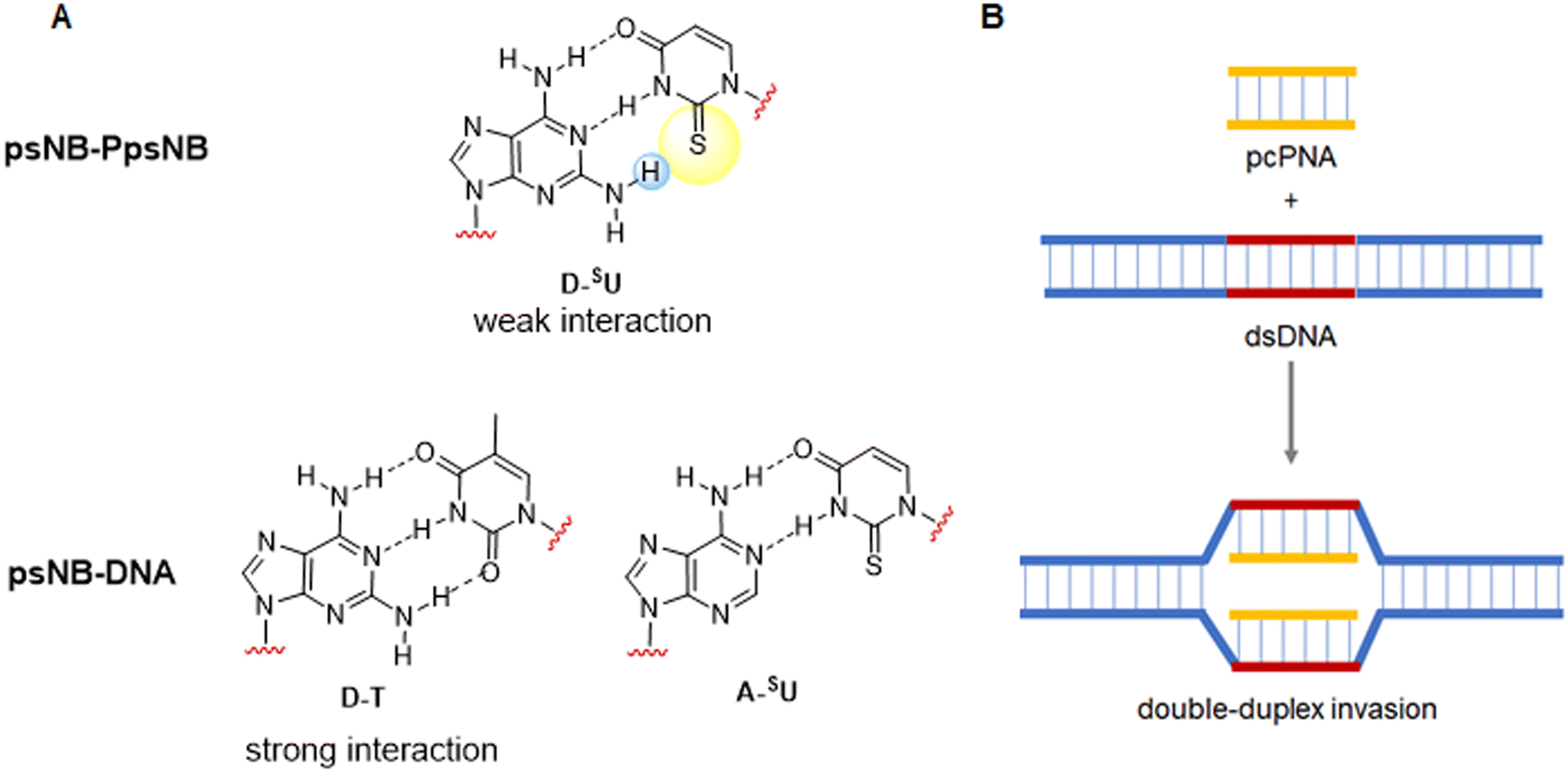 | ||
| Fig. 4 (A) Pattern of H-bonding between D-SU, D-T and A-SUs. (B) Double-duplex invasion of pcPNA into double-stranded DNA. | ||
Although pcPNAs were very promising, their use has not been extended as expected because it is not easy to get the monomers from commercial sources. Initially, U and D monomers were only described for Boc/Z strategy128,144,145 but later, these monomers were also protected in a compatible Fmoc/Bhoc way.84,146 Thus, like the canonical nucleobases, SU and D need to be protected and derivatized with a methyl carboxylic acid group (Fig. 5).
 | ||
| Fig. 5 Protected psNB acetic acid. | ||
The sulfur atom on SU must be protected to avoid alkylation thus, when working with this nucleobase, the sequence of protection followed by alkylation is mandatory (Scheme 19A). The main protecting group used is p-methoxybenzyl (Pmb). This group was thought to be only compatible with a Boc-protected backbone because its high acidic stability. For that reason, SU monomers compatible with Fmoc chemistry were not available for a long time. It was not until 2017 that Sugiyama et al.84 introduced the 2-methyl-4-methoxybenzyl as protecting group. The additional methyl group at position 2 of the phenyl ring conferred higher acidic lability and enabled the removal under the cleavage conditions used at the end of the Fmoc strategy. Interestingly, the ring electron enrichment due to the methyl substituent provoked the competition of O-alkylation with the desired at position N1. This secondary reaction was not observed when SU was protected with Pmb. In a more recently study, Hudson et al.146 examined the need for SU protection. This study explored the use of protecting groups for 2-thiouracil such as trimethoxybenzyl, 2,4-dimethoxybenzyl, and 2-methoxybenzyl. Their synthesis and acidic lability were tested and compared with 2-methyl-4-methoxybenzyl, and 4-methoxybenzyl. The first observation was that the more electron-donating substituent on the ring, the more favored O alkylation vs. N1. Thus, the trimethoxybezyl group resulted not useful. For the other mentioned groups the amount of O alkylation was variable (Scheme 19B). However, the most unexpected finding was that acidolysis of 4-methoxybenzyl-protected thiouracil occurred rapidly under 95% TFA treatment, and by using 2% TFA, 1% TES in CHCl3 in 2 h the deprotection was almost complete. The conclusion of this observation was that the Pmb group would be removed at the same time that Boc, thus the S-protection should be not necessary for the chain elongation. this was demonstrated by synthesizing one PNA oligomer introducing the SU monomer unprotected.
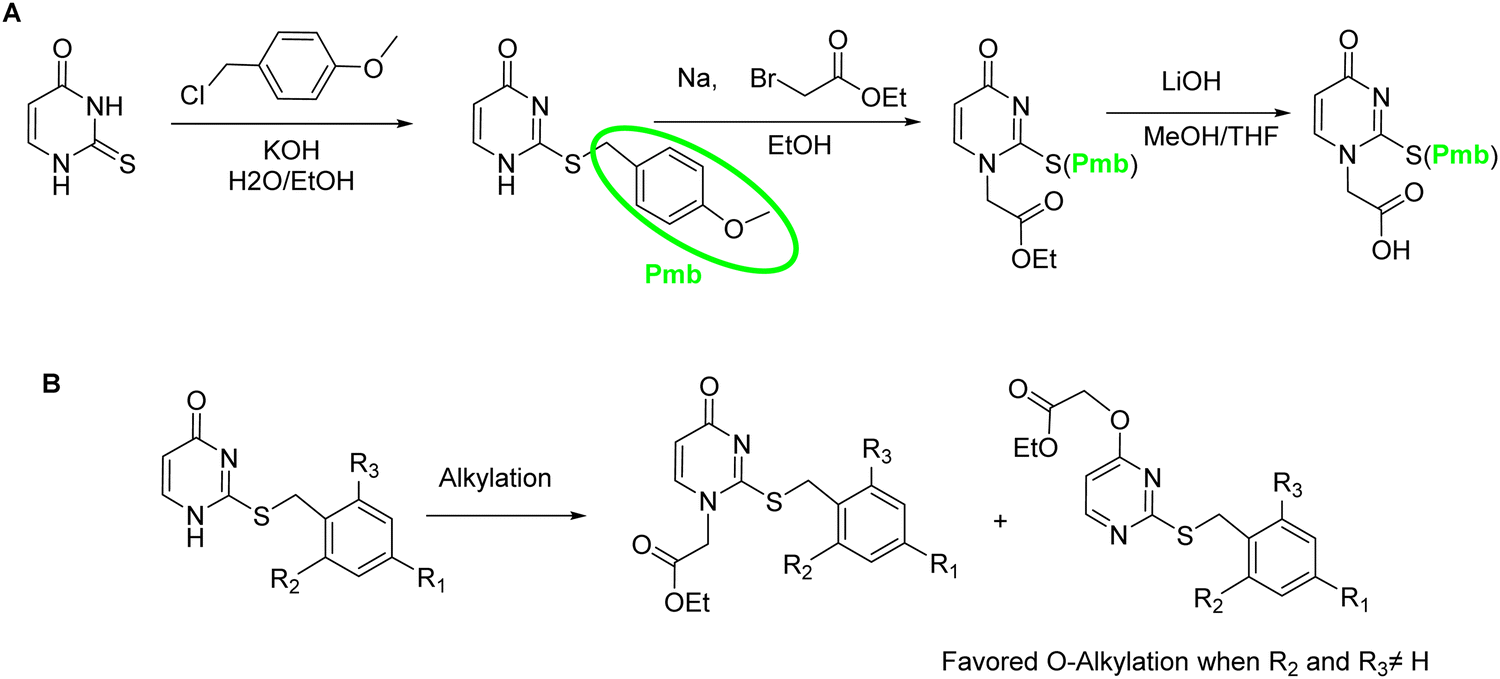 | ||
| Scheme 19 (A) Synthesis of s-protected SUaa. (B) O-Alkylation of protected SU. | ||
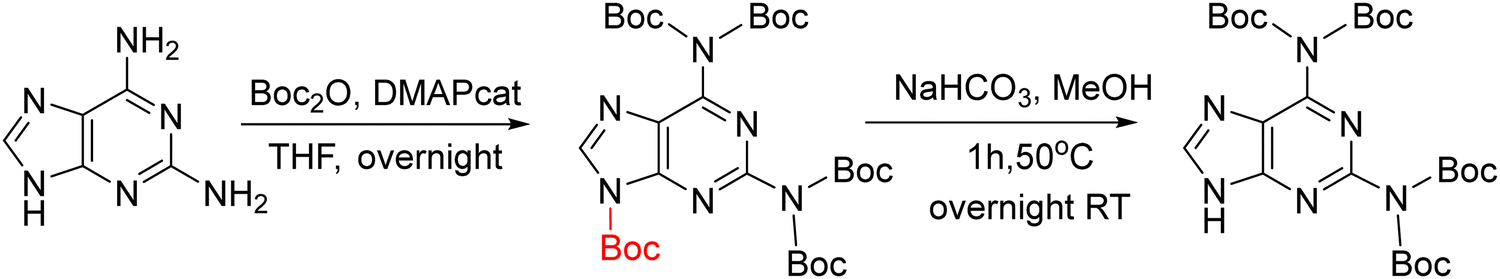
Initially, for the synthesis of the Daa the preferred reaction sequence was alkylation followed by protection as in the case of Aaa (Scheme 20).144,145 However, it is remarkable that only the amino group at position 6 was protected because the lower reactivity of the amino group at position 2. Thus, when the D-PNA monomer is used for PNAs synthesis, once the first unit is incorporated into the sequence, the capping step should be omitted. This protocol was the only available for long time, but in 2013, Ackermann et al.147 proposed a new synthetic protocol (Scheme 20B). The new synthetic scheme instead of using D as starting material, used 2A-6Cl as in the case of G The alkylation of this nucleobase was well known as described in the previous section, but the innovative step of this synthetic route was to perform first the condensation between the nucleobase and the protected AEG backbone and then conversion of Cl into NH2 followed by protection of it.
 | ||
| Scheme 20 (A) Synthesis of protected Daa. (B) Alternative route by condensing the nucleobase and the protected backbone before the protection. | ||
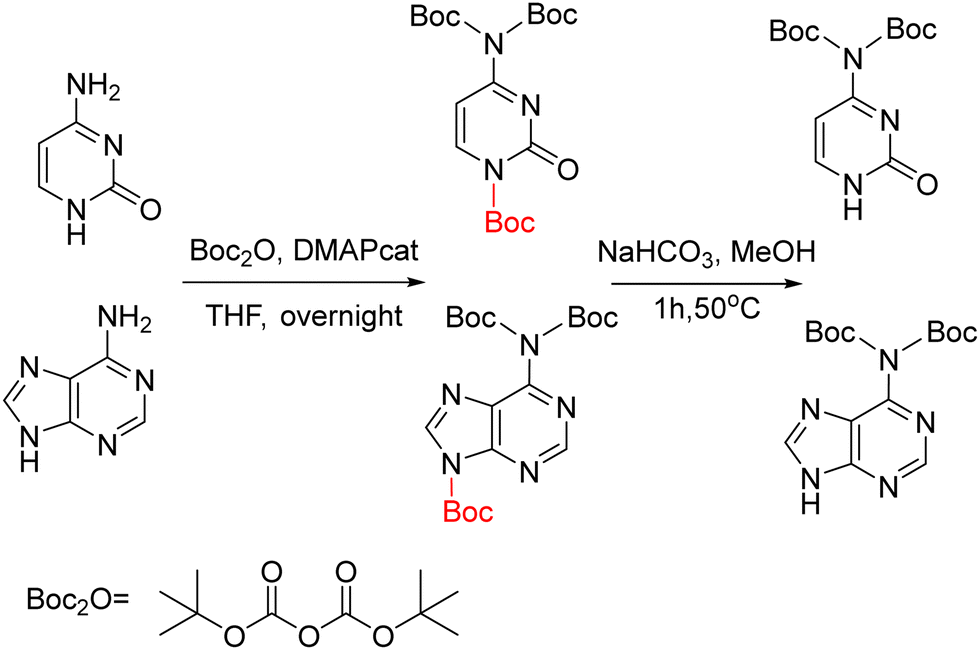
The D protection compatible with Fmoc strategy was developed similarly to protect adenine using bisBoc. Like in the case of adenine, in this strategy an extra Boc group is introduced in position 9 that is easily removed by a treatment with a NaHCO3 solution (Scheme 21).83,84,148 The alkylation had been described using the benzyl ester of bromoacetic acid followed by catalytic hydrogenation83 or ethyl ester of bromoacetic acid followed by NaOH hydrolysis.84,148
 | ||
| Scheme 21 The synthesis of bis-N-Boc protected D. | ||
4.3 Assembly of the two subunits onto the final monomer
Once the two subunits have been synthesized, they have to be linked through an amide bond. For this purpose, a great arsenal of coupling cocktails has been used and summarized in Table 5. The analysis of the data shows that the amide bond formation between the two moieties is not a particularly demanding reaction since the yield values are moderate to good regardless of whether mild derivatives such as N-hydroxysuccinimide (OSu) or pentafluorphenyl (Pfp) mediate the coupling, or stronger ones, such as derivatives of 1-hydroxy-7-azabenzotriazole (HOAt) and 3,4-dihydro-3-hydroxy-4-oxo-1,2,3-benzotriazine (DhbtOH), do. However, the protected nucleobase acetic acid derivative to be introduced seems to be the main factor affecting the yields, being in general higher in the case of pyrimidines than purines.| # | PG1 | PG2 | Coupling conditionsa | Yield | Ref. |
|---|---|---|---|---|---|
| a BOP = benzotriazol-1-yloxytris(dimethylamino)phosphonium hexafluorophosphate; DCC = N,N′-dicyclohexylcarbodiimide; DIC = N,N′-diisopropylcarbodiimide; EDC·HCl = 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide hydrochloride; HBTU = O-(benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate; HOAt = 1-hydroxy-7-azabenzotriazole; HOBt = 1-hydroxybenzotriazole; PyBroP = bromotri(pyrrolidino)phosphonium hexafluorophosphate; TOTU = O-[cyano(ethoxycarbonyl)methyleneamino]-N,N,N′,N′-tetramethyluronium tetrafluoroborate; DhbtOH = 3,4-dihydro-3-hydroxy-4-oxo-1,2,3-benzotriazine; IBC = isobutylchloroformate; Piv-Cl = pivaloyl chloride; NMM = N-methylmorpholine; NEM = N-ethylmorpholine; HOOBt = 3,4-dihydro3-hydroxy-4-oxo-l,2,3benzotriazin and NHS = N-hydroxysuccinimide. | |||||
| 1 | Boc | — | PfpOH/DCC (Taa) | Not reported | 16 |
| — | HBTU/DIEA (Taa) | 69–75% | 109 | ||
| — | EDC·HCl/DhbtOH (Taa) | High yield | 100 | ||
| Z | DhbtOH/DCC (Caa) | Not reported | 17 | ||
| DhbtOH/DCC (Aaa) and PyBroP(Gaa) | 99% for T and C, 63% for G, and 23% for A | 15 and 43 | |||
| Acyl | DhbtOH/DCC (Aaa, Caa) and PyBroP (Gaa) | 65–67% | 75 | ||
| 2 | Fmoc | Z | EDC·HCl (Taa, Caa) and BOP/HOBt (Aaa, Gaa) | 55–80% | 42 |
| Mmt | TOTU/DIEA | 52–78% | 67 | ||
| Acyl | HBTU/HOBt/DIEA | 40–85% | 76 | ||
| Boc | HOBt/DCC | 70–85% | 57 | ||
| BOP/HATU | Not reported | 82 | |||
| EDC·HCl/HOBt | 77% | 83 | |||
| bis-N-boc | DCC/NHS(Aaa), EDC·HCl (Caa) and EDC·HCl/HOBt (Gaa) | High yields | 59 | ||
| IBC/NMM (Aaa) and Piv-Cl/NMM (Caa, Gaa) | 85% | 60 | |||
| EDC·HCl | 70–75% | 58 | |||
| 3 | Mmt | — | PfpOH/TEA (Taa) | 70% | 149 |
| Acyl | DIC/HOOBt/NEM | >80% for T, A, C and 49% for G | 63 | ||
| 50% propylphosphonic anhydride in EtOAc/DIEA (Taa) or TEA (Caa) and TOTU/DIEA (Aaa, Gaa) | 64–91% | 25 | |||
| Boc | HATU/2,6-lutidine/DIEA | 85–95% | 62 | ||
| 4 | Azide | Bhoc | EDCl/4-DMAP (Taa), Piv-Cl/NMM (Aaa, Caa) and TOTU/DIEA (Gaa) | 95% for C and 76–80% for A, T, G | 66 |
| 5 | Bts | Bhoc | DCC/HOBt | 68–82% | 74 |
| 6 | Dde | Mmt | DCC/HOBT/NMM (Taa) and PyBroP/DIEA (Caa, Aaa, Gaa) | 75% for C, and < 50% for A, C, G | 68 |
| 50% propylphosphonic anhydride in DMF/NEM (Taa) and PyBroP/DIEA (Aaa, Caa, Gaa) | 77% for T and < 50% for A, C & G | 26 | |||
| 7 | NVOC | Acyl | 50% propylphosphonic anhydride in EtOAc/DIEA (Taa, Caa) and BOP/HOBT/DIEA (Aaa, Gaa) | 98% for T, 87% for C, A, and 81% for G | 78 |
| 8 | pNZ | Bis-N-boc | DIC/HOAt | 82% for T and 50–64% for G, A, C | 61 |
Upon completion of monomer assembly, the C-terminal protection of the backbone (PG3, Scheme 3) is removed (Table 6) and the monomer is then ready to be used for PNA SPS. The removal of PG3 should be compatible with the N-terminal and nucleobase protection. Thus, tBu esters failed to be used with Boc, thus making this protection scheme unsuitable for the purpose.58,150 Allyl is totally compatible with Fmoc/bis-N-Boc PNA monomers,59 but its use requires Pd(0) catalyst [Pd(Ph3P)4], whose is moisture sensitive, hence is more complex to handle in non-organic chemistry labs. In this context, simple methyl and ethyl esters are the most used as PG3. The removal of these esters in the presence of the Fmoc group is very often accompanied by the removal of Fmoc, which could be re-introduced in situ in the same reaction pot.58,67
| S. no. | C-Terminal esters on PNA monomer | Ester removal conditions | Yielda | Ref. | |
|---|---|---|---|---|---|
| a Quantitative = close to 100%, excellent = >90%, high = >80%, moderate = 60–70%, and low = 50%. | |||||
| 1 | Methyl ester | NaOH | THF/H2O | Excellent | 60 |
| MeOH | High | 100 | |||
| Dioxane/H2O | High | 63, 67 and 149 | |||
| LiOH | Dioxane/H2O | High | 62 | ||
| THF/H2O | Moderate | 15, 43 and 75 | |||
| 1 M KOH in H2O | Moderate | 75 | |||
| 1 M Cs2CO3 in MeOH/H2O | Low | 26 and 68 | |||
| 2 | Ethyl ester | LiOH | THF | High | 58 |
| THF/H2O | High | 17, 61 and 74 | |||
| NaOH/EtOH/H2O | Moderate | 15 | |||
| 3 | tBu ester | HCl | 1,4-Dioxane | Moderate | 42 |
| TFA | DCM/TES | High | 25 and 78 | ||
| DCM/1,3-dimethoxybenzene | Moderate | 76 | |||
| 4 | Benzyl ester | H2, Pd/C | Quantitative | 59, 82 and 83 | |
| NaOH/dioxane/H2O | Excellent | 66 | |||
| 5 | Cyanoethyl ester | 0.5 M DBU in acetonitrile | 64 | ||
| 6 | Allyl ester | Pd(pph3)4, NMM in DCM | High | 59 | |
4.4 Alternative methods of PNA monomer synthesis
As described in the previous sections, the chemistry associated with the preparation of PNA monomers and then the PNAs themselves is complex. Such complexity has fuelled research into alternative methods for the preparation of these intriguing compounds. Herein, the two most representative alternative methods are discussed. The first comprises N-alkylation of the nucleobase by the PG1–AEG–PG3 backbone which was derivatized with a chloroacetyl moiety at the secondary amine. The second one involves a Multi-Component Reaction (MCR) approach, where four components are used in ‘one pot’ to synthesize the PNA monomer.151 MCRs are considered to be green reactions due to the excellent atom economy associated with them.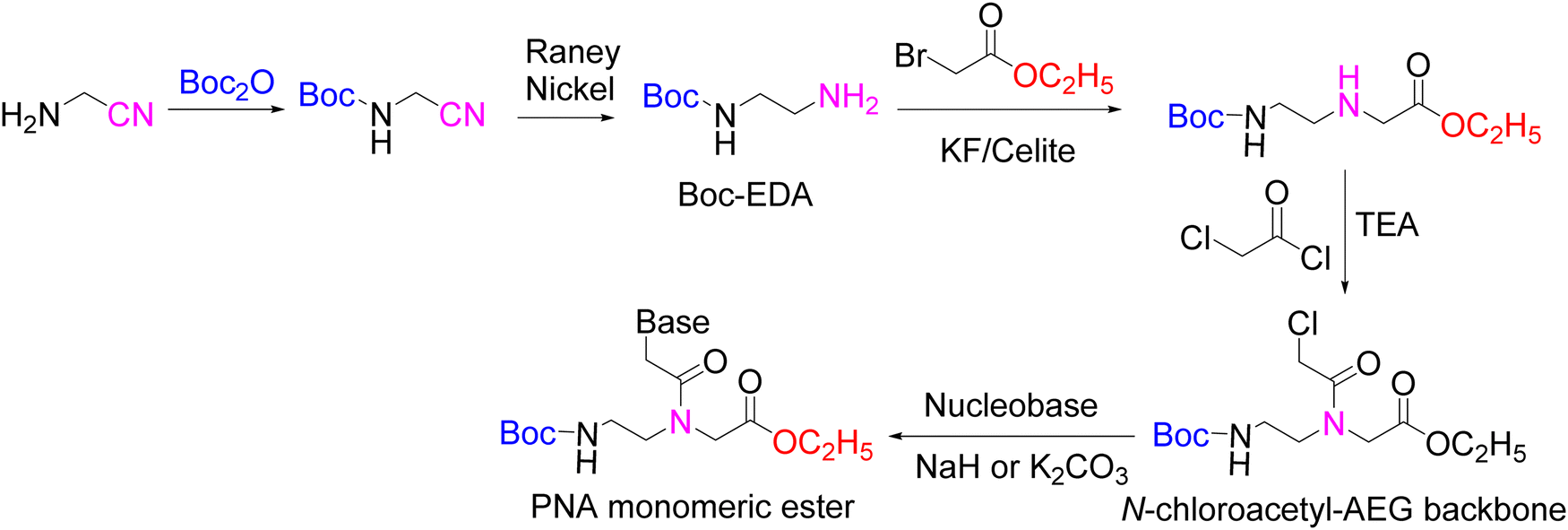 | ||
| Scheme 22 Synthesis of PNA monomeric through N-chloroacetyl-AEG backbone. | ||
Once the backbone is ready, direct N-alkylation is performed with three unprotected nucleobases, T, C, and A, in the presence of NaH/DMF, giving moderate yields. However, 2A-6Cl, used as G precursor, was alkylated in the presence of K2CO3 in DMF with 95% yield (Scheme 22).152 The ester hydrolysis were carried with NaOH, giving variable results, the worse being for G, where, in addition to the hydrolysis, the conversion of the Cl to O derivative occurred. Years later, a similar strategy was also described for T, using K2CO3 in DMF and giving an excellent yield.153 In this second example, Boc-ethylenediamine was used as the starting product.
These monomers with unprotected bases cannot be used for the synthesis of PNA, and although the authors claim that this strategy is also compatible for the preparation of the monomers with protected bases,152 there is no report in the literature showing the applicability of this method.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) R2H) as the oxo component. As formaldehyde is not very friendly for use in the Ugi-4C,157 most of these bis-amide monomers contain chiral centres. Of all the work described in the literature, Xu et al.131 has been the only group to describe a dimer.131
R2H) as the oxo component. As formaldehyde is not very friendly for use in the Ugi-4C,157 most of these bis-amide monomers contain chiral centres. Of all the work described in the literature, Xu et al.131 has been the only group to describe a dimer.131
5. Conclusions and perspectives
PNAs have gained special interest over the last three decades as nucleic acid analogues. While their synthesis resembles the solid-phase synthesis strategies used to prepare peptides, the process is rather more tedious and greatly depends on the choice of a suitable orthogonal/compatible permanent/temporary pair of protecting groups for the nucleobase and backbone, respectively. The suitability of different protecting groups on PNA not only makes the synthesis of this compound convenient but also could have an important impact into the development of PNA–peptide conjugates and PNA–DNA chimeras in SPS. The distinct strategies used to synthesize PNA have advantages and disadvantages. Thus, for example the use of Boc for the N-terminal AEG backbone is subjected to less side-reactions than when Fmoc is used. On the other hand, the use of Boc could require protecting groups for the nucleobases more difficult to be removed. Therefore, there is still considerable scope to explore other closely related options, as shown in Table 2. Regarding solid supports, the conventional polystyrene is the most used, although the use of PEG-based resins such as ChemMatrix and Tenta-Gel use to circumvent some of the synthetic problems associated with the interchain aggregation.Although this review focuses only on the chemistry of the original PNA backbone (AEG backbone) and its monomer preparation, it also helps to understand ongoing research into the chemical modifications of the AEG backbone and nucleobases themselves to improve the properties of the monomers during synthesis. Since PNA is similar to DNA from a structural perspective, modified PNA is likely to drive further research into its capacity as a potential drug candidate in the near future.
Author contributions
All authors have contributed to the preparation of the manuscript and agreed with the final version.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work in the laboratory of the authors is being partially funded by the National Research Foundation of South Africa.Notes and references
- O. Al Musaimi, D. Al Shaer, F. Albericio and B. G. de la Torre, Pharmaceuticals, 2021, 14, 145 CrossRef PubMed.
- H. Adachi, M. Hengesbach, Y. T. Yu and P. Morais, Biomedicines, 2021, 9, 550 CrossRef CAS PubMed.
- F. Eckstein, Antisense Nucleic Acid Drug Dev., 2000, 10, 117–121 CrossRef CAS PubMed.
- F. Eckstein, Nucleic Acid Ther., 2014, 24, 374–387 CrossRef CAS PubMed.
- M. Faria, D. G. Spiller, C. Dubertret, J. S. Nelson, M. R. H. White, D. Scherman, C. Hélène and C. Giovannangeli, Nat. Biotechnol., 2001, 19, 40–44 CrossRef CAS PubMed.
- S. Gryaznov and J.-K. Chen, J. Am. Chem. Soc., 1994, 116, 3143–3144 CrossRef CAS.
- P. S. Miller, J. Yano, E. Yano, C. Carroll, K. Jayaraman and P. O. P. Ts'o, Biochemistry, 1979, 18, 5134–5143 CrossRef CAS PubMed.
- C. Dolain, A. Patwa, G. Godeau and P. Barthélémy, Appl. Sci., 2012, 2, 245–259 CrossRef CAS.
- P. E. Nielsen, M. Egholm, R. H. Berg and O. Buchardt, Science, 1991, 254, 1497–1500 CrossRef CAS PubMed.
- S. Shakeel, S. Karim and A. Ali, J. Chem. Technol. Biotechnol., 2006, 81, 892–899 CrossRef CAS.
- P. E. Nielsen, Origins Life Evol. Biosphere, 1993, 23, 323–327 CrossRef CAS PubMed.
- K. E. Nelson, M. Levy and S. L. Miller, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 3868–3871 CrossRef CAS PubMed.
- S. A. Banack, J. S. Metcalf, L. Jiang, D. Craighead, L. L. Ilag and P. A. Cox, PloS One, 2012, 7, e49043 CrossRef CAS PubMed.
- M. Eriksson and P. E. Nielsen, Q. Rev. Biophys., 1996, 29, 369–394 CrossRef CAS PubMed.
- M. Egholm, C. Behrens, L. Christensen, R. H. Berg, P. E. Nielsen and O. Buchardt, Chem. Commun., 1993, 800–801 RSC.
- M. Egholm, O. Buchardt, P. E. Nielsen and R. H. Berg, J. Am. Chem. Soc., 1992, 114, 1895–1897 CrossRef CAS.
- M. Egholm, P. E. Nielsen, O. Buchardt and R. H. Berg, J. Am. Chem. Soc., 1992, 114, 9677–9678 CrossRef CAS.
- J. D. R. Perera, K. E. W. Carufe and P. M. Glazer, Biopolymers, 2021, 112, e23460 CrossRef CAS PubMed.
- A. B. Armitage, Drug Discovery Today, 2003, 8, 222–228 CrossRef PubMed.
- M. Robaczewska, R. Narayan, B. Seigneres, O. Schorr, A. Thermet, A. J. Podhajska, C. Trepo, F. Zoulim, P. E. Nielsen and L. Cova, J. Hepatol., 2005, 42, 180–187 CrossRef CAS PubMed.
- P. E. Nielsen, Perspect. Drug Discov. Des., 1996, 4, 76–84 CrossRef CAS.
- S. Montazersaheb, M. S. Hejazi and H. Nozad Charoudeh, Adv. Pharm. Bull., 2018, 8, 551–563 CrossRef CAS PubMed.
- Q. Lai, W. Chen, Y. Zhang and Z. Liu, Analyst, 2021, 146, 5822–5835 RSC.
- R. Patel, S. Sarma, A. Shukla, P. Parmar, D. Goswami and M. Saraf, Mol. Biol. Rep., 2020, 47, 8113–8131 CrossRef CAS PubMed.
- G. Breipohl, D. W. Will, A. Peyman and E. Uhlmann, Tetrahedron, 1997, 53, 14671–14686 CrossRef.
- L. Bialy, J. J. Díaz-Mochón, E. Specker, L. Keinicke and M. Bradley, Tetrahedron, 2005, 61, 8295–8305 CrossRef CAS.
- A. Porcheddu and G. Giacomelli, Curr. Med. Chem., 2005, 12, 2561–2599 CrossRef CAS PubMed.
- A. Das and B. Pradhan, Chem. Biol. Drug Des., 2021, 97, 865–892 CrossRef CAS PubMed.
- C. S. Swenson and J. M. Heemstra, Chem. Commun., 2020, 56, 1926–1935 RSC.
- C. S. Swenson, A. Velusamy, H. S. Argueta-Gonzalez and J. M. Heemstra, J. Am. Chem. Soc., 2019, 141, 19038–19047 CrossRef CAS PubMed.
- J. Saarbach, P. M. Sabale and N. Winssinger, Curr. Opin. Chem. Biol., 2019, 52, 112–124 CrossRef CAS PubMed.
- A. Dragulescu-Andrasi, S. Rapireddy, B. M. Frezza, C. Gayathri, R. R. Gil and D. H. Ly, J. Am. Chem. Soc., 2006, 128, 10258–10267 CrossRef CAS PubMed.
- A. Periyalagan, Y.-T. Kim and I. S. Hong, Bull. Korean Chem. Soc., 2021, 42, 1304–1309 CrossRef CAS.
- In Peptide Nucleic Acids: Methods and Protocols, ed. P. E. Nielsen, Springer New York, Totowa, NJ, 2002 Search PubMed.
- In Peptide Nucleic Acids: Methods and Protocols, ed. P. E. Nielsen and D. H. AppellaHumana Press, Totowa, NJ, 2014 Search PubMed.
- In Peptide Nucleic Acids: Methods and Protocols, ed. P. E. Nielsen, Springer US, New York, NY, 2020 Search PubMed.
- In Peptide Nucleic Acids: Protocols and Applications, ed. P. E. Nielsen, Horizon Bioscience, 2004 Search PubMed.
- B. D. Gildea, S. Casey, J. MacNeill, H. Perry-O'Keefe, D. Sørensen and J. M. Coull, Tetrahedron Lett., 1998, 39, 7255–7258 CrossRef CAS.
- E. Uhlmann, A. Peyman, G. Breipohl and D. W. Will, Angew. Chem., Int. Ed., 1998, 37, 2796–2823 CrossRef CAS PubMed.
- J. M. Coull, M. Egholm, R. P. Hodge, M. Ismail and S. B. Rajur, US Pat., 6172226, 2000 Search PubMed.
- A. Y. Shaikh, F. Björkling, P. E. Nielsen and H. Franzyk, Eur. J. Org. Chem., 2021, 2792–2801 CrossRef CAS.
- S. A. Thomson, J. A. Josey, R. Cadilla, M. D. Gaul, C. F. Hassman, M. J. Luzzio, A. J. Pipe, K. L. Reed, D. J. Ricca, R. W. Wiethe and S. A. Noble, Tetrahedron, 1995, 51, 6179–6194 CrossRef CAS.
- K. L. Dueholm, M. Egholm, C. Behrens, L. Christensen, H. F. Hansen, T. Vulpius, K. H. Petersen, R. H. Berg, P. E. Nielsen and O. Buchardt, J. Org. Chem., 1994, 59, 5767–5773 CrossRef CAS.
- J. Tailhades, H. Takizawa, M. J. Gait, D. A. Wellings, J. D. Wade, Y. Aoki and F. Shabanpoor, Front. Chem., 2017, 5, 5–81 Search PubMed.
- R. Pipkorn, M. Wiessler, W. Waldeck, U. Hennrich, K. Nokihara, M. Beining and K. Braun, Int. J. Med. Sci., 2012, 9, 1–10 CrossRef CAS PubMed.
- https://www.sigmaaldrich.com/ES/es/technical-documents/technical-article/chemistry-and-synthesis/peptide-synthesis/fmoc-cleavage-deprotection#tfa .
- P. J. Finn, N. J. Gibson, R. Fallon, A. Hamilton and T. Brown, Nucleic Acids Res., 1996, 24, 3357–3363 CrossRef CAS PubMed.
- D. A. Stetsenko, E. N. Lubyako, V. K. Potapov, T. L. Azliikina and E. D. Sverdlov, Tetrahedron Lett., 1996, 37, 3571–3574 CrossRef CAS.
- M. Inagaki, R. Uematsu, T. Mizutani, D. Unabara, Y. Araki, S. Sakamoto, H. Kashida, M. Nishijima, H. Asanuma, Y. Inoue and T. Wada, Chem. Lett., 2019, 48, 341–344 CrossRef CAS.
- G. Barany and F. Albericio, J. Am. Chem. Soc., 1985, 107, 4936–4942 CrossRef CAS.
- F. Albericio, Biopolymers, 2000, 55, 123–139 CrossRef CAS PubMed.
- D. Ben-Ishai and A. Berger, J. Org. Chem., 1952, 17, 1564–1570 CrossRef CAS.
- A. M. Felix, J. Org. Chem., 1974, 39, 1427–1429 CrossRef CAS.
- A. R. Mitchell and R. B. Merrifield, J. Org. Chem., 1976, 41, 2015–2019 CrossRef CAS.
- Y. Kiso, K. Ukawa and T. Akita, Chem. Commun., 1980, 101–102 RSC.
- A. Isidro-Llobet, M. A. Lvarez and F. Albericio, Chem. Rev., 2009, 109, 2455–2504 CrossRef CAS PubMed.
- O. Seitz and O. Köhler, Chem. – Eur. J., 2001, 7, 3911–3925 CrossRef CAS.
- E. C. Browne, S. J. Langford and B. M. Abbott, Aust. J. Chem., 2012, 65, 539–544 CrossRef CAS.
- F. Wojciechowski and R. H. E. Hudson, J. Org. Chem., 2008, 73, 3807–3816 CrossRef CAS PubMed.
- A. Porcheddu, G. Giacomelli, I. Piredda, M. Carta and G. Nieddu, Eur. J. Org. Chem., 2008, 5786–5797 CrossRef CAS.
- Y.-C. Huang, C. Cao, X.-L. Tan, X. Li and L. Liu, Org. Chem. Front., 2014, 1, 1050–1054 RSC.
- D. Chouikhi, M. Ciobanu, C. Zambaldo, V. Duplan, S. Barluenga and N. Winssinger, Eur. J. Chem., 2012, 18, 12698–12704 CrossRef CAS PubMed.
- D. W. Will, G. Breipohl, D. Langner, J. Knolle and E. Uhlmann, Tetrahedron, 1995, 51, 12069–12082 CrossRef CAS.
- E. Ferrer, M. Eisenhut and R. Eritja, Lett. Pept. Sci., 1999, 6, 209–219 CAS.
- R. D. Viirre and R. H. E. Hudson, Org. Lett., 2001, 3, 3931–3934 CrossRef CAS PubMed.
- F. Debaene and N. Winssinger, Org. Lett., 2003, 5, 4445–4447 CrossRef CAS PubMed.
- G. Breilpohl, J. Knolle, D. Langner, G. O'Malley and E. Uhlmann, Bioorg. Med. Chem. Lett., 1996, 6, 665–670 CrossRef.
- J. J. Díaz-Mochón, L. Bialy and M. Bradley, Org. Lett., 2004, 6, 1127–1129 CrossRef PubMed.
- J. J. Díaz-Mochón, L. Bialy, J. Watson, R. M. Sánchez-Martín and M. Bradley, Chem. Commun., 2005, 3316–3318 RSC.
- E. Vedejs, S. Lin, A. Klapars and J. Wang, J. Am. Chem. Soc., 1996, 118, 9796–9797 CrossRef CAS.
- E. Vedejs, J. Wang, S. Lin and A. Klapars, United States Patent, US Pat., 5900427, 1990 Search PubMed.
- E. Vedejs and C. Kongkittingam, J. Org. Chem., 2000, 65, 2309–2318 CrossRef CAS PubMed.
- E. Marsault, H. R. Hoveyda, M. L. Peterson, C. Saint-Louis, A. Landry, M. Vézina, L. Ouellet, Z. Wang, M. Ramaseshan, S. Beaubien, K. Benakli, S. Beauchemin, R. Déziel, T. Peeters and G. L. Fraser, J. Med. Chem., 2006, 49, 7190–7197 CrossRef CAS PubMed.
- H. Lee, J. H. Jeon, J. C. Lim, H. Choi, Y. Yoon and S. K. Kim, Org. Lett., 2007, 9, 3291–3293 CrossRef CAS PubMed.
- T. Kofoed, H. F. Hansen, H. Ørum and T. Koch, J. Pept. Sci., 2001, 7, 402–412 CrossRef CAS PubMed.
- Z. Timár, L. Kovács, G. Kovács and Z. Schmél, J. Chem. Soc., Perkin Trans., 2000, 19–26 RSC.
- M. Planas, E. Bardaji, K. J. Jensen and G. Barany, J. Org. Chem., 1999, 64, 7281–7289 CrossRef CAS.
- Z. C. Liu, D. S. Shin, K. T. Lee, B. H. Jun, Y. K. Kim and Y. S. Lee, Tetrahedron, 2005, 61, 7967–7973 CrossRef CAS.
- K. R. Bhushan, Synlett, 2006, 2130–2132 CrossRef CAS.
- S. Pothukanuri, Z. Pianowski and N. Winssinger, Eur. J. Org. Chem., 2008, 3141–3148 CrossRef CAS.
- F. Altenbrunn and O. Seitz, Org. Biomol. Chem., 2008, 6, 2493–2498 RSC.
- T. Sugiyama, A. Kittaka, Y. Takemoto, H. Takayama and R. Kuroda, Nucleic Acids Symp. Ser., 2002, 2, 145–146 CrossRef CAS PubMed.
- A. Amant and R. Hudson, Org. Biomol. Chem., 2012, 10, 876–881 RSC.
- T. Sugiyama, G. Hasegawa, C. Niikura, K. Kuwata, Y. Imamura, Y. Demizu, M. Kurihara and A. Kittaka, Bioorg. Med. Chem. Lett., 2017, 27, 3337–3341 CrossRef CAS PubMed.
- Z.-C. Liu, D.-S. Shin, M. Shokouhimehr, K.-N. Lee, B.-W. Yoo, Y.-K. Kim and Y.-S. Lee, Biosens. Bioelectron., 2007, 22, 2891–2897 CrossRef CAS PubMed.
- O. Seitz, Tetrahedron Lett., 1999, 40, 4161–4164 CrossRef CAS.
- L. S. Richter and R. N. Zuckermann, Bioorg. Med. Chem. Lett., 1995, 5, 1159–1162 CrossRef CAS.
- O. Seitz and H. Kunz, J. Org. Chem., 1997, 62, 813–826 CrossRef CAS.
- G. Upert, M. Mehiri, M.-L. Goddard, A. Di Giorgio, R. Benhida, R. Condom and N. Patino, Tetrahedron Lett., 2005, 46, 4081–4085 CrossRef CAS.
- R. H. Hudson and R. D. Viirre, Nucleos. Nucleot. Nucl., 2003, 22, 1017–1022 CrossRef CAS PubMed.
- C. Di Giorgio, S. Pairot, C. Schwergold, N. Patino, R. Condom, A. Farese-Di Giorgio and R. Guedj, Tetrahedron, 1999, 55, 1937–1958 CrossRef CAS.
- J. C. Verheijen, G. M. Grotenbreg, L. Hart de Ruyter, P. A. M. van der Klein, G. A. van der Marel and J. H. van Boom, Tetrahedron Lett., 2000, 41, 3991–3995 CrossRef CAS.
- G. Depecker, C. Schwergold, C. Di Giorgio, N. Patino and R. Condom, Tetrahedron Lett., 2001, 42, 8303–8306 CrossRef CAS.
- C. Schwergold, G. Depecker, C. D. Giorgio, N. Patino, F. Jossinet, B. Ehresmann, R. Terreux, D. Cabrol-Bass and R. Condom, Tetrahedron, 2002, 58, 5675–5687 CrossRef CAS.
- S. Caldarelli, G. Depecker, N. Patino, A. Di Giorgio, T. Barouillet, A. Doglio and R. Condom, Bioorg. Med. Chem. Lett., 2004, 14, 4435–4438 CrossRef CAS PubMed.
- G. Depecker, N. Patino, C. Di Giorgio, R. Terreux, D. Cabrol-Bass, C. Bailly, A.-M. Aubertin and R. Condom, Org. Biomol. Chem., 2004, 2, 74–79 RSC.
- A. Periyalagan and I. S. Hong, Bull. Korean Chem. Soc., 2022, 43, 714–723 CrossRef CAS.
- K. Ogami, Y. Okada and K. Chiba, Chem. Lett., 2018, 47, 138–140 CrossRef CAS.
- G. Tana, S. Kitada, S. Fujita, Y. Okada, S. Kim and K. Chiba, Chem. Commun., 2010, 46, 8219–8221 RSC.
- B. Falkiewicz, A. Kozyra, A. S. Kolodziejczyk, B. Liberek and K. Wisniewski, Nucleic Acids Symp. Ser., 1999, 42, 9–10 CrossRef CAS PubMed.
- R. D. Viirre and R. H. Hudson, J. Org. Chem., 2003, 68, 1630–1632 CrossRef CAS PubMed.
- A. P. Krapcho and C. S. Kuell, Synth. Commun., 1990, 20, 2559–2564 CrossRef CAS.
- J. L. Romera, J. M. Cid and A. A. Trabanco, Tetrahedron Lett., 2004, 45, 8797–8800 CrossRef CAS.
- A. Mastitski, A. Abramov, A. Kruve and J. Järv, Proc. Estonian Acad. Sci., 2017, 66, 10 CrossRef.
- A. Darehkordi and E. Kazemi, Mol. Diver., 2020, 24, 131–139 CrossRef CAS PubMed.
- E. P. Heimer, H. E. Gallo-Torres, A. M. Felix, M. Ahmad, T. J. LamBros, F. Scheidl and J. Meienhofer, Int. J. Pept. Prot. Res, 1984, 23, 203–211 CrossRef CAS PubMed.
- T. A. Feagin, N. I. Shah and J. M. Heemstra, J. Nucleic Acids, 2012, 2012, 354549 Search PubMed.
- A. Y. Shaikh, F. Bjorkling, P. E. Nielsen and H. Franzyk, Eur. J. Org. Chem., 2021, 2792–2801 CrossRef CAS.
- S. R. Malamgari, P. Manikandan, P. Ramani and V. R. Katta, ChemistrySelect, 2018, 3, 3948–3951 CrossRef CAS.
- B. Falkiewicz, L. Lantkiewicz, B. Liberek and K. Wisniewski, Peptides 1996. Proceedings of the 24th Eur. Peptide Symp., 1996.
- K. Wiśniewski, A. S. Kołdziejczyk and B. Falkiewicz, J. Pept. Sci., 1998, 4, 1–14 CrossRef.
- B. Falkiewicz, A. S. Koøodziejczyk, B. Liberek and K. WisÂniewski, Tetrahedron, 2001, 57, 7909–7917 CrossRef CAS.
- S. Emerson William, Org. React., 2011, 61, 174–255 CrossRef.
- A. J. Mancuso, S.-L. Huang and D. Swern, J. Org. Chem., 1978, 43, 2480–2482 CrossRef CAS.
- K. Omura and D. Swern, Tetrahedron, 1978, 34, 1651–1660 CrossRef CAS.
- A. J. Mancuso, D. S. Brownfain and D. Swern, J. Org. Chem., 1979, 44, 4148–4150 CrossRef CAS.
- A. J. Mancuso and D. Swern, Synthesis, 1981, 165–185 CrossRef CAS.
- L. De Luca, G. Giacomelli and A. Porcheddu, J. Org. Chem., 2001, 66, 7907–7909 CrossRef CAS PubMed.
- N. Bischofberger, H. Waldmann, T. Saito, E. S. Simon, W. Lees, M. D. Bednarski and G. M. Whitesides, J. Org. Chem., 1988, 53, 3457–3465 CrossRef CAS.
- H. J. Tobias and P. J. Ziemann, J. Phys. Chem. A, 2001, 105, 6129–6135 CrossRef CAS.
- E. J. Corey and J. W. Suggs, Tetrahedron Lett., 1975, 16, 2647–2650 CrossRef.
- K. L. Dueholm, M. Egholm and O. Buchardt, Org. Prep. Proced. Int., 1993, 25, 457–461 CrossRef CAS.
- K. E. Rittle, C. F. Homnick, G. S. Ponticello and B. E. Evans, J. Org. Chem., 1982, 47, 3016–3018 CrossRef CAS.
- J.-A. Fehrentz, A. Heitz, C. Bertrand, C. Cazaubon and D. Nisato, FEBS Lett., 1984, 167, 273–276 CrossRef CAS PubMed.
- B. Falkiewicz, W. Wiśniowski, A. S. Kołodziejczyk and K. Wiśniewski, Nucleos. Nucleot. Nucl., 2001, 20, 1393–1397 CrossRef CAS PubMed.
- S. Volpi, A. Rozzi, N. Rivi, M. Neri, W. Knoll and R. Corradini, Org. Lett., 2021, 23, 902–907 CrossRef CAS PubMed.
- C. Wang, A. Pettman, J. Bacsa and J. Xiao, Angew. Chem., Int. Ed., 2010, 49, 7548–7552 CrossRef CAS PubMed.
- J. Lohse, O. Dahl and P. E. Nielsen, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 11804–11808 CrossRef CAS PubMed.
- F. Wojciechowski and H. R. E. Hudson, Curr. Top. Med. Chem., 2007, 7, 667–679 CrossRef CAS PubMed.
- M. Chowdhury and R. H. E. Hudson, Chem. Rec., 2023, 23, e202200218 CrossRef CAS PubMed.
- P. Xu, T. Zhang, W. Wang, X. Zou, X. Zhang and Y. Fu, Synthesis, 2003, 1171–1176 CrossRef CAS.
- A. S. Jones, P. Lewis and S. F. Withers, Tetrahedron, 1973, 29, 2293–2296 CrossRef CAS.
- L. Kosynkina, W. Wang and T. C. Liang, Tetrahedron Lett., 1994, 35, 5173–5176 CrossRef CAS.
- Y. Hayakawa, M. Hirose and R. Noyori, J. Org. Chem., 1993, 58, 5551–5555 CrossRef CAS.
- L. Christensen, H. F. Hansen, T. Koch and P. E. Nielsen, Nucleic Acids Res., 1998, 26, 2735–2739 CrossRef CAS PubMed.
- A. Farèse, N. Patino, R. Condom, S. Dalleu and R. Guedj, Tetrahedron Lett., 1996, 37, 1413–1416 CrossRef.
- S. Dey and P. Garner, J. Org. Chem., 2000, 65, 7697–7699 CrossRef CAS PubMed.
- M. Paradís-Bas, J. Tulla-Puche and F. Albericio, Chem. Soc. Rev., 2016, 45, 631–654 RSC.
- D. Datta, Nucleos. Nucleot. Nucl., 2020, 39, 530–541 CrossRef CAS PubMed.
- R. Zou and M. J. Robins, Can. J. Chem., 1987, 65, 1436–1437 CrossRef CAS.
- I. V. Kutyavin, R. L. Rhinehart, E. A. Lukhtanov, V. V. Gorn, R. B. Meyer and H. B. Gamper, Biochem., 1996, 35, 11170–11176 CrossRef CAS PubMed.
- V. V. Demidov, E. Protozanova, K. I. Izvolsky, C. Price, P. E. Nielsen and M. D. Frank-Kamenetskii, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5953–5958 CrossRef CAS PubMed.
- N. Shigi, Y. Mizuno, H. Kunifuda, K. Matsumura and M. Komiyama, Bull. Chem. Soc. Jpn., 2018, 92, 330–335 CrossRef.
- G. Haaima, H. F. Hansen, L. Christensen, O. Dahl and P. E. Nielsen, Nucleic Acids Res., 1997, 25, 4639–4643 CrossRef CAS PubMed.
- M. Komiyama, Y. Aiba, T. Ishizuka and J. Sumaoka, Nat. Prot., 2008, 3, 646–654 CrossRef CAS PubMed.
- R. H. E. Hudson, A. Heidari, T. Martin-Chan, G. Park and J. A. Wisner, J. Org. Chem., 2019, 84, 13252–13261 CrossRef CAS PubMed.
- D. Ackermann and M. Famulok, Nucleic Acids Res., 2013, 41, 4729–4739 CrossRef CAS PubMed.
- Y. Kamiya, F. Sato, K. Murayama, A. Kodama, S. Uchiyama and H. Asanuma, Chem. – Asian J., 2020, 15, 1266–1271 CrossRef CAS PubMed.
- D. A. Stetsenko, E. N. Lubyako, V. K. Potapov, T. L. Azliikina and E. D. Sverdlov, Tetrahedron Lett., 1996, 37, 3571–3574 CrossRef CAS.
- Y.-q Wu, D. C. Limburg, D. E. Wilkinson, M. J. Vaal and G. S. Hamilton, Tetrahedron Lett., 2000, 41, 2847–2849 CrossRef CAS.
- I. Ugi, J. Prakt. Chem., 1997, 339, 499–516 CrossRef CAS.
- P. C. Meltzer, A. Y. Liang and P. Matsudaira, J. Org. Chem., 1995, 60, 4305–4308 CrossRef CAS.
- A. Das and B. Pradhan, J. Chem. Sci., 2020, 132, 32 CrossRef CAS.
- W. Maison, I. Schlemminger, O. Westerhoff and J. Martens, Bioorg. Med. Chem., 2000, 8, 1343–1360 CrossRef CAS PubMed.
- A. Dömling, Nucleos. Nucleot. Nucl., 1998, 17, 1667–1670 CrossRef.
- A. Dömling, K.-Z. Chi and M. Barrère, Bioorg. Med. Chem. Lett., 1999, 9, 2871–2874 CrossRef PubMed.
- C. Liu, W. Huang, J. Zhang, Z. Rao, Y. Gu and F. Jérôme, Green Chem., 2021, 23, 1447–1465 RSC.
| This journal is © The Royal Society of Chemistry 2023 |