
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDihydroindenofluorenes as building units in organic semiconductors for organic electronics
Cyril
Poriel
 and
Joëlle
Rault-Berthelot
and
Joëlle
Rault-Berthelot
UMR CNRS 6226-Université Rennes 1-ISCR-Campus de Beaulieu, 35042 Rennes, France. E-mail: cyril.poriel@univ-rennes1.fr; joelle.rault-berthelot@univ-rennes1.fr
First published on 13th September 2023
Abstract
This review aims to discuss organic semiconductors constructed on dihydroindenofluorene positional isomers, which are key molecular scaffolds in organic electronics. Bridged oligophenylenes are key organic semiconductors that have allowed the development of organic electronic technologies. Dihydroindenofluorenes (DHIFs) belong to the family of bridged oligophenylenes constructed on a terphenyl backbone. They have proven to be very promising building blocks for the construction of highly efficient organic semiconductors for all OE devices, namely organic light emitting diodes (OLEDs), phosphorescent OLEDs, organic field-effect transistors (OFETs), solar cells, etc.
 Cyril Poriel | Cyril Poriel is a CNRS Research Director at “Institut des Sciences Chimiques de Rennes” – UMR 6226 (Rennes 1 University, France). His main research interest deals with the design of π-conjugated molecular architectures for organic electronics. He is particularly interested in the design of blue emitting fluorophores for organic light-emitting diodes (OLEDs), high triplet host materials for phosphorescent OLEDs, electron-deficient semiconductors for n-type organic field-effect transistors and, more recently, in nanoring chemistry. |
 Joëlle Rault-Berthelot | Joëlle Rault-Berthelot is a “CNRS Research Director” at “Institut des Sciences Chimiques de Rennes” – UMR 6226 (Rennes 1 University, France). She has more than 40 years of experience in electrochemistry of π-conjugated systems. She is recognized for her work on electrode surface modification: graphite intercalation compounds (1981–1985) and conducting polymers such as polyfluorene derivatives, polydibenzo-crown-ethers, polyphenylenes, polyarylporphyrines, and so on. She developed the electrochemical synthesis of the polymers and studied their main properties and applications (electrochromism, sensors, catalysis, etc. (1985–2007). Since 2005, she has been working on new materials for organic electronics: design, physico-chemical properties and uses as active layers in electronic devices. Among them: new three-dimensional molecules with ≪ 2π-1spiro ≫ or ≪ 3π-2spiro ≫ architectures in view of their use as active layers in fluorescent OLEDs, as a host for PhOLEDs, and new linear molecules with low LUMO levels and their use as the active layer in n-type OFETs. |
In the DHIF family, there are five positional isomers, with different phenyl linkages (para/meta/ortho) and different ring bridge arrangements (anti/syn) (Scheme 1).
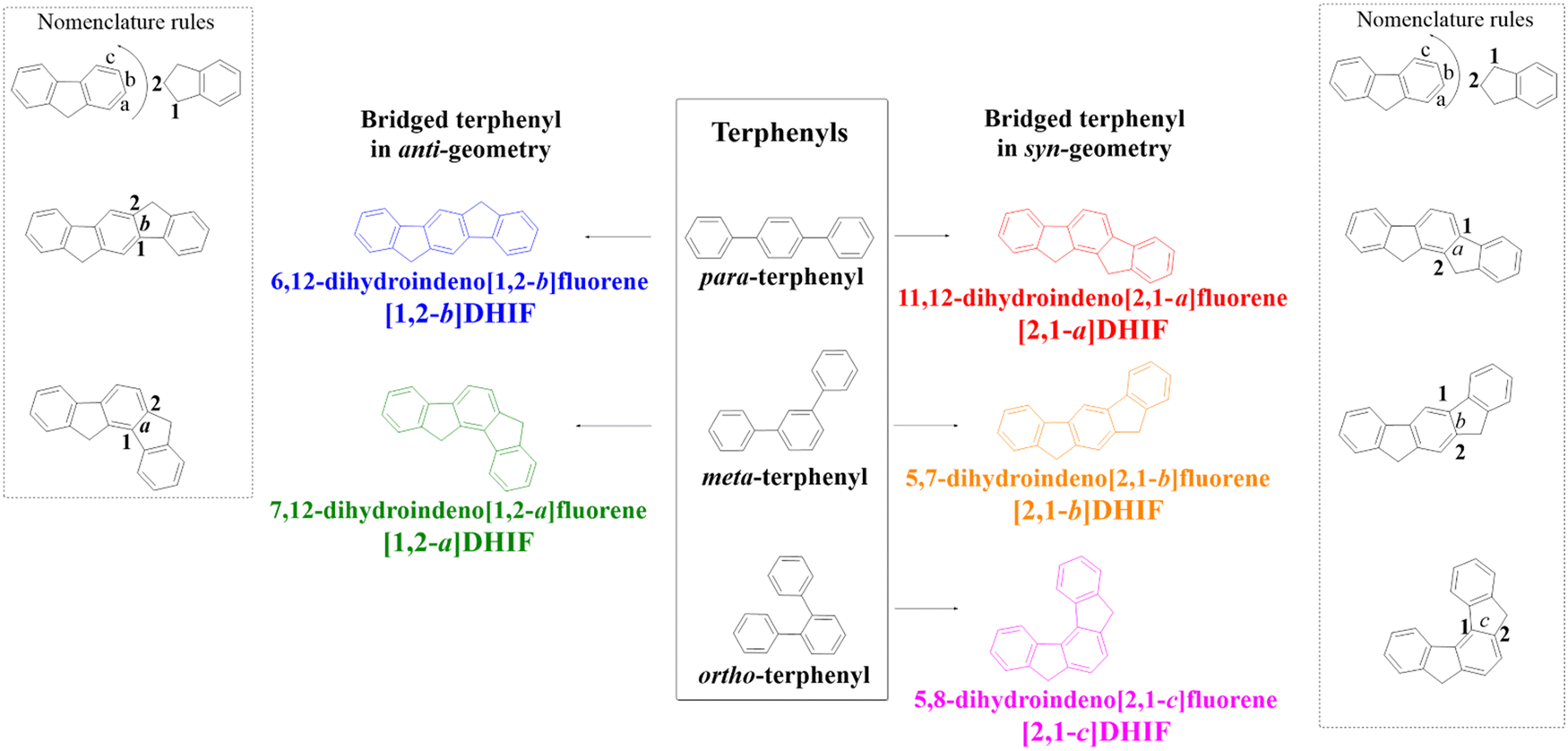 | ||
| Scheme 1 The five DHIFs and their corresponding terphenyl regioisomers. | ||
6,12-Dihydroindeno[1,2-b]fluorene ([1,2-b]-DHIF) is constructed on a para-terphenyl core and possesses anti geometry. This isomer displays many appealing properties such as a high quantum yield and a low oxidation potential and can be easily functionalized at the bridge and/or at the phenyl units to tune its properties. Thus, many functional groups have been attached to this scaffold in order to fit with the requirements of specific applications (OLED, OFET, solar cells…). These design tactics have led, in many cases, to highly efficient materials and high performance devices. This fragment has therefore been widely investigated in organic electronics.
A second isomer in the series, namely 11,12-dihydroindeno[2,1-a]fluorene ([2,1-a]DHIF) is also constructed on a bridged para terphenyl core possessing nevertheless a syn geometry. This specific geometry has been notably used to tune the electronic properties and this isomer has emerged as a promising scaffold to obtain a stable blue emission used to modulate the electronic characteristics and this regioisomer has become an interesting building unit to reach a stable emission from conformationally-controllable intramolecular excimers.
Modification of the phenyl linkages from a para linkage to a meta linkage gives the ‘meta isomers’, namely 7,12-dihydroindeno[1,2-a]fluorene ([1,2-a]DHIF) with an anti geometry and 5,7-dihydroindeno[2,1-b]fluorene ([2,1-b]DHIF) with a syn geometry. The last isomer in this family is 5,8-dihydroindeno[2,1-c]fluorene ([2,1-c]DHIF), which possesses a central ortho terphenyl backbone and a syn geometry. This isomer, due to its ortho linkage, offers a unique example which induces a particular helicoidal turn of the DHIF core. For these three isomers, the strong impact of both the linkage (meta and ortho) and the geometry (syn/anti) on the electronic properties has been particularly highlighted over the years. This review discusses how these fragments have been used in the construction of organic semiconductors for different OE devices. It will be notably shown how the positional isomerism is a powerful tool to tune the electronic properties, which in turn also has huge consequences on the device performance.
1. Introduction
Bridged oligophenylenes are a key class of organic semiconductors in organic electronics (OEs). Fluorene, which is a biphenyl bridged by a carbon atom, is the leader of this class of organic materials. It has led to formidable advances in organic light-emitting diodes (OLEDs, for a brief history of OLEDs, see ref. 1) due to the wide diversity of oligomers,2–7 homo-polymers8–12 or copolymers synthezized.13–16 With a more extended conjugation, dihydroindenofluorenes (DHIFs) belong to the family of bridged terphenyls and can be viewed as the fusion of a fluorene unit with an indene fragment fused at different positions of the fluorene. The DHIF family presents five positional isomers (Scheme 1). They possess three different phenyl linkages (para/meta/ortho) and two different ring bridge arrangements (anti/syn).In the DHIF family,17,18 each isomer is described with a set of numbers and a letter as nomenclature. As depicted in Scheme 1, the letter corresponds to the edge of the indene/fluorene fusion (a, b or c) and the numbers correspond to the orientation of the indene group vs. the fluorene (1,2 or 2,1). Both [1,2-a]DHIF and [1,2-b]DHIF exhibit an anti-relationship between the methylene bridges whereas [2,1-a]DHIF, [2,1-b]DHIF and [2,1-c]DHIF exhibit a syn-relationship.
Thus, 6,12-dihydroindeno[1,2-b]fluorene (para–anti[1,2-b]DHIF in blue, Scheme 1) is built on a para-terphenyl core and possesses an anti-geometry. For the last 20 years, this regioisomer has been largely studied and a lot of organic semiconductors based on this [1,2-b]DHIF core have been synthesized, studied and used as an active material in different OE devices (fluorescent and phosphorescent organic light-emitting diodes, organic field-effect transistors (OFETs) and solar cells). 11,12-Dihydroindeno[2,1-a]fluorene (para–syn[2,1-a]DHIF in red) is also built on a bridged para-terphenyl core but possesses syn-geometry. This core has been significantly less studied despite its singular geometry, which has been notably used to tune the electronic properties.19–22 Modification of the π-system from para- to meta-terphenyl provides the two meta isomers, 7,12-dihydroindeno[1,2-a]fluorene (meta-anti[1,2-a]DHIF in green, Scheme 1) and 5,7-dihydroindeno[2,1-b]fluorene (meta–syn[2,1-b]DHIF in orange, Scheme 1). As for [2,1-a]DHIF, these fragments have been far less used in OE devices than [1,2-b]DHIF. However, the strong impacts of both the linkage and the geometry on the electronic properties of [1,2-a]DHIF and [2,1-b]DHIF derivatives have been particularly highlighted over the years.23–27 There is another positional isomer in the family of DHIF, namely 5,8-dihydroindeno[2,1-c]fluorene ([2,1-c]DHIF in pink, Scheme 1), which possesses a central ortho-terphenyl backbone and a syn-geometry. This isomer has also been scarcely used to construct organic semiconductors despite its interesting ortho linkage, which induces a particular helicoidal turn of the DHIF core.
In addition, in the present review, our interest is focused on the use of organic semiconductors constructed on the five DHIF cores in different electronic devices. In fact, polymers based on DHIF cores are also numerous in the literature;28 however most of them are based on the [1,2-b]-DHIF core. Additionally, the literature also reports a huge number of DHIF derived oligomers and copolymers. Due to the diversity of these materials, their physicochemical properties are broad29 and some of them have been used in different organic electronic devices.30,31 The purpose of this review is to highlight the key synthetic routes, the physicochemical properties and the uses of the molecules belonging to the five DHIF families and not on their oligomers, polymers or copolymers.
As [1,2-b]DHIF is the most investigated isomer in the literature to date, the first part of this review will be dedicated to the description of around 70 [1,2-b]DHIF-derived organic semiconductors and their integration in different OE devices. The second part will be dedicated to the [2,1-a]DHIF-based organic semiconductors. The review will end with a third part describing modification of the DHIF core by different linkages in [1,2-a]DHIF-, [2,1-b]DHIF- and [2,1-c]DHIF-based organic semiconductors. The analysis of the effects of the different linkages on the electronic properties and particularly on the triplet energy will be discussed. A structure–property relationship approach is presented all along the review in order to highlight the impact of structural modification on the physicochemical properties.
It should be noted that several accounts and reviews have discussed DHIFs in recent years.17,18,27,31 In 2022, Usta and Fachetti reported a review on DHIF derivatives for electronics. This review was more centred on OFET and OPV devices and incorporated polymers.31 An account was also dedicated, five years ago, to antiaromatic indenofluorenes by Haley's group.17
2. Dihydroindeno[1,2-b]fluorenyl-based organic semiconductors
2.1. A brief overview of [1,2-b]DHIF synthesis and the main physicochemical properties
To our knowledge, the first synthesis of [1,2-b]DHIF was reported in 1951,32 and since this first synthesis, many different routes have been described (see Scheme 2).33–39 As shown in Scheme 2, the corresponding diketone [1,2-b]IF(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)2 is an important intermediate in these routes. This fragment, due to its electron withdrawing behaviour, is also a very important building unit widely used in electronic devices as detailed below.
O)2 is an important intermediate in these routes. This fragment, due to its electron withdrawing behaviour, is also a very important building unit widely used in electronic devices as detailed below.
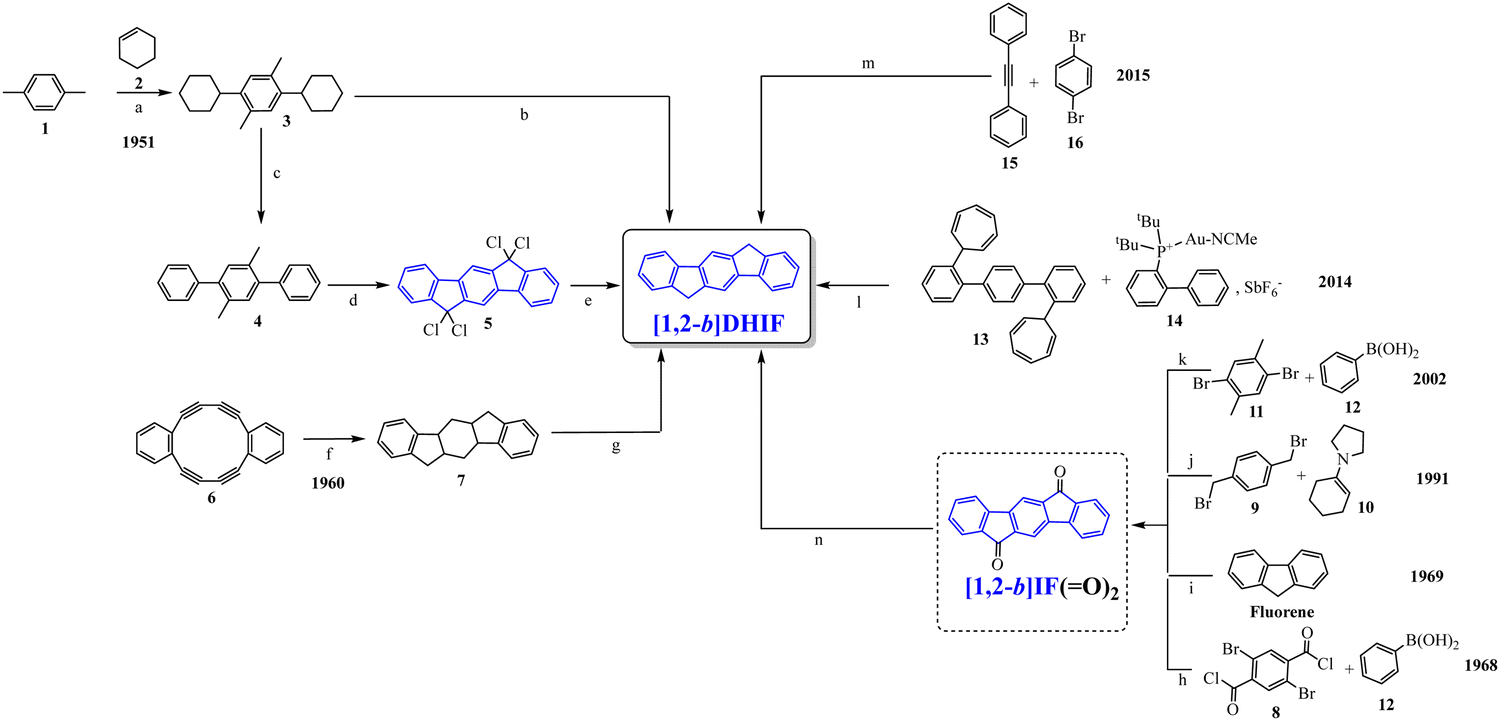 | ||
| Scheme 2 Different approaches to [1,2-b]DHIF described in the literature from 1951 to 2015.28–35 (a) AlCl3, 0 °C; (b) Pd/C, 205 °C; (c) Pd/C cat, 280 °C; (d) Cl2; (e) Zn, pyridine/AcOH; (f) Na, NH2, THF; (g) 15% Pd/C, 320 °C; (h) Pd(II) palladacycle, PhMe, K2CO3, 55 °C then DMA, 120 °C, (i) in 5 steps via the 2-iodo-fluorene, then 2-iodofluorenone, then ortho-tolyl-fluorenone, then ortho-carboxyphenyl-fluorenone; (j) dioxane, reflux under N2 then solvent evaporation; (k) in 3 steps: water, K2CO3, Bu4NBr, Pd(OAc)2, 70 °C leading to 4 then KMnO4, pyridine water leading to terphenyl-2,5-carboxylic acid and finally H2SO4 25 °C; (l) DCE, 120 °C; (m) in 2 steps: PdCl2, DPPE, DBU, CsOPiv, dioxane in a sealed tube, heating for 24 h then KOH, t-BuOK, N2H4/H2O and 18-crown-6, heated for 12 h. | ||
Looking chronologically to the different accesses to [1,2-b]DHIF (Scheme 2), Deuschel described in 1951,32 the condensation of para-xylene 1 and cyclohexene 2 in (2,5-dimethyl-1,4-phenylene)dicyclohexane 3 in 67% yield followed by its direct aromatisation-cyclization in [1,2-b]DHIF (20% yield) or by a three steps aromatization/cyclization/dechlorination sequence (3 → 4 → 5 → [1,2-b]DHIF) (18% yield). In 1960, a novel access to [1,2-b]DHIF from the macrocycle 6 was described in two steps via the hexahydroindeno[1,2-b]fluorene 7.33
The first access to [1,2-b]DHIFvia the reduction of its diketone [1,2-b]IF(O)2 was described in 1968 by Chardonnens and Salamin.34 As the reduction of the dione is not the limiting step (classically the yield is reported between 75 and 95%), the access to [1,2-b]IF(O)2 has been the subject of intense scouting (from 8 + 12, from fluorene, from 9 + 10 or from 11 + 12)34–37 and the yield has been increased from 13% (8 → [1,2-b]IF(O)2) in the initial work of Chardonnens34 to 84% with the more efficient access (11 + 12 → [1,2-b]IF(O)2) described, in 2002, by Wang's group.37
In 2014, direct access to [1,2-b]DHIF was described through double annulation of 2,2′′-di(cyclohepta-2,4,6-trien-1-yl)-1,1′:4′,1′′-terphenyl 13 using a cationic gold complex 1438 with 40% yield. Finally, in 2015, another access to [1,2-b]DHIF was published through a palladium-catalyzed reaction of diphenylethylene 15 and para-dibromobenzene 1639 with 50% yield.
A brief overview of [1,2-b]DHIF physicochemical properties, taken from the literature, is summarized below. This is an important step before describing the different organic semiconductors built on this core. As shown in Scheme 3, [1,2-b]DHIF may be viewed either as (i) the fusion of a fluorene unit with an indene fragment, (ii) the addition of a carbon bridge to a 2-phenyl-fluorene (2-Ph-F) or (iii) the addition of two carbon bridges to a p-terphenyl. Comparing the properties of [1,2-b]DHIF with those of the three fragments (summarized in Table 1) appears then relevant.
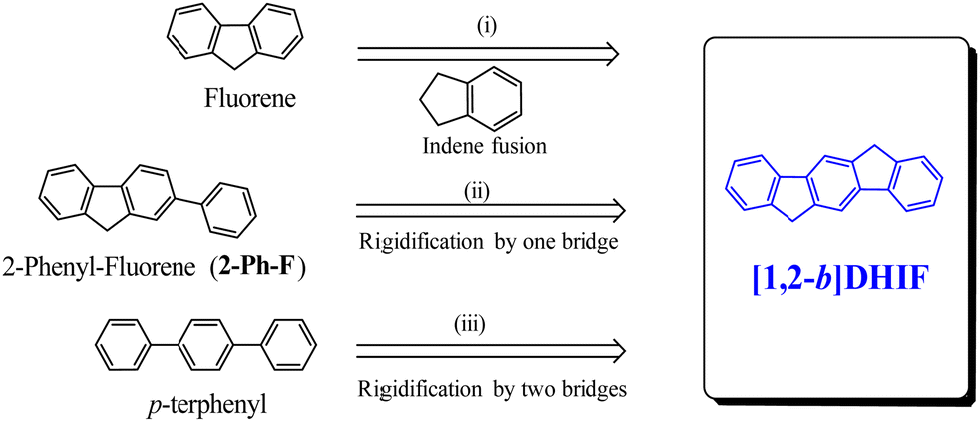 | ||
| Scheme 3 [1,2-b]DHIF and the three model compounds used to discuss its main physicochemical properties. | ||
| p-Terphenyl24 | 2-Ph-F | [1,2-b]DHIF | Fluorene | |
|---|---|---|---|---|
| CHX: cyclohexane, DMF: N,N-dimethylformamide. | ||||
| T d (°C) | 176 | — | 20040 | — |
| λ abs(ε) (nm) (× 104 L mol−1 cm−1) | 277(3.16)(CHX) | 289(3.23), 310(2.68) (CHX)44 | 328, 334 (CH2Cl2)40 | 265, 290, 301 (CH2Cl2)45 |
| 333 (6.76) (Decaline)37 | 272(1.39), 290(0.63), 294(0.53), 301(0.93) (CHX)44 | |||
| λ em (nm) | 325, 339 | 329, 343 (CHX)44 | 339, 347, 356 (CH2Cl2)40 | 310 (CH2Cl2)45 |
| 340, 356 (Decaline)37 | 303, 310 (CHX)44 | |||
| ∅sol | 0.93 (CHX)46 | 0.97 (CHX)44 | 0.61 (Decaline)40 | 0.88 (CHX)44 |
| 0.73 (Decaline)37 | ||||
| 0.91 (CHX)46 | 0.90 (CHX)46 | 0.80 (CHX)46 | ||
| HOMO (eV) | ||||
| from electrochemistry | −5.96 | −5.72 (CH2Cl2)44 | −5.62 (CH2Cl2)40 | −5.91 (CH2Cl2)40,44 |
| from calculation | −6.13 | −5.9044 | −5.4340 | −5.8140 |
| LUMO (eV) | ||||
| from electrochemistry | −2.00 | −1.92 (DMF)44 | −1.99 (DMF)40/−2.16 (DMF)37 | −1.69 (DMF)40 |
| from calculation | −1.40 | −1.5544 | −1.1340 | −0.7740 |
| ΔE (eV) | ||||
| from optical data | 4.03 | 3.7644 | 3.6140/3.71 (Decaline)37 | 3.97 (CH2Cl2)40 |
| from electrochemistry | 3.96 | 3.8044 | 3.6340/3.8537 | 4.1940 |
| from calculation | 4.44 | 4.3544 | 4.3040 | 5.0440 |
| E T1 (eV) | ||||
| from optical data | 2.55 | 2.5744 | − | 2.9344 |
| from calculation | 2.67 | 2.4044 | — | 2.7244 |
The thermal analysis using thermogravimetry (TGA) indicates for [1,2-b]DHIF a decomposition temperature @ 5% mass loss (Td) of 200 °C.40 This Td is higher than that measured for p-terphenyl (176 °C)24 indicating the beneficial effect of the two bridges on the thermal stability. It is known that the rigidification by carbon bridges increases the thermal stability, particularly the Td.24,41,42
The structural characteristics of [1,2-b]DHIF are described in Fig. 1 (CCDC 779934, grown from CH2Cl2/MeOH).43[1,2-b]DHIF possesses an extended central core of 11.05 Å long, a distance of 5.77 Å between its two methylene bridges. The two external phenyl rings are in two parallel planes distant from 0.409 Å and show a small angle of 1.5° with the central phenyl ring. The [1,2-b]DHIF core is therefore almost perfectly flat. The packing of [1,2-b]DHIF occurs only via intermolecular carbon-hydrogen short interactions (Fig. 1-right).
 | ||
| Fig. 1 X-Ray structure of [1,2-b]DHIF from CCDC 779934 (grown from CH2Cl2/MeOH).43 | ||
The absorption spectrum of [1,2-b]DHIF in CH2Cl2,40 presents a low absorption energy band peaking at 334 nm, showing an optical gap of 3.61 eV. Looking at the absorption spectra of the model compounds (recorded in cyclohexane, Fig. 2, left), a 33 nm redshift of the absorbance is observed from 277 nm for p-terphenyl to 310 nm for 2-Ph-F signing the effect of the rigidification of the conjugated terphenyl core by the addition of one bridge. Note that both spectra remain large and ill-defined due to the rotation of one or two phenyl units. The comparison of fluorene and 2-Ph-F clearly shows the impact of the addition of one phenyl unit, namely a loss of structuration and a 9 nm red-shift. The introduction of the second bridge in [1,2-b]DHIF leads to an additional 24 nm red-shift from 2-Ph-F and the spectrum appears to be more defined translating the rigidification of the system.
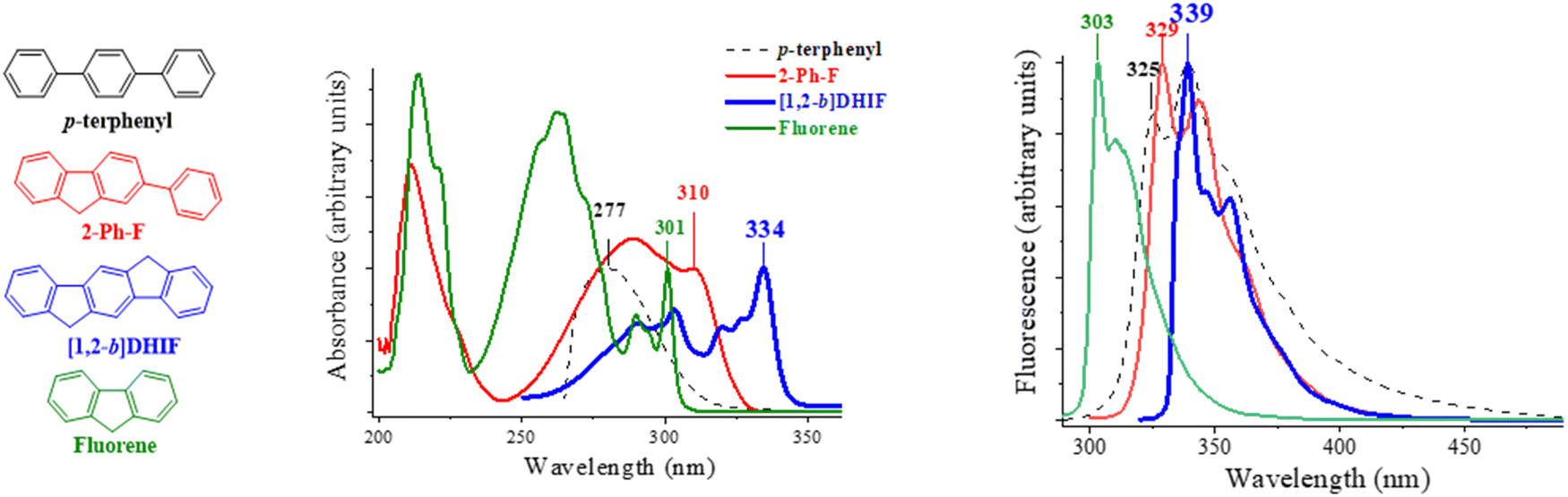 | ||
| Fig. 2 Absorption (left) and emission (right) spectra of [1,2-b]DHIF (absorption/emission recorded in CH2Cl2/decaline) and of model compounds p-terphenyl, 2-Ph-F and fluorene (all recorded in cyclohexane). | ||
The emission spectrum of [1,2-b]DHIF (in decaline or DCM) presents several emission bands peaking at 339, 347 and 356 nm with a fluorescence quantum yield of 0.73 reported by Wang and coworkers37 or of 0.61 reported by Poriel and coworkers.40 A small Stokes shift is observed between the absorption and fluorescence spectra indicating the high rigidity of the molecule with very weak rearrangements at the excited state. Comparing the fluorescent spectrum of the p-terphenyl with the first band peaking at 325 nm, to that of 2-Ph-F observed at 329 nm, one can note a red shift of 4 nm due to the presence of one bridge. Then, a red shift of 10 nm is observed from 329 nm for 2-Ph-F to 339 nm for [1,2-b]DHIF indicating the influence of a second bridge. It is known that there is a planification of the pendant phenyl ring in the first excited state explaining the similitude of the fluorescent spectra of [1,2-b]DHIF and 2-Ph-F.24 The impact of the bridges on the optical properties and particularly on the emission characteristics is discussed in other reports.23,24,47
The electrochemical properties of [1,2-b]DHIF have also been widely investigated in previous studies.48 As shown in Fig. 3, its electrochemical oxidation (CH2Cl2-Bu4NPF6 0.2 M) presents between 0.0 and 2.0 V (vs. Fc/Fc+) two successive oxidation waves with maxima at 0.9 and 1.6 V (Fig. 3A). The first oxidation process is reversible (Fig. 3B). However, the oxidation in a potential range reaching the second oxidation process E2 caused the formation of insoluble electroactive deposits on the working electrode surface visible for example in Fig. 3C by the appearance and the growth of new redox waves (Ian/Icat and IIan/IIcat) in addition to the E1 and E2 waves belonging to [1,2-b]DHIF oxidation. The deposit itself has a high electroactivity over a rather large potential range (0.35–1.25 V vs. Fc/Fc+).48 The polymer formation proceeds by carbon–carbon coupling involving the C2 and C8 carbon atoms of the DHIF core (Fig. 3, right) as classically observed for its shorter analogue fluorene.49–52 To the best of our knowledge, this was, in 2008, the first example of an electrochemical synthesis of poly([1,2-b]DHIF).
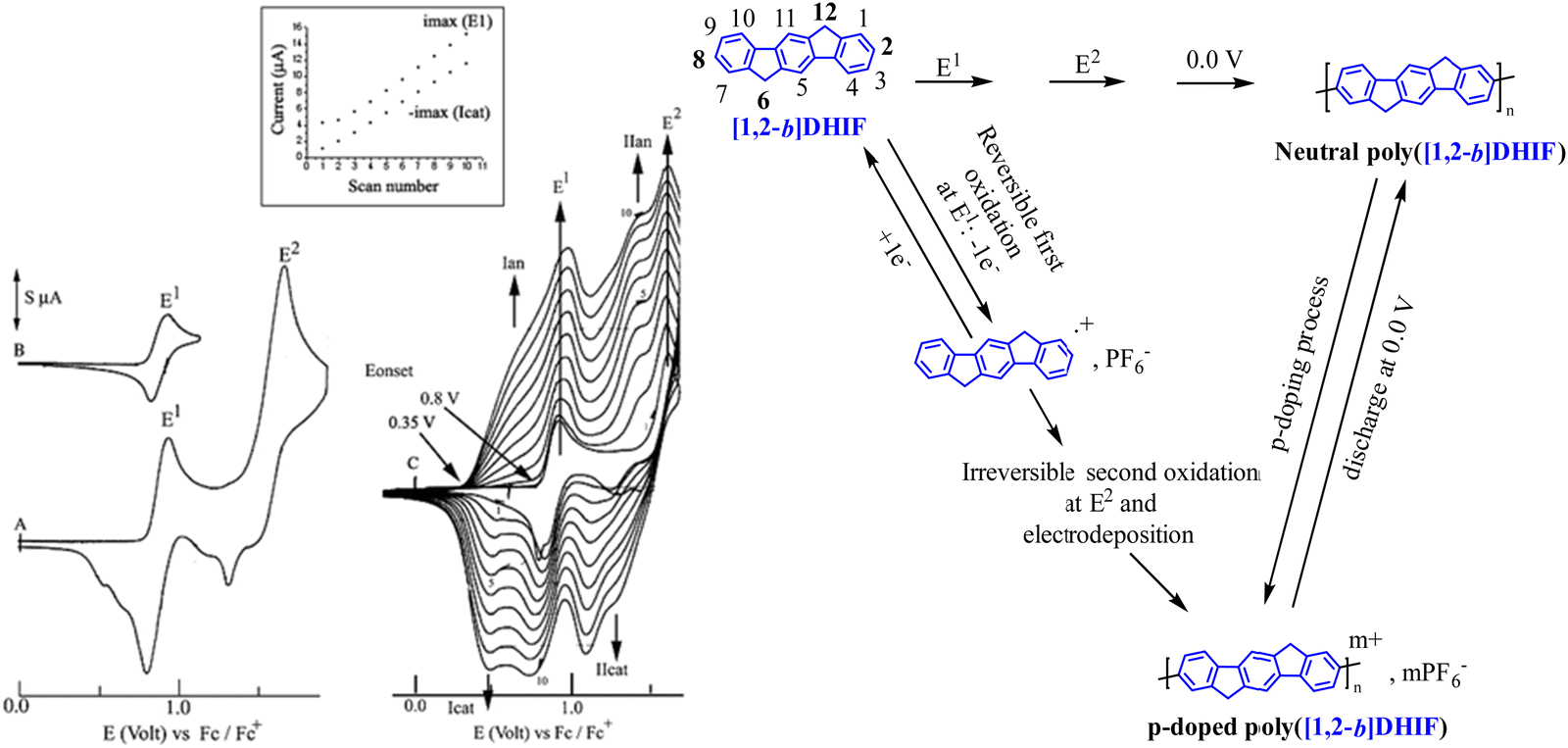 | ||
| Fig. 3 Cyclic voltammetry in the presence of [1,2-b]DHIF (2 × 10−3 M) (A–C) in CH2Cl2 (Bu4NPF6 0.2 M); working electrode: Pt disk diameter 1 mm; sweep-rate 100 mV s−1. The potential limit was of 0.0 to 1.75 V in A; 0.0 to 1.0 V in B; −0.2 to 1.73 V in C. The current scale S is equal to 2 in (A) and to 4 in (B) and in (C). The inset reports the Imax at E1 and −Imax at Icatvs. the scan number. Adapted from ref. 48 with permission from the Royal Society of Chemistry. | ||
2.2. [1,2-b]DHIF-based fluorescent organic light-emitting diodes (OLEDs)
Due to the above-mentioned characteristics and particularly the high quantum yield in the near UV region, [1,2-b]DHIF was first used as a building unit in fluorophores for the first generation of fluorescent OLEDs. Thus, many fluorophores have been constructed on this core through the substitution of the bridges and/or of the π-systems (Scheme 4). | ||
| Scheme 4 [1,2-b]DHIF-based organic semiconductors used in fluorescent OLEDs. | ||
In the following, the syntheses of the organic semiconductors are rather basic starting either from [1,2-b]DHIF or [1,2-b]IF(O)2 and will not be described in detail as the access of the two starting molecules have been discussed above. However, synthetic details will be presented for organic semiconductors obtained by other more sophisticated routes.
[1,2-b]-1 to [1,2-b]-15 have been used as fluorescent emitting layers (or as the hole-transporting layer (HTL) for [1,2-b]-15) in OLED devices, whose performances will be compared in order to draw a structure/properties/device performance map. The basic single-layer devices (SL-OLEDs) are constructed with the simple anode/emitting layer (EML)/cathode structures (with ITO or ITO/ PEDOT:PSS as the anode and Ca, Al or LiF/Al as the cathode). Such simplified devices have been investigated more and more recently.53 The structure of the other devices constructed with additional layers named Multilayer OLED (ML-OLED) are detailed in Table 2 together with all the device performances.
| Device | Device architecture | V on (V) | EQE (%) | CE (cd A−1) | PE (lm W−1) | Luminance (cd m−2) | λ max (nm) CIE (x,y) | Ref. |
|---|---|---|---|---|---|---|---|---|
| 2-(4-biphenylyl)-5-phenyl-1,3,4-oxadiazole (PBD); 4,4′,4′′-tris(carbazol-9-yl)triphenylamine (TCTA); N1,N1′-(biphenyl-4,4′-diyl)bis(N1-phenyl-N4,N4 -di-m-tolylbenzene-1,4-diamine (DNTPD). | ||||||||
| OLED1 | ITO/[1,2-b]-1 (50 nm)/PBD(50 nm)/LiF(1.5 nm)/Al | — | 1 | — | 1400 | — | 54 | |
| (@1600 A m−2) | — | |||||||
| SL-OLED2 | ITO/[1,2-b]-2/Al | 20 | — | — | — | 150 | 465, 500, 530 | 55 |
| (@35 V) | — | |||||||
| SL-OLED3 | ITO/PEDOT:PSS/[1,2-b]-3 (30 nm)/Ca | 7 | — | 0.9 | 0.19 | 200 | 399, 416, 483, 587 | 40 |
| (@15 V & 220 A m−2) | (0.21, 0.16) | |||||||
| OLED4 | ITO/DPAInT2(20 nm)/[1,2-b]-4(20 nm)/TCTA(10 nm)/ DFBTA(60 nm)LiF/Al | 2 | 3.3 | 11.4 | 11.2 | 64![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
— | 67 |
| — | ||||||||
| OLED5 | ITO/DPAInT2(20 nm)/[1,2-b]-4(20 nm)/TCTA(10 nm)/ DFBTA (30 nm)/Alq3(30 nm)/LiF/Al | 2.5 | 3.7 | 12.9 | 11 | 168000 |
— | 67 |
| — | ||||||||
| OLED6 | ITO/NPB(40 nm)/TCTA(20 nm)/[1,2-b]-5(30 nm)/Bphen (30 nm)/LiF(1 nm)/Al | 11.2 | 2.19 | 0.76 | 0.27 | — | 430 | 69 |
| (0.163, 0.073) | ||||||||
| SL-OLED7 | ITO/PEDOT:PSS/[1,2-b]-6 (40 nm)/Ca | 12.7 | — | 0.008 | 0.002 | 31 | 413, 590, 650 | 58 |
| (0.18, 0.08) | ||||||||
| OLED8 | ITO/PEDOT:PSS/NPB(35 nm)/[1,2-b]-6(40 nm)/BCP(10 nm)/ Ca | 8 | — | 1 | 0.3 | 3 740 | 413, 438, 590, 650 | 58 |
| (0.19, 0.14) | ||||||||
| SL-OLED9 | ITO/PEDOT:PSS/[1,2-b]-7(40 nm)/Ca | — | — | — | — | <2 | — | 71 |
| — | ||||||||
| OLED10 | ITO/PEDOT/NPB (35 nm)/[1,2-b]-7 (40 nm)/Ca | 9 | — | 0.005 | — | 25 | 415/436 nm | 71 |
| SL-OLED11 | ITO/PEDOT:PSS/[1,2-b]-8 (40 nm)/LiF(0.8 nm)/Al | 5.2 | — | 0.13 | — | 320 | 470 | 71 |
| OLED12 | ITO/PEDOT:PSS/NPB/[1,2-b]-8(40 nm)/BCP/LiF(0.8 nm)/Al | 4.8 | — | 0.2 | — | 1000 | 465–472 | 72 |
| (0.18, 0.28) | ||||||||
| SL-OLED13 | ITO/PEDOT:PSS/[1,2-b]-9(40 nm)/Ca | 7.2 | — | 0.031 | 0.011 | 52 | 432 | 22 |
| (0.24, 0.24) | ||||||||
| OLED14 | ITO/PEDOT:PSS/NPB(40 nm)/[1,2-b]-9(50 nm)/Ca | 6.8 | — | 0.062 | 0.002 | 230 | 448 | 22 |
| (0.25, 0.27) | ||||||||
| SL-OLED15 | ITO/PEDOT:PSS/[1,2-b]-10(40 nm)/Ca | 10.8 | — | 0.016 | 0.004 | 50 | 452 | 22 |
| OLED16 | ITO/PEDOT:PSS/NPB(40 nm)/[1,2-b]-10(50 nm)/Ca | 6.5 | — | — | — | 65 | 490 | 22 |
| OLED17 | ITO/DNTPD(60 nm)/NPB(30 nm)/[1,2-b]-11 3% in MADN (30 nm)/Alq3(20 nm)/LiF(1 nm)/Al | 4.5 | 5.96 | 8.06 | 4.41 | 10740 |
453, 481 | 76 |
| (0.145, 0.194) | ||||||||
| OLED18 | ITO/DNTPD(60 nm)/NPB(30 nm)/[1,2-b]-11 5% in MADN (30 nm)/Alq3(20 nm)/LiF(1 nm)/Al | 4.5 | 6.17 | 8.44 | 4.62 | 10260 |
454, 482 | 76 |
| (0.145, 0.198) | ||||||||
| OLED19 | ITO/DNTPD(60 nm)/NPB(30 nm)/[1,2-b]-11 7% in MADN (30 nm)/Alq3(20 nm)/LiF(1 nm)/Al | 4.5 | 6.4 | 9.19 | 4.82 | 10480 |
455, 483 | 76 |
| (0.146, 0.212) | ||||||||
| OLED20 | ITO/DNTPD(60 nm)/NPB(30 nm)/[1,2-b]-12 3% in MADN (30 nm)/Alq3(20 nm)/LiF(1 nm)/Al | 5.0 | 5.34 | 7.00 | 3.98 | 7541 | 451, 479 | 76 |
| (0.149, 0.183) | ||||||||
| OLED21 | ITO/DNTPD(60 nm)/NPB(30 nm)/[1,2-b]-12 5% in MADN (30 nm)/Alq3(20 nm)/LiF(1 nm)/Al | 5.0 | 5.73 | 7.84 | 4.11 | 7863 | 451, 474 | 76 |
| (0.150, 0.194) | ||||||||
| OLED22 | ITO/DNTPD(60 nm)/NPB(30 nm)/[1,2-b]-12 7% in MADN (30 nm)/Alq3(20 nm)/LiF(1 nm)/Al | 5.0 | 5.98 | 7.84 | 4.12 | 11580 |
452, 480 | 76 |
| (0.149, 0.193) | ||||||||
| OLED23 | ITO/DNTPD(60 nm)/NPB(30 nm)/[1,2-b]-13 3% in MADN (30 nm)/Alq3(20 nm)/LiF(1 nm)/Al | 5.0 | 8.21 | 12.6 | 6.69 | 15290 |
459, 488 | 76 |
| (0.148, 0.241) | ||||||||
| OLED24 | ITO/DNTPD(6 0 nm)/NPB(30 nm)/[1,2-b]-13 5% in MADN (30 nm)/Alq3(20 nm)/LiF(1 nm)/Al | 4.5 | 7.14 | 11.3 | 5.04 | 13420 |
460, 489 | 76 |
| (0.149, 0.250) | ||||||||
| OLED25 | ITO/DNTPD(60 nm)/NPB(30 nm)/[1,2-b]-13 7% in MADN (30 nm)/Alq3(20 nm)/LiF(1 nm)/Al | 4.5 | 7.55 | 12.2 | 6.18 | 22410 |
461, 491 | 76 |
| (0.151, 0.257) | ||||||||
| OLED26 | ITO/DNTPD(60 nm)/NPB(30 nm)/[1,2-b]-14 3% in MADN (30 nm)/Alq3(20 nm)/LiF(1 nm)/Al | 4.0 | 2.87 | 2.98 | 2.13 | 8767 | 438, 461 | 76 |
| (0.153, 0.121) | ||||||||
| OLED27 | ITO/DNTPD(60 nm)/NPB(30 nm)/[1,2-b]-14 5% in MADN (30 nm)/Alq3(20 nm)/LiF(1 nm)/Al | 4.0 | 2.82 | 3.13 | 1.77 | 8887 | 437, 463 | 76 |
| (0.153, 0.133) | ||||||||
| OLED28 | ITO/DNTPD(60 nm)/NPB(30 nm)/[1,2-b]-14 7% in MADN (30 nm)/Alq3(20 nm)/LiF(1 nm)/Al | 4.0 | 2.96 | 3.25 | 1.82 | 7952 | 437, 462 | 76 |
| (0.153, 0.128) | ||||||||
| OLED29 | ITO/DNTPD(60 nm)/NPB(30 nm)/[1,2-b]-15 5% in MADN (30 nm)/Alq3(20 nm)/LiF(1 nm)/Al | 4.0 | 7.3 | 10.2 | 5.4 | 25100 |
458, 484 | 77 |
| (0.14, 0.21) | ||||||||
The first OLED using a molecular emitter (non-polymeric structure) based on the [1,2-b]DHIF core was described in 2005,54 54 years after the first synthesis of [1,2-b]DHIF32 translating the growing interest for this fragment in OE. [1,2-b]-1 (see the molecular structure in Scheme 4) is based on the central [1,2-b]DHIF core substituted at its C2 and C8 carbon atoms by two 2-thienyl units. [1,2-b]-1 was synthesized among a series of eight 2,8-diaryl-[1,2-b]DHIFs by Suzuki cross-coupling reactions with different arylboronic acids and 2,8-Br2-[1,2-b]DHIF, the latter being synthesized from [1,2-b]DHIF (route A, Scheme 5).54
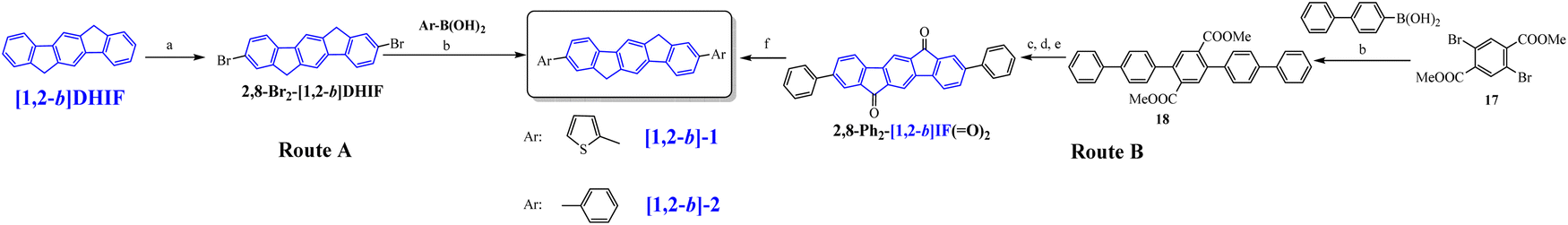 | ||
| Scheme 5 Synthesis of [1,2-b]-1 and [1,2-b]-2. (a) Br2, TCE, RT, (b) Pd(PPh3)4 in 1-methyl-2-pyrrolidinone (NMP), 90 °C, (c) aq. NaOH solution (10%), 1,2-dimethoxyethane, 90 °C, (d) aq. HCl solution (20%), (e) Eaton's reagent, 90 °C, and f) KOH, diethylene glycol, H2NNH2, 200 °C. | ||
The physicochemical properties of this series of 2,8-diaryl-[1,2-b]DHIF show that they all possess thermal stabilities higher than that of [1,2-b]DHIF. An important increase of Td from 200 °C for [1,2-b]DHIF40 to 411 °C for [1,2-b]-154 highlights the huge role played by the substituents on the thermal properties. The UV-vis absorption and emission of the eight compounds were also different to those of the central constituted [1,2-b]DHIF as the presence of the diaryl units induced an extension of the conjugation. In particular, the lowest energy absorption band of [1,2-b]-1 is red shifted by 47 nm, from 334 nm for [1,2-b]DHIF (in CH2Cl2)40 to 381 nm for [1,2-b]-1 (in DMF).54 A similar red shift of 57 nm of the emission spectrum is noted from 339 nm for [1,2-b]DHIF (in decalin)40 to 396 nm for [1,2-b]-1 (in DMF).54
The electroluminescent (EL) properties of [1,2-b]-1 were evaluated in OLED1.54 The architecture and the performance of all the devices presented below are summarized in Table 2. A moderate current efficiency (CE) of 1 cd A−1 and luminance of 1400 cd m−2 was reached. The EL spectrum was independent of the voltage but presented a broad emission with parasite emission bands surely due to oxidation of the bridges. The device emitted a whitish yellow colour.
The performances of OLED1 were nevertheless higher than those of SL-OLED2, a device based on [1,2-b]-2 (Scheme 4), similar to [1,2-b]-1 with two phenyl units instead of two thienyl units, published two years later.55[1,2-b]-2 was prepared using a similar route than that used for [1,2-b]-154 (Scheme 5-route A) but a second route (Scheme 5-route B) was also described. In this second approach,55 the 2,8-Ph2-[1,2-b]IF(O)2 was first synthesized by a coupling reaction between 4-biphenylboronic acid and dimethyl-2,5-dibromoterephthalate 17 in the presence of Pd(PPh3)4 leading to the pentaphenyl 18 followed by a cyclization step leading to 2,8-Ph2-[1,2-b]IF(O)2, which was then reduced to [1,2-b]-2. In this approach, the DHIF core was constructed in the last step, as the phenyl units were already grafted.
T d of [1,2-b]-2, 404 °C,55 was very high and almost identical to that of [1,2-b]-1, 411 °C.54 Its absorption spectrum was blue shifted by 31 nm (λmax: 350 nm) compared to that of [1,2-b]-1 (381 nm) translating a different π-conjugation extension (different aromaticity of phenyl and thienyl units). Similarly, its fluorescence spectrum with a maximum at 375 nm was blue shifted by about 20 nm compared to that of [1,2-b]-1 (396 nm). The HOMO and LUMO reported for [1,2-b]-2, −5.2 and −2.0 eV were similar to those reported for [1,2-b]-1 (the HOMO of [1,2-b]-2 was nevertheless lower by 0.1 eV than that of [1,2-b]-1 in accordance with the substituent effects indicated above). SL-OLED2 based on [1,2-b]-2 emitted a green light with an extremely high threshold voltage (Von) of 20 V and only reached a low luminance of 150 cd m−2 at 35 V, showing very weak OLED performances.
In 2006, our group described a novel route to construct the [1,2-b]DHIF core starting from diiodo-terphenyl derivatives (19a-b) and the 9-fluorenone 20.45 A novel generation of efficient violet emitters called dispirofluorene-indenofluorene (DSF-IF) was then reported.27[1,2-b]-DSF-IF and [1,2-b]-3 belong to this family of emitters (Scheme 4). They possess three π-systems connected via two spiro carbon atoms. Their synthesis, as presented in Scheme 6, starts with diiodo-terphenyls 19a-b involved in a lithium-iodine exchange followed by quenching with the 9-fluorenone 20 to give the corresponding diols, 21a-b, which were finally converted to the corresponding DSF-IFs: [1,2-b]-DSF-IF and [1,2-b]-3. Each fluorenyl unit is in a plane perpendicular to that of the central [1,2-b]DHIF core. The specific architecture of these organic semiconductors was named “3π-2spiro”, the association of 3 π-systems via 2 spiro-bridges.56–62
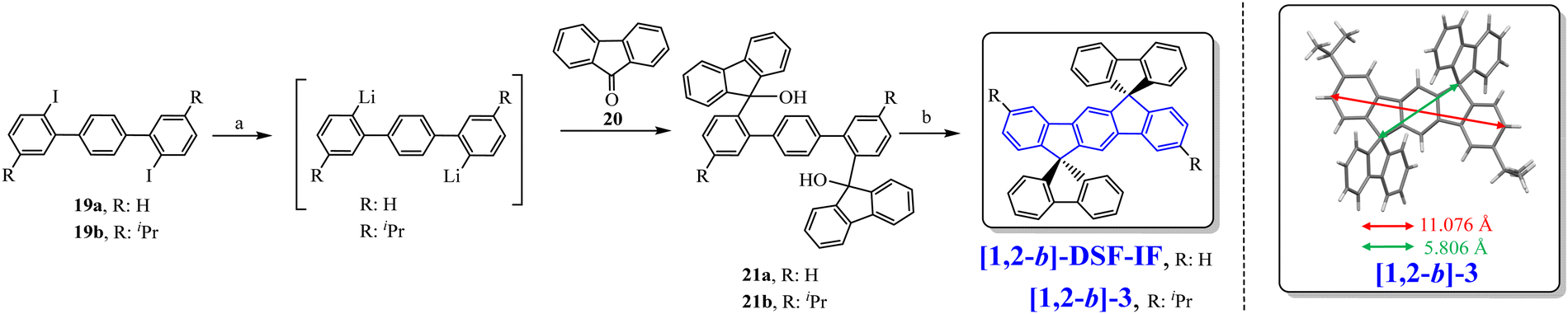 | ||
| Scheme 6 Left: Synthesis of [1,2-b]-DSF-IF and [1,2-b]-3.nBuLi, THF, −78 °C. (b) AcOH/HCl reflux. Right: X-Ray structure of [1,2-b]-3. | ||
The X-ray analysis of [1,2-b]-3,40 presented in Scheme 6, right, reveals that the central [1,2-b]DHIF core is almost perfectly flat and similar to that of [1,2-b]DHIF (Fig. 1) with small distortions probably caused by the two iso-propyl substituents. The angle measured between the central core and the fluorenyl units was ca 89° demonstrating the orthogonality between the central [1,2-b]DHIF core and the external fluorenyl π-systems.
[1,2-b]-3 and [1,2-b]-DSF-IF maintain the main physicochemical properties of [1,2-b]DHIF, with however higher thermal stability (Td: 200 °C for [1,2-b]DHIF, 355 °C for [1,2-b]-DSF-IF and 382 °C for [1,2-b]-3).40 This highlights the strong interest of the incorporation of spiro connected fragments. This feature was introduced by Salbeck et al. in the nineties.41 HOMO and LUMO obtained from electrochemical experiments showed that (i) the HOMO of [1,2-b]-DSF-IF (−5.76 eV) and of [1,2-b]-3 (−5.66 eV) were deepened compared to that of [1,2-b]DHIF (−5.62 eV) and that (ii) their LUMOs were lowered from [1,2-b]DHIF (−1.99 eV), to [1,2-b]-3 (−2.07 eV) and [1,2-b]-DSF-IF (−2.17 eV). These effects may result from the electron-withdrawing impact of the spiro-linked fluorenyl units rendering the [1,2-b]DHIF core more difficult to oxidize and easier to reduce. These moderate effects through the spiro-bridges are induced by the “spiroconjugation”.63–66
Both [1,2-b]-3 and [1,2-b]-DSF-IF absorption spectra (CH2Cl2) present a low energy absorption band at respectively 348 nm and 345 nm and their emission spectra (cyclohexane) show bands at 351 and 347 nm respectively. The small Stokes shifts (2–3 nm) are consistent with the rigidity of the structures and with the high QY (0.66 and 0.62).
[1,2-b]-3 was used as the EML in SL-OLED3.40Von was measured at 7 V and its luminance reached 200 cd m−2 at 15 V for a current density of 220 A m−2 showing the weak efficiency of the device. Its EL spectrum nevertheless possesses two main bands in the blue region (399 and 416 nm) and two others at 483 and 587 nm (CIE 1964 (0.21; 0.16)).
In 2009, the [1,2-b]DHIF-based donor–π–donor (D–π–D) [1,2-b]-467 (Scheme 4) was prepared from the 6,6,12,12-tetratolyl-6,12,dihydroindeno-[1,2-b]DHIF-fluorene 2468 (Scheme 7), itself obtained from the diethyl 2,5-dibromoterephthalate 22 and phenylboronic acid 12via the terphenyl-2′,5′-diester 23. The reaction of 23 with an excess of 4-methylphenyllithium (generated in situ by reacting 1-bromo-4-methylbenzene with n-BuLi at −78 °C) provided a diol, which underwent ring closure to generate 24. [1,2-b]-4 was then obtained through the selective bromination of 24 with 2.2 eq. of Br2 in the presence of a catalytic amount of FeCl3 (0.05 eq.) to afford the dibromo chromophore 25. Pd-catalyzed C–N bond cross-coupling of 25 with diphenylamine gave [1,2-b]-4.
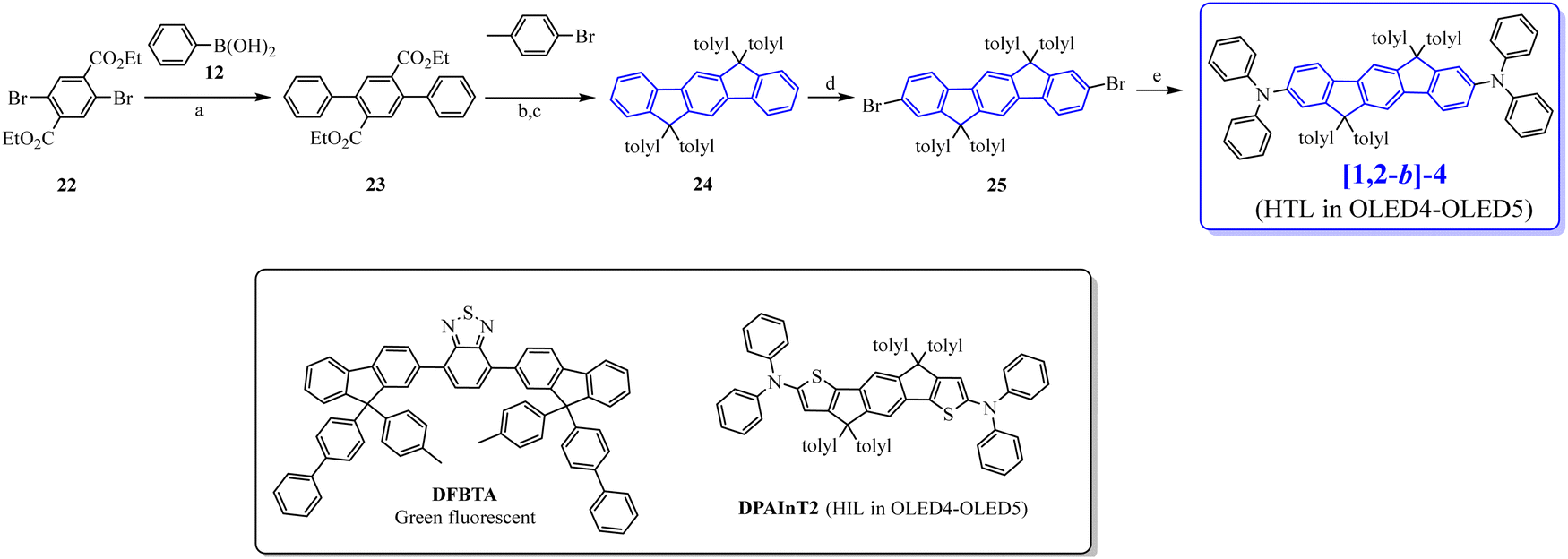 | ||
| Scheme 7 Top: Synthesis of [1,2-b]-4. (a) Pd(PPh3)4, PtBu3, K3PO4, toluene, 60 °C, (b) nBuLi, THF, −78 °C, (c) boiling acetic acid/HCl, (d) Br2, FeCl3, CHCl3, 0 °C, and (e) Pd(OAc)2, NHPh2, NatBuO, PtBu3, toluene, 60 °C. Bottom: The molecular structure of the green fluorescent emitter DFBTA and of the DPAInT2 ETL used in OLED 4 and OLED 5. | ||
Compared to [1,2-b]DHIF, [1,2-b]-4 presented a higher thermal stability with a Td of 420 °C (200 °C for [1,2-b]DHIF40). The substitution at C2 and C8 by two donor units in addition to the substitution at C6 and C12 by four tolyl units led to a gap contraction of 0.69 eV from 3.61 eV for [1,2-b]DHIF40 to 2.92 eV for [1,2-b]-4.67 A high hole mobility (μh) of 10−3 cm2 V−1 s−1 was measured for [1,2-b]-467 using time-of-flight measurements highlighting the significant role of the NPh2 units.
[1,2-b]-4 was then used as the HTL in ML-OLEDs using as the EML the efficient green fluorescent emitter DFBTA (see the molecular structure in Scheme 7). Two devices were constructed (OLED4 and OLED5). Compared to similar OLEDs with a hole injection layer (HIL)/HTL of PEDOT:PSS/N,N′-Di(1-naphthyl)-N,N′-diphenyl-(1,1′-biphenyl)-4,4′-diamine (NPB) instead of DPAInT2/[1,2-b]-4,67 OLED4 and OLED5 were slightly more efficient; the external quantum efficiency (EQE) increases from 2.6% to 3.3% in OLED4 and from 3% to 3.7% in OLED5. OLED5 exhibited the highest efficiency: Von of 2.5 V, a maximum EQE of 3.7% (CE: 12.9 cd A−1) and an extremely high luminance reaching 168 000 cd m−2. This showed that DHIF derivatives could be efficiently used as the HTL when a judicious substitution is chosen.
In 2010, the group of Park69 has prepared an extended [1,2-b]DHIF derivative substituted at C2 and C8 by two terphenyl groups, namely [1,2-b]-5 (Scheme 4). Synthesis of [1,2-b]-5 (Scheme 10, p. 13) starts from the dialkylation of [1,2-b]DHIF in 32 followed by dibromination giving 33 further involved in Suzuki coupling with 34 to provide [1,2-b]-5 (Scheme 10). [1,2-b]-5 showed a high Tg (179 °C), a key point for OLED applications, and also a high Td (418 °C). The UV-vis absorption spectrum (THF) presented a maximum at 366 nm, red-shifted by 32 nm compared to [1,2-b]DHIF.40 The optical gap of [1,2-b]-5 (3.13 eV) was therefore more contracted than that of [1,2-b]DHIF (3.61 eV), indicating an extension of the conjugation in [1,2-b]-5 due to the terphenyl substituents. Compared to the optical gap of [1,2-b]-1 (3.1 eV),54 with two 2-thienyl substituents, the extension of conjugation appears similar for [1,2-b]-5 with two terphenyl groups. The emission spectrum of [1,2-b]-5 presented a non-structured band, due to the rotation of the terphenyl fragments (λmax: 405 nm, Stokes-shift: 39 nm). The authors measured the μh of films of [1,2-b]-5 by the time-of-flight transient photocurrent techniques as 8.3 × 10−5 cm2 V−1 s−1 (one order of magnitude lower than that of [1,2-b]-4).67 This showed the importance of the substituents grafted to the IF core to improve the charge transport. OLED6 with [1,2-b]-5 as the EML and 4,7-diphenyl-1,10-phenanthroline (Bphen) as the electron-transporting layer (ETL) reached an EQEmax of 2.19% and emitted a pure blue light with an ELmax centered at 430 nm with CIE coordinates of (0.163, 0.073) indicating an efficient electron injection from the cathode to the EML via the Bphen ETL.
Following our explorations on three dimensional [1,2-b]DHIF-based organic semiconductors, our group synthesized, in 2010, an organic semiconductor based on xanthenyl π-systems.57[1,2-b]-6 (Scheme 4), possessing also a 3π-2spiro architecture, was constructed following two different routes presented in Scheme 8: the first from 2,2′′-diiodoterphenyl 26 and xanthenone 27, and the second starting from [1,2-b]IF(O)2 in a one-step rearrangement reaction. Both routes provided [1,2-b]-6 in moderate yield (50% from 26 and 35% from [1,2-b]IF(O)2).
 | ||
| Scheme 8 Synthesis of [1,2-b]-6. (a) nBuLi, THF, −78 °C to RT, (b) CH3CO2H/HCl, 100 °C, and (c) MeSO3H, 160 °C. | ||
The main physicochemical properties of [1,2-b]-657 may be first compared to those of [1,2-b]DHIF to investigate the impact of the xanthenyl substituents. Thanks to cyclic voltammetries,70 HOMOel/LUMOel were reported at −5.79/−2.15 eV, [1,2-b]-6 displays almost identical energy levels compared to those of [1,2-b]-DSF-IF (HOMOel/LUMOel: −5.76/−2.17 eV),40 indicating that xanthenyl units or fluorenyl units have similar electronic effects on the central [1,2-b]DHIF core. The gaps remain therefore similar for the three organic semiconductors (3.63/3.59/3.64 eV for [1,2-b]DHIF/[1,2-b]-DSF-IF/[1,2-b]-6) as the main electronic properties in solution are imposed by the [1,2-b]DHIF core with only a moderate effect of the spiro connected units.
In absorption spectroscopy (cyclohexane), [1,2-b]-6 presented a low energy band at 344 nm almost identical to that of [1,2-b]-DSF-IF (345 nm, see above).40,45 The emission of [1,2-b]-6 (cyclohexane) showed the first maximum at 347 nm is also similar to that of [1,2-b]-DSF-IF (347 nm, see above),40,45 indicating that the optical properties of [1,2-b]-6 and [1,2-b]-DSF-IF were both directed by their common [1,2-b]DHIF cores. For the two organic semiconductors, the Stokes shift was very small (3 nm) indicating rigid molecular structures.
[1,2-b]-6 was used as the fluorescent EML in devices of different architectures.57,58 The performances of SL-OLED7 were extremely low (Von: 12.7 V and maximum luminance of only 31 cd m−2). OLED 8 was designed with N,N′-di(1-naphthyl)-N,N′-diphenyl-[1,1′-biphenyl]-4,4′-diamine (NPB) as the HTL and bathocuproine (BCP) as the hole blocking layer (HBL) inserted on both sides of [1,2-b]-6. Thus, for OLED8, Von was measured at 8 V and the luminance reached 3 740 cd m−2, showing the efficiency of this OLED architecture. A comparison of SL-OLED7 and OLED8 indicates that the addition of the HTL and HBL on both sides of the emitting layer ([1,2-b]-6) leads to a lower Von (best charge injection in the device) and to a significant increase in the luminance (by a factor 100) indicating a more important exciton formation (best charges recombination in the EML) and relaxation. The EL spectra of the devices both showed the blue emission arising from [1,2-b]-6.
In 2011, our group synthesized a bipolar [1,2-b]DHIF-based emitter with a donor–π–acceptor (D–π–A) design71 ([1,2-b]-8, Scheme 4) and compared its main physicochemical properties to those of its central π-system [1,2-b]-719 and of its donor–π–donor (D-[1,2-b]IF-D) and acceptor–π–acceptor (A-[1,2-b]IF-A) parent compounds72 (molecular structure of A-[1,2-b]IF-A and of D-[1,2-b]IF-D in Scheme 9). Indeed the design of bipolar D–π–A materials with both electron and hole-transporting capabilities is of high interest for OLEDs, and particularly for simplified SL-devices.25,53,71–73
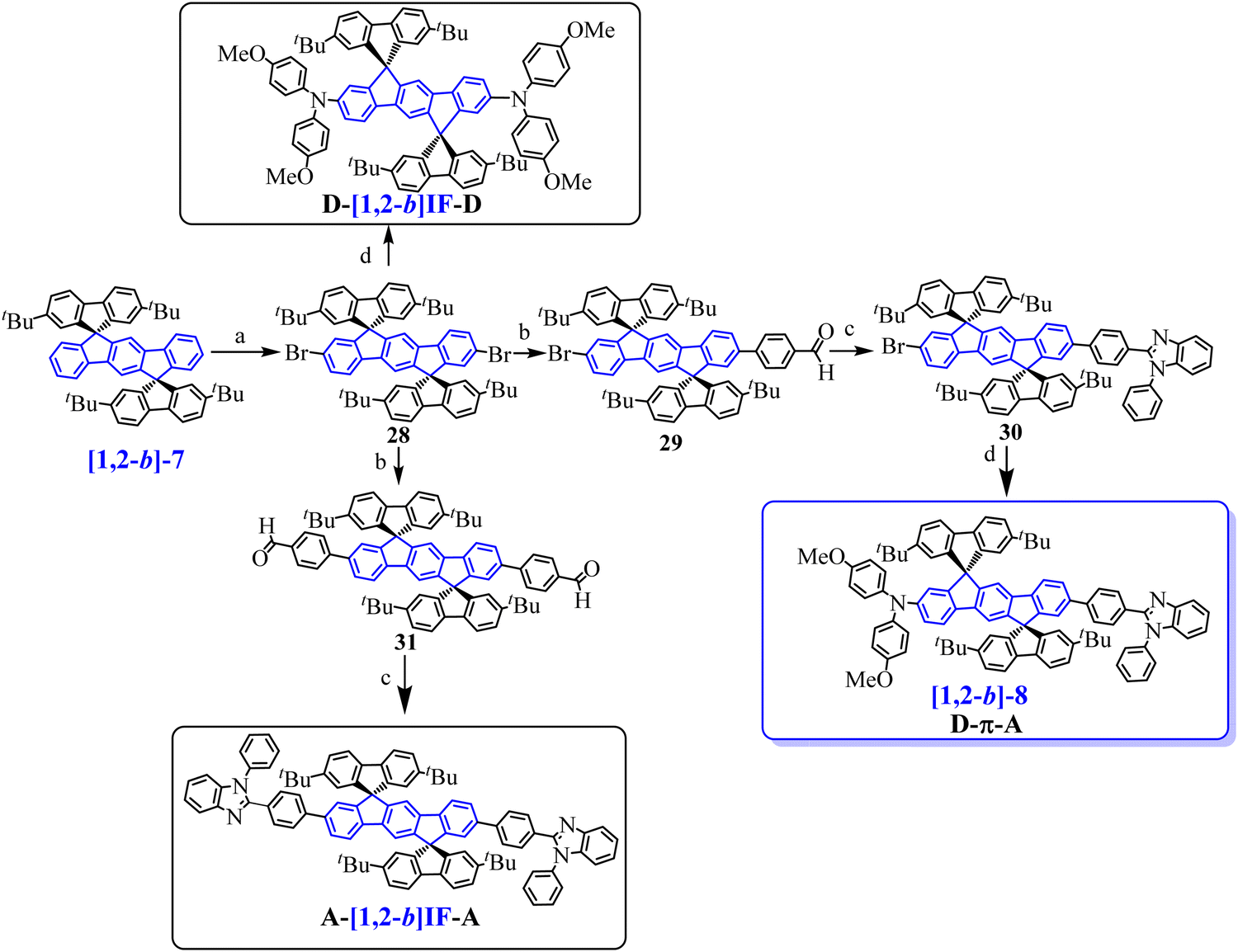 | ||
| Scheme 9 Synthesis of [1,2-b]-8, and of its parent A-[1,2-b]IF-A and D-[1,2-b]IF-D derivatives. (a) Br2, Na2CO3, I2 (catalytic amount), CH2Cl2/water, RT, (90%); (b) Pd2(dba)3/P(tBu)3, Na2CO3, 4-formylphenylboronic acid (1.4 eq. vs.28 for 29 or 2 eq. vs.28 for 31), toluene/water, 100 °C; (c) methoxyethanol, N-phenyl-o-phenylenediamine, 120 °C; (d) Pd(OAc)2, P(tBu)3, tBuOK, 4,4′-dimethoxydiphenylamine, toluene, 100 °C. | ||
[1,2-b]-7 was first synthesized following a protocol similar to that used for [1,2-b]-6 (Scheme 8, left) using 2,7-(tBu)2-fluorenone instead of xanthenone 27. Then [1,2-b]-8 and its D–π–D and A–π–A parent compounds were prepared from [1,2-b]-7 (see Scheme 9).71,72 The D–π–A derivative, [1,2-b]-8, was prepared in four steps, from [1,2-b]-7 by (a) a dibromation of the [1,2-b]DHIF central core leading to the 2′,8′-dibromo-derivative 28, followed by (b) a selective Pd-catalyzed desymmetrization with 4-formylphenylboronic acid leading to 29. The benzimidazolyl group of 30 was then constructed (c) through the condensation with N-phenyl-o-phenylenediamine in methoxyethanol (Scheme 9). Then [1,2-b]-8 was obtained from 30 by the introduction of the dimethoxydiphenylamine unit. The overall yield from [1,2-b]-7 was 10%. Similarly, D-[1,2-b]IF-D was obtained from 28 by the introduction of two dimethoxydiphenylamine units and A-[1,2-b]IF-A was obtained in two steps (28 → 31 → A-[1,2-b]IF-A) following steps b and c.
Extension of the central [1,2-b]DHIF core of [1,2-b]-7 by two electron donating units in D-[1,2-b]IF-D or by two electron accepting units in A-[1,2-b]IF-A leading to (i) an increase of the HOMO (without strong variation of the LUMO level) in D-[1,2-b]IF-D (HOMOel/LUMOel: −4.81/−2.02 eV72) or to (ii) a decrease of the LUMO (without strong variation of the HOMO level) in A-[1,2-b]IF-A (HOMOel/LUMOel: −5.55/−2.44 eV72), Fig. 4. Indeed, in these systems, the HOMO and LUMO levels are governed by the pending substituents and not by the DHIF core. [1,2-b]-8 (HOMOel/LUMOel: −4.87/−2.35 eV72) gathers the characteristics of both A-[1,2-b]IF-A and D-[1,2-b]IF-D. Indeed, the substitution by both the donor and the acceptor units in [1,2-b]-8, leads to a decrease of the LUMO and an increase of the HOMO compared to [1,2-b]-7. It must be noted that [1,2-b]-7 oxidation has been particularly studied both by cyclic voltammetries and spectroelectrochemical methods.74 These studies showed the singular multielectronic oxidation of [1,2-b]-7 with the first monoelectronic oxidation of the indenofluorenyl core followed by a second one electron oxidation and the reorganization of the dicationic charges on the two external fluorenyl units.
 | ||
| Fig. 4 Representation of the HOMO and LUMO energy levels obtained from electrochemical studies of different [1,2-b]-derivatives. Left: Representation of the electron density contour (HOMO and LUMO) obtained from DFT calculations for [1,2-b]-8 (isovalue: 0.03 [e Bohr−3]1/2). | ||
In absorption spectroscopy (cyclohexane), there was a shift of the lowest energy absorption band from 345 nm for [1,2-b]-7 to 410 nm for [1,2-b]-8. Thus, [1,2-b]-8 presented a ΔEopt of 2.83 eV, strongly contracted compared to that of [1,2-b]-7 (ΔEopt = 3.49 eV). The emission spectrum of [1,2-b]-8 showed fluorescence in the blue region (435 nm). This spectrum was significantly red-shifted by 86 nm compared to that of [1,2-b]-7 (349 nm) due to both the intramolecular charge transfer and conjugation extension induced by the substituents. In addition, the emission spectrum of [1,2-b]-8 is more resolved than its absorption one, indicating that, in the excited state, the bond linking the central DHIF core and the benzimidazolyl fragment gains some double bond character and therefore a more rigid/planar molecular structure. Finally, [1,2-b]-8 also presented a very high quantum yield of ca. 0.97, in the blue region, ensuring therefore its potential as an efficient blue emitter.
OLED devices were first constructed with model compound [1,2-b]-7 as the EML.71 Both single (SL-OLED9) and double (OLED10) layer OLEDs displayed a very low performance translating the high difficulty to inject charges within the EML. Two OLEDs were then constructed using [1,2-b]-8 as the EML, SL-OLED11 and OLED 12. Compared to the performance of SL-OLED9, SL-OLED11 appeared more efficient showing the interest of the D–π–A architecture of [1,2-b]-8. The performance of SL-OLED11 remains however low reaching only 320 cd m−2 and a CE max of 0.13 cd A−1. The performance was improved using multilayer stacking in OLED1272 (1 000 cd m−2 and 0.2 cd A−1).
In 2011, [1,2-b]-975 and in 2012, [1,2-b]-1022 (Scheme 4) were synthesized and studied by our group. The HOMO/LUMO energy levels of [1,2-b]-9 and [1,2-b]-10 were compared to those of structurally related compounds: [1,2-b]-DSF-IF and [1,2-b]-7 in order to highlight the key role played by the substituents on the electronic properties of the different organic semiconductors (Fig. 5).
 | ||
| Fig. 5 HOMO/LUMO energy levels and optical gaps of different [1,2-b]-derivatives. HOMO levels were calculated from electrochemical studies and LUMO levels were calculated from the HOMO and the optical gap.22,40,75 | ||
The HOMO level obtained by cyclic voltammetries was increased by 0.27 eV from [1,2-b]-DSF-IF (−5.76 eV) to [1,2-b]-9 (−5.49 eV) due to the incorporation of the 3,4,5-trimethoxyphenyl units, which extends the π-conjugation of the fluorene units. In [1,2-b]-9, the HOMO/LUMO energy levels are therefore governed by the “aryl-fluorene-aryl” core. For the same reason, the LUMO (calculated from the optical gap ΔEopt and the HOMO) was decreased by 0.21 eV from [1,2-b]-DSF-IF (LUMO: −2.25 eV) to [1,2-b]-9 (LUMO: −2.04 eV).
For [1,2-b]-10, the modulation of the HOMO and LUMO levels was associated with the extension of conjugation of the [1,2-b]DHIF central unit by the two 3,4,5-trimethoxyphenyl moieties. For this compound, both HOMO and LUMO were governed by the aryl-[1,2-b]DHIF-aryl core. Thus the HOMO level was increased by 0.18 eV from [1,2-b]-7 to [1,2-b]-10 and the LUMO level was decreased by 0.17 eV from [1,2-b]-7 to [1,2-b]-10 providing a gap contraction of 0.35 eV.
The UV-vis absorption spectrum of [1,2-b]-10 presented a maximum at 345 nm whereas that of [1,2-b]-9 was red-shifted by 17 nm (362 nm). The same trend was observed in emission spectroscopy (381 vs. 391 nm for [1,2-b]-9 and [1,2-b]-10). The optical properties of [1,2-b]-10 are fully governed by the central aryl-[1,2-b]DHIF-aryl moiety whereas those of [1,2-b]-9 are governed by the two spiro-linked aryl-fluorene-aryl units.
SL-OLED13 and SL-OLED15 based on [1,2-b]-9 and [1,2-b]-10 reached very low performances (CE lower than 0.06 cd A−1). The insertion of a NPB layer between the anode and the EML in OLED14 and in OLED16, slightly increases the performances which remain however very low (luminance reaching only 230 or 65 cd m−2 for OLED 14 or OLED16).
Four other non-symmetric [1,2-b]DHIF-derivatives, [1,2-b]-11 to [1,2-b]-14 (Scheme 4), were prepared, in 2012, by Lee and Yoon.76 As shown in Scheme 10, all of them were obtained by a non-symmetric modification of the 2,8-dibromo-6,6,12,12-tetraethyl-6,12-dihydroindeno[1,2-b]fluorene 33 (obtained from [1,2-b]-DHIF as described before for the synthesis of [1,2-b]-569). 33 was then monolithiated, followed by reaction with DMF to afford the monoformylated intermediate 35, which was coupled with 3,5-di-tert-butylphenylboronic acid 36. Finally, [1,2-b]-11-[1,2-b]-14 were obtained by Horner–Wadsworth–Emmons reaction between 37 and the corresponding phosphonates 38a-38d (Scheme 10).
 | ||
| Scheme 10 Synthesis of [1,2-b]-5 and [1,2-b]-11 to [1,2-b]-15. (a) Bromoethane, nBu4NBr, toluene, NaOH/Reflux; (b) Br2, FeCl3, CHCl3; (c) Pd(OAc)2,Et4NOH, Ph3P, toluene; (d) nBuLi, DMF, THF, −78 °C; (e) [Pd(PPh3)4], 2 M Na2CO3, ethanol, toluene; (f) tBuOK, THF; (g) Pd(PPh3)4, Aliquat 336, K2CO3, toluene. | ||
Absorption spectra were only weakly modulated from [1,2-b]-11 to [1,2-b]-13 indicating the weak influence of the phenyl-vinylene/biphenyl-vinylene or fluorenyl-vinylene links between the diphenylamine and the [1,2-b]DHIF core in the three compounds. For [1,2-b]-14, a blue shift of 10 nm of the absorption spectrum related to [1,2-b]-11 was noted due to the different influence of the triphenylamine in [1,2-b]-11 and phenylcarbazole in [1,2-b]-14. The four fluorophores displayed a blue light with maxima at 464, 469, 480 and 429 nm for [1,2-b]-11-[1,2-b]-14 respectively. The maximum was red-shifted from [1,2-b]-11 to [1,2-b]-13 indicating the extension of conjugation from [1,2-b]-11 with one phenyl linker to [1,2-b]-12 with a biphenyl linker and finally to [1,2-b]-13 with one fluorenyl linker. [1,2-b]-14 which contains a C3-substituted carbazole unit (disruption of the conjugation) showed a blue shift relative to [1,2-b]-11–[1,2-b]-13.
The four compounds were employed as emitters dispersed in a matrix of 2-methyl-9,10-bis(2-naphthyl)anthracene (MADN). In these devices (OLED17 to OLED28), the blue fluorophores were used to dope the MADN host in concentrations varying from 3 to 5 and 7%. The highest device performances were obtained with [1,2-b]-13 as the dopant, either in 3% in MADN showing an EQE higher than 8.2% (OLED23) or in 7% in MADN with a luminance reaching 22 410 cd m−2 (OLED25). Performances of OLED17 to OLED28 were significantly higher than the performances of the non-doped OLEDs presented above (OLED1 to OLED16), showing the importance of using a host matrix to improve the device performance. All devices with [1,2-b]-11 to [1,2-b]-14 as the dopant displayed a low Von of 4.0 to 5.0 V with blue emission for all the devices.
The same group also synthesized [1,2-b]-1577 by an asymmetric modification of 28 (Scheme 10). Using this strategy, two different substituents were introduced, namely Ph(tBu)2 and vinyl-TPA. Thus, the first monoformylation led to 29, which was involved in C–C coupling with the boronic acid 36 leading to 39. Then, a Horner-Wadsworth-Emmons reaction of 39 with the phosphonate 38a afforded [1,2-b]-15 with 11% yield from 28.
[1,2-b]-15 physicochemical properties were similar to those of [1,2-b]-11 showing that the main properties are governed by the same central extended [1,2-b]DHIF. [1,2-b]-15 was also used as the dopant of MADN in OLED29. This device was a very high efficiency blue OLED reaching an EQEmax of 7.3% (CEmax/PEmax: 10.2 cd A−1/5.4 lm W−1) with a maximum luminance of 25100 cd m−2 at 8.5 V. Compared to OLED18, OLED29 outperformed with an EQE increasing from 6.17 to 7.3% and a luminance increasing from 10 260 to 25 100 cd m−2. The performance of OLED29 was the highest in the series (OLED17-OLED29) with the lower Von, the highest luminance and an EQE of 7.3%. OLED23-OLED25 based on [1,2-b]-13 were also very efficient (EQE between 7.14 and 8.21%) with however higher Von and lower luminances.
2.3. [1,2-b]DHIF-based organic semiconductors used in organic field-effect transistors
Organic field-effect transistors have attracted great interest in the last 30 years due to their possible incorporation in full OE devices.78–82 In the course of writing the present review, another review also dedicated to [1,2-b]DHIF fragments was published by Facchetti and Usta.31 Some of the organic semiconductors presented in Scheme 11 have been described in this review with however a different view angle.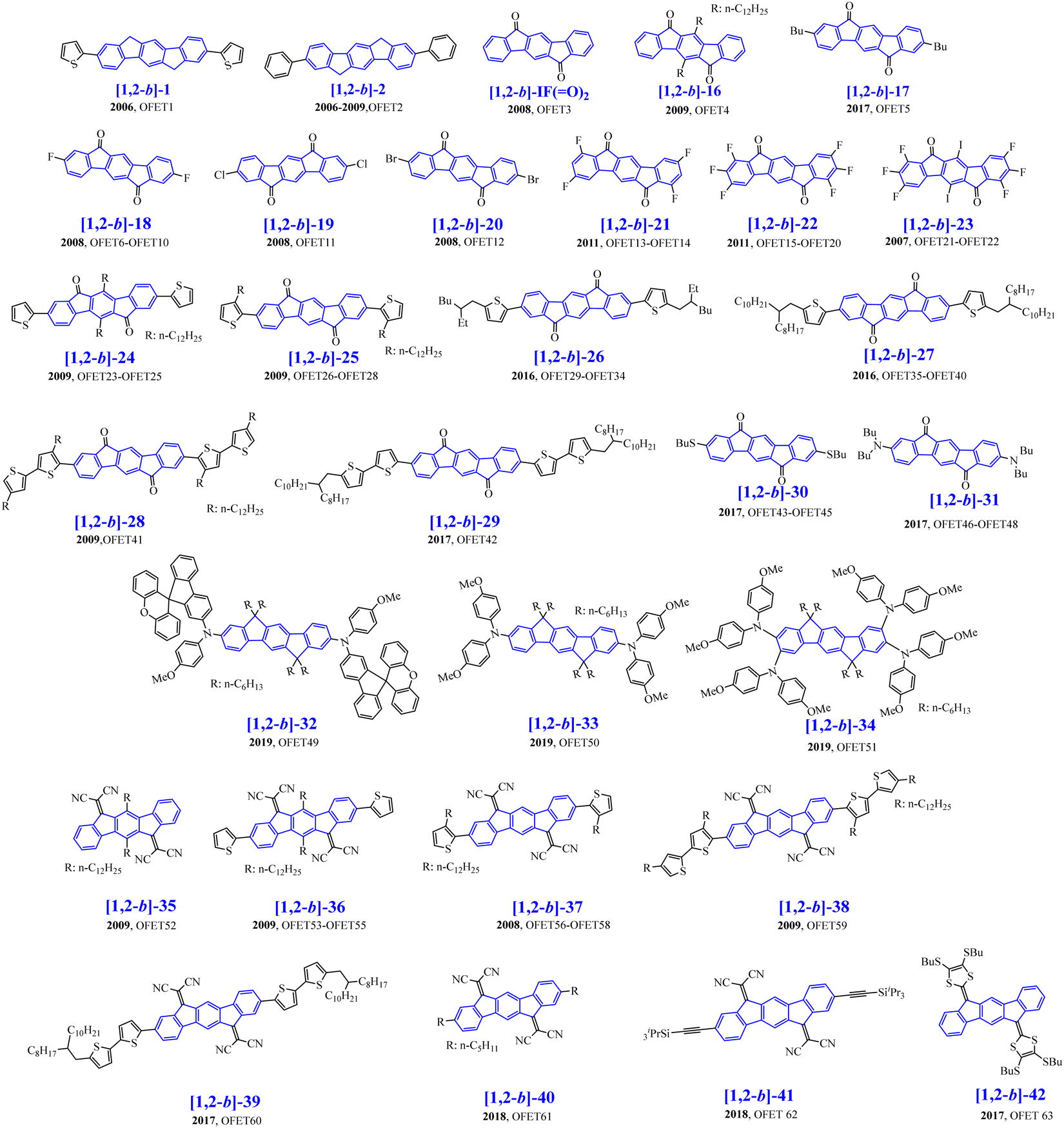 | ||
| Scheme 11 [1,2-b]DHIF-based organic semiconductors used as the active material in OFET ([1,2-b]-16–[1,2-b]-42). | ||
The development of [1,2-b]DHIF derivatives for OFET is described below starting from the first p-type OFET1 based on [1,2-b]-183 to electron-transporting n-type OFETs based on more efficient organic semiconductors ([1,2-b]-16 to [1,2-b]-42) described in Scheme 11. The [1,2-b]DHIF core displays appealing characteristics for charge transport such as its extended π-conjugated backbone, which may favour π–π interactions. As exposed above, it can also be easily functionalizable either on the bridges or on the benzene rings, of great interest to tune the electronic properties such as HOMO/LUMO levels. In this part, we will describe the [1,2-b]DHIF derivatives, which have been used as active materials in OFETs (Table 3). As previously presented above for OLED devices, analysis of both the organic semiconductor physicochemical properties and the OFET performances will allow us to draw a structure/properties/device performance relationship map.
| Device | Device architecture | μ | I on/Ioff | V T (V) | Ref. |
|---|---|---|---|---|---|
| Bis(triméthylsilyl)amine (HMDS); n-Octadecyltrimethoxysilane (OTMS); polystyrene (PS); photosensible resin (SU-8); octadecyltrichlorosilane (OTS). | |||||
| OFET1 | Top-contact/bottom gate device | p-type transistor | — | −55 | 83 |
| Si/SiO2/HMDS/[1,2-b]-1/Au | μ e: 0.012 cm2 V−1 s−1 | ||||
| OFET2 | SCO/metal/SCO permeable-base transistor | p-Type transistor | — | — | 84 |
| GaIn/p-Si/Sn/[1,2-b]-2/PEDOT:PSS/Al | |||||
| OTFT transistor based on [1,2-b]-2 | Weak n-type characteristic | — | — | 55 | |
| SCO/metal/SCO permeable-base transistor | Permeable-base transistors with low operating voltage | — | — | 55 | |
| GaIn/p-Si/Sn/[1,2-b]-2/Au | Instability | ||||
| Vertical permeable-base hybrid transistor based on a multilayer metal base | Common-emitter current gain of 2, independent of collector-emitter voltage and base current, stable device | — | — | 85 | |
| OFET3 | Bottom-contact structure | No-gate effect | — | — | 86 |
| Si/SiO2/HMDS/[1,2-b]IF(O)2)/Au |
|||||
| OFET4 | Top-contact/bottom gate device | No-gate effect | — | — | 87 |
| p+-Si/SiO2/[1,2-b]-16/Au | |||||
| OFET5 | Top-contact/bottom gate device in glove box | No gate effect whatever the temperature | — | — | 88 |
| p-Si/SiO2/OTMS/[1,2-b]-17/Au | |||||
| OFET6 | Bottom-contact structure | n-type μFET | 2 × 107 | 69 | 86 |
| Si/SiO2/HMDS/[1,2-b]-18/Au | μ e: 0.17 cm2 V−1 s−1 | ||||
| OFET7 | Top-contact structure | n-type μFET | 2 × 104 | 75 | 86 |
| Si/SiO2/HMDS/[1,2-b]-18/Au | μ e: 0.06 cm2 V−1 s−1 | ||||
| OFET8 | Bottom-contact structure | n-type μFET | 6 × 103 | 88 | 86 |
| Si/SiO2/[1,2-b]-18/Au | μ e: 1.9 × 10−4 cm2 V−1 s−1 | ||||
| OFET9 | Top-contact/bottom gate device | n-type μFET | 6.9 × 105 | 31.7 | 89 |
| n-Si/SiO2/PS/[1,2-b]-18/Al/LiF | μ e: 0.07 cm2 V−1 s−1 | ||||
| OFET10 | Top-contact/bottom gate device | n-type μFET | 1.2 × 105 | 32.1 | 89 |
| n-Si/SiO2/PS/[1,2-b]-18/Au | μ e: 0.14 cm2 V−1 s−1 | ||||
| OFET11 | Bottom -contact structure | n-type μFET | 1 × 107 | 56 | 86 |
| Si/SiO2/HMDS/[1,2-b]-19/Au | μ e: 0.018 cm2 V−1 s−1 | ||||
| OFET12 | Bottom -contact structure | n-type μFET | 4 × 106 | 61 | 86 |
| Si/SiO2/HMDS/[1,2-b]-20/Au | μ e: 9.9 × 10−3 cm2 V−1 s−1 | ||||
| OFET13 | Top-contact/bottom gate device | n-type μFET | 4.3 × 105 | 26.1 | 89 |
| n-Si/SiO2/PS/[1,2-b]-21/Al/LiF | μ e: 0.05 cm2 V−1 s−1 | ||||
| OFET14 | Top-contact/bottom gate device | n-type μFET | 5.4 × 105 | 22.2 | 89 |
| n-Si/SiO2/PS/[1,2-b]-21/Au | μ e: 0.07 cm2 V−1 s−1 | ||||
| OFET15 | Top-contact/bottom gate device | n-type μFET | 1.8 × 105 | 4.7 | 89 |
| n-Si/SiO2/PS/[1,2-b]-22/Al/LiF | μ e: 0.09 cm2 V−1 s−1 | ||||
| OFET16 | Top-contact/bottom gate device | n-type μFET | 6.8 × 105 | 9.2 | 89 |
| n-Si/SiO2/PS/[1,2-b]-22/Au | μ e: 0.16 cm2 V−1 s−1 | ||||
| OFET17 | Bottom gate-top contact OFET | n-type μFET | 5.1 × 104 | -56.8 | 90 |
| n-Si/SiO2/NH2-alkylsilane/[1,2-b]-22/Au | μ e: 0.05 cm2 V−1 s−1 | ||||
| OFET18 | Bottom gate-top contact OFET | n-type μFET | 4.4 × 105 | -15.5 | 90 |
| n-Si/SiO2/PS/[1,2-b]-22/Au | μ e: 0.18 cm2 V−1 s−1 | ||||
| OFET19 | Bottom gate-top contact OFET | n-type μFET | 5.8 × 104 | 16.2 | 90 |
| n-Si/SiO2/ CH3-alkylsilane /[1,2-b]-22/Au | μ e: 0.02 cm2 V−1 s−1 | ||||
| OFET20 | Bottom gate-top contact OFET | n-type μFET | 0.9 × 103 | 9.2 | 90 |
| n-Si/SiO2/ CF3-alkylsilane /[1,2-b]-22/Au | μ e: 3.7 × 10−4 cm2 V−1 s−1 | ||||
| OFET21 | Top-contact structure | n-type μFET (vacuum) | 105 | 48 | 91 |
| n+-Si/SiO2/[1,2-b]-23/Au | μ e: 2.93 × 10−5 cm2 V−1 s−1 | ||||
| OFET22 | Top-contact structure | n-type μFET (Air) | 103 | 35 | 91 |
| n+-Si/SiO2/[1,2-b]-23/Au | μ e: 6.08 × 10−6 cm2 V−1 s−1 | ||||
| OFET23 | Top-contact/bottom gate device | Bipolar n, p | 87 | ||
| Thermal evaporation (70 °C) | μ e: 2 × 10−3 cm2 V−1 s−1 | 105 | 45 | ||
| p+-Si/SiO2/[1,2-b]-24/Au | μ h: 2 × 10−4 cm2 V−1 s−1 | — | −50 | ||
| OFET24 | Top-contact/bottom gate device | Bipolar n, p | 87 | ||
| Thermal evaporation (110 °C) | μ e: 0.01 cm2 V−1 s−1 | 106 | 60 | ||
| p+-Si/SiO2/[1,2-b]-24/Au | μ h: 6 × 10−4 cm2 V−1 s−1 | 107 | −40 | ||
| OFET25 | Top-contact/bottom gate device | μ h: 10−3 cm2 V−1 s−1 | 104 | −42 | 87 |
| Solution-processed | |||||
| p+-Si/SiO2/[1,2-b]-24/Au | |||||
| OFET26 | Top-contact/bottom gate device | Ambipolar n, p | 104 | 50 | 87 |
| Thermal evaporation (70 °C) | μ e: 2 × 10−3 cm2 V−1 s−1 | ||||
| p+-Si/SiO2/[1,2-b]-25/Au | μ h: 2 × 10−3 cm2 V−1 s−1 | — | −32 | ||
| OFET27 | Top-contact/bottom gate device | Ambipolar n, p | 87 | ||
| Thermal evaporation (110 °C) | μ e: 6 × 10−3 cm2 V−1 s−1 | 104 | 50 | ||
| p+-Si/SiO2/[1,2-b]-25/Au | μ h: 6 × 10−3 cm2 V−1 s−1 | — | −30 | ||
| OFET28 | Top-contact/bottom gate device | μ e: 2 × 10−4 cm2 V−1 s−1 | 104 | 60 | 87 |
| Solution-processed | |||||
| p+-Si/SiO2/[1,2-b]-25/Au | μ h: 2 × 10−4 cm2 V−1 s−1 | — | −35 | ||
| OFET29 | Top-contact/bottom gate device | Bipolar n-type and p-type | 92 | ||
| Drop-casting process measured under vacuum | μ e: 1.6 × 10−3 cm2 V−1 s−1 | 2.8 × 105 | 53 | ||
| Si/SiO2/PS/[1,2-b]-26/Au | μ h: 8.2 × 10−4 cm2 V−1 s−1 | 1.5 × 102 | −22 | ||
| OFET30 | Top-contact/bottom gate device in vacuum | Bipolar n-type and p-type | 92 | ||
| Droplet-pinned crystallization process | μ e: 7.7 × 10−4 cm2 V−1 s−1 | 3.1 × 102 | 46 | ||
| Si/SiO2/PS/[1,2-b]-26/Au | μ h: 2.5 × 10−4 cm2 V−1 s−1 | 5.3 × 104 | −4 | ||
| OFET31 | Top-contact/bottom gate device in vacuum | Bipolar n-type and p-type | 92 | ||
| Solution-shearing process | μ e: 0.12 cm2 V−1 s−1 | 8.5 × 105 | 57 | ||
| Si/SiO2/PS/[1,2-b]-26/Au | μ h: 0.02 cm2 V−1 s−1 | 2.1 × 102 | −62 | ||
| OFET32 | Top-contact/bottom gate device in air | p-type μFET | 6.3 × 106 | −33 | 92 |
| Drop-casting process | |||||
| Si/SiO2/PS/[1,2-b]-26/Au | μ h: 2.7 × 10−4 cm2 V−1 s−1 | ||||
| OFET33 | Top-contact/bottom gate device in air | p-type μFET | 2.6 × 104 | −38 | 92 |
| Droplet-pinned crystallization process | |||||
| Si/SiO2/PS/[1,2-b]-26/Au | μ h: 1.6 × 10−4 cm2 V−1 s−1 | ||||
| OFET34 | Top-contact/bottom gate device in air | p-type μFET | 4.8 × 102 | 2.8 | 92 |
| Solution-shearing process | |||||
| Si/SiO2/PS/[1,2-b]-26/Au | μ h: 2.2 × 10−3 cm2 V−1 s−1 | ||||
| OFET35 | Top-contact/bottom gate device in vacuum | Bipolar n-type and p-type | 92 | ||
| Drop-casting process | μ e: 1.5 × 10−4 cm2 V−1 s−1 | 6.7 × 105 | 47 | ||
| Si/SiO2/PS/[1,2-b]-27/Au | μ h: 5.3 × 10−7 cm2 V−1 s−1 | 5.9 × 103 | −54 | ||
| OFET36 | Top-contact/bottom gate device in vacuum | Bipolar n-type and p-type | 92 | ||
| Droplet-pinned crystallization process | μ e: 0.013 cm2 V−1 s−1 | 1.2 × 104 | 26 | ||
| Si/SiO2/PS/[1,2-b]-27/Au | μ h: 2.7 × 10−4 cm2 V−1 s−1 | 4.2 × 104 | −35 | ||
| OFET37 | Top-contact/bottom gate device in vacuum | Bipolar n-type and p-type | 92 | ||
| Solution-shearing process | μ e: 0.04 cm2 V−1 s−1 | 2.2 × 105 | 10 | ||
| Si/SiO2/PS/[1,2-b]-27/Au | μ h: 3.3 × 10−4 cm2 V−1 s−1 | 1.2 × 105 | −45 | ||
| OFET38 | Top-contact/bottom gate device in air | p-type μFET | 2 × 105 | −12 | 92 |
| Drop-casting process | |||||
| Si/SiO2/PS/[1,2-b]-27/Au | μ h: 2.9 × 10−4 cm2 V−1 s−1 | ||||
| OFET39 | Top-contact/bottom gate device in air | p-type μFET | 8.1 × 102 | −14 | 92 |
| Droplet-pinned crystallization process | |||||
| Si/SiO2/PS/[1,2-b]-27/Au | μ h: 8.0 × 10−4 cm2 V−1 s−1 | ||||
| OFET40 | Top-contact/bottom gate device in air | p-type μFET | 5.2 × 104 | −9.7 | 92 |
| Drop-casting process | |||||
| Si/SiO2/PS/[1,2-b]-27/Au | μ h: 5.4 × 10−4 cm2 V−1 s−1 | ||||
| OFET41 | Top-contact/bottom gate device | μ h: 10−4 cm2 V−1 s−1 | 104 | −30 | 87 |
| Solution-processed | |||||
| p+-Si/SiO2/[1,2-b]-28/Au | |||||
| OFET42 | Top-contact/bottom gate device | Ambipolar n-type and p-type | 1.4 × 105 | 34 | 93 |
| Solution-processed measured under vacuum | μ e: 0.02 cm2 V−1 s−1 | ||||
| p+-Si/SiO2/[1,2-b]-29/Au | μ h: 0.01 cm2 V−1 s−1 | 2.2 × 106 | −55 | ||
| OFET43 | Top-contact/bottom gate device in glove box at RT | Ambipolar n-type and p-type | 88 | ||
| μ e: 0.05 cm2 V−1 s−1 | 104 | 62 | |||
| p-Si/SiO2/OTMS/[1,2-b]-30/Au | μ h: 0.14 cm2 V−1 s−1 | 106 | −36 | ||
| OFET44 | Top-contact/bottom gate device in glove box at 60 °C | Ambipolar n-type and p-type | 88 | ||
| μ e: 0.65 cm2 V−1 s−1 | 104 | 41 | |||
| p-Si/SiO2/OTMS/[1,2-b]-30/Au | μ h: 0.71 cm2 V−1 s−1 | 105 | −48 | ||
| OFET45 | Top-contact/bottom gate device in glove box at 100 °C | Ambipolar n-type and p-type | 88 | ||
| μ e: 0.14 cm2 V−1 s−1 | 105 | 60 | |||
| p-Si/SiO2/OTMS/[1,2-b]-30/Au | μ h: 0.24 cm2 V−1 s−1 | 104 | −32 | ||
| OFET46 | Top-contact/bottom gate device in glove box at RT | p-type μFET | 106 | −19 | 88 |
| p-Si/SiO2/OTMS/[1,2-b]-31/Au | μ h: 0.19 cm2 V−1 s−1 | ||||
| OFET47 | Top-contact/bottom gate device in glove box at 60 °C | p-type μFET | 105 | −27 | 88 |
| p-Si/SiO2/OTMS/[1,2-b]-31/Au | μ h: 1.03 cm2 V−1 s−1 | ||||
| OFET48 | Top-contact/bottom gate device in glove box at 100 °C | p-type μFET | 106 | −14 | 88 |
| p-Si/SiO2/OTMS/[1,2-b]-31/Au | μ h: 0.22 cm2 V−1 s−1 | ||||
| OFET49 | Space-charge-limited current (SCLC) | μ h: 1.41 × 10−4 cm2 V−1 s−1 | 97 | ||
| ITO/PEDOT:PSS/[1,2-b]-32/MoO3 (10 nm)/Al (100 nm) | |||||
| OFET50 | Space-charge-limited current (SCLC) | μ h: 8.40 × 10−5 cm2 V−1 s−1 | 97 | ||
| ITO/PEDOT:PSS/[1,2-b]-33/MoO3 (10 nm)/Al (100 nm) | |||||
| OFET51 | Space-charge-limited current (SCLC) | μ h: 1.96 × 10−5 cm2 V−1 s−1 | 97 | ||
| ITO/PEDOT:PSS/[1,2-b]-34/MoO3 (10 nm)/Al (100 nm) | |||||
| OFET52 | Top-contact/bottom gate device | No-gate effect | — | — | 87 |
| p+-Si/SiO2/[1,2-b]-35/Au | |||||
| OFET53 | Top-contact/bottom gate device | n-type air stable | 104 | 10 | 87 |
| Thermal evaporation (70 °C) | |||||
| p+-Si/SiO2/[1,2-b]-36/Au | μ e: 4 × 10−4 cm2 V−1 s−1 | ||||
| OFET54 | Top-contact/bottom gate device | n-type air stable | 105 | 3 | 87 |
| Thermal evaporation (110 °C) | |||||
| p+-Si/SiO2/[1,2-b]-36/Au | μ e: 10−3 cm2 V−1 s−1 | ||||
| OFET55 | Top-contact/bottom gate device | n-type air stable | 104 | 7 | 87 |
| Solution-processed | |||||
| p+-Si/SiO2/[1,2-b]-36/Au | μ e: 10−4 cm2 V−1 s−1 | ||||
| OFET56 | Top-contact/bottom gate device | n-type air stable | 106 | 2 | 87 |
| Thermal evaporation (70 °C) | |||||
| p+-Si/SiO2/[1,2-b]-37/Au | μ e: 0.03 cm2 V−1 s−1 | ||||
| OFET57 | Top-contact/bottom gate device | n-type air stable | 107 | 5 | 87 |
| Thermal evaporation (110 °C) | |||||
| p+-Si/SiO2/[1,2-b]-37/Au | μ e: 0.16 cm2 V−1 s−1 | ||||
| OFET58 | Top-contact/bottom gate device | n-type air stable | 108 | 5 | 87 |
| Solution-processed | |||||
| p+-Si/SiO2/[1,2-b]-37/Au | μ e: 0.16 cm2 V−1 s−1 | ||||
| OFET59 | Top-contact/bottom gate device | Bipolar n-type and p-type air stable | 105 | 20 | 87 |
| Solution-processed | μ e: 10−3 cm2 V−1 s−1 | ||||
| p+-Si/SiO2/[1,2-b]-38Au | μ h: 10−4 cm2 V−1 s−1 | — | −25 | ||
| OFET60 | Top-contact/bottom gate device | Bipolar n-type and p-type air stable | 93 | ||
| Solution-processed measured in air | μ e: 0.13 cm2 V−1 s−1 | 104 | 13 | ||
| p+-Si/SiO2/[1,2-b]-39/Au | μ h: 0.01 cm2 V−1 s−1 | 1.1 × 103 | −33 | ||
| OFET61 | Bottom-Gate/bottom-contact device in glove box under ambient nitrogen | μ FET Linear: 0.005 cm2 V−1 s−1, SS: 1.3 V dec−1 | 4 × 106 | 26 | 99 |
| Al/SU-8/Au/[1,2-b]-40 | μ FET saturation: 0.02 cm2 V−1 s−1, SS: 1.9 V dec−1 | 5 × 105 | 20 | ||
| OFET62 | Top-contact/bottom gate device | n-type μFET | 107–108 | 2 | 94 |
| Solution processable | |||||
| [1,2-b]-41/PS/ SiO2/n++-Si | μ e: 0.02 cm2 V−1 s−1 | ||||
| OFET63 | Top-contact/bottom gate device-Thermal evaporation | p-OFET (single crystal) | 103 | — | 104 |
| OTS-Si/SiO2/[1,2-b]-42/Au | μ h: 1.44 cm2 V−1 s−1 | ||||
In 2006, a transistor based on [1,2-b]-1 was reported.83[1,2-b]-1 was used as the organic semiconductor in the top-contact/bottom-gate device exhibiting p-type transport with a high μh of 0.012 cm2 V−1 s−1 and a threshold of −55 V (OFET1).
The same year, [1,2-b]-2 was used in semiconductor/metal/semiconductor (SMS) transistors (a thin metal base located between two semiconductor layers, [1,2-b]-2 and the collector). In this work,84 it was shown that hybrid permeable-base transistors, operating with positive charges as the majority carriers can be obtained and possess a nearly ideal base transport factor. In 2007, a thin-film transistor (TFT) and SMS were fabricated and characterized by the same group.55[1,2-b]-2 showed a weak n-type character from the TFT measurement, while SMS transistors using [1,2-b]-2 behave like permeable-base transistors with low operating voltages in both common-base and common-emitter modes and a feature of current amplifications.
Finally, in 2009, the same group presented a permeable-base transistor consisting of [1,2-b]-2 with a Ca/Al/Ca multilayer as the metal base and p-Si as the collector.85 Playing with the thickness of Al, they have shown that the device presents common-base current gain and common-emitter current gain. This last gain of around 2 was independent of collector-emitter voltage and base current. All these first experiments showed the possibility of using [1,2-b]-2 in different types of transistors and may be seen as a proof of concept.
In 2008, OFET3 based on [1,2-b]IF(O)237 was fabricated on SiO2/Si substrates by thermal evaporation (SiO2 gate dielectric was treated with hexamethyldisilane (HMDS)) in bottom-contact geometry.86 However, these OFETs indicated a no-gate effect.
Then, differently substituted [1,2-b]IF(O)2 were synthesized and used in different OFETs. To improve the processability of [1,2-b]IF(O)2, two dodecyl chains were first positioned on the central phenyl unit of [1,2-b]-16. Despite this, OFET4, based on a top-contact/bottom-gate device, showed no gate effect.87 Similarly, the substitution of the C2 and C8 atoms of [1,2-b]IF(O)2 by two n-butyl chains in [1,2-b]-17 was tested88 but no gate effect was observed in OFET5.
In 2008, different [1,2-b]IF(O)2 substituted with two halogen atoms at C2 and C8: fluorine in [1,2-b]-18, chloride in [1,2-b]-19 and bromide in [1,2-b]-2086 were synthesized using the adapted method of Wang et al.37 The idea was to incorporate electron-withdrawing fragments to depress the LUMO energy levels of the corresponding organic semiconductors. OFET devices (OFET6-OFET12) based on these organic semiconductors were fabricated on SiO2/Si substrates by thermal evaporation (the SiO2 gate dielectric was treated with HMDS) with a bottom-contact geometry. As reported above, OFET3 based on [1,2-b]IF(O)2 indicated no-gate effect however those based on [1,2-b]-18 to [1,2-b]-20 successfully showed n-type OFET behaviour increasing from [1,2-b]-20 (OFET12: 9.9 × 10−3 cm2 V−1 s−1, Ion/Ioff: 4 106, +61 V), to [1,2-b]-19 (OFET11: 1.8 × 10−2 cm2 V−1 s−1, Ion/Ioff: 107, +56 V) and to [1,2-b]-18 (OFET6: 0.17 cm2 V−1 s−1, Ion/Ioff: 2 107, +69 V). This work showed that the two terminal halogen groups induced n-type FET behaviour and that the mobility was dependent on the halogen size. The high mobility obtained with OFET6, 0.17 cm2 V−1 s−1, revealed the potential of [1,2-b]-18 as an n-type material.
The top-contact device using [1,2-b]-18, OFET7, was also constructed and showed lower FET mobility (OFET7, μe: 6.6 × 10−2 cm2 V−1 s−1) than the bottom-contact one (OFET6, μe: 0.17 cm2 V−1 s−1)). The treatment of SiO2 by HMDS or by polystyrene (PS) was also highlighted for the bottom-contact device using [1,2-b]-18. Without HMDS or PS (OFET886), the n-type μe was measured at 1.9 × 10−4 cm2 V−1 s−1 and was significantly increased to 0.07 or 0.17 cm2 V−1 s−1 with PS89 (OFET9) or HMDS86 (OFET6) treatment.
In 2011, a novel series of diones substituted on their external phenyls by two (in [1,2-b]-21) or three (in [1,2-b]-22) fluorine atoms was synthesized and used in n-type OFETs (OFET13-OFET20).89 Their properties and efficiencies in OFET were compared to that of [1,2-b]-18, presented above.86 The LUMO energy levels obtained by electrochemical studies were lowered from −3.38 eV for [1,2-b]-18 to −3.53 eV for [1,2-b]-22 showing a decrease in the LUMO in relation to the increase of the number of fluorine atoms on the [1,2-b]DHIF core. Depressing the LUMO energy level is, in this field, an important feature notably to reach air stable OFETs. [1,2-b]-21 and [1,2-b]-22 present similar UV-vis absorption spectra slightly red-shifted compared to [1,2-b]-18.
Top-contact OFETs based on [1,2-b]-21 and [1,2-b]-22 were fabricated on heavily doped n-type Si substrates (devices OFET13-OFET16) and compared to those based on [1,2-b]-18 (OFET9-OFET10). All devices were found to be very efficient as n-type transistors with Ion/Ioff exceeding 105 and μe measured between 0.05 and 0.16 cm2 V−1 s−1 depending on the organic semiconductor and on the nature of the source drain contacts (LiF/Al or Au). These results showed that [1,2-b]indenofluorenones with a different number of fluorine atoms ([1,2-b]-18, [1,2-b]-21 and [1,2-b]-22) are excellent transporters in n-type OFETs. The mobility was increasing consistently with the number of fluorine atoms: 2 in [1,2-b]-18, 4 in [1,2-b]-21 and 6 in [1,2-b]-22, following the decrease in LUMO energy levels (−3.38/−3.43/−3.52 eV for [1,2-b]-18/[1,2-b]-21/[1,2-b]-22) showing the efficiency of the strategy.
In 2012, the electrical performance and stability of n-type OFETs based on [1,2-b]-22 were more deeply investigated.90 Different organic interlayers such as self-assembled monolayers (NH2 in OFET17, CH3 in OFET19 or CF3 in OFET20) and polymeric layers (PS in OFET18) were introduced at the [1,2-b]-22-SiO2 interfaces to control the interfacial properties. Bottom gate-top contact OFET18 showed an excellent device performance with μe of 0.18 cm2 V−1 s−1 and Ion/Ioff of 4.4 × 105. Moreover, OFET18 yielded excellent electrical stability allowing us to fabricate a complementary inverter fabricated using both p-type pentacene and n-type [1,2-b]-22.90
In 2007, a highly halogenated derivative, [1,2-b]-2391 bearing eight fluorine atoms on the external phenyl and two iodine atoms on the central phenyl of the [1,2-b]indenofluorenone core was synthesized. Two top-contact OFET using [1,2-b]-23 on a Si/SiO2 substrate were prepared by vacuum deposition and tested either under vacuum (OFET21) or under air (OFET22). The device performances were low, with μ < 3 × 10−5 cm2 V−1 s−1 and high threshold potentials. Compared to the performances reported above with [1,2-b]-18 (OFET7 and OFET10), with [1,2-b]-21 (OFET14) or [1,2-b]-22 (OFET16–20), the low efficiency of OFET21 may be due to the poor film morphology and to the nature of the semiconductor/SiO2 interface.
Elongation of the [1,2-b]indenofluorenone core has also been an investigated strategy to achieve efficient OFET devices (see [1,2-b]-24 to [1,2-b]-31 in Scheme 11).87,88,92–94[1,2-b]-24 and [1,2-b]-25 were substituted at C2 and C8 positions by two thienyl units whereas [1,2-b]-28 possessed two dithienyl units at these positions. Moreover, in order to increase the processability of these organic semiconductors, solubilising groups were added to the external thienyl units in [1,2-b]-26, [1,2-b]-27 and [1,2-b]-29. Different devices, OFET23-OFET42, were constructed with [1,2-b]-24 - [1,2-b]-29.87,88,92,93 With similar top-contact/bottom-gate architecture but varying their modes of fabrication (thermal evaporation at 70 or 110 °C, drop-casting or droplet pinned crystallization in vacuum or in air) a lot of different performances were reported. The devices based on [1,2-b]-25 (OFET26-OFET28) have shown interesting ambipolar properties with μh equal to μe values (μe = μh = 2 × 10−3/6 × 10−3/2 × 10−4 cm2 V−1 s−1 for OFET26/27/28). Other devices have also shown bipolar properties (OFET29-OFET31, OFET35–37, OFET42) and the highest mobilities were obtained for [1,2-b]-26 (μe: 0.12 cm2 V−1 s−1 and μh: 0.02 cm2 V−1 s−1) in the solution processed OFET31. [1,2-b]-24 and [1,2-b]-27 showed n-type and p-type OFET behaviour with however μe largely higher than μh (OFET23-OFET24 and OFET35-OFET37). From all the devices, those measured in air (OFET32–34 with [1,2-b]-26 and OFET38–40 with [1,2-b]-27) present only a p-type character indicating the instability of the electron-transport in this organic semiconductor due to their too high LUMO value (−3.65 eV for [1,2-b]-26 and −3.63 eV for [1,2-b]-27) (ambient-stable electron-transport is possible only with organic semiconductor possessing a LUMO lower than −4.1 eV87,95).
The use of alkylthio side chains in [1,2-b]-30 or of dibutylamino side chains in [1,2-b]-31 has been tested in order to modulate both the processability and the electronic properties.88[1,2-b]-30 and [1,2-b]-31 were obtained from their respective diketones.88 Compared with [1,2-b]-17, substituted by n-butyl units (HOMO/LUMO: −6.0/−3.5 eV), it was observed that the sulfur linkage, in [1,2-b]-30, lowers the LUMO to −3.7 eV and raises the HOMO to −5.6 eV (the SBu groups are weakly electron-donating meanwhile the sulfur atom has some π-acceptor capability due to the formation of the pπ(C)-dπ(S) orbital overlap96). For [1,2-b]-31, the introduction of the dibutylamino groups results in an increase of the HOMO energy level at −5.0 eV due the strong electron-donating effect of the nitrogen atoms and to a small rise of the LUMO at −3.4 eV. X-Ray analysis has shown that (a) [1,2-b]-17 exhibited one-dimensional column stacking, (b) [1,2-b]-30 exhibited two-dimensional brick wall stacking and (c) [1,2-b]-31 adopted a two-dimensional crossed stacking. Short π–π distances of 3.45, 3.22 and 3.39 Å between the [1,2-b]-indenofluorenone planes were measured in [1,2-b]-17, [1,2-b]-30 and [1,2-b]-31, respectively. This variation in the π–π distances demonstrates that the different side chains induce different molecular packing in the crystals, which in turn modifies the charge transfer.
To assess the charge transport properties of the films for the three semiconductors, the bottom-gate/top-contact OFET devices were produced and studied at different temperatures (RT, 60 °C or 100 °C). Devices based on [1,2-b]-30 (OFET43–45) have shown the bipolar character of this organic semiconductor (μe/μh = 0.05/0.14, 0.65/0.71 and 0.14/0.24 cm2 V−1 s−1 for OFET43/44/45) whereas the ones based on [1,2-b]-31 (OFET46–48) have shown only p-type OFET characteristics (μh = 0.18, 1.03 and 0.22 cm2 V−1 s−1 for OFET46, OFET47 and OFET48). All the mobilities obtained were very high. It must be recalled that OFET5 based on [1,2-b]-17 presents no gate effect due to the large injection barrier and the 1D molecular packing. Oppositely, [1,2-b]-30 has a more appropriate HOMO level due to the alkylthio side chain and also 2D molecular stacking, which provides favourable charge transport channels for hole and electrons. Finally, with [1,2-b]-31 the 2D molecular stacking was also favourable for the charge transport however, the LUMO of [1,2-b]-31 (−3.4 eV) was similar to that of [1,2-b]-17 (−3.5 eV) and electron injection was rather difficult only leading to a p-type transport.
In conclusion, for [1,2-b]-17 with a large injection barrier (HOMO/LUMO: −6.0/−3.5 eV) and a one-dimensional packing, no gate effect was observed (see OFET5). [1,2-b]-30 with appropriate energy levels (HOMO/LUMO: −5.6/−3.7 eV) due to the sulphur linkage and a two-dimensional packing, exhibited high and balanced ambipolar transport behaviour. Finally, the nitrogen linkages in [1,2-b]-31 significantly raised the HOMO level (−5.0 eV) but the LUMO level (−3.4 eV) appeared too high for electron injection. OFET46–48 exhibited high μh but no electron transport properties.
In 2019, [1,2-b]-32-[1,2-b]-34 were synthesized (Scheme 12).97 They were all prepared from the 6,6,12,12-tetrahexyl-6,12-dihydroindeno[1,2-b]fluorene 40, which was di-brominated at C2 and C8 with benzyltrimethylammonium tribromide (BTMABr3) or tetrabrominated at C2, C3, C8 and C9 with Br2 leading to the intermediates 41 and 42. The last step was a classical Buchwald–Hartwig C–N coupling reaction between 41 and/or 42 and the corresponding amines (43 or 44) to reach [1,2-b]-32–[1,2-b]-34 (see Scheme 12). These three organic semiconductors were used as the Hole Transport Material (HTM) in Perovskite Solar Cells (PSC) and the performance of these devices will be discussed below in section 2.4. As will be shown, devices with [1,2-b]-32 as the HTM are more efficient than those based on [1,2-b]-33 and [1,2-b]-34. In order to understand the different performances, μh of the three organic semiconductors have been investigated by using space-charge-limited current (SCLC) techniques (OFET49-OFET51). Although the three organic semiconductors are constructed on the same central core (tetrahexyl-[1,2-b]DHIF), they exhibit different μh of 1.41 × 10−4, 8.4 × 10−5 and 1.96 × 10−5 cm2 V−1 s−1 for [1,2-b]-32, [1,2-b]-33 and [1,2-b]-34 respectively. The enhanced μh for [1,2-b]-32 was attributed by the authors to the introduction of the spirofluorene-xanthenyl unit with a 3D electronic structure, which can improve intermolecular interactions to some extent and increase the transporting channels in spite of causing partial twisting of the whole molecule. For [1,2-b]-34, the introduction of four diphenylamine groups on the [1,2-b]DHIF core can reduce the planarity and disturb intermolecular packing, thus leading to a decrease in intermolecular charge carrier hopping, which in turn decreases the μh value as well (8.4 × 10−5 and 1.96 × 10−5 cm2 V−1 s−1 for [1,2-b]-33 and [1,2-b]-34 respectively).
 | ||
| Scheme 12 Synthesis of [1,2-b]-32, [1,2-b]-33 and [1,2-b]-34. (a) ZnCl2, AcOH, 70 °C; (b) CHCl3, FeCl3, 0 °C; (c) P(tBu)3, Pd(OAc)2, sodium tert-butoxide, toluene, 110 °C. | ||
In order to depress the LUMO energy levels and to increase the stability in air of their related n-type OFETs, organic semiconductors based on the electron-deficient [1,2-b]-indenofluorenebis(dicyanovinylene) ([1,2-b]DHIF(C(CN)2)2) have then been investigated (see [1,2-b]-35 to [1,2-b]-41 in Scheme 11). These molecules were obtained by Knoevenagel condensation of their related diones with malononitrile.87,98 Our group also designed in 2018 another organic semiconductor, [1,2-b]-40,99 also based on the electron-deficient [1,2-b]DHIF(C(CN)2)2) core with two alkyl units (n-C5H11) substituting the external phenyl rings (instead of two n-C12H25 groups in the central phenyl ring in [1,2-b]-35).99 Highly soluble [1,2-b]-41, also based on the electron-deficient [1,2-b]DHIF(C(CN)2)2) core bearing two triisopropylsilyl-ethynyl endgroups was prepared by the Kim and Usta group.94
The main electronic properties modification of [1,2-b]-35 to [1,2-b]-39 compare to their related diones ([1,2-b]-16, [1,2-b]-24-25 and [1,2-b]-28-29) concerns their LUMO energy levels that decreased by ∼0.63 eV (when their HOMO levels increase by ∼0.15 eV). Due to these HOMO/LUMO variations, the gap of [1,2-b]-35 to [1,2-b]-39 was reduced from 0.25 to 0.38 eV compared to that of their related diones. Compared to [1,2-b]-35, in which the LUMO was measured at −4.30 eV,87 our group measured the LUMO of [1,2-b]-40 at the very low energy of −4.1 eV.99 The dicyanovinylene fragment appeared then as a very efficient electron-withdrawing group to strongly depress the LUMO energy level. The dicyanovinylene is currently a widely used fragment in efficient n-type OFET.78,99–103
Top contact FETs (OFET52–60) were fabricated by either vacuum deposition or spin-coating on octadecyltrichlorosilane (OTS)-treated SiO2/p+-Si substrates. No-gate effect was observed for OFET52 based on [1,2-b]-35.
The top-contact devices based on [1,2-b]-36 (OFET53–55) and [1,2-b]-37 (OFET56–58) have shown n-type air stable activity with μe around 10−4/10−3 cm2 V−1 s−1 for [1,2-b]-36 and μe around 0.03/0.16 cm2 V−1 s−1 for [1,2-b]-37. Finally, [1,2-b]-38 in OFET59 and [1,2-b]-39 in OFET60 have also shown bipolar properties with μe (10−3 cm2 V−1 s−1) equal to 10 × μh for [1,2-b]-38 and μe (0.13 cm2 V−1 s−1) equal to 13 × μh for [1,2-b]-39.
A comparison of these performances with those of the related dione-based devices indicates that for [1,2-b]-36, OFET53–55, despite the OFETs being air-stable, the μe was lower than that for [1,2-b]-24 in OFET23–25 (see Table 3).
Oppositely, the comparison of devices based on [1,2-b]-37 (OFET56-OFET58) to those based on [1,2-b]-25 (OFET26-OFET28), shows n-type air-stable devices with the former and a μe increasing from 2 × 10−3 cm2 V−1 s−1 in OFET26 to 0.03 cm2 V−1 s−1 in OFET56, from 6 × 10−3 cm2 V−1 s−1 in OFET27 to 0.16 cm2 V−1 s−1 in OFET57 and from 2 × 10−4 cm2 V−1 s−1 in OFET28 to 0.16 cm2 V−1 s−1 in OFET58, indicating the positive effect of the presence of the dicyanovinylene units in [1,2-b]-37.
It must also be noted that OFET56–58 exhibits very low threshold voltages (around 2 to 5 V) in accordance, among other parameters (morphological or device mode preparation), to its very low LUMO of 4.32 eV.87
The comparison between OFET59 and OFET41, highlights the similarity of μh in both devices (10−4 cm2 V−1 s−1). However, due to the presence of the dicyanovinylene units of [1,2-b]-38, a μe value of 10−3 cm2 V−1 s−1 was measured in the air stable OFET59. Finally, a similar comparison between OFET60 and OFET42, also points to the same μh value in both devices (10−2 cm2 V−1 s−1). However, the presence of the dicyanovinylene units in [1,2-b]-40, increases the μe value to 0.13 cm2 V−1 s−1 which was also measured in the air stable OFET60.
Bottom-gate bottom-contact n-type channel OFETs were also constructed with [1,2-b]-40 as the active layer.99 Contrary to OFET 3 based on [1,2-b]IF(O)2, which demonstrated no-gate effect, an electron mobility of 0.02 cm2 V−1 s−1 was measured in OFET61. The performance of OFET61 remains lower than those of OFET6 (μe: 0.17 cm2 V−1 s−1) based on the difluoro [1,2-b]-18, and higher than that of OFET11 and OFET12 based on the dichloro [1,2-b]-19 or dibromo [1,2-b]-20. This shows how rational design is complicated to establish to reach high performances.
The solution processable OFET62 based on [1,2-b]-41 has shown μe as high as 0.02 cm2 V−1 s−1 with a very high Ion/Ioff ratio of 107–108 and a low threshold voltage of around 2 V under ambient atmosphere.94 These results demonstrate that (triisopropylsilyl)ethynyl substitution is a possible approach to realize new families of solution-processable organic semiconductors.
Finally, OFET63 is a p-type OFET based on the indenofluorene-extended tetrathiafulvalene, [1,2-b]-42 (HOMO/LUMO: −4.64/−2.09 eV).105 OFET63 was constructed using a single crystal of [1,2-b]-42 and carrier mobility was demonstrated to vary with the α- or β-phase of the crystal. With the α-phase [1,2-b]-42 single crystal, the hole mobility, μh, reached the very high value of 1.44 cm2 V−1 s−1 with an Ion/Ioff ratio of 103.104
2.4. [1,2-b]DHIF-based organic semiconductors used as the host for phosphorescent OLEDs
At the end of the twentieth century, the development of phosphorescent organic light-emitting devices (PhOLEDs) attracted a great deal of attention since Baldo, O’Brien, and Forrest106 announced the possibility of harvesting all the singlet and triplet excitons generated in an appropriate organic semiconductor matrix via a heavy metal-containing complex dispersed as the dopant in this organic semiconductor. The design of organic host materials possessing specific properties and notably a triplet energy value higher than that of the phosphorescent emitters then started to be significantly developed and has been the driving force of the field.Different design tactics were studied to gather within a host material all the necessary properties to fit with a phosphorescent emitter in a PhOLED. The ideal host should possess:
(i) a triplet energy (ET1) above that of the phosphor used (ca. 2.2 eV for red, 2.5 eV for green, and 2.7 eV for blue phosphors) to confine the triplet excitons within the phosphorescent guest,
(ii) HOMO/ LUMO energy level fitting with those of the phosphor and with the Fermi levels of the electrodes (or of the adjacent transporting layers) to ensure efficient charge injection,
(iii) good and equilibrated μh and μe values for efficient charge transport and recombination,
(iv) thermal and morphological stabilities to reach a stable electroluminescence and extend the lifetime of the device.
The [1,2-b]DHIF core has been used to construct a high ET1 host material for PhOLEDs. Due to the presence of three phenyl rings, reaching high ET1 values was nevertheless a difficult task (see below). Indeed, high ET1 materials are usually obtained with small π-conjugated units with restricted conjugation.73,107–116
The first organic semiconductor constructed on the [1,2-b]DHIF core used as the host for a phosphorescent emitter was [1,2-b]-43117 (Fig. 6). The [1,2-b]DHIF core was built from a SBF fragment 45 in which was bridged an additional phenyl ring via an intramolecular electrophilic substitution (Scheme 13).
 | ||
| Fig. 6 [1,2-b]DHIF-based organic semiconductors used as the host for phosphorescent dopants in PhOLEDs ([1,2-b]-43 and [1,2-b]-DSF-IF) and green phosphorescent Ir(ppy)2(acac) and Ir(ppy)3. | ||
 | ||
| Scheme 13 Synthesis of [1,2-b]-43. (a) Pd(PPh3)4, PtBu3, K3PO4, toluene, reflux, (b)THF, −78 °C, and (c) HOAc/H2SO4. | ||
Due to the substituents attached on the two bridges, the Td of [1,2-b]-43 was higher (354 °C) than that of [1,2-b]DHIF (200 °C). [1,2-b]-43 presented a Tg of 171 °C suggesting good morphological stability as an active material in a device. The HOMO of [1,2-b]-43 was measured at −5.83 eV, similar to that of [1,2-b]DHIF (−5.81 eV) showing that the first oxidation occurs on the [1,2-b]DHIF core for both compounds. The optical gap was measured at 3.49 eV (in CHCl3) for [1,2-b]-43 slightly more contracted than for [1,2-b]DHIF (3.61 eV in CH2Cl2).
E T1 of [1,2-b]-43 (2.52 eV) was estimated from the first phosphorescent peak (492 nm) measured at 77 K in ethanol. This value was close to that of the parent para-terphenyl (ET1: 2.55 eV)24 and higher than that of the green emitter Ir(ppy)2(acac) (ET1: 2.41 eV).118 In [1,2-b]-43, the ET1 is driven by the [1,2-b]DHIF core but remains relatively high for an extended π-conjugated fragment. Thus, these works have revealed that the bridges (as in [1,2-b]-43 and [1,2-b]DHIF) and the planification induced, do not significantly modify the ET1 of these compounds compared to the para-terphenyl parent.24,27 This was an important finding, which has driven the molecular engineering works of the triplet state of this fragment.24,27,119 Interestingly, μh of [1,2-b]-43 has been measured at a relatively high value for a spiro compound, from 3.9 × 10−3 to 5.6 × 10−3 cm2 V−1 s−1. This value of μh was higher than that measured for [1,2-b]-4 (10−3 cm2 V−1 s−1) and that measured for [1,2-b]-5 (around 10−4 cm2 V−1 s−1).
[1,2-b]-43 was used as the host for the green phosphor Ir(ppy)2(acac) in multi-layer PhosOLED1. With a Von of 2.5 V, an EQEmax of 15.8% (CE: 60 cd A−1, PE: 63 lm W−1) and an EL spectrum in accordance with that of the green dopant without emission of [1,2-b]-43, this first phosphorescent device based on the [1,2-b]DHIF fragment was promising.
In 2015, following a similar strategy, our group showed the efficiency of [1,2-b]-DSF-IF as the host for green PhOLED.24 As the ET1 of [1,2-b]-DSF-IF (2.52 eV24) was similar to that of [1,2-b]-43 (2.52 eV117), it was concluded that ET1 was driven by the [1,2-b]DHIF core and therefore adapted to the Ir(ppy)3 green phosphorescent (ET1: 2.43 eV}). PhosOLED2 emitted light since 3.5 V and reached EQEmax: 9.9% (CEmax: 31.4 cd A−1 and PEmax: 15.5 lm W−1). After this work, modification of the terphenyl linkages appeared to be an efficient strategy to increase the ET1 (Part 4).
2.5. [1,2-b]DHIF-based organic semiconductors used in other electronic devices
The [1,2-b]DHIF core has also been used as the building unit in organic semiconductors for other electronic devices showing the versatility of this fragment. Thus, [1,2-b]-32–[1,2-b]-3497 (Schemes 11 and 14) were used as the HTM in dopant-free perovskite solar cells (PSCs) (Table 5). All devices were prepared by spin-coating processes and the thickness of the HTM layer was optimized to tune the charge transport and device performances. Under the optimized conditions, the PSC1 with [1,2-b]-32 as the HTM exhibited the best performances: a power conversion efficiency (PCE) of 17.6% with an open-circuit voltage (VOC) of 1.05 V, short-circuit current density (JSC) of 21.5 mA cm−2 and fill factor (FF) of 77.3%. The PSC2 device based on [1,2-b]-33 was also efficient (PCE: 14.5%, VOC: 0.96 V, JSC: 20.8 mA cm−2, and FF: 72.5%) contrary to the PSC3 device based on [1,2-b]-34, which exhibited lower values (PCE: 12.7%, VOC: 0.92 V, JSC: 20.8 mA cm−2 and FF: 66.6%). The higher VOC obtained with [1,2-b]-32 as the HTM (compared to devices using [1,2-b]-33 or [1,2-b]-34) was linked to its deeper HOMO energy (HOMO: −5.22, −5.16 and −5.07 eV for [1,2-b]-32, [1,2-b]-33 and [1,2-b]-34 respectively). Moreover, [1,2-b]-33 and [1,2-b]-34 present a stronger tendency toward crystallization, which causes a negative effect on the intermolecular charge transport and reduces the FF values (72.5 or 66.6% with [1,2-b]-33 or [1,2-b]-34 as the HTM) in comparison to [1,2-b]-32 (FF: 77.3%).| Device | Device architecture | V on (V) | EQE (%) | CE (cd A−1) | PE (lm W−1) | CIE (x, y) | Ref. |
|---|---|---|---|---|---|---|---|
| 2,2′,2′′-(1,3,5-Benzinetriyl)-tris(1-phenyl-1-H-benzimidazole) (TPBI); Copper(II) phthalocyanine (CuPc). | |||||||
| PhosOLED1 | ITO/PEDOT:PSS(30 nm)/NPB(15 nm)/TCTA(5 nm)/[1,2-b]-43: Ir(ppy)2(acac) 10% (25 nm)/TPBI(50 nm)/LiF(0.5 nm)/Al(100 nm) | 2.5 | 15.8 | 60 | 63 | (0.32, 0.64) | 117 |
| PhosOLED2 | ITO/CuPc(10 nm)/NPB(40 nm)/TCTA(10 nm)/ [1,2-b]-DSF-IF:Ir(ppy)3 10% (20 nm)/TPBI(40 nm)/LiF(1.2 nm)/Al(100 nm) | 3.5 | 9.9 | 31.4 | 15.5 | (0.33, 0.62) | 24 |
| Device | Device architecture | J SC (mA cm−2) | V OC (V) | FF (%) | PCE (%) | Ref. |
|---|---|---|---|---|---|---|
| Fluorine-doped Tin Oxide (FTO); poly[9,9-bis(3′-(N,N-dimethyl)-propyl-2,7-fluorene)-alt-2,7-(9,9-dioctylfluorene)] (PFN); poly((4,8-bis[(5-(2-ethylhexyl)-2-thienyl]benzo[1,2-b:4,5-b′]dithiophene-2,6-diyl)(3-fluoro-2-[(2-ethylhexyl)carbonyl]thieno[3,4-b]thiophenediyl) (PTB7-Th) | ||||||
| PSC1 | Dopant-free perovskite solar cell | 21.5 | 1.05 | 77.3 | 17.6 | 97 |
| FTO/C60/perovskite (CH3NH3PPbI3−xClx)/ [1,2-b]-32/MoO3/Ag | ||||||
| PSC2 | Dopant-free perovskite solar cell | 20.8 | 0.96 | 72.5 | 14.5 | 97 |
| FTO/C60/perovskite (CH3NH3PbI3−xClx)/ [1,2-b]-33/MoO3/Ag | ||||||
| PSC3 | Dopant-free perovskite solar cell | 20.8 | 0.92 | 66.6 | 12.7 | 97 |
| FTO/C60/perovskite (CH3NH3PPbI3−xClx)/ [1,2-b]-34/MoO3/Ag | ||||||
| DSSC1 | Dye sensitized solar cell | 6.95 | 0.70 | 69 | 3.36 | 120 |
| TiO2/FTO/[1,2-b]-44/Platinized FTO | ||||||
| DSSC2 | Dye sensitized solar cell | 8.10 | 0.70 | 71 | 4.04 | 120 |
| TiO2/FTO/[1,2-b]-45 /Platinized FTO | ||||||
| DSSC3 | Dye sensitized solar cell | 8.20 | 0.71 | 70 | 4.05 | 120 |
| TiO2/FTO/[1,2-b]-46/Platinized FTO | ||||||
| SC1 | ITO/ZnO/PFN/PTB7-Th:[1,2-b]-47(1:1)/MoO3/Al | 11.87 | 0.90 | 45.8 | 4.89 | 121 |
| SC2 | ITO/ZnO/PFN/ PTB7-Th:[1,2-b]-47(1:1.5)/MoO3/Al | 12.71 | 0.92 | 54.2 | 6.33 | 121 |
| SC3 | ITO/ZnO/PFN/ PTB7-Th:[1,2-b]-47(1:2)/MoO3/Al | 11.85 | 0.90 | 53.5 | 5.74 | 121 |
| DSSC4 | Dye sensitized solar cell | 7.3 | 0.81 | 81 | 4.8–5.0 | 122 |
| TiO2/FTO/[1,2-b]-48 + I−/I3−/Platinized FTO | ||||||
| DSSC5 | Dye sensitized solar cell | 10.1 | 0.81 | 81 | 6.4–7.1 | 122 |
| TiO2/FTO/[1,2-b]-49 + I−/I3−/Platinized FTO | ||||||
| DSSC6 | Dye sensitized solar cell | 11.3 | 0.71 | 70.75 | 5.72 | 123 |
| TiO2/FTO/[1,2-b]-50 + I−/I3−/Platinized FTO | ||||||
Other organic semiconductors based on a central [1,2-b]DHIF core were used as organic dyes for dye-sensitized solar cells (DSSCs). As examples, [1,2-b]-44-[1,2-b]-46 (Scheme 14), with tetraethylhexyl-[1,2-b]DHIF as the linker, a diphenylamine unit as the donor and three different acceptor units (cyanoacrylic acid for [1,2-b]-44, thienyl-cyanoacrylic acid for [1,2-b]-45 and furyl-cyanoacrylic acid for [1,2-b]-46) were prepared from [1,2-b]DHIF.120 DSSCs were fabricated using [1,2-b]-44–[1,2-b]-46 and nanocrystalline anatase TiO2. The electrolyte used was composed of 0.05 M I2/0.5 M tert-butylpyridine in acetonitrile. The device performances recorded under standard global AM 1.5 solar conditions are summarized in Table 5 (devices DSSC1-DSSC3). The JSC, VOC and FF of the three devices were reported in the ranges of 6.95–8.20 mA cm−2, 0.70–0.71 V and 69–71%, respectively corresponding to an overall conversion efficiency of 3.36–4.05%. [1,2-b]-46-based DSSC3 has the best PCE among all. Action spectra of the incident photon-to-current conversion efficiency (IPCE) as a function of wavelength were measured to evaluate the photo-response in the whole region. An IPCE performance of ca. 80% was observed from 400 to 550 nm for [1,2-b]-45 and [1,2-b]-46 with maximum values of 80% at 480 nm and 88% at 460 nm. The higher efficiency of [1,2-b]-45 and [1,2-b]-46 was attributed to the better light harvesting due to the extended π-conjugation and/or the higher dye density of [1,2-b]-45 (4.04 × 10−7 mol cm−2) and [1,2-b]-46 (3.84 × 10−7 mol cm−2) than [1,2-b]-44 (3.34 × 10−7 mol cm−2) on the TiO2 surface.
 | ||
| Scheme 14 [1,2-b]DHIF-based organic semiconductors used as the active material in different solar cells. | ||
In 2017, [1,2-b]-47121 based on a central [1,2-b]-tetraoctyl-IF core decorated at C2 and C8 by two electrodeficient units (Scheme 14) was prepared from [1,2-b]DHIF. [1,2-b]-47 showed intense and broad UV-vis absorption in the 470–690 nm wavelength region and suitable energy levels (HOMO: −5.42 eV and LUMO: −3.85 eV). The photovoltaic performances of [1,2-b]-47 were tested with a highly efficient low gap polymer as the donor namely PTB7-Th. Organic solar cells (SC1-SC3) were fabricated by spin-coating from o-dichlorobenzene of PTB7-Th and [1,2-b]-47 solution in an inverted configuration of ITO/ZnO/PFN/PTB7-Th:[1,2-b]-47/MoO3/Al. Three devices with different ratios of PTB7-Th:[1,2-b]-47 (SC1-SC3: 1/1, 1:1.5 and 1:2, respectively) were tested. The best performance was reached with SC2 with the ratio PTB7-Th:[1,2-b]-47 of 1:1.5. The SCLC method was used to measure μe and μh in PTB7-Th:[1,2-b]-47 (1:1.5) blended films showing balanced and moderate charge transport properties (μh: 8.24 × 10−5 cm2 V−1 s−1 and μe: 3.02 × 10−5 cm2 V−1 s−1).
In 2020, two extended [1,2-b]DHIF-TTF based on [1,2-b]-42 (Scheme 11) were synthesized ([1,2-b]-48 and [1,2-b]-49, Scheme 14). The two donor–acceptor molecules possess [1,2-b]-42 as the donor unit and either benzoic acid in [1,2-b]-48 or benzothiadiazole-benzoic acid in [1,2-b]-49 as the acceptor unit. The two molecules were used in dye-sensitized solar cells containing the I−/I3− redox mediator (DSSC4 and DSSC5 in Table 5). The highest power conversion efficiency reached for DSSC5 sensitized by [1,2-b]-49 was 6.4% with a photocurrent density of 10.1 mA cm−2 and photovoltage of 0.81 V. With [1,2-b]-48, the conversion efficiency was 4.8% with a photocurrent density of 7.3 mA cm−2 and photovoltage of 0.81 V (DSSC4). The best results obtained with [1,2-b]-49 were due to better intramolecular charge separation caused by the stronger electron accepting unit on which the LUMO was mainly located.122
More recently, [1,2-b]-50 with extended [1,2-b]-IF moieties as the donor unit and a cyanoacrylic acid moiety as the acceptor unit was designed (Scheme 14).123 The [1,2-b]-50 absorption spectrum in solution showed two sets of bands: the first one in the 300–400 nm range (attributed to the transitions of the π–π* conjugated system) and the second in the 400–600 nm range (assigned to the intramolecular charge transfer (ICT) transition between the donor and the acceptor units). When recorded in the solid state, these two sets of absorption bands merged in a broad absorption band in the 350–700 nm range. In this work, the authors highlight the interest of [1,2-b]-50 with its five hexyl units to control the aggregation and to reduce the exciton quenching. The photocurrent–voltage (J–V) characteristics of the DSSC6 based on [1,2-b]-50 (Table 5) were recorded under simulated AM 1.5G irradiation using the I−/I3− redox couple as the electrolyte to evaluate the photovoltaic parameters. DSSC6 gives a JSC of 11.3 mA cm−2, VOC of 0.71 V, FF of 70.75% and PCE of 5.72%. Compared to DSSC1-DSCC5, DSCC6 presents greater performances. The five hexyl chains present in the [1,2-b]-50 skeleton play an important role in the electron injection efficiency. They act as an hydrophobic barrier to avoid the charge recombination of electrons from TiO2 to oxide electrolyte and they reduce the intermolecular stacking.
Numerous π-conjugated organic molecules have been described as multi-photon absorbing materials due to their versatile applications in two-photon cellular imaging, optical power limiting, multiphoton pumper lasing, three-dimensional optical storage and photodynamic therapy.124 Large two-photon absorption (TPA) cross-sections have often been related to their effective π-delocalization, which leads to an extended charge separation. Some organic semiconductors based on a central [1,2-b]DHIF core have been highlighted in the literature for their 2-photon or 3-photon absorption properties (see [1,2-b]-51–[1,2-b]-61 in Scheme 15).
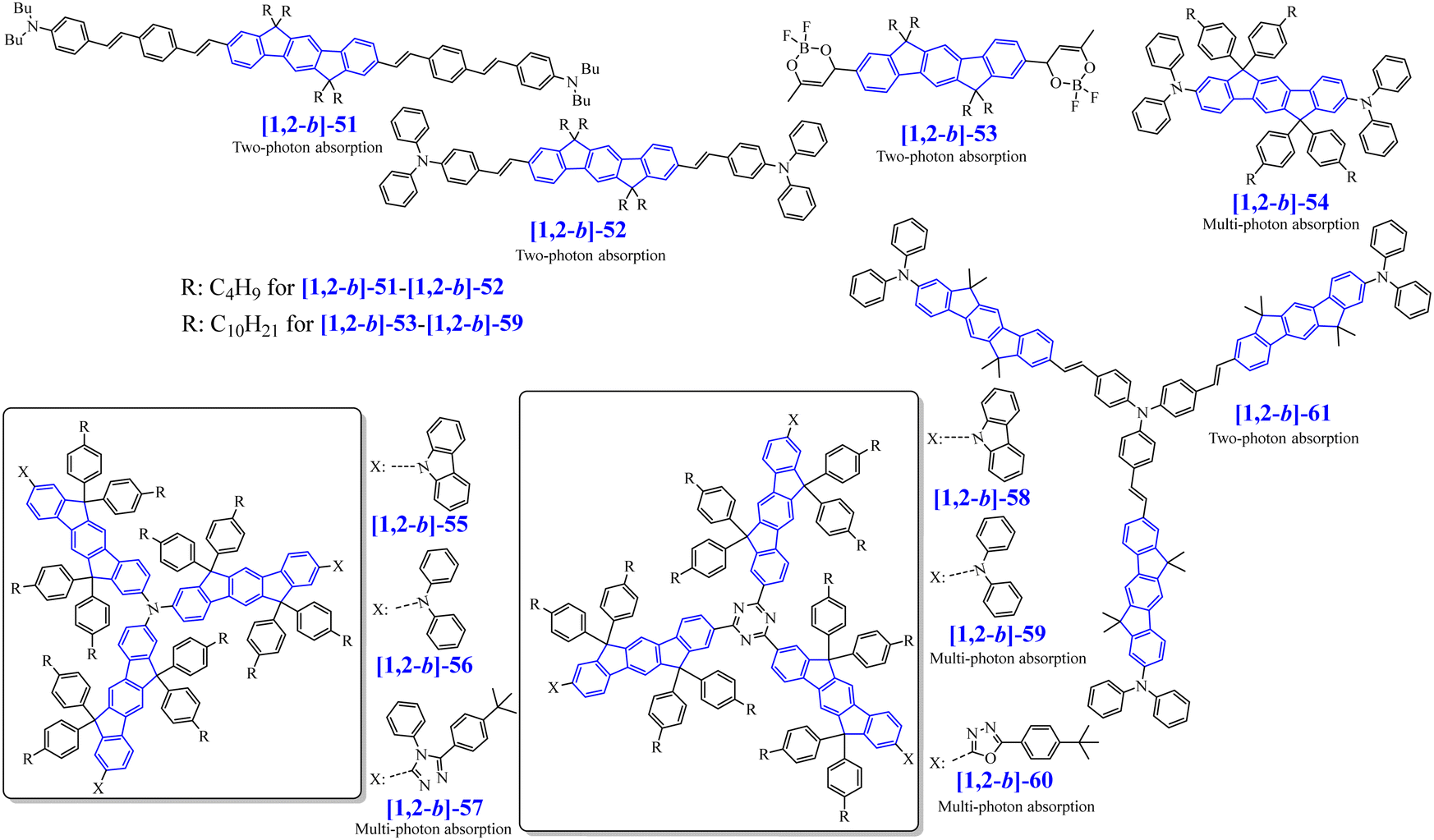 | ||
| Scheme 15 [1,2-b]DHIF-based organic semiconductors presenting NLO properties. | ||
In 2008, [1,2-b]-51125 was used for its 2-photon absorption and optical limiting properties. In [1,2-b]-51, the central [1,2-b]-tetra-butyl-IF was flanked on both sides by two dibutylamino groups with (E)-1-styryl-4-vinylbenzene bridges. Compared to the absorption spectrum of [1,2-b]DHIF,40 that of [1,2-b]-51 (334 vs. 423 nm in cyclohexane) was red-shifted by 89 nm due to the presence of the two arms at C2 and C8. Similarly, the emission was red-shifted from 356 nm for [1,2-b]DHIF to 462 nm for [1,2-b]-51125 with a similar fluorescent quantum yield (around 0.6). Contrary to [1,2-b]DHIF, [1,2-b]-51125 presented a positive solvatochromism in emission with increasing solvent polarity with a red shift from 462 nm in non-polar cyclohexane to 586 nm in more polar dimethylacetamide, showing charge transfer properties.
The TPA value of [1,2-b]-51,125 in CHCl3 at 775 nm, was 890 GM and the value measured by the nonlinear transmission method (CHCl3) was 670 GM. TPA cross-section values at 775 nm, in five different solvents other than CHCl3, were also determined (551 GM in cyclohexane, 853 GM in toluene, 685 GM in THF, 675 GM in CH2Cl2 and 723 GM in dimethylacetamide). The TPA-cross-section appears to be neither monotonically dependent on the polarity of the solvent nor of the optical dielectric constant of the solvents (Table 6).
| Compound | λ abs | λ em | QY | NLO properties | T d (°C) | Ref. |
|---|---|---|---|---|---|---|
| a 3PA cross-section (×10−76 cm6 s2); dimethylacetamide (DMA); Goeppert–Mayer (GM): 10−50 cm4 s photon−1. | ||||||
| [1,2-b]-51 | λ abs: 423 nm (CHX) | λ em: 462 (493) nm (CHX) | QY(fluo): 0.60 (CHX) | 2PA(775 nm): 551 GM (CHX) | — | 125 |
| λ abs: 429 nm (Toluene) | λ em: 477 (508) nm (Toluene) | QY(fluo): 0.70 (Toluene) | 2PA(775 nm): 853 GM (Toluene) | |||
| λ abs: 428 nm (CHCl3) | λ em: 509 nm (CHCl3) | QY(fluo): 0.64 (CHCl3) | 2PA(775 nm): 670 GM (CHCl3) | |||
| λ abs: 428 nm (THF) | λ em: 533 nm (THF) | QY(fluo): 0.68 (THF) | 2PA(775 nm): 685 GM (THF) | |||
| λ abs: 430 nm (CH2Cl2) | λ em: 543 nm (CH2Cl2) | QY(fluo): 0.69 (CH2Cl2) | 2PA(775 nm): 675 GM (CH2Cl2) | |||
| λ abs: 431 nm (DMA) | λ em: 586 nm (DMA) | QY(fluo): 0.28 (DMA) | 2PA(775 nm):723 GM (DMA) | |||
| [1,2-b]-52 | λ abs: 413 nm (Toluene) | λ em: 450 nm (Toluene) | QY(fluo): 0.66 (Toluene) | 2PA(775 nm): 1280 GM (Toluene) | — | 126 |
| [1,2-b]-53 | λ abs: 440 nm (CH3CN) | λ em: 506 nm (CH3CN) | QY(fluo): 0.67 (CH3CN) | 2PA(730 nm): 2800 GM (CH3CN) | — | 127 |
| [1,2-b]-54 | λ abs: 405 nm (Toluene) | λ em: 414, 443 nm (Toluene) | QY(fluo): 0.85 (Toluene) | 2PA(810 nm): 59.6 GM (Toluene) | 435 | 128 and 129 |
| λ abs: 404 nm (CHCl3) | λ em: 421, 444 nm (CHCl3) | 3PAa(1230 nm): 2.02 (Toluene) | ||||
| λ abs: 403 nm (DMF) | λem: 428 nm (DMF) | |||||
| [1,2-b]-55 | λ abs: 423 nm (Toluene) | λ em: 443 nm (Toluene) | QY(fluo): 0.96 (Toluene) | 2PA(700 nm): 1887 GM (Toluene) | 438 | 130 and 131 |
| λ abs: 416 nm (CHCl3) | λ em: 450 nm (CHCl3) | 3PAa (1270 nm): 2.2 (Toluene) | ||||
| λ abs: 419 nm (DMF) | λ em: 469 nm (DMF) | |||||
| [1,2-b]-56 | λ abs: 429 nm (Toluene) | λ em: 443 nm (Toluene) | QY(fluo): 0.86 (Toluene) | 2PA(700 nm): 2579 GM (Toluene) | 463 | 130 and 131 |
| λ abs: 423 nm (CHCl3) | λ em: 451 nm (CHCl3) | 3PAa (1270 nm): 2.45 (Toluene) | ||||
| λ abs: 427 nm (DMF) | λ em: 457 nm (DMF) | |||||
| [1,2-b]-57 | λ abs: 429 nm (Toluene) | λ em: 450 nm (Toluene) | QY(fluo): 0.84 (Toluene) | 2PA(700 nm): 1620 GM (Toluene) | 400 | 129 and 130 |
| λ abs: 431 nm (CHCl3) | λ em: 464 nm (CHCl3) | 3PAa (1270 nm): 3.35 (Toluene) | ||||
| λ abs: 425 nm (DMF) | λ em: 493 nm (DMF) | |||||
| [1,2-b]-58 | λ abs: 400(399129) nm (Toluene) | λ em: 426(430129) nm (Toluene) | QY(fluo): 0.98 (0.87129)(Toluene) | 2PA(770 nm): 5120 GM (Toluene) | 448 | 129 and 131 |
| λ abs: 399 nm (CHCl3) | λ em: 459 nm (CHCl3) | 3PAa (1200 nm): 3.3 (Toluene) | ||||
| Non soluble in DMF | Non soluble in DMF | |||||
| [1,2-b]-59 | λ abs: 428(427129) nm (Toluene) | λ em: 467(461129) nm (Toluene) | QY(fluo): 0.90 (Toluene) | 2PA(820 nm): 3630 GM (Toluene) | 426 | 129 and 131 |
| λ abs: 426 nm (CHCl3) | λ em: 510 nm (CHCl3) | 3PAa (1260 nm): 1.6 (Toluene) | ||||
| λ abs: 427 nm (DMF) | λ em: 591 nm (DMF) | |||||
| [1,2-b]-60 | λ abs: 381, 403 nm (Toluene) | λ em: 415, 440 nm (Toluene) | QY(fluo): 0.89 (Toluene) | 2PA(700 nm): 1420 GM (Toluene) | — | 131 |
| 3PAa (1200 nm): 2 (Toluene) | ||||||
| [1,2-b]-61 | λ abs: 419 nm (THF) | λ em: 479 nm (THF) | QY(fluo): 0.72 (THF) | 2PA(720 nm): 5270 GM (THF) | — | 132 |
In 2010, [1,2-b]-52 was prepared with a series of TPA-active D–π–D fluorophores126 with different central cores. [1,2-b]-52 presents, in toluene, an absorption/emission band at 413/450 nm, both blue shifted compared to those of [1,2-b]-51 (429/477 nm in toluene) and translating to a less extended conjugation in [1,2-b]-52 than in [1,2-b]-51. The TPA cross-section of [1,2-b]-52 was of 1280 GM (@775 nm), 1.5 more important than for [1,2-b]-51 (853 GM @ 775 nm).
[1,2-b]-53 127 belongs to the A–π–A fluorophores family as dioxaborine moieties are strong electron-acceptor units. This dye is a strong absorber with a long-wavelength maximum at 440 nm (in CH3CN) and its blue emission was observed with a maximum at 506 nm and a quantum yield of 0.67. The large Stokes shift of 3200 cm−1 indicates a significant rearrangement in the excited state. The TPA spectrum of [1,2-b]-53 reaches 2800 GM at 730 nm, which was in 2018 beyond the values reported for quadrupolar D–π–D and A–π–A architectures with similar conjugation and low molecular weight.127 This highlights the efficiency of the [1,2-b]DHIF derivatives for such applications.
In 2012, [1,2-b]-54 (Scheme 15), an organic semiconductor similar to [1,2-b]-467 (Scheme 4) (with four decylphenyl units instead of four tolyl units at C6 and C12) was synthesized and investigated for its multiphoton absorption properties.128[1,2-b]-54 was obtained following a route different to that described to prepare [1,2-b]-4 (Scheme 7) as the two diphenylamine donor units were incorporated on the terphenyl intermediate 52 (obtained from 50 and 51) before the formation of the [1,2-b]DHIF core (Scheme 16).
 | ||
| Scheme 16 Synthesis of [1,2-b]-54. (a) Pd(AcO)2, PPh3, K2CO3, toluene/methanol/H2O, 85 °C. (b) nBuLi, THF, −78 °C then RT. (c) CF3SO3H in THF/CH2Cl2. | ||
The Tg and Td of [1,2-b]-54 (200 °C and 435 °C resp.) were slightly higher than those reported for [1,2-b]-467 (183 °C and 420 °C resp.) indicating an increase of the thermal stability due to the presence of the decyl groups in [1,2-b]-54. In toluene, [1,2-b]-54 presented an UV-vis absorption centered at 405 nm (similar to [1,2-b]-467) and two resolved emission bands at 414 and 443 nm with a high fluorescence quantum yield of 0.85 (slightly blue-shifted compared to [1,2-b]-4 (423, 448 nm67)). The absorption and emission spectra showed no significant red/blue shifts in different solvents (CHCl3, DMF) suggesting nonpolar ground and excited states. This is due to its D–π–D structure. [1,2-b]-54 exhibited multiphoton upconverted photoluminescence (two-, three-, four- and five-photon) when excited by near infrared femtosecond laser pulses. The multiphoton-excited photoluminescence spectral features were similar to those of the one-photon-excited showing that the emission energy levels were likely to be identical. [1,2-b]-54 presented a 2PA cross-section of 59.6 GM at 810 nm and a 3PA cross-section of 2.02 GM at 1230 nm. In the same work,128[1,2-b]-54 parent compounds with longer central ladder-type oligophenylenes (from 4 to 7 bridged phenyls instead of 3 bridged phenyls in [1,2-b]-54) were also prepared.128 Their linear and nonlinear optical properties were studied and these longer ladder-type oligomers exhibited very strong multiphoton upconverted photoluminescence as well as blue lasing properties.
[1,2-b]-55-[1,2-b]-57 130 are star-shaped [1,2-b]DHIF based dendrimers (Scheme 15) composed of an amine-based central core, with three [1,2-b]DHIF arms decorated at C6 and C12 by four non-coplanar decyl-phenyl rings. They also possess either electron-donating carbazole ([1,2-b]-55), diphenylamine ([1,2-b]-56) or electron-accepting triazole ([1,2-b]-57) end caps. The three dendrimers exhibit two absorption bands. The strong absorption band between 380 and 440 nm correspond to the π–π* transition of the rigid [1,2-b]DHIF backbones. The weaker absorption around 300–325 nm is due to the n-π* transition of the central triarylamine unit. The emission spectrum presents a blue emission with a quantum yield over 0.84 peaking at 443–450 nm. The three star-shaped organic semiconductors exhibit extremely intense multiphoton induced blue photoluminescence upon excitation at 700, 1270 and 1800 nm corresponding to the two-, three- and four-photon absorption, respectively.
[1,2-b]-58 131 and [1,2-b]-59131 are analogous to [1,2-b]-55 and [1,2-b]-56 respectively, with a central triazine unit instead of a central nitrogen atom (Scheme 15). Finally, [1,2-b]-60131 also belongs to the [1,2-b]-58 and [1,2-b]-59 dendrimer family with a 2-(4-(tert-butyl)phenyl)-1,3,4-oxadiazole unit substituting the C7 atom of each of the three [1,2-b]DHIF units.
All these star-shaped structures based on the [1,2-b]DHIF unit exhibit remarkable efficient multiphoton absorption properties with a 2PA cross-section up to 2579 GM (@700 nm) for [1,2-b]-56 in the nitrogen centered dendrimer series and 2PA cross-section up to 5120 GM (@770 nm) for [1,2-b]-58 in the triazine centered dendrimer series. The comparison of the 2PA cross-section of [1,2-b]-55 and [1,2-b]-58 and that of [1,2-b]-56 and [1,2-b]-59 highlights the advantage of adopting the triazine center ring over the nitrogen atom for the star-shaped skeleton in view of 2PA enhancement. Furthermore, σ3 of [1,2-b]-58 with maximum 3.3 × 10−76 cm6 s2 (@1200 nm) is significantly higher than that of [1,2-b]-55 (σ3: 2.2 × 10−76 cm6 s2 (@1200 nm)) also showing the key role played by the triazine to improve the π-electron delocalization through the π-network and through-space excitonic interactions for large 3PA enhancements. The lower 2PA cross-section in [1,2-b]-55, [1,2-b]-57 and [1,2-b]-60 indicate that the end-groups also play a role in the efficiency of the multiphoton absorption.
Another star-shaped molecule based on the [1,2-b]DHIF core was also described in the literature.132[1,2-b]-61 possesses a central triphenylamine unit linked through a vinyl fragment to three [1,2-b]DHIF units each of them substituted at C8 by a diphenylamine unit. The absorption spectrum of [1,2-b]-61 presents a maximum around 420 nm and its emission spectrum was centered at 479 nm with a quantum yield of 0.72 (THF). With the 2PA cross-section up to 5270 GM (@720 nm), [1,2-b]-61 appears to be a superior 2PA-absorbing molecule than all the previous [1,2-b]-55–[1,2-b]-57.130 With a central nitrogen atom and the external diphenylamine units, the 2PA cross-section of [1,2-b]-61 was twice that of [1,2-b]-56 (2579 GM (@700 nm)).130,131 The presence of less voluminous hexyl units at the bridges in [1,2-b]-61 compared to tolyl units in [1,2-b]-56 may have an influence on the 2PA efficiency of [1,2-b]-61.
2.6. Other [1,2-b]DHIF-based organic semiconductors
In the previous section, all the [1,2-b]DHIF-based organic semiconductors presented were incorporated in electronic devices. In this last part, we rapidly present some [1,2-b]DHIF-derivatives for which no device application was reported but which deserve to be briefly presented due to the singularity of their electronic properties (Scheme 17). This also allows us to show the versatility of the [1,2-b]DHIF core. | ||
| Scheme 17 Molecular structures of [1,2-b]-62-[1,2-b]-71. | ||
In 2005, two rod-like bimetallic ruthenium [1,2-b]-62-Ru and osmium [1,2-b]-62-Os complexes bridged by a “phenyl-[1,2-b]DHIF-phenyl” core have been synthesized.133 For both compounds, the UV-vis absorption spectra (CH3CN) presented first, at 290 nm, the singlet intraligand π–π* transition of the three bipyridine units, then close to 400 nm the absorbance of the “phenyl-[1,2-b]DHIF-phenyl” core. The third absorption centered at 466 nm for [1,2-b]-62-Ru and at 490 nm for [1,2-b]-62-Os corresponded to the MLCT absorption bands of the bimetallic complexes. Additionally, [1,2-b]-62-Os showed a weak absorption band (550–700 nm) that was assigned to spin-forbidden electronic transitions (3MLCT) partly allowed due to strong spin-orbit coupling in the osmium complexes. The PL spectra were structureless with a maximum at 624 nm for [1,2-b]-62-Ru and at 748 nm for [1,2-b]-62-Os. The oxidation of the two compounds occurred in a single reversible bi-electronic wave centered on the metallic units with a peak at 1.28 V vs. SCE for [1,2-b]-62-Ru and at 0.86 V for [1,2-b]-62-Os whereas the reduction occurred in three successive reversible waves corresponding to the bipyridine ligand reductions with the first one localized on the phenylene-substituted pyridine. In [1,2-b]-62-Ru and [1,2-b]-62-Os, only a weak interaction exists between the two metal centers in the ground state. However, in the excited state, the electronic density is delocalized on the central “phenyl-[1,2-b]DHIF-phenyl” spacers.
In 2007, [1,2-b]-63 and a series of extended difluoromethylene-[1,2-b]DHIF derivatives ([1,2-b]-64-[1,2-b]-65) were synthesized and their electronic properties were compared to those of [1,2-b]-63.134 In reduction, [1,2-b]-63 showed a reduction peak potential at −2.46 V vs. Fc/Fc+ whereas that of [1,2-b]DHIF was at −2.96 V vs. Fc/Fc+.40 This positive shift of 0.5 V indicates that the electron affinity increases upon the substitution of hydrogen atoms by fluorine atoms at the two bridged positions of [1,2-b]-63. The UV-vis absorption spectrum of [1,2-b]-63 showed the lowest energy band peaking at 325 nm (THF), blue-shifted compared to [1,2-b]DHIF (334 nm in THF). A different effect was observed for the emission spectrum, which appeared more red-shifted for [1,2-b]-63 (400 nm in THF) compared to [1,2-b]DHIF (341, 353 nm in THF), despite no explanation being provided to explain this feature.
The extension of the [1,2-b]DHIF core by the substitution at C2 and C8 found in [1,2-b]-64–[1,2-b]-68, also leads to modification of the optical properties as exposed above. Compared to the maximum absorption band of [1,2-b]-63 (325 nm) all the absorption maxima were red-shifted upon extension of conjugation with the shift increasing from 22 nm for [1,2-b]-64 with the addition of two phenyl units to 33 nm in [1,2-b]-68 with the addition of two trifluorophenylethanone units. The five extended-[1,2-b]DHIF, [1,2-b]-64–[1,2-b]-68 show blue emission with maxima between 410 nm for [1,2-b]-65 with two pentafluorophenyl units to 436 nm for [1,2-b]-66 with two tolyl units, all red-shifted compared to the maximum highlighted at 400 nm for [1,2-b]-63. Single crystals of the compounds confirmed the planarity of the central tetrafluoro-[1,2-b]DHIF unit and showed different torsion angles between this core and the external phenyl units varying between 7.6° for [1,2-b]-68 and 35.8° for [1,2-b]-64, driving not only the extension of conjugation but also the π–π intermolecular interactions. The introduction of the carbonyl group on the phenyl in [1,2-b]-67 and [1,2-b]-68 improved the planarity of the molecule as well as the face-to-face packing.
In 2014, our group reported the luminescence modulation in liquid crystalline phases containing the extended [1,2-b]-69.135 This organic semiconductor was obtained in a good yield via a palladium(0)-catalyzed cross-coupling reaction between the 2,8-Br2-[1,2-b]-7 (28, Scheme 10) and N-(4-ethynylphenyl)-3,4,5-tris(hexadecyloxy)benzamide in n-propylamine as a base and solvent. The grafting of N-(ethynylphenyl)benzamide fragments carrying hexadecyl carbon chains on the bulky [1,2-b]-7 core has allowed the emergence of strongly luminescent liquid crystalline phases.
In 2019, a planar terphenyl-based o-carboranyl compound, [1,2-b]-70 (Scheme 17) was synthesized. It possesses a central [1,2-b]-tetramethyl-IF core decorated by two electron-withdrawing o-carborane units at C2 and C8.136[1,2-b]-70 exhibits low-energy absorption bands at 340 and 356 nm in THF and demonstrates intense emission at 375 and 548 nm. Compared to [1,2-b]DHIF emission (339, 347, 356 nm),40 that of [1,2-b]-70 was red-shifted and presented an intense ICT transition between o-carborane cages and the central [1,2-b]DHIF core.
Finally, in 2022, the optical properties of the aromatic ketone [1,2-b]-71137 (Scheme 17) were studied in solution and in crystalline nanostructured films. From those studies it was demonstrated that the crystalline films exhibit enhanced light absorption, amplified emission quantum yields and longer excited state lifetimes compared to diluted solutions. These properties, termed as aggregation-induced delayed fluorescence (AIDF) are promising. AIDF materials are promising emitting layers in non-doped OLEDs.
3. Modification of the indenofluorenyl core using the geometry
3.1. Introduction to the 11,12-dihydroindeno[2,1-a]fluorene core: the para–syn isomer of [1,2-b]DHIF
Despite the [1,2-b]DHIF isomer being the most studied isomer, another arrangement of the three phenyl units has also been studied in the literature keeping nevertheless the para-terphenyl linkages. This isomer, the 11,12-dihydroindeno[2,1-a]-fluorene, labelled as [2,1-a]DHIF (colored in red in Scheme 18) presents different properties than its [1,2-b]DHIF counterpart. Despite two different routes to indeno[2,1-a]fluorenone, [2,1-a]IF(O)2, being published in 1934140 and in 1939,141 the first synthesis of [2,1-a]DHIF was described in 1951 by Deuschel32 (Scheme 18). This first approach consisted in the reduction of [2,1-a]IF(O)2 obtained by a Diels-Alder addition and aromatization of 54 and 55 leading to 56 itself cyclized in fluorenone 57, which led to the dione [2,1-a]IF(O)2.
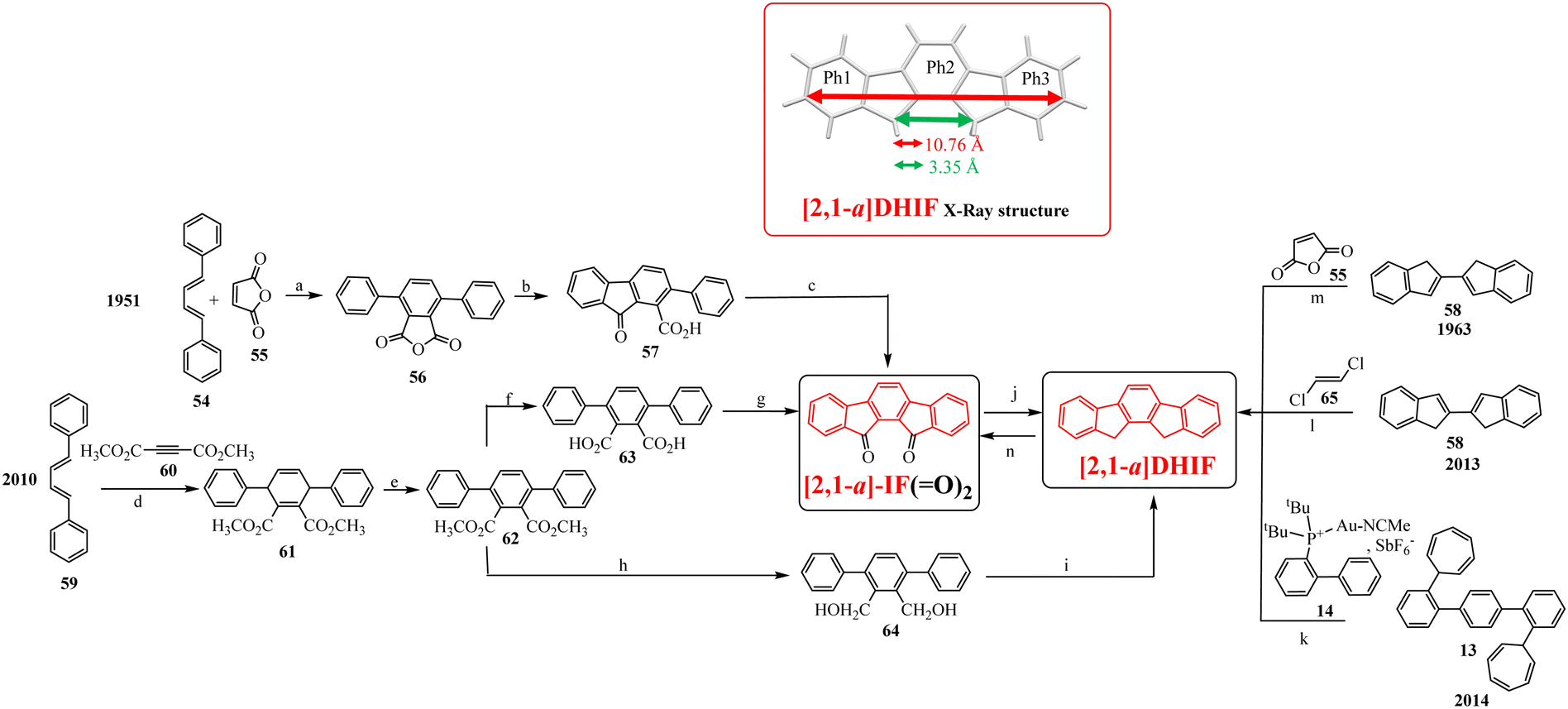 | ||
| Scheme 18 Different routes to [2,1-a]DHIF21,32,38,138,139 or [2,1-a]IF(O)232,140,141 and [2,1-a]DHIF X-ray structures. (a) Experimental conditions not available, (b) AlCl3, CS2, (c) SOCl2, CCl4, H2SO4, (d) toluene, reflux, (e) toluene, Pd/C reflux, (f) NaOH /EtOH, reflux, (g) H2SO4, 140 °C, (h) DIBAL, DCM, (i) PPA, 1,2-dichlorobenzene, 140 °C, (j) NH2-NH2, H2O/KOH, diethyleneglycol 180 °C, (k) DCE, 120 °C, (l) 230 °C in sealed glass tube, (m) not available, and (n) CrO3, acetic anhydride. | ||
In 1963, another access to [2,1-a]DHIF was described by Schroth et al.138 from the 2,2′-diindenyl 58 and maleic anhydride 55. In 2010, our group detailed the access to [2,1-a]DHIF following different routes (Scheme 18).21 These routes both started with a cycloaddition reaction between 1,4-diphenyl-1,3-butadiene 59 and dimethyl-but-2-ynedioate 60 leading to the 1,4-dihydroterphenyl derivative 61, itself dehydrogenated to build the terphenyldicarboxylate 62. The reduction of 62 led to its corresponding diol 64. The last step was the cyclisation of 64 in [2,1-a]DHIF. The second route involved the synthesis of [2,1-a]IF(O)2 by intramolecular cyclisation of 63 in acidic medium, the last step being its reduction in [2,1-a]DHIF. In 2013, a new route to [2,1-a]DHIF was described starting from the 2,2′-diindenyl 58 in the presence of (E)-1,2-dichloroethene 65.139 Finally, in 2014, [2,1-a]DHIF was obtained in a mixture with [1,2-b]DHIF (in a 1:4 ratio) through double annulation of terphenyl 13 using a cationic gold complex 14.38
The main physicochemical properties of [2,1-a]DHIF have been reported by our group in 201021 and compared to those of [1,2-b]DHIF presented above.40 The structural analysis shows that the positioning of the two bridges in a syn arrangement leads to a contraction of the DHIF core compared to [1,2-b]DHIF (10.76 vs. 11.05 Å). This contraction and particularly the short distance between the two bridges in a syn arrangement (3.35 Å) will play a key role in the development of the [2,1-a]DHIF isomer (see below). The UV-vis absorption spectrum of [2,1-a]DHIF (THF) presents well-defined absorption bands with maxima at 307, 315 and 322 nm, blue-shifted by 12 nm compared to that of [1,2-b]DHIF (319, 328, 334 nm) providing a gap expansion (3.76 eV for [2,1-a]DHIFvs. 3.61 eV for [1,2-b]DHIF).40 This indicates a better delocalization of π-electrons in [1,2-b]DHIF than in [2,1-a]DHIF. Similarly, the emission spectrum of [2,1-a]DHIF presents two main bands at 326 and 343 nm, hypsochromically shifted by 13 nm compared to [1,2-b]DHIF. Due to its rigid and planar structure, the Stokes shift was very small (4 nm) and [2,1-a]DHIF was highly fluorescent (∅sol: 0.6 in THF).
The first oxidation of [2,1-a]DHIF (Fig. 7A) is reversible and occurs at a potential of 1.31 V vs. SCE identical to the one reported for [1,2-b]DHIF.48 The HOMO and LUMO levels of [2,1-a]DHIF were of −5.62 and −1.94 eV, quite similar to those reported for [1,2-b]DHIF (−5.61 and −1.99 eV). After this first oxidation, an irreversible wave is observed (1.98 V, Fig. 7B) almost identical to that reported for its positional isomer (2.01 eV).48 Recurrent scans involving the two oxidation waves lead to a regular electropolymerization process (see the arrows in Fig. 7C).21 As described in Fig. 7D, poly[2,1-a]DHIF appears stable in a wide potential range between 0.4 and 2.4 V vs. SCE, larger than that reported for poly[1,2-b]DHIF,48 indicating the higher stability of the polymer and also a lower HOMO level (−5.04 eV vs. −5.15 eV).
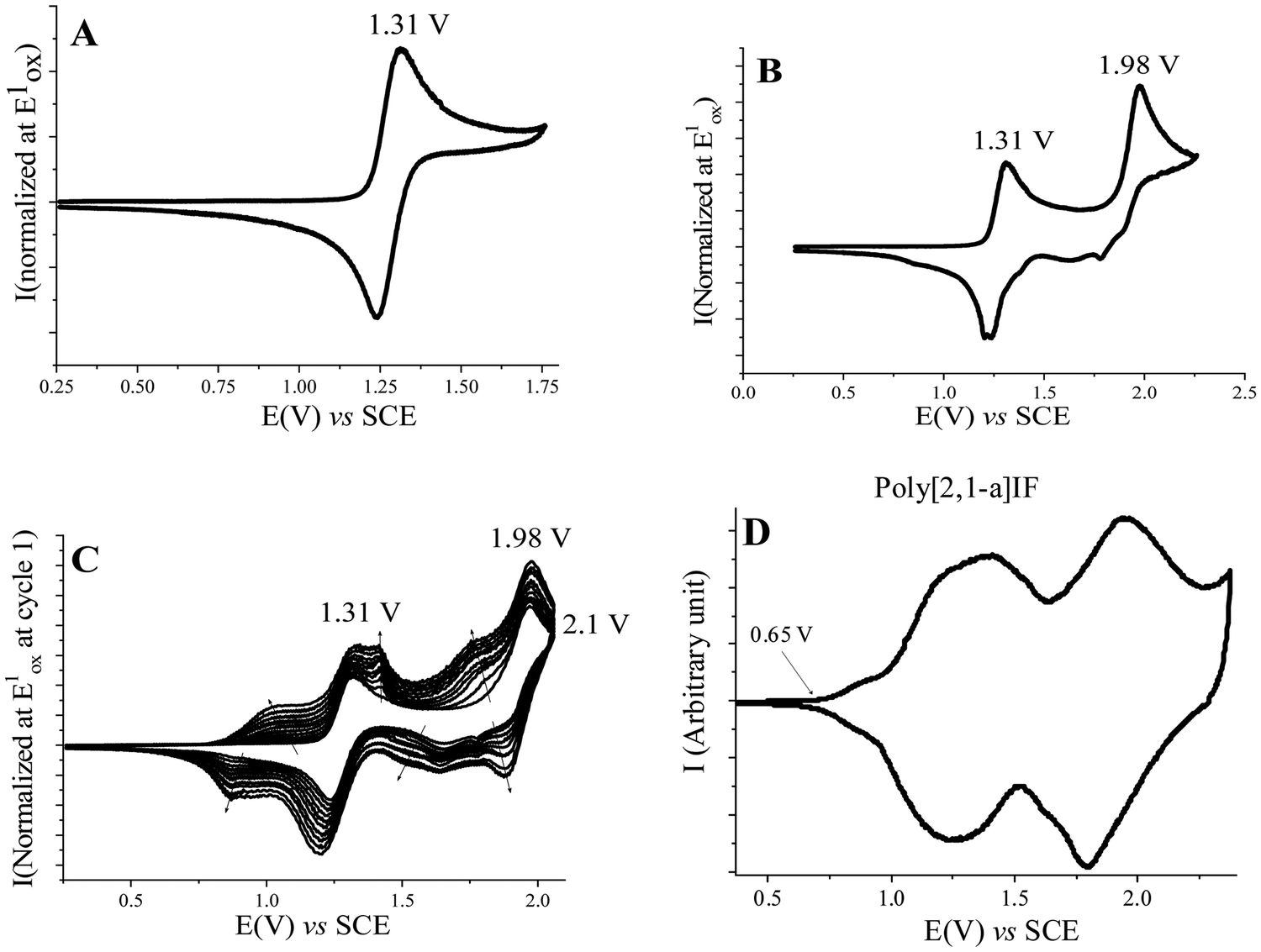 | ||
| Fig. 7 Cyclic voltammetry at 100 mV s−1 in CH2Cl2/[NBu4][]PF6] 0.2 M. A. [2,1-a]DHIF, 1 cycle between 0.25 and 1.76 V, B. [2,1-a]DHIF, 1 cycle between 0.25 and 2.27 V, C. [2,1-a]DHIF, 10 cycles between 0.25 and 2.1 V and D. modified electrode (5 cycles between 0.25 and 2.27 V in the presence of [2,1-a]DHIF) investigated in the absence of [2,1-a]DHIF, with 3 cycles between 0.4 and 2.4 V. | ||
Contrary to [1,2-b]DHIF derivatives, which have been used as active materials in different OE devices (OLED, PhOLED, OFET, OPV, etc., see above), [2,1-a]DHIF derivatives have been principally used as active materials in fluorescent OLEDs. As the synthesis of [2,1-a]DHIF is not even currently widely developed, this could be the reason for this lack of application. The main characteristic used to date is its geometry with the two bridges on the same side of the [2,1-a]DHIF plane. This can allow us to force π–π interactions between two fragments attached at these positions. The examples using such an approach are described below.19,20,22,75,142
3.2. Excimer emission of [2,1-a]DHIF-based organic semiconductors
The first isolation of a [2,1-a]DHIF-based molecule, in our group, was not expected.19 Indeed, in 2008, during the synthesis of [1,2-b]-DSF(tBu)4-IF, starting from the 2,2”-diiodo-terphenyl 26 and the 2,7-ditBu-fluorenone, a second isomer with a [2,1-a]DHIF core was also isolated (Scheme 19).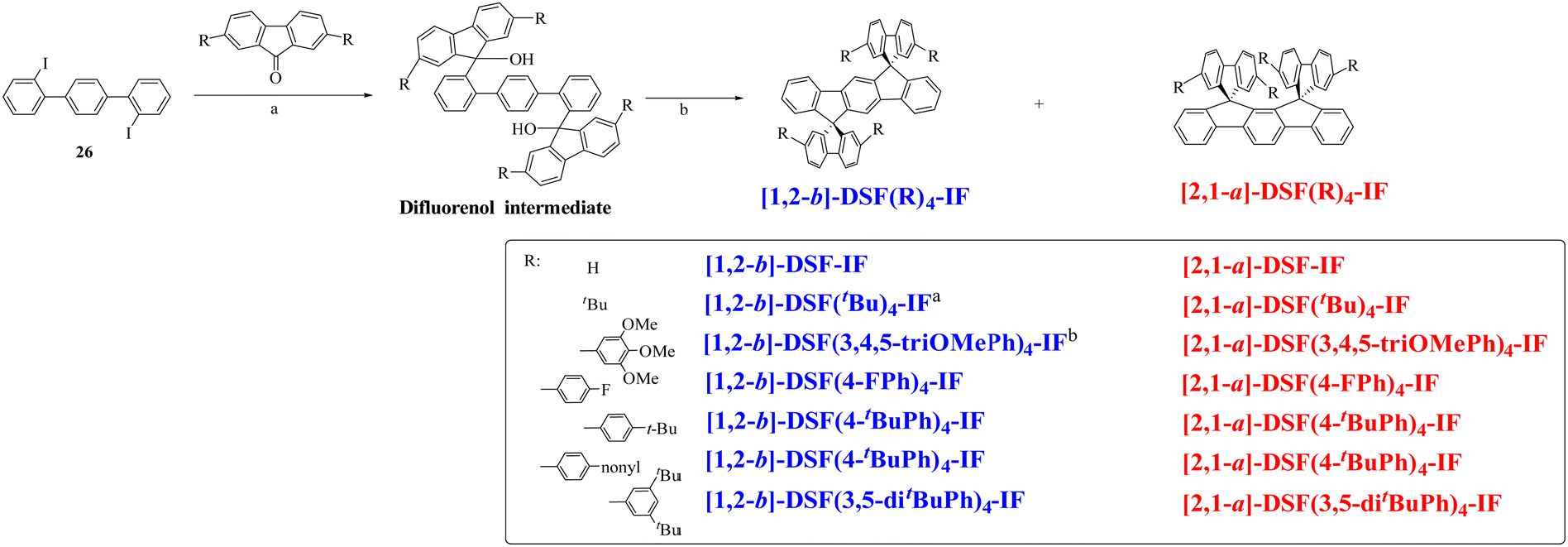 | ||
| Scheme 19 Synthesis of [1,2-b]-DSF(R)4-IF and [2,1-a]-DSF(R)4-IF. (a) nBuLi, THF, −78 °C to RT, (b) CH3CO2H/HCl, 100 °C (a: named [1,2-b]-7 in Scheme 4,b: named [1,2-b]-9 in Scheme 4). | ||
The two isomers were formed during the bicyclization of the difluorenol intermediate. In 2009, our group decided to investigate the regioselectivity of this reaction and we observed that the anti/syn ratio of isomers formed could be modified as a function of different parameters: the temperature, the solvent and the substituents attached on the fluorenone. This reaction and the physicochemical properties of different couples of isomers have been then intensively studied and detailed in different publications.19,20,22,59,75,142,143
We have proposed that the bicyclization of the difluorenol intermediate (Fig. 8, Left) unfolds sequentially.143 The first ring closure gives a 2-substituted spirobifluorene (see pro-conformers in Fig. 8) and the second one provides the corresponding [2,1-a]DHIF and [1,2-b]DHIF core (via a pro-Wheland intermediate).
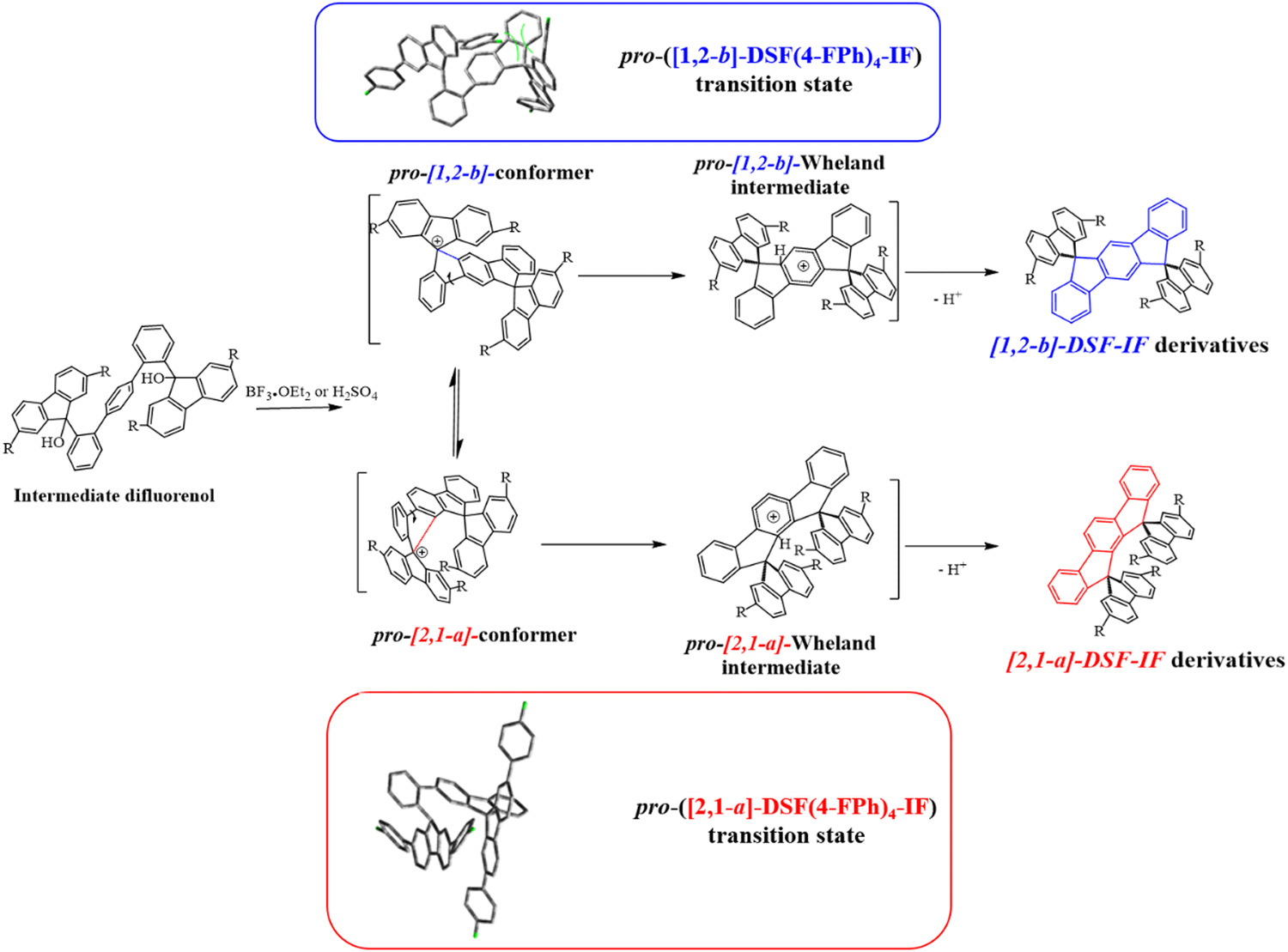 | ||
| Fig. 8 Middle: Cyclization reaction of difluorenols with the corresponding intermediates hypothesized.143 Top and Bottom: Modelization of pro-([1,2-b]-DSF(4-FPh)4-IF) and pro-([2,1-a]-DSF(4-FPh)4-IF) transition states144 showing the postulated steric hindrance at the origin of the selectivity observed. | ||
In this second step, the cyclisation can occur either on the opposite sides leading to the anti isomer with the [1,2-b]DHIF geometry or on the same side of the first cyclisation, leading to the syn isomer with the [2,1-a]DHIF geometry. This powerful reaction was then further investigated to develop structurally related materials for electronics (another mechanistic study was also reported in 2019144).20,22,27,59,75,142
The most influential parameter on the selectivity of this cyclization reaction is the nature of the R groups attached (see some examples in Scheme 19). As R becomes more encumbering (from RH to R3,5-di-tbutylphenyl), the distribution shifts towards the formation of [2,1-a]-isomers at the expense of [1,2-b]-isomers. With a very bulky substituent attached, R = 3,5-di-tbutylphenyl, the reaction becomes regioselective exclusively providing [2,1-a]-DSF(3,5-di-tBuPh)4-IF (see the structure in Scheme 19). Thus, despite the [2,1-a] isomers being less energetically stable than their [1,2-b] counterparts, their formation was favoured by sterically hindered substituents.143 Mechanistic investigations have shed light on this intriguing feature and have revealed that the selectivity towards [2,1-a] isomers is increased by the solvent, substituent and temperature. This involves the relative locking of the sterically hindered rotamers in the more stable pro-[2,1-a]-conformation due to a high energy barrier for the interconversion to the pro-[1,2-b]-conformation (Fig. 8).143,144 Thus, in this reaction, the [2,1-a] isomers are favoured by (i) high polarity solvents, (ii) high temperature and (iii) bulky R groups providing an interesting degree of control.
The photophysical properties of the two families of regioisomers have also appeared to be very different as a function of the substituents attached on the fluorenes. With non-aryl substituents at C2/C7 of the fluorenyl units (H or tBu, see the structures in Scheme 19), the physicochemical properties of the two isomers remain similar (see the emission of [1,2-b]-DSF-IF/[2,1-a]-DSF-IF in blue/red straight lines, Fig. 9A1/A2). However, if aryl units are attached on the fluorenes as in the couples [1,2-b]-DSF(4-tBuPh)4-IF – [2,1-a]-DSF(4-tBuPh)4-IF or [1,2-b]-DSF(4-FPh)4-IF – [2,1-a]-DSF(4-FPh)4-IF, the emission properties of the two regioisomer families become different. Indeed, [1,2-b]-DSF(4-tBuPh)4-IF and [1,2-b]-DSF(4-FPh)4-IF are violet emitters (blue dotted lines in the 350–450 nm wavelength range in Fig. 9A1) whereas [2,1-a]-DSF(4-tBuPh)4-IF and [2,1-a]-DSF(4-FPh)4-IF are blue emitters (red dotted lines in Fig. 9A2). The modulation of the emitted light colour is also highlighted in Fig. 9B for [1,2-b]-DSF(4-FPh)4-IF/[2,1-a]-DSF(4-FPh)4-IF isomers. This feature is due to emission arising in the case of the [2,1-a]-DSF(aryl)4-IF, from the interactions between the two cofacial “aryl-fluorene-aryl” arms. This is directly induced by the particular geometry of the indeno[2,1-a]fluorenyl core.75
 | ||
| Fig. 9 (A) Emission spectra in THF of [1,2-b]-DSF(R)4-IF derivatives (A1 in blue) and [2,1-a]-DSF(R)4-IF derivatives (A2 in red), (B) photograph of the light emitted by [1,2-b]-DSF(4-FPh)4-IF (left) and of [2,1-a]-DSF(4-FPh)4-IF (right) (λexc = 365 nm), (C) X-Ray structure of [2,1-a]-DSF(4-FPh)4-IF, (D) schematic representation of the impact of the substituent on the structural and electronic properties of the [2,1-a]-DSF(aryl)4-IF series, (E) cyclic voltammetry in the oxidation of [1,2-b]-DSF(R)4-IF derivatives (E1 in blue) and [2,1-a]-DSF(R)4-IF derivatives (E2 in red). | ||
The [2,1-a]-DSF(aryl)4-IF family also presents original tuneable emission properties since the emission wavelength can be modulated by the steric hindrance induced between the substituted phenyl rings. When the substituent borne by the phenyl unit is bulky such as a tert-butyl group in [2,1-a]-DSF(4-tBuPh)4-IF, the two “aryl-fluorene-aryl” arms move away from each other lessening the cofacial interaction. This results in an hypsochromic shift of the emission band vs.[2,1-a]-DSF(FPh)4-IF.75
The two DSF(R)4-IF families (R: H; Alkyl or Aryl) also present different electrochemical properties as presented in Fig. 9E. In the case of [1,2-b]-DSF(R)4-IF, the first oxidation occurs either on the [1,2-b]-IF core or on the external “aryl-fluorene-aryl” arms or even on the three π-systems depending on the nature of the substituents borne by the fluorenyl units.59 Due to these effects, the first oxidation potential varies between 1.16 V for [1,2-b]-9 to 1.47 V for [1,2-b]-DSF-IF (Scheme 19). In the case of the [2,1-a]-DSF(R)4-IF family, the specific geometry leads to a face-to-face arrangement of the “aryl-fluorene-aryl” arms and shifts the first oxidation potential to lower values (between 1.07 V for [2,1-a]-DSF(3,4,5-tri-OMePh)4-IF to 1.36 V for [2,1-a]-DSF-IF). Depending on the interaction between the face-to-face “aryl-fluorene-aryl” in the [2,1-a]-DSF(aryl)4-IF family, the first oxidation was assigned to an oxidation centered on the “aryl-fluorene-aryl” cofacial dimer. The intramolecular interaction in [2,1-a]-DSF(4-FPh)4-IF is clearly highlighted in its X-ray structure provided in Fig. 9C.
In 2011 and 2012, our group synthesized two specific blue emitting fluorophores based on the [2,1-a]-IF core, incorporating pending fluorene units [2,1-a]-73142 or trimethoxyphenyl units [2,1-a]-7422 following the route presented above in Scheme 19. These two derivatives, presented in Fig. 10 with their respective [1,2-b]-isomers [1,2-b]-72 and [1,2-b]-9, were used as the EML in fluorescent OLEDs. The idea was to show that intramolecular interactions occurring in [2,1-a]-derivatives can be used to produce blue light in a device.
 | ||
| Fig. 10 [2,1-a]DHIF-based organic semiconductors used as the EML in fluorescent OLEDs ([2,1-a]-73 and [2,1-a]-74) and their respective [1,2-b]-isomers([1,2-b]-72 and [1,2-b]-9), fluorescent spectra of the four compounds. | ||
The two isomers were stable with Td of 215 and 320 °C for [2,1-a]-73 and [1,2-b]-72 respectively. The UV-vis absorption spectrum of [1,2-b]-72 displayed one maximum at 347 nm whereas that of [2,1-a]-73 displayed a maximum at 343 nm (THF). The low energy bands were similar to those of the terfluorene145 or other terfluorenyl oligomers,146–150 showing that the electronic properties were driven by the terfluorenyl fragments. Thus, [2,1-a]-73 and [1,2-b]-72 presented a similar optical gap of 3.15 eV close to that of terfluorenyl derivatives. Both isomers exhibited deep blue emission in solution with however different spectra. The [1,2-b]-72 spectrum was well defined with maxima at 395 and 417 nm fitting the emission spectrum of the terfluorene and indicating an emission from terfluorenyl arms. The [2,1-a]-73 emission spectrum was different. It was larger, less structured and red-shifted (402, 422 nm). The shoulder at ca. 450 nm was also more intense with a tail up to ca. 600 nm. This emission shoulder at 450 nm has been assigned to frustrated intramolecular interactions between the two face-to-face terfluorenyl units (see above, Fig. 9). In this case, the long octyl chains were considered responsible for the sterically hindered environment. Before using the two isomers as the EML in fluorescent OLED, their solid state emission spectra and their stability with the temperature were studied. For [1,2-b]-72, at room temperature, the solid state emission spectrum was similar to that in solution indicating no specific intermolecular interactions involving the π-systems in the film. This spectrum remained stable from RT to 150 °C, however at higher temperature, a green emission band11,56,58,151–155 started to grow and became intense after one night at 200 °C. In contrast, the solid state emission spectrum of [2,1-a]-73 appeared different to its solution spectrum with a non-structured main band centred at 464 nm and weak shoulders at 405 and 437 nm. The main emission was attributed to intramolecular excimer formation arising from stacked face-to-face terfluorenyl units and the shoulder to non-stacked terfluorenyl units.
[2,1-a]-73 was finally used as the EML in a single layer OLED device (SL-OLED30, Table 7).142 Despite very low performances, the EL spectrum was in perfect accordance with the [2,1-a]-73 emission spectrum. The blue emission arose from the electrogenerated intramolecular terfluorenyl excimers.
| Device | Device architecture | V on (V) | CE (cd A−1) | PE (lm W−1) | Luminance (cd m−2) | λ max (nm) CIE (x, y) | Ref. |
|---|---|---|---|---|---|---|---|
| SL-OLED30 | ITO/PEDOT:PSS/[2,1-a]-73 (50 nm)/Ca | 7.0 | 0.05 | — | 100 | 464 nm (0.19, 0.23) | 142 |
| SL-OLED31 | ITO/PEDOT:PSS/[2,1-a]-74 (30 nm)/Ca | 5.0 | 0.037 | 0.018 | 100 | 447 (0.19, 0.19) | 22 |
| OLED32 | ITO/PEDOT:PSS/NPB (40 nm)/[2,1-a]-74 (45 nm)/Ca | 5.8 | 0.1 | 0.002 | 510 | 462 (0.19, 0.18) | 22 |
As another example, [1,2-b]-9 and [2,1-a]-74 both possessing four 3,4,5-trimethoxyphenyl units grafted at C2/C7 of their two fluorene units (Fig. 10) have shown that the emission of such compounds was mainly driven by the “aryl-fluorene-aryl” arms in [1,2-b]-9 and by face-to-face “aryl-fluorene-aryl” dimers in [2,1-a]-74. Interestingly, as for [2,1-a]-73, the emission of [2,1-a]-74 in the solid state was almost unchanged compared to its solution spectrum showing the stability of this type of emission.
SL-OLED31 and OLED32, Table 7, both using [2,1-a]-74 as the EML have been studied and compared to that using [1,2-b]-9 as the EML (SL-OLED13 and OLED14) (Table 1). Comparing SL-OLED13 and SL-OLED31, one notes a lowering of the Von (7.2 vs. 5.0 V) and a global increase of the other characteristics particularly the luminance, which was doubled. Concerning the EL spectra, that of SL-OLED31 (CIE: 0.19, 0.19) appears shifted to a shorter wavelength compared to that of SL-OLED13 (CIE: 0.24, 0.24) indicating the efficiency of excimer emission to achieve purer blue light. A comparison of the performance of OLED14 with [1,2-b]-9 as the EML and OLED32 with [2,1-a]-74 as the EML also shows the interest of the intramolecular excimer emission from [2,1-a]-74 compared to [1,2-b]-9 with a lower Von, a higher luminance and an emission at shorter wavelength.
Beyond the performances, which were very low, these studies have evidenced the possibility to generate stable blue light with intramolecular interactions arising from the [2,1-a]DHIF core.
3.3. Other organic semiconductors based on the [2,1-a]DHIF core
Despite several other organic semiconductors based on the [2,1-a]DHIF core being described in the literature (see the examples in Scheme 20), none of them have been used as the active material in OE devices. The particular geometry of the core has been however beneficially used to construct different molecules with specific properties. For example, in 2011, the electrophilic transannular cyclization of octadehydrodibenzo[12]annulene (66) was examined and revealed the formation of an anti and a syn isomer of the IF-dione. More precisely, the reaction of 66 in the presence of iodine leads to a mixture of [2,1-a]-75 and its [1,2-b]-isomer in one step. On the other hand, the reaction of 66 in the presence of bromide leads to [2,1-a]-76 and its [1,2-b]-isomer in two steps. The ratio of one isomer vs. the other was not reported in the publication. | ||
| Scheme 20 Synthesis of different [2,1-a]DHIF-based organic semiconductors reported in literature.156–159 Synthesis of [2,1-a]-75: (a) I2, O2 in benzene. Synthesis of [2,1-a]-76; (b) Br2 in DCM then, (c) NaOAc, AcOH in DCM. Synthesis of [2,1-a]-77: (d) tBuOK in THF. The inset is the X-ray structure of [2,1-a]-77 from Cambridge Data Base No. 1474507. Synthesis of [2,1-a]-78: (e) trifluoroacetic acid, trifluoroacetic anhydride in MeOH, 75 °C; f) P2O5, MsOH, 75 °C. | ||
In 2016, the polycyclic hydrocarbon [2,1-a]-77 with a twisted cyclooctatetraene (COT) core fused by two [2,1-a]DHIF units was synthesized by stepwise cycloaddition of the in situ generated non-aromatic bromo derivative 11-Br-[2,1-a]DHIF obtained from its dibromo-parent 11,12-Br2-[2,1-a]DHIF by HBr elimination. X-Ray crystallographic analysis of [2,1-a]-77 indicated the twisted conformation of the central COT core (see the inset in Scheme 20). This central core was moderately antiaromatic as indicated by the NICS value (+11.9). [2,1-a]-77 is a highly conjugated molecule which displays an absorption at 634 nm extending to about 850 nm in accordance with the gap of 2.21 eV theoretically estimated by the DFT calculations.157
In 2017, a nanohoop, [2,1-a]-78 incorporating three [2,1-a]IF(O)2 units has been synthesized using the “banana shape” of the [2,1-a]DHIF core. [2,1-a]-78 has been synthesized from a preformed macrocycle 68 constituted of 9 phenyl rings linked in para positions in which three phenyls were disubstituted in the ortho position by two CO2CH3 groups. Different synthetic pathways were described in this work finally leading to [2,1-a]-78 from 68 in a 25% yield in a two-step process. The UV-vis absorption spectrum of [2,1-a]-78 displayed several bands at 288, 379 and 408 nm and the fluorescence spectrum had bands at 448 and 606 nm. Although fluorene160 and other bridged oligophenylenes such as carbazole161 have been integrated in nanohoops, [2,1-a]-78 is to date the only example of this macrocycle incorporating a [2,1-a]DHIF building unit. The presence of three carbonyls in the present ring is of interest for further functionalization of this macrocycle and more generally for the development of nanohoops in electronics.
Finally, in 2019, [2,1-a]-IF-derivatives decorated by thiophene cyclic compounds, [2,1-a]-79-82, were synthesized by superelectrophilic Friedel–Crafts reaction between [2,1-a]-IF(OH)2 and thiophene building units.159 The photophysical properties of the four [2,1-a]-79-82 were studied and compared to that of [2,1-a]-IF(OH)2. The UV-vis absorption and emission spectra of [2,1-a]-79-81 were almost similar with λabs at ca. 310 and 325 nm and λem at ca. 350 nm. Moreover, compared to [2,1-a]-IF(OH)2 and [2,1-a]-82, [2,1-a]-79-81 presented a higher fluorescence quantum yield (2/5% for [2,1-a]-IF(OH)2/[2,1-a]-82vs. 29/47/52% for [2,1-a]-79/80/81) which may be correlated to the rigidity of the molecular structures.
4. Modification of the indenofluorenyl core via terphenyl linkages: dihydroindeno[1,2-a]fluorenyl-, dihydroindeno[2,1-b]fluorenyl- or dihydroindeno[2,1-c]fluorenyl-based organic semiconductors
Modification of the indenofluorenyl core via the arrangement of the phenyl linkages from para-terphenyl (described above in [1,2-b]DHIF derivatives or in [2,1-a]DHIF derivatives) to meta-terphenyl provides the meta isomers, namely, 7,12-dihydroindeno[1,2-a]fluorene (called the meta–anti isomer) and 5,7-dihydroindeno[2,1-b]fluorene (called the meta–syn isomer) (see [2,1-b]DHIF and [1,2-a]DHIF in orange and green in Scheme 1). These two positional isomers have revealed, over the years, the importance of the linkage and the geometry on the electronic properties.23–25 The 5,8-dihydroindeno[2,1-c]fluorene is the last isomer in the DHIF family. This isomer displays a syn geometry induced by the central ortho-terphenyl core. The ortho linkage gives this isomer unique characteristics and notably, from a structural point of view, a helicoidal turn of the DHIF unit (see [2,1-c]DHIF in pink, Scheme 1). Their properties and specific use in OE are described in the last part of this review.4.1. Synthesis of [1,2-a]DHIF, [2,1-b]DHIF and [2,1-c]DHIF
The oldest synthesis to [1,2-a]DHIF involve both the synthesis of the dione [1,2-a]IF(O)2 following two different routes starting from the bis-indene-dione 69 (Radulescu in 1939162) or from the (o-chlorobenzylidene)-9-fluorene 72 (Chardonnens in 1955163), Scheme 21. In 1967, Kemp et al.164 prepared [1,2-a]DHIF directly from the bis-indene 75. The diene system of 75 reacted with maleic anhydride 55 to give 76, which was aromatized in [1,2-a]DHIF following the Altman and Ginsburg method.165 In 1970, Chardonnens166 reported another access to [1,2-a]DHIF also involving [1,2-a]IF(O)2. This synthesis started by the condensation of the α-methylchalcone 77 and the diethylcetone 78 leading to the trimethyl-2,4,6-diphenyl-3,5-cyclohenen-2-one 79, which was then transformed into the triacid 80 following a four steps process. The double cyclisation of 80 led to the dione 71, which was converted in [1,2-a]IF(O)2 as reported in 1939 by Radulescu.162
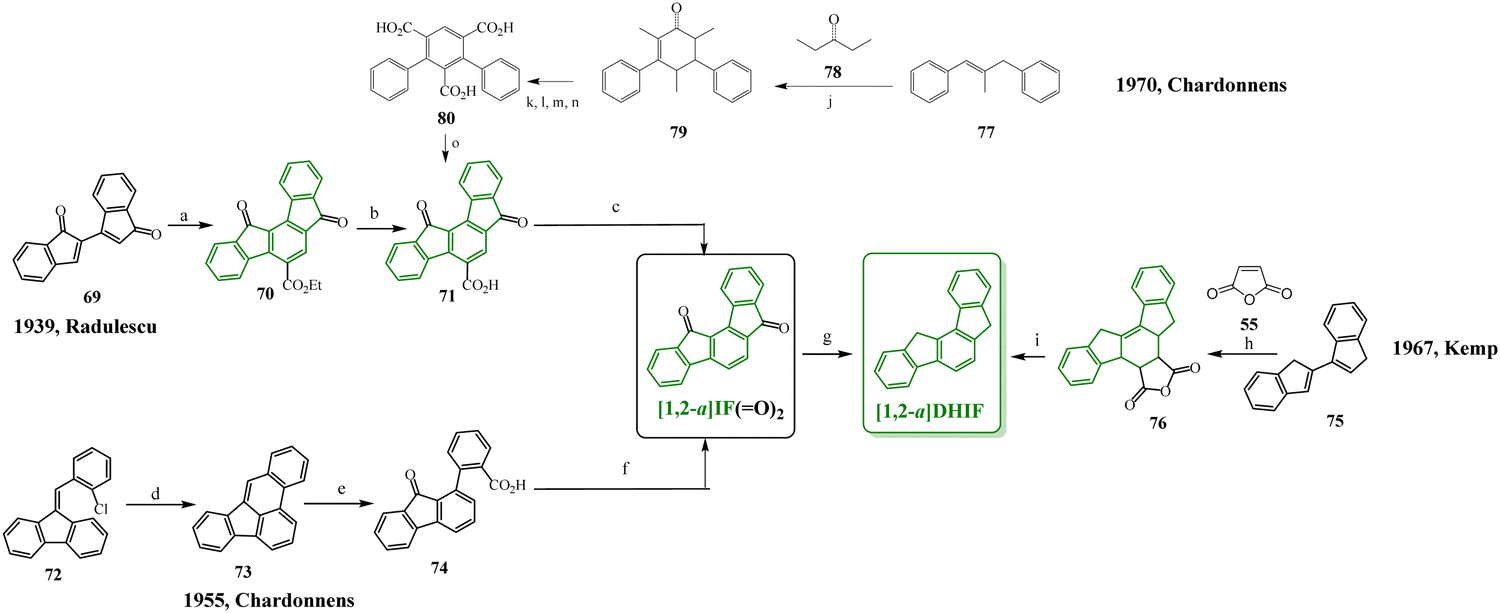 | ||
| Scheme 21 Different routes to [1,2-a]DHIF164 or [1,2-a]IF(O)2.162,163,166 Access to [1,2-a]IF(O)2 from 69: (a) EtOH, NaOEt, ICH2CH2CO2C2H5, (b) KOH, HCl, (c) heating in sublimation apparatus; or from 72: (d) conditions not available, (e) Na2Cr2O7, 60 °C, (f) H2SO4, 140 °C; or from 77: (j) C2H5ONa, (k) Dioxane, 75 °C, NaBH4, (l) H2O, PdC, 295–315 °C, (m) pyridine/H2O, KMnO4, (n) Na2CO3 25%, KMnO4, and (o) H2SO4. Access to [1,2-a]DHIF from [1,2-a]IF(O)2: (g) zinc reduction or from 75: (h) p-xylene, reflux, (i) Cu powder, Ba(OH)2 and soda-glass, heating to 400 °C in an inert atmosphere. | ||
The first synthesis of [2,1-b]DHIF was reported in 1945 (see Scheme 22) from a trinuclear by-product of ellargic acid 81 through zinc dust distillation.167 The name of ellagene was given early to [2,1-b]DHIF. In 1951, Deuschel reported the access to [2,1-b]DHIF168 starting by the condensation of 82 and 83 leading to 84, which was aromatized in 85. The [2,1-b]DHIF core was constructed by double cyclization of 85 giving 86, itself dehydroxylated in [2,1-b]DHIF. Different routes to [2,1-b]DHIF, all involving, in the last step, the hydrogenation of the dione [2,1-b]IF(O)2, were then described starting from different terphenyls168–170 or fluorene derivatives.171
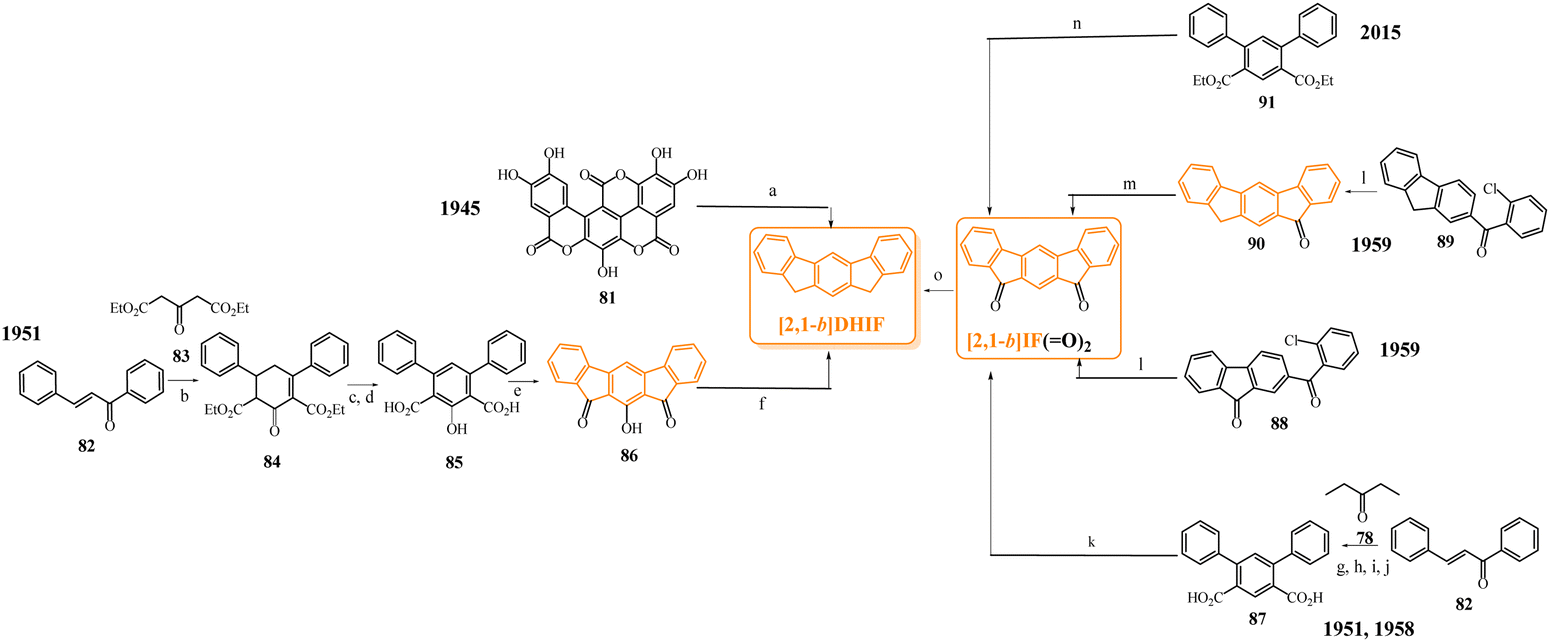 | ||
| Scheme 22 Different routes to [2,1-b]DHIF167,168 or [2,1-b]IF(O)2168–171 described in the literature. Access to [2,1-b]DHIF from 81: (a) zinc dust distillation or from 82: (b) ethanol, piperidin, 40 °C, (c) ethylacetate, N-bromosuccinimide, Br2, (d) KOH 20% in methanol, (e) H2SO4 concentrated, and (f) pyridine, zinc dust, H2SO4. Access to [2,1-b]DHIFvia[2,1-b]IF(O)2, from 82: (g) NaOH/EtOH, reflux H2SO4/H2O, (h) Ether/H2O, Na, cooling to 0 °C then H2SO4 concentrated, (i) Pd/C, 280–300 °C, (j) pyridine/H2O, 90 °C, then KMnO4, (k) H2SO4 concentrated 70–80 °C, or from 88: (l) quinoline, NaOH/H2O, 250 °C, then RT and transfer in HCl 12%, or from 89: (l) quinoline, NaOH/H2O, 250 °C, then RT and transfer in HCl 12%, (m) oxidation or from 91: (n) MsOH 120 °C. Reduction of [2,1-b]IF(O)2 in [2,1-b]DHIF, (o): NH2NH2/KOH. | ||
The condensation of 82 with the diethylcetone 78 leads to the meta-terphenyl 87, of which double cyclization led to [2,1-b]IF(O)2 and to [2,1-b]DHIF.169 Access to the dione was also described, in 1959, from the 2-substituted fluorenone 88 or the 2-substituted fluorene 89 (via the monocetone [2,1-b]IF(O) 90).171 More recently, in 2015, our group prepared the [2,1-b]IF(O)2 from the terphenyl 91.170
Finally, the synthesis of the last positional isomer, [2,1-c]DHIF, also originates from the sixties (Scheme 23) and involves either the condensation of bis-indene 92 and maleic anhydride 55172 or, in a longer process, the syntheses of dione [2,1-c]IF(O)2 from the propiophenone 94.173
 | ||
| Scheme 23 Two routes to [2,1-c]DHIF172 and [2,1-c]IF(O)2.173 Access to [2,1-c]DHIF from 92: (a) p-xylene, reflux, and (b) Cu powder, Ba(OH)2, 400 °C. Access to [2,1-c]DHIFvia[2,1-c]IF(O)2 from 94: (c) Al reduction, (d) H2O, (a) p-xylene, reflux, (e) sulfur, 280 °C, (f) NaOH 0.1 M, 100 °C, (g) quinoline, Cu2Cr2O5, Cu powder, 220 °C then HCl/H2O, (h) pyridine/H2O, KMnO4, (i) concentrated H2SO4, 70–80 °C, and (j) KOH, diethylene glycol, H2NNH2, 180–190 °C. | ||
Other [2,1-c]-IF-based π-conjugated systems, differently substituted, have also been synthesized together with some oligomers and polymers.174–176 Although their photophysical and electrochemical properties have been, in some cases, evaluated, no device applications of the monomers were reported.
The literature only reports a few properties of the five DHIF isomers themselves and no comparative studies of these properties. Concerning the thermal properties, the literature reports a melting point of 140 °C for [2,1-c]DHIF and of 203 °C/212–213 °C or 215–220 °C for [2,1-a]DHIF/[1,2-a]DHIF or [2,1-b]DHIF respectively. The only data available is a high decomposition temperature of 200 °C reported for [1,2-b]DHIF.40
The UV-vis absorption spectra reported in different studies show a red-shift of the lowest energy absorption bands from 314 nm for [2,1-c]DHIF165 to 334 nm for [1,2-b]DHIF40 with intermediate values of 321/322 and 332 nm for [1,2-a]DHIF164/[2,1-a]DHIF21 and [2,1-b]DHIF170 respectively. These values are in accordance with an extension of conjugation in the DHIF core from the less conjugated ortho-DHIF to the more conjugated para-DHIF with a weaker modulation in the para–syn-isomer and in the two meta isomers.
As far as we know, emission properties have only been reported for three isomers (no data found for [2,1-c]DHIF or for [1,2-a]DHIF). The three compounds present structured emission spectra with small Stokes shifts (below 5 nm) and emission maximum increasing from 326 to 337 and 339 nm for [2,1-a]DHIF, [2,1-b]DHIF and [1,2-b]DHIF in accordance with an increase of the π-conjugation. For the three compounds the fluorescent quantum yield was reported around 0.6.
4.2. Impact of the positional isomerism on the electronic properties: the example of the five dispirofluorene-indenofluorene (DSF-IF) isomers
The IF cores display different geometries, which lead to very different and unusual electronic properties, which have been reported by our group since 2006. The five dispirofluorene-indenofluorene (DSF-IF) regioisomers (dihydroindeno-fluorenes substituted on the bridges by fluorenyl units, Fig. 11) have been the investigated compounds for this structure/properties relationship study. In these examples, two parameters drive the properties: the nature of the linkage (ortho, meta, para) and the position of the bridges (syn, anti). Synthesis of the five DSF-IF isomers and the main physicochemical properties, summarized in Table 8, will only be briefly summarized hereafter as they have been described in detail in previous publications21,24,40,45 and in a dedicated account.27 | ||
| Fig. 11 Molecular structure of the five DSF-IF isomers and their respective cyclic voltammetries in oxidation. | ||
| [1,2-b]-DSF-IF | [2,1-a]-DSF-IF | [1,2-a]-DSF-IF | [2,1-b]-DSF-IF | [2,1-c]-DSF-IF | |
|---|---|---|---|---|---|
| a In CH2Cl2 + Bu4NPF6 0.2 M. b From Eoxonset. c In DMF + Bu4NPF6 0.1 M. d In THF + Bu4NPF6 0.1 M. e From Eredonset. f In THF. g Quinine sulfate as the reference. h In methylcyclohexane/2-methylpentane. | |||||
| T d (°C) | 355 | — | 330 | 359 | — |
| E ox (V)avs. SCE | 1.47, 1.87 | 1.36, 1.69, 1.99 | 1.57, 1.88 | 1.53, 1.94 | 1.6(sh), 1.77 |
| HOMOb (eV) | −5.76 | −5.64 | −5.86 | −5.80 | −5.87 |
| E red (V) vs. SCE | −2.32c | −2.48c | −2.86d | −2.84d | −2.59d |
| LUMOe (eV) | −2.17 | −2.03 | −1.70 | −1.75 | −2.00 |
| λ abs (nm) | 298, 310, 328, 336, 344 | 296, 311, 323, 339 | 298, 308, 333, 342 | 298, 310, 326, 334, 342 | 298, 309, 317, 338 |
| λ em (nm) | 343, 358 | 345, 363, 381, 403 | 341, 354 | 346, 354, 363 | 344, 358 |
| ∅solfg | 0.62 | 0.60 | 0.23 | 0.54 | 0.50 |
| E T (eV) | 2.52 | 2.52 | 2.76 | 2.76 | 2.63 |
The five positional isomers display different electrochemical behaviours, highlighting the impact of the positional isomerism on the electron transfers. Comparing first the isomers based on the para-terphenyl core, [1,2-b]-DSF-IF presents two oxidation waves with maxima at 1.47 and 1.87 V vs. SCE, whereas [2,1-a]-DSF-IF possesses three waves at 1.36, 1.69, and 1.99 V vs. SCE. For both isomers, the first monoelectronic oxidation wave has been assigned to the oxidation of the IF core. Compared with [1,2-b]-DSF-IF, two differences are noted for [2,1-a]-DSF-IF: (i) the first oxidation (1.36 V vs. SCE) is shifted toward less anodic potentials and (ii) an additional oxidation process is observed at 1.69 V vs. SCE. The shift between the first oxidation potentials of [1,2-b]-DSF-IF and [2,1-a]-DSF-IF is due to the specific arrangement of the two fluorenyl units. In fact, in [2,1-a]-DSF-IF, the central IF core is subjected to a less important withdrawing effect of the cofacial fluorenyl units which are in strong interaction (vs. the two non-interacting fluorenyl units in [1,2-b]-DSF-IF). The additional oxidation detected at 1.69 V vs. SCE for [2,1-a]-DSF-IF was ascribed to the oxidation of this cofacial fluorene dimer. Thus, the different ring bridge arrangements of [1,2-b]-DSF-IF and [2,1-a]-DSF-IF and the different geometries induced lead to distinct electrochemical properties. As these two molecular systems are built on an identical para-terphenyl fragment this indicates that the linkage is not the only parameter implied in the electronic properties. Indeed, the syn arrangement significantly modified these properties by contracting the [1,2-b]-IF core.
[1,2-a]-DSF-IF and [2,1-b]-DSF-IF isomers provide a different example. Both compounds possess two oxidation waves at 1.57/1.88 V vs. SCE for [1,2-a]-DSF-IF and at 1.53/1.94 V vs. SCE for [2,1-b]-DSF-IF. The first monoelectronic oxidation has been assigned to the oxidation of the IF core, and the second multielectronic wave to the concomitant oxidation of fluorenyl units and of the IF core radical cation. The shift of the first oxidation potential to more positive values from [1,2-b]-DSF-IF/[2,1-a]-DSF-IF to [1,2-a]-DSF-IF/[2,1-b]-DSF-IF has been assigned to a linkage effect (para vs. meta linkages). Linkage effects were particularly studied in the field of phosphorescent OLEDs to design high ET1 materials. Our group has notably investigated the differences induced by ortho, meta and para linkages on the parameters, which drive the OLEDs performances (ie: HOMO/LUMO energy levels, ES1, ET1, …) for many different π-conjugated positional isomers.42,44,177,178
The slight shift in the first oxidation potential from [2,1-b]-DSF-IF to [1,2-a]-DSF-IF, despite their identical meta linkages, has been assigned to the contraction and deformation of the [1,2-a]DHIF core of [1,2-a]-DSF-IF, which induces poorer delocalization of the π electrons and hence a more anodic oxidation potential.
Finally, isomer [2,1-c]-DSF-IF displays a singular behaviour, its first oxidation being detected as a shoulder at 1.6 V. The ortho-terphenyl linkage of [2,1-c]-DSF-IF causes a deformation of the IF core, leading to an inversion of the HOMO and HOMO-1 compared with the other isomers.24
In conclusion, the linkages are strongly involved in the π-delocalization and therefore the electrochemical properties. However, the ring bridging is also of chief importance with two main consequences: (i) it changes the geometry (syn vs. anti) and can force the two cofacial fluorene moieties to interact such as in [2,1-a]-DSF-IF and (ii) it can increase the deformation of the IF core such as in [2,1-c]-DSF-IF.
The positional isomerism of the IF core also has important consequences on the optical properties. In THF, the five isomers display very similar low-energy absorption maxima between 338 and 344 nm (Fig. 12 and Table 8). [1,2-b]-DSF-IF and [2,1-a]-DSF-IF possess similar well-resolved absorption spectra, which is a characteristic of highly rigid structures. Compared to [1,2-b]-DSF-IF, the UV-vis spectrum of [2,1-a]-DSF-IF exhibits a slightly shifted main absorption band (344/339 nm for [1,2-b]-DSF-IF/[2,1-a]-DSF-IF). This blue shift has been assigned to a better delocalization of the π electrons in [1,2-b]-DSF-IF compared to [2,1-a]-DSF-IF. In contrast to the electrochemistry, [1,2-b]-DSF-IF and [2,1-a]-DSF-IF display similar behaviours in absorption spectroscopy, and the cofacial fluorenes of [2,1-a]-DSF-IF have only a limited influence on the electronic transitions. The spectrum of [2,1-b]-DSF-IF was also similar in shape but was red-shifted by 3 nm compared with the para–syn homologue [2,1-a]-DSF-IF. Hence, para-substituted compounds do not always present longer π-conjugated pathways vs. their meta-substituted analogues. This indicates that the linkages are not the only feature driving the electronic properties and that the bridging is also of key importance.
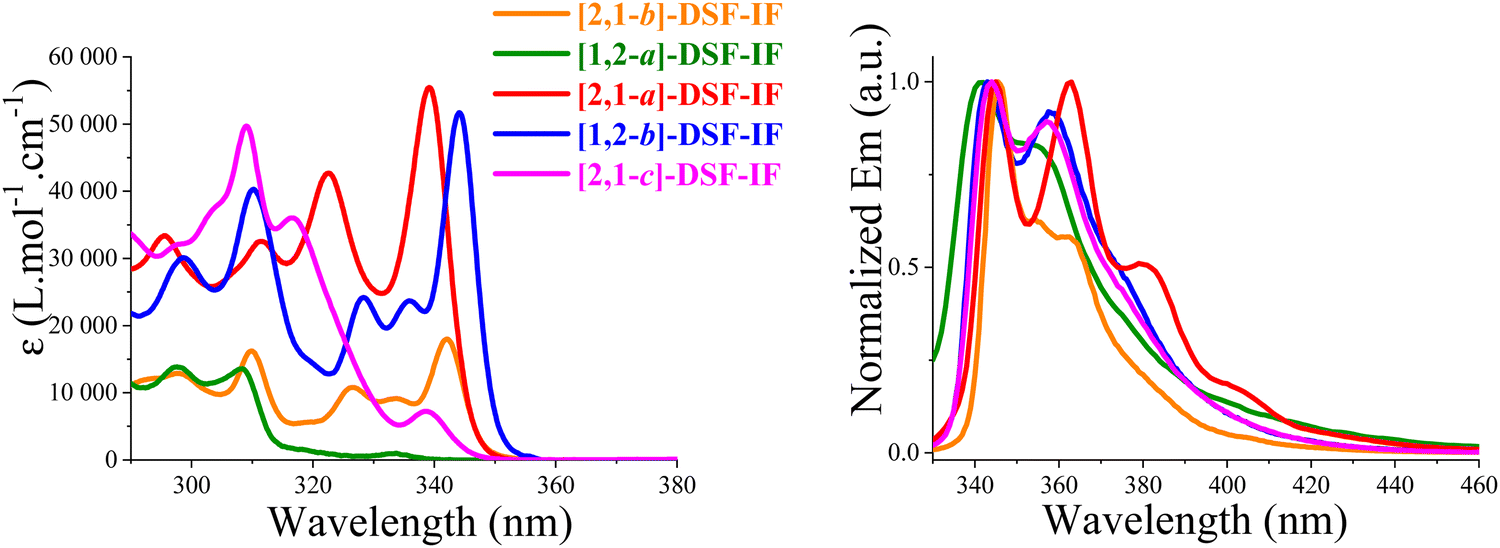 | ||
| Fig. 12 Absorption (left) and emission (right) spectra of the five DSF-IF isomers in THF. Adapted with permission from ref. 27. Copyright 2023 American Chemical Society. | ||
The specific absorption spectrum of [1,2-a]-DSF-IF highlights the role played by the ring bridging (structural deformation) in the transitions. Indeed, isomers [1,2-b]-DSF-IF, [2,1-a]-DSF-IF and [2,1-b]-DSF-IF all possess low energy absorption bands with high ε whereas very weak bands at 333 and 342 nm with low ε were displayed for the isomer [1,2-a]-DSF-IF (Fig. 12). Indeed, [1,2-a]-DSF-IF has no symmetry axis/plane, and its constrained structure leads to a deformation of its [1,2-a]-central core, which modifies this first π–π* transition. [2,1-c]-DSF-IF, which also possesses a highly distorted IF backbone, displays an identical characteristic with a weak band at 338 nm.
With regard to the emission properties, [1,2-b]-DSF-IF, [2,1-a]-DSF-IF and [2,1-b]-DSF-IF possess well-resolved spectra with a very small Stokes shift. [2,1-a]-DSF-IF presents the most important Stokes shift, which has been related by theoretical calculations to structural modifications. Indeed, in the ground state [2,1-a]-DSF-IF shows a staggered conformation of the fluorenes, whereas in the first singlet excited state a cofacial eclipsed arrangement was detected.21 Compared to the other isomers, [1,2-a]-DSF-IF was found to have the lowest quantum yield (0.23) and its emission decay occurred with two lifetimes instead of one. This unusual emission translates to unconventional processes, such as multiple emissive states or partially decoupled excited states and points to the impact of the regioisomerism on the photophysics.
The phosphorescence contribution was also very informative. The two para-isomers ([1,2-b]-DSF-IF and [2,1-a]-DSF-IF) and the two meta-isomers ([1,2-a]-DSF-IF and [2,1-b]-DSF-IF) possess almost identical phosphorescence contributions, indicating that the triplet state is driven by the linkages (para or meta) with a very limited influence of the ring bridging. The ET1 value were therefore identical for [1,2-b]-DSF-IF and [2,1-a]-DSF-IF (2.52 eV) and for [1,2-a]-DSF-IF and [2,1-b]-DSF-IF (2.76 eV). ET1 of the ortho-isomer [2,1-c]-DSF-IF was reported to be 2.63 eV. Thus, it has been shown that ET1 is only very weakly dependent on the bridging. This was an important result, which has driven further studies on this fragment. Thanks to an ET1 above 2.5 eV, all these materials have been incorporated as host materials in green and/or blue PhOLEDs (see below).
4.3. Uses of [1,2-a]-, [2,1-b]- or [2,1-c]DHIF-based organic semiconductors in OLED or PhOLEDs
Despite being largely less developed than the [1,2-b]DHIF- and [2,1-a]DHIF-based organic semiconductors described above in parts 2 and 3, the literature also reports some examples of electronic devices using [1,2-a]-, [2,1-b]- or [2,1-c]-IF-based organic semiconductors. First, [1,2-a]-DSF-IF and [2,1-b]-DSF-IF have been used as the EML in fluorescent multilayer OLED33-OLED34. OLED34 using [2,1-b]-DSF-IF as the EML displays higher performances than OLED33 with [1,2-a]-DSF-IF reaching a CE of 0.7 cd A−1. Interestingly, both devices display a violet emission with CIE coordinates of (0.193, 0.128) for OLED33 and (0.201, 0.188) for OLED34.[2,1-b]-83 76 (Scheme 24) is an isomer of [1,2-b]-11 (Scheme 4) and has been used as a dopant in ML-OLED with host-guest EMLs (see OLED35-OLED37). With similar architectures to those used with [1,2-b]-11 in OLED22-OLED24 (Table 1), OLED35-OLED37 with [2,1-b]-83 doped in MADN as the EML were largely less efficient. In fact, EQEmax was reported close to 6% for OLEDs22–24 whereas EQEmax reached less than 1.5% for OLEDs35–37. The different IF core in [2,1-b]-83 and [1,2-b]-11 leads to a different gap for the two isomers76 (2.93/2.85 eV for [1,2-b]-83/[1,2-b]-11). The shortest conjugation has an influence on the efficiency of the devices. The red shift of the emission of [1,2-b]-11/[1,2-b]-83, from 464 nm to 492 nm was also observed in the EL spectra of the devices, which were red-shifted from 453/455 nm for OLEDs22–24 (blue emission, CIE: 0.14; 0.2) to 507/508 nm for OLEDs35–37 (bluish-green emission, CIE: 0.22, 0.36).
 | ||
| Scheme 24 Molecular structure of organic semiconductors based on [2,1-b]-, [1,2-a]- or [2,1-c]-indenofluorenyl cores used in OLED or PhOLEDs. | ||
[2,1-b]-84 179 is built on the linkage of two [2,1-b]DHIF units via a spiro bridge. This 3D rigid blue emitter was prepared from its dione parent.180 The length of each [2,1-b]DHIF core, measured on the X-Ray structure, was 10.68/10.62 Å similar to that of [2,1-b]-DSF-IF (10.63 Å) and the distance of the spiro bridge in each [1,2-b]DHIF unit was 5.22/5.25 Å, slightly shorter than the one measured for [2,1-b]-DSF-IF (5.27 Å). TGA measurements show that [2,1-b]-84 was highly thermally stable with a Td of 450 °C, nearly 100 °C superior to that of [2,1-b]-DSF-IF (359 °C) which can be seen as “half” of this molecule. Also, except the thermal properties, all the other physicochemical properties (optical, electrochemical) were similar for the two compounds. [2,1-b]-84 was used as the host for the yellow fluorescent emitter 4CzPNPh (3,4,5,6-tetrakis(3,6-diphenyl-carbazol-9-yl)-1,2-dicyanobenzene) in a double EML OLED configuration: 4CzPNPh doped [2,1-b]-84/non-doped [2,1-b]-84. With the first 5% doping level for 4CzPNPh in [2,1-b]-84, OLED38 reaches very high performances CE(cd A−1)/PE(lm W−1)/EQE(%) of 32.5/15.3/11. Blue and yellow emission peaks were observed in the EL spectrum of OLED38 at 400 nm (blue emission of [2,1-b]-84) and at 550 nm (yellow emission of 4CzPNPh). The blue emission was however weak at a low voltage (4.7 V) and increases moderately with the voltage (up to 13.7 V). A second device was tested with a lower 4CzPNPh concentration, 0.5% in OLED39, and both blue and yellow emission were observed however the OLED performances were significantly lower (CE(cd A−1)/PE(lm W−1) /EQE(%) of less than 18/10/7.5). In the EL spectrum of OLED39, the blue/yellow ratio emission was higher than in OLED38 (Table 9).
| Device | Device architecture | V on (V) | EQEa (%) | CEa (cd A−1) | PEa (lm W−1) | Luminance (cd m−2) | λ (EL spectrum) nm CIE(x, y) | Ref. |
|---|---|---|---|---|---|---|---|---|
| a @10 mA cm−2. b Maximum value. | ||||||||
| OLED33 | ITO/CuPc/NPB/TCTA/[1,2-a]-DSF-IF/TPBI/LiF/Al | 4.5 | 0.5 | 0.4 | — | 200b | 390–400 nm (0.193, 0.128) | 23 |
| OLED34 | ITO/CuPc/NPB/TCTA/[2,1-b]-DSF-IF/TPBI/LiF/Al | 4.9 | 0.9 | 0.7 | — | 704b | 390–400 nm (0.201, 0.188) | 23 |
| OLED35 | ITO/DNTPD(60 nm)/NPB(30 nm)/[2,1-b]-83 3% in MADN (30 nm)/Alq3 (20 nm)/LiF(1 nm)/Al | 4.0 | 0.91 | 2.21 | 1.59 | 7 394 | 507 (0.222, 0.376) | 76 |
| OLED36 | ITO/DNTPD(60 nm)/NPB(30 nm)/[2,1-b]-83 5% in MADN (30 nm)/Alq3 (20 nm)/LiF(1 nm)/Al | 3.5 | 0.89 | 1.99 | 1.18 | 9 490 | 508 (0.217, 0.362) | 76 |
| OLED37 | ITO/DNTPD(60 nm)/NPB(30 nm)/[2,1-b]-83 7% in MADN (30 nm)/Alq3 (20 nm)/LiF(1 nm)/Al | 3.5 | 1.37 | 3.68 | 3.71 | 9 114 | 507 (0.216, 0.355) | 76 |
| OLED38 | ITO/MoO3(6 nm)/NPB(70 nm)/mCP(5 nm)/[2,1-b]-84:4CzPNPh 5%(20 nm)/[2,1-b]-76(5 nm)/TPBI(30 nm)/LiF(1 nm)/Al | — | 11 | 32.5 | 15.3 | — | — | 179 |
| OLED39 | ITO/MoO3(6 nm)/NPB(70 nm)/mCP(5 nm)/[2,1-b]-84:4CzPNPh 0.5%(20 nm)/[2,1-b]-76(5 nm)/TPBI(30 nm)/LiF(1 nm)/Al | — | 7.5 | 18 | 10 | — | — | 179 |
| OLED40 | ITO/[1,2-a]-85(60 nm)/Alq3(60 nm)/LiF(0.5 nm)/Al(150 nm) | <3.0 | — | 3.4 | — | 37000 (@14 V) |
530 nm | 181 |
| OLED41 | ITO/PEDOT/PVK/[1,2-a]-86/TPBI/Ba/Al | 5.3 | 2.0 | — | — | 774 (@8.4 V) | 432 nm 0.17, 0.08 | 182 |
| OLED42 | ITO/PEDOT/PVK/[1,2-a]-87/TPBI/Ba/Al | 3.9 | 1.4 | — | — | 1717 (@7.4 V) | 424 nm 0.17, 0.08 | 182 |
| OLED43 | ITO/PEDOT/PVK/[1,2-a]-88/TPBI/Ba/Al | 5.8 | 2.9 | — | — | 480 (@7.8 V) | 442 nm 0.16, 0.09 | 182 |
| OLED44 | ITO/PEDOT/PVK/[1,2-a]-89/TPBI/Ba/Al | 5.0 | 2.4 | — | — | 997 (@6.7 V) | 432 nm 0.17, 0.10 | 182 |
| SL-OLED45 | ITO/PEDOT(40 nm)/[1,2-a]-89(60 nm)/Ba(4 nm)/Al(150 nm) | — | — | 0.01 | — | — | — | 183 |
| SL-OLED46 | ITO/PEDOT(40 nm)/[1,2-a]-90(60 nm)/Ba(4 nm)/Al(150 nm) | 4.7 | — | 0.04 | — | 230 | 0.20, 0.21 | 183 |
| SL-OLED47 | ITO/PEDOT(40 nm)/[1,2-a]-91(60 nm)/Ba(4 nm)/Al(150 nm) | 5.2 | — | 0.10 | — | 150 | 0.17, 0.12 | 183 |
| SL-OLED48 | ITO/PEDOT(40 nm)/[1,2-a]-92(60 nm)/Ba(4 nm)/Al(150 nm) | 5.2 | — | 0.08 | — | 250 | 0.17, 0.11 | 183 |
| OLED49 | ITO/PEDOT(40 nm)/PVK(40 nm)/[1,2-a]-87(60 nm)/Ba(4 nm)/Al (150 nm) | 6.9 | — | 0.39 | — | 1980 | 0.17, 0.07 | 183 |
| OLED50 | ITO/PEDOT(40 nm)/PVK(40 nm)/[1,2-a]-90(60 nm)/Ba(4 nm)/Al (150 nm) | 6.9 | — | 0.43 | — | 1550 | 0.18, 0.10 | 183 |
| OLED51 | ITO/PEDOT(40 nm)/PVK(40 nm)/[1,2-a]-91(60 nm)/Ba(4 nm)/Al (150 nm) | 8.1 | — | 0.80 | — | 463 | 0.17, 0.08 | 183 |
| OLED52 | ITO/PEDOT(40 nm)/PVK(40 nm)/[1,2-a]-92(60 nm)/Ba(4 nm)/Al (150 nm) | 6.6 | — | 0.56 | — | 414 | 0.17, 0.07 | 183 |
In 2005, Takagi's group181 synthesized [1,2-a]-85 based on a central planar truxene core,184 which can be seen as an extended [1,2-a]DHIF core (Scheme 24). Three 2,7-diphenyl-aminofluorenes are spirolinked to the central truxene backbone. This compound was used as the HTL in a green fluorescent OLED using Alq3 as the emitter (OLED40). This OLED emits green light from Alq3 at Von lower than 3 V and reaches luminance as high as 37 000 cd m−2 at 14 V. The lifetime of OLED40 was studied at a luminance of 200 cd m−2 and showed that the device could work for a thousand hours. Therefore, [1,2-a]-85 appears to be a thermally durable HTL in combination with Alq3 in green fluorescent OLEDs.
In 2007, [1,2-a]-86-87 and [1,2-a]-88-89 all based on the [1,2-a]-IF-based truxene core were prepared and used as the blue emitting layer in ML-OLEDs.182 These organic semiconductors were highly stable with Td > 400 °C. Their UV-vis absorption emission spectra were rather similar and appear to be directed by the more conjugated system (terfluorene) surrounding the central truxenyl unit. Indeed, their spectra are similar to those of other organic semiconductors possessing terfluorenyl driving systems as for example [1,2-b]-72 (Fig. 10).142 OLEDs41–44 based on these organic semiconductors exhibit blue light and color stability at different operating voltages. OLED43-OLED44 based on [1,2-a]-88 and [1,2-a]-89 with an EQE of 2.9/2.4% were slightly more efficient than OLED41-OLED42, based on [1,2-a]-86 and [1,2-a]-87 with an EQE of 2.0/1.4%. The presence of external spirobifluorene in [1,2-a]-88-89 instead of dialkylfluorene in [1,2-a]-86-87 may change the packing and therefore the charge transport in the materials.
Then, in 2010, three organic semiconductors based on a similar truxene central core possessing three flexible alkoxy chains ([1,2-a]-90 to [1,2-a]-92183) have been designed. Electron-rich units (carbazole in [1,2-a]-90183) or electron-deficient moieties (1,2,4-triazole in [1,2-a]-91183 or 2,5-diphenyl-1,3,4-oxadiazole in [1,2-a]-92183) were introduced on the flexible alkoxy chains (Scheme 24). The three organic semiconductors display similar optical properties to [1,2-a]-89 mainly governed by the external highly conjugated SBF-fluorenyl-SBF arms. This indicates that the substitution of the central truxene by different electron-rich or electron-deficient units via non-conjugated alkoxy chains does not affect the main photophysical properties of [1,2-a]-90 to [1,2-a]-92. The HOMO/LUMO levels of the three organic semiconductors were determined by cyclic voltammetry on solid films. The HOMO levels of [1,2-a]-90-[1,2-a]-92 were similar (−5.6 eV), but their LUMO levels were different: −2.4/−2.7/−2.6 eV for [1,2-a]-90/[1,2-a]-91/[1,2-a]-92 respectively. The authors ascribed the HOMO to the external highly conjugated SBF-fluorenyl-SBF arms whereas the LUMO was ascribed to the alkoxy-electron-deficient moieties.
SL-OLEDs with [1,2-a]-90 to [1,2-a]-92 as the EML (SL-OLEDs46–48) display higher performances than SL-OLED45 based on [1,2-a]-89 showing that the introduction of the side chains on the truxenyl core has a positive effect on the performances. The performances remained however very low: 0.01 < CE < 0.1 cd A−1 for the four SL-OLED devices but higher device efficiencies (0.3 < CE < 0.8 cd A−1) were reached introducing a thin layer (40 nm) of PVK (working as the HTL/EBL) between the anode and the EML (OLEDs49–52).
The potential of several [1,2-a]DHIF-, [2,1-b]DHIF- or [2,1-c]DHIF-based organic semiconductors have also been then tested as hosts for phosphorescent dopants (Table 10). First, [1,2-a]-DSF-IF, [2,1-b]-DSF-IF and [2,1-c]-DSF-IF (Fig. 11) with an ET1 value of 2.76 eV for the two former and 2.63 eV for the latter were tested as the host for green PhOLEDs. [1,2-a]-DSF-IF as the host for green phosphorescent Ir(ppy)3 has been tested in PhosOLED3 with a similar architecture to PhosOLED2 using [1,2-b]-DSF-IF as the host (Table 4). PhosOLED3 emits light at a low voltage of 2.8 V with a CEmax as high as 57.3 cd A−1, a PEmax of 30.3 lm W−1 and an EQEmax of 14.8%. These performances were higher than that of PhosOLED2 (CEmax/PEmax/EQEmax: 31.4 cd A−1 /15.5 lm W−1 /9.9%) showing that the higher ET1 of [1,2-a]-DSF-IF may help in the efficiency of the device. With an ET1 of 2.76 eV, [1,2-a]-DSF-IF has also been used as the host for the sky-blue phosphor FIrpic, which ET1: 2.64 eV allows the triplet exciton transfer from the host to the guest. With different doping levels (25% in PhosOLED4, 19% PhosOLED5 or 20% in PhosOLED6), the best performance was recorded for PhosOLED4 (CEmax/PEmax/EQEmax: 21.1 cd A−1 /11.9 lm W−1 /7.5% and Von: 3.2 V). These results showed that despite an extended π-conjugated core, the ET1 can be kept sufficiently high in a [1,2-a]-indenofluorenyl core.
| Device | Device architecture | V on (V) | EQEb (%) | CEb (cd A−1) | PEb (lm W−1) | Luminance (cd m−2) | CIE (x, y) | Ref. |
|---|---|---|---|---|---|---|---|---|
| a @10mA cm−2. b Maximum value. | ||||||||
| PhosOLED3 | ITO/CuPc(10 nm)/NPB(40 nm)/TCTA(10 nm)/[1,2-a]-DSF-IF:Ir(ppy)3 10% (20 nm)/TPBI(40 nm)/LiF(1.2 nm)/Al(100 nm) | 2.8 | 14.8 | 57.3 | 30.3 | 31500@240 mA cm−2 |
0.30, 0.63 | 24 |
| PhosOLED4 | ITO/CuPc(10 nm)/NPB(40 nm)/TCTA(10 nm)/[1,2-a]-DSF-IF:FIrpic 25% (20 nm)/TPBI(40 nm nm)/LiF (1.2 nm) /Al(100 nm) | 3.2 | 7.5 | 21.1 | 11.9 | — | 0.18, 0.45 | 24 |
| PhosOLED5 | ITO/CuPc(10 nm)/NPB(40 nm)/TCTA(10 nm)/[1,2-a]-DSF-IF:FIrpic 19% (20 nm)/TPBI(40 nm)/LiF (1.2 nm)/Al(100 nm) | 3.2 | 6.1 | 16.7 | 8.9 | — | 0.18, 0.45 | 24 |
| PhosOLED6 | ITO/CuPc(10 nm)/NPB(40 nm)/TCTA(10 nm)/[1,2-a]-DSF-IF:FIrpic 20% (20 nm)/TPBI(40 nm)/ LiF(1.2 nm)/Al(100 nm) | 4.0 | 5.7 | 15.4 | 8.5 | 4600@100 mA cm−2 | 0.18, 0.44 | 25 |
| PhosOLED8 | ITO/CuPc(10 nm)/NPB(40 nm)/TCTA(10 nm)/[2,1-b]-DSF-IF:Ir(ppy)3 10% (20 nm)/TPBI(40 nm)/ LiF(1.2 nm)/Al(100 nm) | 3.7 | 12.3a | 49.6a | 18.5a | 30000@220 mA cm−2 |
0.32, 0.62 | 25 |
| PhosOLED9 | ITO/CuPc(10 nm)/NPB(40 nm)/TCTA(10 nm)/[2,1-b]-DSF-IF:FIrpic 20% (20 nm)/TPBI(40 nm)/ LiF(1.2 nm)/Al(100 nm) | 3.9 | 5.3 | 14.7 | 8.4 | 3300@60 mA cm−2 | 0.20, 0.44 | 25 |
| PhosOLED10 | ITO/CuPc(10 nm)/NPB(40 nm)/TCTA(10 nm)/[2,1-c]-DSF-IF:Ir(ppy)3 10% (20 nm)/TPBI(40 nm)/ LiF(1.2 nm)/Al(100 nm) | 3.0 | 13.3 | 49.0 | 26.6 | — | 0.31, 0.63 | 24 |
| PhosOLED11 | ITO/CuPc(10 nm)/NPB(40 nm)/TCTA(10 nm)/[2,1-b]-93:Ir(ppy)3 10% (20 nm)/TPBI(40 nm)/LiF (1.2 nm)/Al(100 nm) | 3.8 | 6.5 | 23.3 | 15 | 11200@170 mA cm−2 |
0.32, 0.62 | 25 |
| PhosOLED12 | ITO/CuPc(10 nm)/NPB(40 nm)/TCTA(10 nm)/[2,1-b]-93:FIrpic 20% (20 nm)/TPBI(40 nm)/LiF (1.2 nm)/Al(100 nm) | 3.6 | 2.9 | 7.0 | 4.5 | 1800@90 mA cm−2 |
0.19, 0.41 | 25 |
| PhosOLED13 | ITO/CuPc(10 nm)/NPB(40 nm)/TCTA(10 nm)/[2,1-b]-94:Ir(ppy)3 10% (20 nm)/TPBI(40 nm)/LiF (1.2 nm)/Al(100 nm) | 3.3 | 12.8 | 44.1 | 30.3 | 26100@170 mA cm−2 |
0.33, 0.62 | 25 |
| PhosOLED14 | ITO/CuPc(10 nm)/NPB(40 nm)/TCTA(10 nm)/[2,1-b]-94:FIrpic 20% (20 nm)/TPBI(40 nm)/LiF (1.2 nm)/Al(100 nm) | 4.8 | 3.4 | 9.0 | 4.5 | 2500@70 mA cm−2 | 0.19, 0.43 | 25 |
| PhosOLED15 | ITO/CuPc(10 nm)/NPB(40 nm)/TCTA(10 nm)/[2,1-b]-95:Ir(ppy)3 10% (20 nm)/TPBI(40 nm)/LiF(1.2 nm)/Al(100 nm) | 3.2 | 13.8 | 52.2 | 33.5 | — | 0.33, 0.62 | 26 |
| PhosOLED16 | ITO/CuPc(10 nm)/NPB(40 nm)/TCTA(10 nm)/[2,1-b]-95:FIrpic 20% (20 nm)/TPBI(40 nm)/LiF(1.2 nm)/Al(100 nm) | 3.9 | 4.8 | 15.2 | 8.4 | — | 0.20, 0.47 | 26 |
| PhosOLED17 | ITO/CuPc(10 nm nm)/NPB(40 nm)/TCTA(10 nm)/[1,2-a]-96:Ir(ppy)3 10% (20 nm)/TPBI(40 nm)/LiF (1.2 nm)/Al(100 nm) | 3.9 | 15.9 | 55.7 | 33.3 | 32250@140 mA cm−2 |
0.30, 0.63 | 25 |
| PhosOLED18 | ITO/CuPc(10 nm)/NPB(40 nm)/TCTA(10 nm)/[1,2-a]-97:FIrpic 20% (20 nm)/TPBI(40 nm)/LiF (1.2 nm)/Al(100 nm) | 3.9 | 7.9 | 19.6 | 10.9 | 6400@80 mA cm−2 | 0.18, 0.42 | 25 |
It must be noted, that in 2015, the performance of PhosOLED3 was the third most efficient green device using a pure hydrocarbon (PHC) host (top one: EQE: 17.3% with a trimer of Spirobifluorene as the host185 and top two: EQE: 15.8% with [1,2-b]-43117 (see PhOLED 1 in Table 4)). Since then, thanks to the optimization of the device architectures and to the design of new efficient PHC hosts, EQEmax now surpass 27% for the three colours.186,187 These pioneering works on indenofluorenyl derivatives have significantly contributed to the development of PHC host materials in PhOLEDs, which is now a very promising approach.116,186
With an ET1 of 2.76 eV, [2,1-b]-DSF-IF was also adapted to host both green and blue phosphors. With an architecture similar to that used with both [1,2-b]-DSF-IF and [1,2-a]-DSF-IF, the green PhosOLED8 appears as efficient as PhosOLED3 indicating that the charge injection and the energy transfer from the host to guest are similar for the two isomers. However, the blue PhosOLED9 appears slightly less efficient than PhosOLED4 highlighting the difference between the isomers as a function of the phosphor used.
[2,1-c]-DSF-IF, with an ET1 of 2.63 eV has only been used to host Ir(ppy)3 in PhosOLED10. Compared to PhosOLED2 and PhosOLED8, this device presents the lowest Von (3.0 V) and the highest EQEmax (13.3%) showing the interest of using the [2,1-c]-DHIF core. PhOLED10 with [2,1-c]-DSF-IF as the host is however slightly less efficient than PhOLED3 with [1,2-a]-DSF-IF as the host with a larger Von (3.0 vs. 2.8 V) and the lowest EQE (13.3 vs. 14.8%).
In order to increase the devices efficiency using host materials with a DHIF backbone, our group synthesized, in 2015, the donor–acceptor derivatives [2,1-b]-93 and [1,2-a]-96 (Scheme 24), in which each central IF core was, on one hand, substituted by two electron-acceptor groups CF3 and on the other hand spiro-bridged to two donor moieties (phenylacridine).25 The main idea behind this work was to improve the injection and transport of charges through the incorporation of donor and acceptor units keeping the ET1 controlled by the IF core. Indeed, the development of the Donor/Acceptor host has been an important step to reach high performance PhOLED, particularly single layer devices.188 The oxidation of [2,1-b]-93, Fig. 13, involves first the oxidation of the phenylacridine units whereas its reduction involves the central electron-deficient IF core. Compared to [2,1-b]-DSF-IF, the oxidation and reduction both involve the IF core, the HOMO and LUMO of [2,1-b]-93 were respectively higher and lower leading to a strong contraction of the electrochemical gap from [2,1-b]-DSF-IF (4.05 eV) to [2,1-b]-93 (3.14 eV).
 | ||
| Fig. 13 Anodic oxidation (Left), phosphorescence spectra (Right) and electrochemical HOMO/LUMO levels of [2,1-b]-93 (in orange) and [1,2-a]-96 (in green) and of their related DSF-IF parents. | ||
For the same reasons, the gap of [1,2-a]-96 (3.10 eV) was more contracted than that of [1,2-a]-DSF-IF (4.06 eV). Looking at the phosphorescence spectra, the ET1 values were nevertheless similar (2.76/2.74 eV for [2,1-b]-DSF-IF/[2,1-b]-93 and for [1,2-a]-DSF-IF/[1,2-a]-96) indicating that for both isomers, the incorporation of CF3 groups and of phenylacridine (instead of fluorene) has only weak influence on the ET1 values (0.02 eV). This shows that the HOMO and LUMO energy levels can be controlled and tuned, maintaining the high ET1 of the IF core.
With such high ET, [2,1-b]-93, [2,1-b]-94 and [1,2-a]-96 have been used as hosts for green (Ir(ppy)3) and blue (FIrpic) phosphors. For both blue and green devices, the most efficient devices were those based on [1,2-a]-96 as the host (green PhosOLED17 and blue PhosOLED18) followed by those based on [2,1-b]-94 (green PhosOLED13 and blue PhosOLED14) and finally those based on [2,1-b]-93 (green PhosOLED11 and blue PhosOLED12). Compared to the devices based on the non-substituted core [1,2-a]-DSF-IF or [2,1-b]-DSF-IF, the donor/acceptor design has a positive effect on the device performance for [1,2-a]-96 but a negative effect for [2,1-b]-93. To unravel this intriguing feature, hole mobilities were measured using the SCLC technique for both [2,1-b]-93 and [1,2-a]-96 and the results showed that [2,1-b]-93 possesses a modest μh of about 1.4 × 10−6 cm2 V−1 s−1 whereas that of [1,2-a]-96 was impressively enhanced and reaches about 1.5 × 10−4 cm2 V−1 s−1.25 There are therefore two orders of magnitude difference in μh between the two hosts, which was the arguments made by the authors to explain the different performances.
It should also be mentioned that regioisomers [2,1-b]-93 and [1,2-a]-96 have also been successfully studied by ion mobility mass spectroscopy. As these isomers have been both synthesized in the same chemical reaction, this technique is particularly relevant to readily detect these isomers.189
Keeping the same molecular design strategy, i.e. modulation of the HOMO/LUMO levels without modifying the ET1 value, another organic semiconductor based on the [2,1-b]DHIF core has been designed by our group. In [2,1-b]-95,26 the fluorenyl units, present in [2,1-b]-DSF-IF, have been changed in dioxothioxanthene (TXO2) units (Scheme 24). The electron-withdrawing effect of the TXO2 units deepened the HOMO level from −5.8 eV in [2,1-b]-DSF-IF to −6.04 eV in [2,1-b]-95 and lowered the LUMO from −1.75 eV in [2,1-b]-DSF-IF to −2.06 eV in [2,1-b]-95. These two effects lead to a slight decrease of the gap from 4.05 eV for [2,1-b]-DSF-IF to 3.98 eV for [2,1-b]-95. The ET1 of [2,1-b]-95, 2.76 eV, was nevertheless kept similar to that of [2,1-b]-DSF-IF as imposed by the [2,1-b]-IF core. Therefore, [2,1-b]-95 has been used as the host for green (PhosOLED15) and for blue phosphors (PhosOLED16). A comparison of the performances of these two devices to that using [2,1-b]-DSF-IF as the host (PhosOLED8-PhosOLED9) reveals, for the green PhosOLED15, a decrease of Von (3.2 V) and an increase of the EQEmax showing the efficiency of the design strategy. Indeed, the HOMO/LUMO gap was decreased (and decreases in turn the Von of the PhOLED) but the ET, controlled by the [2,1-b]DHIF core, was kept high.
In conclusion, among the 18 PhOLEDs presented in this part (Table 4 and Table 10), [1,2-a]-96 appears to be the most efficient matrix to host both the green (PhosOLED17) and blue phosphors (PhosOLED18). With its donor/acceptor design, [1,2-a]-96 used as the host for green Ir(ppy)3 and blue FIrpic PhOLEDs has led to an EQE of 15.9 and 7.9% respectively. Despite these performances being far from the best reported to date (EQE surpassing 30% for blue190–194 or green195–197 devices) these works have shown that the meta-substituted indenofluorenyl core can be used to design high ET host materials. With further molecular design consideration, it is clear that these performances could be easily improved.
4.4. Uses of [2,1-b]DHIF-based organic semiconductors in TADF OLEDs
Thermally activated delayed fluorescence (TADF) emitters have demonstrated remarkable potential towards application in organic displays.198 Owing to the efficient reverse intersystem crossing (RISC), TADF emitters could harvest non-emissive triplet excitons, which results in a 100% internal quantum efficiency (IQE). When employed as emitters, external quantum efficiencies (EQEs) of over 30% could be achieved.199–201 Among others, spiroacridine is an important core which has been used to construct donor–acceptor type TADF emitters. For example, the acridine unit has been used to connect the two bridges of the [2,1-b]DHIF core to donor or acceptor units ([2,1-b]-97 to [2,1-b]-102) leading to efficient TADF emitters and interesting TADF-OLED device performances. These OSCs are presented in Scheme 25 with some of their characteristics and the related TADF-OLED performances are summarized in Table 11.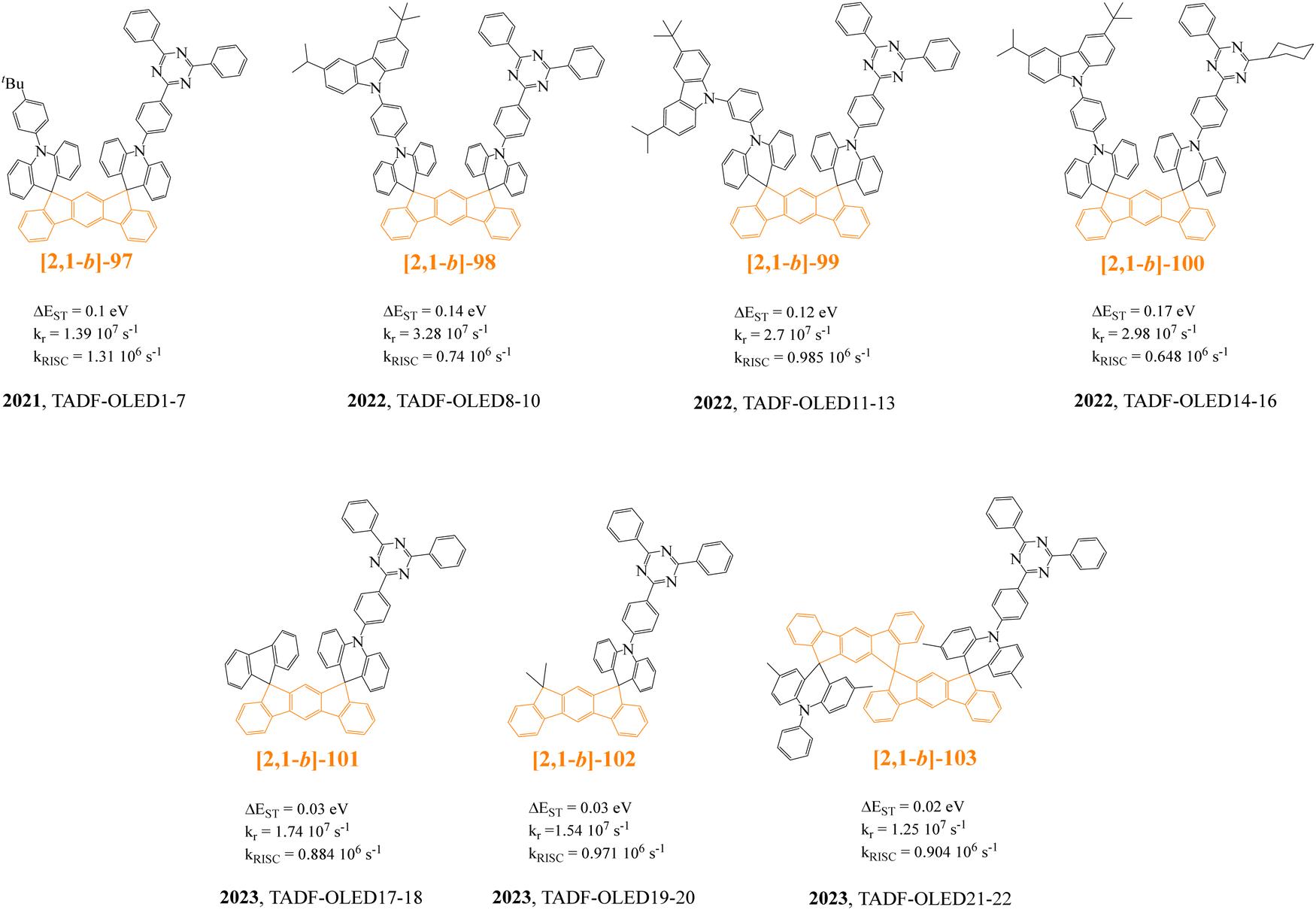 | ||
| Scheme 25 Molecular structure of organic semiconductors based on [2,1-b]DHIF cores used in TADF OLEDs. | ||
| Device | Device architecture | V on (V) | EQEa (%) | CEa (cd A−1) | PEa (lm W−1) | Luminance (cd m−2) | λ (EL spectrum) nm CIE (x, y) | Ref. |
|---|---|---|---|---|---|---|---|---|
| ANT-BIZ: 1-(4-(10-([1,1′-biphenyl]4-yl)anthracene-9-yl)phenyl)-2ethyl-1H-benzo[d]imidazole. | ||||||||
| TADF-OLED1 | ITO/MoO3 (1 nm)/TAPC (40 nm)/TcTa (10 nm)/mCP (10 nm)/DPEPO:20 wt% [2,1-b]-97 (25 nm)/DPEPO (5 nm)/Bphen (30 nm)/LiF (1 nm)/Al (1100 nm) | 3.2 | 27.1 | 69.2 | 63.9 | 14752 |
490 | 202 |
| 0.20, 0.45 | ||||||||
| TADF-OLED2 | ITO/MoO3 (1 nm)/TAPC (40 nm)/TcTa (10 nm nm)/mCP (10 nm)/DPEPO: 10 wt% [2,1-b]-97 (25 nm)/DPEPO (5 nm)/Bphen (30 nm)/LiF (1 nm)/Al (1100 nm) | 4.0 | 22.7 | 52.6 | 33 | 3222 | — | 215 |
| 0.189, 0.402 | ||||||||
| TADF-OLED3 | ITO/MoO3 (1 nm)/TAPC (40 nm)/TcTa (10 nm)/mCP (10 nm)/DPEPO: 20 wt% [2,1-b]-97 (25 nm)/DPEPO (5 nm)/Bphen (30 nm)/LiF (1 nm)/Al (1100 nm) | 3.4 | 27.1 | 69.1 | 63.9 | 10130 |
— | 215 |
| 0.199, 0.448 | ||||||||
| TADF-OLED4 | ITO/MoO3 (1 nm)/TAPC (40 nm)/TcTa (10 nm)/mCP (10 nm)/DPEPO: 30 wt% [2,1-b]-97 (25 nm)/DPEPO (5 nm)/Bphen (30 nm)/LiF (1 nm)/Al (1100 nm) | 3.2 | 24.5 | 64.9 | 54.0 | 14752 |
— | 215 |
| 0.208, 0.473 | ||||||||
| TADF-OLED5 | ITO/MoO3 (1 nm)/TAPC (40 nm)/TcTa (10 nm)/mCP (10 nm)/DPEPO: 40 wt% [2,1-b]-97 (25 nm)/DPEPO (5 nm)/Bphen (30 nm)/LiF (1 nm)/Al (1100 nm) | 3.2 | 24.5 | 66.5 | 65.2 | 18153 |
— | 215 |
| 0.211, 0.488 | ||||||||
| TADF-OLED6 | ITO/MoO3 (1 nm)/TAPC (40 nm)/TcTa (10 nm)/mCP (10 nm)/DPEPO: 70 wt% [2,1-b]-97 (25 nm)/DPEPO (5 nm)/Bphen (30 nm)/LiF (1 nm)/Al (1100 nm) | 3.2 | 22.3 | 60.1 | 55.5 | 18289 |
— | 215 |
| 0.213, 0.492 | ||||||||
| TADF-OLED7 | ITO/MoO3 (1 nm)/TAPC (40 nm)/TcTa (10 nm)/mCP (10 nm)/[2,1-b]-97 (25 nm)/DPEPO (5 nm)/Bphen (30 nm)/LiF (1 nm)/Al (1100 nm) | 3.0 | 12.4 | 31.7 | 32.4 | 8064 | — | 215 |
| 0.197, 0.443 | ||||||||
| TADF-OLED8 | ITO/MoO3 (1 nm)/TAPC (40 nm)/TcTa (10 nm)/mCP (10 nm)/DPEPO: 10 wt% [2,1-b]-98 (25 nm)/DPEPO (5 nm)/Bphen (30 nm)/LiF (1 nm)/Al (1100 nm) | 3.8 | 20.4 | 45.9 | 34.2 | 2296 | 488 | 216 |
| 0.183, 0.384 | ||||||||
| TADF-OLED9 | ITO/MoO3 (1 nm)/TAPC (40 nm)/TcTa (10 nm)/mCP (10 nm)/DPEPO: 20 wt% [2,1-b]-98 (25 nm)/DPEPO (5 nm)/Bphen (30 nm)/LiF (1 nm)/Al (1100 nm) | 3.4 | 22.0 | 54.5 | 42.5 | 6183 | 492 | 216 |
| 0.192, 0.425 | ||||||||
| TADF-OLED10 | ITO/MoO3 (1 nm)/TAPC (40 nm)/TcTa (10 nm)/mCP (10 nm)/DPEPO: 30 wt% [2,1-b]-98 (25 nm)/DPEPO (5 nm)/Bphen (30 nm)/LiF (1 nm)/Al (1100 nm) | 3.4 | 25.6 | 66.6 | 61.6 | 10849 |
496 | 216 |
| 0.200, 0.454 | ||||||||
| TADF-OLED11 | ITO/MoO3 (1 nm)/TAPC (40 nm)/TcTa (10 nm)/mCP (10 nm)/DPEPO: 10 wt% [2,1-b]-99 (25 nm)/DPEPO (5 nm)/Bphen (30 nm)/LiF (1 nm)/Al (1100 nm) | 4.0 | 18.6 | 42.9 | 30.9 | 2174 | 490 | 216 |
| 0.186, 0.388 | ||||||||
| TADF-OLED12 | ITO/MoO3 (1 nm)/TAPC (40 nm)/TcTa (10 nm)/mCP (10 nm)/DPEPO: 20 wt% [2,1-b]-99 (25 nm)/DPEPO (5 nm)/Bphen (30 nm)/LiF (1 nm)/Al (1100 nm) | 3.4 | 22.3 | 55.6 | 50.5 | 6159 | 492 | 216 |
| 0.197, 0.431 | ||||||||
| TADF-OLED13 | ITO/MoO3 (1 nm)/TAPC (40 nm)/TcTa (10 nm)/mCP (10 nm)/DPEPO: 30 wt% [2,1-b]-99 (25 nm)/DPEPO (5 nm)/Bphen (30 nm)/LiF (1 nm)/Al (1100 nm) | 3.4 | 21.4 | 53.9 | 47 | 7447 | 492 | 216 |
| 0.201, 0.445 | ||||||||
| TADF-OLED14 | ITO/MoO3 (1 nm)/TAPC (40 nm)/TcTa (10 nm)/mCP (10 nm)/DPEPO: 10 wt% [2,1-b]-100 (25 nm)/DPEPO (5 nm)/Bphen (30 nm)/LiF (1 nm)/Al (1100 nm) | 4.0 | 22.2 | 43.5 | 32.6 | 1093 | 478 | 216 |
| 0.171, 0.321 | ||||||||
| TADF-OLED15 | ITO/MoO3 (1 nm)/TAPC (40 nm)/TcTa (10 nm)/mCP (10 nm)/DPEPO: 20 wt% [2,1-b]-100 (25 nm)/DPEPO (5 nm)/Bphen (30 nm)/LiF (1 nm)/Al (1100 nm) | 3.4 | 26.3 | 55.3 | 51.5 | 2775 | 484 | 216 |
| 0.173, 0.347 | ||||||||
| TADF-OLED16 | ITO/MoO3 (1 nm)/TAPC (40 nm)/TcTa (10 nm)/mCP (10 nm)/DPEPO: 30 wt% [2,1-b]-100 (25 nm)/DPEPO (5 nm)/Bphen (30 nm)/LiF (1 nm)/Al (1100 nm) | 3.4 | 23.6 | 52.2 | 46.6 | 3950 | 488 | 216 |
| 0.180, 0.375 | ||||||||
| TADF-OLED17 | ITO/MoO3 (1 nm)/TAPC (40 nm)/TcTa (10 nm)/mCP (10 nm)/DPEPO: 10 wt% [2,1-b]-101 (25 nm)/DPEPO (5 nm)/Bphen (30 nm)/LiF (1 nm)/Al (1100 nm) | 3.6 | 32.6 | 95 | — | 42055 |
— | 217 |
| TADF-OLED18 | ITO/MoO3 (1 nm)/TAPC (40 nm)/TcTa (10 nm)/mCP (10 nm)/DPEPO: 20 wt% [2,1-b]-101 (25 nm)/DPEPO (5 nm)/Bphen (30 nm)/LiF (1 nm)/Al (1100 nm) | 3.2 | 35.6 | 108.6 | — | 76964 |
— | 217 |
| TADF-OLED19 | ITO/MoO3 (1 nm)/TAPC (40 nm)/TcTa (10 nm)/mCP (10 nm)/DPEPO: 10 wt% [2,1-b]-102 (25 nm)/DPEPO (5 nm)/Bphen (30 nm)/LiF (1 nm)/Al (1100 nm) | 3.6 | 35.8 | 105.5 | — | 51590 |
— | 217 |
| TADF-OLED20 | ITO/MoO3 (1 nm)/TAPC (40 nm)/TcTa (10 nm)/mCP (10 nm)/DPEPO: 20 wt% [2,1-b]-102 (25 nm)/DPEPO (5 nm)/Bphen (30 nm)/LiF (1 nm)/Al (1100 nm) | 3.2 | 36.1 | 111.6 | — | 105907 |
— | 217 |
| TADF-OLED21 | ITO/HATCN (5 nm)/TAPC (30 nm)/TcTa (15 nm)/mCBP (10 nm)/PPF: 10 wt% [2,1-b]-103 (25 nm)/PPF (10 nm)/ANT-BIZ (30 nm)/LiF (1 nm)/Al (1100 nm) | 3.6 | 25.3 | 70.4 | — | 22400 |
— | 218 |
| TADF-OLED22 | ITO/HATCN (5 nm)/TAPC (30 nm)/TcTa (15 nm)/mCBP (10 nm)/PPF: 20 wt% [2,1-b]-103 (25 nm)/PPF (10 nm)/ANT-BIZ (30 nm)/LiF (1 nm)/Al (1100 nm) | 3.2 | 34.2 | 104.8 | — | 69357 |
— | 218 |
In 2021, the group of Z. Wang and C. Yang synthesized a donor–acceptor derivative, [2,1-b]-97, in which the two units (tBu-Ph as the donor and triphenyltriazine as the acceptor) were linked through two acridine units themselves connected to a [2,1-b]-DHIF core. [2,1-b]-97 was obtained from the dione [2,1-b]IF(O)2 first converted in dispiroacridine-[2,1-b]-IF, itself substituted step by step by a tert-butylphenyl on one acridine and then by the triphenyltriazine unit on the second acridine. The [1,2-b]IF core was used to arrange the donor and acceptor in a face-to-face geometry forcing the acridine units to be perpendicular to the indenofluorenyl core. Calculations show that the LUMO is located on the electron-deficient triphenyl-triazine unit linked to one acridine unit whereas the HOMO is spread on the other acridine unit connected to the tert-butylphenyl. The triazine unit weakens the electron-donating properties of its acridine. The calculated Natural Transition Orbital (NTO) indicates that S1 and T1 originate from the transition between this acridine and the triazine. The energies of S1 and T1 were calculated at 2.595 and 2.586 eV with a small ΔEST of 0.009 eV, which facilitate a fast triplet → singlet up-conversion. A local excited triplet state (3LE) centred on the [2,1-b]-DHIF core is also observed with an energy of 2.658 eV facilitating an RISC process. Photophysical studies confirm the calculations with measurements of the singlet ES1 and triplet ET1 energy levels at 2.86 and 2.76 eV respectively, resulting in a short ΔEST of 0.1 eV (measured from the onset of fluorescence and phosphorescence at 77 K in the spectra). The PL quantum yield was measured at 98% in the solid state in a matrix of bis[2-(diphenylphosphino)phenyl ether oxide (DPEPO). A transient PL decay spectrum confirms the TADF characteristic of [2,1-b]-97 with two components of 28 ns and 2.1 μs which originate from prompt and delayed fluorescence respectively. All these properties were promising for the use of [2,1-b]-97 as a TADF emitter in an OLED.
The first OLED based on a multi-layer architecture was fabricated (TADF-OLED1) with [2,1-b]-97 doped in DPEPO in 20 wt% as the EML. This first TADF-OLED reached a very high EQE of 27.1%,202 which was in 2021 among the best EQE reached for bluish-green doped TADF-OLEDs.200,203–214 The same group studied then the influence of [2,1-b]-97 concentration in DPEPO (from 10 to 70 wt%) on the performance of the devices (TADF-OLED2 to TADF-OLED6).215 This study showed that EQE increases from 22.1 to 27.1% increasing the concentration from 10 to 20 wt% and then decreases up to 22.3% for a concentration of 70 wt%. The efficiencies of these devices were all significantly higher than that using a non-doped [2,1-b]-97 EML (TADF-OLED7, EQE: 12.4%215) showing the interest of the DPEPO doping.
A second series of TADF emitters constructed on the [2,1-b]-DHIF core was synthesized in 2022 ([2,1-b]-98–[2,1-b]-100).216 Compared to [2,1-b]-97, both [2,1-b]-98 and [2,1-b]-99 possess a carbazole donor unit (instead of a tBu group), respectively in the para and meta position of the phenyl acridine core. In [2,1-b]-100 one of the three phenyl units, decorating the triazine group in [2,1-b]-98, is changed in a cyclohexane unit. All these structural modifications influence the photophysical properties of [2,1-b]-98–[2,1-b]-100 compared to [2,1-b]-97. The presence of the carbazole induced a weak blue shift of the absorption spectra from (400 nm) for [2,1-b]-97 to (393/394/387 nm) for [2,1-b]-98/[2,1-b]-99/[2,1-b]-100. In emission, a red shift of 25 nm was measured from [2,1-b]-97 (489 nm) to [2,1-b]-98 and [2,1-b]-99 (514 nm) whereas the red shift is only of 15 nm from [2,1-b]-97 (489 nm) to [2,1-b]-100 (504 nm). A weak increase of ΔEST (measured from the onset of fluorescence and phosphorescence at 77 K in the spectra) from 0.1 eV for [2,1-b]-97 to (0.14/0.12/0.17 eV) for [2,1-b]-98/[2,1-b]-99/[2,1-b]-100 is observed. For each emitter, [2,1-b]-98–[2,1-b]-100, TADF-OLEDs were constructed with the TADF emitter in 10, 20 or 30 wt% concentration in DPEPO. The most efficient OLED reaching an EQE of 26.3% was TADF-OLED15 with [2,1-b]-100 20 wt% in DPEPO as the EML. The two other TADF emitters led to less efficient devices (25.6% for TADF-OLED10 with [2,1-b]-98 30 wt% in DPEPO as the EML and 22.3% for TADF-OLED12 with [2,1-b]-99 20 wt% in DPEPO as the EML) despite these EQE remaining high. The highest performances of [2,1-b]-97vs. the three other TADF OSCs ([2,1-b]-98 - [2,1-b]-100) may be corelated to its lower ΔEST. One should also note an extension of conjugation in those OSCs compared to [2,1-b]-97. This extension of conjugation leads to an increase of the ratio of prompt fluorescence in comparison with the delayed fluorescence (see the kr and kRISC values in Scheme 25) decreasing the TADF emission yield.
Based on these observations and in order to decrease the conjugation, the same group recently synthesized two new TADF emitters also constructed on the [2,1-b]-DHIF core with however only one acridine unit between this core and the acceptor unit (triphenyltriazine) and either a fluorene (in [2,1-b]-101) or two methyl units (in [2,1-b]-102) decorating the second bridge of the [2,1-b]-DHIF core.217 Both OSCs were doped in DPEPO as the EML in TADF-OLEDs. The EQEs surpassed those previously reached and the four devices are in the top 10 of the more efficient bluish green TADF-OLEDs,200,201,210 especially TADF-OLED20 using [2,1-b]-102 as the dopant which reaches an extremely high EQE of 36.1%. Those results show that a well-thought-out structural modification may offer fine control of the photophysical properties which is useful to optimize the TADF emitters and the device performances. As revealed in Scheme 25, the new OSCs possess a lower ΔEST value and kr and kRISC values similar to the one of [2,1-b]-97. The structural modifications from [2,1-b]-97 to [2,1-b]-98 (increase conjugation and decrease electron-donating properties) or from [2,1-b]-97-[2,1-b]-98 to [2,1-b]-101-[2,1-b]-102 (decrease conjugation) or finally from [2,1-b]-101-[2,1-b]-102 (enhance electron-donating properties) have allowed the authors to construct TADF-OLED20 which reaches excellent performances.
Finally, [2,1-b]-103 with a 4π-3spiro architecture (four π-systems connected thanks to 3 spiro-bridges) was very recently designed and tested in TADF-OLED21–22 also with interesting performances.218 As expected, the HOMO and HOMO-1 are spread on the two acridine units and the LUMO on the triphenyltriazine substituent. For this emitter ΔEST is 0.02 eV, the decay profile exhibits two components corresponding to prompt (27 ns) and delayed fluorescence (3.2 μs) and the rate constant of radiative decay and reverse intersystem crossing are similar to those of [2,1-b]-101-[2,1-b]-102 (Scheme 25) suggesting efficient device performances. In fact, TADF-OLED22 with [2,1-b]-103 (10 wt% doped in dibenzo[b,d]furan-2,8-diylbis(diphenylphosphine oxide) (PPF)) as the EML reaches an EQE of 34.2%. Due to the difference in host materials (PPF in TADF-OLED21 and 22 or DPPO in the other TADF-OLEDs, the comparison is not simple, however all these results show the interest of the [2,1-b]-DHIF core to construct efficient TADF bluish-green emitters.
4.5. Uses of [1,2-a]- or [2,1-b]DHIF-based organic semiconductors in OFET, OPV, DSSC, etc.
Chemical modification of the bridges and/or of the DHIF core has allowed us to drastically tune the electronic properties of the resulting organic semiconductors and this strategy has highlighted the potential of the DHIF-based organic semiconductors in other OE devices. This is what we aim to describe in this last part. As presented above in section 3 (Scheme 22), access to [2,1-b]IF(O)2 dates from 1951,168 however its physicochemical properties were only reported in 2015 with those of its parents compounds [2,1-b]-104, [2,1-b]-105 (Scheme 26) and [2,1-b]DHIF.170 Electrochemical data showed that, due to the presence of strong electron-deficient groups (carbonyl or dicyanovinylene), [2,1-b]IF(O)2 (−5.96 eV), [2,1-b]-104 (−5.90 eV) and [2,1-b]-105 (−5.91 eV) possessed HOMO levels deeper than that of [2,1-b]DHIF (−5.71 eV). Their LUMO levels recorded between −3.52 eV for [2,1-b]IF(O)2 and −3.93 eV for [2,1-b]-104 were strongly deepened compared to that of [2,1-b]DHIF (−2.04 eV). Despite the presence of two dicyanovinylene groups in [2,1-b]-105, its LUMO level was surprisingly higher than that of [2,1-b]-104 with only one dicyanovinylene (−3.81 eV vs. −3.93 eV). This unexpected behaviour has been assigned to the deformation of the [2,1-b]DHIF core induced by steric hindrance or to electronic repulsion between the adjacent cyano groups.170
 | ||
| Scheme 26 Molecular structure of the organic semiconductors based on [2,1-b]DHIF or [1,2-a]DHIF cores used in OFETs, OSCs or DSSCs. | ||
The potential of the three electron-deficient organic semiconductors as active layers of n-channel OFETs was estimated with bottom-gate bottom-contact OFETs. As noticed above for [1,2-b]IF(O)286 in OFET 3 (Table 3), no field-effect was observed for OFET64 (Table 12) using [2,1-b]IF(O)2.170 The introduction of one CN group in [2,1-b]-104 has allowed us to measure in OFET65 a modest μe of 4.6 × 10−6 cm2 V−1 s−1, with a threshold voltage (VT) of 18.4 V, a subthreshold slope (SS) of 5.74 V dec−1 and a Ion/Ioff of 103–104. Although low, the performance of OFET65 highlighted the effect of the integration of one dicyanovinylene unit on the [2,1-b]DHIF core. OFET66 based on [2,1-b]-105 has allowed us to reach higher values with μe of 1.02 × 10−3 cm2 V−1 s−1, VT of 7.2 V, SS of 2.16 V dec−1 and a Ion/Ioff of 6.3 × 105. Thus, incorporation of CN units instead of oxygen atoms has allowed us to consistently increase the electron mobility and other OFET parameters.
| OFETs | ||||||
|---|---|---|---|---|---|---|
| Device | Device architecture | μ FET (cm2 V−1 s−1) | V T (V) | SS (V dec−1) | I on/Ioff | Ref. |
| DIO: 1,8-diiodooctane | ||||||
| OFET64 | Bottom-gate bottom-contact structure | No-gate effect | 170 | |||
|
[2,1-b]IF(O)2
|
||||||
| OFET65 | Bottom-gate bottom-contact structure | 4.6 × 10−6 | 18.4 | 5.74 | 103–104 | 170 |
| [2,1-b]-104 | ||||||
| OFET66 | Bottom-gate bottom-contact structure | 1.02 × 10−3 | 7.2 | 2.16 | 6.3 × 105 | 170 |
| [2,1-b]-105 | ||||||
| OFET67 | Bottom-gate/bottom-contact device in a glove box under ambient nitrogen | μ FET linear: 0.0009 cm2 V−1 s−1 | 20 | 2.3 | 8 × 104 | 99 |
| Al/SU-8/Au/[1,2-b]-105 | μ FET saturation: 0.002 cm2 V−1 s−1 | 17 | 3.2 | 7 × 104 | ||
| BHJ OPVs | ||||||
|---|---|---|---|---|---|---|
| Device | Device architecture | V oc (V) | J SC (mA cm−2) | FF | PCE(%) | Ref. |
| BHJ-OPV1 prepared in TCE | ITO/PEDOT:PSS/PTB7:[2,1-b]-106(1:2)/ Ca/Al | 0.87 | 1.13 | 0.49 | 0.64 | 180 |
| BHJ-OPV2 prepared in TCE | ITO/PEDOT:PSS/PTB7:[2,1-b]-106(1:1)/ Ca/Al | 0.89 | 1.41 | 0.48 | 0.80 | 180 |
| BHJ-OPV3 prepared in TCE/DCB | ITO/PEDOT:PSS/PTB7:[2,1-b]-106 (1:1)/ Ca/Al | 0.89 | 0.22 | 0.19 | 0.05 | 180 |
| BHJ-OPV4 prepared in DCB | ITO/PEDOT:PSS/PTB7:[2,1-b]-106 (1:1)/ Ca/Al | 0.09 | 0.10 | 0.09 | 0.01 | 180 |
| BHJ-OPV5 prepared in TCE/3%DIO | ITO/PEDOT:PSS/PTB7:[2,1-b]-106 (1:1)/ Ca/Al | 0.42 | 0.03 | 0.20 | 0.00 | 180 |
Electrical stability of OFET66 has been studied using a gate bias stress and it was shown that (i) the instability of the OFETs was only linked to charges trapped in the insulator and not in the [2,1-b]-105 active layer, and (ii) the SS related to defects in the interface between [2,1-b]-105 and the insulating layer was almost constant under gate bias stress indicating that no defect state was created at this interface. This study provided an interesting finding about the origin of the instability in OFETs using the active layer of DHIF. As the μe of [2,1-b]-105 was constant under applied stress, the electrical stability of OFET66 was then promising for its integration in a circuit. Pseudo CMOS inverters with [2,1-b]-105-based OFETs have been successfully fabricated and characterized.170
Performance improvements of OFETs based on [2,1-b]-105 have been then evaluated by optimization of the fabrication process.219 The μe was improved reaching 2.2 × 10−3 cm2 V−1 s−1. The μFET improvement after post-annealing treatment can be attributed to some reasons: (i) eliminated vacancies and/or lattice defects in [2,1-b]-105, (ii) structural arrangement of [2,1-b]-105 film and (iii) better matching at the SU-8/[2,1-b]-105 interface resulting in higher mobility. The annealing effect on the electrical properties of [2,1-b]-105 was more precisely studied by characterizing the devices at different temperatures from 300 to 380 K, allowing us to evaluate the activation energy of the mobility (Ea) and the density of states DOS.100 After annealing treatment, a reduction of Ea was observed. Defects at the electrodes/organic semiconductor interface, accelerating the charge injection in the active layer, have been involved in this decrease. A reduction in the width of the band tail of the calculated DOS was also observed which could be linked to the increase ordering of [2,1-b]-105 molecules after annealing treatment. In order to understand the charge transport phenomena in the [2,1-b]-105 layer, the temperature dependence mobility and the current characteristics (U–V) were modeled using the VRH model. Good coherence between experimental and theoretical results was obtained, showing the validity of the VRH model to represent the charge conduction in the [2,1-b]-98 layer.
[2,1-b]-105 was also tested in OFET6799 built in similar conditions than OFET61 based on [1,2-b]-40 (Table 3). As shown in Table 12, μe extracted from OFET67 was one order of magnitude lower than that from OFET61. This shows that switching from the syn geometry found in [2,1-b]-105 to the anti geometry in [1,2-b]-40 coupled to the addition of pentyl side chains have beneficial effects on both μFET (2 × 10−3vs. 2.1 × 10−2 cm2 V−1 s−1), Ion/Ioff ratio (7 × 104vs. 5 × 105) and SS (3.2 vs. 1.9 V dec−1) without a negative consequence on VTH (17 vs. 20 V).99 This shows the key role played by isomerism in the design of organic semiconductors. However, despite better electrical parameters, [1,2-b]-40 leads to less stable OFETs than [2,1-b]-105 showing that the most efficient device is not always the most stable one. This is an important consideration for real electronic applications. Comparing OFET61 and OFET67, it has been highlighted that the VTH shift of the OFETs has a different origin. At low gate bias stress, the carrier trapping inside the insulator is at the origin of the instability of OFET61. Oppositely, in OFET67, both the carrier trapping in the insulator and the defect density formation in the active layer are responsible for the instability. At high bias stress, the authors proposed a different situation. This time, only the carrier trapping in the insulator layer is implied for both [1,2-b]-40 and [2,1-b]-105-based OFETs. In this work the authors have also shown that, under the same gate bias stress, the [1,2-b]-40-based OFET displays a larger VTH shift than the [2,1-b]-105-based OFET, which was assigned to a higher gate leakage current in the former. This feature was related to the different insulator/organic semiconductor interfaces. Thus, in this work, it was shown that minor structural modifications of the molecular structure of the organic semiconductor ([1,2-b]-40vs.[2,1-b]-105) can induce electrical instabilities of the corresponding OFET (OFET61 vs. OFET67) arising from different features showing the importance of the DHIF core.
Pseudo CMOS inverters made of n-type [2,1-b]-105-based OFETs have been also constructed (Fig. 14A) and characterized (Fig. 14C).170 Generally, the response efficiency of the logic gate as the inverter is related to on/off states of p type and n type OFETs. The on/off states are defined by the RDSon and RDSoff ratios corresponding to the (IDS, VDS) slope. These studies were performed with n-type OFETs. In a circuit, there is a switch of the OFETs from the saturated to the linear regime. This switch is a very important parameter and is directly connected to the difference between linear RDS (RDSon) and saturated RDS (RDSoff). The RDSon/RDSoff ratio was evaluated in this work at 1.5 × 103. This value is significantly high to discriminate well between the off and the on state.
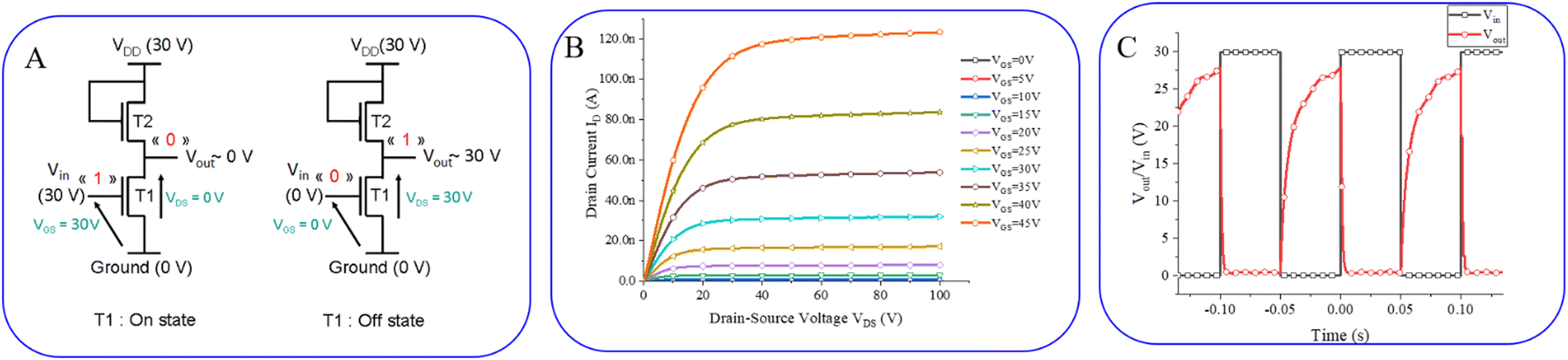 | ||
| Fig. 14 (A) Schematic of the pseudo CMOS inverter and (B) T1 associated output characteristics, (C) inverter made of n-type OFETs with W/L = 1000/2 and 250/20 for the active charge. Adapted from ref. 170 with permission from the Royal Society of Chemistry. | ||
Pseudo CMOS inverters were fabricated and characterized with a 10 Hz square signal applied as the input voltage (Vin). The output voltage Vout followed a classical inverter behaviour with a high value of about 27.5 V when Vin = 0 V and a low value of about 0.5 V when Vin = 30 V (Fig. 14C). In addition, for a VDD of 30 V, the Vout was measured at ca. 27.5 V, which was promising for embedded circuits. These preliminary data show that the [2,1-b]-105 based OFET can be used under real polarization with no degradation of on and off voltage for Vout (12 hours polarization).
[2,1-b]-106 (Scheme 26) is a 3D small molecule acceptor, which has been used as a non-fullerene electron acceptor in bulk-heterojunction (BHJ) solar cells.180 TGA analysis reveals for [2,1-b]-106 a Td of 460 °C, almost identical to the one measured for [2,1-b]-84 (450 °C)179 showing that very good thermal properties were maintained despite different bridge substitution. The LUMO of [2,1-b]-106 was measured by cyclic voltammetry at −3.63 eV, slightly higher than that measured for [2,1-b]-105 under similar conditions (−3.81 eV). This is directly connected to the number of dicyanovinylene fragments linked to each [2,1-b]-DHIF core, one in [2,1-b]-106 and two in [2,1-b]-105.
To demonstrate the potential application of [2,1-b]-106 as an acceptor in photovoltaic devices, [2,1-b]-106 was blended with polythieno[3,4-b]-thiophene-co-bezodithiophene (PTB7) as an electron-donor polymer. PTB7 polymer absorption is partially complementary to the absorption of [2,1-b]-106.180
BHJ organic photovoltaic (OPV) devices with the structure ITO/PEDOT:PSS/PTB7:[2,1-b]-106/Ca/Al were fabricated varying the mode of preparation in modifying (i) the donor–acceptor weight ratio (1:1 or 1:2) and (ii) the solvent used to prepare the device (tetrachloroethane (TCE), dichlorobenzene (DCB) or a mixture TCE/DBE or TCE/3% 1,8-diiodooctane (DIO)). BHJ-OPV1 prepared from TCE with a donor:acceptor ratio, PTB7:[2,1-b]-106 of 1:2 exhibited an open-circuit voltage (VOC) of 0.87 V, a short-circuit current density (JSC) of 1.13 mA cm−2, a fill factor (FF) of 0.49 and a power conversion efficiency (PCE) of 0.64%. In BHJ-OPV2, the donor:acceptor ratio was 1:1 and led to an increase of the whole performances with a PCE reaching 0.8%. Changing the solvent for the three last devices (BHJ-OPV3 to BHJ-OPV5) was not efficient or even did not work. However, BHJ-OPV2 even with modest performances demonstrate that [2,1-b]-106 could be an alternative to fullerene-based acceptors.
To end, [1,2-a]-107, [1,2-a]-108 and [1,2-a]-109 (Scheme 26) all based on a central truxene were used in DSSCs. These three dyes possess thienyl-2-cyanoacrylic acid as the acceptor and starbust diarylamine as the donor. The central truxene was substituted either by ethyl groups in [1,2-a]-107 or by longer hexyl-substituents in [1,2-a]-108 and [1,2-a]-109. In solution in DCM, all organic semiconductors exhibit two prominent absorption bands between 300–360 nm (localized π–π* transitions) and between 400–450 nm (charge transfer transition). The extinction coefficients of [1,2-a]-109 (4.13 × 10−4 L mol−1 cm−1 at 430 nm and 6.65 × 10−4 L mol−1 cm−1 at 365 nm) appear more intense than those of the two others (2.75 × 10−4 L mol−1 cm−1 at 406 nm and 5.42 × 10−4 L mol−1 cm−1 at 361 nm for [1,2-a]-107 and 2.49 × 10−4 L mol−1 cm−1 at 420 nm and 5.44 × 10−4 L mol−1 cm−1 at 360 nm for [1,2-a]-108) due to the stronger electron-donating capability of [1,2-a]-109 with methoxy groups. Thin films were deposited on TiO2 surfaces and the absorption spectra present for each organic semiconductor a large absorption band centered at ca. 425 nm. The maximum absorption spectrum of [1,2-a]-107 in the solid state was shifted from 406 nm (in solution) to 422 nm and the spectrum was broadened indicating a certain degree of π–π aggregation for [1,2-a]-107, which does not exist for the two other dyes possessing longer alkyl substituents on the central truxenyl core (hexyl instead of ethyl).
Due to the four methoxy groups decorating [1,2-a]-109, its HOMO level was higher (−4.98 eV) than that of the two other dyes (−5.22/−5.15eV for [1,2-a]-107/[1,2-a]-108). The LUMOs (calculated from their HOMO and optical gaps) were reported at −2.91/−2.84 and −2.72 eV respectively for the three dyes indicating that the electron injection process from the excited dyes to the TiO2 conduction band (−4.4 eV) was energetically achievable. The performance of DSSCs shows the highest PCE for DSSC8 based on [1,2-a]-108) (4.27%), higher than the one of DSSC7 (3.90%) based on [1,2-a]-107) highlighting the positive effect of the hexyl chains on the electron injection efficiency because of the weak π–π aggregation. The higher VOC for DSSC8 and DSSC9 than for DSCC7 indicate that the charge recombination process was retarded in the devices based on [1,2-a]-108 and [1,2-a]-109 due to the cone-shape sensitizer, which could constitute a compact layer at the TiO2 surface to avoid the approach of the redox couple. The present results remain nevertheless modest. However, modulation of the DSSCs performances by the molecular structural modification remains the more important effect in this work.
5. Conclusion
The development of the dihydroindeno[1,2-b]fluorene molecular fragment as a building unit for the design of organic semiconductors has been important in the last two decades. This fragment has found applications in the three main devices of organic electronics, namely OLEDs, OFETs and OPVs, showing its real versatility. Positional isomerism, more and more used in the design of organic semiconductors, has allowed, over the years, to consider four other positional isomers of dihydroindeno[1,2-b]fluorene with different phenyl connections (ortho, meta or para linkages), which not only provide different π-conjugation pathways but also different geometries. The five positional isomers have been a fertile playground for molecular chemists to construct organic semiconductors. However, it is clear that the structural diversity provided by these isomers has not been fully exploited yet.In this review, the five isomers and more than one hundred of their derivatives have been reviewed and the description of their uses as active components in different devices have highlighted their high interest for use in electronics. For each isomer family, the physicochemical properties were discussed and compared to relevant building units, in order to highlight the importance of molecular design on the electronic and physical properties, i.e. HOMO/LUMO energy levels, singlet/triplet energy, charge carrier mobility, decomposition temperature, etc. We show how substitution of either bridges and/or phenyl units offers many design combinations, which can be fitted for a specific application.
However, the [1,2-b] isomer has been far more developed than the other positional isomers, surely due to synthetic features. Nevertheless, the other isomers present very interesting properties due either to their different geometries or to their intrinsic electronic characteristics. For example, modulation of the linkages from para linkages in the [1,2-b] isomer to meta linkages in [1,2-a] and [2,1-b] isomers has been an efficient tool to tune the electronic properties. This has allowed a significant increase in the triplet state energy allowing the design of host materials for blue PhOLED applications. This was particularly interesting as most of the time high triplet host materials display a very short π-conjugated pathway. However, only a very limited number of examples are described in the literature and in future years, these cores might be decorated for that purpose (incorporating for example high-efficiency functional units such as electron-accepting phosphine oxides and electron-donating phenylacridine). Similarly, although considerable efforts have been made, only a limited number of compounds have reached high mobility values. This shows that significant improvements in terms of molecular design are still possible with these fragments.
Another interesting direction for the future consists of the use of different π-conjugated systems of different isomers to control the ICT between a donor- and an acceptor-fragment. Indeed, if many donor/acceptor molecules based on the [1,2-b] isomer, with functional units attached at each extremity, have been reported, the number of molecules incorporating the other isomers is very limited.
The different geometry of these isomers is also an important point still understudied. The best example is the [2,1-a] isomer in which the arrangement of the two bridges allow us to force interaction between attached substituents, providing singular optical and electrochemical properties. In the [2,1-b] isomer, the two bridges also present a similar arrangement but further apart. This particularity has nevertheless not been exploited yet.
In recent years, other applications of dihydroindenofluorene building units, not linked to electronics, have been reported in the literature, showing the versatility of this fragment. For example, in 2017, Wuest and coworkers have reported predictably ordered materials based on the [1,2-b] isomer.221 These materials crystallized to form open hydrogen-bonded networks with a significant volume available for accepting guest molecules. Using the other isomers may offer very different arrangements, which can tune the materials cavity.
In 2021, an organic cathode material using [1,2-b]IF(O)2 for lithium batteries was reported. Each carbonyl presents a four-electron reduction giving a high energy density of 1392 W h kg−1.222 This organic electrode material is among the most efficient reported to date and show the real potential of the dihydroindenofluorene backbone in the field of batteries.
Dihydroindeno[1,2-b]fluorene has also been integrated, in 2022, in the design of catalysts.223 A ruthenocene bimetallic complex incorporating this fragment was synthesized and tested as a burning rate catalyst on the thermal decomposition of ammonium perchlorate. The performance reached was promising with notably a reduction in the activation energy compared to other ruthenium complexes previously reported in the literature. It is impressive to see how this fragment can be very efficient in very different fields.
However, in these last three examples, the [1,2-b] isomer is again used. To develop more in depth the other positional isomers, improving their synthesis is a mandatory step. This could be achieved by modern synthetic approaches such as those recently develop by Akhmetov and coworkers with Caryl–F bond activation.224 Using alumina-mediated C–F bond activation, they managed to synthesize the [2,1-b]-isomer. Another interesting example is provided by Kotora and coworkers in 2020, who have reported an efficient Rh-catalysed intramolecular [2 + 2 + 2] cyclotrimerization of triyndiols to efficiently provide a series of dihydroindeno[2,1-c]fluorene.225 This approach has also been used to synthesize [7]-helical extended DSF-IFs, which appears as an interesting route to design, in the future, other chiral materials based on the [2,1-c]-DHIF core.226
As discussed herein, the dihydroindeno[1,2-b] fluorene positional isomers, the flagship parent of the dihydroindenofluorene family has become a major player in the field of organic electronics. The other positional isomers have been far less developed but deserve, undoubtedly, particular attention. Future rational designs of these five exciting π-conjugated systems may have the ambition to reach state of the art device performances. This constitutes an exciting journey.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank all their collaborators, who have been involved in the projects and especially the PhD students: Dr Damien Thirion, Dr Nicolas Cocherel, Dr Maxime Romain, Dr Sébastien Thiery, Dr Jean-David Peltier, Dr Lambert Sicard, and Dr Fabien Lucas. The authors also thank the CNRS, the University of Rennes 1, the ANR (11-BS07-020-01/14-CE05-0024/19/CE05-0024, CE07-0041), the Ademe and the Région Bretagne for their respective financial supports.References
- G. Hong, X. Gan, C. Leonhardt, Z. Zhang, J. Seibert, J. M. Busch and S. Bräse, Adv. Mater., 2021, 33, 2005630 CrossRef CAS PubMed.
- D. Xia, C. Duan, S. Liu, D. Ding, M. Baumgarten, M. Wagner, D. Schollmeyer, H. Xu and K. Müllen, New J. Chem., 2019, 43, 3788–3792 RSC.
- C.-F. Wang, W.-Y. Hung, M.-H. Cheng, J.-S. Hwang, M.-K. Leung and K.-T. Wong, Org. Electron., 2013, 14, 1958–1965 CrossRef CAS.
- X. Li, H.-J. Chi, G.-H. Lu, G.-Y. Xiao, Y. Dong, D.-Y. Zhang, Z.-Q. Zhang and Z.-Z. Hu, Org. Electron., 2012, 13, 3138–3144 CrossRef CAS.
- H. Huang, Q. Fu, S. Zhuang, Y. Liu, L. Wang, J. Chen, D. Ma and C. Yang, J. Phys. Chem. C, 2011, 115, 4872–4878 CrossRef CAS.
- H. P. Rathnayake, A. Cirpan, Z. Delen, P. M. Lahti and F. E. Karasz, Adv. Funct. Mater., 2007, 17, 115–122 CrossRef CAS.
- C. Tang, F. Liu, Y. J. Xia, J. Lin, L. H. Xie and G. Y. Zhong, Org. Electron., 2006, 7, 155 CrossRef CAS.
- D. Neher, Macromol. Rapid Commun., 2001, 22, 1365–1385 CrossRef CAS.
- I. Fischer and A. P. H. J. Schenning, in Organic electronics: Emerging concepts and technologies, eds. F. Cicoira and C. Santato, 2013, vol. 1, pp. 1–25 Search PubMed.
- M. Leclerc, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 2867–2873 CrossRef CAS.
- U. Scherf and E. J. W. List, Adv. Mater., 2002, 14, 477–487 CrossRef CAS.
- J. Rault-Berthelot, Curr. Top. Electrochem., 2004, 10, 265–284 CAS.
- Q. Hou, Y. Xu, W. Yang, M. Yuan, J. Peng and Y. Cao, J. Mater. Chem., 2002, 12, 2887–2892 RSC.
- S. Grigalevicius, L. Ma, Z.-Y. Xie and U. Scherf, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 5987–5994 CrossRef CAS.
- K. T. Kamtekar, H. L. Vaughan, B. P. Lyons, A. P. Monkman, S. U. Pandya and M. R. Bryce, Macromolecules, 2010, 43, 4481–4488 CrossRef CAS.
- P. Leclère, M. Surin, P. Brocorens, M. Cavallini, F. Biscarini and R. Lazzaroni, Mater. Sci. Eng., R, 2006, 55, 1–56 CrossRef.
- C. K. Frederickson, B. D. Rose and M. M. Haley, Acc. Chem. Res., 2017, 50, 977–987 CrossRef CAS PubMed.
- A. G. Fix, D. T. Chase and M. M. Haley, in Polyarenes I. Topics in Current Chemistry, eds. J. Siegel and Y. Wu, Springer, Berlin, Heidelberg, 2012, vol. 349, pp. 159–195 Search PubMed.
- C. Poriel, J. Rault-Berthelot, F. Barrière and A. M. Z. Slawin, Org. Lett., 2008, 10, 373–376 CrossRef CAS PubMed.
- D. Thirion, C. Poriel, F. Barrière, R. Métivier, O. Jeannin and J. Rault-Berthelot, Org. Lett., 2009, 11, 4794–4797 CrossRef CAS PubMed.
- D. Thirion, C. Poriel, J. Rault-Berthelot, F. Barrière and O. Jeannin, Chem. – Eur. J., 2010, 16, 13646–13658 CrossRef CAS PubMed.
- D. Thirion, M. Romain, J. Rault-Berthelot and C. Poriel, J. Mater. Chem., 2012, 22, 7149–7157 RSC.
- M. Romain, D. Tondelier, J.-C. Vanel, B. Geffroy, O. Jeannin, J. Rault-Berthelot, R. Métivier and C. Poriel, Angew. Chem., Int. Ed., 2013, 52, 14147–14151 CrossRef CAS PubMed.
- M. Romain, S. Thiery, A. Shirinskaya, C. Declairieux, D. Tondelier, B. Geffroy, O. Jeannin, J. Rault-Berthelot, R. Métivier and C. Poriel, Angew. Chem., Int. Ed., 2015, 54, 1176–1180 CrossRef CAS PubMed.
- M. Romain, D. Tondelier, B. Geffroy, O. Jeannin, E. Jacques, J. Rault-Berthelot and C. Poriel, Chem. – Eur. J., 2015, 21, 9426–9439 CrossRef CAS PubMed.
- M. Romain, C. Quinton, D. Tondelier, B. Geffroy, O. Jeannin, J. Rault-Berthelot and C. Poriel, J. Mater. Chem. C, 2016, 4, 1692–1703 RSC.
- C. Poriel and J. Rault-Berthelot, Acc. Chem. Res., 2018, 51, 1818–1830 CrossRef CAS PubMed.
- J. Jacob, L. Oldridge, J. Zhang, M. Gaal, E. J. W. List, A. C. Grimsdale and K. Müllen, Curr. Appl. Phys., 2004, 4, 339–342 CrossRef.
- A. C. Grimsdale, P. Leclere, R. Lazzaroni, J. D. MacKenzie, C. Murphy, S. Setayesh, C. Silva, R. H. Friend and K. Müllen, Adv. Funct. Mater., 2002, 12, 729–733 CrossRef CAS.
- J. Kim, S. H. Kim, I. H. Jung, E. Jeong, Y. Xia, S. Cho, I.-W. Hwang, K. Lee, H. Suh, H.-K. Shim and H. Y. Woo, J. Mater. Chem., 2010, 20, 1577–1586 RSC.
- A. Can, A. Facchetti and H. Usta, J. Mater. Chem. C, 2022, 10, 8496–8535 RSC.
- W. Deuschel, Helv. Chim. Acta, 1951, 34, 2403–2416 CrossRef CAS.
- O. M. Behr, G. Eglinton, A. R. Galbraith and R. A. Raphael, J. Chem. Soc., 1960, 3614–3625 RSC.
- L. Chardonnens and L. Salamin, Helv. Chim. Acta, 1968, 51, 1095–1102 CrossRef.
- L. Chardonnens and L. Avar, Helv. Chim. Acta, 1969, 52, 1091–1095 CrossRef CAS.
- R. G. Harvey, J. Pataki, C. Cortez, P. Di Raddo and C. X. Yang, J. Org. Chem., 1991, 56, 1210–1217 CrossRef CAS.
- S. Merlet, M. Birau and Z. Y. Wang, Org. Lett., 2002, 4, 2157–2159 CrossRef CAS PubMed.
- Y. Wang, P. R. McGonigal, B. Herlé, M. Besora and A. M. Echavarren, J. Am. Chem. Soc., 2014, 136, 801–809 CrossRef CAS PubMed.
- C.-W. Lee, E.-C. Liu and Y.-T. Wu, J. Org. Chem., 2015, 80, 10446–10456 CrossRef CAS PubMed.
- C. Poriel, J.-J. Liang, J. Rault-Berthelot, F. Barrière, N. Cocherel, A. M. Z. Slawin, D. Horhant, M. Virboul, G. Alcaraz, N. Audebrand, L. Vignau, N. Huby, G. Wantz and L. Hirsch, Chem. – Eur. J., 2007, 13, 10055–10069 CrossRef CAS PubMed.
- T. P. I. Saragi, T. Spehr, A. Siebert, T. Fuhrmann-Lieker and J. Salbeck, Chem. Rev., 2007, 107, 1011–1065 CrossRef CAS PubMed.
- C. Poriel, L. J. Sicard and J. Rault-Berthelot, Chem. Commun., 2019, 55, 14238–14254 RSC.
- C.-C. Hsiao, Y.-K. Lin, C.-J. Liu, T.-C. Wu and Y.-T. Wu, Adv. Synth. Catal., 2010, 352, 3267–3274 CrossRef CAS.
- L. Sicard, C. Quinton, J.-D. Peltier, D. Tondelier, B. Geffroy, U. Biapo, R. Métivier, O. Jeannin, J. Rault-Berthelot and C. Poriel, Chem. – Eur. J., 2017, 23, 7719–7727 CrossRef CAS PubMed.
- D. Horhant, J.-J. Liang, M. Virboul, C. Poriel, G. Alcaraz and J. Rault-Berthelot, Org. Lett., 2006, 8, 257–260 CrossRef CAS PubMed.
- I. B. Berlman, J. Chem. Phys., 1970, 52, 5616–5621 CrossRef CAS.
- G. Heimel, M. Daghofer, J. Gierschner, E. J. List, A. C. Grimsdale, K. Mullen, D. Beljonne, J. L. Bredas and E. Zojer, J. Chem. Phys., 2005, 122, 054501–054511 CrossRef.
- J. Rault-Berthelot, C. Poriel, F. Justaud and F. Barrière, New J. Chem., 2008, 32, 1259–1266 RSC.
- J. Rault-Berthelot and J. Simonet, J. Electroanal. Chem., 1985, 182, 187–192 CrossRef CAS.
- J. Rault-Berthelot, M.-M. Granger and L. Mattiello, Synth. Met., 1998, 97, 211–215 CrossRef CAS.
- C. Poriel, Y. Ferrand, P. Le Maux, J. Rault-Berthelot and G. Simonneaux, Inorg. Chem., 2004, 43, 5086–5095 CrossRef CAS PubMed.
- C. Poriel, Y. Ferrand, P. Le Maux, C. Paul-Roth, G. Simonneaux and J. Rault-Berthelot, J. Electroanal. Chem., 2005, 583, 92–103 CrossRef CAS.
- C. Poriel and J. Rault-Berthelot, Adv. Funct. Mater., 2020, 30, 1910040 CrossRef CAS.
- T. Hadizad, J. Zhang, Z. Y. Wang, T. C. Gorjanc and C. Py, Org. Lett., 2005, 7, 795–797 CrossRef CAS PubMed.
- T. Hadizad, J. Zhang, D. Yan, Z. Y. Wang, J. P. M. Serbena, M. S. Meruvia and I. A. Hümmelgen, J. Mater. Sci.: Mater. Electron., 2007, 18, 903–912 CrossRef CAS.
- N. Cocherel, C. Poriel, J. Rault-Berthelot, F. Barrière, N. Audebrand, A. M. A. Slawin and L. Vignau, Chem. – Eur. J., 2008, 14, 11328–11342 CrossRef CAS PubMed.
- N. Cocherel, C. Poriel, L. Vignau, J.-F. Bergamini and J. Rault-Berthelot, Org. Lett., 2010, 12, 452–455 CrossRef CAS PubMed.
- C. Poriel, N. Cocherel, J. Rault-Berthelot, L. Vignau and O. Jeannin, Chem. – Eur. J., 2011, 17, 12631–12645 CrossRef CAS PubMed.
- C. Poriel, J. Rault-Berthelot and D. Thirion, J. Org. Chem., 2013, 78, 886–898 CrossRef CAS PubMed.
- L.-H. Xie, X.-Y. Hou, C. Tang, Y.-R. Hua, R.-J. Wang, R.-F. Chen, Q.-L. Fan, L.-H. Wang, W. Wei, B. Peng and W. Huang, Org. Lett., 2006, 8, 1363–1366 CrossRef CAS PubMed.
- J.-D. Peltier, B. Heinrich, B. Donnio, O. Jeannin, J. Rault-Berthelot and C. Poriel, Chem. – Eur. J., 2017, 23, 17290–17303 CrossRef CAS PubMed.
- Z. Jiang, H. Yao, Z. Liu, C. Yang, C. Zhong, J. Qin, G. Yu and Y. Liu, Org. Lett., 2009, 11, 4132–4135 CrossRef CAS PubMed.
- M. Lv, X. Lu, Y. Jiang, M. E. Sandoval-Salinas, D. Casanova, H. Sun, Z. Sun, J. Xu, Y. Yang and J. Chen, Angew. Chem., Int. Ed., 2022, 61, e202113190 CrossRef CAS PubMed.
- C.-C. Wu, W.-G. Liu, W.-Y. Hung, T.-L. Liu, Y.-T. Lin, H.-W. Lin, K.-T. Wong, Y.-Y. Chien, R.-T. Chen, T.-H. Hung, T.-C. Chao and Y.-M. Chen, Appl. Phys. Lett., 2005, 87, 052103 CrossRef.
- P. Maslak, Adv. Mater., 1994, 6, 405–407 CrossRef CAS.
- H. E. Simmons and T. Fukunaga, J. Am. Chem. Soc., 1967, 89, 5208–5215 CrossRef CAS.
- S.-Y. Ku, L.-C. Chi, W.-Y. Hung, S.-W. Yang, T.-C. Tsai, K.-T. Wong, Y.-H. Chen and C.-I. Wu, J. Mater. Chem., 2009, 19, 773–780 RSC.
- K.-T. Wong, L.-C. Chi, S.-C. Huang, Y.-L. Liao, Y.-H. Liu and Y. Wang, Org. Lett., 2006, 8, 5029–5032 CrossRef CAS PubMed.
- Y. Park, J.-H. Lee, D. H. Jung, S.-H. Liu, Y.-H. Lin, L.-Y. Chen, C.-C. Wu and J. Park, J. Mater. Chem., 2010, 20, 5930–5936 RSC.
- A. P. Kulkarni, C. J. Tonzola, A. Babel and S. A. Jenekhe, Chem. Mater., 2004, 16, 4556–4573 CrossRef CAS.
- D. Thirion, J. Rault-Berthelot, L. Vignau and C. Poriel, Org. Lett., 2011, 13, 4418–4421 CrossRef CAS PubMed.
- D. Thirion, M. Romain, J. Rault-Berthelot and C. Poriel, Mater. Adv., 2021, 2, 1271–1283 RSC.
- C. Poriel and J. Rault-Berthelot, Adv. Funct. Mater., 2021, 31, 2010547 CrossRef CAS.
- F. Barrière, C. Poriel and J. Rault-Berthelot, Electrochim. Acta, 2013, 110, 735–740 CrossRef.
- D. Thirion, C. Poriel, R. Métivier, J. Rault-Berthelot, F. Barrière and O. Jeannin, Chem. – Eur. J., 2011, 17, 10272–10287 CrossRef CAS PubMed.
- K. H. Lee, S. O. Kim, S. Kang, J. Y. Lee, K. S. Yook, J. Y. Lee and S. S. Yoon, Eur. J. Org. Chem., 2012, 2748–2755 CrossRef CAS.
- K. H. Lee, S. O. Kim, J. N. You, S. Kang, J. Y. Lee, K. S. Yook, S. O. Jeon, J. Y. Lee and S. S. Yoon, J. Mater. Chem., 2012, 22, 5145–5154 RSC.
- Q. Meng and W. Hu, Phys. Chem. Chem. Phys., 2012, 14, 14152–14164 RSC.
- S. Kola, J. Sinha and H. E. Katz, J. Polym. Sci., Part B: Polym. Phys., 2012, 50, 1090–1120 CrossRef CAS.
- C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS PubMed.
- K. Zhou, H. Dong, H.-I. Zhang and W. Hu, Phys. Chem. Chem. Phys., 2014, 16, 22448–22457 RSC.
- X. Gao and Z. Zhao, Sci. China: Chem., 2015, 58, 947–968 CrossRef CAS.
- C. Py, T. C. Gorjanc, T. Hadizad, J. Zhang and Z. Y. Wang, J. Vac. Sci. Technol., A, 2006, 24, 654–656 CrossRef CAS.
- J. P. M. Serbena, I. A. Hümmelgen, T. Hadizad and Z. Y. Wang, Small, 2006, 2, 372–374 CrossRef CAS PubMed.
- J. P. M. Serbena, J. Y. Huang, D. Ma, Z. Y. Wang and I. A. Hümmelgen, Org. Electron., 2009, 10, 357–362 CrossRef CAS.
- T. Nakagawa, D. Kumaki, J.-I. Nishida, S. Tokito and Y. Yamashita, Chem. Mater., 2008, 20, 2615–2617 CrossRef CAS.
- H. Usta, C. Risko, Z. Wang, H. Huang, M. K. Deliomeroglu, A. Zhukhovitskiy, A. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2009, 131, 5586–5608 CrossRef CAS PubMed.
- Z.-P. Fan, X.-Y. Li, X.-E. Luo, X. Fei, B. Sun, L.-C. Chen, Z.-F. Shi, C.-L. Sun, X. Shao and H.-L. Zhang, Adv. Funct. Mater., 2017, 27, 1702318 CrossRef.
- Y.-I. Park, J. S. Lee, B. J. Kim, B. Kim, J. Lee, D. H. Kim, S.-Y. Oh, J. H. Cho and J.-W. Park, Chem. Mater., 2011, 4038–4044 CrossRef CAS.
- B. J. Kim, Y.-I. Park, H. J. Kim, K. Ahn, D. R. Lee, D. H. Kim, S.-Y. Oh, J.-W. Park and J. H. Cho, J. Mater. Chem., 2012, 22, 14617–14623 RSC.
- Y. Miyata, T. Minari, T. Nemoto, S. Isoda and K. Komatsu, Org. Biomol. Chem., 2007, 5, 2592–2598 RSC.
- M. Ozdemir, D. Choi, G. Kwon, Y. Zorlu, H. Kim, M.-G. Kim, S. Seo, U. Sen, M. Citir, C. Kim and H. Usta, RSC Adv., 2016, 6, 212–226 RSC.
- R. Ozdemir, D. Choi, M. Ozdemir, G. Kwon, H. Kim, U. Sen, C. Kim and H. Usta, J. Mater. Chem. C, 2017, 5, 2368–2379 RSC.
- R. Ozdemir, S. Park, İ. Deneme, Y. Park, Y. Zorlu, H. A. Alidagi, K. Harmandar, C. Kim and H. Usta, Org. Chem. Front., 2018, 5, 2912–2924 RSC.
- H. Usta, A. Facchetti and T. J. Marks, Acc. Chem. Res., 2011, 44, 501–510 CrossRef CAS PubMed.
- Y.-J. Cheng, J. Luo, S. Huang, X. Zhou, Z. Shi, T.-D. Kim, D. H. Bale, S. Takahashi, A. Yick, B. M. Polishak, S.-H. Jang, L. R. Dalton, P. J. Reid, W. H. Steier and A. K. Y. Jen, Chem. Mater., 2008, 20, 5047–5054 CrossRef CAS.
- F. Liu, F. Wu, Z. Tu, Q. Liao, Y. Gong, L. Zhu, Q. Li and Z. Li, Adv. Funct. Mater., 2019, 29, 1901296 CrossRef.
- H. Usta, A. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2008, 130, 8580–8581 CrossRef CAS PubMed.
- S. Bebiche, P. Cisneros-Perez, T. Mohammed-Brahim, M. Harnois, J. Rault-Berthelot, C. Poriel and E. Jacques, Mater. Chem. Front., 2018, 2, 1631–1641 RSC.
- N. Arfaoui, M. Mahdouani, I. Bouhadda, C. Poriel, R. Bourguiga, E. Jacques, M. Chevrier and S. Bebiche, Superlattices Microstruct., 2018, 123, 286–296 CrossRef CAS.
- E. Jacques, M. Romain, A. Yassin, S. Bebiche, M. Harnois, T. Mohammed-Brahim, J. Rault-Berthelot and C. Poriel, J. Mater. Chem. C, 2014, 2, 3292–3302 RSC.
- X. Gao and Y. Hu, J. Mater. Chem. C, 2014, 2, 3099–3117 RSC.
- Y. Zhao, Y. Guo and Y. Liu, Adv. Mater., 2013, 25, 5372–5391 CrossRef CAS PubMed.
- L. Feng, H. Dong, Q. Li, W. Zhu, G. Qiu, S. Ding, Y. Li, M. A. Christensen, C. R. Parker, Z. Wei, M. B. Nielsen and W. Hu, Sci. China Mater., 2017, 60, 75–82 CrossRef CAS.
- M. A. Christensen, C. R. Parker, T. J. Sørensen, S. de Graaf, T. J. Morsing, T. Brock-Nannestad, J. Bendix, M. M. Haley, P. Rapta, A. Danilov, S. Kubatkin, O. Hammerich and M. B. Nielsen, J. Mater. Chem. C, 2014, 2, 10428–10438 RSC.
- M. A. Baldo, D. F. O'Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151–154 CrossRef CAS.
- A. Chaskar, H.-F. Chen and K.-T. Wong, Adv. Mater., 2011, 23, 3876–3895 CrossRef CAS PubMed.
- H. Sasabe and J. Kido, Chem. Mater., 2011, 23, 621–630 CrossRef CAS.
- Y. Tao, C. Yang and J. Qin, Chem. Soc. Rev., 2011, 40, 2943–2970 RSC.
- L. Xiao, Z. Chen, B. Qu, J. Luo, S. Kong, Q. Cong and J. Kido, Adv. Mater., 2011, 23, 926–952 CrossRef CAS PubMed.
- K. S. Yook and J. Y. L. Lee, Adv. Mater., 2012, 24, 3169–3190 CrossRef CAS PubMed.
- H. Sasabe and J. Kido, Eur. J. Org. Chem., 2013, 7653–7663 CrossRef CAS.
- D. Chen, S.-J. Su and Y. Cao, J. Mater. Chem. C, 2014, 2, 9565 RSC.
- K. S. Yook and J. Y. Lee, Adv. Mater., 2014, 26, 4218–4233 CrossRef CAS PubMed.
- K. S. Yook and J. Y. Lee, Chem. Rec., 2016, 16, 159–172 CrossRef CAS PubMed.
- C. Poriel and J. Rault-Berthelot, Acc. Mater. Res., 2022, 3, 379–390 CrossRef CAS.
- L.-C. Chi, W.-Y. Hung, H.-C. Chiu and K.-T. Wong, Chem. Commun., 2009, 3892–3894 RSC.
- F. Lucas, D. Tondelier, B. Geffroy, T. Heiser, O. A. Ibraikulov, C. Quinton, C. Brouillac, N. Leclerc, J. Rault-Berthelot and C. Poriel, Mater. Chem. Front., 2021, 5, 8066–8077 RSC.
- C. Poriel, R. Métivier, J. Rault-Berthelot, D. Thirion, F. Barrière and O. Jeannin, Chem. Commun., 2011, 47, 11703–11705 RSC.
- S. Chaurasia, Y.-C. Chen, H.-H. Chou, Y.-S. Wen and J. T. Lin, Tetrahedron, 2012, 68, 7755–7762 CrossRef CAS.
- J. Zhang, B. Zhao, Y. Mi, H. Liu, Z. Guo, G. Bie, W. Wei, C. Gao and Z. An, Dyes Pigm., 2017, 140, 261–268 CrossRef CAS.
- J. Mogensen, H. Michaels, R. Roy, L. Broløs, M. D. Kilde, M. Freitag and M. B. Nielsen, Eur. J. Org. Chem., 2020, 6127–6134 CrossRef CAS.
- Z.-E. Chen, Q.-L. Qi and H. Zhang, Synth. Met., 2020, 267, 116473 CrossRef CAS.
- M. Pawlicki, H. A. Collins, R. G. Denning and H. L. Anderson, Angew. Chem., Int. Ed., 2009, 48, 3244–3266 CrossRef CAS PubMed.
- Q. Zheng, S. K. Gupta, G. S. He, L.-S. Tan and P. N. Prasad, Adv. Funct. Mater., 2008, 18, 2770–2779 CrossRef CAS.
- Z. Liu, D. Cao, Y. Chen and Q. Fang, Dyes Pigm., 2010, 86, 63–67 CrossRef CAS.
- Á. M. Marín, J. P. Telo, D. Collado, F. Nájera, E. Pérez-Inestrosa and U. Pischel, Chem. – Eur. J., 2018, 24, 2929–2935 CrossRef PubMed.
- H. H. Fan, L. Guo, K. F. Li, M. S. Wong and K. W. Cheah, J. Am. Chem. Soc., 2012, 134, 7297–7300 CrossRef CAS PubMed.
- D. An and Z. Ye, J. Chil. Chem. Soc., 2015, 60, 2971–2974 CrossRef CAS.
- L. Guo, K. F. Li, M. S. Wong and K. W. Cheah, Chem. Commun., 2013, 49, 3597–3599 RSC.
- L. Guo, X. Liu, T. Zhang, H.-B. Luo, H. H. Fan and M. S. Wong, J. Mater. Chem. C, 2020, 8, 1768–1772 RSC.
- T.-C. Lin, C.-S. Hsu, C.-L. Hu, Y.-F. Chen and W.-J. Huang, Tetrahedron Lett., 2009, 50, 182–185 CrossRef CAS.
- S. Welter, N. Salluce, A. Benetti, N. Rot, P. Belser, P. Sonar, A. C. Grimsdale, K. Müllen, M. Lutz, A. L. Spek and L. De Cola, Inorg. Chem., 2005, 44, 4706–4718 CrossRef CAS PubMed.
- Y. Ie, M. Nitani and Y. Aso, Chem. Lett., 2007, 36, 1326–1327 CrossRef CAS.
- S. Thiery, B. Heinrich, B. Donnio, C. Poriel and F. Camerel, J. Mater. Chem. C, 2014, 2, 4265–4275 RSC.
- H. So, J. H. Kim, J. H. Lee, H. Hwang, D. K. An and K. M. Lee, Chem. Commun., 2019, 55, 14518–14521 RSC.
- K.-W. Park and T. L. Andrew, J. Phys. Chem. C, 2022, 126, 17663–17669 CrossRef CAS.
- W. Schroth and K. Schmidt, Chem, 1963, 3, 309 CAS.
- J.-H. Ho, Y.-C. Lin, L.-T. Chou, Y.-Z. Chen, W.-Q. Liu and C.-L. Chuang, Tetrahedron Lett., 2013, 54, 1991–1993 CrossRef CAS.
- W. Dilthey, I. Thewalt and O. Trösken, Ber. Dtsch. Chem. Ges., 1934, 67, 1959–1964 CrossRef.
- C. Weizmann, E. Bergmann and L. Haskelberg, J. Chem. Soc., 1939, 391–397 RSC.
- C. Poriel, J. Rault-Berthelot, D. Thirion, F. Barrière and L. Vignau, Chem. – Eur. J., 2011, 17, 14031–14046 CrossRef CAS PubMed.
- C. Poriel, F. Barrière, D. Thirion and J. Rault-Berthelot, Chem. – Eur. J., 2009, 15, 13304–13307 CrossRef CAS PubMed.
- C. Poriel, F. Barrière, J. Rault-Berthelot and D. Thirion, Chem. – Eur. J., 2019, 25, 10689–10697 CrossRef CAS PubMed.
- P. Hapiot, C. Lagrost, F. Le Floch, E. Raoult and J. Rault-Berthelot, Chem. Mater., 2005, 17, 2003–2012 CrossRef CAS.
- K.-T. Wong, Y.-Y. Chien, R.-T. Chen, C.-F. Wang, Y.-T. Lin, H.-H. Chiang, P.-Y. Hsieh, C.-C. Wu, C. H. Chou, Y. O. Su, G.-H. Lee and S.-M. Peng, J. Am. Chem. Soc., 2002, 124, 11576–11577 CrossRef CAS PubMed.
- S. Tang, M. Liu, C. Gu, Y. Zhao, P. Lu, D. Lu, L. Liu, F. Shen, B. Yang and Y. Ma, J. Org. Chem., 2008, 73, 4212–4218 CrossRef CAS PubMed.
- G. Klaerner and R. D. Miller, Macromolecules, 1998, 31, 2007–2009 CrossRef CAS.
- C.-C. Wu, T.-L. Liu, Y.-T. Lin, W.-Y. Hung, T.-H. Ke, K.-T. Wong and T.-C. Chao, Appl. Phys. Lett., 2004, 85, 1172–1174 CrossRef CAS.
- H. Li, A. S. Batsanov, K. C. Moss, H. L. Vaughan, F. B. Dias, K. T. R. Kamtekar, M. R. Bryce and A. P. Monkman, Chem. Commun., 2010, 46, 4812–4814 RSC.
- E. J. W. List, R. Guentner, P. Scandiucci de Freitas and U. Scherf, Adv. Mater., 2002, 14, 374–378 CrossRef CAS.
- X. Chen, H.-T. Tseng, J.-L. Liao and S.-A. Chen, J. Phys. Chem. B, 2005, 109, 17496–17502 CrossRef CAS PubMed.
- F. Montilla and R. Mallavia, Adv. Funct. Mater., 2007, 17, 71–78 CrossRef CAS.
- S. Kappaun, H. Scheiber, R. Trattnig, E. Zojer, E. J. W. List and C. Slugovc, Chem. Commun., 2008, 5170–5172 RSC.
- Y.-F. Bo, Y.-Y. Liu, H. Soleimaninejad, M.-N. Yu, L.-H. Xie, T. A. Smith, K. P. Ghiggino and W. Huang, J. Phys. Chem. A, 2019, 123, 2789–2795 CrossRef CAS PubMed.
- T. Takeda, K. Inukai, K. Tahara and Y. Tobe, J. Org. Chem., 2011, 76, 9116–9121 CrossRef CAS PubMed.
- S. Nobusue and Y. Tobe, Synlett, 2016, 2140–2144 CAS.
- S. Li, M. Aljhdli, H. Thakellapalli, B. Farajidizaji, Y. Zhang, N. G. Akhmedov, C. Milsmann, B. V. Popp and K. K. Wang, Org. Lett., 2017, 19, 4078–4081 CrossRef CAS PubMed.
- Y. Wei, X. Zheng, D. Lin, H. Yuan, Z. Yin, L. Yang, Y. Yu, S. Wang, L.-H. Xie and W. Huang, J. Org. Chem., 2019, 84, 10701–10709 CrossRef CAS PubMed.
- L. Sicard, O. Jeannin, J. Rault-Berthelot, C. Quinton and C. Poriel, ChemPlusChem, 2018, 83, 874–880 CrossRef CAS PubMed.
- F. Lucas, L. Sicard, O. Jeannin, J. Rault-Berthelot, E. Jacques, C. Quinton and C. Poriel, Chem. – Eur. J., 2019, 25, 7740–7748 CrossRef CAS PubMed.
- D. Radulescu and M. Alexa, Bul. Chim., Soc. Chim. Romania, 1939, 1, 25–29 CAS.
- L. Chardonnens and R. Ritter, Helv. Chim. Acta, 1955, 38, 393–396 CrossRef CAS.
- W. Kemp and J. Spanswick, J. Chem. Soc. C, 1967, 2544–2545 RSC.
- Y. Altman and D. Ginsburg, J. Chem. Soc., 1961, 1498–1505 RSC.
- L. Chardonnens and J. Häger, Helv. Chim. Acta, 1970, 53, 843–847 CrossRef CAS.
- M. Nierenstein and C. W. Webster, J. Am. Chem. Soc., 1945, 67, 691–692 CrossRef CAS.
- W. Deuschel, Helv. Chim. Acta, 1951, 34, 168–185 CrossRef CAS.
- L. Chardonnens and H. Chardonnens, Helv. Chim. Acta, 1958, 41, 2109–2111 CrossRef CAS.
- M. Romain, M. Chevrier, S. Bebiche, T. Mohammed-Brahim, J. Rault-Berthelot, E. Jacques and C. Poriel, J. Mater. Chem. C, 2015, 3, 5742–5753 RSC.
- L. Chardonnens and J. Rody, Helv. Chim. Acta, 1959, 42, 1328–1331 CrossRef CAS.
- W. Deuschel, Helv. Chim. Acta, 1952, 35, 1774–1776 CrossRef CAS.
- L. Chardonnens and H. Chardonnens, Helv. Chim. Acta, 1966, 49, 1931–1934 CrossRef CAS.
- B. Du, L. Wang, S.-C. Yuan, T. Lei, J. Pei and Y. Cao, Polymer, 2013, 54, 2935–2944 CrossRef CAS.
- Y.-Y. Li, H.-Y. Lu, M. Li, X.-J. Li and C.-F. Chen, J. Org. Chem., 2014, 79, 2139–2147 CrossRef CAS PubMed.
- R. P. Kaiser, D. Nečas, T. Cadart, R. Gyepes, I. Císařová, J. Mosinger, L. Pospíšil and M. Kotora, Angew. Chem., Int. Ed., 2019, 58, 17169–17174 CrossRef CAS PubMed.
- J.-D. Peltier, B. Heinrich, B. Donnio, O. Jeannin, J. Rault-Berthelot, E. Jacques and C. Poriel, J. Mater. Chem. C, 2018, 6, 13197–13210 RSC.
- C. Poriel, C. Quinton, F. Lucas, J. Rault-Berthelot, Z.-Q. Jiang and O. Jeannin, Adv. Funct. Mater., 2021, 31, 2104980 CrossRef CAS.
- L. Zhao, C. Duan, D. Ding, S. Liu, D. Xia, Y. Guo, H. Xu and M. Baumgarten, Chin. Chem. Lett., 2021, 32, 397–400 CrossRef CAS.
- D. Xia, D. Gehrig, X. Guo, M. Baumgarten, F. Laquai and K. Müllen, J. Mater. Chem. A, 2015, 3, 11086–11092 RSC.
- M. Kimura, S. Kuwano, Y. Sawaki, H. Fujikawa, K. Noda, Y. Taga and K. Takagi, J. Mater. Chem., 2005, 15, 2393–2398 RSC.
- J. Luo, Y. Zhou, Z.-Q. Niu, Q.-F. Zhou, Y. Ma and J. Pei, J. Am. Chem. Soc., 2007, 129, 11314–11315 CrossRef CAS PubMed.
- T. Lei, J. Luo, L. Wang, Y. Ma, J. Wang, Y. Cao and J. Pei, New J. Chem., 2010, 34, 699–707 RSC.
- F. Goubard and F. Dumur, RSC Adv., 2015, 5, 3521–3551 RSC.
- C. Fan, Y. Chen, P. Gan, C. Yang, C. Zhong, J. Qin and D. Ma, Org. Lett., 2010, 12, 5648–5651 CrossRef CAS PubMed.
- C. Poriel, J. Rault-Berthelot and Z.-Q. Jiang, Mater. Chem. Front., 2022, 6, 1246–1252 RSC.
- J. Ma, M. Idris, T. Y. Li, D. S. M. Ravinson, T. Fleetham, J. Kim, P. I. Djurovich, S. R. Forrest and M. E. Thompson, Adv. Opt. Mater., 2022, 10, 2101530 CrossRef CAS.
- F. Lucas, C. Brouillac, S. Fall, N. Zimmerman, D. Tondelier, B. Geffroy, N. Leclerc, T. Heiser, C. Lebreton, E. Jacques, C. Quinton, J. Rault-Berthelot and C. Poriel, Chem. Mater., 2022, 34, 8345–8355 CrossRef CAS.
- Q. Duez, M. Romain, C. Tonneaux, J. De Winter, V. Lemaur, J. Cornil, C. Poriel and P. Gerbaux, Anal. Methods, 2018, 10, 2303–2306 RSC.
- D. Li, J. Li, D. Liu, W. Li, C.-L. Ko, W.-Y. Hung and C. Duan, ACS Appl. Mater. Interfaces, 2021, 13, 13459–13469 CrossRef CAS PubMed.
- K. Udagawa, H. Sasabe, C. Cai and J. Kido, Adv. Mater., 2014, 26, 5062–5066 CrossRef CAS PubMed.
- M. Kim and J. Y. Lee, Adv. Funct. Mater., 2014, 24, 4164–4169 CrossRef CAS.
- S.-y Chang, G.-T. Lin, Y.-C. Cheng, J.-J. Huang, C.-L. Chang, C.-F. Lin, J.-H. Lee, T.-L. Chiu and M.-k Leung, ACS Appl. Mater. Interfaces, 2018, 10, 42723–42732 CrossRef CAS PubMed.
- C. W. Lee and J. Y. Lee, Adv. Mater., 2013, 25, 5450–5454 CrossRef CAS PubMed.
- Q.-L. Xu, X. Liang, S. Zhang, Y.-M. Jing, X. Liu, G.-Z. Lu, Y.-X. Zheng and J.-L. Zuo, J. Mater. Chem. C, 2015, 3, 3694–3701 RSC.
- G. Lu, R. Wu, N. Li, X. Wang, L. Zhou and C. Yang, J. Mater. Chem. C, 2022, 10, 17303–17308 RSC.
- M. Kim and J. Y. Lee, ACS Appl. Mater. Interfaces, 2014, 6, 14874–14880 CrossRef CAS PubMed.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234 CrossRef CAS PubMed.
- Y. Fu, H. Liu, B. Z. Tang and Z. Zhao, Nat. Commun., 2023, 14, 2019 CrossRef CAS PubMed.
- T.-A. Lin, T. Chatterjee, W.-L. Tsai, W.-K. Lee, M.-J. Wu, M. Jiao, K.-C. Pan, C.-L. Yi, C.-L. Chung, K.-T. Wong and C.-C. Wu, Adv. Mater., 2016, 28, 6976–6983 CrossRef CAS PubMed.
- X. Hong, D. Zhang, C. Yin, Q. Wang, Y. Zhang, T. Huang, J. Wei, X. Zeng, G. Meng, X. Wang, G. Li, D. Yang, D. Ma and L. Duan, Chem, 2022, 8, 1705–1719 CAS.
- H. Liu, Z. Liu, G. Li, H. Huang, C. Zhou, Z. Wang and C. Yang, Angew. Chem., Int. Ed., 2021, 60, 12376–12380 CrossRef CAS PubMed.
- C.-C. Peng, S.-Y. Yang, H.-C. Li, G.-H. Xie, L.-S. Cui, S.-N. Zou, C. Poriel, Z.-Q. Jiang and L.-S. Liao, Adv. Mater., 2020, 32, 2003885 CrossRef CAS PubMed.
- D. H. Ahn, J. S. Moon, S. W. Kim, S. Y. Lee, D. Karthik, J. Y. Lee and J. H. Kwon, Org. Electron., 2018, 59, 39–44 CrossRef CAS.
- S.-J. Woo, Y. Kim, S.-K. Kwon, Y.-H. Kim and J.-J. Kim, ACS Appl. Mater. Interfaces, 2019, 11, 7199–7207 CrossRef CAS PubMed.
- B. Li, Z. Li, T. Hu, Y. Zhang, Y. Wang, Y. Yi, F. Guo and L. Zhao, J. Mater. Chem. C, 2018, 6, 2351–2359 RSC.
- M. Godumala, S. Choi, S. Y. Park, M. J. Cho, H. J. Kim, D. H. Ahn, J. S. Moon, J. H. Kwon and D. H. Choi, Chem. Mater., 2018, 30, 5005–5012 CrossRef CAS.
- X.-Q. Wang, S.-Y. Yang, Q.-S. Tian, C. Zhong, Y.-K. Qu, Y.-J. Yu, Z.-Q. Jiang and L.-S. Liao, Angew. Chem., Int. Ed., 2021, 60, 5213–5219 CrossRef CAS PubMed.
- X. Tang, L. S. Cui, H. C. Li, A. J. Gillett, F. Auras, Y. K. Qu, C. Zhong, S. T. E. Jones, Z. Q. Jiang, R. H. Friend and L. S. Liao, Nat. Mater., 2020, 19, 1332–1338 CrossRef CAS PubMed.
- Z. Wang, D. Li, W. Li, J. Zhang, M. Luo, S. Du, X. Zhang, S. Xu and Z. Ge, Adv. Opt. Mater., 2023, 2300017 CrossRef CAS.
- J. Jayakumar, T.-L. Wu, M.-J. Huang, P.-Y. Huang, T.-Y. Chou, H.-W. Lin and C.-H. Cheng, ACS Appl. Mater. Interfaces, 2019, 11, 21042–21048 CrossRef CAS PubMed.
- P. Rajamalli, N. Senthilkumar, P. Y. Huang, C. C. Ren-Wu, H. W. Lin and C. H. Cheng, J. Am. Chem. Soc., 2017, 139, 10948–10951 CrossRef CAS PubMed.
- P. Rajamalli, V. Thangaraji, N. Senthilkumar, C.-C. Ren-Wu, H.-W. Lin and C.-H. Cheng, J. Mater. Chem. C, 2017, 5, 2919–2926 RSC.
- Y. Chen, Q. Sun, Y. Dai, D. Yang, X. Qiao and D. Ma, J. Mater. Chem. C, 2020, 8, 13777–13785 RSC.
- Z. Liu, G. Li, H. Liu, C. Zhou, K. Li, Z. Wang and C. Yang, Adv. Opt. Mater., 2021, 9, 2101410 CrossRef CAS.
- H. Peng, J. Lou, G. Li, C. Zhou, Z. Wang and H. Liu, J. Mater. Chem. C, 2022, 10, 5813–5820 RSC.
- H. Peng, Y. Xu, C. Zhou, R. Pei, J. Miao, H. Liu and C. Yang, Adv. Funct. Mater., 2023, 33, 2211696 CrossRef CAS.
- Y. Liu, Y. Xu, H. Peng, J. Miao, H. Liu and C. Yang, Chem. Commun., 2023, 59, 9255–9258 RSC.
- S. Bebiche, I. Bouhadda, T. Mohammed-Brahim, N. Coulon, J. F. Bergamini, C. Poriel and E. Jacques, Solid-State Electron., 2017, 130, 49–56 CrossRef CAS.
- Z. Ning, Q. Zhang, H. Pei, J. Luan, C. Lu, Y. Cui and H. Tian, J. Phys. Chem. C, 2009, 113, 10307–10313 CrossRef CAS.
- D. Beaudoin, J.-N. Blair-Pereira, S. Langis-Barsetti, T. Maris and J. D. Wuest, J. Org. Chem., 2017, 82, 8536–8547 CrossRef CAS PubMed.
- Z. Chen, P. Sun, P. Bai, H. Su, J. Yang, Y. Liu, Y. Xu and Y. Geng, Chem. Commun., 2021, 57, 10791–10794 RSC.
- Y. Dibdalli, J. Gaete, C. Valdebenito, J. L. Arroyo, I. Martínez, G. Abarca and C. Morales-Verdejo, J. Organomet. Chem., 2022, 973–974, 122408 CrossRef CAS.
- M. Feofanov, A. Förtsch, K. Amsharov and V. Akhmetov, Chem. Commun., 2021, 57, 12325–12328 RSC.
- I. Caivano, Z. Tošner, I. Císařová, D. Nečas and M. Kotora, ChemPlusChem, 2020, 85, 2010–2016 CrossRef CAS PubMed.
- T. Cadart, D. Nečas, R. P. Kaiser, L. Favereau, I. Císařová, R. Gyepes, J. Hodačová, K. Kalíková, L. Bednárová, J. Crassous and M. Kotora, Chem. – Eur. J., 2021, 27, 11279–11284 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2023 |