
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
13C pNMR shifts of MOFs based on Cu(II)-paddlewheel dimers – DFT predictions for spin–1/2 defects†
Edoardo
Fusco
,
Sharon E.
Ashbrook
 and
Michael
Bühl
*
and
Michael
Bühl
*
EaStCHEM School of Chemistry and Centre of Magnetic Resonance, University of St Andrews, Fife KY16 9ST, UK. E-mail: buehl@st-andrews.ac.uk
First published on 7th November 2023
Abstract
We present DFT predictions (CAM-B3LYP/II level) for the paramagnetic Nuclear Magnetic Resonance (pNMR) spectra of small molecular models based on the Cu(II)-paddlewheel dimer motif that is present in metal–organic frameworks (MOFs, notably the HKUST and STAM families). We explore potential point defects with spin–1/2 discovered through electron paramagnetic resonance (EPR) experiments. We consider defects through substitution of one Cu(II) centre in the dimer with protons, or through one-electron reduction, affording a mixed-valence dimer. While most of the defects have predicted pNMR shifts at room temperature in the range of those for the non-defective MOFs, their detection and assignment should be possible based on their distinct temperature dependence.
Introduction
Metal organic frameworks (MOFs) are crystalline materials, also known as coordination polymers. They have two components: organic ligands (such as phosphonates, carboxylates, sulfonates or imidazolates) and a metal centre (such as Cu2+, Zn2+, Al3+ or Cr3+), and variation in both of these can result in different structural forms or changes in the physical or chemical properties.1 MOFs are widely studied and often a centre of attention for the scientific community due to their versatility and tunability. In fact, MOFs find application in several sectors such as medicine, catalysis and gas storage.2–7 For this work, the interest lies in MOFs that are based on copper paddlewheel dimer building blocks. This motif is composed of two copper atoms in the +2 oxidation state, and the specific MOFs considered here contain 1,3,5-benzene tricarboxylic acid (BTC) and derivatives thereof as the organic linker(s). In these cases, four O atoms, from four different acetate groups of BTC, coordinate the copper atoms in the equatorial plane, affording the characteristic “paddlewheel” structure and leaving one axial coordination site free for each metal centre (see Fig. 1). These sites are the key to the chemistry of the material as they act as anchor points for small ligands, such as gasses which are stored in the MOF pores. | ||
| Fig. 1 Schematic showing a paddlewheel dimer with axial ligand site, L. | ||
HKUST-1 was one the first MOFs discovered, being synthesized for the first time in 1999.8 The material is characterised by having pores with three different sizes (ranging from 5 to 13.5 Å) to which several polar and non-polar molecules (including Ar, N2, O2, CH4, H2O, MeOH and EtOH) have access.9 Porous materials such as HKUST-1 and other related MOFs are interesting for gas storage applications and therefore understanding the intimate connection between function and structure is crucial in order to fine tune their properties. Moreover, when considering the impact of small chemical changes at the molecular level it is clear that an analytical technique that is able to report on such changes is necessary. To this end, experimental solid-state NMR spectroscopy paired with theoretical prediction can aid in unravelling the structure of such paramagnetic MOFs and aid in the optimisation of their properties. Ke et al.10,11 have established a DFT-based methodology to compute the paramagnetic NMR (pNMR) chemical shifts of Cu-based MOFs using suitable model complexes. The paddlewheel type of system is special due to the coupling of the spins within each dimer. Experimentally observed 13C pNMR shifts could be reproduced by assuming a Boltzmann equilibrium between a diamagnetic ground state and thermally populated, excited paramagnetic states of molecular models with one or more dimers present, and also giving rise to the unusual temperature dependence of the pNMR shifts seen experimentally. Usually, the pNMR shift has an inverse linear relationship with temperature (because of the macroscopic magnetic susceptibility of paramagnetic materials). However, for HKUST-1 and select members of the related STAM family12,13 (based on different BTC derivatives) the relationship is non-linear.11,14 Ref. 10 proved that such behaviour arises from the thermal population of the accessible excited paramagnetic spin states in addition to a diamagnetic ground state. The smallest models used in ref. 10 were based on a single paddlewheel dimer (as shown in Fig. 1) with antiparallel and parallel electron spins within the dimer pair (corresponding to a total electron spin of 0 or 1, respectively). However, analysis of magnetic measurements (from Electron Paramagnetic Resonance, EPR, spectroscopy) has provided evidence for the presence of a sizeable amount of spin−1/2 centres, accounting for up to 10% of spin centres.15 Although the presence of such defects is appreciated in Cu-paddlewheel MOFs,16 the precise nature of these is still unclear and so far they do not appear to have been identified in solid-state NMR spectra, which could either be due to their low abundance (i.e., low signal intensity), excessive line widths, or occurrence of the signals in a spectral area that has not so far been considered (i.e., outside the spectral range usually acquired even for pNMR spectra). This raises the question, what is the nature of the spin–1/2 defects and in what region of the 13C (and 1H) NMR spectra would these be expected to appear?
Assuming the overall MOF structure remains intact around the defect sites (as there are no indications to the contrary from X-ray diffraction11), a single Cu(II) site surrounded by four carboxylate moieties would carry two negative charges. In reality, some charge balancing would be expected to occur. In this work, we have considered two possible situations; one where electroneutrality is maintained by substitution of one Cu centre in a dimer with two bridging H atoms, and one where the paddlewheel is negatively charged, with both Cu atoms remaining in place but with one Cu adopting a +1 oxidation state, resulting in a mixed valence defect.
Results and discussion
Proton-containing defect
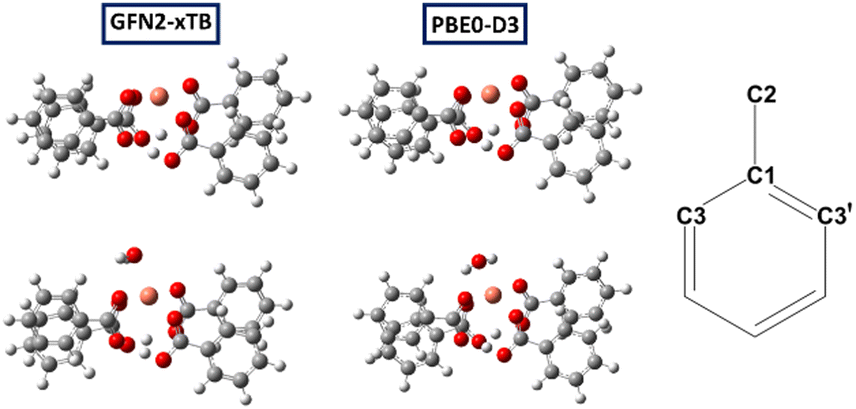 | ||
| Fig. 2 Molecular models of the Cu(H)2 (1a, top) and hydrated Cu(H)2 defects (bottom), optimised at xTB (left) and PBE0-D3 level (middle); right: labelling scheme (C3 is the ortho carbon nearest to the Cu centre). | ||
This model corresponds to model M1 in ref. 10, with four Ph groups around a single paddlewheel dimer, and gives a simple model for the MOF. Most results in the previous work for models containing a single dimer were based on a structure with one Ph and three Me groups (model M4 in ref. 10) to reduce computational cost. However, owing to the reduced symmetry of models containing protonated defects we decided to retain the four phenyl groups in this study.
To effectively investigate the protonated defect, we consider two states of each structural model, “activated” (i.e., no axial guest bound to Cu), and “hydrated” (i.e., one axial water bound to Cu). The latter would correspond to the STAM and HKUST-1 MOFs as obtained from hydrothermal synthesis, the former to the dehydrated (or “activated”) form of HKUST-1. Using the same methodology as in ref. 10, we have computed the 13C and 1H chemical shifts for these models in their doublet (spin–1/2) states. Because the actual linkers in the MOFs have carboxylate substituents in the meta position we only report data for C1, C2, C3 and C3′ (cf. labelling scheme in Fig. 2 – note that for paddlewheel motifs with two Cu atoms C3 and C3′ sites become equivalent).
 | ||
| Fig. 3 Schematic plots of predicted pNMR 13C peak positions of the Cu(H)2 defect models (blue points for xTB and orange for PBE0-D3 optimised structures) and of the experimental HKUST-1 (grey) and computed non-defective MOF models for (a) activated defect and (b) hydrated defect. | ||
Because all shifts are expected in a similar range in the NMR spectrum, this could make the unambiguous experimental identification of signals resulting from this defect challenging, particularly given the expected lower intensities. However, the temperature behaviour of the signals should be different, as the defective material is expected to have a linear relationship with inverse temperature,17–19 whereas it is known to be non-linear for the pristine material, as discussed above.10 Therefore, in principle, it should be possible to distinguish between signals from defective and pristine materials using variable temperature pNMR experiments. Considering such an experiment over the 250–350 K range, the expected pNMR shift of the defect would evolve as shown in Table S2 (ESI†). The C1 signal is predicted to be the most sensitive to temperature changes as shown in Fig. 4a and b (although perhaps the hardest signal to see experimentally owing, at least in the pristine material, to the much larger linewidths and extreme shift, typically requiring a change in the carrier frequency to ensure efficient excitation). Moreover, for the non-defective material a decrease in chemical shift for C1 and an increase for C2 is predicted with increasing T, which is the opposite of the behaviour predicted for the defect, making identification of spectral signals easier. Lastly, valuable information can be gathered from the chemical shift of the protons involved in the metal substitution (with predicted chemical shifts between ca. 10–13 ppm at room temperature, see Table S3, ESI†) if they are indeed present to balance the charge as assumed here.
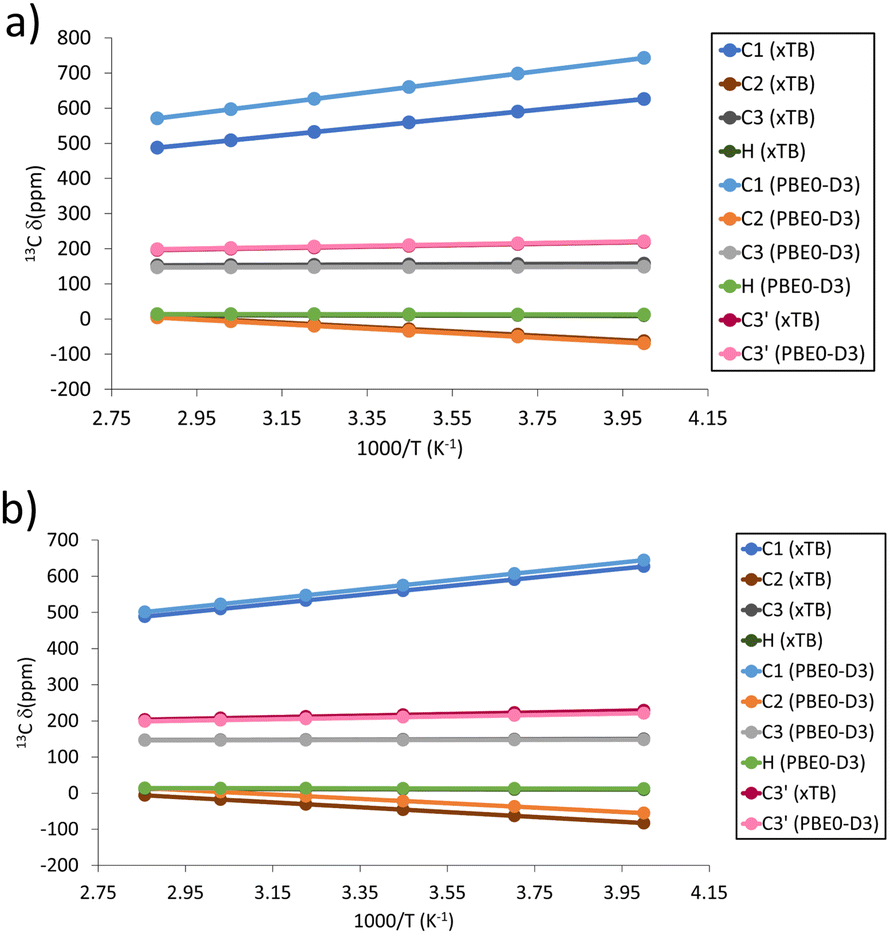 | ||
| Fig. 4 Predicted temperature dependence of pNMR shifts for 13C and 1H signals in the Cu(H)2 defect models for (a) activated and (b) hydrated models. | ||
It should be noted that all defect models considered so far, being activated or hydrated, have been fully optimised, resulting in large distortions from the ideal paddlewheel building block with Ph groups protruding radially from a four-fold axis. In most cases minima with a “folded” structure are obtained due to π-stacking interactions between adjacent Ph groups (see Fig. 5 left). It is unlikely that such distortions can be sustained in the extended MOF structure where most paddlewheel dimers will have the conventional structure. We have attempted to model defects more compatible with the extended MOF by imposing constraints during the geometry optimisation that prevent intramolecular π-stacking interactions from occurring. These models restrict the phenyl rings to be fixed to their positions in the non-defect dimers (xTB and PBE0-D3 geometry optimised positions, respectively), and ensure that the geometry remains compatible with the extended network. Such models could be a valid alternative to the global folded minimum structure. Some differences were found in terms of the predicted pNMR shifts between the constrained and unconstrained models, as shown in Fig. 6 and Table S7 (ESI†). When the models are optimised using xTB the biggest changes were for C1 and C2, whereas the C3 shift and the proton shifts are mostly conserved. However, models optimised at the PBE0-D3 level retain the pNMR parameters predicted previously to a higher degree once constraints are applied. There remains a significant shift of C1 between constrained and unconstrained models; however, C2, C3 and H differ by less than 60 and 4 ppm, respectively. Given the ppm range investigated, and the typical magnitude of paramagnetic NMR shifts, we deem a change over 200 ppm for the C sites significant. If the overall shift change is considered (defined as the average shift difference for each C atom with respect to the unconstrained model), all of the models, irrespective of the optimisation method used, show changes below 200 ppm (see Fig. 6a and b and Table S7, ESI†).
 | ||
| Fig. 5 Left: Fully optimised (unconstrained) model for the activated proton defect (1a); right: partially optimised after imposing constraints to ensure compatibility with the MOF structure. Both models were optimised at the PBE0-D3 level. | ||
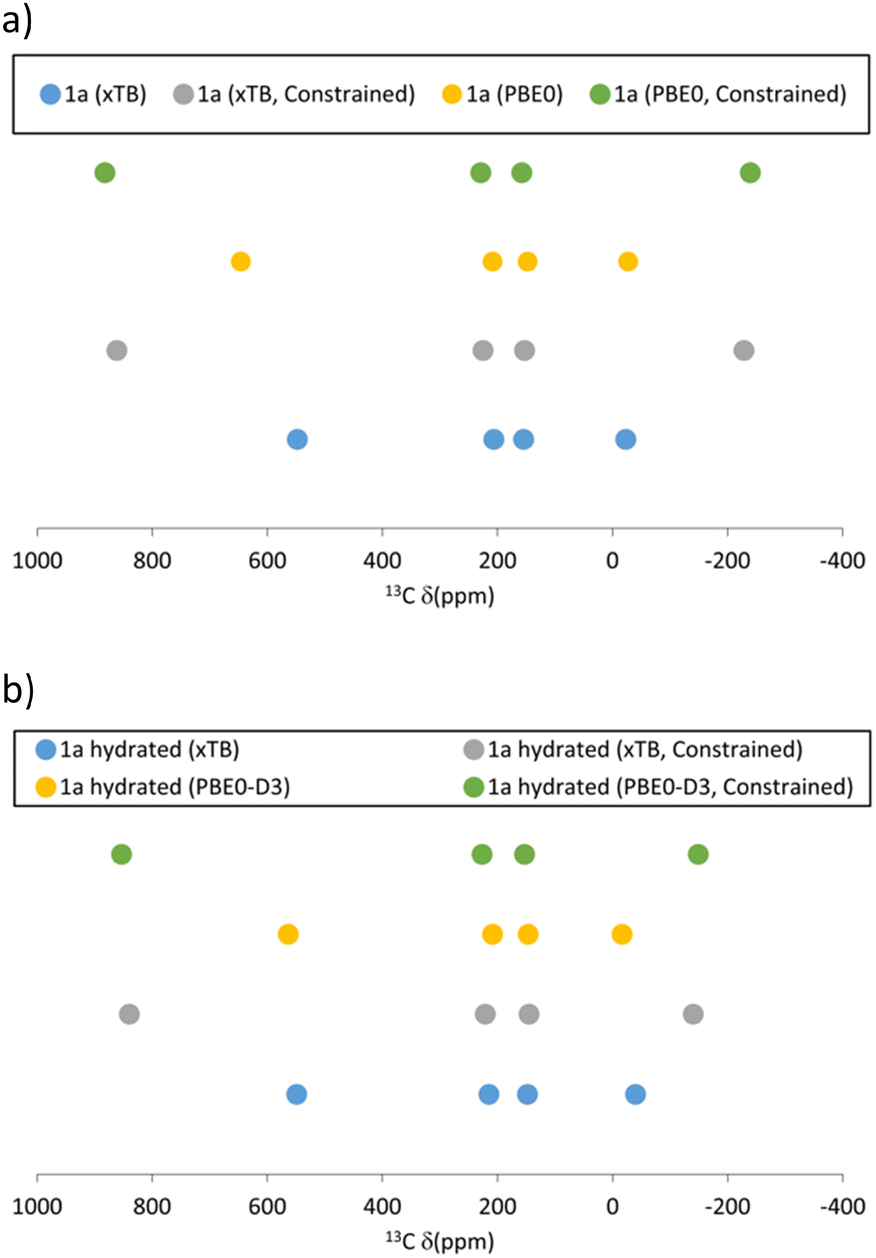 | ||
| Fig. 6 Schematic plot of predicted 13C pNMR peak positions of the constrained and unconstrained Cu(H)2 defect models optimised at the xTB and PBE0-D3 level for (a) activated and (b) hydrated models. | ||
Structurally, the constrained models afford a more symmetric coordination site (see Fig. 5 right), emphasized by the O⋯H–O distance. The model becomes more symmetric with the constraints, as apparent in the Δd(OH), defined as the difference between the O⋯H and H–O distances, which decreases from Δd ≈ 0.6 Å in the fully optimised minima to Δd ≈ 0.5 Å in the constrained structures.
Notable distortion energies (i.e., energy differences between constrained and fully optimised models) are computed at the CAM-B3LYP-D3 level, especially for the PBE0-D3 optimised structures where the deformation energies exceed 10 kcal mol−1 (see Table S8, ESI†). It is unclear at this point to what extent deformation energies of individual molecules would translate into resistance toward defect incorporation into the extended MOF frameworks, and we would not refute the existence of defects modelled by 1a based on these energetic arguments. Further study of this aspect could be rewarding.
Mixed valence defect
 | ||
| Fig. 7 Molecular models with corresponding spin density of the activated Cu(I)Cu(II) (top) and hydrated Cu(I)Cu(II) defects (bottom), optimised at xTB (left) and PBE0-D3 level (right). | ||
For the localised static structures (favoured by PBE0-D3) we expect a dynamic delocalisation through rapid interconversion between two isomers on the NMR time scale with the spin localised at either metal centre (and have averaged the computed pNMR shifts in the static minima accordingly – this would correspond to a type II complex in the Robin–Day classification, though we do not imply such an assignment, which will need further analysis, see ref. 22). At the DFT level, a delocalised structure can be enforced by imposing symmetry. For the activated model a structure with D2d symmetry (a higher-order saddle point) is 10 kcal mol−1 above the asymmetric minimum at the CAM-B3LYP-D3//PBE0-D3 level, fully in line with the expected fluxionality.
 | ||
| Fig. 8 Schematic plot of predicted 13C pNMR peak positions of the Cu(I)Cu(II) defect models (blue and orange points for structure optimised with xTB and PBE0-D3) and of the experimental HKUST-1 (grey points) and non-defective (yellow) HKUST-1 models; for (a) activated and (b) hydrated models. | ||
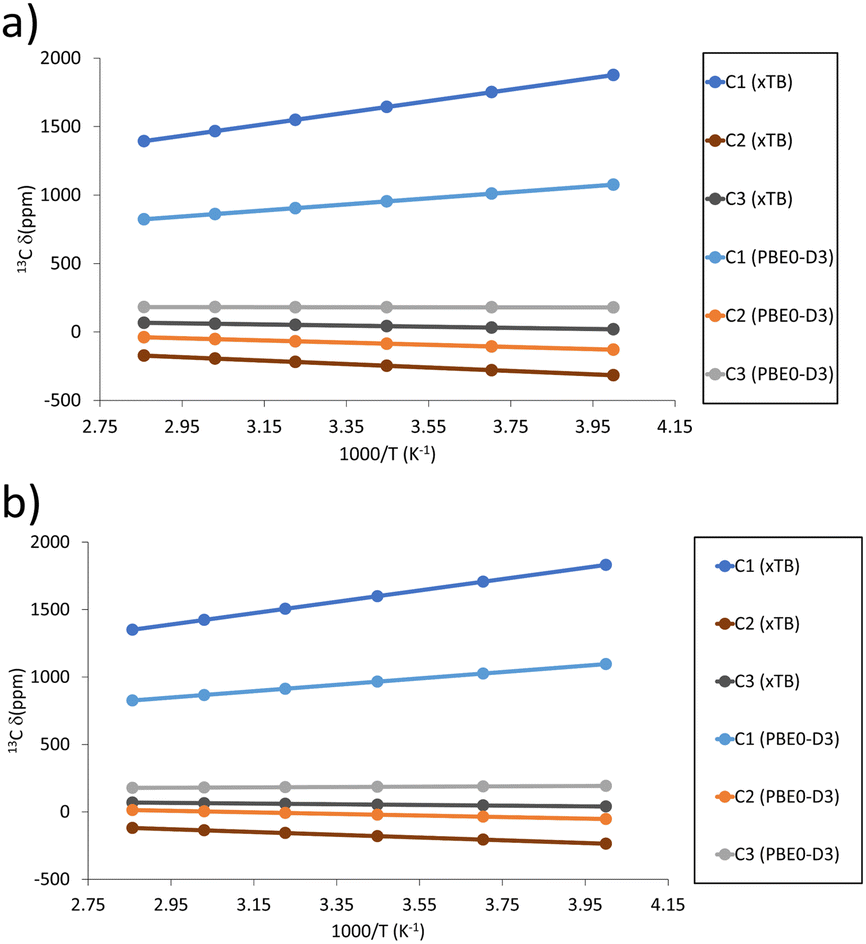 | ||
| Fig. 9 Predicted temperature dependence of 13C pNMR shifts, δ, in the mixed valence defects for (a) activated and (b) hydrated models. | ||
Note that the previously mentioned D2d symmetric saddle point shows similar spin and magnetic properties as the xTB optimised structure. Such a D2d symmetric structure captures the transition between the two spin-localised isomers and shows a full spin delocalisation and a strongly deshielded C1 and shielded C2 (see Table 1 and Table S7 for the temperature dependence, ESI†).
| Site | TS (DFT) | Min (DFT) | Min (xTB) |
|---|---|---|---|
| a Higher-order saddle point, to be precise. | |||
| C1 | 1521 | 933 | 1605 |
| C2 | −265 | −78 | −235 |
| C3 | 47 | 180 | 46 |
Based on the computed pNMR data, the presence of the spin-delocalised defect should be easier to identify than the proton defect discussed above, as the separation of its signals from those of the pristine material are stark for C1 and C3, while for the spin-localised defect only C2 can be used to distinguish between a defective and non-defective material. Nevertheless, employing a variable-temperature approach would still provide the best analytical method to distinguish between defective and non-defective signals, as the temperature dependence is different10 (see the strictly linear dependence on 1/T for the defects in Fig. 9a and b and Table S10 (ESI†), as opposed to the nonlinear dependencies in the pristine material, reported in ref. 10).
In terms of their pNMR properties, both activated and hydrated constrained models do not afford significant changes when compared to their unconstrained counterpart (see Fig. 10). The xTB optimised models predict greater deshielding of C1, whereas the PBE0 optimised models display the opposite behaviour, i.e., larger shielding, while C2 and C3 are more shielded in the constrained model, irrespective of the optimisation method used. It must be pointed out that given the magnitude of the explored chemical shift range, such changes are perhaps not significant as they are below 200 ppm.
 | ||
| Fig. 10 Schematic plot of predicted 13C pNMR peak positions for the constrained and unconstrained mixed valence defect models optimised at the xTB and PBE0-D3 level for (a) activated and (b) hydrated models. | ||
The spin density follows a similar trend as the pNMR shifts just discussed, since no significant changes are found between the constrained and unconstrained models. The models optimised with PBE0 retain a spin localised nature, while xTB optimisation produces full spin delocalisation between metal centres (see Fig. 11).
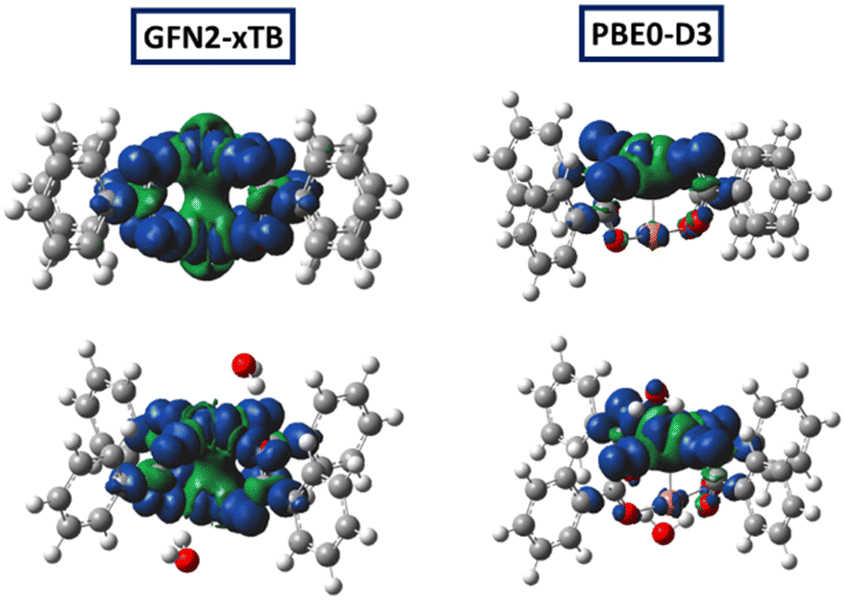 | ||
| Fig. 11 Molecular models with corresponding spin density of the activated Cu(I)Cu(II) (top) and hydrated Cu(I)Cu(II) (bottom) defect models constrained during optimisation, optimised at xTB (left) and PBE0-D3 levels (right). | ||
| Model (fully optimised/constrained)a | Optimisation method | ΔEb (kcal mol−1) |
|---|---|---|
| a Fully optimised (i.e., unconstrained) or constrained (see text). b Energies are calculated relative to the unconstrained model. | ||
| Activated (fopt) | xTB | 0.00 |
| activated (cons) | xTB | 4.03 |
| Hydrated (fopt) | xTB | 0.00 |
| hydrated (cons) | xTB | 6.95 |
| Activated (fopt) | PBE0-D3 | 0.00 |
| activated (cons) | PBE0-D3 | 4.80 |
| Hydrated (fopt) | PBE0-D3 | 0.00 |
| hydrated (cons) | PBE0-D3 | 11.74 |
Given their magnetic properties and their framework-compatible geometry, these models could be a valid alternative to the fully optimised ones.
In view of the abovementioned uncertainty as to the practical viability of such mixed-valence dimers as possible candidates for the defects seen in MOFs, no attempt was made to estimate driving forces for their formation (which would come down to the prediction of redox potentials, which has its own methodological challenges). From the distortion energies in Table 2 it appears that if one-electron reduction of individual dimers were possible during MOF formation, their incorporation into the resulting porous framework would not be prohibitive.
Conclusions
Defects based on the copper paddlewheel motif have been explored for small model complexes in order to gain insight into previous EPR findings for HKUST-1 and the STAM family of MOFs,11 both in their hydrated and activated forms, and the viability of their observation using pNMR spectroscopy. The neutral protonated defect, Cu(H)2, affords a predicted pNMR spectrum in the same spectral region as that for the non-defective material. Nevertheless, we suggest that variable temperature NMR measurements would be an effective strategy to distinguish between defective and non-defective signals, owing to the linearly inverse relationship with temperature of the defect signals. Regarding the negatively charged, mixed valence defect, the results have a strong method dependence. Irrespective of applying structural constraints to accommodate the defect in the extended MOF, or of using hydrated or activated models, for PBE0-D3 optimised structures the predicted pNMR spectra are close to that of the non-defective material, whereas xTB-optimised structures show strong differences for C1 and C2, i.e., the more deshielded and shielded carbon atoms in the system. Once more, the temperature dependence is indicated to be the key to assign signals belonging to the defect.In summary, we have used an established protocol to predict the pNMR spectral characteristics of potential spin–1/2 defects in MOFs based on Cu paddlewheel dimers. We expect this information to be useful for a more complete spectroscopic characterisation of such defects, eventually leading to an improved understanding of the structure (geometric and electronic) of these important materials.
Methods
Geometry optimisation was performed by using the semi-empirical method GFN2-xTB23–28 (here referred to as xTB for short) and DFT with the functional PBE024 with D3 dispersion corrections25 as implemented in Gaussian09.29 The 6-31G* basis set was used for all atoms except for Cu, for which an augmented Wachters basis set30,31 was used (62111111/3311111/3111). For the constrained optimisations, all 24 atoms of the phenyl rings were frozen in their positions in the non-defective paddlewheel dimers optimised at the respective level of theory, and all other atoms were relaxed. Amongst the existing methodologies for computing pNMR shifts,19,32–34 we adopted that from Hrobárik and Kaupp,32 where the isotropic shielding, σiso, is computed according to eqn (1): | (1) |
For the formation energies of the defect, single point calculations were performed at the CAM-B3LYP-D3/IGLO-II level detailed above (augmented with Grimme dispersion corrections) using Gaussian09. For the Cu2(O2CPh)4 dimer, the triplet state was used for these energy evaluations. Lastly, to account for the polar environment the single-point energies were also calculated with a simple polarisable continuum model (PCM)41 employing the parameters of water (ε = 80). All Gaussian calculations employed the default convergence criteria (maximum change in density matrix elements 10−8 a.u., maximum change in energy 10−6 a.u. for SCF, maximum and RMS gradient 4.5 × 10−4 a.u. and 3 × 10−4 a.u., respectively, for geometry optimisations), whereas very tight convergence criteria were used for the xTB geometry optimisation (maximum energy change 10−7 a.u. for SCF, and maximum gradient 2 × 10−4 a.u. for optimisations) and the ORCA properties calculations (maximum energy change 10−8 a.u.).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. B. thanks the EaStCHEM School of Chemistry for support. We thank Dr Hassane El Mkami for helpful discussions. Computations were performed on a local HPC cluster maintained by Dr H. Früchtl. The research data supporting this publication can be accessed at https://doi.org/10.17630/f9ce99be-957f-43d4-b289-d6654b2a9323.References
- H. C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673–674 CrossRef CAS PubMed
.
- J. Han, B. Xiao, P. K. Le and C. Mangwandi, Materials, 2023, 16, 1554 CrossRef CAS PubMed
- S. Gautam, J. Singhal, H. K. Lee and K. H. Chae, Mater. Today Chem., 2022, 23, 2468–5194 Search PubMed
- D. Sharma, S. Rasaily, S. Pradhan, K. Baruah, S. Tamang and A. Pariyar, Inorg. Chem., 2021, 60, 7794–7802 CrossRef CAS PubMed
- X. Ma, L. Wang, H. Wang, J. Deng, Y. Song, Q. Li, X. Li and A. M. Dietrich, J. Hazard. Mater., 2022, 424, 0304–3894 Search PubMed
- A. Rosado, A. Borrás, J. Fraile, J. A. R. Navarro, F. Suarez-Garcia, K. C. Stylianou, A. M. López-Periago, J. G. Planas, C. Domeingo and A. Yazdi, ACS Appl. Nano Mater., 2021, 4, 12712–12725 CrossRef CAS
- Y. Cui, B. Li, H. He, W. Zhou, B. Chen and G. Qian, Acc. Chem. Res., 2016, 49, 483–493 CrossRef CAS PubMed
- S. S. Chui, S. M. Lo, J. P. Charmant, A. G. Orpen and I. D. Williams, Science, 1999, 283, 1148–1150 CrossRef CAS PubMed
- J. J. Gutiérrez, J. M. Vicent-Luna, D. Dubbeldam and S. Calero, J. Phys. Chem. C, 2013, 117, 11357–11366 CrossRef
- Z. Ke, D. M. Dawson, S. E. Ashbrook and M. Bühl, Chem. Sci., 2022, 13, 2674–2685 RSC
- D. M. Dawson, C. E. F. Sansome, L. N. McHugh, M. J. McPherson, L. J. McCormick McPherson, R. E. Morris and S. E. Ashbrook, Solid State Nucl. Magn. Reson., 2019, 101, 44–50 CrossRef CAS PubMed
- M. I. H. Mohideen, B. Xiao, P. S. Wheatley, A. C. McKinlay, Y. Li, A. M. Z. Slawin, D. W. Aldous, N. F. Cessford, T. Düren, X. Zhao, R. Gill, J. M. Griffin, S. E. Ashbrook and R. E. Morris, Nat. Chem., 2011, 3, 304–310 CrossRef CAS PubMed
- L. N. McHugh, M. J. McPherson, L. J. McCormick, S. A. Morris, P. S. Wheatley, S. J. Teat, D. McKay, D. M. Dawson, C. E. F. Sansome, S. E. Ashbrook, C. A. Stone, M. W. Smith and R. E. Morris, Nat. Chem., 2018, 10, 1096–1102 CrossRef CAS PubMed
- D. M. Dawson, L. E. Jamieson, M. I. H. Mohideen, A. C. McKinlay, I. A. Smellie, R. Cadou, N. S. Keddie, R. E. Morris and S. E. Ashbrook, Phys. Chem. Chem. Phys., 2012, 15, 919–929 RSC
- H. E. Mkami, M. I. Mohideen, C. Pal, A. McKinlay, O. Scheimann and R. E. Morris, Chem. Phys. Lett., 2012, 544, 17–21 CrossRef
- M. D. Allendorf, V. Stavila, M. Witman, C. K. Brozek and C. H. Hendon, J. Am. Chem. Soc., 2021, 143, 6705–6723 CrossRef CAS PubMed
- M. Bühl, S. E. Ashbrook, D. M. Dawson, R. A. Doyle, P. Hrobárik, M. Kaupp and I. A. Smellie, Chem. – Eur. J., 2016, 22, 15328–15339 CrossRef PubMed
- A. J. Pell, G. Pintacuda and C. P. Grey, Prog. Nucl. Magn. Reson. Spectrosc., 2019, 111, 1–271 CrossRef CAS PubMed
-
S. Moon and S. Patchkovskii, in Calculation of NMR and EPR Parameters: Theory and Applications, ed. M. Kaupp, M. Bühl and V. G. Malkin, Wiley, Weinheim (Germany), 2004, pp. 325–340 Search PubMed
- I. G. Crivelli and C. G. Andrade, Inorg. Chim. Acta, 1993, 203, 115–120 CrossRef CAS
- M. Šimėnas, M. Kobalz, M. Mendt, P. Eckold, H. Krautscheid, J. Banys and A. Pöppl, J. Phys. Chem. C, 2015, 119, 4898–4907 CrossRef
- M. Parthey and M. Kaupp, Chem. Soc. Rev., 2014, 43, 5067–5088 RSC
- J. P. Perdew and Y. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 45, 13244–13249 CrossRef PubMed
- J. P. Perdew, M. Ernzerhof and K. Burke, J. Chem. Phys., 1996, 105, 9982–9985 CrossRef CAS
- A. D. Becke and E. R. Johnson, J. Chem. Phys., 2005, 123, 154101 CrossRef PubMed
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed
- E. R. Johnson and A. D. Becke, J. Chem. Phys., 2006, 124, 174104 CrossRef PubMed
- C. Bannwarth, S. Ehlert and S. Grimme, J. Chem. Theory Comput., 2019, 15, 1652–1671 CrossRef CAS PubMed
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Revision D.01, Gaussian Inc., 2013 Search PubMed
- A. J. Wachters, J. Chem. Phys., 1970, 52, 1033–1036 CrossRef CAS
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS
- P. Hrobárik, R. Reviakine, A. V. Arbuznikov, O. L. Malkina, V. G. Malkin, F. H. Köhler and M. Kaupp, J. Chem. Phys., 2007, 126, 024107 CrossRef PubMed
- T. O. Pennanen and J. Vaara, Phys. Rev. Lett., 2008, 100, 133002 CrossRef PubMed
- B. Martin and J. Autschbach, Phys. Chem. Chem. Phys., 2016, 18, 21051–21068 RSC
- M. Bühl, C. Reimann, D. A. Pantazis, T. Bredow and F. Neese, J. Chem. Theory Comput., 2008, 4, 1449–1459 CrossRef PubMed
- M. Bursch, H. Neugebauer and S. Grimme, Angew. Chem., Int. Ed., 2019, 58, 11078–11087 CrossRef CAS PubMed
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS
- M. Munzarova and M. Kaupp, J. Phys. Chem. A, 1999, 103, 9966–9983 CrossRef CAS
-
U. F. W. Kutzelnigg and M. Schindler, NMR Basic Principles and Progress, Springer, Berlin, 1990, vol. 23, pp. 165–262 Search PubMed
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS
- S. Miertu, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117–129 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp04618d |
| This journal is © the Owner Societies 2023 |