
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotoactivation of the orange carotenoid protein requires two light-driven reactions mediated by a metastable monomeric intermediate†
Justin B.
Rose
a,
José A.
Gascón
 b,
Markus
Sutter
cde,
Damien I.
Sheppard
c,
Cheryl A.
Kerfeld
cde and
Warren F.
Beck
*a
b,
Markus
Sutter
cde,
Damien I.
Sheppard
c,
Cheryl A.
Kerfeld
cde and
Warren F.
Beck
*a
aDepartment of Chemistry, Michigan State University, East Lansing, Michigan 48824-1322, USA. E-mail: beckw@msu.edu
bDepartment of Chemistry, University of Connecticut, Storrs, Connecticut 06269-3060, USA
cMSU–DOE Plant Research Laboratory, Michigan State University, East Lansing, Michigan 48824-1322, USA
dEnvironmental Genomics and Systems Biology Division, Lawrence Berkeley National Laboratory, Berkeley, California 94720, USA
eMolecular Biophysics and Integrated Bioimaging Division, Lawrence Berkeley National Laboratory, Berkeley, California 94720, USA
First published on 30th November 2023
Abstract
The orange carotenoid protein (OCP) functions as a sensor of the ambient light intensity and as a quencher of bilin excitons when it binds to the core of the cyanobacterial phycobilisome. We show herein that the photoactivation mechanism that converts the resting, orange-colored state, OCPO, to the active red-colored state, OCPR, requires a sequence of two reactions, each requiring absorption of a single photon by an intrinsic ketocarotenoid chromophore. Global analysis of absorption spectra recorded during continuous illumination of OCPO preparations from Synechocystis sp. PCC 6803 detects the reversible formation of a metastable intermediate, OCPI, in which the ketocarotenoid canthaxanthin exhibits an absorption spectrum with a partial red shift and a broadened vibronic structure compared to that of the OCPO state. While the dark recovery from OCPR to OCPI is a first-order, unimolecular reaction, the subsequent conversion of OCPI to the resting OCPO state is bimolecular, involving association of two OCPO monomers to form the dark-stable OCPO dimer aggregate. These results indicate that photodissociation of the OCPO dimer to form the monomeric OCPO intermediate is the first step in the photoactivation mechanism. Formation of the OCPO monomer from the dimer increases the mean value and broadens the distribution of the solvent-accessible surface area of the canthaxanthin chromophore measured in molecular dynamics trajectories at 300 K. The second step in the photoactivation mechanism is initiated by absorption of a second photon, by canthaxanthin in the OCPO monomer, which obtains the fully red-shifted and broadened absorption spectrum detected in the OCPR product state owing to displacement of the C-terminal domain and the translocation of canthaxanthin more than 12 Å into the N-terminal domain. Both steps in the photoactivation reaction of OCP are likely to involve changes in the structure of the C-terminal domain elicited by excited-state conformational motions of the ketocarotenoid.
Introduction
The orange carotenoid protein (OCP) mediates nonphotochemical quenching (NPQ) in cyanobacteria by serving as a sensor of the ambient light intensity and by quenching bilin excited states in the core of the phycobilisome.1–12 Photoactivation of the OCP from the resting, orange-colored state, OCPO, to the NPQ-active, red-colored state, OCPR, is driven by absorption of blue-green light by a ketocarotenoid, such as 3′-hydroxyechinenone or canthaxanthin, which spans the interface between the N-terminal and C-terminal domains (NTD and CTD, respectively) of the protein. Formation of the active OCPR product translocates the ketocarotenoid more than 12 Å and allows the CTD to rotate away (Fig. 1).10,12 This reorganization of the protein structure of OCP requires dissociation of a dark-stable homodimer complex.2,8,13,14 The OCPO dimer is stabilized by salt bridges and hydrogen bonds principally between the parallel facing α helices from the NTD of the two interacting monomers.2,8 The absorption spectrum of the ketocarotenoid shifts more than 50 nm to the red in response to the structural changes that accompany the translocation of the ketocarotenoid.9,10 The recent cryo-EM structure of the complex of OCPR with the phycobilisome shows that the exposed NTD surface associates with allophycocyanin-containing segments in the core, allowing the exposed length of the ketocarotenoid to be inserted near to bilin chromophores along the excitation energy transfer pathway to the terminal emitter bilins.12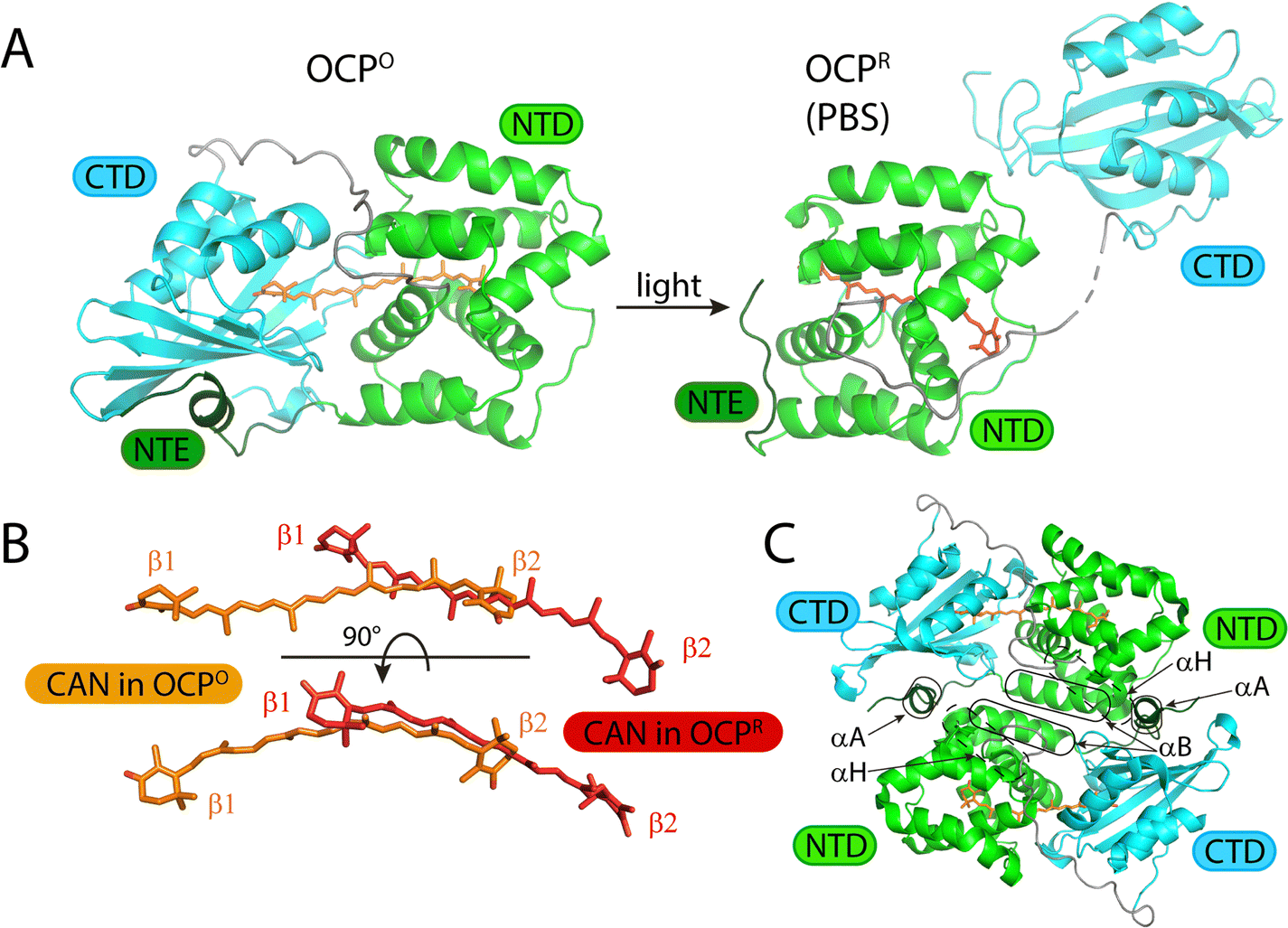 | ||
| Fig. 1 Structures of OCPO, OCPR, and the OCPO dimer. (A) Structures of the OCPO (left, pdb id 4xb5) with canthaxanthin (CAN, orange) and the phycobilisome (PBS)-bound form of OCPR (right, from the OCP-PBS structure, pdb id 7sc9) with CAN (red). C-terminal domains (CTD) are cyan, N-terminal domains (NTD) green, the N-terminal extension (NTE) dark green, and the connecting linkers grey. (B) Detailed view showing the translocation of CAN in OCPO (orange) and OCPR (red) in two orientations, indicating the β-ionone rings in the CTD and NTD, β1 and β2, respectively. (C) Overview of the OCPO dimer as observed in the crystal structure of Syncechocystis sp. PCC 6803 OCP (pdb id 4xb5), with the α helices labeled that form the major interaction surface between the two monomers. | ||
The photochemical mechanism that initiates photoactivation of OCP following optical excitation of the ketocarotenoid to its second excited singlet state, S2, remains elusive because the quantum yield of the OCPR product state is only a few percent at best. Currently available evidence supports the idea that the ketocarotenoid undergoes a change in its conformation during nonradiative decay from the S2 state that initiates photoactivation in a small number of events. Wilson et al.6 assigned a non-decaying signal they observed in femtosecond pump–probe spectra to OCPR with the ketocarotenoid in the electronic ground state, S0, with a yield of ∼1%. Šlouf et al.15 subsequently detected light-induced changes in the conformation of the ketocarotenoid in OCP using the magnitude of a S1-state intramolecular charge transfer (ICT) signal they observed in the near-IR to assess the torsional angles of the β-ionone end rings. Konald et al.16 then used information from time-resolved IR spectroscopy to conclude that nonradiative decay from S2 and S1 in OCP produces the low-energy S1 intermediate called S*,17 which initiates the translocation of the ketocarotenoid by breaking the hydrogen bonds between the carbonyl substituent on the ketocarotenoid's β1-ionone ring and the side chains of two conserved amino-acid residues in the CTD, Y201 and W288 (in Synechocystis sp. PCC 6803).2,18 A subsequent study by Yaroshevich et al.19 of OCP with the Y201W mutation led them to propose that this process involves protonation of the carbonyl substituent. However, a recent study by Niziński et al.20 of pump–probe spectra with coverage of the near-IR region has suggested instead that the first intermediate P1 in the ketocarotenoid translocation mechanism arises not from S* but from the S1 state proper. Based on evidence from time-resolved X-ray crystal structures, Chukhutsina et al.21 raised the alternative proposal that a “bicycle pedal” configuration22 of the ketocarotenoid's isoprenoid backbone is produced during the photoactivation reaction.
Although the photochemistry of retinal in rhodopsin23,24 should not be directly compared to that of the ketocarotenoid in OCP,6 it is similarly assumed in most studies that absorption of a single photon by the ketocarotenoid in OCP initiates the entire sequence of events in the photoactivation mechanism that produces the OCPR state.25 The observations by Maksimov et al.26 and by Niziński et al.25 that the rate of formation of OCPR depends nonlinearly on the incident light intensity, however, suggest that the mechanism incorporates a sequence of at least two light-driven reactions. In this contribution, we have investigated this further by using the vibronic structure and red shift of the absorption spectrum of the bound ketocarotenoid canthaxanthin (CAN) as intrinsic probes of the surrounding protein environment during the photoactivation of OCP preparations from Synechocystis sp. PCC 6803. Absorption spectroscopy measurements during continuous illumination show that absorption of light by CAN first causes dissociation of the dark-stable OCPO dimer, which results in partial exposure of the CAN binding site region between the CTD and NTD to the surrounding aqueous medium.2 We then find that the monomeric intermediate of OCP persists in the dark until a second light-driven reaction triggers the formation of OCPR. We propose that both steps in the photoactivation reaction are triggered by excited-state conformational motions of CAN that are launched in the photoexcited S2 electronic state.
Experimental
Sample preparation
OCPO preparations from Synechocystis sp. PCC 6803 with the ketocarotenoid canthaxanthin (CAN) were isolated as described previously.18 Briefly, the gene for OCP was expressed in BL21(DE3) competent Escherichia coli cells with a C-terminal 6× His tag added and cloned in a pCDFDuet-1 vector (Novagene). The BL21(DE3) cells were transformed with the pCDFDuet-1 vector and the pAC-CANTHipi plasmid (Addgene plasmid #53301)27 containing the genes for biosynthesis of CAN in E. coli. The OCP-CAN holoprotein was isolated using the method described previously.10 Ni-NTA affinity chromatography (HisTrap Affinity column, GE Healthcare) was used to obtain a mixture of the apo- and holoproteins, and then the OCP-CAN holoprotein was isolated by hydrophobic interaction chromatography (HiTrap HIC phenyl column, GE Healthcare).As a stock solution for use in the photoactivation and dark recovery experiments, the OCP-CAN holoprotein was suspended in a buffer solution containing 50 mM tris(hydroxymethyl)aminomethane-NaOH, pH 8.0, and 200 mM NaCl, with 10% (v/v) glycerol present as a cryoprotectant to allow storage in a freezer. The stock protein solution was diluted 30-fold with the buffer solution chosen for a particular experiment, usually to prepare samples with an absorbance of 0.13 at 500 nm. In several experiments, 60% (v/v) glycerol was present in the diluent buffer solution. The working OCP-CAN samples were prepared in the dark and then allowed to further rest in a dark refrigerator at 3 °C for as long as one week prior to use in an experiment. 1.5 mL aliquots were transferred in total darkness to a 1 cm square fluorescence cuvette and then placed in the cuvette holder in the sample temperature controller in the photoactivation spectrometer. Prior to illumination, the sample was incubated in total darkness for 10 min in situ at the temperature chosen for an experiment.
CAN (Sigma-Aldrich 11775) was used as received for measurement of the absorption spectrum in 2-methyltetrahydrofuran (Sigma-Aldrich 673277).
Photoactivation assays
Absorption spectra during continuous illumination and dark recovery of OCP preparations were recorded with a home-built multichannel absorption spectrophotometer. Continuous excitation light for the photoactivation reaction was routed from a 430 nm LED (Thorlabs M430L5) to the sample by a fiber-bundle light guide (Thorlabs OSL2FB) after passing through a computer-controlled shutter (Thorlabs SH-1), a 430 nm interference filter (Thorlabs FB430-10, 10 nm bandpass), and then a stack of neutral density filters, which were varied to adjust the illumination power for a particular experiment. The diameter of the excitation beam was 1 cm across the sample cuvette. A Newport Model 835 power meter equipped with a 1 cm silicon photodiode detector was used to measure the power of the excitation beam after it passed through a sample cuvette containing a blank solution. The continuous white-light probe beam for the absorption measurement was obtained from a tungsten halogen lamp in an Ocean Optics DH-2000 fiber light source. An Ocean Optics QP400-1-VIS-NIR fiber and a Thorlabs FS240SMA-A fiber collimator were used to direct the unfiltered probe beam (3 mm diameter) across the sample cuvette at 90° incidence from the excitation beam and centered inside its profile across the sample. The power of the probe beam incident on the sample was 450 μW; the integrated power over the absorption spectrum of CAN was estimated to be 165 μW. The transmitted probe beam was collected by a fiber collimator and then routed by an optical fiber (as above) to an Ocean Optics Flame-T grating spectrometer/CCD detector, which was controlled by a LabVIEW (National Instruments) program. During illumination and during the subsequent dark recovery periods, the CCD detector was operated with a 4 ms integration time per scan; 25 successive spectra were integrated to obtain a total acquisition time of 0.1 s per spectrum. The OCP samples were held in 1 cm square fluorescence cuvettes in a Peltier-type thermoelectric sample temperature controller (Quantum Northwest Luma 40), which was also used to stir the sample continuously with a magnetic stir bar. Singular-value decomposition (SVD) factor analysis of the time-resolved spectra was performed using the GlobalWorks software package (On-Line Instrument Systems). Global and target modeling employed the GlobalWorks and CarpetView (Light Conversion) software packages.Results and discussion
In line with the results reported by Niziński et al.,25 our recordings of the photoactivation of OCP under continuous illumination (Fig. 2) exhibit more than one kinetic phase as OCPO is converted into OCPR. The approach we used to determine the mechanism behind this phenomenon was to record snapshots of the absorption spectra of OCP solutions during continuous illumination with blue light (wavelength of 430 nm), which we find results in a more rapid conversion to the OCPR product than with light at longer wavelengths. Further, we chose initially to record photoactivation assays at 273 K because the dark recovery of OCPR is slowed by a factor of at least 55 compared to that of the forward photoactivation rate (Fig. S1, ESI†).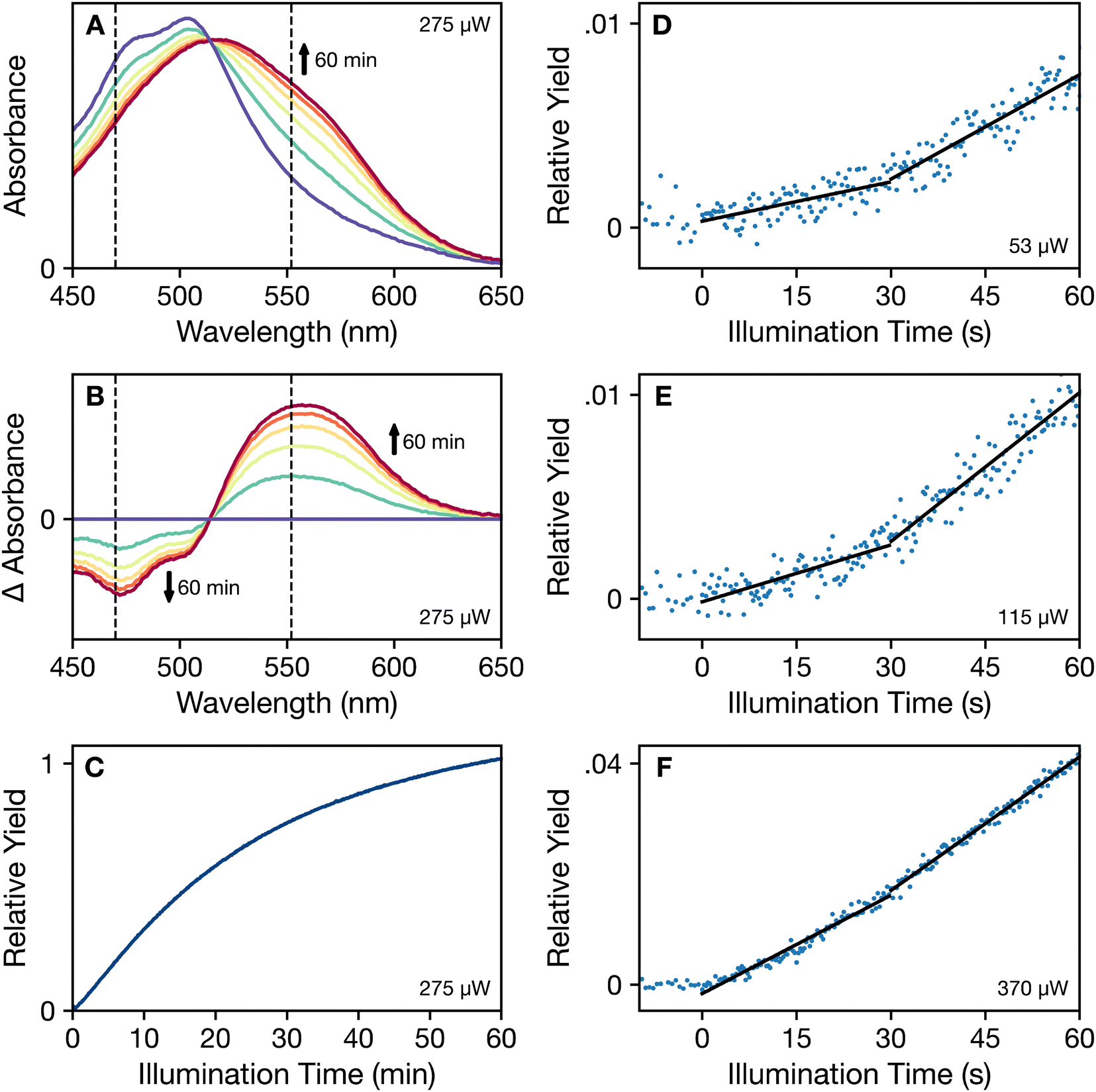 | ||
| Fig. 2 Photoactivation assays of dark-adapted OCP at 273 K in the presence of 60% (v/v) glycerol under continuous 430 nm illumination. (A) Time evolution of the absorption spectrum from zero illumination (purple) to 60 min of 275 μW illumination (dark red), with intermediate spectra spaced by 12 min of illumination. (B) Difference absorption spectra (light – dark adapted) for the set of spectra shown in panel (a). The vertical dashed lines shown in (a) and (b) mark the wavelengths used for the transients in Fig. 3. (C) Time course of the relative yield of the OCPR product, determined from the integral of the absolute value of the difference spectrum. (D)–(F) Time courses of the relative yields of OCPR at short illumination times with increasing illumination power: 53 μW, 115 μW, and 370 μW, respectively. The y-axis scaling is consistent with that used in panel (c). Approximately linear segments in these time courses are indicated with superimposed black lines. | ||
The series of absorption spectra in Fig. 2a show the pattern of spectral changes commonly observed as OCPO is converted to OCPR under continuous illumination. Extending to bluer wavelengths from the peak maximum at 505 nm, the absorption spectrum of OCPO exhibits a progression of partially resolved vibronic transitions due to excited-state stretching displacements of the C–C and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds of the isoprenoid backbone17,28 of the bound CAN chromophore in these samples from Synechocystis sp. PCC 6803. With illumination, however, the absorption spectrum shifts to longer wavelengths and broadens, largely eliminating any resolution of vibronic peaks. Although the overall absorption maximum of the spectrum shifts only to 520 nm, the 50% absorbance wavelength shifts from 535 nm in OCPO to 580 nm in OCPR. The corresponding series of light – dark-adapted difference spectra in Fig. 2b decrease in intensity in the 470 nm region as the vibronic structure broadens and increase in intensity in the 560 nm region.
C bonds of the isoprenoid backbone17,28 of the bound CAN chromophore in these samples from Synechocystis sp. PCC 6803. With illumination, however, the absorption spectrum shifts to longer wavelengths and broadens, largely eliminating any resolution of vibronic peaks. Although the overall absorption maximum of the spectrum shifts only to 520 nm, the 50% absorbance wavelength shifts from 535 nm in OCPO to 580 nm in OCPR. The corresponding series of light – dark-adapted difference spectra in Fig. 2b decrease in intensity in the 470 nm region as the vibronic structure broadens and increase in intensity in the 560 nm region.
Fig. 2c–f show time courses of the relative yield of the photoactivated OCPR state, as determined from the integral of the absolute value of the light – dark difference spectrum. This approach would be appropriate if the photoactivation mechanism involves a direct conversion from OCPO to OCPR. Fig. 2c indicates that the rate of change of the yield slows with increasing illumination time; similar trends were reported by Niziński et al.28 using single-wavelength absorption traces. The decreasing rate of change accompanies the net accumulation of OCPR at long illumination times. At short illumination times, the detailed views in Fig. 2d–f show that a period of induction precedes the onset of the maximum rate of change, which is here observed after about 30 s of illumination. At the lowest illumination power (53 μW in Fig. 2d), two time regions are marked by linear segments at short illumination times to indicate that the initial rate increases as the illumination power increases to approach that reached at longer times. The trace shown in Fig. 2f (370 μW) is almost linear over the entire 60 s time range. Despite the observation that the absorption spectra exhibit an isosbestic point, where the spectra at all illumination times appear to intersect at 510 nm in Fig. 2a, this kinetic behavior indicates directly that an intermediate is formed at short illumination times and that conversion of the intermediate to the OCPR product is also light induced.
The kinetics for the formation and decay of the intermediate during the photoactivation assays can be retrieved using global and target modeling29 of the time evolution of the absorption spectrum. This approach allows us to model simultaneously the three-dimensional absorption vs. wavelength vs. time response surface. The absorption spectrum observed at any time from the ensemble of OCP molecules in a sample is modeled as the sum of that from each of the population compartments in a kinetic model, which are here treated as distinct chemical species. The total signal from each compartment is computed as the linear combination of the population (fraction of molecules in the ensemble) with the corresponding evolution-associated spectrum, the absorption spectrum of a pure sample of a given kinetic compartment. A preliminary singular-value decomposition (SVD) analysis (Fig. S2, ESI†) indicates that there are at least three spectrokinetic species with significant populations in the ensemble of OCP molecules under illumination. Global and target modeling then employed a linear, first-order kinetic model with three compartments,  , with the boundary conditions setting the initial dark concentrations of the intermediate OCPI and the product OCPR to zero. In order to apply the simplest possible model, we assume here that OCPI and OCPR are stable in the dark, which is in line with the finding that the rate of dark recovery at 273 K is very slow (Fig. S1, ESI†).
, with the boundary conditions setting the initial dark concentrations of the intermediate OCPI and the product OCPR to zero. In order to apply the simplest possible model, we assume here that OCPI and OCPR are stable in the dark, which is in line with the finding that the rate of dark recovery at 273 K is very slow (Fig. S1, ESI†).
Fig. 3 summarizes the results of the three-compartment global model for the photoactivation assay carried out with 220 μW illumination. The evolution-associated spectra shown in Fig. 3a resolve the absorption spectrum of the intermediate, labeled here as OCPI, which exhibits a partially broadened vibronic structure and a partial red shift relative to those of the absorption spectrum of the product OCPR. The time constant τ1 for the formation of OCPI is about ten times longer than that for its conversion to OCPR, τ2, so its presence in the ensemble is most evident at short illumination times. At the end of the 60 min exposure to light, the population ratio of OCPR to OCPO is about 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, and OCPI is present with perhaps an order of magnitude smaller proportion. The need for OCPI in the global model is made apparent, however, by a comparison with the results of a direct, two-compartment global model,
:1, and OCPI is present with perhaps an order of magnitude smaller proportion. The need for OCPI in the global model is made apparent, however, by a comparison with the results of a direct, two-compartment global model, 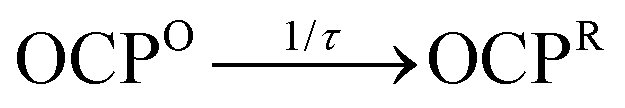 , which is included as ESI,† Fig. S3. The residuals (data – model) for this model exhibit systematic oscillatory trends, with large deviations from zero noted especially at short illumination times. In contrast, the residuals shown in Fig. 3c and d for the three-compartment model including OCPI are flat and not significantly different from zero for the entire illumination period.
, which is included as ESI,† Fig. S3. The residuals (data – model) for this model exhibit systematic oscillatory trends, with large deviations from zero noted especially at short illumination times. In contrast, the residuals shown in Fig. 3c and d for the three-compartment model including OCPI are flat and not significantly different from zero for the entire illumination period.
 | ||
Fig. 3 Global modeling of the time evolution of the absorption spectrum of OCP at 273 K in the presence of 60% (v/v) glycerol under continuous 430 nm illumination (220 μW), applying a three-compartment kinetic model: 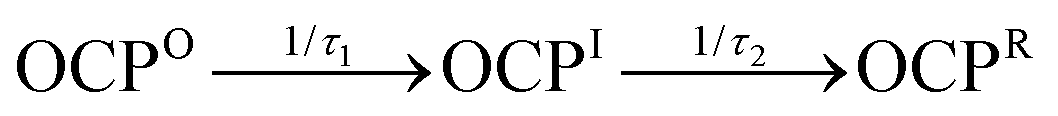 . (A) Evolution-associated absorption spectra for the three kinetic compartments: dark-adapted OCPO reactant (black), the OCPI intermediate (blue), and the final OCPR product (red). (B) Time evolution of the populations for the OCPO, OCPI, and OCPR kinetic compartments. (C) and (D) Fit of the global model (black) to the observed absorption (blue dots) at two detection wavelengths, 470 nm and 552 nm, as marked in Fig. 2a and b, showing the single-wavelength absorption transients (top panel) and residuals (bottom panel). The residuals (Δ = data − global model) are shown with a 20× zoom factor for the absorbance ordinate. . (A) Evolution-associated absorption spectra for the three kinetic compartments: dark-adapted OCPO reactant (black), the OCPI intermediate (blue), and the final OCPR product (red). (B) Time evolution of the populations for the OCPO, OCPI, and OCPR kinetic compartments. (C) and (D) Fit of the global model (black) to the observed absorption (blue dots) at two detection wavelengths, 470 nm and 552 nm, as marked in Fig. 2a and b, showing the single-wavelength absorption transients (top panel) and residuals (bottom panel). The residuals (Δ = data − global model) are shown with a 20× zoom factor for the absorbance ordinate. | ||
The time constants τ1 and τ2 exhibit a linear dependence on the power of the excitation light beam, at least under the low intensity conditions where OCPI can be resolved with global modeling. The linear plots shown in Fig. 4 of the rates 1/τ as a function of illumination power retrieve estimates for the intrinsic rate constants k1 and k2 from the slopes and for the zero-intensity rate constants k01 and k02 from the y-intercepts. The y-intercepts are nonzero due to the use of the continuous white-light measurement beam to measure the photoactivation reaction's progress. The possibility that the rate constants k1 and k2 depend on the wavelength of incident light could be addressed in future work to obtain additional information on how each step in the mechanism is triggered by the ketocarotenoid photochemistry.
 | ||
Fig. 4 Dependence of the photoactivation rate constants observed at 273 K in the presence of 60% (v/v) glycerol on the incident total power of the 430 nm excitation beam for (A)  and (B) and (B)  , as determined by two-step global models. The error bars show the 95% confidence limits for the measured rates at a given illumination power. The dashed lines are least-squares fits to the linear model 1/τ = kP + k0, where k is the rate constant, P is the illumination power, and k0 is the rate due to the white-light probe beam alone. For (A), k1 = 0.000169 ± 6.9 × 10−5 ms−1 μW−1 and k01 = 0.176 ± 0.016 ms−1. For (B), k2 = 0.0170 ± 0.011 ms−1 μW−1 and k02 = 1.05 ± 2.16 ms−1. , as determined by two-step global models. The error bars show the 95% confidence limits for the measured rates at a given illumination power. The dashed lines are least-squares fits to the linear model 1/τ = kP + k0, where k is the rate constant, P is the illumination power, and k0 is the rate due to the white-light probe beam alone. For (A), k1 = 0.000169 ± 6.9 × 10−5 ms−1 μW−1 and k01 = 0.176 ± 0.016 ms−1. For (B), k2 = 0.0170 ± 0.011 ms−1 μW−1 and k02 = 1.05 ± 2.16 ms−1. | ||
The dark recovery following illumination of OCPR also follows a two-step mechanism,  , with the prime used here in the label OCPI′ for the intermediate to distinguish it formally from the OCPI intermediate present during illumination for the forward reaction. The spectral evolution shown in Fig. 5a and b for the set of time-resolved absorption spectra recorded at 293 K in the presence of 60% (v/v) glycerol following 30 min of illumination resembles the reverse of that shown in Fig. 2a and b for an OCP sample under illumination. The vibronic structure in the 450–500 nm region of the spectrum recovers and the broad, red-shifted region over the 510–600 nm range decreases as the OCPO state is formed. SVD analysis of the set of time-resolved spectra (Fig. S4, ESI†) supports the conclusion that a kinetic intermediate is present during the nearly three-hour recovery back to the dark-stable OCPO state. In support of this conclusion, a poor fit to the overall spectral response is obtained by a global model for the direct recovery in the absence of the intermediate (Fig. S5, ESI†).
, with the prime used here in the label OCPI′ for the intermediate to distinguish it formally from the OCPI intermediate present during illumination for the forward reaction. The spectral evolution shown in Fig. 5a and b for the set of time-resolved absorption spectra recorded at 293 K in the presence of 60% (v/v) glycerol following 30 min of illumination resembles the reverse of that shown in Fig. 2a and b for an OCP sample under illumination. The vibronic structure in the 450–500 nm region of the spectrum recovers and the broad, red-shifted region over the 510–600 nm range decreases as the OCPO state is formed. SVD analysis of the set of time-resolved spectra (Fig. S4, ESI†) supports the conclusion that a kinetic intermediate is present during the nearly three-hour recovery back to the dark-stable OCPO state. In support of this conclusion, a poor fit to the overall spectral response is obtained by a global model for the direct recovery in the absence of the intermediate (Fig. S5, ESI†).
 | ||
Fig. 5 Dark recovery of photoactivated OCP at 293 K in the presence of 60% (v/v) glycerol following continuous 430 nm illumination (275 μW) for 30 min. (A) Time evolution of the absorption spectrum from zero dark time (red) to 150 min of darkness (purple), with intermediate spectra spaced by 30 min of darkness. (B) Difference absorption spectra (increasing dark time – initial spectrum after illumination) for the set of spectra shown in panel (a). (C) and (D) Evolution-associated spectra and populations for a two-step kinetic model:  . ESI,† Fig. S5 and S6, respectively, show the fitted transients and residuals for the one-step and two-step global models, respectively. Panel (D) indicates the initial populations (boundary conditions) for the dark recovery, which were determined from a global model for the forward, photoactivation response: OCPR(0) = 0.94, OCPI(0) = 0.06, and OCPO(0) = 0.00. The rates and the associated populations have not been compensated for the forward rates due to the white light measurement beam. . ESI,† Fig. S5 and S6, respectively, show the fitted transients and residuals for the one-step and two-step global models, respectively. Panel (D) indicates the initial populations (boundary conditions) for the dark recovery, which were determined from a global model for the forward, photoactivation response: OCPR(0) = 0.94, OCPI(0) = 0.06, and OCPO(0) = 0.00. The rates and the associated populations have not been compensated for the forward rates due to the white light measurement beam. | ||
The two-step global model reported in Fig. 5c (and ESI,† Fig. S6) returns estimates for the time constants τ−1 and τ−2 for the dark recovery of 7800 s and 1200 s, respectively, at 293 K in the presence of 60% (v/v) glycerol. These time constants are not compensated for the forward rate due to the continuous white-light probe beam, but it is clear that the intermediate OCPI′ is metastable. It persists in the dark for certainly an hour under these conditions, whereas OCPR back-reacts to form OCPI′ several times faster. Maksimov et al.29 previously observed a biexponential kinetic response during the dark recovery of OCP preparations from Arthrospira maxima, which they attributed to the presence of the sub-populations of OCPR from isoforms containing 3′-hydroxyechinenone and echinenone. The results shown here also evidence a biexponential response, but the global model supports the conclusion that it arises from the sequential kinetics of the populations from OCPR and OCPI′. Recall that we are using here preparations of OCP expressed in E. coli. using the gene from Synechocystis sp. PCC 6803 along with the gene for biosynthesis of CAN, so only a single type of ketocarotenoid is present. Further, modeling of the photoactivation and dark recovery responses with the assumption of an inhomogeneous resting OCP ensemble with two components proves to be inadequate especially at short illumination times even though such a model is kinetically overdetermined compared to the three-compartment, two-step global models discussed above.
Effects of temperature and glycerol
As noted above, the choice we made to characterize initially the photoactivation of OCP at 273 K was intended to simplify the global modeling of the response by suppressing as much as possible the rate of the dark recovery reaction. Inclusion of 60% (v/v) glycerol as a cryoprotectant further ensured that the samples remained liquid and bubble free while being stirred continuously during a photoactivation experiment at temperatures above 260 K.We performed additional experiments at 293 K and in the absence of glycerol to test further the relevance of the intermediates OCPI and OCPI′ in the two-step photoactivation and dark recovery models. Fig. 6 shows a global model for the two-step photoactivation response of OCP at 293 K in the absence of glycerol, where a nearly complete conversion to OCPR can be obtained in only a few minutes of illumination. Fig. S7 and S8 (ESI†) present the SVD analysis and a one-step global model for the direct photoactivation to OCPR, without inclusion of the intermediate, which provides a much poorer description of the response. The dark recovery response at 293 K in the absence of glycerol also requires a two-step model; the SVD analysis and global models are provided in ESI,† Fig. S9–S11. Comparison of the evolution-associated spectra for the photoactivation and dark recovery responses (Fig. S12, ESI†) indicates that the intermediates OCPI and OCPI′ have very similar spectra under these conditions, in line with a conclusion that the forward and reverse mechanisms are reversible. A subsequent study designed to avoid continuous exposure of the sample to a white-light probe beam during dark recovery would be required to decide definitively whether or not the OCPI and OCPI′ intermediates should be considered different structures.
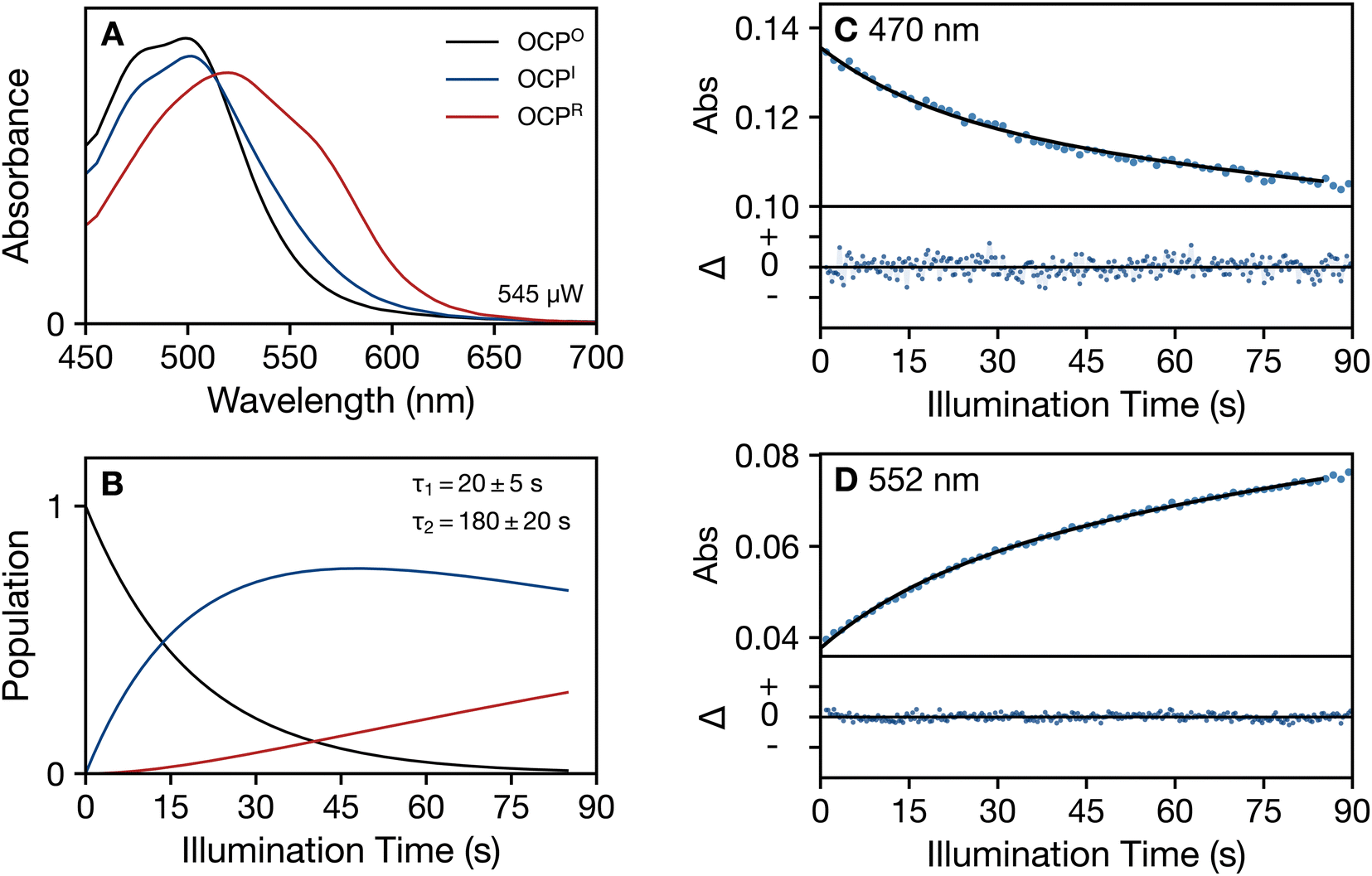 | ||
Fig. 6 Global modeling of the time evolution of the absorption spectrum of OCP at 293 K in the absence of glycerol under continuous 430 nm illumination (545 μW), applying a three-compartment kinetic model: 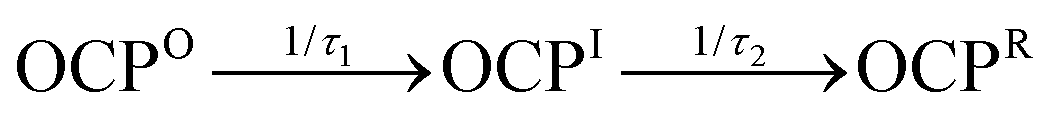 . (A) Evolution-associated absorption spectra for the three kinetic compartments: dark-adapted OCPO reactant (black), the OCPI intermediate (blue), and the final OCPR product (red). (B) Time evolution of the populations for the OCPO, OCPI, and OCPR kinetic compartments. (C) and (D) Fits of the global model (black) to the observed absorption (blue dots) at two detection wavelengths, 470 nm and 552 nm, showing the single-wavelength absorption transients (top panel) and residuals (bottom panel). The residuals (Δ = data − global model) are shown with a 20× zoom factor for the absorbance ordinate. . (A) Evolution-associated absorption spectra for the three kinetic compartments: dark-adapted OCPO reactant (black), the OCPI intermediate (blue), and the final OCPR product (red). (B) Time evolution of the populations for the OCPO, OCPI, and OCPR kinetic compartments. (C) and (D) Fits of the global model (black) to the observed absorption (blue dots) at two detection wavelengths, 470 nm and 552 nm, showing the single-wavelength absorption transients (top panel) and residuals (bottom panel). The residuals (Δ = data − global model) are shown with a 20× zoom factor for the absorbance ordinate. | ||
The summary of results provided in Table 1 indicates that increasing the temperature from 273 K to 293 K and/or removing the glycerol cryoprotectant markedly increases the rates of the photoactivation and dark recovery processes. Fig. S13–S15 (ESI†) present the additional results for the photoactivation response at 293 K in the presence of glycerol. Under all of the conditions studied, a two-step global model with an intermediate is required to obtain an adequate description of the photoactivation and dark recovery responses. The first step of the photoactivation reaction and both steps of the dark recovery reaction at 293 K are slowed, by factors of three and five, respectively, by the addition of glycerol. In contrast, the rate of the second step of the photoactivation mechanism, from OCPI to OCPR, is enhanced in the presence of glycerol by a factor of three. These findings suggest that the rates of the molecular mechanisms of the two steps are oppositely affected by the exchange of water molecules in the hydration shell with glycerol molecules from the bulk.30 Note that the presence of sucrose, methylpentanediol, or glycerol in the medium is required for crystallization of OCP due to binding in a surface pocket, in the NTD–CTD interface near the linker polypeptide segment (Fig. 1a).2,31
| T (K) | Glycerol (% v/v) | k 1 (ms−1 μW−1) | k 2 (ms−1 μW−1) | k −1 (ms−1) | k −2 (ms−1) |
|---|---|---|---|---|---|
| 273 | 60 | 0.00169 ± 6.9 × 10−5 | 0.0170 ± 1.1 × 10−2 | ∼0 | ∼0 |
| 293 | 60 | 0.00333 ± 6.2 × 10−5 | 0.0300 ± 5.0 × 10−3 | 0.128 ± 0.0012 | 0.833 ± 0.14 |
| 293 | <0.3 | 0.0917 ± 2.3 × 10−2 | 0.0102 ± 1.1 × 10−3 | 0.714 ± 0.10 | 3.57 ± 0.26 |
Molecularity of the dark recovery
The three-compartment global models treat both steps of the photoactivation and dark recovery reactions as unimolecular reactions with first-order kinetic responses. Zhang et al.13 had concluded previously on the basis of mass spectrometric evidence, however, that OCPO is a dimer aggregate and that photoactivation yields OCPR monomers. The results presented so far show that the two steps in the photoactivation and dark recovery are well described by reversible first-order processes with respect to the concentration of CAN, as detected in terms of its absorption spectrum, but conversion of the monomer to the dimer of OCP during one of the steps in the dark recovery would be bimolecular and second-order with respect to the protein concentration. We can identify which of the two steps is bimolecular by determining the concentration dependence of the rate constants obtained by the global models for the dark-recovery reaction. To carry this out, we performed a series of dark recovery experiments at 293 K in the absence of glycerol over a range of dilutions of the stock OCPO solution. In each experiment, the sample was first illuminated for 30 min at 430 nm to generate the OCPR photoproduct. The initial concentrations of the three OCP species for the subsequent dark recovery were obtained from the populations determined by a global model for the forward photoactivation response. Fig. S16 and S17 (ESI†) show the global models for dark recoveries for the lowest and highest OCP concentrations used in the series of experiments.
Fig. 7 shows that the rate constant k−2 measured at 293 K in the absence of glycerol for the dark recovery from OCPR to OCPI′ is independent of the concentration of OCP, which confirms that this step of the mechanism is unimolecular, involving a reorganization of the structure of OCPR to obtain the OCPI′ intermediate. In contrast, the rate constant k−1 for the conversion of OCPI′ to OCPO is linearly dependent on the OCP concentration. As discussed in the kinetic analysis presented in the ESI,† these results prove that the conversion of OCPI′ to OCPR is a bimolecular reaction, combining two monomeric proteins to yield a dimer aggregate in the OCPO state. In the simplest picture, the OCPI and OCPI′ intermediates correspond to two OCPO monomers. The dark recovery mechanism,  , exhibits first-order kinetics in the global models because each of the kinetic compartments contains two CAN chromophores, each of which contributes identical absorption spectra. The forward photoactivation process would be the reverse of the dark recovery,
, exhibits first-order kinetics in the global models because each of the kinetic compartments contains two CAN chromophores, each of which contributes identical absorption spectra. The forward photoactivation process would be the reverse of the dark recovery,  , but the linear illumination power dependences (Fig. 4) indicate that photoexcitation of only one of the two CAN chromophores in the OCPO dimer is required to drive the photodissociation reaction yielding the two OCPO monomers, which are then converted independently upon absorption of a second photon.
, but the linear illumination power dependences (Fig. 4) indicate that photoexcitation of only one of the two CAN chromophores in the OCPO dimer is required to drive the photodissociation reaction yielding the two OCPO monomers, which are then converted independently upon absorption of a second photon.
 | ||
| Fig. 7 Concentration dependence of the first-order rate constants determined by global modeling of the dark recovery of OCP at 293 K in the absence of glycerol following illumination at 430 nm. The absorption (A) of the dark adapted OCPO sample was measured at 500 nm prior to exposing the sample to light. Fig. S16 and S17 (ESI†) show the global models for the dark recovery measured for the OCP samples with absorbances of 0.06 and 0.6. The dashed lines show fitted linear models: k1 = (1.48 ± 0.10) × A + (0.060 ± 0.050) ms−1, and k2 = (−0.17 ± 0.36) × A + (2.95 ± 0.15) ms−1. | ||
Absorption spectral line shapes
A structural interpretation of the finding that the evolution associated spectrum from OCPI is broadened and red shifted compared to that of the resting OCPO state is aided by numerical simulations of the absorption spectra of CAN using the multimode Brownian oscillator (MBO) model.32 We discussed this theory previously in our analyses of the absorption and fluorescence spectra from carotenoids in solution33,34 and in the OCP.18 In the MBO model, the reorganization energy, λ,35 controls the broadening of the vibronic transitions due to the fluctuating electrostatic interactions of the chromophore with groups in motion in the surrounding protein and aqueous medium.As an example, the MBO model fits the absorption spectrum of CAN in 2-methyltetrahydrofuran (2-MTHF, Fig. 8a) very well except at the shortest wavelengths. The lack of any resolved vibronic structure is consistent with solvation of CAN in a polar solvent, which results in a fairly large value for λ. In comparison, the partial resolution of the vibronic structure in the OCPO spectrum at 293 K and the absence of glycerol (Fig. 8b) corresponds to a lower value for λ, which is consistent with CAN being more protected from the surrounding aqueous medium by the surrounding protein structure. The successive increase in the values for λ and the shifting of the ground-to-excited-state energy gap to lower energies in the spectra for OCPI (Fig. 8c) and OCPR (Fig. 8d) at 293 K and the absence of glycerol indicate that the CAN chromophore is more exposed as the photoactivation reaction proceeds, especially for the OCPI-to-OCPR step in the mechanism. The optimized value for λ for the spectrum of OCPI is similar to that for CAN in 2-MTHF, and that for OCPR is still larger. These values are tabulated in the ESI,† along with the other parameters for the MBO models in Table S1. As the vibronic structure broadens, the MBO models provide a much better fit to the low-frequency onset of the absorption spectrum, which has been discussed in terms of an inhomogeneous distribution of CAN conformations.36,37
 | ||
| Fig. 8 Numerical simulations (dotted spectra) using the multimode Brownian oscillator (MBO) model of the experimental absorption oscillator strength spectra (solid spectra) for (A) canthaxanthin (CAN) in 2-methyltetrahydrofuran solvent and (B)–(D) of the evolution-associated spectra for three components in the photoactivation global model at 293 K in the absence of glycerol (from Fig. 6). The absorption spectra are plotted here as the oscillator strength, ε(ν)3/ν, where ε gives the absorbance and ν stands for the frequency. The MBO model parameters for each of the simulated spectra are listed in the ESI,† in Table S1. The legends for panels (a)–(d) give the values for the vertical ground-to-excited-state energy gap, ΔE, and the reorganization energy, λ, in wavenumbers (cm−1). | ||
Conclusions
The main finding of this study, that the photoactivation and dark recovery of OCP involve two-step mechanisms mediated by metastable monomeric intermediates, OCPI and OCPI′, leads to a new picture for the protein structural response in OCP that starts with absorption of light by the bound ketocarotenoid. In short, the bimolecular dark recovery kinetics indicate that the resting OCPO spectrum arises from a dimer aggregate of OCPO,35 which very likely corresponds to the homodimer (Fig. 1c) observed in the crystal structures of the orange form of all the structures of the OCP1 family to date, either in the asymmetric unit (e.g. pdb IDs 3mg1, 5ui2, 5hgr, 7ekr, 7qd1) or via crystallographic symmetry (e.g. pdb IDs 4xb5, 6pq1). The amount of surface area buried at the dimer interface (∼1100 Å2) suggests that it is a biologically relevant form of the OCPO. Photodissociation of the OCPO dimer would yield two OCPO monomers, which are assigned to the OCPI and OCPI′ intermediates. Dissociation of the OCPO dimer is a precondition for the subsequent separation of the NTD and CTD interface and has been proposed to be a possible mode of OCP regulation.38 The events associated with translocation of the ketocarotenoid into the NTD and the displacement of the CTD in the reaction that produces the OCPR product state9,10,16,39 are triggered by absorption of light by a OCPO monomer. The finding that the addition of 60% (v/v) glycerol enhances the rate of formation of OCPR in the second step is new information, very likely reporting on the dynamics of hydration of the NTD–CTD interface as the NTD is rotated away.The present global model results and the light intensity dependence of the rates for the two steps in the photoactivation mechanism build on the prior conclusions of Zhang et al.13 to show that the conversion of the OCPO dimer to yield two OCPO monomers requires absorption of a single photon and precedes the light-driven formation of OCPR from the resulting OCPO monomers. Further, owing to the mixture of OCPO and OCPR monomers that persists for a long time during illumination, global modeling is clearly required to isolate the OCPR spectrum for spectroscopic analysis. Similarly, the global model for the dark recovery from OCPR shows that the OCPO monomer is present during most of the measured response but anticipates that the OCPO dimer will be exclusively present after prolonged dark adaptation. Šlouf et al.26 attributed the absorption spectra observed in their OCP preparations to two dark monomeric variants of OCPO, a main form and a form with a red-shifted absorption spectrum that may correspond to OCPO monomers. Maksimov et al.37 also considered the possibility that the OCP dimer and monomer would account for their observations of a biexponential absorption response during dark recovery from OCPR. In a subsequent paper, Maksimov et al.40 observed using fluorescence anisotropy measurements that the effective hydrodynamic radius of their tetramethylrhodamine-labeled OCP preparations increased substantially when the ketocarotenoid is photoexcited, as would be expected for the conversion of the OCPO monomer to OCPR. This result is consistent with the very low protein concentrations used in the fluorescence experiments, which Fig. 7 indicates would substantially slow the dark reassociation of the OCPO monomers.
The requirement that formation of the product OCPR state requires a sequence of two one-photon reactions may be unique in photobiology. Owing to the use of very low light intensities to drive the forward photoactivation process and the long lifetime of the OCPO monomer in the dark, we can conclude that the conversion of the OCPO monomer to OCPR in the second step involves photoexcitation of the CAN chromophore in its singlet ground state, S0. In comparison, Niziński et al.25 reported that an intermediate in the photoactivation reaction was observed with a ∼0.1 s lifetime following absorption of a single laser pulse by their OCPO samples. Given the rate at which the absorption spectra were recorded in the present study, we would not expect to have detected any ground-state photophysical intermediates from CAN with lifetimes in the dark of less than a few seconds.
The structure of the homodimer of OCPO (Fig. 1c) provides a starting point for a discussion of the partial red shift and the reduced resolution of vibronic structure that would be associated with formation of the monomeric OCPO intermediate. Molecular dynamics (MD) simulations at 300 K indicate that the solvent accessible surface area (SASA) of CAN is larger in the OCPO monomer than in the OCPO dimer (Fig. 9). This would be consistent with the expectation that the absorption spectrum of CAN in the OCPO monomer exhibits a partial red shift and a modest broadening of the vibronic structure, respectively, compared to that of the dimer. This effect would arise from an overall change in polarity and an increase in λ due to a stronger interaction with the electrostatic fluctuations in the surroundings. The large surface cavity leading to the ketocarotenoid-binding cleft is not fully protected from the surrounding solvent upon formation of the OCP dimer.38 However, metadynamics simulations by Sharawy et al.41 have revealed a thermally accessible local minimum in the OCPO monomer characterized by the detachment of the αA helix in the N-terminal extension (NTE, Fig. 1a and c) from the CTD domain. Several of the NTE residues (D6, R9, G10, P13, N14, T15, L16) are involved in the dimer interface. This finding suggests that the monomer might undergo additional fluctuations compared to the dimer, where detachment of the αA helix is hindered. The SASA distribution observed in the MD simulations is, in fact, about ten percent broader for the monomer than the dimer.
 | ||
| Fig. 9 Distribution of the solvent accessible surface area (SASA) for the canthaxanthin chromophore in the OCPO monomer (black) and dimer (red) structures from molecular dynamics (MD) trajectories at 300 K. The dotted vertical lines mark the average values, which are labeled with 95% confidence intervals due to the standard deviation of the mean for the 1000 samples in the MD trajectory. The distributions are characterized by the standard deviations 5.4 Å2 for the dimer and 6.0 Å2 for the monomer. See the ESI,† for details about the MD simulations used to calculate the SASA. | ||
Given that hydrogen bonds and salt bridges between the NTDs of the two monomers stabilize the dimer assembly, it is intriguing to consider the possibility that similar photophysical responses by the bound ketocarotenoid are responsible for the triggering of both steps of the forward photoactivation mechanism. As noted in the Introduction, the current perspective for the photoactivation of the OCPO monomer is that absorption of light by the ketocarotenoid is followed by nonradiative decay processes leading to the breaking of the hydrogen-bonding interactions between the β1-ionone ring of the ketocarotenoid and the surrounding binding site in the CTD, which would somehow initiate the translocation of the ketocarotenoid (Fig. 1a and b). Photodissociation of the dimer would be a low-probability event, given that only one of the two ketocarotenoids in a given complex would be triggered by absorption of a photon, but light-induced conformational changes of one of the NTDs due to the photophysics of its associated ketocarotenoid might well destabilize the dimer assembly enough to trigger its dissociation in a small fraction of events. The analysis by Pigni et al.39 of the intermediate states along the translocation of the ketocarotenoid in the photoactivation mechanism using QM/MM calculations further suggests that the partially red-shifted absorption spectrum of the OCPO monomer may require at least a partial translocation of the ketocarotenoid. Given this suggestion, one should consider the possibility that the OCPI and OCPI′ spectra report the contributions of more than one intermediate state16,39 in the translocation of the ketocarotenoid in an OCPO monomer.
Author contributions
CAK and WFB conceived the study, and WFB, JAG, and CAK obtained funding for the project. DS contributed the OCP sample preparations. JBR performed photoactivation experiments and MBO simulations and analyzed the results. JAG performed MD simulations and analyzed the results. JBR, JAG, MS, CAK, and WFB contributed the overall interpretation of the experimental results. The manuscript was written by JBR and WFB with additional contributions by JAG, MS, and CAK.Data availability
The datasets required to perform the analyses reported in this paper are provided in an archive at the following DOI: https://doi.org/10.5281/zenodo.8306513.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Work in the laboratory of W. F. B. was supported by grant awards 1904655 and 2203577 and grant award 2203567 in the laboratory of J. A. G. from the Chemistry of Life Processes program of the U.S. National Science Foundation. Work in the laboratory of C. A. K. was supported by grant award DE-SC0020606 from the Photosynthetic Systems program of the Office of Basic Energy Sciences, U.S. Department of Energy.References
- Y. P. Wu and D. W. Krogmann, The orange carotenoid protein of Synechocystis PCC 6803, Biochim. Biophys. Acta, 1997, 1322, 1–7 CrossRef CAS PubMed.
- C. A. Kerfeld, M. R. Sawaya, V. Brahmandam, D. Cascio, K. K. Ho, C. C. Trevithick-Sutton, D. W. Krogmann and T. O. Yeates, The crystal structure of a cyanobacterial water-soluble carotenoid binding protein, Structure, 2003, 11, 55–65 CrossRef CAS PubMed.
- C. A. Kerfeld, Water-soluble carotenoid proteins of cyanobacteria, Arch. Biochem. Biophys., 2004, 430, 2–9 CrossRef CAS PubMed.
- A. Wilson, G. Ajlani, J. M. Verbavatz, I. Vass, C. A. Kerfeld and D. Kirilovsky, A soluble carotenoid protein involved in phycobilisome-related energy dissipation in cyanobacteria, Plant Cell, 2006, 18, 992–1007 CrossRef CAS PubMed.
- C. Boulay, L. Abasova, C. Six, I. Vass and D. Kirilovsky, Occurrence and function of the orange carotenoid protein in photoprotective mechanisms in various cyanobacteria, Biochim. Biophys. Acta, 2008, 1777, 1344–1354 CrossRef CAS PubMed.
- A. Wilson, C. Punginelli, A. Gall, C. Bonetti, M. Alexandre, J. M. Routaboul, C. A. Kerfeld, R. van Grondelle, B. Robert, J. T. Kennis and D. Kirilovsky, A photoactive carotenoid protein acting as light intensity sensor, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 12075–12080 CrossRef CAS PubMed.
- R. Berera, I. H. M. van Stokkum, S. d’Haene, J. T. M. Kennis, R. van Grondelle and J. P. Dekker, A mechanism of energy dissipation in cyanobacteria, Biophys. J., 2009, 96, 2261–2267 CrossRef CAS PubMed.
- A. Wilson, J. N. Kinney, P. H. Zwart, C. Punginelli, S. D’Haene, F. Perreau, M. G. Klein, D. Kirilovsky and C. A. Kerfeld, Structural determinants underlying photoprotection in the photoactive orange carotenoid protein of cyanobacteria, J. Biol. Chem., 2010, 285, 18364–18375 CrossRef CAS PubMed.
- S. Gupta, M. Guttman, R. L. Leverenz, K. Zhumadilova, E. G. Pawlowski, C. J. Petzold, K. K. Lee, C. Y. Ralston and C. A. Kerfeld, Local and global structural drivers for the photoactivation of the orange carotenoid protein, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E5567–E5574 CrossRef CAS PubMed.
- R. L. Leverenz, M. Sutter, A. Wilson, S. Gupta, A. Thurotte, C. B. de Carbon, C. J. Petzold, C. Ralston, F. Perreau, D. Kirilovsky and C. A. Kerfeld, A 12 Å carotenoid translocation in a photoswitch associated with cyanobacterial photoprotection, Science, 2015, 348, 1463–1466 CrossRef CAS PubMed.
- D. Kirilovsky and C. A. Kerfeld, Cyanobacterial photoprotection by the orange carotenoid protein, Nat. Plants, 2016, 2, 16180 CrossRef CAS PubMed.
- M. A. Domínguez-Martín, P. V. Sauer, H. Kirst, M. Sutter, D. Bína, B. J. Greber, E. Nogales, T. Polívka and C. A. Kerfeld, Structures of a phycobilisome in light-harvesting and photoprotected states, Nature, 2022, 609, 835–845 CrossRef PubMed.
- H. Zhang, H. Liu, D. M. Niedzwiedzki, M. Prado, J. Jiang, M. L. Gross and R. E. Blankenship, Molecular mechanism of photoactivation and structural location of the cyanobacterial orange carotenoid protein, Biochemistry, 2014, 53, 13–19 CrossRef CAS PubMed.
- H. Liu, H. Zhang, G. S. Orf, Y. Lu, J. Jiang, J. D. King, N. R. Wolf, M. L. Gross and R. E. Blankenship, Dramatic Domain Rearrangements of the Cyanobacterial Orange Carotenoid Protein upon Photoactivation, Biochemistry, 2016, 55, 1003–1009 CrossRef CAS PubMed.
- V. Šlouf, V. Kuznetsova, M. Fuciman, C. B. de Carbon, A. Wilson, D. Kirilovsky and T. Polívka, Ultrafast spectroscopy tracks carotenoid configurations in the orange and red carotenoid proteins from cyanobacteria, Photosynth. Res., 2017, 131, 105–117 CrossRef PubMed.
- P. E. Konold, I. H. M. van Stokkum, F. Muzzopappa, A. Wilson, M.-L. Groot, D. Kirilovsky and J. T. M. Kennis, Photoactivation Mechanism, Timing of Protein Secondary Structure Dynamics and Carotenoid Translocation in the Orange Carotenoid Protein, J. Am. Chem. Soc., 2019, 141, 520–530 CrossRef CAS PubMed.
- T. Polívka and V. Sundström, Dark excited states of carotenoids: consensus and controversy, Chem. Phys. Lett., 2009, 477, 1–11 CrossRef.
- J. K. Gurchiek, H. Bao, M. A. Domínguez-Martín, S. E. McGovern, C. E. Marquardt, J. D. Roscioli, S. Ghosh, C. A. Kerfeld and W. F. Beck, Fluorescence and Excited-State Conformational Dynamics of the Orange Carotenoid Protein, J. Phys. Chem. B, 2018, 122, 1792–1800 CrossRef CAS PubMed.
- I. A. Yaroshevich, E. G. Maksimov, N. N. Sluchanko, D. V. Zlenko, A. V. Stepanov, E. A. Slutskaya, Y. B. Slonimskiy, V. S. Botnarevskii, A. Remeeva, I. Gushchin, K. Kovalev, V. I. Gordeliy, I. V. Shelaev, F. E. Gostev, D. Khakhulin, V. V. Poddubnyy, T. S. Gostev, D. A. Cherepanov, T. Polívka, M. Kloz, T. Friedrich, V. Z. Paschenko, V. A. Nadtochenko, A. B. Rubin and M. P. Kirpichnikov, Role of hydrogen bond alternation and charge transfer states in photoactivation of the Orange Carotenoid Protein, Commun. Biol., 2021, 4, 539 CrossRef CAS PubMed.
- S. Niziński, A. Wilson, L. M. Uriarte, C. Ruckebusch, E. A. Andreeva, I. Schlichting, J.-P. Colletier, D. Kirilovsky, G. Burdzinski and M. Sliwa, Unifying Perspective of the Ultrafast Photodynamics of Orange Carotenoid Proteins from Synechocystis: Peril of High-Power Excitation, Existence of Different S* States, and Influence of Tagging, JACS Au, 2022, 2, 1084–1095 CrossRef PubMed.
- V. U. Chukhutsina, J. M. Baxter, A. Fadini, R. M. Morgan, M. A. Pope, K. Maghlaoui, C. M. Orr, A. Wagner and J. J. van Thor, Light activation of Orange Carotenoid Protein reveals bicycle-pedal single-bond isomerization, Nat. Commun., 2022, 13, 6420 CrossRef CAS PubMed.
- A. Warshel, Bicycle-pedal model for the first step in the vision process, Nature, 1976, 260, 679–683 CrossRef CAS PubMed.
- Q. Wang, R. W. Schoenlein, L. A. Peteanu, R. A. Mathies and C. V. Shank, Vibrationally coherent photochemistry in the femtosecond primary event of vision, Science, 1994, 266, 422–424 CrossRef CAS PubMed.
- R. A. Mathies, Photochemistry: A coherent picture of vision, Nat. Chem., 2015, 7, 945–947 CrossRef CAS PubMed.
- S. Niziński, I. Schlichting, J.-P. Colletier, D. Kirilovsky, G. Burdzinski and M. Sliwa, Is orange carotenoid protein photoactivation a single-photon process?, Biophys. Rep., 2022, 2, 100072 Search PubMed.
- E. G. Maksimov, E. A. Shirshin, N. N. Sluchanko, D. V. Zlenko, E. Y. Parshina, G. V. Tsoraev, K. E. Klementiev, G. S. Budylin, F.-J. Schmitt, T. Friedrich, V. V. Fadeev, V. Z. Paschenko and A. B. Rubin, The Signaling State of Orange Carotenoid Protein, Biophys. J., 2015, 109, 595–607 CrossRef CAS PubMed.
- F. X. Cunningham and E. Gantt, A portfolio of plasmids for identification and analysis of carotenoid pathway enzymes: Adonis aestivalis as a case study, Photosynth. Res., 2007, 92, 245–259 CrossRef CAS PubMed.
- T. Polívka and V. Sundström, Ultrafast dynamics of carotenoid excited states—from solution to natural and artificial systems, Chem. Rev., 2004, 104, 2021–2071 CrossRef PubMed.
- I. H. M. van Stokkum, D. S. Larsen and R. van Grondelle, Global and target analysis of time-resolved spectra, Biochim. Biophys. Acta, 2004, 1657, 82–104 CrossRef CAS PubMed.
- M. Hirai, S. Ajito, M. Sugiyama, H. Iwase, S.-I. Takata, N. Shimizu, N. Igarashi, A. Martel and L. Porcar, Direct Evidence for the Effect of Glycerol on Protein Hydration and Thermal Structural Transition, Biophys. J., 2018, 115, 313–327 CrossRef CAS PubMed.
- C. A. Kerfeld, Y. P. Wu, C. Chan, D. W. Krogmann and T. O. Yeates, Crystals of the carotenoid protein from Arthrospira maxima containing uniformly oriented pigment molecules, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1997, 53, 720–723 CrossRef CAS PubMed.
- G. R. Fleming and M. Cho, Chromophore-solvent dynamics, Annu. Rev. Phys. Chem., 1996, 47, 109–134 CrossRef CAS.
- S. Ghosh, M. M. Bishop, J. D. Roscioli, J. J. Mueller, N. C. Shepherd, A. M. LaFountain, H. A. Frank and W. F. Beck, Femtosecond Heterodyne Transient-Grating Studies of Nonradiative Decay of the S2 (11Bu+) State of β-Carotene: Contributions from Dark Intermediates and Double-Quantum Coherences, J. Phys. Chem. B, 2015, 119, 14905–14924 CrossRef CAS PubMed.
- S. Ghosh, M. M. Bishop, J. D. Roscioli, A. M. LaFountain, H. A. Frank and W. F. Beck, Femtosecond heterodyne transient grating studies of nonradiative deactivation of the S2 (11Bu+) state of peridinin: detection and spectroscopic assignment of an intermediate in the decay pathway, J. Phys. Chem. B, 2016, 120, 3601–3614 CrossRef CAS PubMed.
- R. A. Marcus, On the theory of oxidation-reduction reactions involving electron transfer. I, J. Chem. Phys., 1956, 24, 966–989 CrossRef CAS.
- V. Balevičius Jr, A. G. Pour, J. Savolainen, C. N. Lincoln, V. Lukeš, E. Riedle, L. Valkunas, D. Abramavicius and J. Hauer, Vibronic energy relaxation approach highlighting deactivation pathways in carotenoids, Phys. Chem. Chem. Phys., 2015, 17, 19491–19499 RSC.
- E. Kish, M. M. Pinto, D. Kirilovsky, R. Spezia and B. Robert, Echinenone vibrational properties: From solvents to the orange carotenoid protein, Biochim. Biophys. Acta, 2015, 1847, 1044–1054 CrossRef CAS PubMed.
- C. A. Kerfeld, M. R. Melnicki, M. Sutter and M. A. Dominguez-Martin, Structure, function and evolution of the cyanobacterial orange carotenoid protein and its homologs, New Phytol., 2017, 215, 937–951 CrossRef CAS PubMed.
- N. B. Pigni, K. L. Clark, W. F. Beck and J. A. Gascón, Spectral Signatures of Canthaxanthin Translocation in the Orange Carotenoid Protein, J. Phys. Chem. B, 2020, 124, 11387–11395 CrossRef CAS PubMed.
- E. G. Maksimov, N. N. Sluchanko, K. S. Mironov, E. A. Shirshin, K. E. Klementiev, G. V. Tsoraev, M. Moldenhauer, T. Friedrich, D. A. Los, S. I. Allakhverdiev, V. Z. Paschenko and A. B. Rubin, Fluorescent labeling preserving OCP photoactivity reveals its reorganization during the photocycle, Biophys. J., 2017, 112, 827 CrossRef CAS PubMed.
- M. Sharawy, N. B. Pigni, E. R. May and J. A. Gascón, A favorable path to domain separation in the orange carotenoid protein, Protein Sci., 2022, 31, 850–863 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Global modeling and SVD analyses of additional photoactivation and dark recovery responses, multimode Brownian oscillator model parameters for numerically simulated absorption spectra of canthaxanthin in solution and in OCP, analysis of the kinetics of the dark recovery response, and computational methods. See DOI: https://doi.org/10.1039/d3cp04484j |
| This journal is © the Owner Societies 2023 |