
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photodissociative decay pathways of the flavin mononucleotide anion and its complexes with tryptophan and glutamic acid†
Kelechi O.
Uleanya
 ,
Cate S.
Anstöter
and
Caroline E. H.
Dessent
*
,
Cate S.
Anstöter
and
Caroline E. H.
Dessent
*
Department of Chemistry, University of York, Heslington, York YO10 5DD, UK. E-mail: caroline.dessent@york.ac.uk
First published on 3rd November 2023
Abstract
Flavin mononucleotide (FMN) is a highly versatile biological chromophore involved in a range of biochemical pathways including blue-light sensing proteins and the control of circadian rhythms. Questions exist about the effect of local amino acids on the electronic properties and photophysics of the chromophore. Using gas-phase anion laser photodissociation spectroscopy, we have measured the intrinsic electronic spectroscopy (3.1–5.7 eV) and accompanying photodissociative decay pathways of the native deprotonated form of FMN, i.e. [FMN-H]− complexed with the amino acids tryptophan (TRP) and glutamic acid (GLU), i.e. [FMN-H]−·TRP and [FMN-H]−·GLU, to investigate the extent to which these amino acids perturb the electronic properties and photodynamics of the [FMN-H]− chromophore. The overall photodepletion profiles of [FMN-H]−·TRP and [FMN-H]−·GLU are similar to that of the monomer, revealing that amino acid complexation occurs without significant spectral shifting of the [FMN-H]− electronic excitations over this region. Both [FMN-H]−·TRP and [FMN-H]−·GLU are observed to decay by non-statistical photodecay pathways, although the behaviour of [FMN-H]−·TRP is closer to statistical fragmentation. Long-lived FMN excited states (triplet) are therefore relatively quenched when TRP binds to [FMN-H]−. Importantly, we find that [FMN-H]−, [FMN-H]−·TRP and [FMN-H]−·GLU all decay predominantly via electron detachment following photoexcitation of the flavin chromophore, with amino acid complexation appearing not to inhibit this decay channel. The strong propensity for electron detachment is attributed to excited-state proton transfer within FMN, with proton transfer from a ribose alcohol to the phosphate preceding electron detachment.
1. Introduction
Electron-transfer processes within peptides and proteins are key steps in many biochemical reactions, including respiration and photosynthesis.1 These events involve cofactors as the redox centres, but can also use amino acid radicals as intermediates.2 Such events are challenging to characterize due to their short-lived nature, but are thought to be of particular importance in light-induced electron-transfer reactions.3,4 Flavins are common cofactors in these chains, where their photoreduction by electron transfer from proximal tryptophan or tyrosine to an electronically excited flavin is believed to act as an efficient fluorescence quenching pathway.5 Examples of such pathways include those active in cryptochrome, BLUF domain proteins and DNA photolyases.6 Given the wide biochemical importance of these systems, they have been extensively investigated by static and ultrafast photochemical methods.7 However, interpretation of the results obtained is still highly challenging, particularly given the difficulties of applying computational chemistry to a system that is conformationally complex, alongside the demands of modelling coupled electron-transfer processes and electronic excited states.7In this work, we present the first in vacuo electronic laser spectroscopy of a flavin molecule bound directly in a molecular complex to the amino acid tryptophan (TRP), which is a key amino acid in photoredox active centres in flavin containing proteins.8 We aim to investigate the potential of such systems as prototypes for obtaining novel biochemical insights into flavin photochemistry, and obtain benchmarking data to evaluate the applicability of high-level computational chemistry approaches to flavin–amino acid structures. We have selected flavin mononucleotide (FMN) as the flavin molecule for this work, and study it as its native, singly-deprotonated form, where deprotonation is expected to occur at the phosphoric acid group (pka ∼ 2).9,10 In addition, we have chosen to study a complex of FMN with glutamic acid (GLU), as a prototype simple, non-aromatic amino acid that is not generally involved in flavin photoredox proteins. Scheme 1 illustrates the individual structures of FMN, TRP and GLU. Since negatively charged systems are studied in this work, we are able to investigate how electron detachment competes with molecular fragmentation following photoexcitation. This is an important aspect of the fundamental photochemistry of flavins given their role as photoredox species. By studying the FMN monomer, and the amino acid complexes, we are able to measure the extent to which a bound amino acid can quench electron detachment from the flavin and determine whether photoinitiated electron-transfer from the flavin to these amino acids occurs.
 | ||
| Scheme 1 Schematic diagram of the structures of flavin mononucleotide (FMN), tryptophan (TRP) and glutamic acid (GLU). | ||
Experiments in this work were performed via laser-interface mass spectrometry (LIMS), which provides the gas-phase absorption spectra via photodepletion action spectroscopy, along with the spectroscopic production profiles of any ionic photofragments produced following photoexcitation.11,12 We combine our LIMS measurements with higher-energy collisional dissociation (HCD) measurements that reveal the ionic fragmentation pattern for the ground electronic state of the system under study.13 This provides information on whether photofragmentation follows a statistical or non-statistical route, and hence provides insight into the photodynamics of decay.14,15 (Further information on the insight that can be obtained from identifying statistical versus non-statistical routes in photodissociation can be found in ref. 16.)16
Giacomozzi and co-workers have studied the non-statistical fragmentation of photoactivated flavin mononucleotide anions recently using a custom-built instrument, the SepI accelerator mass spectrometer at Aarhus University.17 They found that [FMN-H]− decomposes largely through non-statistical fragmentation pathways following photoexcitation between 300–500 nm. Preliminary measurements were also presented on the complex of [FMN-H]− with betaine to probe whether the electronic transitions display intramolecular charge-transfer character. The measurements we present here complement this earlier study as our instrument measures the production of photofragments over an extended timescale (typically up to 0.1 ms)11 that is longer than biochemical decay processes to provide a complete picture of the photofragments produced.12 Our accompanying HCD measurements provide further fundamental information for understanding the nature of the photodecay mechanisms displayed by the complexes studied here.13 Furthermore, since our experiment allows us to measure the electron detachment profile of a system we can obtain insight into the propensity for an anion to undergo electron detachment versus fragmentation following photoexcitation.18,19
Over the last five years, a growing number of flavin molecule ions and complexes have been investigated in vacuo using laser (IR and UV-VIS) photodissociation spectroscopy, including alkali metal-flavin (e.g. riboflavin and lumiflavin) clusters,20,21 and protonated and deprotonated flavin molecules.17,22–26 These studies have provided important new fundamental information on the spectroscopy and photochemistry of relatively simple flavin systems. We aim here to build on these recent studies by increasing the complexity and biological relevance of system to be investigated. Such measurements are part of wider advances employing gas-phase laser spectroscopy to advance our fundamental understanding of biomolecular systems.27–33
A very wide range of physical methods have been applied to study the condensed phase photophysics of flavins, both for free flavin moieties and flavoprotein photoreceptors.7,34 A recent review by Kar et al. provides a useful overview of the state of knowledge on the broad field of flavin electronic structure that provides further background to the work presented here.35 Readers may also be interested in the role of solvent in the dynamical process linked to flavin photoexcitation; ref z provides for further background on that point.36
2. Experimental method
FMN (70% purity), TRP (98% purity) and GLU (99% purity) were purchased from Sigma Aldrich and was used without further purification. HPLC-grade ethanol was purchased from Fisher Scientific, Inc. (Pittsburgh, PA, USA) and again used as received.2.1 Laser-interfaced mass spectrometry
The experiment has been described in-full previously.11,12 Briefly, gas-phase UV photodissociation experiments were conducted in an AmaZon SL dual funnel electrospray ionization quadrupole ion trap (ESI-QIT) mass spectrometer (Bruker Daltonics Inc.), which was modified to allow for laser-interfaced mass spectrometry. Solutions of FMN (1.0 × 10−5 mol dm−3 with NH3 (0.4%) added to aid ionization) in EtOH were introduced to the mass spectrometer using typical instrumental parameters: nebulizing gas pressure of 10.0 psi, an injection rate of 0.33 mL h−1, a drying gas flow rate of 8.0 L min−1, a capillary temperature of 160 °C. To form the amino acid–flavin complexes, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixtures of FMN and the amino acids were combined, with both solutions being 1.0 × 10−5 mol dm−3.
:1 mixtures of FMN and the amino acids were combined, with both solutions being 1.0 × 10−5 mol dm−3.
Ions were mass selected and isolated in the ion trap prior to UV laser irradiation. UV photons were produced with a 10 Hz Nd:YAG (Surelite™) pumped OPO laser (Horizon™), providing ∼0.2–1.0 mJ across the range 400–218 nm. Laser step sizes of 2 nm were used. Photofragmentation experiments were conducted with a set ion accumulation time of 10 ms and a corresponding fragmentation time of 100 ms, allowing for each mass-selected ion packet to interact with one laser pulse. The laser beam diameter is ∼2 mm in the ion interaction region of the ion trap. When fluorescence is negligible, UV-excited gaseous ions fragment upon excited-state relaxation, yielding an action absorption spectrum by photodepletion.
2.2 Analysis
Photodepletion (PD) was measured as a function of scanned wavelength, with the photofragment production (PF) recorded simultaneously at each corresponding wavelength.37 As described by eqn (1a) and (1b), IntOFF and IntON represent the parent ion intensities with laser off and on, respectively; IntFRAG is the photofragment intensity with the laser on; λ is the excitation wavelength (nm); and P is the tuneable laser pulse energy (mJ). | (1a) |
 | (1b) |
2.3 Higher-energy collisional dissociation
Experiments were performed in a Thermo Fisher Orbitrap FusionTM mass spectrometer, as described previously.13 (We are not able to provide a center-of-mass collision energy that corresponds to a percentage HCD in our experiments, as we perform the experiments in a commercial mass spectrometer, which does not allow sufficient collision cell pressure variability to allow calibration. In our earlier work,13 we were able to link our results on deprotonated adenosine monophosphate to those of Ho and Kebarle,38 who had obtained an absolute measurement of center of mass collision energy through absolute calibration of their system for the same ion. This allowed us to obtain an approximate calibration of the HCD fragmentation curves and conclude that the 4–6 eV energy range (photon energy) corresponds to the 20–38% range on the HCD fragmentation plots.)The following settings were used for the HCD experiments: the syringe was operated at a flow rate of 3 μL min−1 and with the following settings: MS2 scan isolation mode, ion trap; detector type, ion trap; positive ion spray voltage (3500 V); RF lens (60%); normalized AGC target (100%); maximum injection time (100 ms); ion transfer tube temperature (275 °C); and vaporizer temperature (20 °C). For the MS scan in this instrument, the settings were as follows: detector type, Orbitrap; positive ion spray voltage (3200 V); RF lens (45%); normalized AGC target (100%); and maximum injection time (100 ms).
2.4 Calculations
Schrodinger's MacroModel with mixed Monte Carlo torsional and low-mode sampling parameters were employed to conduct conformational molecular dynamics searches to generate possible conformers of the [FMN-H]−·TRP and [FMN-H]−·GLU clusters.39 Possible conformers were energy gradient minimized with the OPLS3e force-field. Single-point energies were then performed at the PBE0/6-311++G level of theory.3. Results and discussion
3.1 Photodepletion spectra
Fig. 1a displays the photodepletion (gas-phase absorption) spectra of singly deprotonated FMN, i.e. [FMN-H]−, acquired from 3.1–5.7 eV (400–218 nm). While related data has been published by Giacomozzi et al. recently,17 the [FMN-H]− spectrum was reacquired here to allow a direct comparison with the cluster spectra that are the main focus of this work.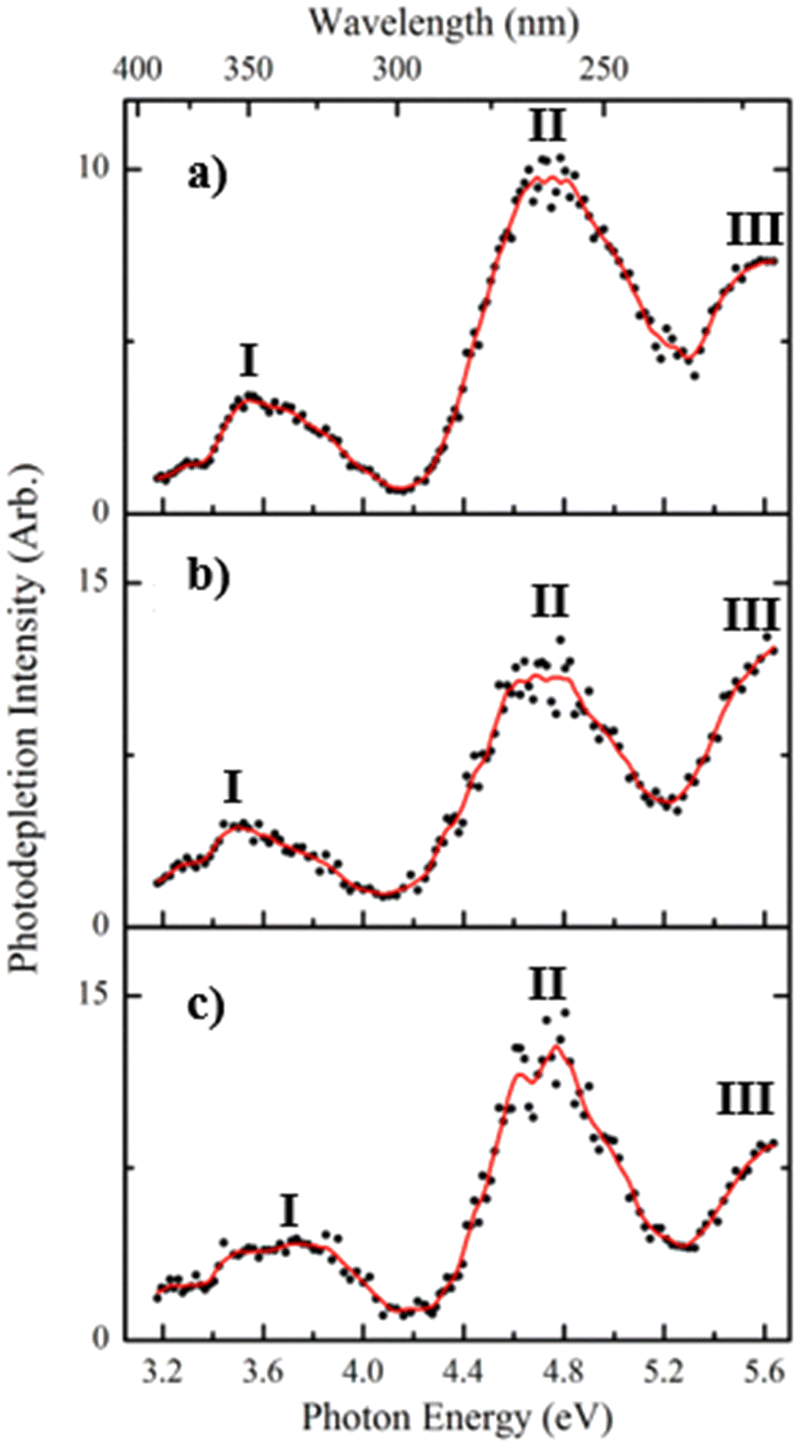 | ||
| Fig. 1 Gas-phase photodepletion (action absorption) spectra of (a) [FMN-H]−, (b) [FMN-H]−·TRP and (c) [FMN-H]−·GLU across the range 3.1–5.7 eV. The solid lines are five point adjacent averages of the data points. | ||
Three regions of absorption for [FMN-H]− are evident over the spectral region (labelled I–III), with the absorption onset occurring around 3.2 eV. Band I is a relatively weak broad band and is observed across the visible region, centred around 3.6 eV. This band decreases in intensity above 3.9 eV and approaches a baseline level by 4.1 eV. Band II appears strongly between 4.2–5.2 eV, displaying a maximum at 4.75 eV, with the rising edge of band III is evident above 5.3 eV. The photodepletion spectra of the complexes (Fig. 1b and c), [FMN-H]−·TRP and [FMN-H]−·GLU, are similar to that of the monomer with the λmax of bands I and II occurring at 3.52 and 4.71 eV, respectively for [FMN-H]−·TRP, and at 3.73 and 4.76 eV for [FMN-H]−·GLU. The fact that the photodepletion spectra of the two complexes, and the monomer, all appear very similar is striking, as complexation of a molecule to a chromophore will generally result in a significant spectral shift. One explanation of the absence of spectral shifts in the spectra shown in Fig. 1 may be that the amino acids bind to the phosphate chain of FMN and are therefore distant from the alloxazine chromophore. This will be discussed in more detail below. Finally, we note that neutral tryptophan has an absorption band between 300–250 nm, so that [FMN-H]−·TRP photodepletion observed across that range may include depletion associated with excitation of the tryptophan chromophore.
The bands observed in the spectra presented in Fig. 1 can be related to the known electronic states of flavins. In brief, flavins display singlet excited states at approximately 2.85, 3.76 and 4.7, with triplet states occurring at around 3.1 and 4.3 eV. Bands I and II can therefore be assigned to excitation of the second and third singlet states of [FMN-H]−, with the first and second triplet states, respectively, being accessible via intersystem crossing from these bands, respectively.40
Laser-energy dependent photodepletion measurements were conducted for [FMN-H]−, with laser energies above ∼0.4 mJ being associated with multiphoton processes (Section S1, ESI†). The observation of multiphoton processes at higher laser energies is consistent with similar measurements conducted on this system previously.17 Laser-energy dependent photodepletion measurements were also attempted for the clusters, but were challenging due to the relatively lower ion intensities of the precursor complexes. It is reasonable to assume that at sufficiently high laser energies, similar multiphoton effects will be present for the clusters. For the spectra presented here, we reduced the laser energy to 0.20 mJ to conduct the measurements on [FMN-H]− in a single-photon regime, obtaining photodepletion ∼35% of the precursor ion intensity. A similar photodepletion intensity was employed for the measurements on the complexes (35% of precursor ion intensity) so that the cluster spectra also predominantly result from single-photon excitations.
While the photodepletion spectra of [FMN-H]−, [FMN-H]−·TRP and [FMN-H]−·GLU are evidently very similar, the photodepletion cross section masks the detail of the excited state decay pathways. This is revealed much more clearly by measuring the spectral profiles of the photofragments produced following excited-state decay.12 We therefore turn to exploring the [FMN-H]−, [FMN-H]−·TRP and [FMN-H]−·GLU photofragment ions and their production spectra in the next section.
3.2 Photofragmentation
Fig. 2 displays the photofragment ions produced for each of these species following photoexcitation at 4.73 eV, the region of the band II peak (maximum photodepletion). Photofragmentation of [FMN-H]− occurs into numerous channels, with the most intense fragments being m/z 213, 199, and 241. These ions correspond to the following three dissociative pathways, where LC and LF are the smaller flavin molecules, lumichrome and lumiflavin, respectively:| [FMN-H]− + hν → m/z 213 (C5H10PO7−) + LC | (2a) |
| → m/z 199 (C4H8PO7−) + LF | (2b) |
| → m/z 241 (C5H11PO7) + [LC-H]− | (2c) |
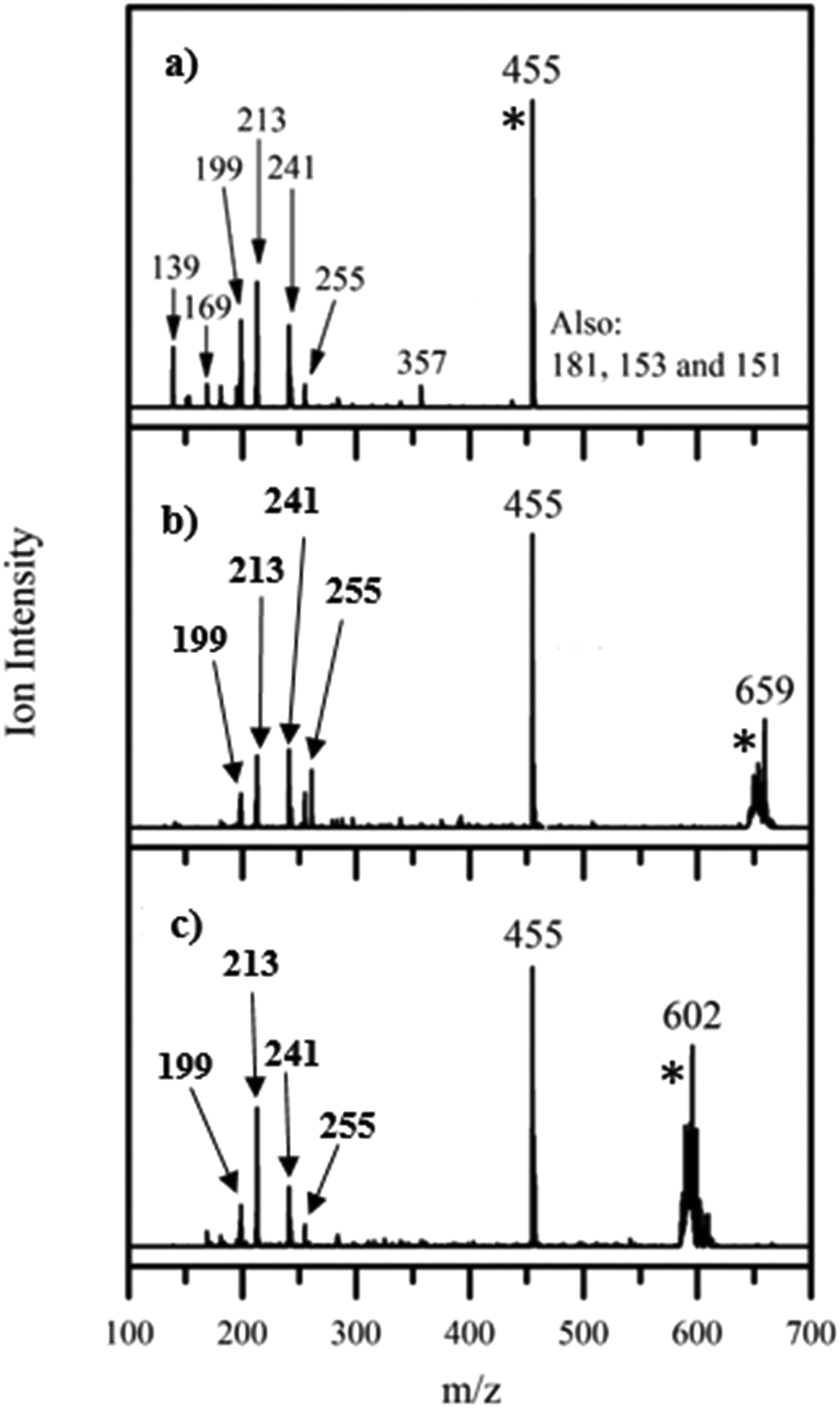 | ||
| Fig. 2 Photofragment difference (laserON − laserOFF) mass spectra for (a) [FMN-H]−, (b) [FMN-H]−·TRP and (c) [FMN-H]−·GLU excited at 4.73 eV. [*] indicates the photodepleted parent ion. | ||
A list of the most intense photofragments of [FMN-H]− observed at 4.73 eV is given in Table 1, with their photofragmentation production spectra being displayed in Fig. S3 (ESI†). There is good agreement between the photofragments we observe and those seen by Giacomozzi et al. upon 450 nm photoexcitation, particularly with respect to the major intensity photofragments.17 The major photofragments are also structurally similar to those observed upon photoexcitation of the related, deprotonated riboflavin ion across 2.0–5.74 eV.22 (The riboflavin structure lacks the phosphate group of FMN.)
| Fragments | HCD | Photo fragments | Neutral loss | Fragment identity | Neutral identity |
|---|---|---|---|---|---|
| a RF = riboflavin, LC = lumichrome, LF = lumiflavin, FMFH2 = formylmethylflavin. | |||||
| 79 | ✓ | X | 376 | PO3− | RF |
| 97 | ✓ | X | 358 | H2PO4− | RF-H2O |
| 139 | ✓ | ✓ | 316 | C2H4PO5− | LF + 2(H2CO) |
| 159 | ✓ | X | 296 | C2H8PO6− | LC + C3H2O |
| 169 | X | ✓ | 286 | C3H6PO6− | FMFH2 |
| 181 | ✓ | ✓ | 274 | C4H6PO6− | LF + H2O |
| 199 | ✓ | ✓ | 256 | C4H8PO7− | LF |
| 213 | ✓ | ✓ | 242 | C5H10PO7− | LC |
| 241 | ✓ | ✓ | 214 | [LC-H]− | C5H11PO7 |
| 255 | ✓ | ✓ | 200 | [LF-H]− | C4H9PO7 |
| 357 | ✓ | ✓ | 98 | [RF-H2O-H]− | H3PO4 |
| 375 | ✓ | X | 80 | [RF-H]− | HPO3 |
| 412 | ✓ | X | 43 | [FMN-HCNO]− | HCNO |
| 437 | ✓ | ✓ | 18 | FMN-H2O | H2O |
Fig. 2b displays the photofragments observed upon photoexcitation of the [FMN-H]−·TRP complex at 4.73 eV (peak of band II), with a list of the photofragments being given in Table 2 along with assignments. The most intense photofragment observed is m/z 455, i.e. [FMN-H]−, which is the product of upon cluster fission, with the other major fragments (m/z 241, 213, 199 and 255) having been observed for the [FMN-H]− monomer. The photofragmentation mass spectrum for the [FMN-H]−·GLU complex, again obtained at 4.73 eV, is displayed in Fig. 2c, and is similar to that of [FMN-H]−·TRP, with m/z 455 being the dominant photofragment (i.e. [FMN-H]−) along with m/z 241, 213, 199 and 255. Table 3 lists the fragments observed for [FMN-H]−·GLU. We note that the deprotonated amino acid is not observed as a photofragment from either complex.
| Fragments | HCD | Photo fragments | Neutral loss | Fragment identity | Neutral identity |
|---|---|---|---|---|---|
| a RF = riboflavin, LC = lumichrome, LF = lumiflavin, FMFH2 = formylmethylflavin. | |||||
| 139 | X | ✓ | 520 | C2H4PO5− | LF + 2(H2CO) + TRP |
| 169 | X | ✓ | 490 | C3H6PO6− | FMFH2 + TRP |
| 199 | X | ✓ | 460 | C4H8PO7− | LF + TRP |
| 213 | X | ✓ | 446 | C5H10PO7− | LC + TRP |
| 241 | X | ✓ | 418 | [LC-H]− | C5H11PO7 + TRP |
| 255 | X | ✓ | 404 | [LF-H]− | C4H9PO7 + TRP |
| 375 | X | ✓ | 384 | [RF-H]− | HPO3 + TRP |
| 392 | X | ✓ | 267 | [FMN-H2O]− | H2O + TRP |
| 455 | ✓ | ✓ | 204 | [FMN-H]− | TRP |
| Fragments | HCD | Photo fragments | Neutral loss | Fragment identity | Neutral identity |
|---|---|---|---|---|---|
| a LC = lumichrome, LF = lumiflavin. | |||||
| 199 | X | ✓ | 403 | C4H8PO7− | LF + GLU |
| 213 | X | ✓ | 389 | C5H10PO7− | LC + GLU |
| 241 | X | ✓ | 361 | [LC-H]− | C5H11PO7 + GLU |
| 255 | X | ✓ | 347 | [LF-H]− | C4H9PO7 + GLU |
| 455 | ✓ | ✓ | 147 | [FMN-H]− | GLU |
Fig. S4 and S5 (ESI†) display the full photofragmentation spectra of [FMN-H]−·TRP and [FMN-H]−·GLU across 3.1–5.7 eV, and reveal how photofragment production varies for the two complexes across the spectral range investigated. For [FMN-H]−·TRP, the m/z 241, 213 and 199 fragments’ production profiles closely match that of band I. The m/z 455 fragment is produced most strongly through band I, but with a profile that differs from those of the m/z 241, 213 and 199 group. For [FMN-H]−·GLU, the m/z 241, 213 and 199 group are again produced with a profile that follows band I. However, the fragment production across band II is more complicated, with m/z 455 being the most intense fragment, but not being produced with a profile that matches the photodepletion band. These variations reflect differences in the excited states and available decay pathways of the three systems studied, and will be discussed further in Section 3.5.
3.3 Photofragmentation versus electron detachment and electron transfer
For simple anionic systems, i.e. ones that are rigid, photofragmentation will occur in competition with electron detachment at photon energies above the electron detachment threshold.41,42 In our experiment, we can estimate the extent of electron detachment by assuming that any photodepletion that is not associated with ionic-fragment production is associated with electron loss.18,19 The electron production spectra for [FMN-H]−, [FMN-H]−·TRP and [FMN-H]−·GLU (Fig. S6, ESI†) show that electron detachment is the dominant decay channel with >80% of all observed photodecay mechanisms channelling into electron detachment. This is intriguing since the electron detachment energy of the phosphate moiety of [FMN-H]− is expected to be above ∼5 eV,43,44 so that electron loss cannot be attributed to direct detachment below such energies. (However, the electron detachment yield can be seen to increase significantly above 5.4 eV where direct detachment is likely to occur.) Furthermore, the electron production spectra for [FMN-H]−, and the complexes (Fig. S6, ESI†) show that the electron detachment profiles closely match the photodepletion spectra. This is important as it allows us to conclude that electron detachment is strongly coupled to electronic excitation of the flavin chromophore.41 We will discuss this point further in Section 4.No photoinitiated electron transfer is observed from [FMN-H]− to either of the amino acids studied here. We have been able to observe intracluster electron transfer from halide ions to a range of molecules in previous work,41,45–47 where dipole-bound excited states have acted as doorway states for electron transfer within the cluster. The absence of electron transfer for the flavin clusters via such mechanisms here may be attributed to a low (conformational) dipole moment of the amino acid within the cluster, or to relatively weak Frank–Condon factors from the relatively delocalized phosphate anion to any existing dipole-bound excited state.48
3.4 Higher-energy collisional dissociation
To probe the collisional activated fragmentation pathways of [FMN-H]−, [FMN-H]−·TRP and [FMN-H]−·GLU in their electronic ground states, higher-energy collisional dissociation (HCD) was employed. Fig. 3 displays the HCD curves for the major fragments observed for each of these species. HCD measurements are important for interpreting ionic photodissociation measurements as they allow for the identification of primary and secondary products, formed when a precursor species fragments at high internal energy.13 They are also important as any photofragment ions not observed in HCD can be identified as purely photochemical products.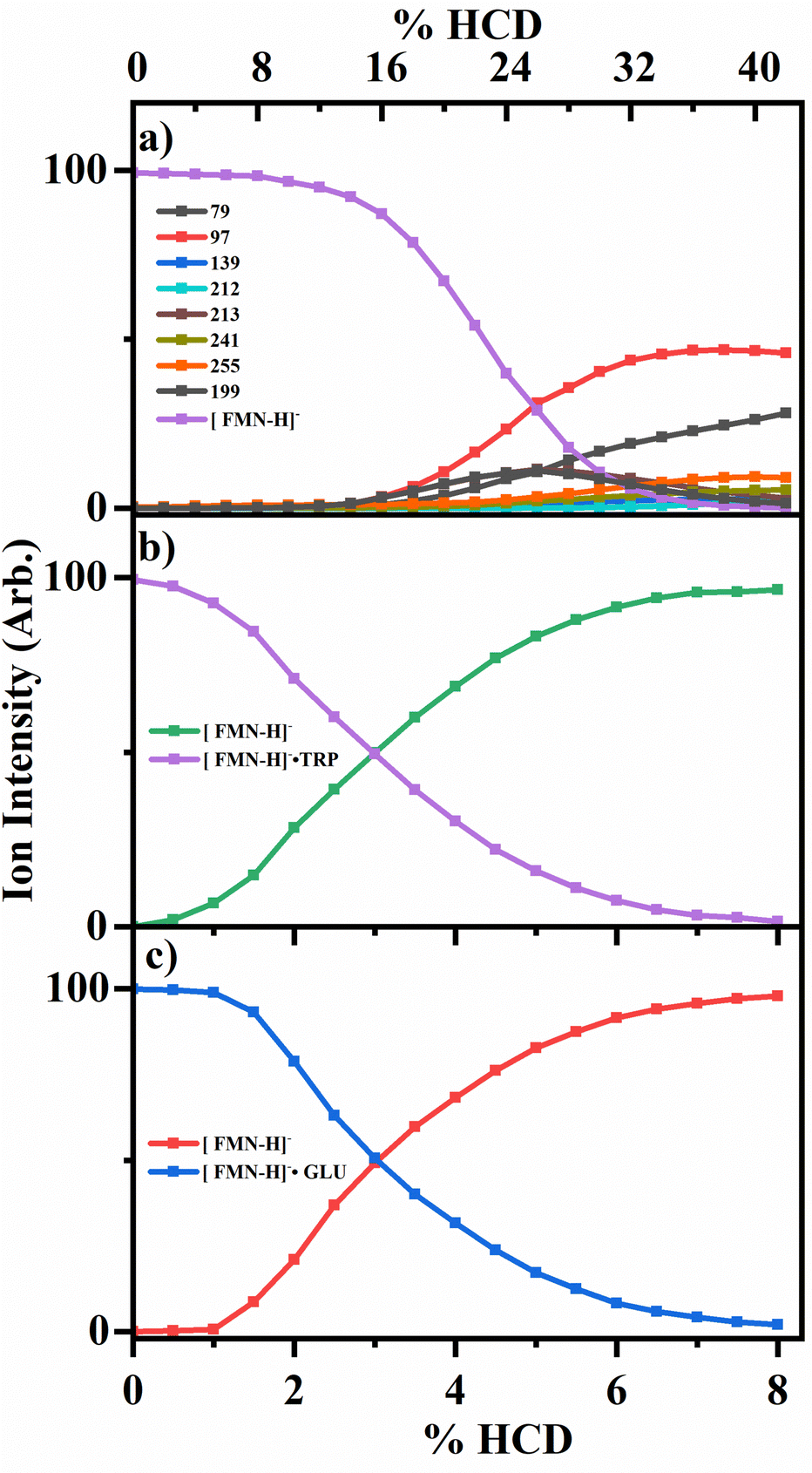 | ||
| Fig. 3 Percent ion intensity of the HCD fragments of (a) [FMN-H]− (from 0–42% HCD energy) (b) [FMN-H]−·TRP and (c) [FMN-H]−·GLU (from 0–8.5% HCD energy). The solid lines are three point averages of the data points. | ||
The HCD results for [FMN-H]− are presented in Fig. 3a, clearly showing that the m/z 97 ion (H2PO4−) is the most intense fragment across the majority of the range studied. (At the very lowest energies, the m/z 255 fragment is most intense, albeit with very low intensity.) The next most intense fragments are m/z 79, 199 and 255. At higher collisional energies (>28%), the relative intensity of m/z 79 (PO3−) can be seen to increase significantly, indicating that this fragment is a secondary fragment, particularly of the higher mass fragments. Giacomozzi et al. have previously measured collisional dissociation of [FMN-H]− in the SepI accelerator mass spectrometer at low and high collision energies.17 In their high-energy collision measurement, they observe the m/z 97 ion as the dominant fragment ion, with only m/z 79 and m/z 241 as additional ionic fragments. At low collision energy (multiple few-eV collisions in ion trap), they observe the m/z 241 ion as the dominant fragment, with the m/z 97 ion as the second most intense fragment, with m/z 255, 213, and 79 also present in significant quantities. This is consistent with their high-collision energy measurement being more energetic than the maximum HCD energy studied in this work. While the two measurements are complementary, the HCD data presented in Fig. 3 give a fuller picture of how fragment ions vary continuously with respect to one another over the laser excitation energies employed here.13
Fig. 3b and c display the HCD results for the two complexes, [FMN-H]−·TRP and [FMN-H]−·GLU, showing that the clusters undergo cluster fission with ejection of the amino acid as the dominant process at all collisional energies, i.e.
| [FMN-H]−·TRP → [FMN-H]− + TRP | (3a) |
| [FMN-H]−·GLU → [FMN-H]− + GLU | (3b) |
In summary, the HCD measurements indicate that ionic fragmentation of the ground electronic states of [FMN-H]−, [FMN-H]−·TRP and [FMN-H]−·GLU are straightforward. [FMN-H]− fragments predominantly with loss of the H2PO4− ion, with the m/z 79, 199 and 255 ions also being observed at significant intensities, and m/z 79 (PO3−) growing in intensity as collisional energy increases. Both complexes dissociate solely via cluster fission and production of [FMN-H]−. We next turn to comparing the photofragmentation and HCD fragmentation patterns to gain insight into the photodissociation dynamics, and assess whether complexation affects the photodynamics.
3.5 Comparison of photofragmentation and higher-energy collisional dissociation of [FMN-H]−, [FMN-H]−·TRP and [FMN-H]−·GLU
From the data presented in Sections 3.2 and 3.3, it is evident that the ionic fragments observed upon photoexcitation of [FMN-H]− at 4.73 eV do not match those observed in the HCD measurements with photofragmentation being dominated by production of m/z 213, 199, and 241 while collisional dissociation leads dominantly to production of m/z 97, along with m/z 79, 199 and 255. Furthermore, the photofragment profiles vary significantly with photon energy (Fig. S2, ESI†). These observations are all consistent with non-statistical photodecay dynamics operating for the [FMN-H]− monomer. (Non-statistical decay is associated with dissociation occurring directly from the excited state without the involvement of a conical intersection to return the system to a near-starting point geometry. In non-statistical decay, the photofragments obtained will be notably different in their identities and relative intensities from the ground electronic state fragments seen from HCD.14,15,50 Further information on the insight that can be obtained from identifying statistical versus non-statistical routes in photodissociation can be found in ref. 50.)For ease of comparison, Fig. 4 presents a selection of plots which compare HCD fragment production with photofragment production for the [FMN-H]− monomer. In these plots, we have selected HCD energies that should be approximately equivalent to the photon energies employed. We emphasise that this is an approximation, as it is not able to straightforward to calibrate the HCD energies, but our previous work on deprotonated adenosine triphosphate allows us to make the approximate qualitative comparison we employ here.13 The data displayed in Fig. 4 very clearly illustrate that the photofragment intensities do not mirror those obtained from HCD. Focusing on the key data for the band I and band II regions (Fig. 4A and B), the photofragments m/z 199, 213 and 241 can be seen to be significantly more intense than would be expected from the HCD fragments over the band I region (Fig. 4A) while production of m/z 241 ion in particular increasing for the band II region. Strikingly, the energy channelling into the dominant HCD fragments, m/z 79 and 97, is largely minimised in the photofragmentation process, even at the highest photon energy (Fig. 4C).
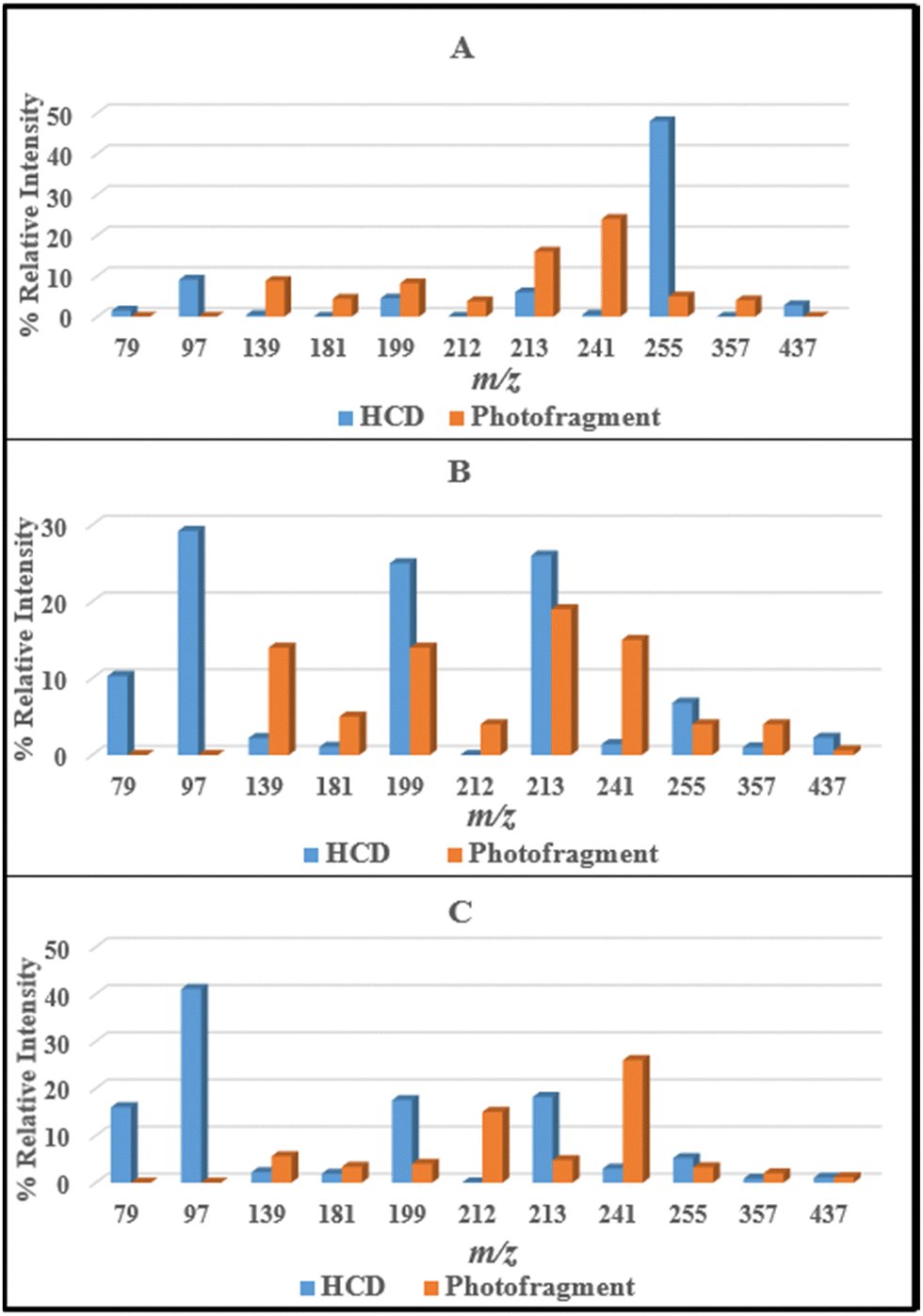 | ||
| Fig. 4 Comparison of the HCD and photofragments ions produced by [FMN-H]− at (A) 8% and 3.6 eV, (B) 18% and 4.7 eV, and (C) 26% and 5.6 eV HCD energy and photon energy, respectively. | ||
Similar overall conclusions about the dominance of non-statistical decay behaviour can be drawn for the [FMN-H]−·TRP and [FMN-H]−·GLU complexes. For both clusters, photochemical production of the [FMN-H]− does not follow the profile of the overall photodepletion spectra (Section S2, ESI†) as would be expected if decay of the clusters was predominantly statistical. For example, both [FMN-H]−·TRP and [FMN-H]−·GLU show enhanced production of the m/z 213 and 241 photofragments in the lower-energy regions of band II, reflecting strong non-statistical behaviour in this region.
Fig. 5 presents a comparison of the production of the m/z 455 ([FMN-H]−) fragment from HCD against the photofragment for the [FMN-H]−·TRP and [FMN-H]−·GLU complexes. It is only possible to present this straightforward comparison for the m/z 455 fragment for the complexes as this is the only fragment observed in HCD. Notably, the patterns of HCD vs. photofragmentation intensity differ for the two complexes. This reveals that distinctive photodynamics are present in the two clusters, with tryptophan binding appearing to enhance the photochemical production of the [FMN-H]− fragment compared to glutamic acid across the spectral range studied. Intriguingly, this suggests that the [FMN-H]−·TRP complex is displaying photodynamics closer to statistical decay than either [FMN-H]− or [FMN-H]−·GLU, consistent with TRP quenching long-lived triplet states of FMN.51
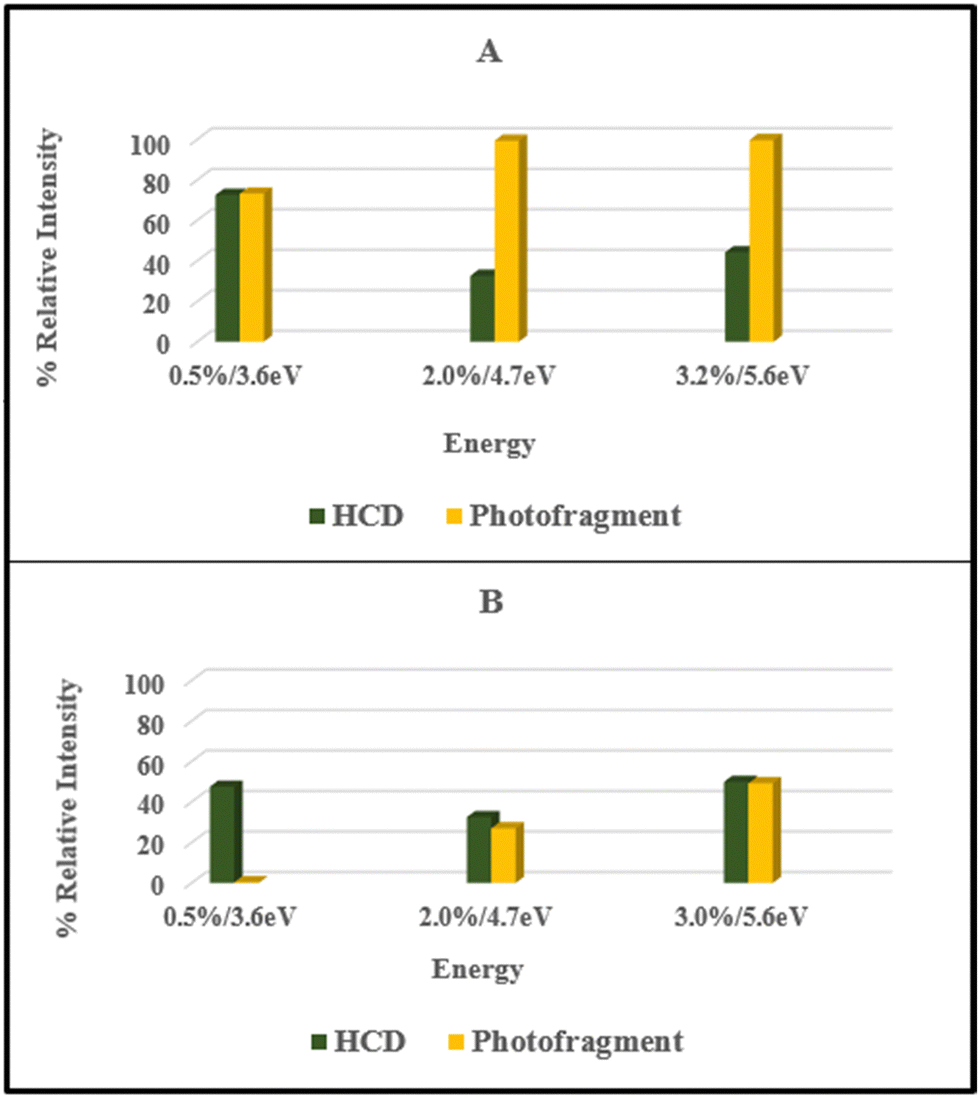 | ||
| Fig. 5 Comparison of the HCD and photofragment intensities for production of the m/z 455 ion (i.e. [FMN-H]−) at different % HCD and photon energy (hν) respectively for (A) [FMN-H]−·TRP and (B) [FMN-H]−·GLU. | ||
3.6 Comparison of the geometric structures of [FMN-H]−·TRP and [FMN-H]−·GLU
One of the immediate questions to be answered in understanding the differences in the photodynamics of [FMN-H]−·TRP and [FMN-H]−·GLU is the extent to which the two complexes are structurally distinct. Therefore, a series of calculations were performed to gain an initial insight into the clusters’ geometric structures.Fig. 6 displays the lowest-energy structures obtained for [FMN-H]−·TRP and [FMN-H]−·GLU clusters. Section S4 (ESI†) presents the results for other low-energy structures of the complexes. All of the calculated structures show a strong propensity to form hydrogen-bonding interactions, as expected for such gas-phase anionic molecular systems. (We note that Molano-Arevalo et al. have studied the gas-phase structure of the related molecule, flavin adenine dinucleotide, using ion mobility spectrometry, and obtained candidate structures using computational chemistry as part of this work.52 Those structures incorporate similar hydrogen bonding patterns to the ones observed here.) However, comparison of the [FMN-H]−·TRP and [FMN-H]−·GLU lowest-energy structures shows that the two clusters display distinctive conformational preferences. While the [FMN-H]−·TRP complex exhibits a preference for adopting a folded structure where the two aromatic functional groups are able to interact via favorable π–π interactions, the [FMN-H]−·GLU cluster adopts a more open structure where the glutamic acid hydrogen bonds to the deprotonated phosphate group away from the isoalloxazine chromophore. The folded structure of [FMN-H]−·TRP is notable in the context of the modified photodynamics evident for this system compared to bare [FMN-H]− and [FMN-H]−·GLU and reflects TRP-flavin geometric arrangements seen in flavin proteins.2
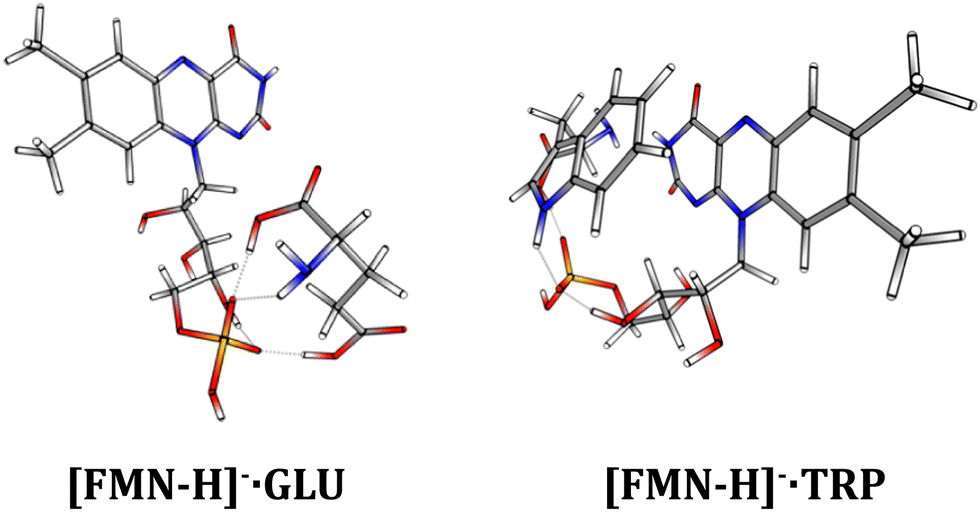 | ||
| Fig. 6 The lowest-energy calculated structures of the [FMN-H]−·GLU and [FMN-H]−·TRP complexes. See text for details. | ||
4. Further discussion of the mechanism of electron detachment and excited state dynamics
Electron detachment can occur above the electron-detachment threshold of an anionic system either via direct detachment (4a) or indirectly from an excited state of the cluster (4b). It can also occur from hot photofragments, e.g.(4c). Eqn (4a)–(4c) illustrate these pathways for [FMN-H]−:| [FMN-H]− + hν → [FMN-H] + e− | (4a) |
| [FMN-H]− + hν → [FMN-H]−* → [FMN-H]* + e− | (4b) |
| [FMN-H]− + hν → [FMN-H]−* → [LC-H]− + C5H11PO7 → [LC-H] + C5H11PO7 + e− | (4c) |
Wang et al. used anion photoelectron spectroscopy to determine that the H2PO4− anion has an adiabatic electron affinity of 4.57 eV, with a vertical detachment energy of 5.06 eV. Similarly, Nobel et al. have found that the adiabatic electron affinity of the phenylphosphonate anion as 4.60 eV.44 The electron detachment energy of [FMN-H]− has not been determined to date, but would be expected to be similar to, or greater than, H2PO4− due to intramolecular hydrogen bonding stabilising the flavin anion. Similarly, the detachment energies of the complexes would be likely to be higher that of the [FMN-H]− monomer as a result of intermolecular hydrogen bonding.
These considerations lead us to conclude that the direct detachment, pathway (4a), is unlikely to be important across the majority of the photoexcitation region studied here. To fully rule out decay pathways of the type illustrated in eqn (4c) would require estimates of the electron detachment energies of the possible anionic photofragments. However, from the photofragments we observe, many correspond to ions that are phosphate containing (e.g. C5H10PO7−), and would hence again have an associated high detachment energies that would be energetically inaccessible. This leads to the conclusion that the dominant electron loss pathway is (4b) and indicates that photoexcitation of the flavin chromophore of [FMN-H]− leads to efficient electron loss from the excited states accessed across the range 3.1–5.7 eV. While the phosphate anion displays a relatively high electron detachment of ∼5 eV, alkoxide anions have significantly lower detachment energies of ∼2 eV.53 Therefore, one explanation of the strong detachment propensity observed here is that proton transfer occurs on the excited state surface of the flavin from the ribose alcohol to the phosphate group, with subsequent detachment from the resulting alkoxide. It is also possible that proton transfer occurs from the isoalloxazine group, with resulting electron detachment from the deprotonated isoalloxazine ring (detachment energy ∼3.6 eV).23
Stockett and co-workers have suggested that photo-induced proton transfer occurs in the related molecule, flavin adenine dinucleotide.25 In that work it was argued that proton transfer occurred from the isoalloxazine group to a phosphate group, although a definitive structural assignment could not be made. Taken together, the measurements on the flavin adenine dinucleotide and the systems studied here provide evidence for a strong propensity for photo-activated proton transfer in these biochemically relevant flavin molecules. In future work, it should be possible to directly probe this process using time-resolved photoelectron spectroscopy.27,54,55 Currently there are no published photoelectron spectroscopy studies of flavin anions. Such measurements are highly desirable to determine the detachment energies and provide further key information on the coupling between photoexcitation and photodetachment.56 They would also be important in the context of the known behaviour of lumiflavin in solution, where presolvated electrons have been observed following photoexcitation of lumiflavin under conditions where the deprotonated is present.57
Finally, we note that the propensity for electron detachment is equally strong for [FMN-H]−·TRP and [FMN-H]−·GLU, as for the [FMN-H]− monomer. It is striking that complexation with the TRP and GLU amino acids does not reduce the intrinsic propensity for electron detachment from [FMN-H]−, reflecting the dominance of the intrinsic chromophore dynamics.
5. Conclusions
Previous work on [FMN-H]− revealed that a substantial fraction of photoactivated ions decay by non-statistical fragmentation pathways, which occur prior to the complete conversion and redistribution of electronic excitation energy across the vibrational degrees of freedom.17 Our studies of the [FMN-H]−·TRP and [FMN-H]−·GLU complexes reveal that similar non-statistical decay pathways persist upon complexation of [FMN-H]− with these amino acids. This behaviour contrasts with that of nucleobase clusters we have studied previously, where statistical photodecay pathways dominate as a result of ultrafast excited state decay by the nucleobase chromophore.49,58 While both complexes display non-statistical decay pathways, there are clear differences in the photodynamics displayed. The [FMN-H]−·TRP complex shows behavior that is closer to statistical photodissociation compared to [FMN-H]−·GLU, which in turn displays behavior that is closer to bare [FMN-H]−. These differences in photodynamics align with structural differences between the two complexes, where [FMN-H]−·TRP adopts a more closed structure due to π–π stabilization between the TRP and FMN. Despite the importance of key photoredox flavin systems including cryptochromes, many questions remain in relation to their detailed mechanism of action. Within such systems, three highly conserved TRP residues occupy sites adjacent to the flavin centre and form the electron-transfer chain.8 Over recent years, a number of model systems have been developed to simplify functional studies, including flavin-TRP dyads and flavin-TRP containing unilamella vesicles.8,59 These approaches are challenging in terms of the synthetic techniques involved. In vacuo cluster studies provide an alternative approach, with the results presented herein demonstrating the potential of cluster studies for addressing aspects of flavin photodynamics as a function of proximity to selected amino acids.Data availability
Data is available from the authors on request.Author contributions
Conceptualization, C. E. H. D.; methodology, C. E. H. D.; formal analysis, K. O. U., C. S. A.; investigation, K. O. U., C. S. A.; resources, C. E. H. D.; writing—original draft preparation, C. E. H. D.; writing—review and editing, C. E. H. D., K. O. U., C. S. A.; supervision, C. E. H. D.; funding acquisition, C. E. H. D. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
Acknowledgment is made to the Donors of the American Chemical Society Petroleum Research Fund for support or partial support of this research, through the award of grant ACS PRF 56174-ND6. We (KOU and CED) also thank the Royal Society of Chemistry for providing further funding to support this work through the award of a Research Enablement Grant (E22-9265726124). We thank the University of York and the Department of Chemistry for provision of funds for the OPO laser system. The York Centre of Excellence in Mass Spectrometry, used for the HCD work, was created thanks to a major capital investment through Science City York, supported by Yorkshire Forward with funds from the Northern Way Initiative, and has more recently received additional support from the EPSRC and BBSRC. We also thank Dr Edward Matthews and Dr Jacob Berenbeim for early contributions to this work.References
- H. B. Gray and J. R. Winkler, Annu. Rev. Biochem., 1996, 65, 537–561 CrossRef CAS PubMed.
- J. L. Dempsey, J. R. Winkler and H. B. Gray, Chem. Rev., 2010, 110, 7024–7039 CrossRef CAS PubMed.
- L. Zanetti-Polzi, M. Aschi, A. Amadei and I. Daidone, J. Phys. Chem. Lett., 2017, 8, 3321–3327 CrossRef CAS PubMed.
- J. Zelenka, R. Cibulka and J. Roithová, Angew. Chem., Int. Ed., 2019, 58, 15412–15420 CrossRef CAS PubMed.
- N. Mataga, H. Chosrowjan, S. Taniguchi, F. Tanaka, N. Kido and M. Kitamura, J. Phys. Chem. B, 2002, 106, 8917–8920 CrossRef CAS.
- B. Zhuang, U. Liebl and M. H. Vos, J. Phys. Chem. B, 2022, 126, 3199–3207 CrossRef CAS PubMed.
- K. S. Conrad, C. C. Manahan and B. R. Crane, Nat. Chem. Biol., 2014, 10, 801–809 CrossRef CAS PubMed.
- S. Paul, L. Meng, S. Berger, G. Grampp, J. Matysik and X. Wang, ChemPhotoChem, 2017, 1, 12–16 CrossRef CAS.
- W. D. Kumler and J. J. Eiler, J. Am. Chem. Soc., 1943, 65, 2355–2361 CrossRef CAS.
- G. Li and K. D. Glusac, J. Phys. Chem. A, 2008, 112, 4573–4583 CrossRef CAS PubMed.
- E. Matthews, A. Sen, N. Yoshikawa, E. Bergström and C. E. H. Dessent, Phys. Chem. Chem. Phys., 2016, 18, 15143–15152 RSC.
- E. Matthews and C. E. H. Dessent, J. Phys. Chem. A, 2016, 120, 9209–9216 CrossRef CAS PubMed.
- R. Cercola, E. Matthews and C. E. H. Dessent, J. Phys. Chem. B, 2017, 121, 5553–5561 CrossRef CAS PubMed.
- S. B. Nielsen, J. U. Andersen, J. S. Forster, P. Hvelplund, B. Liu, U. V. Pedersen and S. Tomita, Phys. Rev. Lett., 2003, 91, 048302 CrossRef PubMed.
- J. C. Marcum, A. Halevi and J. M. Weber, Phys. Chem. Chem. Phys., 2009, 11, 1740 RSC.
- G. Grégoire, H. Kang, C. Dedonder-Lardeux, C. Jouvet, C. Desfrançois, D. Onidas, V. Lepere and J. A. Fayeton, Phys. Chem. Chem. Phys., 2006, 8, 122–128 RSC.
- L. Giacomozzi, C. Kjær, S. Brøndsted Nielsen, E. K. Ashworth, J. N. Bull and M. H. Stockett, J. Chem. Phys., 2021, 155, 044305 CrossRef CAS PubMed.
- N. G. K. Wong, J. A. Berenbeim and C. E. H. Dessent, ChemPhotoChem, 2019, 3, 1231–1237 CrossRef CAS.
- R. Cercola, K. O. Uleanya and C. E. H. Dessent, Mol. Phys., 2020, 118, e1662128 CrossRef.
- P. Nieto, A. Günther, G. Berden, J. Oomens and O. Dopfer, J. Phys. Chem. A, 2016, 120, 8297–8308 CrossRef CAS PubMed.
- D. Müller and O. Dopfer, J. Phys. Chem. A, 2021, 125, 3146–3158 CrossRef PubMed.
- N. G. K. Wong, C. Rhodes and C. E. H. Dessent, Molecules, 2021, 26, 6009 CrossRef CAS PubMed.
- E. Matthews and C. E. H. Dessent, J. Phys. Chem. Lett., 2018, 9, 6124–6130 CrossRef CAS PubMed.
- E. Matthews, R. Cercola and C. Dessent, Molecules, 2018, 23, 2036 CrossRef PubMed.
- J. N. Bull, E. Carrascosa, L. Giacomozzi, E. J. Bieske and M. H. Stockett, Phys. Chem. Chem. Phys., 2018, 20, 19672–19681 RSC.
- M. H. Stockett, Phys. Chem. Chem. Phys., 2017, 19, 25829–25833 RSC.
- A. Henley and H. H. Fielding, Int. Rev. Phys. Chem., 2019, 38, 1–34 Search PubMed.
- S. Bakels, E. M. Meijer, M. Greuell, S. B. A. Porskamp, G. Rouwhorst, J. Mahé, M.-P. Gaigeot and A. M. Rijs, Faraday Discuss., 2019, 217, 322–341 RSC.
- E. K. Ashworth, M.-H. Kao, C. S. Anstöter, G. Riesco-Llach, L. Blancafort, K. M. Solntsev, S. R. Meech, J. R. Verlet and J. N. Bull, Phys. Chem. Chem. Phys., 2023, 25, 23626–23636, 10.1039/D3CP03250G.
- C. S. Anstöter, B. F. E. Curchod and J. R. R. Verlet, Phys. Chem. Chem. Phys., 2022, 24, 1305–1309 RSC.
- W. Zagorec-Marks, L. G. Dodson, P. Weis, E. K. Schneider, M. M. Kappes and J. M. Weber, J. Am. Chem. Soc., 2021, 143, 17778–17785 CrossRef CAS PubMed.
- R. Antoine and P. Dugourd, Phys. Chem. Chem. Phys., 2011, 13, 16494 RSC.
- T. R. Rizzo, J. A. Stearns and O. V. Boyarkin, Int. Rev. Phys. Chem., 2009, 28, 481–515 Search PubMed.
- E. F. Yee, S. Chandrasekaran, C. Lin and B. R. Crane, Methods in Enzymology, Elsevier, 2019, vol. 620, pp. 509–544 Search PubMed.
- R. K. Kar, A. Miller and M. Mroginski, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1541 Search PubMed.
- Y. El Khoury, L. J. G. W. Van Wilderen and J. Bredenbeck, J. Chem. Phys., 2015, 142, 212416 CrossRef PubMed.
- E. Matthews, PhD thesis, University of York, 2018.
- Y. Ho and P. Kebarle, Int. J. Mass Spectrom. Ion Processes, 1997, 165–166, 433–455 CrossRef.
- F. Mohamadi, N. G. J. Richards, W. C. Guida, R. Liskamp, M. Lipton, C. Caufield, G. Chang, T. Hendrickson and W. C. Still, J. Comput. Chem., 1990, 11, 440–467 CrossRef CAS.
- T. Climent, R. González-Luque, M. Merchán and L. Serrano-Andrés, J. Phys. Chem. A, 2006, 110, 13584–13590 CrossRef CAS PubMed.
- K. O. Uleanya and C. E. H. Dessent, Phys. Chem. Chem. Phys., 2021, 23, 1021–1030 RSC.
- J. Simons, J. Phys. Chem. A, 2008, 112, 6401–6511 CrossRef CAS PubMed.
- X.-B. Wang, E. R. Vorpagel, X. Yang and L. Wang, J. Phys. Chem. A, 2001, 105, 10468–10474 CrossRef CAS.
- J. A. Noble, E. Marceca, C. Dedonder, I. Carvin, E. Gloaguen and C. Jouvet, Eur. Phys. J. D, 2021, 75, 95 CrossRef CAS.
- E. Matthews, R. Cercola, G. Mensa-Bonsu, D. M. Neumark and C. E. H. Dessent, J. Chem. Phys., 2018, 148, 084304 CrossRef PubMed.
- W.-L. Li, A. Kunin, E. Matthews, N. Yoshikawa, C. E. H. Dessent and D. M. Neumark, J. Chem. Phys., 2016, 145, 044319 CrossRef PubMed.
- A. J. A. Harvey, N. Yoshikawa, J.-G. Wang and C. E. H. Dessent, J. Chem. Phys., 2015, 143, 101103 CrossRef PubMed.
- R. Cercola, E. Matthews and C. E. H. Dessent, Mol. Phys., 2019, 117, 3001–3010 CrossRef CAS.
- A. Sen, T. F. M. Luxford, N. Yoshikawa and C. E. H. Dessent, Phys. Chem. Chem. Phys., 2014, 16, 15490 RSC.
- B. Lucas, M. Barat, J. A. Fayeton, C. Jouvet, P. Çarçabal and G. Grégoire, Chem. Phys., 2008, 347, 324–330 CrossRef CAS.
- B. Zhuang, D. Seo, A. Aleksandrov and M. H. Vos, J. Am. Chem. Soc., 2021, 143, 2757–2768 CrossRef CAS PubMed.
- J. C. Molano-Arevalo, D. R. Hernandez, W. G. Gonzalez, J. Miksovska, M. E. Ridgeway, M. A. Park and F. Fernandez-Lima, Anal. Chem., 2014, 86, 10223–10230 CrossRef CAS PubMed.
- S. T. Stokes, J. E. Bartmess, A. Buonaugurio, Y. Wang, S. N. Eustis and K. H. Bowen, Chem. Phys. Lett., 2019, 732, 136638 CrossRef.
- C. S. Anstöter, J. N. Bull and J. R. R. Verlet, Int. Rev. Phys. Chem., 2016, 35, 509–538 Search PubMed.
- A. Kunin and D. M. Neumark, Phys. Chem. Chem. Phys., 2019, 21, 7239–7255 RSC.
- A. Sen, G.-L. Hou, X.-B. Wang and C. E. H. Dessent, J. Phys. Chem. B, 2015, 119, 11626–11631 CrossRef CAS PubMed.
- P. F. Heelis, R. F. Hartman and S. D. Rose, Photochem. Photobiol., 1993, 57, 1053–1055 CrossRef CAS PubMed.
- A. Sen and C. E. H. Dessent, J. Phys. Chem. Lett., 2014, 5, 3281–3285 CrossRef CAS PubMed.
- Y. Oka, T. Miura and T. Ikoma, J. Phys. Chem. B, 2021, 125, 4057–4066 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Section S1: Laser energy measurements of [FMN-H]−. Section S2: PF scans of [FMN-H]− and complexes. Section S3: Electron production spectra. Section S4: Cluster structures. See DOI: https://doi.org/10.1039/d3cp04359b |
| This journal is © the Owner Societies 2023 |