
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRefining the thermochemical properties of CF, SiF, and their cations by combining photoelectron spectroscopy, quantum chemical calculations, and the Active Thermochemical Tables approach
Ugo
Jacovella
 *a,
Branko
Ruscic
*b,
Ning L.
Chen
a,
Hai-Linh
Le
a,
Séverine
Boyé-Péronne
a,
Sebastian
Hartweg
c,
Madhusree Roy
Chowdhury
c,
Gustavo A.
Garcia
c,
Jean-Christophe
Loison
d and
Bérenger
Gans
*a
*a,
Branko
Ruscic
*b,
Ning L.
Chen
a,
Hai-Linh
Le
a,
Séverine
Boyé-Péronne
a,
Sebastian
Hartweg
c,
Madhusree Roy
Chowdhury
c,
Gustavo A.
Garcia
c,
Jean-Christophe
Loison
d and
Bérenger
Gans
*a
aUniversité Paris-Saclay, CNRS, Institut des Sciences Moléculaires d'Orsay, 91405 Orsay, France. E-mail: ugo.jacovella@universite-paris-saclay.fr; berenger.gans@universite-paris-saclay.fr
bChemical Sciences and Engineering Division, Argonne National Laboratory, Lemont, Illinois 60439, USA. E-mail: ruscic@anl.gov
cSynchrotron SOLEIL, L’Orme des Merisiers, 91192 Saint Aubin, Gif-sur-Yvette, France
dUniversité Bordeaux, CNRS, Bordeaux INP, ISM, UMR 5255, F-33400 Talence, France
First published on 10th October 2023
Abstract
Fluorinated species have a pivotal role in semiconductor material chemistry and some of them have been detected beyond the Earth's atmosphere. Achieving good energy accuracy on fluorinated species using quantum chemical calculations has long been a challenge. In addition, obtaining direct experimental thermochemical quantities has also proved difficult. Here, we report the threshold photoelectron and photoion yield spectra of SiF and CF radicals generated with a fluorine reactor. The spectra were analysed with the support of ab initio calculations, resulting in new experimental values for the adiabatic ionisation energies of both CF (9.128 ± 0.006 eV) and SiF (7.379 ± 0.009 eV). Using these values, the underlying thermochemical network of Active Thermochemical Tables was updated, providing further refined enthalpies of formation and dissociation energies of CF, SiF, and their cationic counterparts.
Introduction
The first group 14 monofluoride radicals, namely fluoromethylidyne (CF) and fluorosilylidyne (SiF), along with their cations fluoromethyliumylidene (CF+) and fluorosilyliumylidene (SiF+), are commonly found in plasmas employed for the purpose of silicon-containing semiconductor material surface etching.1–3 In addition to being one of the most abundant ions present in semiconductor processing,3 CF+ has been detected in diverse regions of outer space.4–7 It has thus been identified as a key intermediate in the chemistry of fluorine-bearing molecules in diffuse and dense interstellar gas clouds.8,9 Although we are still awaiting the astronomical detection of SiF and SiF+, their presence in outer space as well as their role in the chemistry of silicon in the gas phase and on grains is strongly suspected. Indeed, reactions involving SiF+ are already incorporated in astrochemical models.8Thermodynamic properties such as enthalpies of formation, dissociation energies, and adiabatic ionisation energies are of utmost importance for refining chemical models of fluorinated chemistry in plasmas and in space. Furthermore, accurate thermodynamic properties are very well suited to benchmarking the accuracy of existing and upcoming state-of-the-art quantum chemical calculations, particularly with respect to pushing the limits of the long-standing challenge of obtaining accurate thermochemical data for fluorinated compounds.10–14
Dyke and coworkers have published two papers presenting the He I photoelectron spectra of CF15,16 from which they determined the adiabatic ionisation energies of the X+ 1Σ+ ← X 2Π and a+ 3Π ← X 2Π ionising transitions of CF to be 9.11 ± 0.02 eV and 13.94 ± 0.02 eV, respectively. From photoelectron spectra, the ωe+ value of CF+ was derived initially to be 1840 ± 30 cm−1,15 and later revised to 1810 ± 30 cm−1.16
The CF+ cation has also been investigated using direct infrared (IR) diode laser spectroscopy.17,18 Preliminary work from Hirota and coworkers17 yielded precise structural information on CF+ from its vibronic ground state (re+ = 1.154272(35) Å). Subsequently, Saykally and coworkers18 have extended the IR study of CF+ to the seven lowest vibrational levels of the electronic ground state, providing precise vibrational constants such as ωe+ = 1792.6654(18) cm−1 and ωexe+ = 13.229668(54) cm−1, as well as re+ = 1.1542551(25) Å, the latter being in good agreement with the results of Hirota et al. However, of the two photoelectron spectroscopic studies, only the most recent one16 reported a vibrational frequency (1810 ± 30 cm−1) congruent with the accurate constants of Saykally and coworkers.18 The spectroscopic molecular constants were also well predicted using multireference configuration interaction (MRCI) calculations for the lowest singlet and triplet electronic states of CF+.19 Subsequently, the landscape of the CF+ electronic states has been further explored along with several Rydberg states.20,21
The dissociation energy of the CF+ ground state has been estimated16 as D0(CF+) = 7.82 ± 0.12 eV, based on combining the ionisation energies of CF and carbon atom with the dissociation energy of CF of 5.67 ± 0.10 eV from electron-ionisation mass spectrometric studies.22 This is just within the combined error bars compared to the value obtained in the work of Saykally,18D0(CF+) = 7.698 ± 0.004 eV, extracted from the fit based on a crude Morse potential model. Theodorakopoulos and coworkers20 calculated the dissociation energy of CF+ using MRCI and obtained a significantly underestimated value (D0(CF+) = 6.73 eV) compared to those extracted from experimental studies. Later, Letelier and coworkers23 made the first step toward reconciliation of the experimental results with theory by using MRCI+Q/aug-cc-pV5Z calculations. They obtained a D0 value of 7.54 eV.
There is a remarkable paucity of photoelectron spectroscopic data available for SiF. No direct measurement of the adiabatic ionisation energy of SiF has been obtained, and the indirectly derived values span a rather large energy range. Using ion chemistry, one can estimate 7.54 ± 0.16 eV24 and 7.08 ± 0.10 eV,25 whereas extrapolation from Rydberg spectroscopy leads to 7.26 eV,26 7.31 eV,27 and 7.31 ± 0.02 eV.28 The two calculations of the adiabatic ionisation energy available in the literature report 7.36 eV at the coupled cluster single, double, and perturbative triple level and complete basis set extrapolation (CCSD(T)/CBS)29 and 7.41 eV at the G3(CC)/B2df+ level,30 although neither of the two is sufficiently accurate to confidently provide a value within chemical accuracy (with uncertainty less than ±1 kcal mol−1 or ±0.04 eV). However, the SiF+ structural information and vibrational properties in the electronic ground state have been precisely determined using microwave (re+ = 1.5264950(2) Å; ωe+ = 1050.7(2) cm−1)31 and IR spectroscopy (re+ = 1.5265(8) Å; ωe+ = 1050.3757(13) cm−1; ωexe+ = 4.9462(4) cm−1).32 Several values for the SiF+ dissociation energy have been extracted from experimental studies, D0(SiF+) = 6.3 eV,33 6.46 ± 0.14 eV,34 and 6.32 ± 0.11 eV,24 all of which are lower than the most accurate theoretical values obtained either using CCSD(T) with CBS extrapolation,35D0 = 6.92 ± 0.04 eV, the configuration-interaction method,36D0 = 6.60 eV, or MP4SDTQ,19D0 = 6.85 eV. It is worth noting that, while high-level calculations appear to underestimate the experimentally derived dissociation energy of CF+, they seem to overestimate that of SiF+.
This paper presents the threshold photoelectron and photoion yield spectra of SiF and CF radicals in the vicinity of their first ionisation threshold. The spectra are analysed with the support of ab initio calculations that aid the extraction of relevant spectroscopic parameters. The new experimental values were added to the thermochemical network underlying the Active Thermochemical Tables (ATcT) approach,37–39 thus producing further improved thermochemical parameters of SiF, CF and the corresponding cations.
Methodologies
Experimental details
The experiments were performed at the SOLEIL synchrotron facility, on the DESIRS beamline.40 The experimental setup combining the DELICIOUS3 spectrometer41,42 and the flow-tube reactor has been described elsewhere.43,44The experimental spectra recorded for CF and SiF have been obtained using the conditions described in ref. 45 and 46, respectively, in which CF and SiF were side products of the reaction of F with CH4 (or C2H4) or SiH4, respectively. The output of the flow-tube reactor was sampled through two consecutive skimmers before reaching the interaction region at the centre of DELICIOUS3. From CF+ and SiF+ photoion images, mean velocities were measured along the molecular beam at 915 and 1010 m s−1, corresponding to translational temperatures in the 200–220 K range.
In the present paper, only details concerning the resolutions of the presented spectra are summarised. The spectral resolutions of the ion yields are 0.72 Å (5.8 meV at 10 eV) for CF and 2.16 Å (17.4 meV at 10 eV) for SiF, which correspond to the photon spectral resolution. The final spectral resolutions δE of the slow photoelectron spectra (SPES) are estimated to be 14 meV at 9.5 eV and 24 meV at 7.5 eV for the CF and SiF spectra, respectively. The energy scale has been calibrated using the He third order and Si atomic transitions, with an energy scale uncertainty of ±3 meV for CF and ±6 meV for SiF, respectively. Note that the extraction field (F) used to extract the ions and the electrons (88.7 V cm−1) leads to a field-induced downshift of the ionisation energies of approximately 7 meV, following the well-established formula 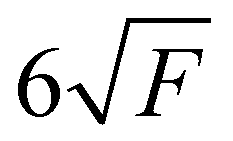 (in cm−1 with F in V cm−1).47
(in cm−1 with F in V cm−1).47
Although the universality of the Stark shift in polyatomic systems has been previously questioned,48,49 we have checked in our experiment using absorption lines from the gas filter (not subject to the Stark shift) and known adiabatic ionisation energies of molecules that the above-mentioned Stark shift applied within the error bars.
Calculation details
Supporting ab initio calculations on the electronic states of CF, CF+, SiF, and SiF+ were carried out using the internally contracted multireference configuration interaction method with Davidson correction (icMRCI+Q) with complete active space self-consistent field (CASSCF) wave-functions. The CASSCF and MRCI calculations were performed at full valence, i.e., 11 electrons for CF and SiF (10 for CF+ and SiF+). These electrons were distributed in 8 orbitals keeping the 1s orbital of carbon and fluorine atoms and the 1s, 2s, and 2p orbitals of silicon atom doubly occupied. All calculations were performed using the MOLPRO 2016 package.50 Single point calculations were performed at the aug-cc-pV nZ (n = T, Q, 5, and 6) basis set51 for the equilibrium geometries and anharmonic frequencies (using the DIATOMIC routine in MOLPRO where the potential curves were fitted with a polynomial of 8th order) allowing the spectroscopic constants to be calculated (ωe, ωexe, and ωeye) for the CF(X![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 2Π), CF+ (X+ 1Σ+), SiF(X 2Π) and SiF+ (X+ 1Σ+) states. CBS extrapolations were carried out using the aug-cc-pV nZ (n = T, Q, 5, and 6) basis set series. The CASSCF and dynamical correlation (EMRCI+Q − ECASSCF) energies were extrapolated using the ECASSCF(CBS) + A × exp(−Bn) and ECorr(CBS) + C × n−3 functions, respectively. The calculated energies reported in this paper correspond to ECASSCF(CBS) + Ecorr(CBS). The anharmonic constants (ωexe and ωeye) could not be confidently extrapolated to CBS because of their weak dependence on the basis used; instead, the values obtained with the largest basis set (aug-cc-pV6Z) were used. Our calculated values are reported in Tables 1 and 2 and discussed in the following section.
2Π), CF+ (X+ 1Σ+), SiF(X 2Π) and SiF+ (X+ 1Σ+) states. CBS extrapolations were carried out using the aug-cc-pV nZ (n = T, Q, 5, and 6) basis set series. The CASSCF and dynamical correlation (EMRCI+Q − ECASSCF) energies were extrapolated using the ECASSCF(CBS) + A × exp(−Bn) and ECorr(CBS) + C × n−3 functions, respectively. The calculated energies reported in this paper correspond to ECASSCF(CBS) + Ecorr(CBS). The anharmonic constants (ωexe and ωeye) could not be confidently extrapolated to CBS because of their weak dependence on the basis used; instead, the values obtained with the largest basis set (aug-cc-pV6Z) were used. Our calculated values are reported in Tables 1 and 2 and discussed in the following section.
| r e/Å | ω e/cm−1 | ω e x e/cm−1 | ω e y e/cm−1 | Ref. | |
|---|---|---|---|---|---|
| CF | 1.2731 | 1303.06 | 10.76 | 0.08 | Calc. |
| CF+ | 1.1558 | 1773.21 | 13.16 | 0.05 | Calc. |
| 1786.5 | 11.89 | Exp. | |||
| 1.154 | 1810 | 16 | |||
| 1.1542551 | 1792.6654 | 13.229668 | 18 | ||
| SiF | 1.6143 | 833.16 | 4.45 | 0.02 | Calc. |
| SiF+ | 1.5394 | 1024.44 | 4.69 | 0.01 | Calc. |
| 1056.8 | 5.17 | Exp. | |||
| 1.5264950 | 1050.7 | 31 | |||
| 1.52652 | 1050.3757 | 4.9462 | 32 |
Results
The first ionising transitions of CF and SiF arise from one-electron removal from the 2π orbital, which is the first anti-bonding (π*) orbital. This leads to a X+ 1Σ+ ← X 2Πr photoionising transition. Looking at the molecular orbital diagrams, one can assume that the ionisation process removes mainly one of the unpaired p-electrons on C (3P) and Si (3P) atoms for CF and SiF, respectively. In the analogous cases of SiH and CH, the p orbitals are rather weakly perturbed, as witnessed by the modest difference in the ionisation energies (IEs) of Si (8.15166 ± 0.00003 eV)52 and SiH (7.934 ± 0.005 eV),46 or those of C (11.260288 ± 0.000002 eV)53 and CH (10.640 ± 0.004 eV).54 In CF and SiF radicals, the interactions between the p-orbitals of C/Si atoms and the doubly occupied p-orbital of the fluorine atom are greater, which leads to a more pronounced decrease of IEs for CF (9.128 ± 0.006 eV) and SiF (7.381 ± 0.009 eV).Photoelectron and photoion yield spectra of CF
In Fig. 1, the mass-selected ion yield of CF is depicted on top of the photoelectron signal (PES) matrix, i.e. the coincidence signal of the electrons (resolved by kinetic energy) as a function of photon energy. The vertical lines (resonant with photon energy) observed above 11 eV correspond to autoionisation of neutral CF Rydberg states converging to several vibronic states of CF+.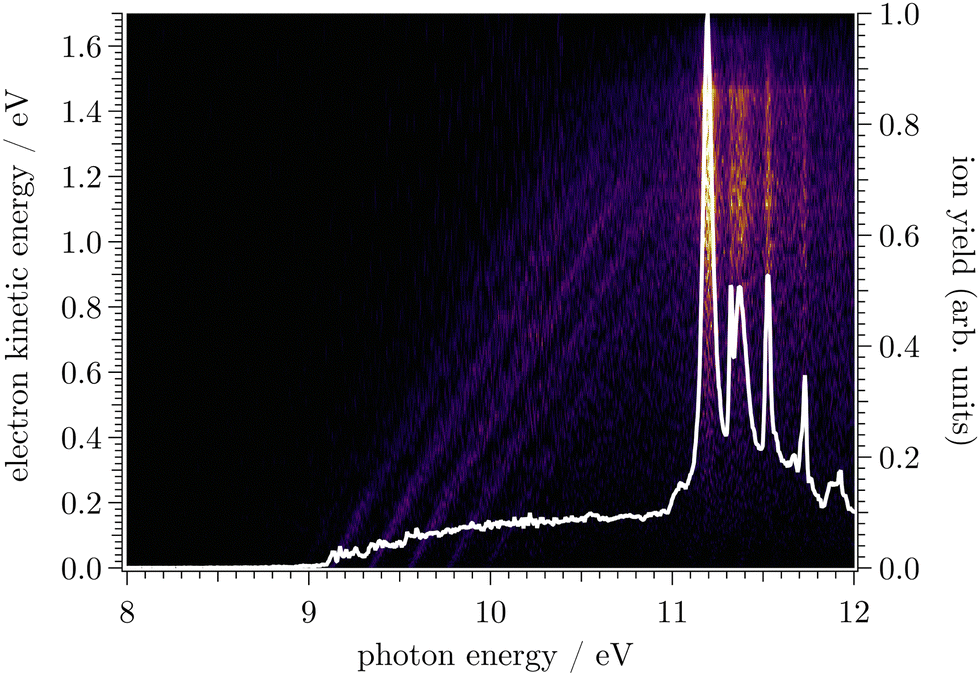 | ||
| Fig. 1 Mass-selected photoelectron signal of CF as a function of the photon energy (horizontal axis) and electron kinetic energy (vertical axis) for m/z 31. The corresponding total ion yield obtained by integrating over all electron energies is represented by the white curve. | ||
In Fig. 2, the corresponding CF threshold photoelectron spectrum is displayed in black and compared with our harmonic Franck–Condon (FC) simulation in red. The experimental spectrum exhibits a progression up to v+ = 6, as well as one hot band around 9 eV arising from the v+ = 0 ← v = 1 transition. While the agreement is satisfactory at low energy, the calculated spectrum starts to progressively deviate from the experimental spectrum above 9.75 eV due to the absence of the anharmonic component in the simulation. Based on the vibrational FC simulation, the origin band is unambiguously assigned. From the prominent vibrational progression, one can extract the harmonic frequency ωe+ and the corresponding anharmonic constant ωexe+. Fig. 3 shows a plot of the observed peak apexes as a function of ( ). The obtained data set was fitted using a second-order polynomial expression. Note that, owing to the aforementioned autoionisation processes, the transition to v+ = 11 was observed, from which the quality of the fit and thus of evaluation of the anharmonicity was greatly improved. Indeed, autoionisation resonances allow sampling of the molecular potential beyond the Franck–Condon region that is not accessible by one-photon direct ionisation.55 The fitted spectroscopic constants are ωe+ = 1786.5 ± 5.5 cm−1 and ωexe+ = 11.89 ± 0.44 cm−1, in reasonable agreement with the very accurate constants obtained by Saykally,18 as well as with the frequency of 1810 ± 30 cm−1 reported by Dyke et al.16 (see Table 1).
). The obtained data set was fitted using a second-order polynomial expression. Note that, owing to the aforementioned autoionisation processes, the transition to v+ = 11 was observed, from which the quality of the fit and thus of evaluation of the anharmonicity was greatly improved. Indeed, autoionisation resonances allow sampling of the molecular potential beyond the Franck–Condon region that is not accessible by one-photon direct ionisation.55 The fitted spectroscopic constants are ωe+ = 1786.5 ± 5.5 cm−1 and ωexe+ = 11.89 ± 0.44 cm−1, in reasonable agreement with the very accurate constants obtained by Saykally,18 as well as with the frequency of 1810 ± 30 cm−1 reported by Dyke et al.16 (see Table 1).
 | ||
| Fig. 2 Experimental threshold photoelectron spectrum of CF in the vicinity of the X+ 1Σ+ ← X 2Π photoionising transition (in black) compared with our harmonic Franck–Condon calculation convolved with a Gaussian line shape (FWHM = 26 meV) at a vibrational temperature of 600 K (in red). The red assignment corresponds to our harmonic calculation whereas the black comb follows the anharmonicity which allows an unambiguous attribution of the ionising transition towards v+ = 11 (observed through autoionisation). See the text for details. | ||
 | ||
Fig. 3 Energies of the lowest vibrational levels of the electronic ground state of CF+ with respect to its v+ = 0 level as a function of ( ). The red crosses represent our harmonic calculations and the black circles our measurements. The blue line corresponds to a second-order polynomial fit of our experimental values. See the text for details. ). The red crosses represent our harmonic calculations and the black circles our measurements. The blue line corresponds to a second-order polynomial fit of our experimental values. See the text for details. | ||
The apex position of the origin band in Fig. 2 can be determined as 9.121 eV, which, after a correction for the Stark shift, becomes 9.128 ± 0.004 eV. It is important to note here that the adiabatic ionisation energy, which is a thermodynamically-relevant quantity, by definition corresponds to the energy difference between the lowest actually existing rovibronic level of the neutral and that of the ion.56 (Mutatis mutandis, the same is true for all thermodynamically-relevant quantities, such as dissociation energies and other reaction energies at 0 K.56) Thus, in CF the adiabatic ionisation energy corresponds to the X+ 1Σ+(v+ = 0, J+ = 0) ← X 2Π1/2(v = 0, J = 1/2) transition.
In regular photoelectron spectroscopy, when individual rotational transitions are not resolved, the standard practice is to report the apex of the vibrational peak that was identified as the v+ = 0 ← v = 0 transition as the adiabatic ionisation energy. This tacitly implies the assumption that the underlying rotational envelope is not shaded either to the red or to the blue, i.e. that the Q branch is rather compact and that the lateral branches (P and R, as well as higher branches, such as O and S, if they happen to be allowed) are symmetric, which may or may not be correct.
However, the nature of the ground state of neutral CF introduces an additional complication. Namely, X 2Πr undergoes spin–orbit coupling (as quantified by the corresponding coupling constant57,58A0 = 77.11 ± 0.01 cm−1), with 2Π1/2 as the lower and 2Π3/2 as the higher component. Consequently, the v+ = 0 ← v = 0 spectral peak in Fig. 2 almost certainly contains an unresolved hot band originating from the upper 2Π3/2 component of the neutral. Prima facie, one would expect that the presence of such an unresolved hot band will cause a shift of the apparent apex to the red, the amount of which depends, inter alia, on the thermal population of the additional component.
To that end, an approximate simulation of the rotational envelope has been performed to estimate the potential shift of the peak apex relative to the adiabatic ionisation energy. While the energy terms of 1Σ+ rotational levels are straightforward, the rotational levels of the 2Π state were calculated using the Hamiltonian that correctly describes the coupling of the spin with the orbital and the rotational angular momenta, as appropriate for partial spin uncoupling pertinent to a transition from Hund's case (a) to case (b),59,60 following the elegantly re-derived formulation by Hougen.61 The spectroscopic constants of CF and CF+ were taken from ref. 58 and 17 respectively. With these, the Hamiltonian for the X 2Πr state of CF produces the term values of F1 = −37.15 cm−1 for 2Π1/2, J = 1/2 and F2 = 41.46 cm−1 for 2Π3/2, J = 3/2. Notably, the lowest rotational levels of each of the two spin–orbit components of 2Πr of CF appear to be almost symmetrically split by roughly ±A0/2 below and above the reference level (vibrationless/rotationless energy level before any spin–orbit consideration). This is in rather stark contrast to the recently discussed situation of spin–orbit-rotation coupling in the otherwise analogous case of X 2Πr of CH,62 where both F1 = 0.15 cm−1 and F2 = 17.97 cm−1 are positive quantities, or, for that matter, to the quite asymmetric splitting in the well-known case of X 2Πi of OH,63–65 neither of which conforms to a symmetric ±A0/2 splitting of the spin–orbit components that is frequently presumed in theoretical calculations.
For the spectral simulation of the rotational envelope, the rotational temperature of 200 K was used as it corresponds to the translational temperature of the beam at the output of the flowtube reactor (see the “Experimental details” subsection). The simulated envelope strongly suggests that the adiabatic ionisation energy (X+ 1Σ+(v+ = 0, J+ = 0) ← X 2Π1/2(v = 0, J = 1/2)) is, in fact, quite close to the actual maximum of the observed band, primarily because of two opposing tendencies: the unresolved hot band originating from X 2Π3/2 indeed tends to shift the apex of the experimental peak slightly toward the red, but this is nearly entirely counteracted by the shading toward the blue of the rotational envelope belonging to transitions that originate from X 2Π1/2.
After the field-induced Stark-shift correction of 7 meV and an estimation of the uncertainty from our rotational contour simulation, the resulting adiabatic ionisation energy is IEad(CF) = 9.128 ± 0.006 eV. This can be compared to the value of 9.120 ± 0.005 eV obtained from the publicly available version of ATcT (ver. 1.124).39 A variance decomposition analysis of the provenance66 indicates that the two top contributors to the ATcT value are the experimental ionisation energy of 9.11 ± 0.01 eV determined by Dyke et al.15 and the difference between the adiabatic ionisation energies of CF3 and CF of 0.055 ± 0.03 eV determined by Asher and Ruscic.67 It is also of note that our calculated adiabatic ionisation energy, 9.059 eV (see Table 2), slightly underestimates the experimental value (by 0.07 eV), but is otherwise in quite reasonable agreement with the experiment, particularly if one considers that the species in question is fluorinated.
Photoelectron and photoion yield spectra of SiF
Similarly as for CF, Fig. 4 represents the mass-selected ion yield of SiF superimposed on the PES matrix, where autoionisation features are observed. Fig. 5 displays the threshold photoelectron spectrum (black trace) and its harmonic FC simulation (red trace). The spectrum consists of one hot band, a progression in SiF vibration from the origin band to v+ = 3, followed at higher energy by a long high v+ series from 20 to 25 caused by resonant autoionisation processes. From our ab initio calculations, we checked that no other electronic states of SiF+ were located in this energy region. Indeed, the first triplet (3Π) and singlet (1Σ+) states are located around 11.83 eV and 13.71 eV above the neutral ground state, respectively. The harmonic FC calculation reproduces satisfactorily the experimental spectrum. The observed energies in the vibrational progression are plotted as a function of ( ) in Fig. 6. The blue line results from a second-order polynomial fit, leading to ωe+ = 1056.8 ± 5.2 cm−1 and ωexe+ = 5.17 ± 0.20 cm−1 in good agreement with the literature values, see Table 1. As in the case of CF, the inclusion of high v+ states accessible via autoionisation processes significantly improves the obtained fit.
) in Fig. 6. The blue line results from a second-order polynomial fit, leading to ωe+ = 1056.8 ± 5.2 cm−1 and ωexe+ = 5.17 ± 0.20 cm−1 in good agreement with the literature values, see Table 1. As in the case of CF, the inclusion of high v+ states accessible via autoionisation processes significantly improves the obtained fit.
 | ||
| Fig. 4 Mass-selected photoelectron signal of SiF as a function of the photon energy (horizontal axis) and electron kinetic energy (vertical axis) for m/z 47. The corresponding total ion yield obtained by integrating over all electron energies is represented by the white curve. | ||
 | ||
| Fig. 5 Experimental threshold photoelectron spectrum of SiF in the vicinity of the X+ 1Σ+ ← X 2Π photoionising transition (in black) compared with our harmonic Franck–Condon calculation convolved with a Gaussian line shape (FWHM = 40 meV) at a vibrational temperature of 600 K (in red). The above red assignment corresponds to our harmonic calculation whereas the black comb follows the anharmonicity which allows an unambiguous attribution of the ionising transitions towards v+ = 20–25 (observed through autoionisation). See the text for details. | ||
 | ||
Fig. 6 Energies of the lowest vibrational levels of the electronic ground state of SiF+ with respect to its v+ = 0 level as a function of ( ). The red crosses represent our harmonic calculations and the black circles our measurements. The blue line corresponds to a second-order polynomial fit of our experimental values. ). The red crosses represent our harmonic calculations and the black circles our measurements. The blue line corresponds to a second-order polynomial fit of our experimental values. | ||
From our measurement displayed in Fig. 5, we can extract the band maximum of the origin band of the SiF photolectron spectrum to be 7.372 ± 0.007 eV. Similarly to the case of CF, the rotational envelope was estimated using the spectroscopic constants for SiF and SiF+ from ref. 68 and 31 respectively. Once again, it was found that the adiabatic ionisation energy (X+ 1Σ+(v+ = 0, J+ = 0) ← X 2Π1/2(v = 0, J = 1/2)) is very close to the band maximum, for essentially the same reasons as in the case of CF. The adiabatic ionisation energy is obtained after correcting for field-induced Stark shift (+7 meV) and estimating the uncertainty in the position of the apex of the vibrational peak from our rotational contour simulation. The corresponding final value of the adiabatic ionisation energy IEad(SiF) = 7.379 ± 0.009 eV. Again, our calculated adiabatic ionisation energy, 7.337 eV (see Table 2), slightly underestimates the experimental value (by 0.04 eV), but is otherwise in reasonable agreement.
Thermochemical considerations
The usual approach to developing new or improved thermodynamic quantities in studies like the present one is to use traditional sequential thermochemistry (A begets B, B begets C, etc.). For instance, since the adiabatic ionisation energy defines the thermodynamic difference between the 0 K enthalpies of formation of the cation and the neutral, the presently determined IEad(CF) = 9.128 ± 0.006 eV can be combined with the 0 K enthalpy of formation of CF, such as that currently available from ATcT,39 ΔfH0°(CF) = 58.107 ± 0.029 kcal mol−1, to derive ΔfH0°(CF+) = ΔfH0°(CF) + IEad(CF) = 268.60 ± 0.14 kcal mol−1 (which can be in turn compared to the 0 K value for the ion of 268.42 ± 0.11 kcal mol−1 available directly from ATcT39; notably, the corresponding IEad(CF), as reported by ATcT, is 9.116 ± 0.005 eV, the difference from the current experiment being just about contained – within a round-off error – by the combined uncertainties). After setting the 0 K enthalpies of formation, the corresponding 298.15 K values can be derived as ΔfH298°(CF) = 58.968 ± 0.029 kcal mol−1 and ΔfH298°(CF+) = 269.37 ± 0.14 kcal mol−1 (the latter within the stationary electron convention56) with the aid of the 298.15 K enthalpy increments of CF (2.167 kcal mol−1),69 CF+ (2.074 kcal mol−1),39 graphite (0.251 kcal mol−1),70 and F2 (2.109 kcal mol−1).70Going further with the sequential thermochemistry approach, one can take the 0 K dissociation energy of CF, D0(CF) = 5.654 ± 0.002 eV from ATcT39 and combine it with the currently determined IEad(CF) and the accurately known IE(C)53 to obtain D0(CF+) = D0(CF) + IE(C) − IEad(CF) = 7.786 ± 0.007 eV (which can again be compared to the publicly available ATcT value of 7.794 ± 0.005 eV).39 The corresponding 298.15 K bond dissociation enthalpies are BDE298(CF) = 5.695 ± 0.002 eV and BDE298(CF+) = 7.833 ± 0.007 eV (after utilising the aforementioned enthalpy increments for CF and CF+ together with those of the C atom, C+ ion, and F atom, of 1.562, 1.589, and 1.558 kcal mol−1, respectively).69
However, the derivations delineated above are entirely dependent not only on the accuracy of the newly determined IEad(CF), but also on the tacit assumption that auxiliary thermochemical quantities, in this case ΔfH0°(CF) and D0(CF), have been a priori firmly established, and that thus the new IEad(CF) leads simply to a revision of ΔfH0°(CF+) and D0(CF+). In this particular case the putative robustness of the available values for ΔfH0°(CF) and D0(CF) does not appear unreasonable, given that the provenances of the ATcT values include, inter alia, state-of-the-art electronic structure computations capable of sub-kJ mol−1 accuracies, such as several flavors of W4,71–74 HEAT,65,75 and FPD,76 all of which appear mutually entirely consistent.
Nonetheless, in a general case the assumptive premise of robustness of the auxiliary thermochemistry may or may not be warranted. In fact, the new measurement may very well also imply the need for revisions or adjustments of the auxiliary thermochemistry. One of the additional potential problems of sequential thermochemistry is that while it tends to produce an improvement in the derived thermodynamic quantity of the targeted chemical species, it also frequently introduces new inconsistencies with previously established thermochemistry of other related species. Namely, their thermochemistry may have been obtained via sequential derivations that were pegged to the old value of the just revised species and should therefore be also revised: an essentially impossible task, since in tabulations obtained by sequential thermochemistry the intricate progenitor-progeny relationships are concealed. In addition, by virtue of pairing down competitive determinations by selecting what subjectively appears to be the “best” determination, traditional sequential thermochemistry chronically under-utilises the available thermochemically-relevant knowledge.
In fact, while in the case of CF and CF+ the situation is relatively benign, there being available thermochemical values for CF that arguably appear reliable, things are veritably more complicated in the case of SiF and SiF+. Namely, the values for the enthalpy of formation of SiF available from the literature24,25,29,69,71,77–88 encompass a spectacularly wide range, differing on the extremes by more than 16 kcal mol−1, as demonstrated in Table 3. In principle, one might venture assuming that the values reported from the three most accurate electronic structure approaches71,81,82 are likely the most reliable. However, all three enthalpies of formation of SiF were derived from computations of the dissociation energy D0(SiF) and thus directly depend on the selected enthalpy of formation of Si atom. Notably, the recent introduction of key Si-containing chemical species in ATcT39 has produced a significantly revised (and improved) value ΔfH0°(Si) = 107.63 ± 0.14 kcal mol−1, which can be compared with the generally accepted, but 1 kcal mol−1 lower (and substantially less accurate) value of 106.5 ± 1.9 kcal mol−1 derived by Gurvich et al.,69 recommended by CODATA,70 and used in JANAF88 and other tabulations. The older (and higher) value of 107.6 ± 0.1 kcal mol−1 found in the NBS Tables86 is, in retrospect, in excellent agreement with the new ATcT value, as is the revised value of 107.2 ± 0.2 kcal mol−1 recently proposed by Karton and Martin.71
| ΔfH0°(SiF) | ΔfH298°(SiF) | Ref. |
|---|---|---|
| −14.2 ± 2.0 | 78 (theory) | |
| −12.70 ± 3.0 | −12.42 ± 3.0 | 79 (theory) |
| −5 ± 3 | 24 (exp) | |
| −11.7 ± 2.1 | −11.2 ± 2.1 | 25 (exp) |
| −13.4 | 80 (theory) | |
| −14.0 ± 2.0 | 29, 77 (theory) | |
| −14.8 ± 0.4 | 81 (theory) | |
| −14.9 ± 0.3 | 71 (theory) | |
| −13.90 ± 0.35 | 82 (theory) | |
| −14.79 | −14.36 | 83 (theory) |
| −15.25 ± 2.00 | −14.95 ± 2.00 | 84 (theory) |
| −14.4 | 85 (theory) | |
| 1.0 ± 1.0 | 1.7 ± 1.0 | 86 (eval) |
| −5 ± 6 | 87 (eval) | |
| −5.2 ± 3.0 | −4.8 ± 3.0 | 88 (eval) |
| −6.5 ± 2.6 | −6.0 ± 2.6 | 69(eval) |
As opposed to the sequential thermochemistry approach, Active Thermochemical Tables rely on a different paradigm, rooted in constructing, statistically analysing, and solving a thermochemical network (TN) that contains all available determinations relevant to the thermochemistry of the included species, irrespective of whether they originate from experiment or high-level electronic structure theory.37,38,89
The current public version of ATcT (ATcT TN ver. 1.124),39 which contains nearly 2800 chemical species, has been developed within the context of several studies, such as the report on thermophysical and thermochemical properties of CH2 and CH3 using nonrigid rotor anharmonic oscillator (NRRAO) partition functions,90 the development and benchmarking of a state-of-the-art computational approach that aims to reproduce total atomisation energies of small molecules within 10–15 cm−1,91 as well as the study of the reversible reaction C2H3 + H2 ⇄ C2H4 + H ⇄ C2H5.92
Since then, the underlying ATcT TN has been successively expanded to fulfil the needs of subsequent studies, such as those involving enthalpies of formation of key bromo- and iodo-methanes, ethenes and ethynes,93 the ring-opening dynamics of cyclopropyl and cyclopropylium (ATcT TN versions 1.128 and 1.130),94 the bond dissociation energy and the adiabatic ionisation energy of CH (ATcT TN ver. 1.140),62 the role of methanediol in the atmosphere (ATcT ver. 1.148),95 as well as a number of ongoing studies, eventually leading to the current developmental version ATcT TN 1.156. The latter contains more than 3150 species interconnected by over 30000 thermochemically relevant determinations, but does not (yet) include the present experimental adiabatic ionisation energies.
Although ATcT TN ver. 1.156 includes more than 350 new species and several thousand new determinations (a few of which do include CF or CF+ as reactant or product), the thermochemistry of CF and CF+ has remained nearly the same: ΔfH0°(CF) = 58.124 ± 0.026 kcal mol−1 and ΔfH0°(CF+) = 268.41 ± 0.11 kcal mol−1, the differences in comparison with the already discussed ATcT TN ver. 1.12439 (less than 0.02 kcal mol−1) comfortably contained within the associated (and slightly tighter) uncertainties. The resulting IEad(CF) = 9.119 ± 0.005 eV is in agreement (within the combined uncertainties) with the current direct experimental value of IEad(CF) = 9.128 ± 0.006 eV.
However, of particular relevance to the current study is that this version includes the SiFn, n = 1–4 group of species and their ions. Thus, ATcT TN ver. 1.156 produces ΔfH0°(SiF) = −14.40 ± 0.15 kcal mol−1 and ΔfH298°(SiF) = −13.97 ± 0.15 kcal mol−1 (where the 0 to 298.15 conversion exploits the 298.15 K enthalpy increments of SiF, 2.261 kcal mol−1 – slightly updated from Gurvich et al.69 using the NASA PAC program96 and spectroscopic constants from Huber and Herzberg,57 crystal silicon, 0.769 kcal mol−1,70 and 1/2 of the already mentioned enthalpy increment for F2, 2.109 kcal mol−1).70 The matching 0 K dissociation energy of SiF is D0(SiF) = 6.093 ± 0.005 eV, and the corresponding BDE298(SiF) = 6.140 ± 0.005 eV. We note here that the ATcT value for D0(SiF) is significantly influenced by highly accurate state-of-the-art W471 and FPD81 calculations of the same quantity extant in the ATcT TN, and that the corresponding ΔfH0°(SiF) makes use of the newly revised enthalpy of formation for gas-phase silicon atom, as already mentioned earlier, and which in this version of ATcT results amounts to ΔfH0°(Si) = 107.63 ± 0.14 kcal mol−1.
For the cation, ATcT TN ver. 1.156 produces ΔfH0°(SiF+) = 154.91 ± 0.39 kcal mol−1 and ΔfH298°(SiF+) = 155.18 ± 0.39 kcal mol−1 (where the conversion peruses the 298.15 K enthalpy increment of SiF+ of 2.094 kcal mol−1, as obtained from available spectroscopic constants31,32,57 by using the NASA PAC program96). A variance decomposition analysis indicates that the primary contributors to ΔfH°(SiF+) are the literature values for IEad(SiF) included in the ATcT TN, combined with ΔfH°(SiF), and only to a minor degree the theoretical D0(SiF+), such as 159.58 (±1.5, estimated) kcal mol−1.77
Noting again that ATcT TN ver. 1.156 does not (yet) include the current experimental ionisation energies of CF and SiF, it is interesting to inspect the difference in ATcT 0 K enthalpies of formation of SiF+ and SiF, which implies an adiabatic ionisation energy of SiF of 7.342 ± 0.016 eV. Curiously, while standard in-house mid-level composite methods extant in the TN suggest adiabatic ionisation energies ranging between 7.31 ± 0.10 eV (CBS-QB3) and 7.41 ± 0.08 eV (G4), with 7.36 ± 0.04 eV (W1) perhaps as a midpoint, the resulting ionisation energy is pushed toward the lower end of this range by the ostensibly accurate experimental value of 7.310 ± 0.011 eV27,28 extracted from VUV absorption spectra, in spite of the fact that, based on a critical evaluation of those studies, the uncertainty initially assigned (a.k.a. ‘prior’) in the TN to this determination was double the uncertainty declared by the authors, and that the subsequent ATcT statistical analysis, which evaluates the internal consistency of the determinations in the TN, has further increased its uncertainty to ±0.032 eV.
Clearly, the above IEad of 7.342 ± 0.016 eV is appreciably less accurate than the current experimental IEad(SiF) = 7.379 ± 0.009 eV, and it also appears inconsistent, given that the difference (0.037 eV) is significantly larger than the combined uncertainties. The normal course of action within the ATcT approach is to add the new results to the TN, and let the ATcT statistical analysis arbitrate between potentially inconsistent determinations.
The addition of the two experimental ionisation energies creates a new version, ATcT TN 1.158. The resulting thermochemical values of relevance to the present study are given in Table 4.
| Quantity | 0 K | 298.15 K | Uncert. | Units |
|---|---|---|---|---|
| a Current experimental adiabatic ionisation energy. b BDE0(AB) is synonymous with D0(AB). c Recently revised value from ATcT, significantly improved over the CODATA70 value of ΔfH0°(Si) = 106.5 ± 1.9 kcal mol−1 and ΔfH298°(Si) = 107.6 ± 1.9 kcal mol−1; see the text. | ||||
| ΔfHT°(CF) | 58.123 | 58.984 | ±0.026 | kcal mol−1 |
| ΔfHT°(CF+) | 268.522 | 269.290 | ±0.089 | kcal mol−1 |
| IEad(CF) | 9.124 | ±0.004 | eV | |
| (9.128)a | (±0.006)a | eV | ||
| BDET(CF)b | 5.653 | 5.695 | ±0.001 | eV |
| BDET(CF+)b | 7.790 | 7.836 | ±0.004 | eV |
| ΔfHT°(SiF) | −14.42 | −13.98 | ±0.15 | kcal mol−1 |
| ΔfHT°(SiF+) | 155.62 | 155.89 | ±0.24 | kcal mol−1 |
| IEad(SiF) | 7.373 | ±0.008 | eV | |
| (7.379)a | (±0.009)a | eV | ||
| BDET(SiF)b | 6.093 | 6.141 | ±0.005 | eV |
| BDET(SiF+)b | 6.872 | 6.924 | ±0.009 | eV |
| ΔfH0°(C) | 170.029 | 171.340 | ±0.010 | kcal mol−1 |
| ΔfH0°(C+) | 429.697 | 431.035 | ±0.010 | kcal mol−1 |
| ΔfH0°(F) | 18.464 | 18.967 | ±0.004 | kcal mol−1 |
| ΔfH0°(Si)c | 107.63 | 108.67 | ±0.14 | kcal mol−1 |
| ΔfH0°(Si+) | 295.62 | 296.60 | ±0.14 | kcal mol−1 |
While the introduction of IEad(CF) has not particularly affected the thermochemistry of neutral CF, with ΔfH°(CF) and D0(CF) remaining essentially identical, it does affect and improve the thermochemistry of CF+, adjusting ΔfH0°(CF+) upward (by 0.113 kcal mol−1) and becoming slightly more accurate (±0.089 kcal mol−1, cf. to earlier ±0.11 kcal mol−1). Importantly, the updated ATcT thermochemistry of the ion corresponds to D0(CF+) = 7.790 ± 0.004 eV and to IEad(CF) = 9.124 ± 0.004 eV, the latter being in excellent agreement with the very slightly higher experimental value of 9.128 ± 0.006 eV.
As one might have expected, the introduction of the experimental IEad(SiF) into the ATcT TN has a more pronounced effect on the relevant species than the introduction of IEad(CF). Also not surprisingly, the thermochemistry of neutral SiF is affected less than that of the ion, given that the former is constrained by D0(SiF) obtained from high-level electronic structure computations.71,81 Thus, D0(SiF) = 6.093 ± 0.005 eV, corresponding to ΔfH0°(SiF) = −14.42 ± 0.15 kcal mol−1, which is only marginally different than the value of −14.40 ± 0.15 kcal mol−1 from the preceding version of ATcT TN. However, the new and improved value for ΔfH0°(SiF+) is now 155.62 ± 0.24 kcal mol−1, rather significantly different than the value of 154.91 ± 0.39 kcal mol−1 from the previous version. The new thermochemistry of SiF+ corresponds to D0(SiF+) = 6.872 ± 0.009 eV and to IEad(SiF) = 7.373 ± 0.008 eV, in superb agreement with the experimental value of 7.379 ± 0.009 eV. For reference purposes, Table 4 also includes the current ATcT values for the constituent atoms. Enthalpies of formation of atoms were considered as ‘key’ quantities already by CODATA,70 but since then they have gained additional importance, because they are needed to convert theoretically computed total atomisation energies to practical enthalpies of formation. While the genesis of the ATcT revision of the enthalpy of formation of C atom was discussed elsewhere,97 the new and improved ATcT enthalpy of formation of Si atom is formally given here for the first time.
Conclusion
In this study we presented photoelectron spectroscopic measurements of the first two group 14 monofluorides, CF and SiF, together with supporting quantum chemical calculations, further amplified by utilising the new experimental ionisation energies in conjunction with the Active Thermochemical Tables approach. The latter combination results in significant refinements of the related thermochemical properties, particularly those of SiF+, as well as those of CF+. The recommended new enthalpies of formation and dissociation energies of the neutral CF and SiF and their cationic counterparts, together with the resulting adiabatic ionisation energies, are summarised in Table 4.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was performed on the DESIRS Beamline at SOLEIL synchrotron under project numbers 20200996 and 20140832. The authors are grateful to the whole staff of SOLEIL for running the facility. The work at Argonne National Laboratory (B.R.) was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Chemical Sciences, Geosciences and Biosciences Division, under Contract No. DE-AC02-06CH11357, through the Gas-Phase Chemical Physics Program. Ning L. Chen acknowledges the support from the Paris Ile-de-France Region (DIM ACAV+) for her PhD grant. This work has also received financial support from the French “Agence Nationale de la Recherche” (ANR) under Grant No. ANR- 12-BS08-0020-02 (Project SYNCHROKIN). This work was supported by the Programme National “Physique et Chimie du Milieu Interstellaire” (PCMI) of CNRS/INSU with INC/INP cofunded by CEA and CNES. The Active Thermochemical Tables are a U.S. Department of Energy Office of Science Public Reusable Research (DOE SC PuRe) Data Resource.98References
- P. Ho, J. E. Johannes, R. J. Buss and E. Meeks, J. Vac. Sci. Technol., A, 2001, 19, 2344–2367 CrossRef CAS.
- K. Williams and E. Fisher, J. Vac. Sci. Technol., A, 2003, 21, 1024–1032 CrossRef CAS.
- J. M. Stillahn, K. J. Trevino and E. R. Fisher, Annu. Rev. Anal. Chem., 2008, 1, 261–291 CrossRef CAS PubMed.
- D. A. Neufeld, P. Schilke, K. M. Menten, M. G. Wolfire, J. H. Black, F. Schuller, H. S. Müller, S. Thorwirth, R. Güsten and S. Philipp, Astron. Astrophys., 2006, 454, L37–L40 CrossRef CAS.
- H. Liszt, J. Pety, M. Gerin and R. Lucas, Astron. Astrophys., 2014, 564, A64 CrossRef.
- H. Liszt, V. Guzmán, J. Pety, M. Gerin, D. Neufeld and P. Gratier, Astron. Astrophys., 2015, 579, A12 CrossRef.
- S. Muller, K. Kawaguchi, J. Black and T. Amano, Astron. Astrophys., 2016, 589, L5 CrossRef.
- D. A. Neufeld, M. G. Wolfire and P. Schilke, Astrophys. J., 2005, 628, 260 CrossRef CAS.
- K. Acharyya and E. Herbst, Astrophys. J., 2017, 850, 105 CrossRef.
- L. A. Curtiss, K. Raghavachari, P. C. Redfern and J. A. Pople, J. Chem. Phys., 1997, 106, 1063–1079 CrossRef CAS.
- B. Ruscic, J. V. Michael, P. C. Redfern, L. A. Curtiss and K. Raghavachari, J. Phys. Chem. A, 1998, 102, 10889–10899 CrossRef CAS.
- Á. Ganyecz, M. Kállay and J. Csontos, J. Phys. Chem. A, 2018, 122, 5993–6006 CrossRef PubMed.
- E. Paulechka and A. Kazakov, J. Chem. Eng. Data, 2019, 64, 4863–4874 CrossRef CAS PubMed.
- Y. Si, Y. Liu, W. Lai, Y. Ma, J. Shi, B. Wang, M. Liu and T. Yu, Adv. Theory Simul., 2022, 2200093 CrossRef CAS.
- J. Dyke, A. Lewis and A. Morris, J. Chem. Phys., 1984, 80, 1382–1386 CrossRef CAS.
- J. Dyke, N. Hooper and A. Morris, J. Electron Spectrosc. Relat. Phenom., 2001, 119, 49–56 CrossRef CAS.
- K. Kawaguchi and E. Hirota, J. Chem. Phys., 1985, 83, 1437–1439 CrossRef CAS.
- M. Gruebele, M. Polak and R. J. Saykally, Chem. Phys. Lett., 1986, 125, 165–169 CrossRef CAS.
- K. A. Peterson, R. C. Woods, P. Rosmus and H.-J. Werner, J. Chem. Phys., 1990, 93, 1889–1894 CrossRef CAS.
- I. D. Petsalakis and G. Theodorakopoulos, Chem. Phys., 2000, 254, 181–186 CrossRef CAS.
- I. D. Petsalakis and G. Theodorakopoulos, Chem. Phys. Lett., 2011, 508, 17–21 CrossRef CAS.
- D. Hildenbrand, Chem. Phys. Lett., 1975, 32, 523–526 CrossRef CAS.
- N. Inostroza, J. Letelier, M. L. Senent and P. Fuentealba, Spectrochim. Acta, Part A, 2008, 71, 798–802 CrossRef CAS PubMed.
- M. Weber and P. Armentrout, J. Chem. Phys., 1988, 88, 6898–6910 CrossRef CAS.
- E. R. Fisher, B. L. Kickel and P. Armentrout, J. Phys. Chem., 1993, 97, 10204–10210 CrossRef CAS.
- J. Johns and R. Barrow, Proc. Phys. Soc., 1958, 71, 476 CrossRef CAS.
- F. Remy, E. Mahieu, D. Macau-Hercot, I. Dubois, H. Bredohl, J. Breton, F. Launay and M. Benharrous, J. Mol. Spectrosc., 1992, 152, 131–136 CrossRef CAS.
- H. Bredohl, J. Breton, I. Dubois, J. Esteva, D. Macau-Hercot and F. Remy, J. Mol. Spectrosc., 1999, 195, 281–283 CrossRef CAS PubMed.
- A. Ricca and C. W. Bauschlicher, J. Phys. Chem. A, 1998, 102, 876–880 CrossRef CAS.
- L. Wang and Y.-L. He, Int. J. Mass Spectrom., 2008, 276, 56–76 CrossRef CAS.
- R. H. Petrmichl, K. A. Peterson and R. C. Woods, J. Chem. Phys., 1988, 89, 5454–5459 CrossRef CAS.
- Y. Akiyama, K. Tanaka and T. Tanaka, Chem. Phys. Lett., 1989, 155, 15–20 CrossRef CAS.
- P. Wilkinson, Astrophys. J., 1963, 138, 778 CrossRef CAS.
- R. Reddy, T. Rao and R. Viswanath, Astrophys. Space Sci., 1992, 189, 29–38 CrossRef CAS.
- A. Ricca and C. W. Bauschlicher Jr, Chem. Phys. Lett., 1998, 287, 239–242 CrossRef CAS.
- S. Karna and F. Grein, J. Mol. Spectrosc., 1987, 122, 28–40 CrossRef CAS.
- B. Ruscic, R. E. Pinzon, M. L. Morton, G. von Laszewski, S. J. Bittner, S. G. Nijsure, K. A. Amin, M. Minkoff and A. F. Wagner, J. Phys. Chem. A, 2004, 108, 9979–9997 CrossRef CAS.
- B. Ruscic, R. E. Pinzon, G. von Laszewski, D. Kodeboyina, A. Burcat, D. Leahy, D. Montoya and A. F. Wagner, J. Phys. Conf. Ser., 2005, 16, 561–570 CrossRef CAS.
- B. Ruscic and D. H. Bross, Active Thermochemical Tables (ATcT) values based on ver. 1.124 of the Thermochemical Network, Argonne National Laboratory, Lemont, Ill. 2022, https://atct.anl.gov/, accessed August 2023.
- L. Nahon, N. D. Oliveira, G. A. Garcia, J. F. Gil, B. Pilette, O. Marcouillé, B. Lagarde and F. Polack, J. Synchrotron Radiat., 2012, 19, 508–520 CrossRef CAS PubMed.
- G. A. Garcia, B. K. de Miranda, M. Tia, S. Daly and L. Nahon, Rev. Sci. Instrum., 2013, 84, 053112 CrossRef CAS PubMed.
- X. Tang, G. A. Garcia, J.-F. Gil and L. Nahon, Rev. Sci. Instrum., 2015, 86, 123108 CrossRef PubMed.
- G. A. Garcia, X. Tang, J.-F. Gil, L. Nahon, M. Ward, S. Batut, C. Fittschen, C. A. Taatjes, D. L. Osborn and J.-C. Loison, J. Chem. Phys., 2015, 142, 164201 CrossRef PubMed.
- G. A. Garcia, B. Gans, X. Tang, M. Ward, S. Batut, L. Nahon, C. Fittschen and J. C. Loison, J. Electron Spectrosc. Relat. Phenom., 2015, 203, 25–30 CrossRef CAS.
- G. A. Garcia, J.-C. Loison, F. Holzmeier, B. Gans, C. Alcaraz, L. Nahon, X. Wu, X. Zhou, A. Bodi and P. Hemberger, Mol. Phys., 2020, 119, e1825851 CrossRef.
- N. L. Chen, B. Gans, S. Hartweg, G. A. Garcia, S. Boyé-Péronne and J.-C. Loison, J. Chem. Phys., 2022, 157, 014303 CrossRef CAS PubMed.
- W. A. Chupka, J. Chem. Phys., 1993, 98, 4520–4530 CrossRef CAS.
- P. Hemberger, Z. Pan, X. Wu, Z. Zhang, K. Kanayama and A. Bodi, J. Phys. Chem. C, 2023, 127, 16751–16763 CrossRef CAS PubMed.
- J. Harvey, P. Hemberger, A. Bodi and R. P. Tuckett, J. Chem. Phys., 2013, 138, 124301 CrossRef PubMed.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby, M. Schütz, P. Celani, T. Korona, R. Lindh, A. Mitrushenkov, G. Rauhut, K. R. Shamasundar, T. B. Adler, R. D. Amos, A. Bernhardsson, A. Berning, D. L. Cooper, M. J. O. Deegan, A. J. Dobbyn, F. Eckert, E. Goll, C. Hampel, A. Hesselmann, G. Hetzer, T. Hrenar, G. Jansen, C. Köppl, Y. Liu, A. W. Lloyd, R. A. Mata, A. J. May, S. J. McNicholas, W. Meyer, M. E. Mura, A. Nicklass, D. P. O'Neill, P. Palmieri, D. Peng, K. Pflüger, R. Pitzer, M. Reiher, T. Shiozaki, H. Stoll, A. J. Stone, R. Tarroni, T. Thorsteinsson and M. Wang, MOLPRO, version 2016, a package of ab initio programs, 2016, see https://www.molpro.net Search PubMed.
- R. A. Kendall, T. H. Dunning and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS.
- C. Brown, S. Tilford, R. Tousey and M. Ginter, J. Opt. Soc. Am., 1974, 64, 1665–1682 CrossRef CAS.
- K. Haris and A. Kramida, Astrophys. J., Suppl. Ser., 2017, 233, 16 CrossRef.
- B. Gans, F. Holzmeier, J. Krüger, C. Falvo, A. Röder, A. Lopes, G. Garcia, C. Fittschen, J.-C. Loison and C. Alcaraz, J. Chem. Phys., 2016, 144, 204307 CrossRef CAS PubMed.
- M. Briant, L. Poisson, M. Hochlaf, P. de Pujo, M.-A. Gaveau and B. Soep, Phys. Rev. Lett., 2012, 109, 193401 CrossRef PubMed.
- B. Ruscic and D. H. Bross, Comput.-Aided Chem. Eng., 2019, 45, 3–114 CAS.
- K. P. Huber and G. Herzberg, Molecular Spectra and Molecular Structure. IV. Constants of Diatomic Molecules, Reinhold, New York, 1979 Search PubMed.
- T. Porter, D. Mann and N. Acquista, J. Mol. Spectrosc., 1965, 16, 228–263 CrossRef CAS.
- E. Hill and J. H. Van Vleck, Phys. Rev., 1928, 32, 250–272 CrossRef CAS.
- G. Herzberg, Molecular Spectra and Molecular Structure. I. Spectra of Diatomic Molecules, D. Van Nostrand, N. J. Princeton, 2nd edn, 1950 Search PubMed.
- J. T. Hougen, The calculation of rotational energy levels and rotational line intensities in diatomic molecules, US National Bureau of Standards, 1970, vol. NIST MN-115.
- J. H. Thorpe, D. Feller, D. H. Bross, B. Ruscic and J. F. Stanton, Phys. Chem. Chem. Phys., 2023, 25, 21162–21172 RSC.
- B. Ruscic, A. F. Wagner, L. B. Harding, R. L. Asher, D. Feller, D. A. Dixon, K. A. Peterson, Y. Song, X. Qian, C.-Y. Ng, J. Liu, W. Chen and D. W. Schwenke, J. Phys. Chem. A, 2002, 106, 2727–2747 CrossRef CAS.
- B. Ruscic, D. Feller, D. A. Dixon, K. A. Peterson, L. B. Harding, R. L. Asher and A. F. Wagner, J. Phys. Chem. A, 2001, 105, 1–4 CrossRef CAS.
- M. E. Harding, J. Vázquez, B. Ruscic, A. K. Wilson, J. Gauss and J. F. Stanton, J. Chem. Phys., 2008, 128, 114111 CrossRef PubMed.
- B. Ruscic, D. Feller and K. A. Petrson, Theor. Chem. Acc., 2014, 133, 1415 Search PubMed.
- R. L. Asher and B. Ruscic, J. Chem. Phys., 1997, 106, 210–221 CrossRef CAS.
- Y. Houbrechts, I. Dubois and H. Bredohl, J. Phys. B: At., Mol. Opt. Phys., 1980, 13, 3369 CrossRef CAS.
- L. V. Gurvich, I. V. Veyts and C. B. Alcock, Thermodynamic Properties of Individual Substances, Hemisphere, New York, 4th edn, 1990, vol. 2 Search PubMed.
- J. D. Cox, D. D. Wagman and V. A. Medvedev, CODATA Key Values for Thermodynamics, Hemisphere, New York, 1989 Search PubMed.
- A. Karton and J. M. L. Martin, J. Phys. Chem. A, 2007, 111, 5936–5944 CrossRef CAS PubMed.
- A. Karton, A. Tarnopolsky, J.-F. Lamere, G. C. Schatz and J. M. L. Martin, J. Phys. Chem. A, 2008, 112, 12868–12886 CrossRef CAS PubMed.
- A. Karton, S. Daon and J. M. L. Martin, Chem. Phys. Lett., 2011, 510, 165–178 CrossRef CAS.
- A. Karton, E. Rabinovich, J. M. L. Martin and B. Ruscic, J. Chem. Phys., 2006, 125, 144108 CrossRef PubMed.
- A. Tajti, P. G. Szalay, A. G. Császár, M. Kállay, J. Gauss, E. F. Valeev, B. A. Flowers, J. Vázquez and J. F. Stanton, J. Chem. Phys., 2004, 121, 11599–11613 CrossRef CAS PubMed.
- D. Feller, K. A. Peterson and D. A. Dixon, J. Chem. Phys., 2008, 129, 204105 CrossRef PubMed.
- A. Ricca and C. W. Bauschlicher, J. Phys. Chem. A, 1998, 102, 876–880 CrossRef CAS.
- E. W. Ignacio and H. B. Schlegel, J. Chem. Phys., 1990, 92, 5404–5416 CrossRef CAS.
- P. Ho and C. F. Melius, J. Phys. Chem., 1990, 94, 5120–5127 CrossRef CAS.
- H. H. Michels and R. H. Hobbs, Chem. Phys. Lett., 1993, 207, 389–396 CrossRef CAS.
- D. Feller and D. A. Dixon, J. Phys. Chem. A, 1999, 103, 6413–6419 CrossRef CAS.
- B. P. Prascher, R. M. Lucente-Schultz and A. K. Wilson, Chem. Phys., 2009, 359, 1–13 CrossRef CAS.
- P. Sukkaew, L. Ojamäe, O. Kordina, E. Janzén and Ö. Danielsson, ECS J. Solid State Sci. Technol., 2015, 5, P27 CrossRef.
- A. Burcat and E. Goos, Int. J. Chem. Kinet., 2018, 50, 633–650 CrossRef CAS.
- J. Guo, S. Tang, L. Chen, D. Wei and A. Tang, Int. J. Quantum Chem., 2023, 123, e27063 CrossRef CAS.
- D. D. Wagman, W. H. Evans, V. B. Parker, R. H. Schumm, I. Halow, S. M. Bailey, K. L. Churney and R. L. Nuttall, J. Phys. Chem. Ref. Data, 1982, 11, 2 Search PubMed.
- R. Walsh, J. Chem. Soc., Faraday Trans. 1, 1983, 79, 2233–2248 RSC.
- M. W. Chase Jr, J. Phys. Chem. Ref. Data, 1985, 14(Suppl. 1) Search PubMed.
- B. Ruscic, R. E. Pinzon, M. L. Morton, N. K. Srinivasan, M.-C. Su, J. W. Sutherland and J. V. Michael, J. Phys. Chem. A, 2006, 110, 6592–6601 CrossRef CAS PubMed.
- B. Ruscic and D. H. Bross, Mol. Phys., 2021, 119, e1969046 CrossRef.
- J. H. Thorpe, J. L. Kilburn, D. Feller, P. B. Changala, D. H. Bross, B. Ruscic and J. F. Stanton, J. Chem. Phys., 2021, 155, 184109 CrossRef CAS PubMed.
- T. L. Nguyen, D. H. Bross, B. Ruscic, G. B. Ellison and J. F. Stanton, Faraday Discuss., 2022, 238, 405–430 RSC.
- D. H. Bross, G. B. Bacskay, K. A. Peterson and B. Ruscic, J. Phys. Chem., 2023, 127, 704–723 CrossRef CAS PubMed.
- N. Genossar, P. B. Changala, B. Gans, J.-C. Loison, S. Hartweg, M.-A. Martin-Drumel, G. A. Garcia, J. F. Stanton, B. Ruscic and J. H. Baraban, J. Am. Chem. Soc., 2022, 144, 18518–18525 CrossRef CAS PubMed.
- T. L. Nguyen, J. Peeters, J.-F. Müller, A. Perera, D. H. Bross, B. Ruscic and J. F. Stanton, Proc. Natl. Acad. Sci. U. S. A., 2023 DOI:10.1073/pnas.2304650120.
- B. J. McBride and S. Gordon, Properties and Coefficients: Computer Program for Calculating and Fitting Thermodynamic Functions, National Aeronautics and Space Administration, 1999.
- P. B. Changala, T. L. Nguyen, J. H. Baraban, J. F. Stanton, D. H. Bross and B. Ruscic, J. Phys. Chem. A, 2017, 121, 8799–8806 CrossRef CAS PubMed.
- PuRe Data Resources at a Glance, https://science.osti.gov/Initiatives/PuRe-Data/Resources-at-a-Glance, accessed August 2023.
| This journal is © the Owner Societies 2023 |