
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotoelectron spectroscopic study of 2-naphthylnitrene and its thermal rearrangement to cyanoindenes†
Mayank
Saraswat
 a,
Adrian
Portela-Gonzalez
a,
Enrique
Mendez-Vega
a,
Ginny
Karir
a,
Wolfram
Sander
*a and
Patrick
Hemberger
*b
a,
Adrian
Portela-Gonzalez
a,
Enrique
Mendez-Vega
a,
Ginny
Karir
a,
Wolfram
Sander
*a and
Patrick
Hemberger
*b
aLehrstuhl für Organische Chemie II, Ruhr-Universität Bochum, Bochum 44780, Germany. E-mail: wolfram.sander@rub.de
bLaboratory for Synchrotron Radiation and Femtochemistry, Paul Scherrer Institut (PSI), Villigen CH-5232, Switzerland. E-mail: patrick.hemberger@psi.ch
First published on 10th November 2023
Abstract
2-Cyanoindene has recently been identified in the interstellar medium, however current models cannot fully account for its formation pathways. Herein, we identify and characterize 2-naphthylnitrene, which is prone to rearrange to 2- and 3-cyanoindene, in the gas phase using photoion mass-selective threshold photoelectron spectroscopy (ms-TPES). The adiabatic ionization energies (AIE) of triplet nitrene (3A′′) to the radical cation in its lowest-energy doublet ![[X with combining tilde]](https://www.rsc.org/images/entities/char_0058_0303.gif) +(2A′) and quartet ã+(4A′) electronic states were determined to be 7.72 ± 0.02 and 8.64 ± 0.02 eV, respectively, leading to a doublet–quartet energy splitting (ΔED–Q) of 0.92 eV (88.8 kJ mol−1). A ring-contraction mechanism yields 3-cyanoindene, which is selectively formed under mild pyrolysis conditions (800 K), while the lowest-energy isomer, 2-cyanoindene, is also observed under harsh pyrolysis conditions at 1100 K. The isomer-selective assignment was rationalized by Franck–Condon spectral modeling and by measuring the AIEs at 8.64 ± 0.02 and 8.70 ± 0.02 eV for 2- and 3-cyanoindene, respectively, in good agreement with quantum chemical calculations.
+(2A′) and quartet ã+(4A′) electronic states were determined to be 7.72 ± 0.02 and 8.64 ± 0.02 eV, respectively, leading to a doublet–quartet energy splitting (ΔED–Q) of 0.92 eV (88.8 kJ mol−1). A ring-contraction mechanism yields 3-cyanoindene, which is selectively formed under mild pyrolysis conditions (800 K), while the lowest-energy isomer, 2-cyanoindene, is also observed under harsh pyrolysis conditions at 1100 K. The isomer-selective assignment was rationalized by Franck–Condon spectral modeling and by measuring the AIEs at 8.64 ± 0.02 and 8.70 ± 0.02 eV for 2- and 3-cyanoindene, respectively, in good agreement with quantum chemical calculations.
Introduction
Arylnitrenes (Ar–N) are key reactive intermediates with widespread applications in synthetic organic chemistry and photoaffinity labelling.1 Investigation of the fundamental properties of these electron-deficient neutral species is challenging due to rapid inter- and/or intramolecular reactions.2,3 Electron loss of arylnitrenes leads to the corresponding nitrene radical cations, which also exhibit a rich chemistry involving ring expansions, contractions, and opening reactions.4 In particular, 2-naphthylnitrene 2 has extensively been studied in organic glasses,5–7 solution,8,9 and low temperature matrices,10–13 as well as using quantum chemical calculations.3,11,14,15 Nitrene 2 is initially formed in its lowest-energy open-shell singlet state upon pyrolysis of the azide precursor 3 (see Scheme 1), and can undergo ring expansion, ring contraction or intersystem crossing (ISC) depending on the experimental conditions. Wentrup et al. reported that the ring contraction mechanism leading to the formation of cyanoindenes is the most favorable and exothermic pathway under mild pyrolytic conditions.3 In this work, we investigate the photoionization of nitrene 2 and its thermal rearrangements products using photoelectron photoion coincidence (PEPICO) spectroscopy.16 Our motivation also stems from the thermal products, cyanoindenes, which have recently been highlighted in the astrochemistry community since their detection in the cold Taurus molecular cloud (TMC-1).17The presence of polyaromatic hydrocarbons (PAHs) in the interstellar medium has been postulated since late 1980s based on the detection of diffuse infrared emission bands. Chemical identification of these compounds, has proven difficult with the first individual detection of a PAH, indene (c-C9H8),18 just reported in 2022.18,19 Over the past decade, there has been a plethora of aromatic and cyclic molecules, observed in the cold interstellar medium (ISM).20–26 These observations highlight a very rich chemistry in interstellar environments, involving PAHs and potentially their nitrogen-containing analogues (PANHs).27,28 Cyanoindene (c-C10H7-CN) 1 has been proposed to form via the bimolecular reaction of the CN radical and indene.17 Interestingly, only the positional isomer 2-cyanoindene 1b was observed whereas 3- and 1-cyanoindenes 1a and 1c were not detected. Isomers 1a and 1b are found to be nearly degenerate in energy, while 1c is a high energy isomer. Hence, the abundance of the cyanoindene isomers in this source, cannot solely be explained by modelling the indene + CN˙ reaction, indicating that additional pathways must also play a role.17
Our understanding of PAH/aromatic/cyclic hydrocarbon chemistry in these extreme environments remains limited due to lack of experimental studies. Formation of PAHs has been proposed to occur either through top-down or bottom-up approaches,29,30 nevertheless, neither model has been completely justified, and it is plausible that both operate under different conditions.28 For the top-down approach, PAH photochemistry has been hypothesized as a competition between fragmentation and isomerization under the “grand PAH” hypothesis.31 Isomerization of PAHs and other cyclic species in the context of astrochemistry, however, remains largely unexplored. Feasible isomerization pathways could also be relevant in other high temperature interstellar and circumstellar environments, like exoplanetary atmospheres.32
Moreover in diffuse environments like photodissociation regions (PDRs), cosmic rays can ionize molecules.33 Ions can further undergo fast reactions which are crucial in the synthesis of complex organic molecules.34 Nitrile ions and protonated nitriles are also abundant in the ISM but also in the upper atmosphere of Titan, the largest satellite of Saturn.35 In this regard, accurate determination of gas-phase ion energetics data, particularly, vertical, and adiabatic ionization energies (VIE and AIE) and dissociative photoionization (DPI) thresholds of closed- and open-shell species reported in this work are crucial values for the astronomers and astrochemical models. Thermochemical data like heat of formation and bond strengths in neutral molecules and ions are important for a mechanistic understanding of astrochemical environments.36,37
Photoelectron photoion coincidence (PEPICO) spectroscopy is an excellent approach to detect and unveil the electronic, geometrical, and vibrational structure of neutral and charged elusive species isomer-selectively, allowing to record vibrationally-resolved photoion mass-selective threshold photoelectron (ms-TPE) spectra.38–41 This suit of techniques has proven to be vital in identifying elusive and stable molecules to unravel reaction mechanisms relevant to combustion,42 catalysis,43,44 and astrochemical environments.45 So far, most of the studies have been dedicated to smaller aliphatic nitrile ions such as: CN, CH3CN, C2H3CN, and i-C3H7CN.46,47
In this work, we study the thermal generation of 2-naphthylnitrene 2 from the azide precursor 3 and investigate its threshold photoelectron spectrum, which gives rise to D-2˙+ and Q-2˙+ cation states and enables determination of the doublet quartet splitting. Furthermore, we study the ring contraction pathways of 2 to form cyanoindenes 1a–c in the gas phase using ms-TPE spectroscopy. Our study may have implication for understanding the chemistry of aromatic compounds in photodissociation regions.
Methodologies
Experimental and computational details
Experiments were performed at the Swiss Light Source (SLS) of the Paul Scherrer Institute using vacuum ultraviolet (VUV) synchrotron radiation.48 2-Naphthylazide 3 was synthesized following a literature procedure49 and employed as precursor to generate 2-naphthylnitrene 2via flash vacuum pyrolysis (FVP). The reactive intermediate 2, further isomerizes to cyanoindenes 1a–c at higher temperatures. During experiments, 2-naphthylazide was sublimed at 45–50 °C and expanded through a 200 μm diameter nozzle along with 30 sccm of He into a silicon carbide tube (1 mm inner diameter and 15 mm heated length), electrically heated in the range of 10–35 W heating power, which corresponds to temperatures of 700–1100 K.50 The pressure and the residence time inside the reactor is estimated to 9–20 mbar and ∼25–50 μs.51 After skimming (2 mm), the molecular beam containing the pyrolysis products reaches the PEPICO spectrometer chamber and is ionized by tunable VUV synchrotron radiation. The pressure in the source and spectrometer chamber is about 3 × 10−5 and 1.5 × 10−6 mbar, respectively. Detection of electrons and ions in delayed coincidence is the basis of the PEPICO method which has been reported in detailed elsewhere and will be only briefly summarized here.52,53 Both photoelectrons and photoions are vertically accelerated in opposite directions using velocity focusing optics and a constant extraction field. Position-sensitive delay-line anode detectors (ROENTDEK DLD40) were used to obtain velocity map images (VMI) for both electrons and ions. The time-of-flight (TOF) mass spectrum of the photoion is recorded by determining the time difference between the coincident photoelectron and photoion.54 The photon energy is scanned and threshold electrons with less than 5 meV kinetic energy are selected in the electron VMI. The coincident photoion signal of interest in a fixed m/z range is plotted in the photoion mass-selected threshold photoelectron spectrum (ms-TPES).54 The spectra are corrected for false coincidences and hot electrons.55 Quantum chemical calculations were performed using the Gaussian 16 program package.56 Geometry optimization and vibrational frequencies were performed with density functional theory using the B3LYP functional and def2-TZVP basis set including the D3 dispersion correction. Additionally, AIEs for nitrene 2 and cyanoindenes 1a–c were computed with the CBS-QB3, CBS-APNO and G4 composite methods, respectively.57 Franck–Condon factors were simulated using optimized geometries and vibrational frequencies calculated at the B3LYP-D3/def2-TZVP level of theory.Results and discussion
Mass spectra of FVP products
The thermal decomposition of 2-naphthylazide 3 (m/z 169) was studied by photoionization mass spectrometry (MS). Mass spectra were recorded at different photon energies and pyrolysis temperatures (Fig. 1). At room temperature (RT) (pyrolysis off) and a fixed photon energy of 9.0 eV, an intense peak appears at m/z 169 that corresponds to azide 3 (Fig. 1, trace a). Upon increasing the photon energy to 10.5 eV, a strong signal at m/z 141 appears due to dissociative photoionization (DPI) of precursor 3via N2 loss (Fig. 1, trace b). Ion images reveal kinetic energy release perpendicular to the molecular beam axis, indicative of DPI (Fig. S1 in the ESI†). The peak at m/z 141 is tentatively assigned to the nitrene radical cation 2˙+, since nitrene 2 is the primary photoproduct of azide 2. Rearrangements of phenyl nitrene radical ion do not typically occur at low photon energies of 8–11 eV,39 instead have been observed upon 70 eV electron ionization.4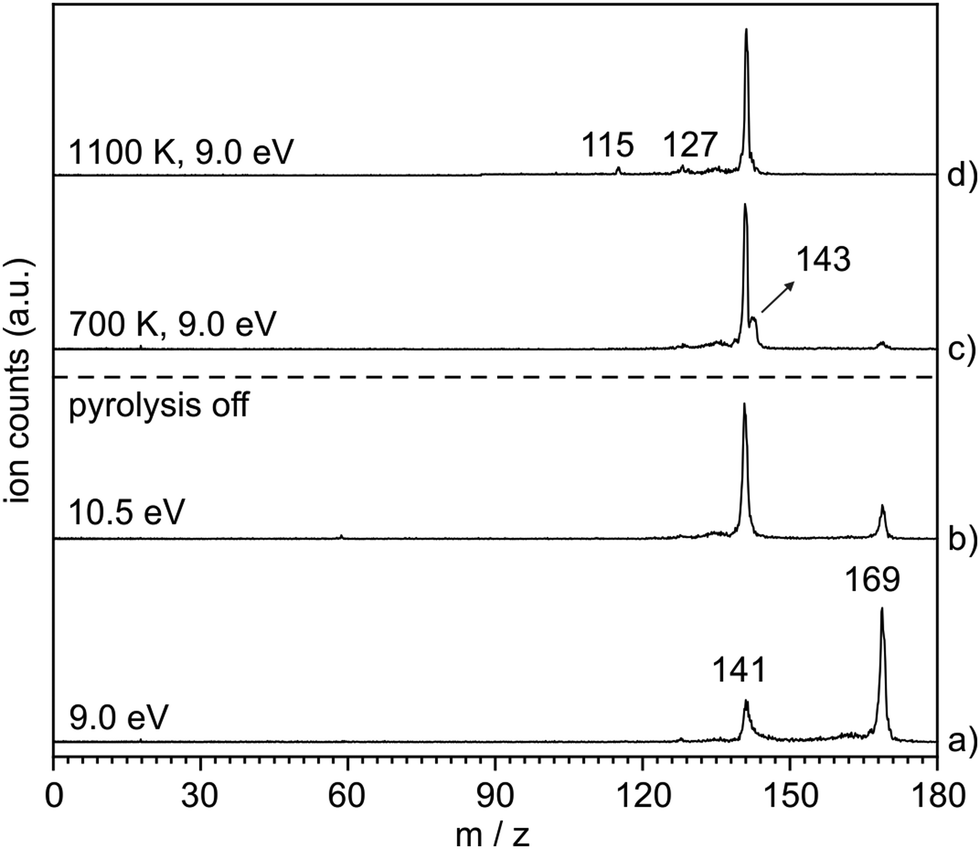 | ||
| Fig. 1 Mass spectra of 3 at RT (pyrolysis off) recorded at photon energies of (a) 9.0 eV and (b) 10.5 eV. (c) and (d) Spectra obtained at 9.0 eV after FVP of 3 at 700 and 1100 K. Spectra also contains acetone (m/z 58) and water (m/z 18) impurities. | ||
The pyrolysis temperature was optimized at a fixed photon energy of 9.0 eV to achieve the maximum yield of nitrene 2 (m/z 141), the primary product of azide 3. Upon FVP at 700 K, a significant conversion from precursor 3 into nitrene 2 is achieved (Fig. 1, trace c), whereas at 1100 K, thermal decomposition of precursor 3 is basically complete (Fig. 1, trace d). Ion velocity map images (Fig. S1 in ESI†) show a narrow speed distribution of the molecular beam component of m/z 141, indicating a clean formation of the nitrene 2 without contributions of DPI. At 1100 K, additional small peaks are also observed at m/z 127 and 115, which are assigned to naphthyl 4 and indenyl 5 radicals, respectively. A similar fragmentation pattern through CN loss was reported upon thermal decomposition of picolyl radicals.58 Moreover, a substantial amount of the H-abstraction product, 2-aminonaphthalene 6 (m/z 143), was noticed at 700 K (Fig. 1, trace c). Interestingly, ion velocity map images of m/z 143 (Fig. S2 in ESI†) show a strong background component, indicative for formation via collisions of the triplet nitrene T-2 with the chamber walls, which was also reported for the parent triplet phenylnitrene.39 This is in agreement with the ion image of nitrene 2m/z 141 (Fig. S1 in ESI†), which shows a negligible background component proofing its high reactivity and quenching on the chamber walls to afford 6. So far, only resonance stabilized and less reactive allyl,59 phenoxy60, lutidyl61, or iodobenzyl62 radicals could be detected after wall collisions and can thus produce room temperature cooled spectra.59 At 1100 K, the peak at m/z 141 is still intense, while the related peak at m/z 143 does not appear anymore in the spectrum (Fig. 1, trace d). The absence of H-abstraction product 6 indicates that nitrene 2 is not present at 1100 K, but it rather thermally converts into a closed-shell isomer with m/z 141 and is thus less reactive towards H atoms. Identification and interconversion of the species of mass m/z 141 will be discussed in the next section by means of isomer-specific ms-TPE spectroscopy.
ms-TPE spectrum of 2-naphthylnitrene
The ms-TPE spectrum of the signal at m/z 141, recorded upon FVP at 700 K, shows two bands with maxima at 7.72, and 8.64 eV (Fig. 2 and 3). The bands at 7.72 and 8.64 eV proportionally decrease upon increasing the FVP temperature, indicating that these belong to the same species and are assigned to the primary thermal product nitrene 2. The ms-TPE spectrum obtained at 700 K arises from contributions from the ionization of nitrene T-2(3A′′) to the radical cation in its doublet D-2˙+(2A′) and quartet Q-2˙+(4A′) electronic states. The lowest-energy band in the ms-TPE spectra at 7.72 eV is assigned to the vibronic transition between the ZPE levels of triplet nitrene(3A′′) and doublet radical cation +(2A′) (Fig. 2). The experimental adiabatic ionization energy (AIE) of 7.72 eV is reproduced by DFT (7.62 eV) and highly-correlated composite method (7.70–7.78 eV) calculations (Table 1). In this transition, photoionization takes place from the triplet ground state nitrene T-2 and involves the removal of an electron at the out-of-plane singly occupied p orbital at the nitrene center. In contrast, the higher-energy band at 8.64 eV is assigned to the vibronic transition ã+(4A′) ← (3A′′), leading to the quartet radical cation Q-2˙+ (4A′) (Fig. 3). The electronic structure of the quartet radical cation Q-2˙+ resembles a triradical, by keeping the two unpaired electrons at the N atom (nitrene) and taking out one electron from the aromatic π system. This transition is blue-shifted as the electron is removed from a lower-energy doubly occupied orbital on the aromatic naphthalene ring. The difference between the AIEs of the doublet and quartet radical cations leads to the quartet-doublet energy gap (ΔED–Q) for 2˙+ and is experimentally determined to be 0.92 eV (88.8 kJ mol−1), in perfect agreement with G4 calculations (Table 1). This value is considerably lower than the ΔED–Q of the analogue phenyl nitrene radical cation (1.44 eV) due to the stabilization of the quartet triradical Q-2˙+(4A′) by extended ring conjugation.39
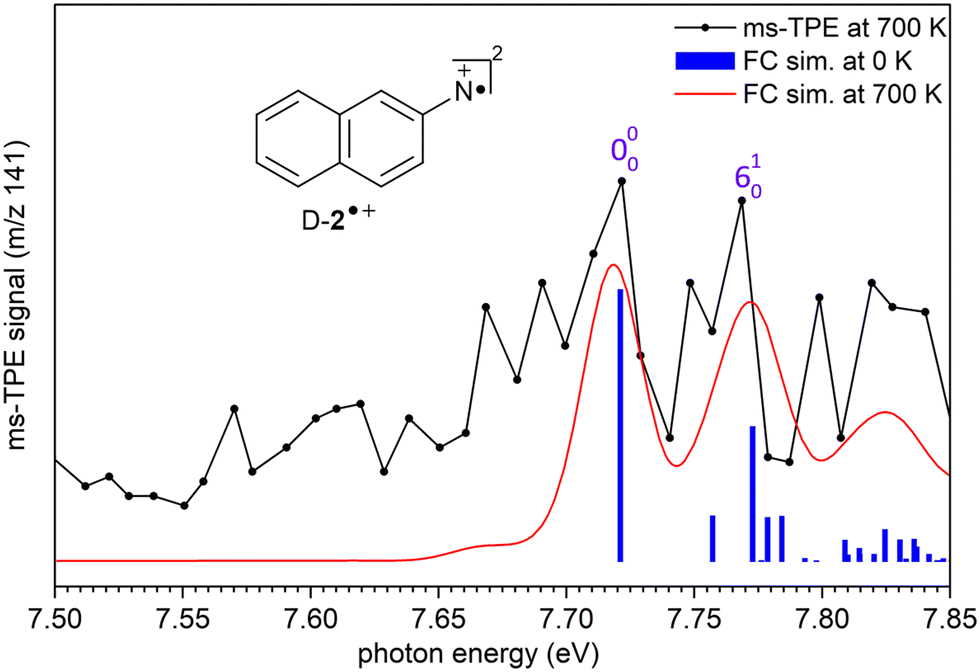 | ||
| Fig. 2 Comparison of the ms-TPE spectrum of m/z 141 in the range of 7.50–7.85 eV recorded after FVP of 2-naphthylazide 3 at 700 K (black trace) with Franck–Condon (FC) simulations of the +(2A′) ← (3A′′) transition of nitrene 2. Spectral simulations were performed at 0 K (blue sticks) and at 700 K (red trace) by convolution with 25 meV fwhm Gaussians. | ||
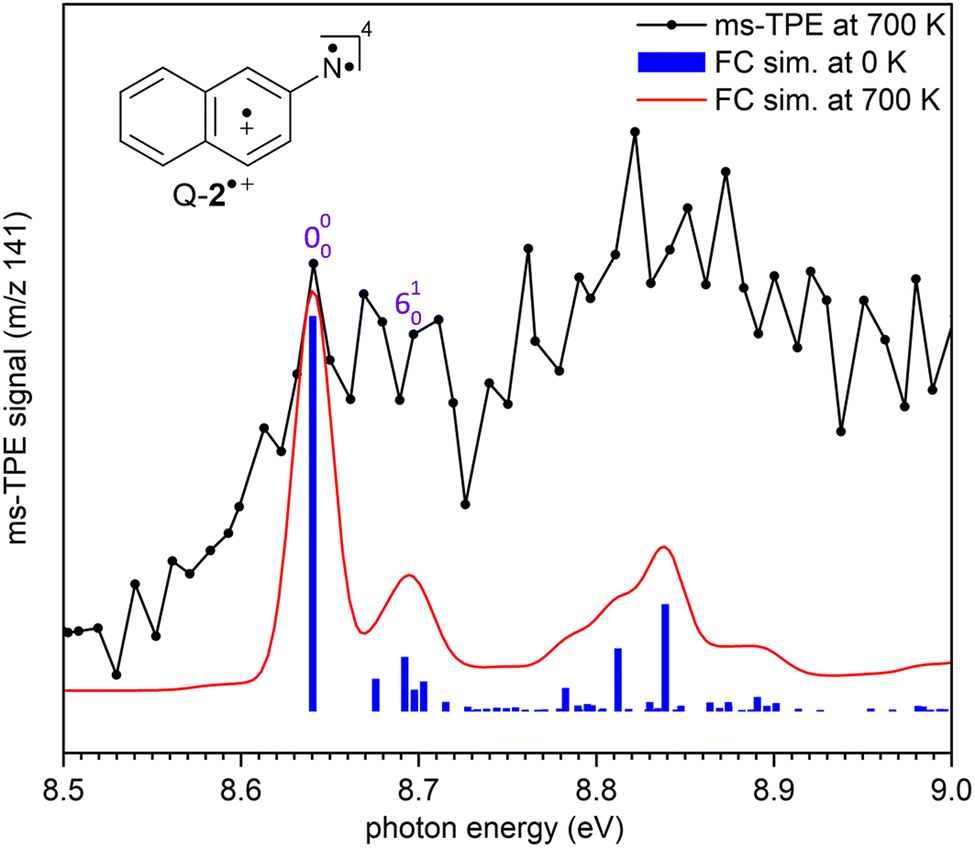 | ||
| Fig. 3 Comparison of the ms-TPE spectrum of m/z 141 in the range of 8.50–9.00 eV, recorded after FVP of 2-naphthylazide 3 at 700 K (black trace) with Franck–Condon (FC) simulations of the ã+(4A′) ← (3A′′) transition of nitrene 2. Spectral simulations were performed at 0 K (blue sticks) and at 700 K (red trace) by convolution with 25 meV fwhm Gaussians. | ||
In addition, a vibrational progression is observed for both doublet and quartet states allowing to gain vibrational information of the cations D-2˙+(2A′) and Q-2˙+(4A′). Transitions +(2A′) ← (3A′′), and ã+ (4A′)← (3A′′) show an origin band (0–0) and excitations of the ring breathing mode (ν6) with a spacing of 50 ± 10 meV (400 ± 80 cm−1), which is reproduced by the calculated (ν6) mode of D-2˙+(2A′) and Q-2˙+(4A′) cations (417 cm−1) at the B3LYP-D3/def2-TZVP level of theory. Due to the insignificant cooling in the molecular beam expansion and high reactivity of nitrene 2, no room temperature cooled spectra could be obtained by using ion velocity imaging. The spectra are thus suffered from hot- and sequence band transitions, which cannot be well reproduced by the FC simulations and explain the intensity mismatch between simulations and experiment. This was also observed for the ms-TPES for hot benzene, most recently.59
The optimized geometries of nitrene T-2, doublet D-2˙+, and quartet Q-2˙+ radical cations are shown in Fig. 4. The C–N bond distance in D-2˙+ (1.27 Å) is shorter than that in T-2 (1.31 Å), whereas slight elongation is found in Q-2˙+ (1.33 Å). Such contrastingly structural response correlates to the extent of the conjugation between the electron-deficient nitrene moiety and the electron-rich aromatic rings. Electron detachment from the nitrene center to yield D-2˙+ makes the nitrogen atom more electronegative and increases the polarity of the C–N bond, whereas the opposite effect is found in Q-2˙+ upon electron removal from the ring. Nevertheless, the overall geometrical change from nitrene T-2 to the radical cation in its ground state doublet D-2˙+ and the quartet Q-2˙+ state is found to be small, resulting in well visible 0–0 vibronic transitions in the experimental spectrum.
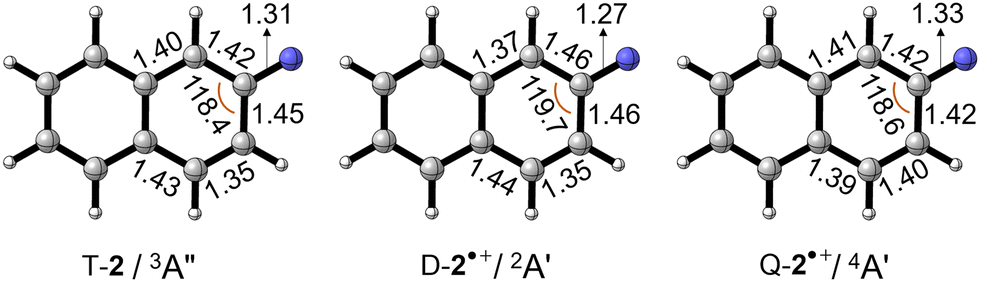 | ||
| Fig. 4 Optimized geometries of T-2, D-2˙+, and Q-2˙+ at the B3LYP-D3/def2-TZVP level of theory. Selected bond lengths and angles are given in Å and °, respectively. | ||
Thermal rearrangement of 2-naphthylnitrene to cyanoindenes
ms-TPE spectra were recorded upon FVP at 700, 800, and 1100 K, under the same experimental conditions (Fig. 5). As mentioned before, the ms-TPE spectrum at 700 K exhibits bands at 7.72 and, 8.64 eV which are assigned to nitrene 2 (Fig. 2 and 3). However, upon increasing the FVP temperature to 800 K, the bands corresponding to nitrene 2 completely disappear (below 8.5 eV), and a new band at ca. 8.70 eV shows up (Fig. 5, trace b). Upon further increasing the pyrolysis temperature to 1100 K, another intense band appeared at 8.64 eV along with the existent band at 8.70 eV (Fig. 5, trace c). The large blue-shift in the lowest-energy bands from 7.72 eV to 8.70 eV indicates that nitrene 2 thermally rearranges into a structurally and electronically different compound. This species undergoes subsequent isomerization, that is associated with the revival of the peak at 8.64 eV. Since the D-2˙+ band is absent at lower photon energies, the bands and 8.64 and 8.70 eV are solely assigned to 2-cyanoindene 1b and 3-cyanoindene 1a, respectively (Fig. 6), evidencing isomerization of nitrene 2 as viable pathway to yield the cyanoindenes.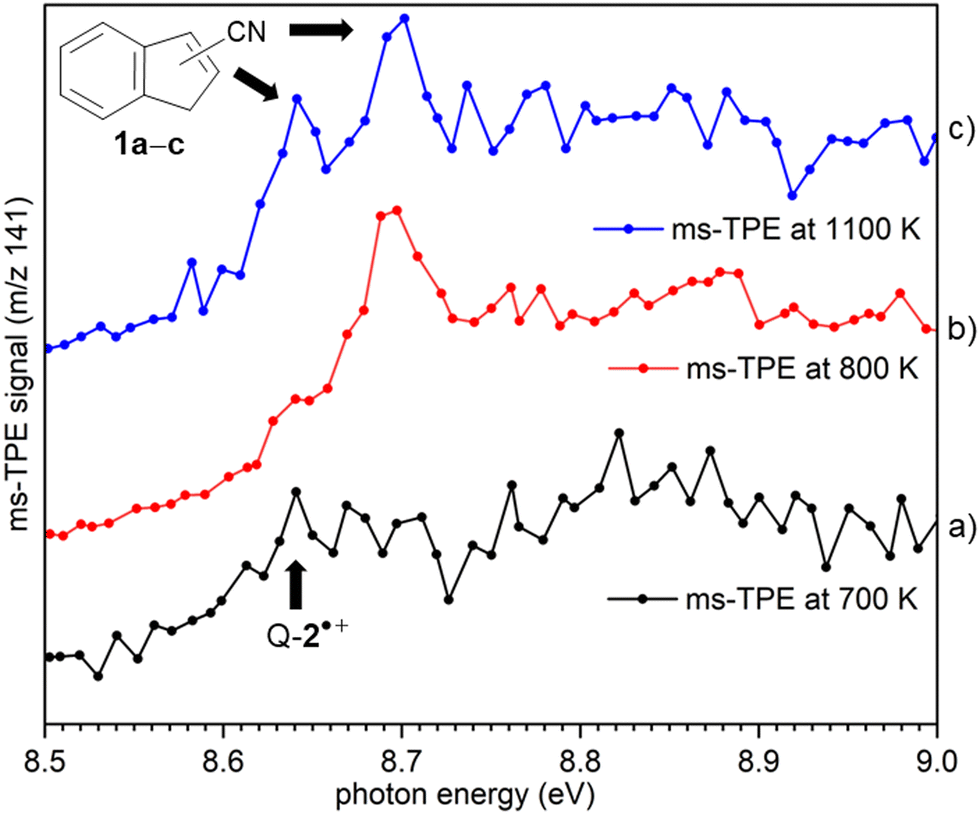 | ||
| Fig. 5 ms-TPE spectra of m/z 141 at different FVP temperatures: 700 K (black trace), 800 K (red trace), and 1100 K (blue trace). Nitrene 2 gradually converts into cyanoindenes 1 upon increasing the temperature. | ||
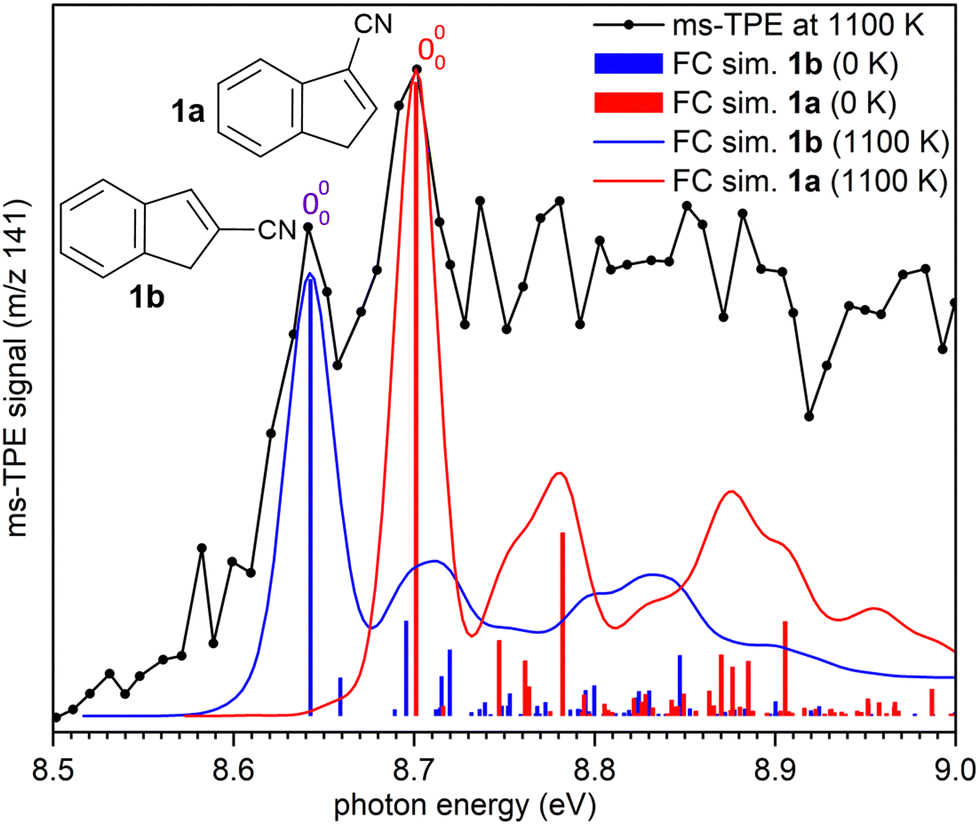 | ||
| Fig. 6 Comparison of the ms-TPE spectrum obtained at 1100 K (black trace) with FC simulations of vibronic transitions of 2-cyanoindene 1b (in blue) and 3-cyanoindene 1a (in red). FC simulations performed at 1100 K by convolution with 25 meV fwhm Gaussians. | ||
AIEs are calculated for 1b and 1a to 8.67 and 8.71 eV, respectively, with the G4 method (Table 2), which very well agree with the experimental features at 8.64 ± 0.02 eV and 8.70 eV ± 0.02. Moreover, FC simulations reasonably match to the experimental bands (Fig. 6). 2-Cyanoindene 1b is predicted to be the lowest-energy isomer, with 1a being nearly degenerate in energy at 2.5 kJ mol−1, whereas isomer 1c lies 21.7 kJ mol−1 higher in energy (G4, Table 2). The higher energy isomer 1c seems to be absent, although it cannot completely be ruled out, since the AIE is very similar to that of the isomer 1a. Other isomeric species such as: ketenimines (7 and 8) were disregarded based on calculated AIE values obtained at G4 level of theory (Table S1 in the ESI†). The thermal rearrangements of 2-naphthylnitrene (2) to 3- and 2-cyanoindene (1a and 1b) are strongly exothermic and the complete mechanism of this ring contraction step has been reported by Wentrup et al. using 13C labelling experiments as well as quantum chemical calculations.3 In these studies, it is proposed that the open-shell singlet 2-naphthylnitrene undergo a highly exothermic ring contraction to 1-cyanoindene that subsequently isomerizes to 3- and 2-cyanoindene via H and CN 1,5-shifts.
| Method | 1c | 1b | 1a |
|---|---|---|---|
| ms-TPES | — | 8.64 ± 0.02 | 8.70 ± 0.02 |
| B3LYP-D3/def2-TZVP | 8.38 (+31.4) | 8.31 (0.0) | 8.38 (+5.8) |
| CBS-QB3 | 8.71 (+22.2) | 8.64 (0.0) | 8.74 (+2.9) |
| CBS-APNO | 8.72 (+23.0) | 8.66 (0.0) | 8.76 (+2.5) |
| G4 | 8.69 (+21.7) | 8.67 (0.0) | 8.71 (+2.5) |
Overall, the experimental spectra show that the kinetic product, isomer 1a, is initially formed (band at 8.70 eV) at 800 K, but partially converts to the thermodynamic product, isomer 1b at 1100 K, via sigmatropic H and CN shifts (Scheme 2). At 1100 K, the gas mixture should be under thermodynamic equilibrium conditions. These findings agree well with the reports by Wentrup et al. using preparative FVP.14 Moreover, collisional deactivation using He as the carrier gas (∼25–50 mbar) prevents the formation of the high energy isomer 1c at 1100 K. A similar product distribution was obtained under comparable experimental conditions for the parent cyanocyclopentadiene.39 Unfortunately, the low yields of the indenyl radical 5 obtained at 1100 K prevents a proper structural identification by ms-TPE spectroscopy. Nevertheless, thermal cleavage of the C–CN bond in any of the isomers 1a–c followed by facile H-shift should yield the resonance-stabilized 1-indenyl 5, since it is the most stable positional isomer by more than 165 kJ mol−1 (Scheme 1).
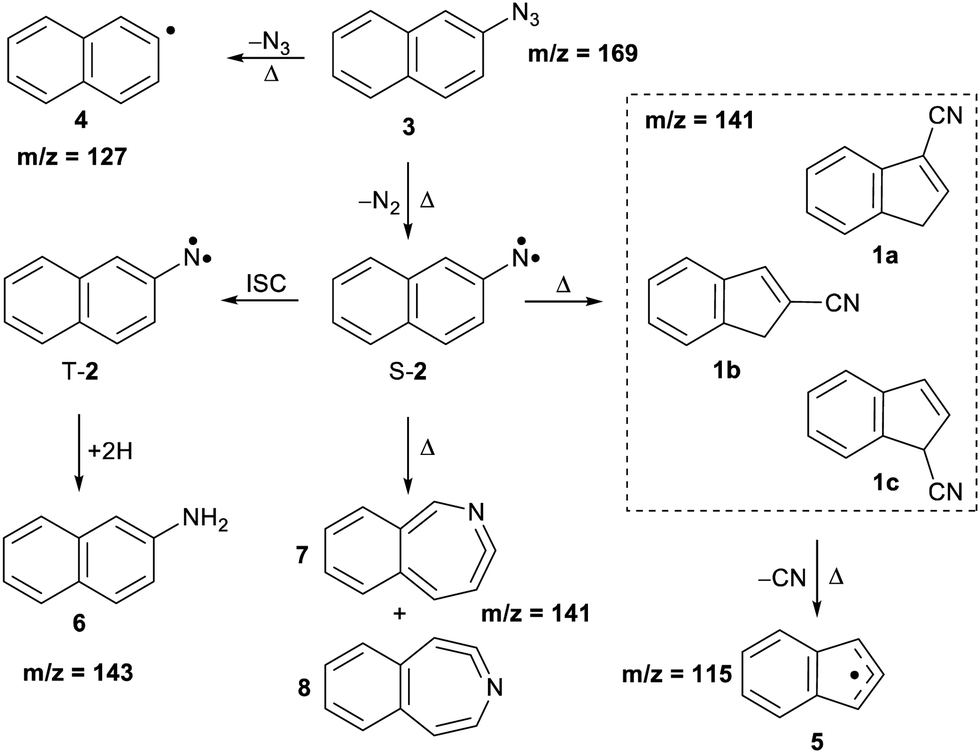 | ||
| Scheme 1 Thermal generation and reactions of naphthylnitrene 2 and cyanoindenes 1a–c. | ||
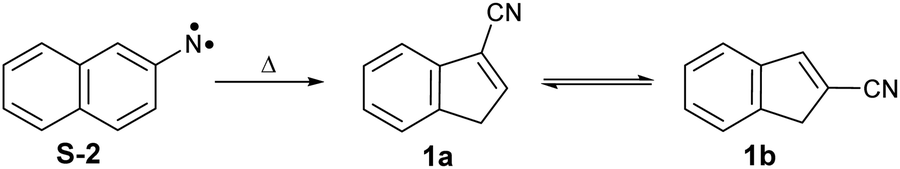 | ||
| Scheme 2 Thermal interconversion of cyanoindenes 1a–c. | ||
Conclusions
Acquiring gas-phase energetics data on neutral and ion species is of great importance to support astrochemical observations but also to rationalize the photochemical and thermal formation of complex organic molecules in extreme environments. A variety of PAHs and PANHs, particularly cyano-compounds, have been recently detected in the ISM.Pyrolysis of 2-naphthylazide 3 in the gas-phase yields the primary product, 2-naphthylnitrene 2. The vibrationally-resolved ms-TPES of nitrene 2 was recorded allowing to measure adiabatic ionization energies (AIEs) of 7.72 ± 0.02 and 8.64 ± 0.02 eV for the +(2A′) ← (3A′′) and ã+(4A′) ← (3A′′) vibronic transitions, respectively. The quartet-doublet energy gap (ΔED–Q) of 2˙+ was experimentally determined to 0.92 eV (88.8 kJ mol−1), and it is perfectly reproduced by G4 calculations (0.92 eV).
The thermal rearrangement of nitrene 2 results in a mixture of cyanoindenes 1a and 1b. Initially, at a relatively low pyrolysis temperature, only the kinetic product, 3-cyanoindene 1a, was observed. However, further increase in the FVP temperature results in the formation of the thermodynamically more stable isomer 2-cyanoindene 1b. Franck–Condon spectral modeling along with an excellent match of experimental AIEs at 8.64 ± 0.02 and 8.70 ± 0.02 eV for 2- and 3-cyanoindene, with the calculations rationalize our assignment. Such isomer-specific spectroscopic and thermodynamic data on nitrene 2 and cyanoindenes 1, are valuable not only for thermochemical models, but also for benchmarking theoretical methods. In addition, such experimental ion energetic data allows obtaining the heat of formation of nitrile ions and its reactions with other small molecules under the extreme conditions of the ISM, in analogy to the reaction of CN+ + CO, recently studied with TPE spectroscopy.46
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 801459 – FP-RESOMUS and was funded by the Deutsche Forschungsgemeinschaft (DFG) under Germany's Excellence Strategy – EXC 2033 – 390677874 – RESOLV. Patrick Ascher (PSI) is thankfully acknowledged for technical assistance. The experiments were carried out at the VUV (x04db) beamline of the Swiss Light Source (SLS), located at the Paul Scherrer Institute in Villigen, Switzerland.References
- Nitrenes and Nitrenium Ions, ed. D. E. Falvey and A. D. Gudmundsdottir, John Wiley & Sons, Inc., Hoboken, NJ, 2013 Search PubMed.
- R. K. Smalley and H. Suschitz, Chem. Ind., 1970, 1338–1345 CAS.
- C. Wentrup, Chem. Rev., 2017, 117, 4562–4623 CrossRef CAS PubMed.
- D. Begue, A. Dargelos and C. Wentrup, J. Phys. Chem. A, 2018, 122, 8490–8496 CrossRef CAS PubMed.
- H. B. Zhai and M. S. Platz, J. Phys. Chem., 1996, 100, 9568–9572 CrossRef CAS.
- M. L. Tsao and M. S. Platz, J. Phys. Chem. A, 2003, 107, 8879–8884 CrossRef CAS.
- M. L. Tsao and M. S. Platz, J. Phys. Chem. A, 2004, 108, 1169–1176 CrossRef CAS.
- J. Wang, J. Kubicki, G. Burdzinski, J. C. Hackett, T. L. Gustafson, C. M. Hadad and M. S. Platz, J. Org. Chem., 2007, 72, 7581–7586 CrossRef CAS PubMed.
- E. Leyva, M. S. Platz, B. Niu and J. Wirz, J. Phys. Chem., 1987, 91, 2293–2298 CrossRef CAS.
- A. Maltsev, T. Bally, M. L. Tsao, M. S. Platz, A. Kuhn, M. Vosswinkel and C. Wentrup, J. Am. Chem. Soc., 2004, 126, 237–249 CrossRef CAS PubMed.
- N. Aylward, D. Kvaskoff, J. Becker and C. Wentrup, J. Org. Chem., 2016, 81, 4609–4615 CrossRef CAS PubMed.
- J. Wang, G. Burdzinski, Z. D. Zhu, M. S. Platz, C. Carra and T. Bally, J. Am. Chem. Soc., 2007, 129, 8380–8388 CrossRef CAS PubMed.
- M. Kuzaj, H. Luerssen and C. Wentrup, Angew. Chem., Int. Ed. Engl., 1986, 25, 480–482 CrossRef.
- N. M. Lan, R. Burgard and C. Wentrup, J. Org. Chem., 2004, 69, 2033–2036 CrossRef CAS PubMed.
- N. P. Gritsan and M. S. Platz, Chem. Rev., 2006, 106, 3844–3867 CrossRef CAS.
- T. Baer and R. P. Tuckett, Phys. Chem. Chem. Phys., 2017, 19, 9698–9723 RSC.
- M. L. Sita, P. B. Changala, C. Xue, A. M. Burkhardt, C. N. Shingledecker, K. L. K. Lee, R. A. Loomis, E. Momjian, M. A. Siebert, D. Gupta, E. Herbst, A. J. Remijan, M. C. McCarthy, I. R. Cooke and B. A. McGuire, Astrophys. J., Lett., 2022, 938, L12 CrossRef.
- J. Cernicharo, M. Agundez, C. Cabezas, B. Tercero, N. Marcelino, J. R. Pardo and P. de Vicente, Astron. Astrophys., 2021, 649, L15 CrossRef CAS PubMed.
- A. M. Burkhardt, K. L. K. Lee, P. B. Changala, C. N. Shingledecker, I. R. Cooke, R. A. Loomis, H. J. Wei, S. B. Charnley, E. Herbst, M. C. McCarthy and B. A. McGuire, Astrophys. J., Lett., 2021, 913, L18 CrossRef CAS.
- B. A. McGuire, Astrophys. J., Suppl. Ser., 2022, 259, 30 CrossRef.
- J. Cernicharo, M. Agundez, R. I. Kaiser, C. Cabezas, B. Tercero, N. Marcelino, J. R. Pardo and P. de Vicente, Astron. Astrophys., 2021, 652, L9 CrossRef CAS.
- K. L. K. Lee, P. B. Changala, R. A. Loomis, A. M. Burkhardt, C. Xue, M. A. Cordiner, S. B. Charnley, M. C. McCarthy and B. A. McGuire, Astrophys. J., Lett., 2021, 910, L2 CrossRef CAS.
- M. C. McCarthy, K. L. K. Lee, R. A. Loomis, A. M. Burkhardt, C. N. Shingledecker, S. B. Charnley, M. A. Cordiner, E. Herbst, S. Kalenskii, E. R. Willis, C. Xue, A. J. Remijan and B. A. McGuire, Nat. Astron., 2021, 5, L10 Search PubMed.
- B. A. McGuire, A. M. Burkhardt, S. Kalenskii, C. N. Shingledecker, A. J. Remijan, E. Herbst and M. C. McCarthy, Science, 2018, 359, 202–205 CrossRef CAS PubMed.
- B. A. McGuire, R. A. Loomis, A. M. Burkhardt, K. L. K. Lee, C. N. Shingledecker, S. B. Charnley, I. R. Cooke, M. A. Cordiner, E. Herbst, S. Kalenskii, M. A. Siebert, E. R. Willis, C. Xue, A. J. Remijan and M. C. McCarthy, Science, 2021, 371, 1265 CrossRef CAS.
- J. Cernicharo, M. Agundez, R. I. Kaiser, C. Cabezas, B. Tercero, N. Marcelino, J. R. Pardo and P. de Vicente, Astron. Astrophys., 2021, 655, L15 CrossRef PubMed.
- M. Rapacioli, F. Calvo, C. Joblin, P. Parneix, D. Toublanc and F. Spiegelman, Astron. Astrophys., 2006, 460, 519–531 CrossRef CAS.
- M. C. McCarthy and B. A. McGuire, J. Phys. Chem. A, 2021, 125, 3231–3243 CrossRef CAS.
- M. Rapacioli, F. Calvo, C. Joblin, P. Parneix, D. Toublanc and F. Spiegelman, Astron. Astrophys., 2006, 460, 519–531 CrossRef CAS.
- B. A. McGuire, R. A. Loomis, A. M. Burkhardt, K. L. K. Lee, C. N. Shingledecker, S. B. Charnley, I. R. Cooke, M. A. Cordiner, E. Herbst, S. Kalenskii, M. A. Siebert, E. R. Willis, C. Xue, A. J. Remijan and M. C. McCarthy, Science, 2021, 371, 1265–1269 CrossRef CAS PubMed.
- A. A. G. M. Tielens, The Molecular Universe, The Molecular Universe. Proceedings of the International Astronomical Union. IAU Symposium 280, ed. J. Cernicharo, and R. Bachiller, Cambridge University Press, Cambridge, United Kingdom, 2011, 280, pp. 3–18 Search PubMed.
- N. Madhusudhan, M. Agundez, J. Moses and Y. Y. Hu, Space Sci. Rev., 2016, 205, 285–348 CrossRef CAS PubMed.
- H. R. Hrodmarsson and E. F. van Dishoeck, Astron. Astrophys., 2023, 675, A25 CrossRef CAS.
- E. Herbst, Chem. Soc. Rev., 2001, 30, 168–176 RSC.
- E. Vigren, J. Semaniak, M. Hamberg, V. Zhaunerchyk, M. Kaminska, R. D. Thomas, M. A. Ugglas, M. Larsson and W. D. Geppert, Planet. Space Sci., 2012, 60, 102–106 CrossRef CAS.
- J. M. Dyke, Phys. Chem. Chem. Phys., 2019, 21, 9106–9136 RSC.
- I. Fischer and P. Hemberger, Chem. Phys. Chem., 2023, 24, e202300334 CrossRef CAS PubMed.
- D. Schleier, P. Hemberger, A. Bodi and J. Bouwman, J. Phys. Chem. A, 2022, 126, 2211–2221 CrossRef CAS PubMed.
- E. Mendez-Vega, W. Sander and P. Hemberger, J. Phys. Chem. A, 2020, 124, 3836–3843 CrossRef CAS PubMed.
- B. Sztaray, K. Voronova, K. G. Torma, K. J. Covert, A. Bodi, P. Hemberger, T. Gerber and D. L. Osborn, J. Chem. Phys., 2017, 147, 013944 CrossRef PubMed.
- I. Fischer and S. T. Pratt, Phys. Chem. Chem. Phys., 2022, 24, 1944–1959 RSC.
- T. Bierkandt, P. Hemberger, P. Osswald, N. Gaiser, M. Hoener, D. Kruger, T. Kasper and M. Kohler, Proc. Combust. Inst., 2023, 39, 1699–1708 CrossRef.
- Z. H. Zhang, J. Perez-Ramirez, J. A. van Bokhoven, A. Bodi and P. Hemberger, Chimia, 2023, 77, 132–138 CrossRef CAS.
- A. Bodi, P. Hemberger and J. Perez-Ramirez, Nat. Catal., 2022, 5, 850–851 CrossRef CAS.
- J. Bouwman, M. N. McCabe, C. N. Shingledecker, J. Wandishin, V. Jarvis, E. Reusch, P. Hemberger and A. Bodi, Nat. Astron., 2023, 7, 423–430 CrossRef.
- B. Gans, S. Boye-Peronne, G. A. Garcia, A. Roder, D. Schleier, P. Halvick and J. C. Loison, J. Phys. Chem. Lett., 2017, 8, 4038–4042 CrossRef CAS PubMed.
- F. D. Ribeiro, G. C. Almeida, W. Wolff, H. M. Boechat-Roberty, M. L. M. Rocco and E. F. da Silveira, Mon. Not. R. Astron. Soc., 2020, 492, 2140–2150 CrossRef CAS.
- M. Johnson, A. Bodi, L. Schulz and T. Gerber, Nucl. Instrum. Meth. A, 2009, 610, 597–603 CrossRef CAS.
- J. Michalak, H. B. Zhai and M. S. Platz, J. Phys. Chem., 1996, 100, 14028–14036 CrossRef CAS.
- D. W. Kohn, H. Clauberg and P. Chen, Rev. Sci. Instrum., 1992, 63, 4003–4005 CrossRef CAS.
- Q. Guan, K. N. Urness, T. K. Ormond, D. E. David, G. B. Ellison and J. W. Daily, Int. Rev. Phys. Chem., 2014, 33, 447–487 Search PubMed.
- P. Hemberger, J. A. van Bokhoven, J. Perez-Ramirez and A. Bodi, Catal. Sci. Technol., 2020, 10, 1975–1990 RSC.
- P. Hemberger, A. Bodi, T. Bierkandt, M. Kohler, D. Kaczmarek and T. Kasper, Energy Fuels, 2021, 35, 16265–16302 CrossRef CAS.
- A. Bodi, B. Sztáray, T. Baer, M. Johnson and T. Gerber, Rev. Sci. Instrum., 2007, 78, 084102 CrossRef PubMed.
- B. Sztáray and T. Baer, Rev. Sci. Instrum., 2003, 74, 3763–3768 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson and H. Nakatsuji, Gaussian 16, 2016 Search PubMed.
- J. A. Montgomery, M. J. Frisch, J. W. Ochterski and G. A. Petersson, J. Chem. Phys., 1999, 110, 2822–2827 CrossRef CAS.
- E. Reusch, F. Holzmeier, M. Gerlach, I. Fischer and P. Hemberger, Chem. – Eur. J., 2019, 25, 16652–16659 CrossRef CAS PubMed.
- P. Hemberger, X. K. Wu, Z. Y. Pan and A. Bodi, J. Phys. Chem. A, 2022, 126, 2196–2210 CrossRef CAS PubMed.
- C. Fernholz, A. Bodi and P. Hemberger, J. Phys. Chem. A, 2022, 126, 9022–9030 CrossRef CAS PubMed.
- K. Kanayama, C. Fernholz, H. Nakamura, K. Maruta, A. Bodi and P. Hemberger, Chem. Phys. Chem., 2023, e202300359, DOI:10.1002/cphc.202300359.
- M. Saraswat, A. Portela-Gonzalez, G. Karir, E. Mendez-Vega, W. Sander and P. Hemberger, J. Phys. Chem. A, 2023 DOI:10.1021/acs.jpca.3c04688.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp04064j |
| This journal is © the Owner Societies 2023 |