
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA local point of view of the Cu(100) → NiTPP charge transfer at the NiTPP/Cu(100) interface†
Silvia
Carlotto
 ab,
Alberto
Verdini
*c,
Giovanni
Zamborlini
d,
Iulia
Cojocariu
ef,
Vitaliy
Feyer
gh,
Luca
Floreano
i and
Maurizio
Casarin
*a
ab,
Alberto
Verdini
*c,
Giovanni
Zamborlini
d,
Iulia
Cojocariu
ef,
Vitaliy
Feyer
gh,
Luca
Floreano
i and
Maurizio
Casarin
*a
aUniversity of Padova, Department of Chemical Sciences, via F. Marzolo 1, 35131, Padova, Italy. E-mail: maurizio.casarin@unipd.it
bICMATE - CNR c/o University of Padova, Department of Chemical Sciences, via F. Marzolo 1, via F. Marzolo 1, 35131, Padova, Italy
cIOM – CNR c/o University of Perugia, Department of Physics and Geology, via A. Pascoli, 06123, Perugia, Italy
dTU Dortmund University, Department of Physics, Otto-Hahn-Straβe 4, 44227 Dortmund, Germany
eUniversity of Trieste, Department of Physics, Via A. Valerio 2, 34127 Trieste, Italy
fElettra-Sincrotrone, S.C.p.A., S.S. 14 - km 163.5, 34149 Trieste, Italy
gForschungszentrum Jülich GmbH, Peter Grünberg Institute (PGI-6), Leo-Brandt-Straβe, 52428 Jülich, Germany
hDuisburg-Essen University, Department of Physics and Center for Nanointegration Duisburg-Essen (CENIDE), 47048 Duisburg, Germany
iCNR – IOM, Lab. TASC, Basovizza S.S. 14, km 163.5, 34149 Trieste, Italy
First published on 13th September 2023
Abstract
A precise understanding, at the molecular level, of the massive substrate → adsorbate charge transfer at the NiTPP/Cu(100) interface has been gained through the application of elementary symmetry arguments to the structural determination of the NiTPP adsorption site by photoelectron diffraction (PED) measurements and Amsterdam density functional calculations of the free D4h NiTPP electronic structure. In particular, the PED analysis precisely determines that, among the diverse NiTPP chemisorption sites herein considered (fourfold hollow, atop, and bridge), the fourfold hollow one is the most favorable, with the Ni atom located at 1.93 Å from the surface and at an internuclear distance of 2.66 Å from the nearest-neighbors of the substrate. The use of elementary symmetry considerations enabled us to provide a convincing modeling of the NiTPP–Cu(100) anchoring configuration and an atomistic view of the previously revealed interfacial charge transfer through the unambiguous identification of the adsorbate π* and σ* low-lying virtual orbitals, of the substrate surface atoms, and of the linear combinations of the Cu 4s atomic orbitals involved in the substrate → adsorbate charge transfer. In addition, the same considerations revealed that the experimentally reported Ni(II) → Ni(I) reduction at the interface corresponds to the fingerprint of the chemisorption site of the NiTPP on Cu(100).
Introduction
The incipit of Ghosh's contribution to the collective volume Letters to a Young Chemist is worded as follows: “Porphyrins are everywhere, as far as the living world is concerned”.1 Although detectable in abiotic systems, the pivotal role played by porphyrin-related molecules in fundamental biological processes such as oxygen transport and storage (haemoglobin and myoglobin), photosynthesis (chlorophyll), and electron transport during cellular respiration and photosynthesis (cytochromes)2,3 supports their evocative classification: “pigments of life”.4,5 Additionally, porphyrin derivatives have been extensively used in cancer treatment due to their therapeutic and diagnostic properties.6 Besides the living world, the relevance of this class of molecules extends nowadays to technological fields ranging from electronics,7 to solar cells,8 and sensors,9 thus justifying the constantly rising interest towards them and motivation to develop new porphyrin-like species, whose electronic and optical properties can be tuned through molecular engineering.10–13 Therefore, surface-supported supramolecular porphyrin arrays may be thought of as functional components in nanodevices.14 In addition, adsorbate–substrate and adsorbate–adsorbate interactions often lead to charge transfer between the adsorbate frontier molecular orbitals (MO)15 and the substrate,16,17 resulting in the addition of spin freedom degrees,18,19 when using magnetic substrates, through the generation of spin-polarized hybrid interface states.20,21 The electronic and magnetic properties of surface-supported metal (M) porphyrin arrays can be further customized through the coordination of axial ligands to M,19,22–25 which can modify the M oxidation number, its spin state, and then its magnetic moment, thus affecting the magnetic anisotropy. Indeed, small molecules such as NO, NH3, and NO2 have been shown to bind to the central M of porphyrins, inducing changes in its spin state. The bonding with the surface and the additional ligands increases the M coordination number, and usually reduces the magnetic anisotropy because of the symmetry reduction of the M environment, which lowers the energy barrier among different magnetic configurations. It is also noteworthy that electronic and structural factors including the occupation number of M-based 3d atomic orbitals (AOs), the M oxidation number, and the complex conformational flexibility,26–29 influence reactivity towards axial ligands.Among the numerous M-porphyrin/metal interfaces studied so far,13 the NiTPP/Cu(100) interface (the free NiTPP corresponds to Ni(II) tetraphenyl-porphyrin)30 has received great attention because of its important role in the development of molecular materials for cutting-edge applications in spintronics, memory storage, and quantum computing.26,31–33
What is known so far about the NiTPP/Cu(100) interface? The experimental evidence for NiTPP/Cu(100) indicates that (i) both the adsorbate and the substrate have a local fourfold (ff) symmetry, and two NiTPP rotational domains, only slightly misaligned with respect to the Cu(100) [001] direction, are present on the surface;31 (ii) a massive Cu(100) → NiTPP charge transfer takes place at the interface, which leads to the partial occupation of low-lying unoccupied NiTPP MOs up to LUMO+3 (LUMO stands for the lowest unoccupied MO);31 (iii) the pristine macrocycle core (pmcNiP) of the chemisorbed NiTPP has a flat geometry32 and is characterized by the presence of the highly reactive 3d9 Ni(I) species.33 Moving from experimental knowledge to the theoretical scenario, periodic density functional theory (DFT) calculations31 indicate that the chemisorbed NiTPP assumes a bowl-shaped arrangement,34 whose base corresponds to the pmcNiP core, which maintains its planarity and lies ∼2 Å above the Cu(100) surface. In addition, the numerical experiments carried out by Zamborlini et al.31 confirm the substantial substrate → NiTPP charge transfer implying the partial occupation of the low-lying NiTPP virtual orbitals. Therefore, numerical experiments carried out with the Amsterdam density functional (ADF)35 package on free D4h CuTPP (isoelectronic with the chemisorbed NiTPP typical for the presence of the 3d9 Ni(I) species) indicate that the empty, low-lying, highly delocalized pmcCuP-based π orbitals (pmcπ*) are the doubly degenerate 13eg and the 9b1u MOs, while the half-occupied pmcσ* orbital, accounting for the antibonding Cu–N σ interaction and strongly localized on the 3d9 Cu(II) 3dx2−y2 AO, corresponds to the 12b1g MO.36 All these data lead us to assume that the experimentally revealed Cu(100) → NiTPP massive charge transfer up to LUMO+3 involves, besides the empty Ni(II)-based 12b1gpmcσ* LUMO+2, the whole set of low-lying pmcπ* MOs (LUMO, LUMO+1, and LUMO+3).37
Despite such a huge amount of electronic and structural information, a precise understanding, at the molecular level, of the Cu(100) → NiTPP charge transfer is still lacking. As a matter of fact, neither the recognition of the adsorbate and substrate MOs involved in such a process nor the identification of the Cu surface atoms (CuS) involved in the partial filling of the low-lying NiTPP virtual orbitals is unambiguously reported in the literature. In this paper, elementary symmetry and geometry arguments were used in combination with experimental literature reports and novel structural photoelectron diffraction (PED) results to provide a convincing molecular picture of NiTPP grafting to the substrate and then a local point of view of the Cu(100) → NiTPP charge transfer. Moreover, it is shown that the experimentally reported Ni(II) → Ni(I) reduction at the interface33 provides the fingerprint of the chemisorption site of the NiTPP on Cu(100).
Experimental and computational details
PED experiments have been performed at the ALOISA beamline of the Elettra synchrotron radiation facility in Trieste38 and in an ultra-high vacuum (UHV) experimental chamber with a base pressure of 10–11 mbar. Measurements have been accomplished with photon energies ranging from 850 to 1440 eV and the intensity of the Ni 2p core level was monitored with the detector (acceptance angle of 2°, FWHM) aligned along the surface normal and the surface oriented at 4° grazing angle (in close to p-polarization). Prior to the intensity integration of the Ni 2p lines, a Shirley-like background has been subtracted from each spectrum. Then, the intensity modulation curve as a function of the kinetic energy of the photoelectron has been normalized to the photon flux and the calculated Ni 2p cross-section in the measured photon energy range.39 Reference high-resolution X-ray photoelectron spectroscopy (XPS) spectra of the N and C 1s regions have been collected at a photon energy of 515 eV by using linearly p-polarized light at a grazing incidence of 4° on the sample surface and by maintaining the hemispherical electron energy analyzer in the normal emission geometry. The binding energies in the XPS data have been referred to the Fermi level of the Cu(100) substrate. The clean Cu(100) surface has been prepared by cycles of Ar+ ion sputtering at 2.0 keV, followed by annealing at 800 K. The absence of contaminants on the surface has been verified by XPS measurements at 650 eV (O 1s) and 515 eV (N and C 1s). The ordering of the surface and the molecular layer has been performed using the reflective high-energy electron diffraction technique. A few dozen mg of NiTPP powder (Porphyrin Systems) have been loaded in a quartz crucible of a homemade Knudsen cell-type evaporator. Before the measurements, the NiTPP sample has been carefully degassed at 480 K for several days while the base pressure of the UHV system was monitored. NiTPP molecules have been then thermally evaporated at ∼550 K onto the copper substrate maintained at room temperature. The molecular coverage has been calibrated with a quartz micro-balance, and the resulting deposition rate was 15 min ML−1. The nominal coverage for all experiments herein presented is a saturated monolayer. Therefore, it is well known that organic molecules tend to decompose when exposed to ionizing radiation. XPS spectra have been then monitored over time to exclude any possible radiation-induced damage. No spectral changes were observed after 1 h, so we confirm that NiTPP molecules are reasonably stable under the adopted experimental conditions.The ground state electronic and structural properties of the free NiTPP are herein investigated by using the ADF package35 within the assumption of an idealized D4h symmetry40 (see Fig. S1 in the ESI†) and by performing nonrelativistic DFT calculations whose set up (generalized gradient corrections self-consistently included through the Becke–Perdew formula,41,42 a triple-ζ with a polarization function Slater-type basis set for all the atoms, Ni 1s–2p AOs and N and C 1s AO kept frozen throughout the calculations) has been provided by the possibility of comparing the results of our calculations with homogeneous theoretical outcomes pertinent to the free CuTPP.36
Results and discussion
Chemically selected quantitative structural information about the local adsorption geometry of NiTPP on Cu(100) can be obtained by monitoring the intensity of the Ni 2p core level line as a function of the energy of the incoming photon. In the PED process, the photoelectron can undergo a series of scattering processes from the scatter atoms (Cu) surrounding the emitter (Ni) along its path to the detector. The final state results from the interference between the direct and the coherently scattered components of the photoelectron signal. Thus, the resulting interference is determined by the path-length differences and the scattering phase shifts between the electron waves. These in turn depend on the kinetic energy of the electron, the detector geometry, and the position of the emitter atom relative to the scatters. By varying the kinetic energy of the emitted electrons, the interference conditions can be changed, and the modulations of the photoemission intensity may be used to extract geometrical information, which allows determination of the position of the Ni atom with respect to the copper substrate. As already mentioned, the oscillations of the core level line intensity as a function of the wave vector k of the outgoing electron (I(k)) contain information relative to the diffraction processes at the interface. The scattering anisotropy χ(k) can be described by eqn (1):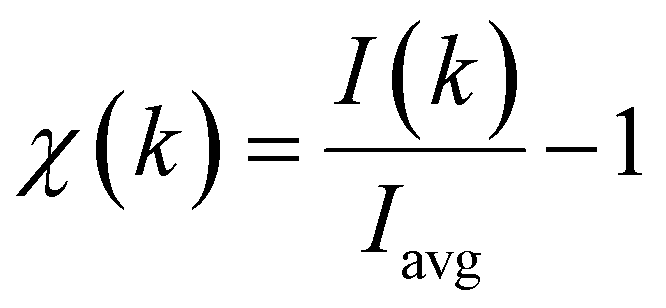 | (1) |
Numerical simulations of PED data have been carried out using the MSCD package43 (multiple scattering up to the eighth order and a Rehr–Albers order44 of 2 have been used) and by considering three diverse chemisorption sites (atop, bridge, and ff hollow) shown schematically in Fig. 1. The obtained results have been then tested by means of the usually adopted reliability factor Rf (see eqn (2)):
 | (2) |
 | ||
| Fig. 1 Schematic representation of the atop, bridge (b), and ff hollow (ffh) chemisorption sites on the bulk terminated Cu(100) topmost layer. | ||
 | ||
| Fig. 2 PED anisotropy χ measured with the detector along the surface normal for the Ni 2p core level. The corresponding simulation, which includes a coarse optimization of the inner potential (namely 10 eV), performed for the Ni atom placed at the atop (top), bridge (middle), and ff hollow (bottom) sites are superimposed on the spectrum. | ||
R f corresponding to the Ni species placed on an ff hollow site (Rffhf) has, among the diverse chemisorption positions herein considered, the minimum value (Rffhf ∼ 0.54, Ratopf ∼ 0.7, and Rbridgef ∼ 0.84; see Fig. 3). In addition, the analysis of the PED data provides the following structural information: the adsorption height of Ni from the outermost layer of the substrate and the internuclear distance between Ni and the nearest-neighbors of the substrate (nnCuS) are 1.95/2.66, 2.6/2.6, and 2.5/2.8 Å at the ffh, atop, and bridge chemisorption sites, respectively.
 | ||
| Fig. 3 Calculated R-factor (Rf) for the three different adsorption sites herein considered. | ||
The most favored ffh site has been further refined by varying the height of Ni from the substrate and the inner potential (V0). The resulting best fit provides the following values: Rffhf = 0.52, a Ni–CuS height of 1.93 Å, and V0 = 10.7 eV (see the corresponding map in Fig. 4).
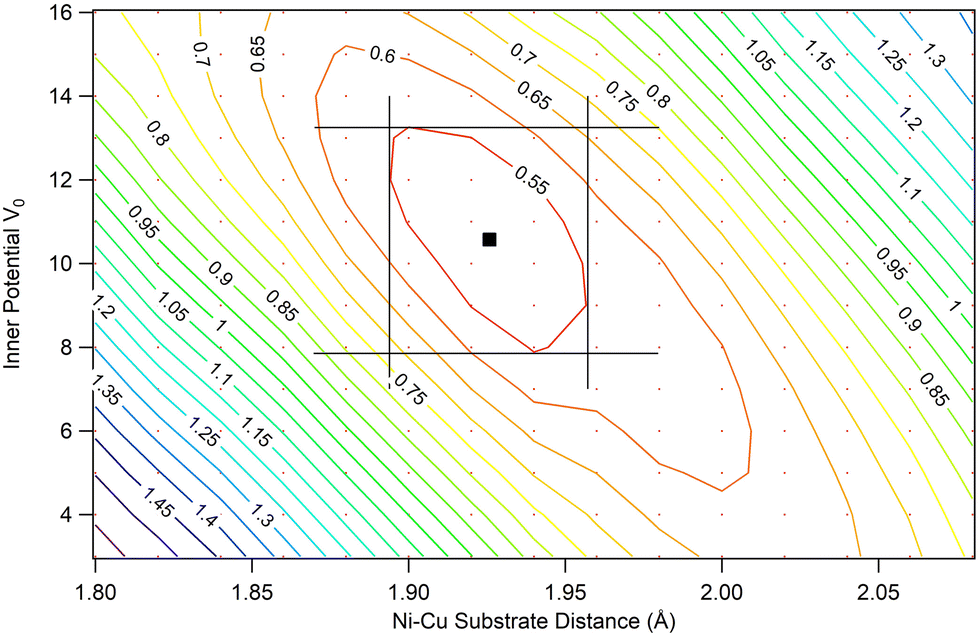 | ||
| Fig. 4 Contour plot of the MSCD simulation Rffhf for the inner potential V0 and the Ni–Cu substrate distance. Black lines correspond to the region where Rffhf is ≤ 0.55 and the lowest Rffhf = 0.52 corresponds to a Ni–Cu substrate distance of 1.93 Å and V0 = 10.7 eV. | ||
PED measurements at normal emission are best suited for determining the vertical distance of the emitter from the surface and the structural information herein obtained perfectly agrees with experimental and theoretical literature data pertaining not only to the NiTPP/Cu(100) system,31 but also to similar MTPP adsorbed on Cu(111) and Cu(110) surfaces.45,46 Nevertheless, we cannot be silent about the aptitude, substantially phenomenological, characterizing all this matter. In other words, neither experimental nor theoretical literature results provide an answer to the question of why the ffh chemisorption site of the NiTPP/Cu(100) interface is more stable than those on the atop and bridge ones. Let us see how the exploitation of elementary symmetry considerations allows us to gain further insights into this issue.
The ff quantization axis, characteristic of the free NiTPP (see Fig. S1 in the ESI†), lifts the fivefold degeneracy of the Ni(II) 3d AOs to generate five MOs transforming as the a (z2), b (x2 − y2 and xy), and e (xz, yz) irreducible representations (IRs) of the C4 point group.40 All of these MOs, except the pmcσ* one, are fulfilled; hence, only the 3dx2−y2 AO of the pristine 3d8 Ni(II) can accept charge regardless of the NiTPP chemisorption site. Upon NiTPP deposition on Cu(100), the N-chelated Ni species is effectively reduced to the oxidation state +1 thus assuming a 3d9 configuration, as measured by X-ray absorption spectroscopy at the Ni L3-edge.33 Since the ff quantization axis is locally preserved in the adsorbate as well as in the substrate,31 we can assess that: (i) a single CuS atom lying on the ff axis may transfer electronic charge only through its half-occupied 4s AO (of symmetry a in the C4 point group),40 and (ii) the symmetry adapted linear combinations (SALC) of 4s AOs localized on four equivalent CuS atoms transform as a + b + e IRs of the C4 point group.40
These elementary symmetry considerations exclude the possibility that the Ni(II) → Ni(I) reduction is driven by a charge transfer from a CuS atom beneath Ni when NiTPP is chemisorbed on Cu(100) atop a ff surface atom (Cusffa). More specifically, the single nnCusffa located at 2.6 Å from Ni (see above) could transfer electronic charge to Ni(II) only through its half-occupied 4s AO, which transforms as the IR a of the C4 group. This AO is orthogonal to the empty MO localized on the Ni atom (namely, on Ni(II) 3dx2−y2 AO, transforming as the IR b of the C4 group40) reminiscent of the NiTPP pmcσ* 12b1g LUMO+2 (see Fig. 5),47 and then the transfer is symmetry forbidden.
 | ||
| Fig. 5 3D contour plot (CP) of the free D4h NiTPP pmcσ* 12b1g LUMO+2.47 Displayed isosurfaces correspond to ±0.02 e1/2 × Å−3/2 values. The pmcNiP core lies in the xy plane in the adopted framework. | ||
Before applying analogous symmetry arguments to the chemisorption of NiTPP at a Cu(100) ffh site,48 it deserves to be noticed that the internuclear distance between Ni and each of the four nnnCusffa (nnn stands for next nearest neighbor) in the bulk terminated Cu(100) surface amounts to 3.646 Å;49 too far to have an effective overlap between the empty pmcσ* 12b1g MO and the SALC of symmetry b of the nnnCusffa 4s AOs shown in Fig. 6.
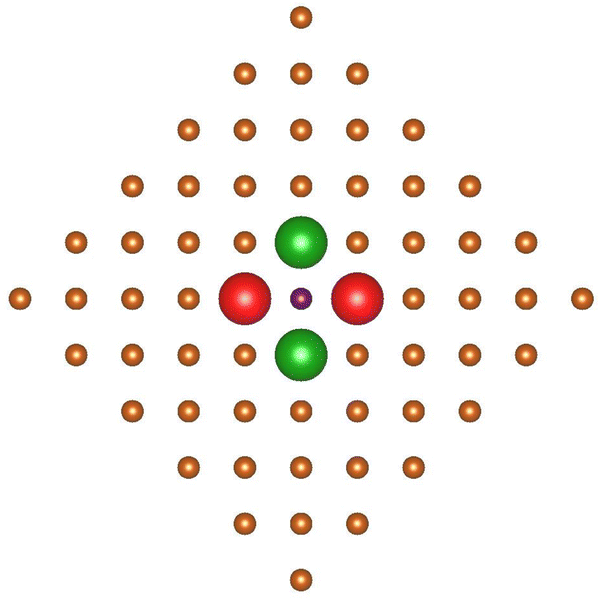 | ||
| Fig. 6 Schematic representation of the nnnCusffa 4s AOs SALC of symmetry b. Large red and green spheres represent CuS 4s AOs and corresponding different phases. | ||
Examination of Fig. 7 clearly shows that the ffh chemisorption site is much more suitable to foster the Ni(II) → Ni(I) reduction.
 | ||
| Fig. 7 Schematic representation of the pmcNiP core superimposed to the bulk terminated Cu(100) at a ffh site. Only atoms of the Cu(100) topmost layer are displayed and the planar pmcNiP core31 of the optimized free D4h NiTPP is placed 1.93 Å above it. In the left (right) panel, the Ni–N bonds are aligned with the 〈011〉 (〈001〉) directions. | ||
In fact, (i) at the ffh chemisorption site, the internuclear distance between Ni and its four nearest neighbors (nnCuSffh) is 2.644 Å;50 (ii) the SALC of symmetry b of the nnCuSffh 4s AOs (see Fig. S2 in the ESI†) perfectly overlaps with the empty Ni(II)-based pmcσ* MO when the Ni–N bonds are aligned with the 〈001〉 directions (see the right panel of Fig. 7 and 8); (iii) periodic DFT calculations carried out by some of us31 ultimately indicated that, among the atop, bridge, and ffh chemisorption sites, the ffh one is more stable than the atop and bridge sites by 2 and 1 eV, respectively. Therefore, it is noteworthy that the higher stability of the ffh site for NiTPP on Cu(100) agrees well with the experimental evidence pertaining to M(II) phthalocyanine (Fe, Co, Cu, and Zn) deposited on the same substrate:51–54 a fulfilled M 3dz2 AO (M = Cu and Zn) favours the ffh site,53,54 while a partially occupied M 3dz2 AO (M = Fe and Co) fosters the ffa one.51,52 This is not surprising if we consider that, at the ffa site, the antibonding combination of the M 3dz2 and nnCuSffh 4s AOs (both of them transforming as the IR a of the C4 point group)40 would be populated (vacant) if the M 3dz2 AO is fulfilled (half-filled).
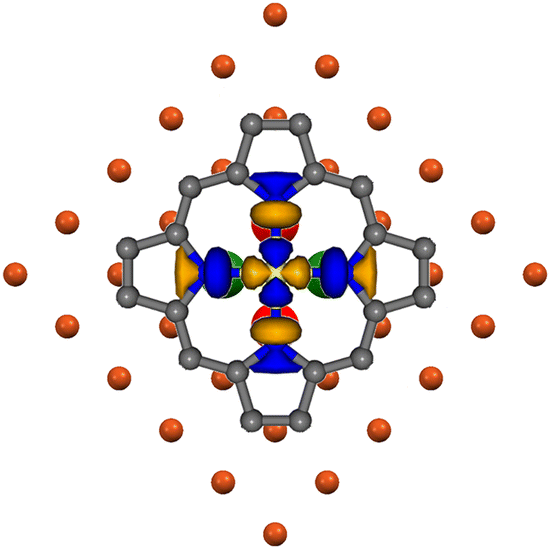 | ||
| Fig. 8 Superposition of the pmcσ* 12b1g-based MO (LUMO+2)37 3D CP to the nnCuSffh 4s AOs SALC of symmetry b of the bulk terminated Cu(100). Ni–N bonds are aligned with the 〈001〉 directions. Large red and green spheres represent CuS 4s AOs and corresponding different phases. | ||
Besides a rationale for the experimentally reported Ni(II) → Ni(I) reduction,33 a real fingerprint of the NiTPP chemisorption site on Cu(100), the same simple arguments can be used to gain insights into the substrate → adsorbate charge transfer involving the doubly degenerate pmcπ* orbitals reminiscent of the NiTPP 13eg LUMO (see the left panel in Fig. 9). Examination of the figure reveals that the 13eg-based MOs, significantly localized on the N 2pπ AOs of the pmcNiP core, can constructively overlap with the nnCuSffh 4s AOs SALC of symmetry e of the bulk terminated Cu(100) (see Fig. S2 in the ESI† and the right panel of Fig. 9).
 | ||
| Fig. 9 (left panel) 3D CP of one component of the free D4h NiTPP pmcπ* 13eg LUMO;37 (right panel) superposition of the same component of the doubly degenerate pmcπ* 13eg-based MO to the nnCuSffh 4s AOs SALC of symmetry e of the bulk terminated Cu(100). Ni–N bonds are aligned with the 〈001〉 directions. Large red and green spheres represent CuS 4s AOs and corresponding different phases. | ||
As such, it is noteworthy that the nnCuSffh–N distance with the pmcNiP core of the optimized D4h NiTPP at 1.93 Å from the bulk terminated Cu(100) surface and the Ni–N bonds aligned with the 〈001〉 directions is, as pointed out by Zamborlini et al.,31 “extremely short” (1.938 Å in the model herein considered).55
Angle-resolved photoemission tomography experiments at the NiTPP/Cu(100) interface revealed that the massive substrate → adsorbate charge transfer takes place through the partial filling of pmcNiP low-lying unoccupied MOs up to LUMO+3.31 The last one is reminiscent of the D4h NiTPP pmcπ* 9b1u MO, which, for symmetry reasons,56 bears no density of states on the N atoms; hence, its nodal properties prevent any possibility to receive charge from the nnCuSffh 4s AOs SALC of symmetry b (see Fig. S2 of the ESI†), while the opposite is true when the eight nnnCuSffh are considered. The SALCs of the nnnCuSffh 4s AOs transform as 2a + 2b + 2e and are sketched in Fig. S3 of the ESI.† Inspection of this figure reveals that the nodal properties of the SALC labeled 2b in Fig. S3 (ESI†) make it very well suited for transferring electronic charge into the pmcπ* 9b1u-like MO (see Fig. 10).
 | ||
| Fig. 10 (left panel) 3D CP of the free D4h NiTPP pmcπ* 9b1u MO (LUMO+3); (right panel) superposition of the same pmcπ* MO to the nnnCuSffh 4s AOs SALC of symmetry b of the bulk terminated Cu(100). Ni–N bonds are aligned with the 〈001〉 directions. Large red and green spheres represent CuS 4s AOs and corresponding different phases. | ||
Analogous considerations hold for the nnnCuSffh SALCs labeled 1e in Fig. S3 in the ESI,† which may contribute to the Cu(100) → pmcNiP charge transfer by involving the Cmeso 2pπ AOs (see Fig. 11) of the pmcπ* 13eg-based MOs (LUMO/LUMO+1).
 | ||
| Fig. 11 Superposition of one component of the doubly degenerate pmcπ* 13eg-based MO to the nnnCuSffh 4s AOs SALC of symmetry e of the bulk terminated Cu(100). Ni–N bonds are aligned with the 〈001〉 directions. Large red and green spheres represent CuS 4s AOs and corresponding different phases. | ||
As such, it can be useful to mention that, with the pmcNiP core of the optimized D4h NiTPP at 1.93 Å from the surface, and the highly reactive species Ni(I) located at the ffh site, the internuclear distances of the eight nnnCuSffh from the pmcNiP α and β pyrrolic C atoms are 2.204 and 2.311 Å, respectively, while the internuclear distance between nnnCuSffh and Cmeso atoms is 2.348 Å.57
Conclusions
The experimentally reported Ni(II) → Ni(I) reduction of NiTPP/Cu(100) controls the molecular adsorption at an ffh site, as well as the alignment of the Ni–N bonds along the substrate 〈001〉 directions. Therefore, the Ni reduction is driven by the charge transfer into the Ni 3dx2−y2-based 12b1g MO (LUMO+2) from the four nnCuSffh underneath the N atoms. The same nnCuSffh are responsible for the charge transfer into the pmcπ* orbitals reminiscent of the NiTPP 13eg LUMO/LUMO+1 strongly localized on the N atoms, whereas the partial filling of the pmcπ* 9b1u-based LUMO+3 (solely localized on the C atoms of the pmcNiP core) is driven by the charge transfer from the eight nnnCuSffh. The latter ones may additionally contribute to the partial filling of the LUMO/LUMO+1 through the Cmeso atoms. In a few words, symmetry and geometry considerations herein presented provide the modeling of the NiTPP–Cu(100) anchoring configuration from an atomistic view, as dictated by the interfacial charge transfer.Author contributions
We strongly encourage authors to include author contributions and recommend using CRediT for standardised contribution descriptions. Please refer to our general author guidelines for more information about authorship.Conflicts of interest
There are no conflicts to declare.Notes and references
- A. Ghosh, Letters to a young Chemist, JohnWiley & Sons, Hoboken, NJ, USA, 2011; p. 34 Search PubMed.
- D. L. Nelson, M. M. Cox and A. A. Hoskins, Lehninger Principles of Biochemistry, Freeman, W.H. Macmillan learning, New York, NY, USA, 8th edn, 2021 Search PubMed.
- J. M. Berg, J. L. Tymoczko and L. Stryer, Biochemistry, W. H. Freeman, New York, NY, USA, 5th edn, 2002 Search PubMed.
- A. R. Battersby, C. J. R. Fookes, G. W. J. Matcham and E. McDonald, Nature, 1980, 285, 17–21 CrossRef CAS PubMed.
- A. R. Battersby, Nat. Prod. Rep., 1987, 4, 77–87 RSC.
- X. Xue, A. Lindstrom and Y. Li, Bioconjugate Chem., 2019, 30, 1585–1603 CrossRef CAS PubMed.
- A. Tsuda and A. Osuka, Science, 2001, 293, 79–82 CrossRef CAS PubMed.
- M. Planells, A. Forneli, E. Martínez-Ferrero, A. Sánchez-Díaz, M. A. Sarmentero, P. Ballester, E. Palomares and B. C. O’Regan, Appl. Phys. Lett., 2008, 92, 153506 CrossRef.
- N. A. Rakow and K. S. Suslick, Nature, 2000, 406, 710–713 CrossRef CAS PubMed.
- The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, New York, NY, USA, 2000 Search PubMed.
- D. Dini and M. Hanack, J. Porphyrins Phthalocyanines, 2004, 8, 915–933 CrossRef CAS.
- C. Di Natale, D. Monti and R. Paolesse, Mater. Today, 2010, 13, 46–52 CrossRef CAS.
- J. M. Gottfried, Surf. Sci. Rep., 2015, 70, 259–379 CrossRef CAS.
- C. M. Drain, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5178–5182 CrossRef CAS PubMed.
- G. L. Miessler, P. J. Fischer and D. A. Tarr, Inorganic Chemistry, Pearson, New York, NY, USA, 5th edn, 2013, p. 137 Search PubMed.
- M. Dyer, A. Robin, S. Haq, R. Raval, M. Persson and J. Klime, ACS Nano, 2011, 5, 1831–1838 CrossRef CAS PubMed.
- F. Bischoff, K. Seufert, W. Auwärter, S. Joshi, S. Vijayaraghavan, D. Écija, K. Diller, A. C. Papageorgiou, S. Fischer, F. Allegretti, D. A. Duncan, F. Klappenberger, F. Blobner, R. Han and J. V. Barth, ACS Nano, 2013, 7, 3139–3149 CrossRef CAS PubMed.
- H. Wende, M. Bernien, J. Luo, C. Sorg, N. Ponpandian, J. Kurde, J. Miguel, M. Piantek, X. Xu, P. Eckhold, W. Kuch, K. Baberschke, P. M. Panchmatia, B. Sanyal, P. M. Oppeneer and O. Eriksson, Nat. Mater., 2007, 6, 516–520 CrossRef CAS PubMed.
- C. Wäckerlin, D. Chylareck, A. Kleibert, K. Müller, C. Iacovita, F. Nolting, T. A. Jumg and N. Ballav, Nat. Commun., 2010, 61, 1–7 Search PubMed.
- S. Lach, A. Altenhof, K. Tarafder, F. Schmitt, M. E. Ali, M. Vogel, J. Sauther, P. M. Oppeneer and C. Ziegler, Adv. Funct. Mater., 2012, 22, 989–997 CrossRef CAS.
- S. Jakobs, A. Narayan, B. Stadtmüller, A. Droghetti, I. Rungger, Y. S. Hor, S. Klyatskaya, D. Jungkenn, J. Stöckl, M. Laux, O. L. A. Monti, M. Aeschlimann, R. J. Cava, M. Ruben, S. Mathias, S. Sanvito and M. Cinchetti, Nano Lett., 2015, 15, 6022–6029 CrossRef CAS.
- P. Gambardella, S. Stepanow, A. Dmitriev, J. Honolka, F. M. F. De Groot, M. Lingenfelder, S. Sen Gupta, D. D. Sarma, P. Bencok, S. Stanescu, S. Clair, S. Pons, N. Lin, A. P. Seitsonen, H. Brune, J. V. Barth and K. Kern, Nat. Mater., 2009, 8, 189–193 CrossRef CAS.
- C. Wäckerlin, K. Tarafder, J. Girovsky, J. Nowakowski, T. Hählen, A. Shchyrba, D. Siewert, A. Kleibert, F. Nolting, P. M. Oppeneer, T. A. Jung and N. Ballav, Angew. Chem., Int. Ed., 2013, 52, 4568–4571 CrossRef.
- J. Miguel, C. F. Hermanns, M. Bernien, A. Krüger and W. Kuch, J. Phys. Chem. Lett., 2011, 2, 1455–1459 CrossRef CAS.
- I. Cojocariu, S. Carlotto, H. M. Sturmeit, G. Zamborlini, M. Cinchetti, A. Cossaro, A. Verdini, L. Floreano, M. Jugovac, P. Puschnig, C. Piamonteze, M. Casarin, V. Feyer and C. M. Schneider, Chem. – Eur. J., 2021, 27, 3526–3535 CrossRef CAS PubMed.
- H. M. Sturmeit, I. Cojocariu, A. Windischbacher, P. Puschnig, C. Piamonteze, M. Jugovac, A. Sala, C. Africh, G. Comelli, A. Cossaro, A. Verdini, L. Floreano, M. Stredansky, E. Vesselli, C. Hohner, M. Kettner, J. Libuda, C. M. Schneider, G. Zamborlini, M. Cinchetti and V. Feyer, Small, 2021, 17, 2104779 CrossRef CAS.
- I. Cojocariu, S. Carlotto, G. Zamborlini, M. Jugovac, L. Schio, L. Floreano, M. Casarin, V. Feyer and C. M. Schneider, J. Mater. Chem. C, 2021, 9, 12559–12565 RSC.
- I. Cojocariu, S. Carlotto, N. Jugovac, L. Floreano, M. Casarin, V. Feyer and C. M. Schneider, J. Mater. Chem. C, 2022, 10, 9748–9757 RSC.
- P. Knecht, J. Reichert, P. S. Deimel, P. Feulner, F. Haag, F. Allegretti, M. Garnica, M. Schwarz, W. Auwärter, P. T. P. Ryan, T. L. Lee, D. A. Duncan, A. P. Seitsonen, J. V. Barth and A. C. Papageorgiou, Angew. Chem., Int. Ed., 2021, 60, 16561–16567 CrossRef CAS PubMed.
- Optimized Cartesian coordinates of the free D4h NiTPP are reported in Table S1 of the ESI while its structure is displayed in Fig. S1 of the ESI†.
- G. Zamborlini, D. Lüftner, Z. Feng, B. Kollmann, P. Puschnig, C. Dri, M. Panighel, G. Di Santo, A. Goldoni, G. Comelli, M. Jugovac, V. Feyer and C. M. Schneider, Nat. Commun., 2017, 8, 1–8 CrossRef PubMed.
- I. Cojocariu, H. M. Sturmeitb, G. Zamborlini, A. Cossaro, A. Verdini, L. Floreano, E. D'Incecco, M. Stredansky, E. Vesselli, M. Jugovaca, M. Cinchetti, V. Feyer and C. M. Schneider, Appl. Surf. Sci., 2020, 504, 144343 CrossRef CAS.
- G. Zamborlini, M. Jugovac, A. Cossaro, A. Verdini, L. Floreano, D. Lüftner, P. Puschnig, V. Feyer and C. M. Schneider, Chem. Commun., 2018, 54, 13423–13426 RSC.
- The NiTPP Ph rings bonded to Cm are upwards tilted. The tilt angle ϕ, defined as the angle between the pcmNiP plane and the C–C bond connecting Ph rings to Cm atoms, amounts to ∼140° (see Fig. 2d of ref. 31).
- ADF2014, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands, https://www.scm.com.
- G. Mangione, M. Sambi, S. Carlotto, A. Vittadini, G. Ligorio, M. Timpel, L. Pasquali, A. Giglia, M. V. Nardi and M. Casarin, Phys. Chem. Chem. Phys., 2016, 18, 24890–24904 RSC.
- To avoid any ambiguity, it must be kept in mind that the free D4h NiTPP LUMO and LUMO+1 correspond to the two components of the degenerate 13eg orbital.
- L. Floreano, G. Naletto, D. Cvetko, R. Gotter, M. Malvezzi, L. Marassi, A. Morgante, A. Santaniello, A. Verdini, F. Tommasini and G. Tondello, Rev. Sci. Instrum., 1999, 70, 3855–3864 CrossRef CAS.
- (a) J. J. Yeh, Atomic calculation of photoionization cross-sections and asymmetry parameters, Gordon and Breach, Langhorne, PA, USA, 1993 Search PubMed; (b) J. J. Yeh and I. Lindau, Atomic Subshell Photoionization Cross Section and Asymmetry Parameters: 1 ≤ Z ≤ 103, At. Data Nucl. Data Tables, 1985, 32, 1–155 CrossRef CAS.
- B. E. Douglas and C. A. Hollingsworth, Symmetry in Bonding and Spectra, an Introduction, Academic Press, Orlando, FL, USA, 1985 Search PubMed.
- A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS.
- J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822–8824 CrossRef PubMed.
- Y. Chen, F. J. García de Abajo, A. Chassé, R. X. Ynzunza, A. P. Kaduwela, M. A. Van Hove and C. S. Fadley, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 13121–13131 CrossRef CAS.
- J. J. Rehr, R. C. Albers and S. I. Zabinsky, High-Order Multiple-Scattering Calculations of X-Ray-Absorption Fine Structure, Phys. Rev. Lett., 1992, 69, 3397–3400 CrossRef CAS PubMed.
- M. Schwarz, M. Garnica, D. A. Duncan, A. P. Paz, J. Ducke, P. S. Deimel, P. K. Thakur, T.-L. Lee, A. Rubio, J. V. Barth, F. Allegretti and W. Auwärter, J. Phys. Chem. C, 2018, 122, 5452–5461 CrossRef CAS.
- P. Donovan, A. Robin, M. S. Dyer, M. Persson and R. Raval, Chem. – Eur. J., 2010, 16, 11641–11652 CrossRef CAS PubMed.
- The empty pmcσ* 12b1g orbital (the LUMO+2) of the free D4h NiTPP is undistinguishable both in shape and localization from the half-occupied CuTPP 12b1g MO.36 3D CPs have been obtained by using the open-source multi-platform molecular visualization program Molekel 5.4.
- At the bridge chemisorption site, the local twofold symmetry (see Fig. 1) determines a very poor interaction between the pmcσ* 12b1g-based MO and the in-phase combination of the two 4s AOs localized on the two nnCuS, whose internuclear distance from Ni is 2.8 Å. This is why the bridge site will not be taken into consideration further.
- The nnnCuffaS–Ni distance of 3.646 Å is obtained by using the Ni–CuS height of 2.6 Å and a Cu cell parameter of 3.6149 Å https://www.webelements.com.
- The nnCuffhS–Ni distance of 2.644 Å is obtained by using the Ni–CuS height of 1.93 Å and a Cu cell parameter of 3.6149 Å https://www.webelements.com/.
- Q. Guo, Z. Qin, K. Zang, C. Liu, Y. Yu and G. Cao, Langmuir, 2010, 26, 11804–11808 CrossRef CAS PubMed.
- P. H. Lippel, R. J. Wilson, M. D. Miller, Ch Wöll and S. Chiang, Phys. Rev. Lett., 1989, 62, 171–174 CrossRef CAS PubMed.
- H. Okuyama, S. Kuwayama, Y. Nakazawa, S. Hatta and T. Aruga, Surf. Sci., 2022, 723, 1–7 CrossRef.
- F. Chen, X. Chen, L. Liu, X. Song, S. Liu, J. Liu, H. Ouyang, Y. Cai, X. Liu, H. Pan, J. Zhu and L. Wang, Appl. Phys. Lett., 2012, 100, 081602 CrossRef.
- The nnCuffhS–N distance of 1.938 Å is obtained by using the Ni–CuS height of 1.93 Å, the herein optimized Ni–N bond length of 1.980 Å and a Cu cell parameter of 3.6149 Å https://www.webelements.com/.
- In the free D4h MTPP, with the MP pristine macrocycle plane corresponding to the xy one (σh) and the pyrrolic N atoms lying in the xz and yz planes (the two σv), the N 2pz SALCs transform as the eg + a2u + b2u IRs of the D4h point group.40 The b1u IR is antisymmetric with respect to σv40 and then pz AOs of atoms lying in these two planes cannot contribute to MOs of b1u symmetry.
- The pmcNiP α(β) pyrrolic C atoms correspond to C1, C4, C6, C9, C11, C14, C16, and C19 (C2, C3, C7, C8, C12, C13, C17, and C18) in Fig. S1 of the ESI†.
Footnote |
| † Electronic supplementary information (ESI) available: Optimized coordinates of the free D4h NiTPP; schematic representation of the free D4h NiTPP; schematic representation of nnCuSffh 4s AOs SALC of symmetry a, b and e; schematic representation of the nnnCuSffh 4s AOs SALC of symmetry a, b and e. See DOI: https://doi.org/10.1039/d3cp04021f |
| This journal is © the Owner Societies 2023 |