
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cryogenic infrared spectroscopy reveals remarkably short NH+⋯F hydrogen bonds in fluorinated phenylalanines†
Marc
Safferthal‡
 ab,
Kim
Greis‡
ab,
Rayoon
Chang‡
ab,
Carla
Kirschbaum
ab,
Waldemar
Hoffmann
ab,
Gerard
Meijer
b,
Gert
von Helden
b and
Kevin
Pagel
*ab
ab,
Kim
Greis‡
ab,
Rayoon
Chang‡
ab,
Carla
Kirschbaum
ab,
Waldemar
Hoffmann
ab,
Gerard
Meijer
b,
Gert
von Helden
b and
Kevin
Pagel
*ab
aDepartment of Biology, Chemistry, Pharmacy, Freie Universität Berlin, Altensteinstraße 23a, 14195 Berlin, Germany. E-mail: kevin.pagel@fu-berlin.de
bFritz Haber Institute of the Max Planck Society, Faradayweg 4-6, 14195 Berlin, Germany
First published on 29th August 2023
Abstract
In past decades, hydrogen bonds involving organic fluorine have been a highly disputed topic. Obtaining clear evidence for the presence of fluorine-specific interactions is generally difficult because of their weak nature. Today, the existence of hydrogen bonds with organic fluorine is widely accepted and supported by numerous studies. However, strong bonds with short H⋯F distances remain scarce and are primarily found in designed model compounds. Using a combination of cryogenic gas-phase infrared spectroscopy and density functional theory, we here analyze a series of conformationally unrestrained fluorinated phenylalanine compounds as protonated species. The results suggest proximal NH+⋯F hydrogen bonds with an exceptionally close H⋯F distance (1.79 Å) in protonated ortho-fluorophenylalanine.
Introduction
Decades ago, the existence of hydrogen bonds involving organic fluorine triggered a controversial discussion. Due to the lack of evidence and examples, Dunitz and Taylor concluded in 1997 in a statistical analysis that “organic fluorine hardly ever accepts hydrogen bonds”.1 However, in 2002 Desiraju provided early hints for C–H⋯F–C hydrogen bonds in fluorobenzene crystals.2 However, due to the inherent weakness of these attractive forces, it is challenging to confirm their existence experimentally. Facilitated by the development of new analytical techniques and instruments, numerous examples with computational as well as experimental evidence for hydrogen bonds involving organic fluorine have been reported.3 In addition to classical neutral–neutral interactions, charged NH+⋯F hydrogen bonding has been described and characterized in great detail in the past decade.4 The intra- and intermolecular H⋯F distances reported in the literature usually range between 2 and 3 Å;3 strong hydrogen bonds involving organic fluorine with significantly closer distances below 2 Å are rare. Remarkable examples of very close H⋯F–C hydrogen bonds (as low as 1.58 Å) were found by Lectka and co-workers in conformationally locked “cage” compounds.5,6The establishment of gas-phase infrared (IR) action spectroscopy paved the way for the structural analysis of intramolecular hydrogen bonds in amino acids7,8 and short peptides.9,10 Ion-dip spectroscopy allows unambiguous assignment of selected amino acid conformers.11 A recent study using infrared multiple photon dissociation (IRMPD) spectroscopy provided hints for close NH+⋯F interactions (1.81 Å and 1.91 Å) in conformationally unrestrained phenylalanine derivatives.12 However, IRMPD spectroscopy suffers from line broadening and redshift of vibrational bands, which can prevent an unambiguous assignment of low-energy conformers.
Cryogenic gas-phase infrared action spectroscopy provides high-resolution IR spectra, which can be used to unravel structural details of various biomolecules such as glycans,13,14 peptides,15,16 RNA,17 glycolipids,18 or fluorinated glycosyl cations.19 Using a combination of gas-phase IR spectroscopy in superfluid helium nanodroplets and density functional theory (DFT), we here provide a systematic study on fluorinated phenylalanine derivatives in positive ion mode. The data suggest the existence of an exceptionally short NH+⋯F hydrogen bond with organic fluorine.
Experimental
Cryogenic gas-phase IR spectroscopy in superfluid helium droplets
The custom-built instrument used in this work has been described in detail previously.20,21 The ions of interest are generated by nanoelectrospray ionization (nESI) and selected by their mass-to-charge ratio (m/z) using a quadrupole mass filter. The m/z-selected analyte ions are collected and stored in a liquid nitrogen-cooled hexapole ion trap at approximately 90 K. A pulsed beam of superfluid helium droplets is generated by the release of pressurized helium into the vacuum at cryogenic temperatures using an Even–Lavie valve.22 This beam of helium droplets passes through the ion trap, where it picks up and cools the ions to 0.4 K. The helium droplets transfer the embedded ions to the detection region, where they interact with IR photons generated by the Fritz Haber Institute free-electron laser (FHI FEL23). After the sequential absorption of resonant IR photons, the analyte ions are released from the helium droplets and detected by a time-of-flight mass analyzer. High-resolution IR spectra are generated by plotting the ion signal against the tunable wavenumber of the laser. IRMPD spectra were recorded on a different instrument, which is described in detail in the ESI.† All IR spectra were recorded in the wavenumber region between 1000 and 1800 cm−1.Computational methods
The conformational space of the fluorinated phenylalanine derivatives was explored using the genetic algorithm (GA) FAFOOM (flexible algorithm for optimization of molecules).24 The GA generates a pool of structures by randomly adjusting all freely rotatable bonds in the molecule. Each thereby sampled geometry is optimized by an external software (FHI-aims)25 for DFT optimization at the PBE+vdWTS![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 26,27 level of theory using light basis set settings. The lowest-energy structures will then be used as parent structures to generate child structures by genetic crossing. These child structures will then be optimized at the previously mentioned DFT level and replace the parent structure if it is lower in energy. Then, new child structures are generated. Additionally, mutations can occur, leading to random adjustments of dihedral angels of flexible bonds. The conformer generation stops after a predefined threshold or until no new lowest-energy structures can be found anymore. The structures of the low-energy conformers were reoptimized followed by harmonic frequency calculations at the PBE0+D3/6-311+G(d,p)28,29 level of theory in Gaussian 16.30 Additionally, selected structures were reoptimized and their harmonic frequencies computed at the DSD-PBEP86+D3/Def2-TZVPP,31,32 CAM-B3LYP+D3/6-311+G(d,p),33 ωB97XD/6-311+G(d,p),34 and M06-2X+D3/6-311+G(d,p)35 levels of theory (see ESI†). The frequencies were scaled by an empirical factor of 0.966. Low-energy conformers with different spatial orientations of the fluorine, NH3+, and COOH functional groups were generated.
26,27 level of theory using light basis set settings. The lowest-energy structures will then be used as parent structures to generate child structures by genetic crossing. These child structures will then be optimized at the previously mentioned DFT level and replace the parent structure if it is lower in energy. Then, new child structures are generated. Additionally, mutations can occur, leading to random adjustments of dihedral angels of flexible bonds. The conformer generation stops after a predefined threshold or until no new lowest-energy structures can be found anymore. The structures of the low-energy conformers were reoptimized followed by harmonic frequency calculations at the PBE0+D3/6-311+G(d,p)28,29 level of theory in Gaussian 16.30 Additionally, selected structures were reoptimized and their harmonic frequencies computed at the DSD-PBEP86+D3/Def2-TZVPP,31,32 CAM-B3LYP+D3/6-311+G(d,p),33 ωB97XD/6-311+G(d,p),34 and M06-2X+D3/6-311+G(d,p)35 levels of theory (see ESI†). The frequencies were scaled by an empirical factor of 0.966. Low-energy conformers with different spatial orientations of the fluorine, NH3+, and COOH functional groups were generated.
Materials
2-Fluoro-L-phenylalanine (oF-Phe), 3-fluoro-L-phenylalanine (mF-Phe), 4-fluoro-L-phenylalanine (pF-Phe) and pentafluoro-L-phenylalanine (F5-Phe) were purchased from Tokyo Chemical Industry (purity >95%). oF-Phe, mF-Phe, pF-Phe, and F5-Phe were dissolved in deionized water to yield 100 μM solutions for the cryogenic IR spectroscopy experiments and 5 mM solutions for the IRMPD experiments. The samples were ionized by nESI using Pd/Pt coated glass capillaries (Sputter Coater HR 208, Cressington) that were pulled to a tip with an inner diameter of 1–2 μm using a micropipette puller (Model P-1000, Sutter Instrument).Results and discussion
First, we compare the room-temperature IRMPD and cryogenic IR spectra of the protonated ortho-fluorophenylalanine (oF-Phe) that were recorded on different instruments (Fig. 1). The IRMPD spectrum shows two broad features in the functional group region (1400–1800 cm−1) and one band in the fingerprint region (1000–1400 cm−1). In the cryogenic IR spectrum, the three features split up into multiple resolved bands. The frequency and intensity of vibrational bands are in general highly sensitive to the spatial position and environment of the oscillators. Generally, for conformers of small molecules, only subtle differences in the IR frequencies are expected. The comparison between the IRMPD and the cryogenic IR spectra clearly shows that an assignment of conformers of the fluorinated phenylalanine cations is generally not possible by IRMPD spectroscopy. Cryogenic IR spectroscopy in helium droplets on the other hand provides sufficient spectral resolution to assign molecular structures – a crucial requirement to probe the rather weak fluorine-specific interactions addressed here.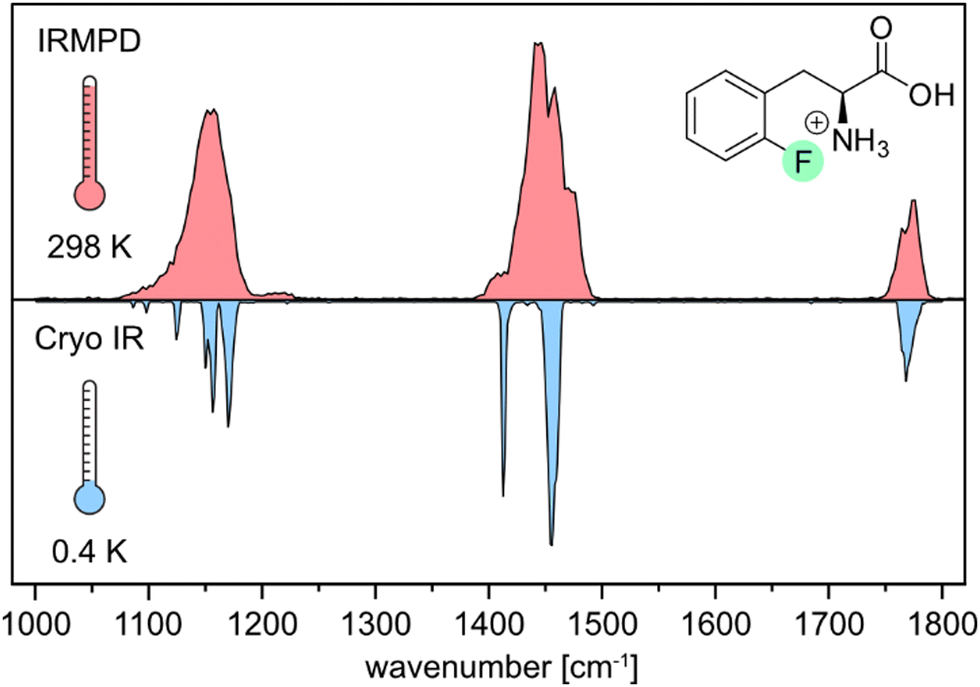 | ||
| Fig. 1 Comparison of experimental IR spectra of the protonated ortho-fluorophenylalanine (oF-Phe) cation generated by room-temperature IRMPD spectroscopy (red) and cryogenic infrared spectroscopy in helium droplets (0.4 K, blue). | ||
Next, we show and define geometrical parameters using the example of protonated oF-Phe to compare and further interpret the non-covalent interactions found in the computational results (Fig. 2). The distance between the hydrogen atom from the NH3+ group and the fluorine atom on the phenyl ring d(NH+⋯F) describes the hydrogen bond length. The C–F⋯H angle σ represents the hydrogen bond angle. An important parameter to compare cation–π interactions is the distance between the nitrogen atom and the center of the phenyl ring d(N⋯X). The torsion angle θ(Cγ–Cβ–Cα–N) describes the rotation around the Cα–Cβ bond and is a good indicator for the strength of cation–π interactions. A θ angle of 0° represents the ideal case with the strongest cation–π interactions.
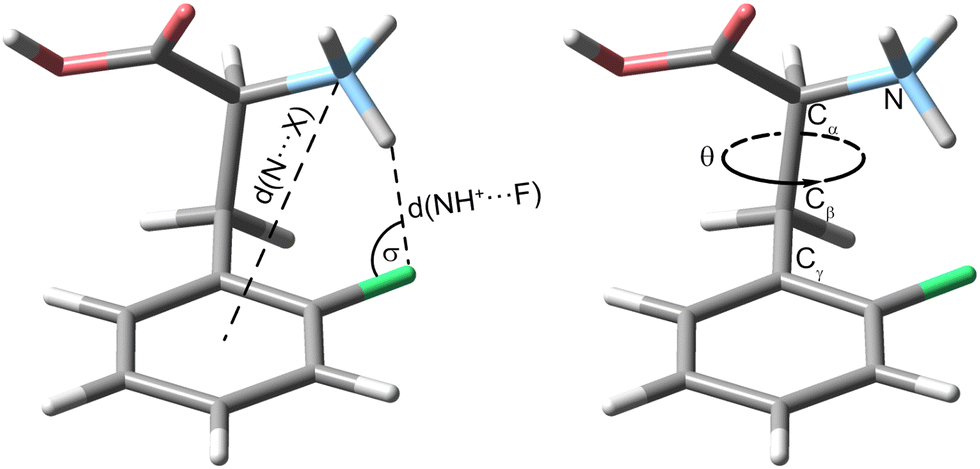 | ||
| Fig. 2 Visualization of important geometrical parameters using the example of the protonated ortho-fluorophenylalanine (oF-Phe). The torsion angle θ(Cγ–Cβ–Cα–N) describes a rotation around the Cα–Cβ bond. | ||
In the following, the IR signature of the protonated oF-Phe as well as computed structures and spectra of low-energy conformers (Fig. 3a) are shown. The isolated band observed at 1768 cm−1 corresponds to the carbonyl stretching vibration. The absorption bands for the C–O–H bending and the symmetric bending of NH3+ can be found between 1400 and 1550 cm−1. IR features between 1000 and 1300 cm−1 originate from C–H deformations, C–O stretching, C–C stretching and coupled modes. The C–F stretching band typically found between 1100 and 1300 cm−1 cannot be identified due to the low intensity of the vibration. The computed spectra are ordered by ascending relative free energy, with conformer A representing the global minimum structure and conformer D being the highest-energy conformer. Generally, the sampled structures show very little difference in energy as they can be converted into each other by the rotation of covalent bonds. For oF-Phe, the spectrum of the global minimum structure matches the best with the experimental spectrum, due to the overlapping absorption bands at 1454 cm−1 and the two adjacent bands at 1156 and 1170 cm−1. The other computed spectra match less well. Therefore, conformer A is likely dominant in the probed ensemble of ions. The global minimum structure reveals an intramolecular hydrogen bond between the NH3+ group and the organic fluorine with an exceptionally short NH+⋯F distance of 1.79 Å in the oF-Phe cation. The directionality of the hydrogen bond is not optimal as indicated by the slightly decreased H⋯F-C angle (σ), which is strongly influenced by the competing cation–π interaction between the NH3+ group and the phenyl ring. However, bending of this angle is a common phenomenon for intramolecular hydrogen bonds due to the presence of constraints from the molecular scaffold.36
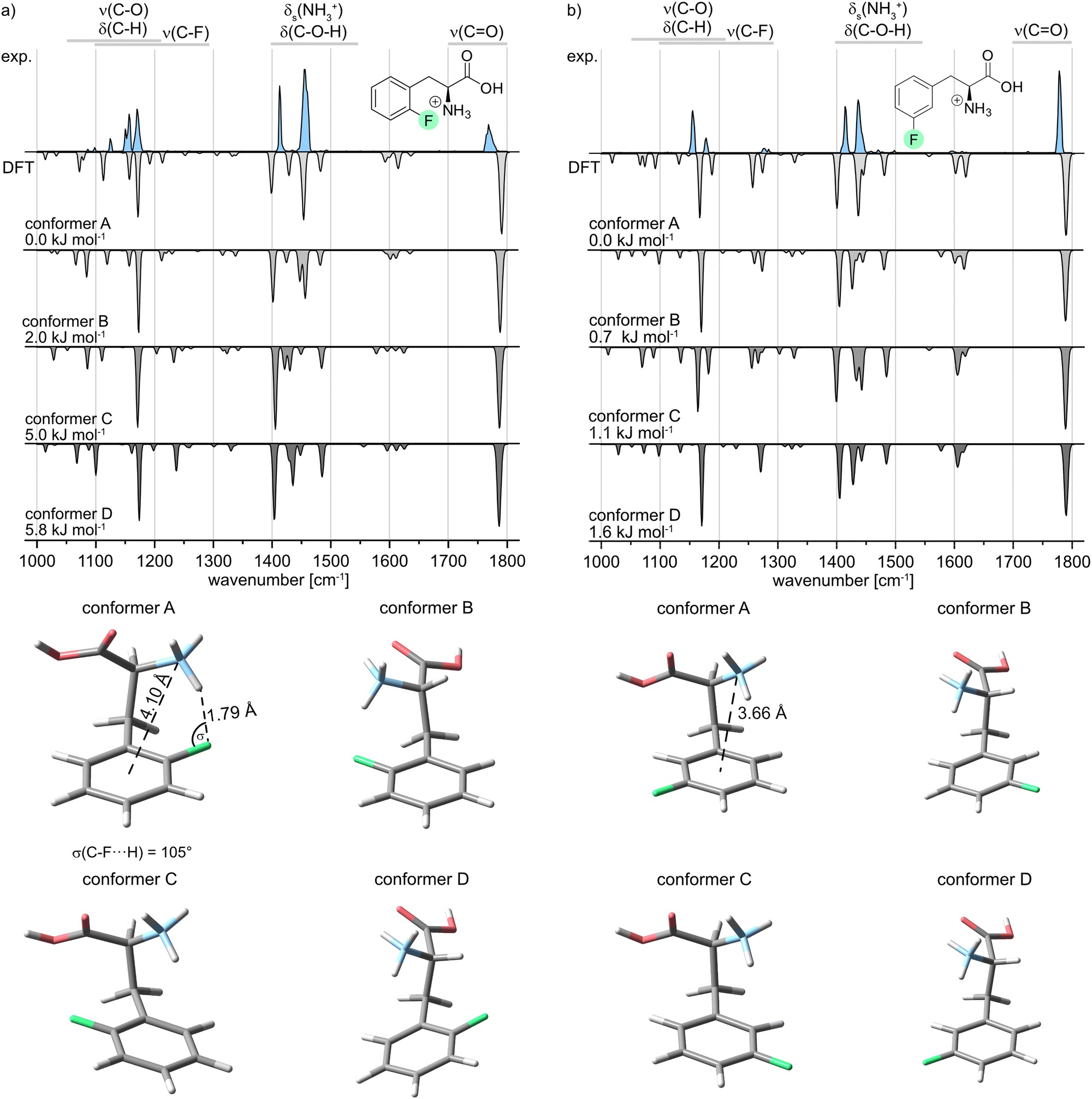 | ||
| Fig. 3 Infrared spectra of protonated (a) ortho- (oF-Phe) and (b) meta-fluorophenylalanine (mF-Phe). Experimental IR spectra are depicted as light blue traces. Computed spectra of low-energy conformers are shown as gray inverted traces. Relative free energies at 90 K are indicated. The structures of the low-energy conformers are depicted below the IR spectra. | ||
In order to validate our computational results, the geometry and harmonic frequencies of the lowest-energy structure of oF-Phe at the PBE0 level of theory were compared to those reoptimized at other levels of theory, including the double-hybrid functional DSD-PBEP86. These results can be found in the ESI† (Fig. S6). The survey confirms the occurrence of a very short NH+⋯F distance of 1.77–1.80 Å. Only the structure computed at the M06-2X level of theory showed no non-covalent interactions within the NH+⋯F moiety (2.22 Å) and the corresponding IR signatures are significantly altered. This discrepancy might be due to the M06-2X functional failing to properly describe long-range electron correlation, which is generally important for non-covalent interactions and hydrogen bonds.37
The IR spectrum of the protonated meta-fluorophenylalanine (mF-Phe) and the computed structures and vibrational frequencies are presented in Fig. 3b. In comparison with the IR signature of the oF-Phe cation, most of the vibrational bands are only slightly shifted and can be found in similar wavenumber regions. However, the symmetric bending of the NH3+ can be found at 1434 cm−1. This vibrational band is significantly blue-shifted by 20 cm−1 in the spectrum of the oF-Phe cation. Two new features can be identified at 1276 and 1284 cm−1, which correspond to coupled modes that include the C–F stretching vibrations. These differences suggest changes of the molecular structure for the NH3+ and C–F moieties. For mF-Phe, the computed spectra of conformer A and C match the experiment. However, due to the absorption band at 1434 cm−1, the global minimum structure A matches slightly better. The assigned structure shows no interaction involving the organic fluorine and the fluorine atom is placed even further away from the NH3+ cation by a rotation of the phenyl ring. The distance d(N⋯X) shows that the NH3+ cation is much closer to the center of the phenyl ring for the protonated mF-Phe, compared to the structure of the protonated oF-Phe, implicating that the former is mainly stabilized by cation–π interactions.
Further, the IR signature of the protonated para-fluorophenylalanine (pF-Phe) is presented in Fig. 4a, accompanied by the computed structures and frequencies. Similar IR features as in the spectrum of mF-Phe can be observed. However, a new strong feature arises at 1516 cm−1, which is caused by a coupled mode corresponding to aromatic C–H in-plane bending and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibrations of the phenyl ring.
C stretching vibrations of the phenyl ring.
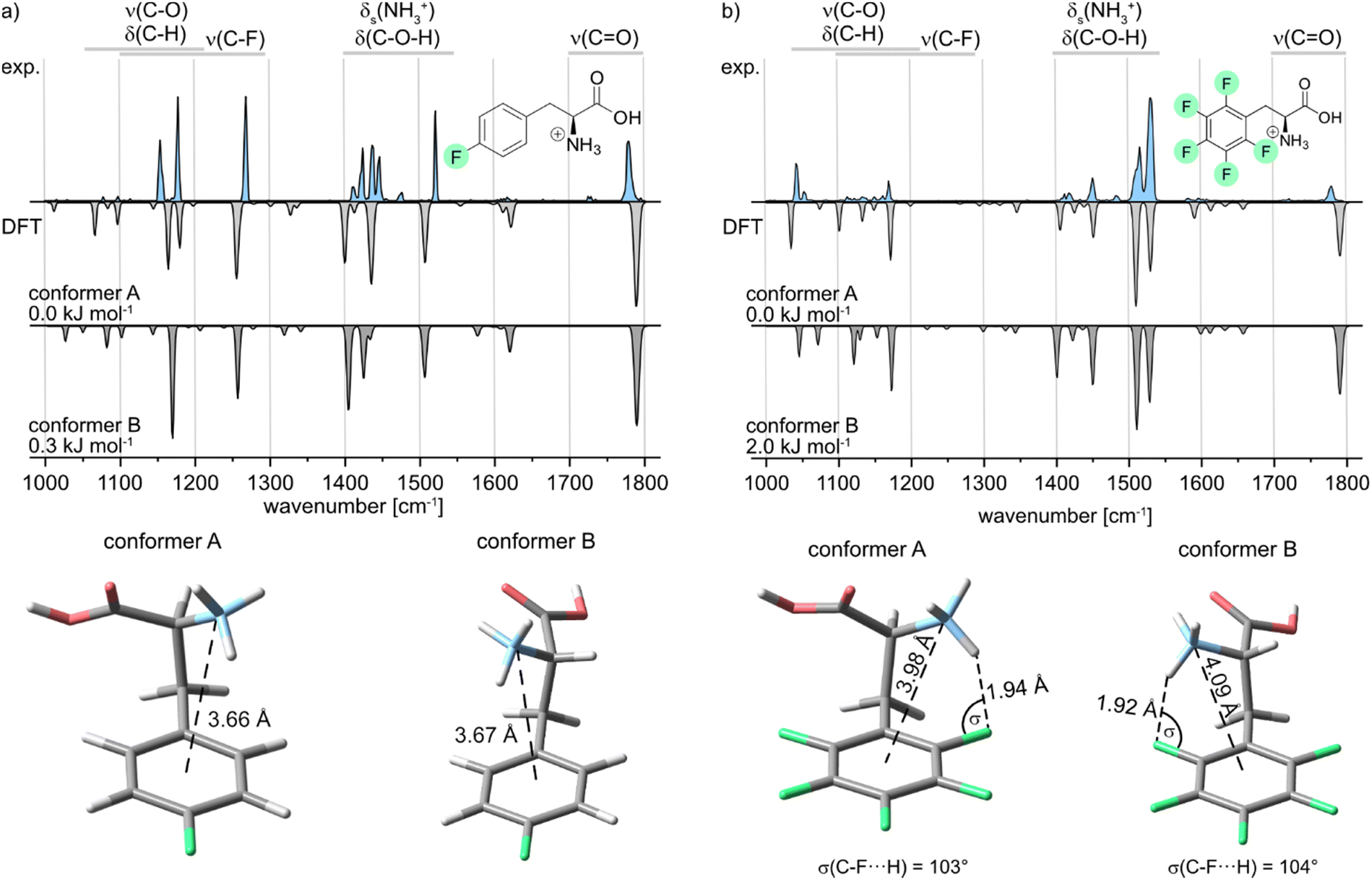 | ||
| Fig. 4 Infrared spectra of the protonated (a) para- (pF-Phe) and (b) penta-fluorophenylalanine (F5-Phe). Experimental IR spectra are depicted as light blue traces. Computed spectra of low-energy conformers are shown as gray inverted traces. Relative free energies at 90 K are indicated. The structures of the low-energy conformers are depicted below the IR spectra. | ||
The bands reflecting the NH3+ and C–O–H bending vibrations split up into four distinct signals between 1400 and 1500 cm−1. Moreover, the intensity of the band at 1268 cm−1, which displays the C–F stretching vibration, increased significantly. For the computational results, it can be pointed out that only two low-energy conformers could be obtained due to the symmetry of the fluorinated phenyl ring in pF-Phe. The two conformers A and B can be transformed into each other by the simple rotation of the Cα–Cβ bond, which further explains the low difference of 0.3 kJ mol−1 in relative free energy. For pF-Phe, the spectrum of the global minimum structure A matches better than B due to the overlapping signal at 1148 cm−1. However, the population of multiple conformers in the ion trap cannot be clearly ruled out based on the experimental and computational results. Both conformers show almost identical d(N⋯X) distances and no interactions involving the organic fluorine. As for mF-Phe, this observation suggests that cation–π interaction is the prevailing attractive force in pF-Phe directing the NH3+ group towards the center of the π-system.
Finally, the IR signature of the pentafluorophenylalanine (F5-Phe) cation is shown in Fig. 4b. Compared to the monofluorinated phenylalanine cations, the experimental spectrum is slightly more congested in the wavenumber region between 1000 and 1400 cm−1, while the relative intensity of the signals in this region is rather weak. New strong features arise at 1515 and 1531 cm−1, which correspond to different coupled modes with C–F and CC stretching vibrations. The band, which corresponds to the symmetric bending stretching mode of the NH3+ group is shifted to 1451 cm−1 and is low in intensity compared to monofluorinated phenylalanine. The computed spectra of both low-energy conformers are very similar and subtle differences between the theoretical spectra are predominantly found between 1000 and 1400 cm−1. However, both computed spectra match the experimental spectrum equally well. As a consequence, based on the experimental results, it is not apparent which of the sampled conformers are populating the ion trap. Both sampled structures show an NH+⋯F hydrogen bond involving the organic fluorine at the ortho-position. Hints for specific trends regarding the non-covalent interactions can be found by comparison of the geometrical parameters summarized in Table 1. In the conformer A structure of F5-Phe, the NH+⋯F distance is significantly longer (1.94 Å) and the σ angle is slightly smaller compared to the structure of the oF-Phe cation. This difference indicates that perfluorination of the aromatic system lowers the electron density of the fluorine at the ortho-position, which weakens the hydrogen bond. The distance between the cation and the center of the phenyl ring is significantly shorter and the torsion angle is smaller for the F5-Phe cation implying that due to the weaker NH+⋯F hydrogen bond, the competing cation–π interaction becomes more dominant. Generally, the countervailing effects of the competing cation-π and NH+⋯F interactions are further supported by the torsion angles and d(N⋯X) distances of the mF-Phe and pF-Phe structures. Due to their inability to form NH+⋯F hydrogen bonds at the ortho-position, they show the strongest cation-π interactions, attributable to their short d(N⋯X) distances and low torsion angles.
| Derivative | Conformer | d(N⋯X) (Å) | θ(Cγ–Cβ–Cα–N) (°) | d(NH+⋯F) (Å) | σ(C–F⋯H) (°) |
|---|---|---|---|---|---|
| oF-Phe | A | 4.10 | 65 | 1.79 | 105 |
| mF-Phe | A | 3.66 | 48 | — | — |
| pF-Phe | A | 3.66 | 47 | — | — |
| pF-Phe | B | 3.67 | −50 | — | — |
| F5-Phe | A | 3.98 | 57 | 1.94 | 103 |
| F5-Phe | B | 4.09 | −65 | 1.92 | 104 |
Conclusions
In conclusion, we have shown that the gas-phase structures of side-chain fluorinated phenylalanine cations strongly depend on the position and number of fluorine atoms. Structures fluorinated in the meta- or para-position of the ring feature a strong cation–π interaction between the protonated amine and the center of the phenyl ring. In protonated oF-Phe and F5-Phe, on the other hand, these cation–π interactions compete with defined and remarkably short NH+⋯F hydrogen bonds. Especially in the oF-Phe cation, this H-bond was found to be exceptionally short (1.79 Å).Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. S., R. C., and K. P. thank the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) for support under project number 387284271-SFB 1349. K. G. thanks the Fonds National de la Recherche (FNR) Luxembourg, for funding the project GlycoCat (13549747). C. K. acknowledges funding by the Fonds der Chemischen Industrie in the form of a Kekulé fellowship. Open Access funding provided by the Max Planck Society.Notes and references
- J. D. Dunitz and R. Taylor, Chem. – Eur. J., 1997, 3, 89–98 CrossRef CAS.
- G. R. Desiraju, Acc. Chem. Res., 2002, 35, 565–573 CrossRef CAS PubMed.
- J.-F. Paquin, P. Champagne and J. Desroches, Synthesis, 2014, 306–322 CrossRef.
- M. T. Scerba, C. M. Leavitt, M. E. Diener, A. F. DeBlase, T. L. Guasco, M. A. Siegler, N. Bair, M. A. Johnson and T. Lectka, J. Org. Chem., 2011, 76, 7975–7984 CrossRef CAS PubMed.
- M. T. Scerba, S. Bloom, N. Haselton, M. Siegler, J. Jaffe and T. Lectka, J. Org. Chem., 2012, 77, 1605–1609 CrossRef CAS PubMed.
- M. D. Struble, C. Kelly, M. A. Siegler and T. Lectka, Angew. Chem., 2014, 53, 8924–8928 CrossRef CAS PubMed.
- A. Simon, L. MacAleese, P. Maître, J. Lemaire and T. B. McMahon, J. Am. Chem. Soc., 2007, 129, 2829–2840 CrossRef CAS PubMed.
- J. Oomens, J. D. Steill and B. Redlich, J. Am. Chem. Soc., 2009, 131, 4310–4319 CrossRef CAS PubMed.
- B. Yan, S. Jaeqx, W. J. van der Zande and A. M. Rijs, Phys. Chem. Chem. Phys., 2014, 16, 10770–10778 RSC.
- A. Masson, M. Z. Kamrath, M. A. Perez, M. S. Glover, U. Rothlisberger, D. E. Clemmer and T. R. Rizzo, J. Am. Soc. Mass Spectrom., 2015, 26, 1444–1454 CrossRef CAS PubMed.
- J. M. Bakker, L. M. Aleese, G. Meijer and G. von Helden, Phys. Rev. Lett., 2003, 91, 203003 CrossRef PubMed.
- W. Fu, P. J. Carr, M. J. Lecours, M. Burt, R. A. Marta, V. Steinmetz, E. Fillion, T. B. McMahon and W. S. Hopkins, Phys. Chem. Chem. Phys., 2016, 19, 729–734 RSC.
- E. Mucha, A. I. Gonzalez Florez, M. Marianski, D. A. Thomas, W. Hoffmann, W. B. Struwe, H. S. Hahm, S. Gewinner, W. Schöllkopf, P. H. Seeberger, G. von Helden and K. Pagel, Angew. Chem., Int. Ed., 2017, 56, 11248–11251 CrossRef CAS PubMed.
- C. Masellis, N. Khanal, M. Z. Kamrath, D. E. Clemmer and T. R. Rizzo, J. Am. Soc. Mass Spectrom., 2017, 28, 2217–2222 CrossRef CAS PubMed.
- F. S. Menges, E. H. Perez, S. C. Edington, C. H. Duong, N. Yang and M. A. Johnson, J. Am. Soc. Mass Spectrom., 2019, 30, 1551–1557 CrossRef CAS PubMed.
- A. Y. Pereverzev, V. N. Kopysov and O. V. Boyarkin, J. Phys. Chem. Lett., 2018, 9, 5262–5266 CrossRef CAS PubMed.
- K. Greis, C. Kirschbaum, M. I. Taccone, M. Götze, S. Gewinner, W. Schöllkopf, G. Meijer, G. von Helden and K. Pagel, Angew. Chem., Int. Ed., 2022, 61, e202115481 CrossRef CAS PubMed.
- C. Kirschbaum, K. Greis, E. Mucha, L. Kain, S. Deng, A. Zappe, S. Gewinner, W. Schöllkopf, G. von Helden, G. Meijer, P. B. Savage, M. Marianski, L. Teyton and K. Pagel, Nat. Commun., 2021, 12, 1201 CrossRef CAS PubMed.
- K. Greis, C. Kirschbaum, G. Fittolani, E. Mucha, R. Chang, G. Helden, G. Meijer, M. Delbianco, P. H. Seeberger and K. Pagel, Eur. J. Org. Chem., 2022, e202200255 CrossRef CAS PubMed.
- D. A. Thomas, E. Mucha, M. Lettow, G. Meijer, M. Rossi and G. von Helden, J. Am. Chem. Soc., 2019, 141, 5815–5823 CrossRef CAS PubMed.
- D. A. Thomas, R. Chang, E. Mucha, M. Lettow, K. Greis, S. Gewinner, W. Schöllkopf, G. Meijer and G. von Helden, Phys. Chem. Chem. Phys., 2020, 22, 18400–18413 RSC.
- U. Even, EPJ Tech. Instrum., 2015, 2, 17 CrossRef.
- W. Schöllkopf, S. Gewinner, H. Junkes, A. Paarmann, G. von Helden, H. Bluem and A. M. M. Todd, Proc. SPIE-Int. Soc. Opt. Eng., 2015, 9512, 95121L CrossRef.
- A. Supady, V. Blum and C. Baldauf, J. Chem. Inf. Model., 2015, 55, 2338–2348 CrossRef CAS PubMed.
- V. Blum, R. Gehrke, F. Hanke, P. Havu, V. Havu, X. Ren, K. Reuter and M. Scheffler, Comput. Phys. Commun., 2009, 180, 2175–2196 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- A. Tkatchenko and M. Scheffler, Phys. Rev. Lett., 2009, 102, 073005 CrossRef PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian Inc., Wallingford CT, 2016 Search PubMed.
- S. Kozuch and J. M. L. Martin, Phys. Chem. Chem. Phys., 2011, 13, 20104–20107 RSC.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- D. Herschlag and M. M. Pinney, Biochemistry, 2018, 57, 3338–3352 CrossRef CAS PubMed.
- E. G. Hohenstein, S. T. Chill and C. D. Sherrill, J. Chem. Theory Comput., 2008, 4, 1996–2000 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp03776b |
| ‡ These authors contributed equally to this work. |
| This journal is © the Owner Societies 2023 |