
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStyrylpyrimidine chromophores with bulky electron-donating substituents: experimental and theoretical investigation†
Maxime
Hodée
 a,
Julien
Massue
*b,
Sylvain
Achelle
*a,
Arnaud
Fihey
*a,
Denis
Tondelier
cd,
Gilles
Ulrich
b,
Françoise Robin-le
Guen
a and
Claudine
Katan
a
a,
Julien
Massue
*b,
Sylvain
Achelle
*a,
Arnaud
Fihey
*a,
Denis
Tondelier
cd,
Gilles
Ulrich
b,
Françoise Robin-le
Guen
a and
Claudine
Katan
a
aUniv Rennes, ENSCR, CNRS, ISCR (Institut des Sciences Chimiques de Rennes) – UMR 6226, F-35000 Rennes, France. E-mail: sylvain.achelle@univ-rennes1.fr; arnaud.fihey@univ-rennes1.fr
bInstitut de Chimie et Procédés pour l’Energie, l’Environnement et la Santé (ICPEES), UMR CNRS 7515, Equipe Chimie Organique pour la Biologie, les Matériaux et l’Optique (COMBO) 25 Rue Becquerel, 67087 Strasbourg, Cedex 02, France. E-mail: massue@unistra.fr
cLaboratoire de Physique des Interfaces et des Couches Minces (LPICM), CNRS, Ecole Polytechnique, IP Paris, Palaiseau Cedex, France
dUniversité Paris-Saclay, CEA, CNRS, NIMBE, LICSEN, Gif-sur-Yvette, France
First published on 15th November 2023
Abstract
Styrylpyrimidines with bulky 9,9-dimethylacridan, phenoxazine and phenothiazine electron-donating fragments were designed. Thermally activated delayed fluorescence (TADF) properties were expected for these structures. These chromophores exhibit peculiar emission properties. For 9,9-dimethylacridan and phenoxazine derivatives, a single emission highly sensitive to the polarity is observed in solution whereas for phenothiazine derivative a dual emission is observed in solution and is attributed to the coexistence of quasi-axial (Qax) and quasi-equatorial (Qeq) conformers. This study intends to understand through theoretical and experimental works, why the studied chromophores do not exhibit TADF properties, contrary to what was expected. The absence of phosphorescence both at room temperature and 77 K tends to indicate the impossibility to harvest triplet states in these systems. Wave-function based calculations show that for both conformers of the three chromophores the S1–T1 splitting is significantly larger than 0.2 eV. The second triplet state T2 of Qeq conformers is found very close in energy to the singlet S1 state, but S1 and T2 states possess similar charge transfer characters. This prevents efficient spin–orbit coupling between the states, which is consistent with the absence of TADF.
Introduction
Styrylpyrimidine derivatives with electron-donating groups have been extensively studied in the last two decades as push–pull chromophores.1 This family of chromophores encompasses in particular linear 4-styrylpyrimidines,2 V-shaped quadrupolar 4,6-distyrylpyrimidines,2a,3 tripodal 2,4,6-tristyrylpyrimidines4 and other arylvinylpyrimidines.5 Many of these compounds exhibit photoluminescence properties that can be tuned by external stimuli such as polarity or pH.2–4,6 Styrylpyrimidines have found a large variety of applications ranging from fluorescent (bio)sensors (metal cations,7 nitroaromatics,8 β-amyloid plaques,9 double-stranded DNA,10etc.), panchromatic white light emission,11 luminescent liquid crystals,12 two-photon absorption chromophores for bioimaging,13 3D data storage14 or microfabrication,15 dyes for dye-sensitized solar cells16 as well as second order nonlinear optical chromophores for second harmonic generation.2b,17In recent years, thermally activated delayed fluorescence (TADF) chromophores have been subject to intensive research due to their application as emitters for organic light emitting diodes (OLEDs).18 The pioneering work of Adachi,19 based on pure organic compounds displaying TADF has emerged as a cutting-edge technology to achieve 100% internal quantum efficiency (IQE) and high external quantum efficiency (EQE), paving the way for a third generation of OLEDs. In addition to high stability and luminous efficiency, metal-free TADF emitters allow the production of a stable deep-blue light, which has always been problematic for previous generations of OLEDs.20 In addition, TADF chromophores have also found applications in photocatalysis21 and biomedical applications.22,23
The TADF process involves the population of a triplet state after electrical charge injection, followed by the thermally activated conversion to a singlet state by reverse intersystem crossing (RISC) (Fig. 1).24 The involvement of metastable triplet state in the TADF process induces longer fluorescence lifetimes, typically in the microsecond range but the spectral distribution of the emitted light is identical to prompt fluorescence.18a
 | ||
| Fig. 1 Principle of TADF. | ||
In order to observe TADF, it is necessary to combine an emissive singlet excited state S1, a sufficiently stable triplet excited state (e.g. T1) and a small singlet–triplet energy splitting (ΔEST) generally around 0.1 eV.18b,20b,25,26 In the case of donor–acceptor compounds, TADF is favored when the spin–orbit coupling is sizeable, which is the case when the singlet excited state exhibits a charge transfer character (1CT) and the triplet corresponds to a local exciton (3LE).6,27–29 A small energy difference between 3LE and 1CT (ΔEST) will increase the thermally activated upconversion.30
For all organic TADF chromophores, the typical design consists in push–pull chromophores with twist/spiro/bulky31 connection between donor (D) and acceptor (A) parts, permitting a reduction in the overlap between the HOMO and LUMO orbitals.18a,32 Indeed, spatially separated HOMO and LUMO orbitals are required to observe sufficiently small ΔEST values.33 Recently, after the pioneering work of Kido and Sasabe,34 numerous pyrimidine-based TADF emitters have been described for OLED applications with high EQE values.35 In all cases, bulky amino electron-donating groups such as carbazole,36 9,9-dimethylacridan,37 9,9-diphenylacridan,38 phenoxazine39 or phenothiazine40 are used. Some typical structures are presented in Fig. 2. To the best of our knowledge, in all these examples, the phenylene unit has almost always been used as the π-linker between the pyrimidine core and the electron-donating fragments. The triplet states of some stilbene derivatives are known to be unstable41 and have been described as potential transitional states for cis–trans isomerization.42 Nevertheless, styrylpyrimidine with the above-mentioned bulky amino electron-donating group remains unknown and appears as potential TADF chromophores.
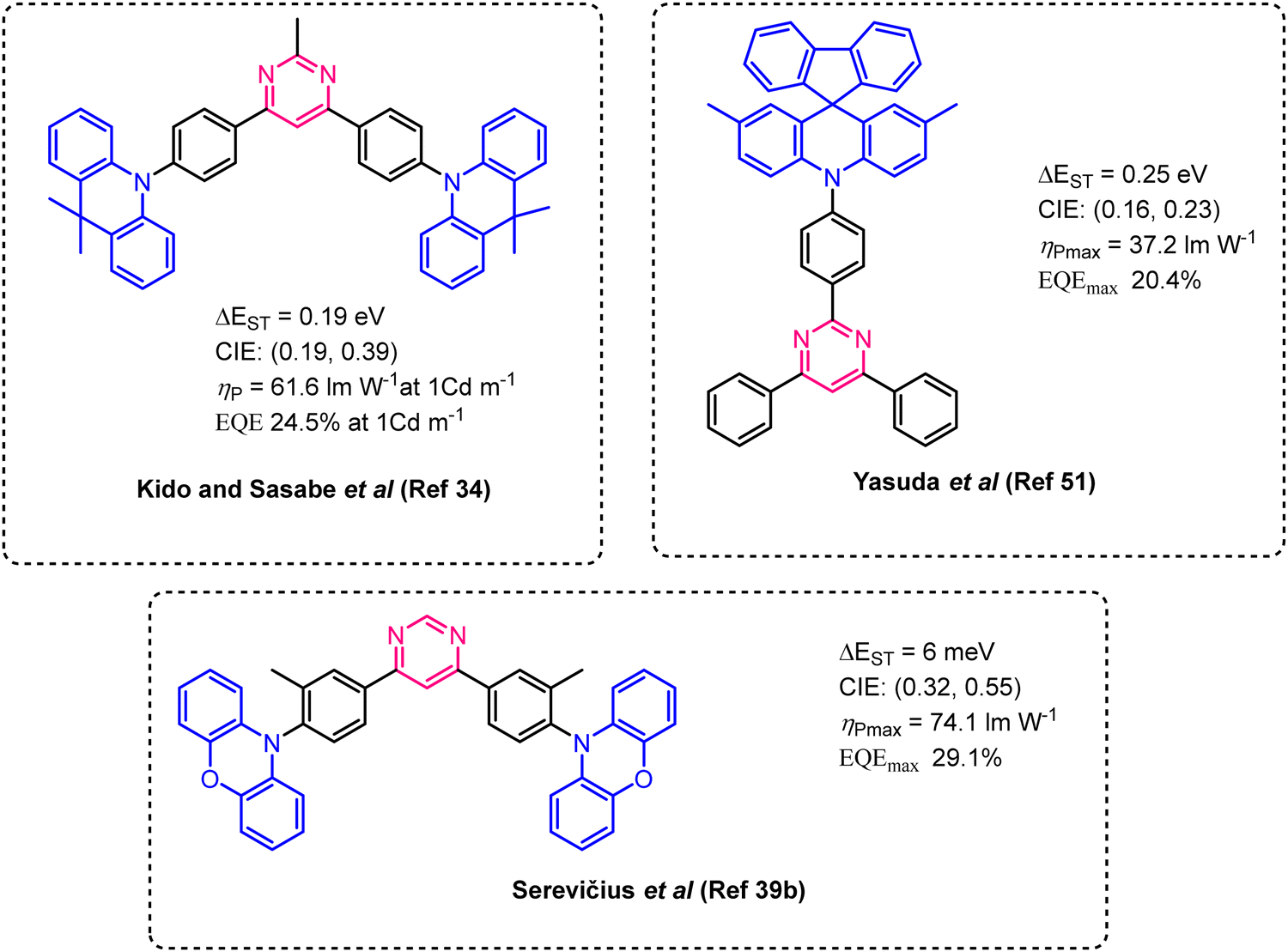 | ||
| Fig. 2 Examples of TADF pyrimidine chromophores34,39b,51 with characteristics of corresponding OLED devices. | ||
In the course of designing organic chromophores for OLED applications, and in particular for TADF applications, density functional theory (DFT) and its time dependent variant (TD-DFT) have demonstrated their usefulness in rationalizing structure–property relationships, and have been successfully used in the case of substituted heterocyclic systems.43 However, the known deficiencies of TD-DFT for the treatment of CT electronic transitions or triplet states makes it rather unsuitable to quantitatively predict TADF key properties such as ΔEST.43a,44 Wavefunction-based methodologies such as approximate singles and doubles coupled cluster (CC2)45,46 have recently been used for extended organic systems to study TADF properties.47 This level of theory proved to be more reliable to investigate the intrinsic nature of the electronic states relevant to the RISC (namely CT and LE)48 as well as their relative energies.49 Typically, for the TADF phenomenon to be theoretically expected in organic fluorophores, the singlet–triplet energy splitting ΔEST should be smaller than ca. 0.2 eV, and the spin–orbit coupling matrix element (SOCME) between these two states should be around 1 cm−1.18b,50
In this contribution, we designed a series of three 4-styrylpyrimidine derivatives substituteed by dimethylacridan, phenoxazine and phenothiazine bulky electron-donating groups. While maintaining an orthogonal conformation between the bulky donor group and the rest of the molecules, the styryl linker is expected to increase intramolecular CT and bathochromically shift the emission spectra with regard to phenylene analogues. With the help of experimental and CC2-based theoretical investigation, the emissive behavior of these derivatives and their potential for TADF properties are discussed.
Results and discussion
Synthesis
The target compounds 1–3 were obtained with moderate yields (Scheme 1) through Pd-catalyzed Buchwald–Hartwig cross-coupling reaction starting from the reported (E)-4-bromostyrylpyrimidine2b with 9,9-dimethyl-9,10-dihydroacridine, 10H-phenoxazine and 10H-phenothiazine respectively. The coupling reaction does not alter the E stereoisomery of the vinylene linker as evidenced by the characteristic coupling constant (3J(H–H) ∼16 Hz) of the doublet of the hydrogen of the double bond. | ||
| Scheme 1 Synthesis of target compounds 1–3 and corresponding yields. | ||
Photophysical properties
The photophysical properties for styrylpyrimidine derivatives 1–3 were recorded in solution in toluene and n-heptane and in the solid-state, as 1% wt doped in poly(methyl methacrylate) (PMMA) films at room temperature (298 K) and 77 K. The photophysical data are compiled in Table 1, while the absorption and emission spectra are displayed in Fig. 3.| Dye | Solvent/matrix | λ abs (nm) | ε (M−1 cm−1) |
λ
em
298![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K
(nm) K
(nm) |
λ
em
77K
(nm) |
Δsse (cm−1) | Φ f |
τ
298K
(ns) |
τ
77K
(ns) |
|---|---|---|---|---|---|---|---|---|---|
| a Maximum absorption wavelength at 298K. b Absorption coefficient at 298 K. c Maximum emission wavelength at 298 K in deaerated solutions. d Maximum emission wavelength at 77 K in deaerated solutions. e Stokes shift. f Relative quantum yield determined in solution at 298 K using Rhodamine 6G as a reference (λexc = 488 nm, Φ = 0.88 in ethanol). g Lifetime recorded at 298 K. h Lifetime recorded at 77 K. i Doped in poly(methylmethacrylate) (PMMA) 1 wt%. j Excitation wavelength. k λ exc = 380 nm. l λ exc = 400 nm. m λ exc = 375 nm. | |||||||||
| 1 | Toluene | 386 | 2300 | 512k | 464k | 6400 | 0.12 | 6.3 | 4.4/21.0 |
| Heptane | 386 | 2300 | 441k | 465k | 3200 | 0.10 | 5.9 | 5.5/20.0 | |
| PMMAi | 370j | — | 473 | 464 | — | 0.40 | 2.8/11.2 | 2.6/11.0 | |
| 2 | Toluene | 410 | 2600 | 571l | 507l | 4700 | 0.07 | 5.3 | 0.7/15.7 |
| Heptane | 410 | 2600 | 499l | 507l | 4700 | 0.05 | 4.6 | 10.6 | |
| PMMAi | 360j | — | 514 | 514 | — | 0.25 | 3.4/10.3 | 3.4/10.2 | |
| 3 | Toluene | 376 | 5700 | 438/584m | 439m | 3800 | 0.02 | 4.8 | 1.5 |
| Heptane | 376 | 5700 | 415/480m | 446m | 2500 | 0.01 | 0.1/6.1 | 1.6 | |
| PMMAi | 360j | — | 468 | 452 | — | 0.14 | 0.6/4.2 | 0.5/4.5 | |
 | ||
| Fig. 3 Absorption spectrum (plain black), emission spectra at 298 K (blue) and at 77 K (dotted black) of toluene (left) and n-heptane (center) solutions, emission spectra of the molecular dispersions in PMMA at 298 K (plain red) and 77 K (dotted red) (right) for dyes (a) 1, (b) 2 and (c) 3. Insets: Photographs of solution of dyes 1–3 in toluene and heptane under irradiation using a UV bench lamp (λexc = 365 nm). | ||
In solution, styrylpyrimidine dyes 1–3 which all show a push–pull structure with sterically hindered donor groups (dimethylacridan, phenoxazine and phenothiazine, respectively) display very similar absorption profiles, i.e., intense bands below 360 nm corresponding to π–π* transitions (vide infra). Additional weakly intense absorption bands whose maxima peak at 376 nm, 386 nm and 410 nm for 1–3, respectively, are also observed and might be attributed at first sight to CT. The lower energy CT band for phenoxazine derivative 2 is similar to what is observed with other series of chromophores52 and is related to the stronger electron-donating character of this group due to the presence of the electron-donating oxygen at the ortho position of the nitrogen atom. The molar absorption coefficients of these bands range between 2300 and 5700 M−1 cm−1. It is interesting to note that phenoxazine substituted dye 2 has the most red-shifted absorption band in the series.
Photoexcitation in the lowest-energy absorption band (370–400 nm) leads to emission in the visible range in both toluene and n-heptane for all dyes at room temperature. In toluene, at 298 K, a single emission band is observed for 1 and 2 at 512 and 571 nm, respectively, while for 3 dual emission is observed (λem = 438/584 nm). In n-heptane, a similar behavior is observed, i.e. substituted pyrimidines 1 and 2 display a single emission at 441 and 499 nm, respectively, while for 3, dual emission is observed (λem = 415/480 nm). The dual emission might be tentatively attributed to the coexistence of Quasi-Equatorial (Qeq) and Quasi-axial (Qax) conformers, as already observed for phenothiazine and dimethylacridan derivatives.53,54 The position of bands is highly dependent on the nature of the solvent and in particular its dipole moment. Going from toluene to heptane leads to a strong hypsochromic shift from 512 to 441 nm for 1, from 571 to 499 nm for 2 and from 584 to 480 nm for 3. It should be mentioned that the dual emission observed for compound 3 is dependent on the excitation wavelength: with excitation at the more energetic absorption band, only one emission band is observed at 586 nm in toluene (Fig. S1, ESI†). Other solvents (THF, CH2Cl2) were tested in order to further prove the charge transfer nature of the emission bands but fluorescence appeared to be quenched, as polarity increased.
To further study the nature of the emission band, fluorescence was also recorded at 77 K for all dyes. In all cases, a single band was observed which was attributed to the decay of the excited 1LE species. In heptane, it is worth mentioning that in the case of 1 and 2, the emission bands recorded at 298 K and 77 K are quasi superimposable, which would suggest a similar energy level for 1LE and 1CT in this solvent at room temperature. In toluene, a strong hypsochromic shift occurs for 1 and 2 when freezing the solution, indicating the sole presence of 1CT at room temperature and 1LE at 77 K. The case of 3 seems to be more elusive. In toluene, a single band is observed at 77 K, whose maximum emission wavelength is reminiscent of a high energy band at room temperature (λem = 439 nm vs. 438 nm for 3 at 77 K and 298 K, respectively). In heptane, however, a significant red-shift is observed for the 1LE at 77 K vs. 298 K (λem = 446 nm vs. 415 nm for 3 at 77 K and 298 K, respectively). It is worth noting that for all dyes studied herein, attempts to record phosphorescence spectra at 298 K or 77 K remained unsuccessful, highlighting the impossibility to harvest triplet states in these systems.
Emission in the solid-state was also studied for 1–3, as 1 wt% doped in PMMA films. The maximum emission wavelengths remained similar, regardless of temperature, and span the range 468–514 nm at 298 K and 452–514 nm at 77 K with highly identical shapes.
The luminescent lifetimes, recorded at both temperatures for 1–3 showed in the majority of the cases a dual exponential decay, comprising of a short and a significantly longer value (in the range of 10 ns), consistent with the short-lived fluorescence of organic dyes and tend to indicate the absence of TADF of these dyes in the specific studied matrixes.
Theoretical study
Among others,54,55 Penfold and co-workers56 described for phenothiazine-substituted push–pull dyes, the existence of two conformers: Qeq and Qax (see Fig. 4 for a representation). Ground state equilibrium geometries of Qeq conformers of 1, 2 and 3 show a nearly 90° dihedral angle (schematically shown as θ in Fig. 4) between the donor group and the styrylpyrimidine acceptor moiety (Table S1, ESI†). At room temperature, thermal energy is sufficient to induce a change of this angle in the 70°–110° range, as it corresponds to an energy difference of only ca. 2 kcal mol−1 (the corresponding energy profile is plotted in Fig. S2, ESI†). This is expected to influence the D–A character of the ground-state, as well as the CT character of the lowest energy transition. | ||
| Fig. 4 Schematic representation of the Quasi-equatorial (Qeq) and Quasi-axial (Qax) conformers, illustrated with the structure of compound 3 in both conformations at the ground state. | ||
In their Qax conformation, the compounds show a bending angle of the donor unit towards the plane of the styrylpyrimidine moiety (schematically shown as angle ϕ in Fig. 4). For both Qeq and Qax, the phenoxazine and phenothiazine unit in compound 2 and 3, respectively, exhibits an out of plane conformation, which is more pronounced for the phenothiazine derivative. The styrylpyrimidine moiety itself is in all cases totally planar. Within the series, the Qeq and Qax conformers appear almost equally stable for 3 (Fig. S5, ESI†), while the Qeq is slightly favoured in the case of 1 and 2, by 1.5 and 3.2 kcal mol−1, respectively (Fig. S3 and S4, ESI†). Energetic barriers of ca. 10 kcal mol−1 are obtained for the three compounds (Fig. S3–S5 for a representation of the transition state, ESI†). Because of their possible coexistence, both Qeq and Qax conformers will be considered in the following sections.
From these ground states, the relevant computed excitation energies and oscillator strengths for compounds 1, 2 and 3 are provided in Table 2. In all cases the Qeq conformers present a low energy (S0 to S1) transition with a near zero oscillator strength, ranging from 3.07 eV (403 nm) for 1 to 3.33/3.31 eV (372/375 nm) for 2/3. The most intense transition for the Qeq conformers that dominates the UV-Visible spectrum is the higher energy S0 to S5 transition for 1, and the S0 to S6 transition for 2 and 3, peaking around 4.20 eV (295 nm). The natural transition orbitals (NTO) plotted in Fig. S6 (ESI†) show for each of the Qeq conformers a strong charge transfer character of the S0 to S1 transition, with the hole located on the dimethylacridan/phenoxazine/phenotiazine donor and the particle on the styrylpyrimidine acceptor, with negligible overlap between the two. This leads to a low transition probability and a vanishing computed oscillator strength. On the opposite side, the NTOs for the S0 to S5/S6 excitation show a clear π–π* localized character on the styrylpyrimidine moiety, explaining the high oscillator strength value computed for these transitions. The Qax conformers exhibit different photophysical properties, with intense S0 to S1 transitions, peaking between 3.39 eV (366 nm) for 1 and 3.63 eV (341 nm) for 3. Indeed, in the Qax conformation, the orientation between the acridine group and the styrylpyrimidine allows for non-negligible π delocalization along the molecular backbone, thus promoting an intense first π–π* excitation channel (Fig. S6, ESI†).
| Compound | Conformation | E abs (eV)/λabs (nm) | f abs | Statesabs | E em (eV)/λem (nm) | f em | ΔEST (eV) | SOCME (cm−1) |
|---|---|---|---|---|---|---|---|---|
| 1 | Qeq | 3.07 (404) | 0 | S1 | 2.64 (470) | 0.00 | 0.49 | 0.72 |
| 4.18 (297) | 1.38 | S5 | ||||||
| Qax | 3.39 (366) | 1.35 | S1 | 3.01 (412) | 1.49 | 0.89 | 0.06 | |
| 2 | Qeq | 3.33 (372) | 0 | S1 | 2.31 (546) | 0.00 | 0.33 | 0.62 |
| 4.19 (296) | 1.19 | S6 | ||||||
| Qax | 3.58 (346) | 1.27 | S1 | 2.98 (406) | 1.37 | 0.92 | 0.13 | |
| 3 | Qeq | 3.31 (374) | 0 | S1 | 2.27 (537) | 0.00 | 0.35 | 0.18 |
| 4.20 (296) | 1.26 | S6 | ||||||
| Qax | 3.63 (341) | 1.35 | S1 | 3.04 (416) | 1.37 | 0.90 | 0.15 |
Theoretically predicted co-existence of both Qeq and Qax conformers in solution are consistent with the absorption spectra observed experimentally. Indeed, in all three compounds, the intense high energy transition measured in solution can be unambiguously attributed to the S0 to S5/S6 π–π* computed transition of the Qeq conformer. Meanwhile, the low energy and lower intensity absorption band may correspond to the S0 to S1 excitation of any of the two conformers. In fact, whereas the S0 to S1 transition of Qeq is computed with a null oscillator strength at equilibrium geometry, it gains sizeable oscillator strength as a result of the above mentioned donor–acceptor angular modulation at finite temperature (θ angle deviating from 90° in Fig. 4), especially for compounds 1 and 2 (Table S8, ESI†). For compound 3, variation of the dihedral angle hardly increases the oscillator strength, and coexistence of both Qeq and Qax conformers is likely to be at the origin of the low-energy band in the experimental absorption spectrum (Fig. 3).
After geometry optimization of the first singlet excited state of each Qeq conformer, the donor group becomes more planar, particularly for 3. The donor part and the styrylpyrimidine groups of 2/3 slightly deviate from orthogonality (angle θ), whereas compound 1 does show very limited geometry reorganisation between S0 and S1. In the Qax conformation, the angle between the donor and the styrylpyrimidine groups (angle ϕ) increases for all compounds, indicating a flattening of the donor group that comes closer to the plan of the acceptor. All the angle values are presented in Table S1 (ESI†). In Table 2 are presented the computed transition energies from the optimized S1 states to the ground states, quantifying their emission properties. In their Qeq conformations, 2 and 3 lead to de-excitation energies of 2.27 (546 nm) and 2.31 eV (536 nm). 1 has a slightly different behaviour with a higher energy at 2.64 eV (470 nm), in line with the smaller reorganization of the molecular structure in the excited state. The lowest lying experimental emission bands recorded at 298 K are consistent with the computed data. However, NTOs presented in Fig. 5 for 1 (see Fig. S7 for 2 and 3, ESI†) show that the S1 to S0 transition of the Qeq conformer has a clear CT character, which leads to near-zero oscillator strengths (Table 2) due to the lack of overlap between orbitals. Thus, if related to Qeq conformations, the non-negligible quantum yields experimentally measured at 298 K should stem from the conformations deviating from the perfect orthogonal geometry, that may be populated at finite temperature. Experimental data measured for 1 and 2 at 77 K, including in PMMA matrixes, remain in line with the computed trends for Qeq conformers, but 3 clearly steps out. In parallel, the optimized S1 states in the Qax conformation lead to higher computed de-excitation energies that are found similar for all three compounds, at ca. 3 eV (410–415 nm), and bear significant oscillator strength (Table 2). This is related to the delocalized π–π* character of the transition as illustrated for compound 1 in Fig. 5 (Fig. S7 for 2 and 3, ESI†). From the dual emission observed for 3 in toluene and heptane, as well as the good energy match with the higher lying band observed at room temperature and the single emission line recorded at 77 K, or in a PMMA matrix, we assign this higher lying emission band to the LE state of the Qax conformer. In short, the observed LE and CT experimental bands in the dual emission of 3 may arise from the presence of both Qeq (CT, low energy emission) and Qax (LE, high energy emission) conformers. For 1 and 2, comparison between experimental and computed data suggests that the emission may stem from Qeq conformers deviating from perfect orthogonal conformations, leading to a reduced CT character and in turn non-zero oscillator strength.
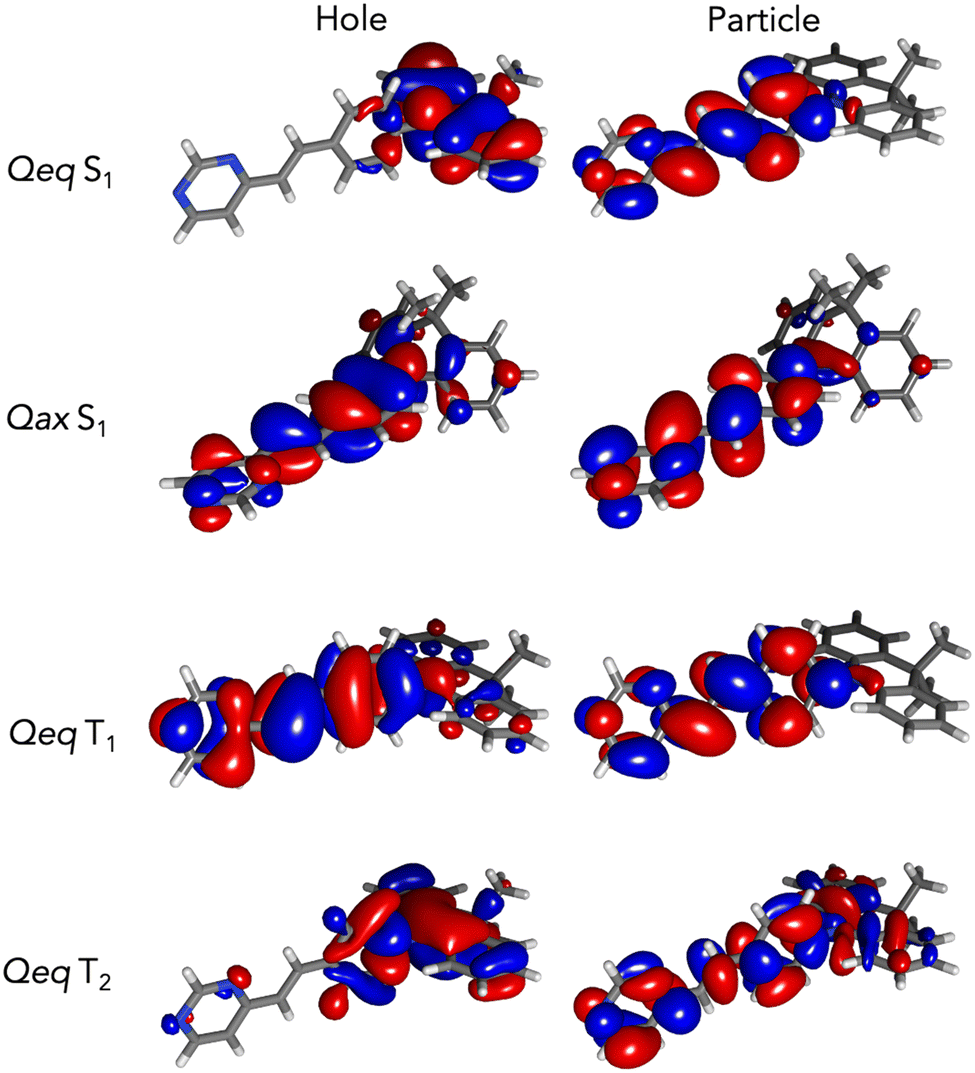 | ||
| Fig. 5 NTOs computed for compound 1 at the S1 geometry of Qeq and Qax conformers, and at the T1 and T2 geometries of Qeq conformer (isovalue = 0.02). | ||
The geometry of the first triplet state of 1–3 (E)-styrylpyrimidines is found to be similar to the ground state one (see angles for Qeq and Qax conformers in Table S1, ESI†). The NTOs describing the T1 to S0 transition show a strong localized character with extended overlap between hole and particle, as illustrated for compound 1 in Fig. 5. Table 2 reports the ΔEST and SOCME values for Qeq conformers. For the three compounds, SOCME values are typical of TADF compounds, ranging between 0.18 cm−1 and 0.72 cm−1. But, the kinetic rate constant of reverse intersystem crossing and the likelihood of this mechanism also depends on the value of ΔEST. Here, in all cases, the S0–T1 splitting is larger than 0.2 eV; 0.46 eV, 0.33 eV and 0.35 eV for the Qeq conformer of 1, 2 and 3, respectively. For Qax conformers, the triplet is found significantly higher in energy leading to large ΔEST values, around 0.9 eV. These values are not in favor of any RISC from the T1 triplet. The second triplet state, T2, of Qeq conformers is found very close in energy to the singlet S1 state, respectively at 0.025 eV and 0.005 eV above for 1 and 2, and 0.013 eV below for 3. However, the S1 and T2 states possess a very similar CT nature as illustrated for compound 1 in Fig. 5, ruling out any efficient coupling. These theoretical results suggest that TADF is mostly unlikely in these series of styrylpyrimidine based chromophores, in line with experimental findings.
Conclusion
In this work we have investigated the photophysical properties of original push–pull chromophores consisting of styrylpyrimidine functionalized with bulky 9,9-dimethylacridan, phenoxazine and phenothiazine electron-donating fragments. Their potential as TADF emitters has been carefully evaluated both from an experimental and theoretical perspective. No evidence of TADF nor phosphorescence was observed. The phenothiazine derivative shows dual emission, with a low-energy band tentatively attributed to a CT transition of the Qeq conformer and a high-energy band related to a LE transition of the Qax conformer. For dimethylacridan and phenoxazine derivatives, the comparison between experimental and computed data suggests that the emission may stem from the Qeq conformers only, thanks to imperfect orthogonal conformation that reduces the CT character and increases the oscillator strength of the transition. The S1–T1 energy splittings were found to be significantly larger than 0.2 eV, which is not in favor of TADF. This is not the case for the second triplet state T2 of Qeq conformers, which is almost isoenergetic with S1. Nevertheless, both bear a similar CT character, leading to poor spin–orbit coupling, which is also unfavourable to TADF.Experimental and computational details
General information
All solvents were of reagent grade for synthesis. The starting materials were purchased from Sigma-Aldrich or TCI and were used without further purification. Thin layer chromatography (TLC) was conducted on pre-coated aluminum sheets with 0.20 mm Merck Alugram SIL G/UV254 with fluorescent indicator UV254 and 0.25 mm Merck silica gel (60-F254). Column chromatography was carried out using Acros silica gel 60 (particle size 63–200 μm). NMR spectra were recorded in CDCl3 on a Bruker AC-300 spectrometer. The chemical shifts δ are reported in ppm and are referenced to the appropriate solvent signals of CDCl3 (1H, δ = 7.27 ppm; 13C, δ = 77.0 ppm) or DMSO-d6 (1H, δ = 2.50 ppm; 13C, δ = 39.5 ppm). The coupling constants J are given in Hz. In the 1H NMR spectra, the following abbreviations are used to describe the peak patterns: s (singlet), d (doublet), t (triplet), q (quadruplet), and m (multiplet). Acidic impurities in CDCl3 were removed through treatment with solid K2CO3. High-resolution mass analyses were carried out at the ‘Centre Régional de Mesures Physiques de l’Ouest’ (CRMPO, Université de Rennes 1) using a Bruker MicroTOF-Q II instrument.Synthesis
:1 mixture of CH2Cl2 and water (50 mL) and the organic layer was separated. The aqueous layer was extracted with CH2Cl2 (2 × 25 mL). The organic phases were combined, dried and evaporated. The residue was purified by column chromatography (SiO2, AcOEt/petroleum ether, 1:1) followed by recrystallization from DCM/n-heptane.
Steady state spectroscopy
Absorption spectra were recorded using a dual-beam grating Schimadzu UV-3000 absorption spectrometer with a quartz cuvette of 1 cm of optical path length. The steady-state fluorescence emission and excitation spectra were recorded by using a Horiba S2 Jobin Yvon Fluoromax 4. All fluorescence and excitation spectra were corrected. Solvents for spectroscopy were of spectroscopic grade.The fluorescence quantum yields (Φexp) were measured in diluted solution with an absorption value below 0.1 at the excitation wavelength using the following equation:
 | (1) |
Luminescence lifetimes were measured on a Horiba Scientific TCSPC system equipped with a nanoLED 370. Lifetimes were deconvoluted with FS-900 software using a light-scattering solution (LUDOX) for instrument response. The excitation source was a laser diode (λexc = 320 nm).
The fluorescence emission spectra in the solid-state were recorded, as 1 wt% doped in poly(methylmethacrylate) (PMMA) films. The quantum yields were calculated as absolute values, using an integration sphere fitted to the spectrofluorimeter.
Computational methods
Ground and excited singlet and triplet geometries, total electronic energies, corresponding transition (absorption and emission) energies and oscillator strengths presented in the main text were obtained at the CC2 level of theory.45 CC2 calculations were conducted with the TURBOMOLE 7.5.1 package,57 using the triple valence def2-TZVP basis set, in the gas phase, with the spin-component scaling (SCS) approximation.46 ΔEST values presented in this work are obtained by computing the CC2 electronic energy difference between the S1 and T1 states in their respective equilibrium geometry37 in the gas phase.For comparison purposes (see the ESI,† Tables S2 to S7), DFT and its time-dependent variant (TD-DFT, in the Tamm–Dancoff approximation58) were also performed with Gaussian 16.59 In the specific case of triplet states, B3LYP, M06-2X hybrid functionals, as well as ωB97X-D range-separated hybrid functional were compared to obtain ground and excited (singlet and triplet) states geometries, vibration calculations, vertical absorption and emissions, using the triple valence 6-311+G(d,p) basis set in all cases. All DFT calculations were made in gas phase or with the linear-response polarizable continuum model (LR-PCM) of toluene. From the comparison between the results obtained in the gas phase and toluene, we conclude that solvent effects do not change our conclusions based on more accurate CC2 calculations in the gas phase, except for an expected shift of the electronic transition energies.
Energetic barriers between Qeq and Qax conformers were determined at the TD-DFT ωB97X-D/6-311+G(d,p) level of theory using the Synchronous Transit-Guided Quasi Newton (STQN) QST3 method.
SOCME values presented in this work were computed at the TD-DFT level (M06-2X/TZVP) on CC2 T1 geometries in the gas phase. The M06-2X functional was preferred in this step as this functional returns the closest transition energies values to CC2. These SOCME values were obtained with the ADF software.60
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
S. A., A. F., C. K. and M. H. are grateful to the EUR LUMOMAT project and the Investments for the Future program ANR-18-EURE-0012. M. H. is thankful to Dr Manon Bousquet for the frequent discussion on CC2 models.References
- S. Achelle, J. Rodríguez-López and F. Robin-le Guen, Org. Biomol. Chem., 2023, 21, 39–52 RSC.
- (a) L. Pascal, J.-J. Vaden Eynde, Y. Van Haverbeke, P. Dubois, A. Michel, U. Rant, E. Zojer, G. Leising, L. O. Van Dorm, N. E. Gruhn, J. Cornil and J.-L. Brédas, J. Phys. Chem. B, 2002, 106, 6442–6450 CrossRef CAS; (b) S. Achelle, A. Barsella, C. Baudequin, B. Caro and F. Robin-le Guen, J. Org. Chem., 2012, 77, 4087–4096 CrossRef CAS PubMed.
- S. Achelle, I. Nouira, B. Pfaffinger, Y. Ramondenc, N. Plé and J. Rodríguez-López, J. Org. Chem., 2009, 74, 3711–3717 CrossRef CAS PubMed.
- M. Fecková, P. le Poul, F. Robin-le Guen, T. Roisnel, O. Pytela, M. Klikar, F. Bureš and S. Achelle, J. Org. Chem., 2018, 83, 11712–11726 CrossRef PubMed.
- S. Achelle, J. Rodríguez-López and F. Robin-le Guen, ChemistrySelect, 2018, 3, 1852–1886 CrossRef CAS.
- A. J. Gillett, A. Pershin, R. Pandya, S. Feldmann, A. J. Sneyd, A. M. Alvertis, E. W. Evans, T. H. Thomas, L.-S. Cui, B. H. Drummond, G. D. Scholes, Y. Olivier, A. Rao, R. H. Friend and D. Beljonne, Nat. Mater., 2022, 21, 1150–1157 CrossRef CAS PubMed.
- C. Hadad, S. Achelle, I. López-Solera, J. C. Garía-Martínez and J. Rodríguez-López, Dyes Pigm., 2013, 97, 230–237 CrossRef CAS.
- J.-P. Malval, M. Cranney, S. Achelle, H. Akdas-Kiliç, J.-L. Fillaut, N. Cabon, F. Robin-le Guen, O. Soppera and Y. Molard, Chem. Commun., 2019, 55, 14331–14334 RSC.
- A. Boländer, D. Kiesser, C. Voss, S. Bauer, X. Schön, S. Burgold, T. Bittner, J. Hölzer, R. Heyny-von Haußen, G. Mall, V. Goetschy, C. Czech, H. Knust, R. Berger, J. Herms, I. Hilger and B. Schmidt, J. Med. Chem., 2012, 55, 9170–9180 CrossRef PubMed.
- A. I. Aranda, S. Achelle, F. Hammerer, F. Mahuteau-Betzer and M.-P. Teulade-Fichou, Dyes Pigm., 2012, 95, 400–407 CrossRef CAS.
- S. Achelle, J. Rodríguez-López, C. Katan and F. Robin-le Guen, J. Phys. Chem. C, 2016, 120, 26986–26995 CrossRef CAS.
- H. Akdas-Kiliç, M. Godfroy, J.-L. Fillaut, B. Donnio, B. Heinrich, P. Kedziora, J.-P. Malval, A. Spangenberg, S. Cleuvenbergen, K. Clays and F. Camerel, J. Phys. Chem. C, 2015, 119, 3697–3710 CrossRef.
- (a) B. Liu, H.-L. Zhang, J. Liu, Y.-D. Zhao, Q.-M. Luo and Z.-L. Huang, J. Mater. Chem., 2007, 17, 2921–2929 RSC; (b) L. Li, J. Ge, H. Wu, Q.-H. Xu and S. Q. Yao, J. Am. Chem. Soc., 2012, 134, 12157–12167 CrossRef CAS PubMed; (c) L. Hu, S. Husain, T. Liu, Y. Yue, J. Liu, Y. Tian and X. Tian, New J. Chem., 2018, 42, 14725–14728 RSC.
- L. Li, Y. Tian, J.-X. Yang, P.-P. Sun, J.-Y. Wu, H.-P. Zhou, S.-Y. Zhang, B.-K. Jin, X.-J. Xing, C.-K. Wang, M. Li, G.-H. Chen, H.-H. Tang, W.-H. Huang, X.-T. Tao and M.-H. Jiang, Chem. – Asian J., 2009, 4, 668–680 CrossRef CAS PubMed.
- J.-P. Malval, S. Achelle, L. Bodiou, A. Spangenberg, L. C. Gomez, O. Soppera and F. Robin-le Guen, J. Mater. Chem. C, 2014, 2, 7869–7880 RSC.
- I. Duesto, S. Sarasa, D. Barrios, J. Orduna, B. Villacampa and M.-J. Blesa, Dyes Pigm., 2022, 203, 110310 CrossRef.
- (a) S. van Cleuvenbergen, P. Kedziora, J.-L. Fillaut, T. Verbiest, K. Clays, H. Akdas-Kiliç and F. Camerel, Angew. Chem. Int., Ed., 2017, 56, 9564 CrossRef PubMed; (b) R. J. Durand, S. Gauthier, S. Achelle, T. Groazard, S. Kahlal, J.-Y. Saillard, A. Barsella, N. le Poul and F. Robin-le Guen, Dalton Trans., 2018, 47, 3965–3975 RSC.
- (a) Y. Tao, K. Yuang, T. Chen, P. Xu, H. Li, R. Chen, C. Zheng, L. Zhang and W. Huang, Adv. Mater., 2014, 26, 7631–7958 Search PubMed; (b) M. Y. Wong and E. Zysman-Colman, Adv. Mater., 2017, 29, 1605444 CrossRef PubMed; (c) J. M. Teng, Y.-F. Wang and C.-F. Chen, J. Mater. Chem. C, 2020, 8, 11340–11353 RSC; (d) W. Che, Y. Xie and Z. Li, Asian J. Org. Chem., 2020, 9, 1262–1276 CrossRef CAS; (e) M. Cai, D. Zhang and L. Duan, Chem. Rec., 2019, 19, 1611–1623 CrossRef CAS PubMed.
- (a) A. Endo, K. Sato, K. Yoshimura, T. Kai, A. Kawada, H. Miyazaki and C. Adachi, Appl. Phys. Lett., 2011, 98, 083302 CrossRef; (b) H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed; (c) Q. Zhang, B. Li, S. Huang, H. Nomura, H. Tanaka and C. Adachi, Nat. Photonics, 2014, 8, 326–332 CrossRef CAS.
- (a) T.-T. Bui, F. Goubard, M. Ibrahim-Ouali, D. Gigmes and F. Dumur, Beilstein J. Org. Chem., 2018, 14, 282–308 CrossRef CAS PubMed; (b) S. Scholz, D. Kondakov, B. Lüssem and K. Leo, Chem. Rev., 2015, 115, 8449–8503 CrossRef CAS PubMed.
- M. A. Bryden and E. Zysman-Colman, Chem. Soc. Rev., 2021, 50, 7587–7680 RSC.
- V.-N. Nguyen, A. Kumar, M. H. Lee and J. Yoon, Coord. Chem. Rev., 2020, 425, 213545 CrossRef CAS.
- W. Liu, C. Zhang, R. Alessandri, B. T. Diroll, Y. Li, H. Liang, X. Fan, K. Wang, H. Cho, Y. Liu, Y. Dai, Q. Su, N. Li, S. Li, S. Wai, Q. Li, S. Shao, L. Wang, J. Xu, X. Zhang, D. V. Talapin, J. J. De Pablo and S. Wang, Nat. Mater., 2023, 22, 737–745 CrossRef CAS PubMed.
- P. Pander and F. B. Dias, Disp. Imaging, 2017, 2, 249–263 Search PubMed.
- (a) L. Gan, K. Gao, X. Cai, D. Chen and S. J. Su, J. Phys. Chem. Lett., 2018, 9, 4725–4731 CrossRef CAS PubMed; (b) P. L. Santos, J. S. Ward, P. Data, A. S. Bartsanov, M. R. Bryce, F. B. Dias and A. P. Monkman, J. Mater. Chem. C, 2016, 4, 3815–3824 RSC.
- N. Aizawa, Y.-J. Pu, Y. Harabuchi, A. Nihonyanagi, R. Ibuka, H. Inuzuka, B. Dhara, Y. Koyama, K. Nakayama, S. Maeda, F. Araoka and D. Miyajima, Nature, 2022, 609, 502–506 CrossRef CAS PubMed.
- F. B. Dias, K. N. Bourdakos, V. Jankus, K. C. Moss, K. T. Kamtekar, V. Bhalla, J. Santos, M. R. Bryce and A. P. Monkman, Adv. Mater., 2013, 25, 3707–3714 CrossRef CAS PubMed.
- Z. Zhao, S. Yan and Z. Ren, Acc. Chem. Res., 2023, 56, 1942–1952 CrossRef CAS PubMed.
- J. Li, M. Zhang, T. Li, D. Guo, T. Tian and H. Zhang, J. Mater. Chem. C, 2022, 10, 13124–13136 RSC.
- (a) B. Li, Z. Wang, S.-J. Su, F. Guo, Y. Cao and Y. Zhang, Adv. Opt. Mater., 2019, 7, 1801496 CrossRef; (b) M. K. Etherington, J. Gibson, H. F. Higginbotham, T. J. Penfold and A. P. Monkman, Nat. Commun., 2016, 7, 13680 CrossRef CAS PubMed; (c) F. B. Dias, J. Santos, D. R. Graves, P. Data, R. S. Nobuyasu, M. A. Fox, A. S. Batsanov, T. Palmeira, M. N. Berberan-Santos, M. R. Bryce and A. P. Monkman, Adv. Sci., 2016, 3, 1600080 CrossRef PubMed.
- (a) N. Sharma, M. Maciejczyk, D. Hall, W. Li, V. Liégeois, D. Beljonne, Y. Olivier, N. Robertson, I. D. W. Samuel and E. Zysman-Colman, ACS Appl. Mater. Interfaces, 2021, 13, 44628–44640 CrossRef CAS PubMed; (b) M. Hempe, N. A. Kukhta, A. Danos, M. A. Fox, A. S. Batsanov, A. P. Monkman and M. R. Bryce, Chem. Mater., 2021, 33, 3066–3080 CrossRef CAS PubMed.
- (a) T. Nakagawa, S. Y. Ku, K. T. Wong and C. Adachi, Chem. Commun., 2012, 48, 9580–9582 RSC; (b) G. Mehes, H. Nomura, Q. S. Zhang, T. Nakagawa and C. Adachi, Angew. Chem., Int. Ed., 2012, 51, 11311–11315 CrossRef CAS PubMed; (c) K. Nasu, T. Nakagawa, H. Nomora, C. Lin, C. Cheng, M. Tseng, T. Yasuda and C. Adachi, Chem. Commun., 2013, 49, 10385–10387 RSC.
- (a) M. Liu, Y. Seino, D. Chen, S. Inomata, S.-J. Su, H. Sasabe and J. Kido, Chem. Commun., 2015, 51, 16353–16356 RSC; (b) Y. Tao, K. Yuan, T. Chen, P. Xu, H. H. Li, R. F. Chen, C. Eheng, L. Zhang and W. Huang, Adv. Mater., 2014, 26, 7931–7958 CrossRef CAS PubMed; (c) H. Wang, L. S. Xie, Q. Peng, L. Q. Meng, Y. Wang, Y. P. Yi and P. F. Wang, Adv. Mater., 2014, 26, 5198–5204 CrossRef CAS PubMed.
- R. Komatsu, H. Sasabe, Y. Seino, K. Nakao and J. Kido, J. Mater. Chem. C, 2016, 4, 2274–2278 RSC.
- (a) R. Komatsu, H. Sasabe and J. Kido, J. Photon. Energy, 2018, 8, 032108 Search PubMed; (b) S. Achelle, M. Hodée, J. Massue, A. Fihey and C. Katan, Dyes Pigm., 2022, 200, 110157 CrossRef CAS; (c) L. Yu and C. Yang, J. Mater. Chem. C, 2021, 9, 17265–17286 RSC; (d) U. Tsiko, O. Bezvikonnyi, G. Sych, R. Keruckiene, D. Volyniuk, J. Simokaitiene, I. Danyliv, Y. Danyliv, A. Bucinskas, X. Tan and J. V. Grazulevicius, J. Adv. Res., 2021, 33, 41–51 CrossRef CAS PubMed; (e) U. Tsiko, D. Volyniuk, V. Andruleviciene, K. Leitonas, G. Sych, O. Bezvikonnyi, V. Jasinskas, V. Gulbinas, P. Stakhira and J. V. Grazulevicius, Mater. Today Chem., 2022, 25, 100955 CrossRef CAS.
- (a) M. Cai, D. Zhang, J. Xu, X. Hong, C. Zhao, X. Song, Y. Qiu, H. Kaji and L. Duan, ACS Appl. Mater. Interfaces, 2019, 11, 1096–1108 CrossRef CAS PubMed; (b) J. S. Jang, H. L. Lee, K. H. Lee and J. Y. Lee, J. Mater. Chem. C, 2019, 7, 12695–12703 RSC; (c) T. Serevičius, J. Dodonova, R. Skaisgiris, D. Banevičius, K. Kazlauskas, S. Juršėnas and S. Tumkevičius, J. Mater. Chem. C, 2020, 8, 11192–11200 RSC.
- (a) K.-C. Pan, S.-W. Li, Y.-Y. Ho, Y.-J. Shiu, W.-L. Tsai, M. Jiao, W.-K. Lee, C.-C. Wu, C. L. Chung, T. Chatterjee, Y.-S. Li, K.-T. Wong, H.-C. Hu, C.-C. Chen and M. T. Lee, Adv. Funct. Mater., 2016, 26, 7560–7571 CrossRef CAS; (b) K. Nakao, H. Sasabe, R. Komatsu, Y. Hayasaka, T. Ohsawa and J. Kido, Adv. Opt. Mater., 2017, 5, 1600843 CrossRef.
- I. S. Park, J. Lee and T. Yasuda, J. Mater. Chem. C, 2016, 4, 7911–7916 RSC.
- (a) K. Wu, T. Zhang, L. Zhan, C. Zhong, S. Gong, N. Jiang, Z.-H. Lu and C. Yang, Chem. Eur. J., 2016, 22, 10860–10866 CrossRef CAS PubMed; (b) T. Serevičius, R. Skaisgiris, J. Dodonova, L. Jagintavičius, D. Banevičius, K. Kazlauskas, S. Tumkevičius and S. Juršėnas, ACS Appl. Mater. Interfaces, 2020, 12, 10727–10736 CrossRef PubMed; (c) Y. Xiang, P. Li, S. Gong, Y.-H. Huang, C.-Y. Wang, C. Zhong, W. Zeng, Z. Chen, W.-K. Lee, X. Yin, C.-C. Wu and C. Yang, Sci. Adv., 2020, 6, eaba7855 CrossRef CAS PubMed.
- T. Serevičius, R. Skaisgiris, J. Dodonova, K. Kazlauskas, S. Juršėnas and S. Tumkevičius, Phys. Chem. Chem. Phys., 2020, 22, 265–272 RSC.
- H. Görner, J. Phys. Chem., 1989, 93, 1826–1832 CrossRef.
- T. Nevesely, M. Wienhold, J. J. Molloy and R. Gilmour, Chem. Rev., 2002, 122, 2650–2694 CrossRef PubMed.
- (a) Q. Zhu, X. Guo and J. Zhang, J. Comput. Chem., 2019, 40, 1578–1585 CrossRef CAS PubMed; (b) B. Li, Z. Wang, S. Su, F. Guo, Y. Cao and Y. Zhang, Adv. Opt. Mater., 2019, 7, 1801496 CrossRef.
- (a) A. D. Laurent and D. Jacquemin, Intern. J. Quantum Chem., 2013, 113, 2019–2039 CrossRef CAS; (b) D. Jacquemin, I. Duchemin and X. Blase, J. Chem. Theory Comput., 2015, 11, 5340–5359 CrossRef CAS PubMed.
- O. Christiansen, H. Koch and P. Jorgensen, Chem. Phys. Lett., 1995, 243, 409–418 CrossRef CAS.
- A. Hellweg, S. A. Grün and C. Hättig, Phys. Chem. Chem. Phys., 2008, 10, 4119 RSC.
- (a) A. Pershin, D. Hall, V. Lemaur, J.-C. Sancho-Garcia, L. Muccioli, E. Zysman-Colman, D. Beljonne and Y. Olivier, Nat. Commun., 2019, 10, 597 CrossRef CAS PubMed; (b) D. Hall, S. M. Suresh, P. L. dos Santos, E. Duda, S. Bagnich, A. Pershin, P. Rajamalli, D. B. Cordes, A. M. Z. Slawin, D. Beljonne, A. Köhler, I. D. W. Samuel, Y. Olivier and E. Zysman-Colman, Adv. Opt. Mater., 2020, 8, 1901627 CrossRef CAS.
- P. K. Samanta, D. Kim, V. Coropceanu and J.-L. Brédas, J. Am. Chem. Soc., 2017, 139, 4042–4051 CrossRef CAS PubMed.
- M. A. El-Sayed, J. Chem. Phys., 1963, 38, 2834–2838 CrossRef CAS.
- (a) Y. J. Cho, B. D. Chin, S. K. Jeon and J. Y. Lee, Adv. Funct. Mater., 2015, 25, 6786–6792 CrossRef CAS; (b) T. Hatakeyama, K. Shiren, K. Nakajima, S. Nomura, S. Nakatsuka, K. Kinoshita, J. Ni, Y. Ono and T. Ikuta, Adv. Mater., 2016, 28, 2777–2781 CrossRef CAS PubMed.
- I. S. Park, H. Komiyama and T. Yasuda, Chem. Sci., 2017, 8, 953–960 RSC.
- C. H. Lee, S. H. Choi, S. J. Oh, J. H. Lee, J. W. Shim, C. Adachi and S. Y. Lee, RSC Adv., 2020, 10, 42897–42902 RSC.
- T. Serevičius, R. Skaigiris, J. Dodonova, L. Jagintavičius, J. Bucevičius, K. Kazlauskas, S. Juršėnas and S. Tumkevičius, Chem. Commun., 2019, 55, 1975–1978 RSC.
- K. Stavrou, L. G. Franca, T. Böhmer, L. M. Duben, C. M. Marian and A. P. Monkman, Adv. Funct. Mater., 2023, 2300910 CrossRef CAS.
- H. Tanaka, K. Shizu, H. Nakanotani and C. Adachi, J. Phys. Chem. C, 2014, 118, 15985–15994 CrossRef CAS.
- T. J. Penfold, F. B. Dias and A. P. Monkman, Chem. Commun., 2018, 54, 3926–3935 RSC.
- (a) S. G. Balasubramani, G. P. Chen, S. Coriani, M. Diedenhofen, M. S. Frank, Y. J. Franzke, F. Furche, R. Grotjahn, M. E. Harding, C. Hättig, A. Hellweg, B. Helmich-Paris, C. Holzer, U. Huniar, M. Kaupp, A. Marefat Khah, S. Karbalaei Khani, T. Müller, F. Mack, B. D. Nguyen, S. M. Parker, E. Perlt, D. Rappoport, K. Reiter, S. Roy, M. Rückert, G. Schmitz, M. Sierka, E. Tapavicza, D. P. Tew, C. van Wüllen, V. K. Voora, F. Weigend, A. Wodyński and J. M. Yu, J. Chem. Phys., 2020, 152, 184107 CrossRef CAS PubMed; (b) TURBOMOLE V7.5.1 2021, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989-2007, TURBOMOLE GmbH, since 2007, available from https://www.turbomole.org.
- M. J. G. Peach, M. J. Williamson and D. J. Tozer, J. Chem. Theor. Comput., 2011, 7, 3578–3585 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, F. Williams, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision A.03, Gaussian Inc., Wallingford CT, 2016 Search PubMed.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp03705c |
| This journal is © the Owner Societies 2023 |