
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Intramolecular locking and coumarin insertion: a stepwise approach for TADF design†
S.
Paredis
 abc,
T.
Cardeynaels
abcd,
S.
Brebels
abc,
J.
Deckers
abc,
S.
Kuila
e,
A.
Lathouwers
abc,
M.
Van Landeghem
bcf,
K.
Vandewal
bcf,
A.
Danos
*e,
A. P.
Monkman
e,
B.
Champagne
d and
W.
Maes
*abc
abc,
T.
Cardeynaels
abcd,
S.
Brebels
abc,
J.
Deckers
abc,
S.
Kuila
e,
A.
Lathouwers
abc,
M.
Van Landeghem
bcf,
K.
Vandewal
bcf,
A.
Danos
*e,
A. P.
Monkman
e,
B.
Champagne
d and
W.
Maes
*abc
aHasselt University, Institute for Materials Research (IMO-IMOMEC), Design & Synthesis of Organic Semiconductors (DSOS), Agoralaan 1, Diepenbeek 3590, Belgium. E-mail: wouter.maes@uhasselt.be
bIMOMEC Division, IMEC, Wetenschapspark 1, Diepenbeek 3590, Belgium
cEnergyville, Thorpark, Genk 3600, Belgium
dUniversity of Namur, Laboratory of Theoretical Chemistry, Theoretical and Structural Physical Chemistry Unit, Namur Institute of Structured Matter, Rue de Bruxelles 61, Namur 5000, Belgium
eDurham University, Department of Physics, OEM Group, South Road, Durham DH1 3LE, UK. E-mail: andrew.danos@durham.ac.uk
fHasselt University, Institute for Materials Research (IMO-IMOMEC), Organic Opto-Electronics (OOE), Wetenschapspark 1, Diepenbeek 3590, Belgium
First published on 18th October 2023
Abstract
Three novel TADF (thermally activated delayed fluorescence) emitters based on the well-studied Qx-Ph-DMAC fluorophore are designed and synthesized. The photophysical properties of these materials are studied from a theoretical and experimental point of view, demonstrating the cumulative effects of multiple small modifications that combine to afford significantly improved TADF performance. First, an extra phenyl ring is added to the acceptor part of Qx-Ph-DMAC to increase the conjugation length, resulting in BQx-Ph-DMAC, which acts as an intermediate molecular structure. Next, an electron-deficient coumarin unit is incorporated to fortify the electron accepting ability, affording ChromPy-Ph-DMAC with red-shifted emission. Finally, the conjugated system is further enlarged by ‘locking’ the molecular structure, generating DBChromQx-DMAC with further red-shifted emission. The addition of the coumarin unit significantly impacts the charge-transfer excited state energy levels with little effect on the locally excited states, resulting in a decrease of the singlet–triplet energy gap. As a result, the two coumarin-based emitters show considerably improved TADF performance in 1 w/w% zeonex films when compared to the initial Qx-Ph-DMAC structure. ‘Locking’ the molecular structure further lowers the singlet–triplet energy gap, resulting in more efficient reverse intersystem crossing and increasing the contribution of TADF to the total emission.
Introduction
Thermally activated delayed fluorescence (TADF) emitters have gained significant attention as active materials for organic light-emitting diodes (OLEDs) as they can harvest both singlet and triplet excitons, realizing theoretical internal quantum efficiencies (IQE) up to 100%.1–4 In contrast to the strong spin–orbit coupling (SOC) that enables phosphorescence in organometallic emitters,5,6 high IQEs for all-organic TADF materials are achieved by reverse intersystem crossing (rISC), up-converting the 75% of triplet excitons arising from electrical excitation to the emissive singlet excited state.7–10 Typically such TADF materials consist of covalently linked electron-donating (D) and electron-accepting (A) units that have near-perpendicular electronic subsystems, spatially limiting the overlap of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) and generating a small energy difference (ΔEST) between the first excited singlet (S1) and triplet (T1) state, which allows the desired rISC up-conversion to occur.1,11Despite the promising properties of D–A TADF materials, their charge-transfer (CT) emissive states lead to rather broad emission spectra, resulting in desaturated emission colours which limits the supported colour spaces in OLED displays.12 The emission can be narrowed by rigidifying the molecular structure though, restricting rotational and vibrational motions that otherwise broaden the range of exciton energies in individual molecules (and hence also the overall spectrum).13 Moreover, rigid structures can also suppress vibrationally-coupled non-radiative decay pathways, improving the photoluminescence quantum yield (PLQY) and OLED efficiency.14 Furthermore, expanding the conjugation length of a molecular structure will typically lower the S1 energy level,15 resulting in a red-shifted emission and, depending on the energy of the relevant triplet state, a smaller ΔEST.16 A downside of increasing the rigidity of the active material is that this is frequently accompanied by a higher degree of planarity and resulting tendency to aggregate, which is particularly common in D–A TADF compounds.17–19 This aggregation behaviour can be resolved, either by ensuring a near-perpendicular orientation between the D and A to sterically frustrate strong intermolecular π–π-interactions,20 or by implementing bulky substituents.21–24
In this work we demonstrate two parallel rational design strategies to enhance the emission properties of D–A TADF molecules. The design of these novel materials starts with Qx-Ph-DMAC,25 a previously reported emitter consisting of a 2,3-diphenylquinoxaline (Qx-Ph) acceptor unit coupled with two 9,10-dihydro-9,9-dimethylacridine (DMAC) donating moieties that exhibits green TADF emission when doped in a non-polar 4,4′-bis(N-carbazolyl)-1,1′-biphenyl (CBP) host. The first modification strategy involves incorporation of an electron-deficient coumarin unit into the molecular structure to tune the acceptor strength and hence the alignment of the excited state energies, i.e.ChromPy-Ph-DMAC. An intermediate molecular structure with an expanded π-system but no coumarin fragment is also prepared, i.e.BQx-Ph-DMAC, and studied to better understand the stepwise effects of coumarin insertion into the molecular structure. A second strategy, recently reported elsewhere,16 is here also applied to the final coumarin-based dyad to ‘lock’ its phenyl substituents into a phenanthrene unit, generating a more rigid molecular structure with a larger conjugation length to potentially improve both colour purity and PLQY.
Comparing the different materials, Qx-Ph-DMAC shows very weak TADF at room temperature in a 1 w/w% zeonex host, while π-expanded BQx-Ph-DMAC exhibits a combination of weak TADF and room temperature phosphorescence (RTP) in zeonex. We confirm that the incorporation of an electron-deficient coumarin unit into the structure drastically lowers the energy of the excited CT states, while the locally excited (LE) state energies remain similar. As a result, ΔEST decreases, enabling efficient TADF for both ChromPy-Ph-DMAC and its ‘locked’ counterpart DBChromQx-DMAC. ‘Locking’ the molecular structure enlarges the conjugated system in the latter, also further reducing the energy gap. This leads to more efficient up-conversion and increases the contribution of TADF to the overall emission efficiency in this final molecular structure of the stepwise series.
Results and discussion
Phenyl-1,2-diamine and naphthalene-1,2-diamine were commercially available, while 3,4-diamino-2H-chromen-2-one (2) was synthesized according to literature.26 Starting from 4-chloro-3-nitro-2H-chromen-2-one the chlorine group was first substituted for an amine and then the nitro substituent was reduced (Scheme 1). Thereafter, a condensation reaction between the diamines and the ‘locked’ or ‘unlocked’ diketone yielded the desired brominated coumarin-quinoxaline products (3–6). The brominated acceptor units were then coupled to 9,10-dihydro-9,9-dimethylacridine (DMAC) using a Buchwald–Hartwig cross-coupling reaction, affording Qx-Ph-DMAC, BQx-Ph-DMAC, ChromPy-Ph-DMAC, and DBChromQx-DMAC. Details on the synthetic procedures and characterization data are provided in the ESI.† | ||
Scheme 1 Synthesis procedure for Qx-Ph-DMAC, BQx-Ph-DMAC, ChromPy-Ph-DMAC, and DBChromQx-DMAC, and their intermediates: (i) NH4OH, 1,4-dioxane, room temperature, 2 h; (ii) Na2S2O4, EtOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water (2:3), 60 °C, 1 h; (iii) glacial acetic acid, 125 °C, 16 h (iv); Pd(OAc)2, [(t-Bu)3PH]BF4, NatBuO, toluene, reflux, 16 h. :water (2:3), 60 °C, 1 h; (iii) glacial acetic acid, 125 °C, 16 h (iv); Pd(OAc)2, [(t-Bu)3PH]BF4, NatBuO, toluene, reflux, 16 h. | ||
To better understand their electronic structures and excited states, the molecular geometries of Qx-Ph-DMAC, BQx-Ph-DMAC, ChromPy-Ph-DMAC, and DBChromQx-DMAC were optimised using density functional theory (DFT) calculations (M06/6-311G(d)). The singlet and triplet energies were determined using additional time-dependent DFT (TDDFT) calculations using a modified LC-BLYP (ω = 0.17 Bohr−1) exchange–correlation (XC) functional.27,28 The TDDFT calculations were performed under the Tamm–Dancoff approximation on the optimised ground state geometries and using the polarizable continuum model (PCM) in cyclohexane to simulate a non-polar environment (similar to zeonex films) in the Gaussian16 package.29 The orbital spatial distributions were obtained from single-point calculations using the same LC-BLYP/6-311G(d) method. The CT character of the involved states was investigated by looking at the differences between ground and excited state electron densities. These CT characters are described by the distance over which the electronic charge is transferred (dCT) and the related change in dipole moment (Δμ), calculated as described by Le Bahers and coworkers.30
DFT geometry optimisations illustrated that the ‘unlocked’ phenyl rings of Qx-Ph-DMAC, BQx-Ph-DMAC, and ChromPy-Ph-DMAC are twisted with respect to the acceptor core (θ ≈ 50°), as shown in Fig. 1. Fusing these two phenyl rings creates a larger planar core (DBChromQx-DMAC), as expected based on prior studies.16 The donating DMAC unit is also twisted for all four dyads, sitting at θ ≈ 37° relative to the bridging phenyl rings for the ‘unlocked’ emitters and at θ = 90° relative to the acceptor core for the ‘locked’ structure. These twisted orientations result in a strong separation of the HOMO and LUMO, i.e. the HOMO and HOMO−1 are localized on the donor, while the LUMO and LUMO+1 are localized on the acceptor part in all cases (Fig. 1). Consequently, for these dyads strong CT character is expected for the lowest energy excited singlet states.31,32 These expectations are fully in line with the theoretical calculations as displayed in Table S1 (showing the CT distance and change in dipole moment of the relevant excited states, ESI†) and Fig. S1 (illustrating the ground–excited state electron density differences, ESI†).
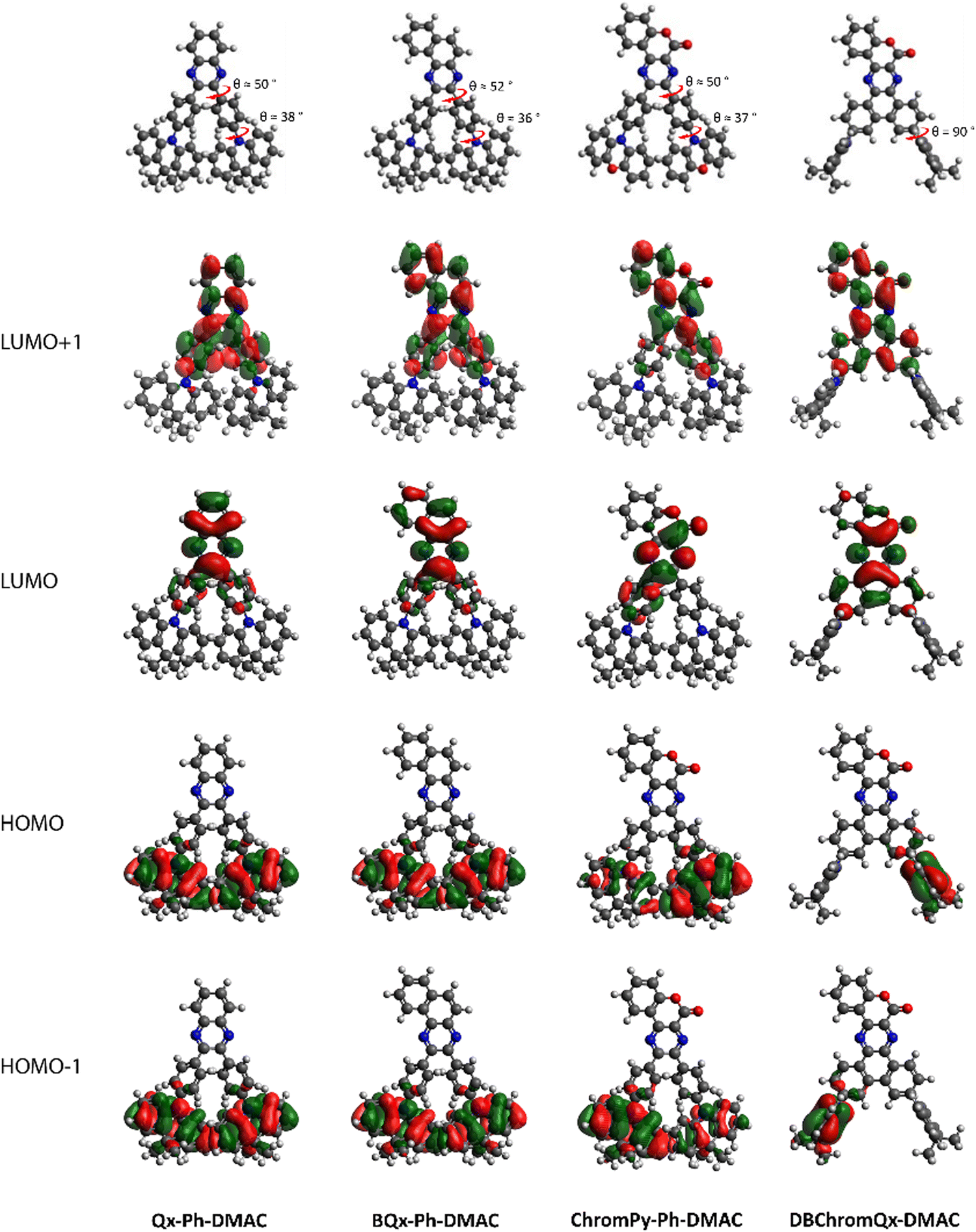 | ||
| Fig. 1 HOMO–LUMO spatial distributions for Qx-Ph-DMAC, BQx-Ph-DMAC, ChromPy-Ph-DMAC, and DBChromQx-DMAC. Isocontour values of 0.02 (a.u.) were used for all orbitals. | ||
Steady-state absorption and emission spectra in toluene and methylcyclohexane (MCH) solution are shown in Fig. 2. In all four cases the absorption spectra consist of a major peak at higher energy, at λ = 270–310 nm, and a minor peak at λ = 340–410 nm, which can be assigned to the absorption of the donor and the acceptor, respectively.25 For Qx-Ph-DMAC, ChromPy-Ph-DMAC, and DBChromQx-DMAC, an additional small absorption peak is observed at λ > 390 nm corresponding to direct CT absorption. These assignments are supported by simulated TDDFT absorption spectra, shown in Fig. S2 (ESI†). The emission spectra in toluene consist of a single broad peak for the four dyads, suggesting CT nature. Some vibronic structure is apparent in the blue-shifted MCH emission spectra though (especially for ChromPy-Ph-DMAC and DBChromQx-DMAC), indicating LE singlet states are the lowest energy emissive states in MCH, with the CT states becoming stabilised (red-shifted) and coming to dominate the emission in the more polar solvent toluene. For all four emitters a clear red-shift of the emission is observed when going from MCH to toluene, providing extra evidence for the CT character of these excited states. These trends in emission wavelengths across the four materials are consistent with the theoretical vertical excitation energies for S1 shown in Table S2 (ESI†). Moreover, the theoretical ΔEST can be estimated by calculating the difference between this S1 value and the calculated vertical excitation energy for T1, which is also tabulated (Table S2, ESI†). The theoretical ΔEST values for both Qx-Ph-DMAC and BQx-Ph-DMAC are considered too large to generate up-conversion from T1 to S1,20 and thus no TADF emission is expected in this environment. In contrast, the calculated energy gap for ChromPy-Ph-DMAC and DBChromQx-DMAC (0.12 and 0.08 eV, respectively) becomes sufficiently small for rISC and up-conversion to occur (which is observed experimentally, vide infra).
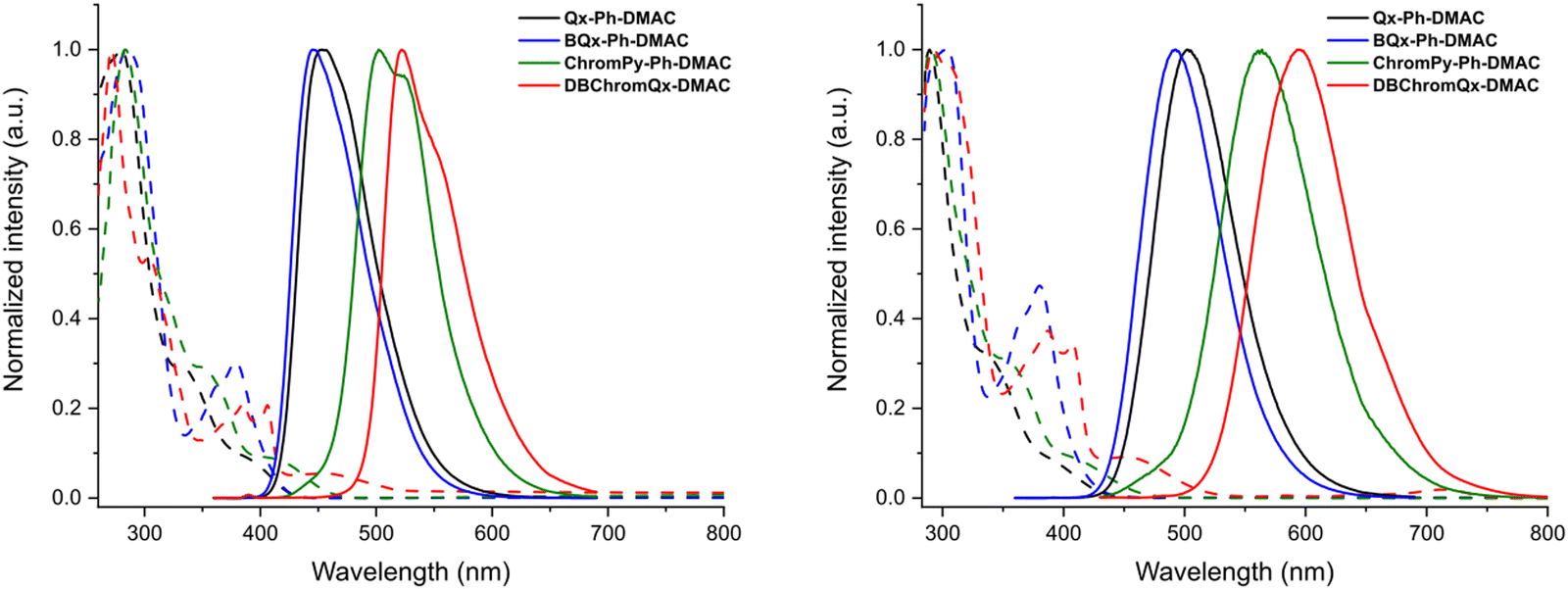 | ||
| Fig. 2 Normalized steady-state absorption spectra (dashed lines) and normalized steady-state emission spectra (solid lines) for the emitters in MCH (left) and in toluene (right) at an excitation wavelength of 350 nm. | ||
Time-resolved emission spectroscopy was performed for all four compounds doped in 1 w/w% zeonex films (photographs are shown in Fig. S3, ESI†) to examine the delayed emission properties. Fig. 3 shows normalized contour plots of the time-resolved emission spectra after pulsed excitation of the four dyads at room temperature, and at 80 K. These plots reveal prompt fluorescence (PF), presence/absence of delayed fluorescence (DF), and phosphorescence (PH) for each of the synthesized compounds. The PF among the series shows similar spectral trends as observed in the steady-state measurements, indicating an enhanced acceptor strength for ChromPy-Ph-DMAC and DBChromQx-DMAC. A spectral redshift is also observed within the first 100 nanoseconds for all four emitters, both at room temperature and at 80 K. This spectral shift could be attributed to either aggregation and/or a distribution of dihedral angles between donor and acceptor units,33,34 each exhibiting a specific emission wavelength and emission lifetime. Furthermore, it can be seen that the coumarin-based compounds exhibit detectable DF throughout the entire time window of the measurement, whereas this is not the case for Qx-Ph-DMAC and BQx-Ph-DMAC where the much weaker DF falls below the noise floor of the instrument in the 1–100 μs range. This contrast is further visualized in the combined decay plot shown in Fig. 4A, which plots the total emission intensity versus time.
 | ||
| Fig. 3 Normalized time-resolved emission spectra for Qx-Ph-DMAC, BQx-Ph-DMAC, ChromPy-Ph-DMAC, and DBChromQx-DMAC in zeonex at room temperature (left) and at 80 K (right). The noise arising after the prompt fluorescence for Qx-Ph-DMAC and BQx-Ph-DMAC represents the noise floor of the hardware. | ||
 | ||
| Fig. 4 (A) Decay of the total emission for Qx-Ph-DMAC, BQx-Ph-DMAC, ChromPy-Ph-DMAC, and DBChromQx-DMAC in zeonex (1 w/w%) at room temperature (open circles) and at 80 K (solid squares). Data points for which the emission signal falls below the hardware noise baseline have been omitted from the decays. (B) Normalized steady-state emission spectra (solid lines), and phosphorescence spectra (dashed lines; degassed, 80 K and 80 ms delay time) for Qx-Ph-DMAC (black), BQx-Ph-DMAC (blue), ChromPy-Ph-DMAC (green), and DBChromQx-DMAC (red) in a 1 w/w% zeonex film. | ||
Delayed emission does nonetheless eventually reappear at room temperature for both Qx-Ph-DMAC and BQx-Ph-DMAC in the ms regime. For Qx-Ph-DMAC this emission originates from the S1 state as the wavelength is similar to the PF at room temperature. BQx-Ph-DMAC on the other hand shows a very broad and red-shifted ms emission, suggesting a combination of TADF from S1 alongside room temperature phosphorescence (RTP) from T1.31,32 These emission properties are further illustrated by the individual time-resolved spectra for each dyad at both temperatures presented in Fig. S4 (ESI†). The claimed dual emission and RTP for BQx-Ph-DMAC is further supported by Fig. S5 (ESI†), which compares the room temperature emission both for the ns (PF) and ms (claimed TADF and RTP) regimes to that at 80 K (unambiguous PH).
ChromPy-Ph-DMAC shows a red-shifted emission in the early μs DF regime, which can be assigned to various different molecular geometries that result from the rotational freedom of this ‘unlocked’ structure as films are formed.34 This red-shift is indeed less pronounced for the ‘locked’ analogue DBChromQx-DMAC, with more restricted D–A geometries. These red-shifts are in both cases small though, and the DF observed in the coumarin-based dyads appears to originate from the same excited state as the PF, i.e. S1, suggesting TADF as the delayed emission mechanism and supported by the small theoretical ΔEST (Table S2, ESI†).
At 80 K, no DF is observed in the μs regime for Qx-Ph-DMAC and BQx-Ph-DMAC, which is similar to the observation at room temperature. In both cases the contour plots of the time-resolved emission spectra (Fig. 3, time points without emission contribution are removed in the decays in Fig. 4) show the normalized shape of the detector baseline. However, red-shifted and unambiguous PH emission later appears in the ms regime. For ChromPy-Ph-DMAC and DBChromQx-DMAC DF from the S1 state is observed throughout the entire time window, even at 80 K. The ‘unlocked’ ChromPy-Ph-DMAC eventually shows red-shifted PH in the ms regime. For DBChromQx-DMAC, no similar red-shift occurs, suggesting that either no phosphorescence is observed or that phosphorescence originates from a triplet state which is nearly isoenergetic to S1 (Fig. 4).
Additional calculations on the higher excited state energy levels were then performed (Fig. S1 and Tables S3, S4, ESI†), showing that the lowest 3CT levels (T4 for Qx-Ph-DMAC and BQx-Ph-DMAC; T3 for ChromPy-Ph-DMAC and DBChromQx-DMAC) are nearly isoenergetic with the S1 states. Furthermore, a triplet state with LE character is also located nearby, resulting in vibronic coupling between the two triplet states.8 The relative position of these excited state energy levels facilitates ISC (kISC values in Table 1), generating triplet excitons which were also quantified by the relatively high singlet oxygen quantum yields in toluene (ΦΔ, Table 1 and Fig. S6, ESI†). Moreover, in all four materials a significant increase is observed for the PLQY in degassed toluene (Φf,inert) compared to the quantum yield in the presence of dissolved oxygen (Φf,atm) (Fig. S7, ESI†). In zeonex films, the differences between Φf,inert and Φf,atm are less pronounced for Qx-Ph-DMAC and BQx-Ph-DMAC, indicating little to no triplet harvesting properties, consistent with the minimal DF in time-resolved spectroscopy (Fig. 3 and 4A). ChromPy-Ph-DMAC and DBChromQx-DMAC on the other hand do show a significant increase in PLQY for zeonex films under inert atmosphere (Table 1), consistent with their stronger TADF activity. Additionally, locking the molecular structure results in an almost 10-fold increase in intersystem crossing rate (krISC; Fig. S8, ESI†) and a 20% reduction in the full width at half maximum (FWHM) in zeonex, nicely confirming the success of the stepwise molecular design strategy.16
| Compound | Toluene | Zeonex | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Φ f,atm | Φ f,inert | Φ Δ | Φ f,atm | Φ f,inert | FWHM (eV) | k ISC (s−1)f | k rISC (s−1)f | ΔEST (eV)g | |
| a Photoluminescence quantum yield in toluene solution under normal atmosphere, determined vs. quinine (Φf = 0.58, λexc = 347 nm in 0.1 M H2SO4). b Photoluminescence quantum yield in toluene solution under inert atmosphere, determined vs. quinine. c Singlet oxygen quantum yield in toluene solution, determined vs coronene (ΦΔ = 0.90, λexc = 325 nm in toluene) by monitoring the absorbance of 1,3-diphenylisobenzofuran at 414 nm. d Photoluminescence quantum yield of 1 w/w% zeonex films measured in an integrating sphere under air. e Photoluminescence quantum yield of 1 w/w% zeonex films measured in an integrating sphere under nitrogen flow. f kISC and krISC were determined using kinetic fitting of the prompt and delayed fluorescence according to literature.35 g Calculated as ES1 − ET1. | |||||||||
| Qx-Ph-DMAC | 0.15 | 0.23 | 0.55 | 0.22 | 0.21 | 0.36 | / | / | 0.32 |
| BQx-Ph-DMAC | 0.22 | 0.32 | 0.54 | 0.31 | 0.35 | 0.36 | / | / | 0.39 |
| Chrom-Py-Ph-DMAC | 0.13 | 0.40 | 0.67 | 0.40 | 0.55 | 0.40 | 1.76 × 107 | 9.16 × 104 | 0.19 |
| DBChrom-Qx-DMAC | 0.15 | 0.34 | 0.35 | 0.41 | 0.60 | 0.32 | 1.94 × 107 | 7.83 × 105 | 0.01 |
Using the difference in onset between the (steady-state) fluorescence and phosphorescence (80 K; 80 ms delay time) as shown in Fig. 4B, the experimental ΔEST can be determined (Table 1). These values are similar but large for Qx-Ph-DMAC and BQx-Ph-DMAC (ΔEST = 0.32 and 0.39 eV, respectively), aligning with theoretical calculations and consistent with their poor TADF activity. Incorporating the coumarin unit lowers this energy gap (ΔEST = 0.19 eV), while ‘locking’ the molecular structure results in nearly isoenergetic S1 and T1 states (ΔEST = 0.01 eV). A schematic representation of the excited state energy levels from both calculations and experiments is given in Fig. 5.
 | ||
| Fig. 5 Schematic representation of the excited state energy levels (in eV) with their corresponding LE/CT (green/red) character as obtained using TDDFT calculations. The values obtained experimentally in 1 w/w% zeonex films are added in brackets. | ||
TDDFT calculations (Fig. S1 and Tables S2–S4, ESI†) illustrate that the lowest 1CT and 3CT states are nearly isoenergetic to each other in each of the four materials. By incorporating the coumarin unit and ‘locking’ the molecular structure, both the singlet and triplet CT states are stabilized and brought closer to the low-lying 3LE states. This analysis of the interplay between excited state energies makes it possible to gain a deeper insight into the differences in the emission mechanisms, and consequently the PLQYs of the emitters in zeonex. For Qx-Ph-DMAC and BQx-Ph-DMAC, the triplet excitons that are formed likely become trapped in the low-lying 3LE states after efficient ISC and internal conversion on the triplet manifold. These trapped triplet excitons can be harvested (though inefficiently) by rISC in both materials, but also by RTP for BQx-Ph-DMAC in which both the S1–T1 and T2–T1 energy gaps are smaller.20 On the other hand, for ChromPy-Ph-DMAC and especially for DBChromQx-DMAC, vibronic coupling between the 3LE and 3CT states (with much smaller ΔEST) enables efficient up-conversion to the singlet state, supporting efficient TADF as the delayed emission mechanism. The improved TADF kinetics of DBChromQx-DMAC are also supported by its smaller experimental ΔEST and evidenced by larger krISC values compared to ChromPy-Ph-DMAC.
Conclusions
In this work we demonstrate the combined impact of two design strategies for improved TADF properties. A first approach involves integrating a coumarin unit into the acceptor part of the molecule (ChromPy-Ph-DMAC and DBChromQx-DMAC), which increases their accepting ability compared to the (benzo)quinoxaline counterparts (Qx-Ph-DMAC and BQx-Ph-DMAC). This leads to the decrease of the excited CT state energies and opens the TADF pathway for the coumarin-based emitters due to their smaller singlet–triplet gaps. In parallel, we also implement a “locking” strategy by fusing the two phenyl pendants, which expands the conjugated system and further reduces the energy of the excited CT states. This approach results in more efficient rISC and thus a larger contribution of TADF to the total emission in zeonex films for the final ‘locked’ DBChrom-Qx-DMAC. Simultaneously, the enhanced rigidity also narrows the inherently broad CT emission from TADF emitters.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors thank the Research Foundation – Flanders (FWO Vlaanderen) for financial support through projects G087718N, G0D1521N, I006320N, GOH3816NAUHL, the Scientific Research Community ‘Supramolecular Chemistry and Materials’ (W000620N), postdoctoral fellowships 1284623N (T. Cardeynaels) and 1270123N (M. Van Landeghem), and PhD scholarship of S. Paredis. The calculations were performed on the computers of the ‘Consortium des équipements de Calcul Intensif (CÉCI)’ (https://www.ceci-hpc.be), including those of the ‘UNamur Technological Platform of High-Performance Computing (PTCI)’ (https://www.ptci.unamur.be), for which we gratefully acknowledge financial support from the FNRS-FRFC, the Walloon Region, and the University of Namur (Conventions No. GEQ U.G006.15, U.G018.19, U.G011.22, RW/GEQ2016, RW1610468, and RW2110213). S. Kuila and A. P. Monkman are supported by EPSRC grant EP/T02240X/1.References
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 CrossRef CAS PubMed.
- X. Yin, Y. He, X. Wang, Z. Wu, E. Pang, J. Xu and J. Wang, Front. Chem, 2020, 8, DOI:10.3389/fchem.2020.00725.
- W. Chen and F. Song, Chin. Chem. Lett., 2019, 30, 1717–1730 CrossRef CAS.
- H. Nakanotani, Y. Tsuchiya and C. Adachi, Chem. Lett., 2021, 50, 938–948 CrossRef CAS.
- G. Hong, X. Gan, C. Leonhardt, Z. Zhang, J. Seibert, J. M. Busch and S. Bräse, Adv. Mater., 2021, 33, 2005630 CrossRef CAS PubMed.
- A. Sarto Polo and K. Passalacqua Morelli Frin, Springer Handbook of Inorganic Photochemistry, 2022, pp. 27–48 Search PubMed.
- J. Singh, U. Shakeel, D. Ompong and K. S. Ram, J. Phys. Chem. Lett., 2022, 13, 6177–6180 CrossRef CAS PubMed.
- M. K. Etherington, J. Gibson, H. F. Higginbotham, T. J. Penfold and A. P. Monkman, Nat. Commun., 2016, 7, 13680 CrossRef CAS PubMed.
- J. Gibson, A. P. Monkman and T. J. Penfold, Chem. Phys. Chem., 2016, 17, 2956–2961 CrossRef CAS PubMed.
- J. Gibson and T. J. Penfold, Phys. Chem. Chem. Phys., 2017, 19, 8428–8434 RSC.
- F.-M. Xie, H.-Z. Li, G.-L. Dai, Y.-Q. Li, T. Cheng, M. Xie, J.-X. Tang and X. Zhao, ACS Appl. Mater. Interfaces, 2019, 11, 26144–26151 CrossRef CAS PubMed.
- J. Adachi and H. Okada, Advanced Display Technology, 2021, pp. 39–65 Search PubMed.
- H. J. Kim and T. Yasuda, Adv. Opt. Mater., 2022, 10, 2201714 CrossRef CAS.
- J.-M. Teng, Y.-F. Wang and C.-F. Chen, J. Mater. Chem. C, 2020, 8, 11340–11353 RSC.
- I. A. Wright, A. Danos, S. Montanaro, A. S. Batsanov, A. P. Monkman and M. R. Bryce, Chem. – Eur. J., 2021, 27, 6545–6556 CrossRef CAS PubMed.
- S.-C. Ji, T. Zhao, Z. Wei, L. Meng, X.-D. Tao, M. Yang, X.-L. Chen and C.-Z. Lu, J. Chem. Eng., 2022, 435, 134868 CrossRef CAS.
- Y. Xiao, H. Wang, Z. Xie, M. Shen, R. Huang, Y. Miao, G. Liu, T. Yu and W. Huang, Chem. Sci., 2022, 13, 8906–8923 RSC.
- T. Cardeynaels, M. K. Etherington, S. Paredis, A. S. Batsanov, J. Deckers, K. Stavrou, D. Vanderzande, A. P. Monkman, B. Champagne and W. Maes, J. Mater. Chem. C, 2022, 10, 5840–5848 RSC.
- M. K. Etherington, N. A. Kukhta, H. F. Higginbotham, A. Danos, A. N. Bismillah, D. R. Graves, P. R. McGonigal, N. Haase, A. Morherr, A. S. Batsanov, C. Pflumm, V. Bhalla, M. R. Bryce and A. P. Monkman, J. Phys. Chem. C, 2019, 123, 11109–11117 CrossRef CAS PubMed.
- M. Mahmoudi, E. Urbonas, D. Volyniuk, D. Gudeika, K. Dabrovolskas, J. Simokaitiene, A. Dabuliene, R. Keruckiene, K. Leitonas, M. Guzauskas, L. Skhirtladze, M. Stanitska and J. V. Grazulevicius, Molecules, 2023, 28, 5999 CrossRef CAS PubMed.
- L. Salah, M. K. Etherington, A. Shuaib, A. Danos, A. A. Nazeer, B. Ghazal, A. Prlj, A. T. Turley, A. Mallick, P. R. McGonigal, B. F. E. Curchod, A. P. Monkman and S. Makhseed, J. Mater. Chem. C, 2021, 9, 189–198 RSC.
- E. Aksoy, A. Danos, C. Li, A. Monkman and C. Varlikli, J. Phys. Chem. C, 2021, 125, 13041–13049 CrossRef CAS.
- J. Royakkers, K. Guo, D. T. W. Toolan, L. Feng, A. Minotto, D. G. Congrave, M. Danowska, W. Zeng, A. D. Bond, M. Al-Hashimi, T. J. Marks, A. Facchetti, F. Cacialli and H. Bronstein, Angew. Chem., Int. Ed., 2021, 60, 25005–25012 CrossRef CAS PubMed.
- D. G. Congrave, B. H. Drummond, V. Gray, A. D. Bond, A. Rao, R. H. Friend and H. Bronstein, Polym. Chem., 2021, 12, 1830–1836 RSC.
- T. Huang, D. Liu, J. Jiang and W. Jiang, Chem. – Eur. J., 2019, 25, 10926–10937 CrossRef CAS PubMed.
- V. L. Savel’ev, O. L. Samsonova, V. S. Troitskaya, V. G. Vinokurov, V. P. Lezina and L. D. Smirnov, Chem. Heterocycl. Compd., 1988, 24, 805–810 CrossRef.
- T. J. Penfold, J. Phys. Chem. C, 2015, 119, 13535–13544 CrossRef CAS.
- T. Cardeynaels, S. Paredis, J. Deckers, S. Brebels, D. Vanderzande, W. Maes and B. Champagne, Phys. Chem. Chem. Phys., 2020, 22, 16387–16399 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, revision A.03, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- T. Le Bahers, C. Adamo and I. Ciofini, J. Chem. Theory Comput., 2011, 7, 2498–2506 CrossRef CAS PubMed.
- S. Paredis, T. Cardeynaels, J. Deckers, A. Danos, D. Vanderzande, A. P. Monkman, B. Champagne and W. Maes, J. Mater. Chem. C, 2022, 10, 4775–4784 RSC.
- S. Paredis, T. Cardeynaels, S. Kuila, J. Deckers, M. Van Landeghem, K. Vandewal, A. Danos, A. P. Monkman, B. Champagne and W. Maes, Chem. – Eur. J., 2023, 29, e202301369 CrossRef CAS PubMed.
- D. Kelly, L. G. Franca, K. Stavrou, A. Danos and A. P. Monkman, J. Phys. Chem. Lett., 2022, 13, 6981–6986 CrossRef CAS PubMed.
- K. Stavrou, L. G. Franca, T. Böhmer, L. M. Duben, C. M. Marian and A. P. Monkman, Adv. Funct. Mater., 2023, 33, 202300910 CrossRef.
- N. Haase, A. Danos, C. Pflumm, A. Morherr, P. Stachelek, W. Brütting and A. P. Monkman, J. Phys. Chem. C, 2018, 122, 29173–29179 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp03695b |
| This journal is © the Owner Societies 2023 |