
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnalysis of the bonding in tetrahedrane and phosphorus-substituted tetrahedranes
Daniel
Del Angel Cruz
a,
Jorge L.
Galvez Vallejo
ab and
Mark S.
Gordon
 *a
*a
aDepartment of Chemistry, Iowa State University, Ames, Iowa 50011, USA. E-mail: mark@si.msg.chem.iastate.edu
bSchool of Computing, Australian National University, Canberra, ACT 2601, Australia
First published on 26th September 2023
Abstract
The bonding structures of tetrahedrane, phosphatetrahedrane, diphosphatetrahedrane and triphosphatetrahedrane are studied by employing an intrinsic quasi-atomic orbital analysis. Ethane, cyclopropane and tetrahedral P4 are employed as reference systems. The orbital analysis is paired with the computation of strain energies via isodesmic reactions. The results show that the increase in geometric strain upon transition from ethane to cyclopropane to tetrahedrane weakens the CC bonds, despite leading to shorter C–C interatomic distances. With the increase in strain, the orbitals centered on C and involved in the bonding of the cage structure are observed to have elevated p-character, and the orbital structure of C deviates from sp3 hybridization. The systematic substitution of CH groups by P atoms in the cage structure of tetrahedrane leads to stronger CC bonds, larger angles in the cage structures of the resulting phosphatetrahedranes, lower p-character in the orbitals involved in the bonding of the cages, and lower strain energies. It is found that P is more amenable to strained molecular arrangements than is C, and that the propensity of a given atom to hybridize s and p functions, or the lack thereof, has implications in the stability of molecules with strained geometries. The combination of the calculations presented here with the existing literature provides insight into the apparent propensity of tetrahedrane and P4 to ‘break’ their tetrahedral structures. Trends in the bonding interactions, such as bond strengths, s- and p-orbital characters and charge transfer are identified and related to the strain energy observed in each of the analyzed systems.
1. Introduction
Strained molecules such as cyclopropane and tetrahedrane have been of great interest due to their unstable nature, unusual bonding, and challenging, frequently elusive, experimental behavior. Theoretical studies have been conducted to evaluate properties derived from the electronic structure of such systems, notably equilibrium geometries, vibrational frequencies, and strain energies.1–4 Tetrahedrane has been the subject of several theoretical studies,5–8 but has not yet been isolated experimentally. Other analog systems with tetrahedral structures, like P4, Si4H4 and N4 have also been studied with computational methods.9–14 Interest in tetrahedrane arises from its unusual molecular geometry, in which the CCC angles are all much smaller than the approximately tetrahedral angles found in most organic molecules, and because of its potential applications as energetic materials, such as explosives, propellants, and pyrotechnics,15–17 as well as the new insight it could provide into the nature of unconventional organic structures.Previous reports on the bonding in tetrahedrane have noted that the CC bond path length, defined by the path of Maximum Electron Density (MED) between two atoms, is longer than the C–C internuclear distance.17,18 That is, the bonds in tetrahedrane have been observed to bend away from the center of the molecule. Theoretical studies have identified bent bonds as a common trait of strained molecules,18–22 and crystallographic studies have shown experimental evidence of bent bonds in small-ring systems.23,24 Bending has also been related to a weakening of the CC bonds in strained molecules.18,21 The work of Coulson and Moffit20 showed that molecular geometries with small angles lead to orbitals with elevated p-type character, and a recent study reported this observation for tetrahedrane.15 In general, the common traits in the bonding of strained molecules22,26,29 can be presumed to apply to tetrahedrane, as will be discussed in Sections 4.2.2 and 4.2.3.
The first bulky group-substituted derivative of tetrahedrane, tetra-tert-butyltetrahedrane, was reported in 1978 by Maier and collaborators,25 and other substituted analogs have since been reported.26–28 The synthesis reported by Maier and collaborators was carried out employing tetra-tert-butylcyclopentadienone as a precursor.25,29 In 2019, Hierlmeier and collaborators reported the synthesis of the di-phosphorous analog of tetra-tert-butyltetrahedrane, di-tert-butyldiphosphatetrahedrane, in which two carbon atoms and the attached tert-butyl groups have been replaced by P atoms. This synthesis was done through dimerization of a tert-butyl-substituted phosphaalkyne (tBuCP) employing various nickel tricarbonyl complexes as catalysts.30 Later, in 2020, Riu and collaborators reported the synthesis of the mono-phosphorous analog of tetra-tert-butyltetrahedrane (Tri-tert-butylphosphatetrahedrane),31 in which a carbon atom and the attached tert-butyl substituent have been replaced by a single phosphorous atom. The synthesis of tri-tert-butylphosphatetrahedrane, P(CtBu)3, was carried out employing a P-containing anthracene derivative and the tri-tert-bytulcyclopropenium ion under an inert atmosphere of purified N2. More recently, in 2021, Riu and coauthors reported the synthesis of triphosphatetrahedrane (P3CH) employing chloroform and an anionic niobium triphosphatetrahedrane complex, which contained an all-phosphorous triangle in its structure.32 The tendency of the phosphorous-substituted tetrahedrane derivatives to polymerize has been noted: tri-tert-butylphosphatetrahedrane was observed to undergo dimerization in the presence of Lewis acids,31 di-tert-butyldiphosphatetrahedrane was reported to dimerize above its melting point of −32 °C,30 and the isolation of triphosphatetrahedrane was not achieved due to its instability and apparent tendency to undergo thermally-driven polymerization.32
As mentioned in ref. 31, the white phosphorous molecule (P4) has a tetrahedral structure analogous to tetrahedrane, which suggests that a P atom could replace a CR group in tetrahedrane. Theoretical studies on the P4 molecule predict that it is a very slightly strained system,10 accounting for its structural stability, and previous computational studies have shown that the substitution of a C atom by a P atom can relieve strain in cyclic molecules.21,32 In ref. 21 Boatz and Gordon used homodesmic reactions to compute strain energies for cyclopropane and phosphirane (phosphacyclopropane) at the restricted Hartree–Fock (RHF) and second order perturbation (MP2) levels of theory. Their results showed that the strain energy was 4.4 (RHF) and 8.2 (MP2) kcal mol−1 lower in phosphirane than in cyclopropane. The cyclopropane strain energies were calculated to be 28.8 (RHF) and 30.2 (MP2) kcal mol−1, respectively.
A recent study by Schaefer and collaborators33 investigated the strain energy and bonding structures of phosphatetrahedrane and other pnictogen-substituted tetrahedranes, in terms of Natural Bond Orbitals (NBOs).34 Their results suggested that the substitution of a CH group in tetrahedrane by increasingly heavier atoms of the pnictogen group has the effect of successively lowering the strain energy of the resulting structures and increasing the partial negative charge on the C atoms. Their analysis of the bonding structures found elevated p-orbital contributions to the NBOs involved in pnictogen–carbon bonds, in line with the findings of the previous report by Riu and collaborators on the bonding of tri-tert-butylphosphatetrahedrane.31 The work of Riu and collaborators analyzed the electron density of tri-tert-butylphosphatetrahedrane computed at the B3LYP-D3/6-31G(d,p) level of theory.31 Their results showed that the C–P MED bond paths deviated from the C–P interatomic axis, in line with the observations for tetrahedrane and other strained molecules discussed above. Riu, Ye and Cummins analyzed the effect of the substitution of CH groups by P atoms in tetrahedrane on the strain energy of the resulting structures32 and predicted that the introduction of P atoms into the tetrahedral structures has the effect of lowering the strain energy of the resulting molecules.
The recent experimental and theoretical studies discussed above indicate a growing interest in the P-substituted analogs of tetrahedrane. However, to the best of the authors’ knowledge, no direct comparison of the bonding structures of these molecules and tetrahedrane has been reported to date. Therefore, a systematic comparison between the parent hydrocarbon molecules and their phosphorus analogs phosphatetrahedrane, diphosphatetrahedrane, and triphosphatetrahedrane is of interest. The present work provides such a study of the parent molecule tetrahedrane (td) and the recently reported phosphatetrahedrane (ptd), diphosphatetrahedrane (p2td) and triphosphatetrahedrane (p3td), to analyze and compare their structures, ring strain, and bonding motifs. Conventional systems such as ethane, cyclopropane and P4 are used as reference molecules. The current analysis combines strain energy predictions based on isodesmic reactions35 with a comparative analysis of the bonding in both strained and unstrained molecules using a unique approach that was developed by Ruedenberg and co-workers.36–43
In isodesmic reactions35 a strained molecule is transformed to an unstrained analog in such a way that all bond types are conserved from reactants to products. Isodesmic reactions have been employed to predict strain energies for cyclopropane, its derivatives, inorganic three-membered rings,22,44–46 as well as for tetrahedrane.1,47 On the other hand, isodesmic reactions have been employed to investigate the unusual stability in delocalized cyclic systems48–50 such as benzene. In the present work, isodesmic reactions are used to evaluate the relative ring strain in cyclopropane, td, ptd, p2td, p3td, and P4.
In addition to the analysis of ring strain in these five molecules, an important goal of the present study is to analyze the nature of the bonding in td, ptd, p2td, and p3td. This is achieved by employing the quasi-atomic orbital (QUAO) analysis developed by Ruedenberg and co-workers36–43 to assess the bonding interactions in all systems. An important aspect of the QUAO analysis is that it is based on the actual molecular wave function and does not employ inherently biased information based on “mathematical expedience”. Moreover, as noted in ref. 38, quasi-atomic orbitals are the ab initio counterparts of the early qualitative concept of “hybridized atomic bond orbitals”. The QUAO analysis can elucidate the nature of the bonding in a molecule in a way that is consistent with the chemical intuition of the reader. In the present study, the QUAO analysis is employed to understand the fundamental aspects of bonding in td, ptd, p2td and p3td.
The most relevant aspects of the theory of the quasi-atomic orbital analysis and isodesmic reactions are outlined in Section 2. The computational details of the present work are provided in Section 3. In Section 4 the nature of the bonding in ethane, cyclopropane, td, ptd, p2td, p3td and P4 is analyzed in terms of QUAOs. The bonding analysis is combined with the assessment of strain energies using isodesmic reactions. It is demonstrated in Section 4 how the QUAO analysis is able to provide insightful interpretations of the computed strain energies. Finally, the conclusions of the present study are given in Section 5.
2. Methods
In this section, the basic concepts and components of the QUAO analysis, available in the GAMESS electronic structure program,51–53 are briefly summarized, as well as the basics of isodesmic reactions. The QUAO analysis has been presented in detail in ref. 37, 38, 41, 54–57, and isodesmic reactions have been applied to many molecular species.2.1 Oriented quasi atomic orbitals
For all molecules in the present study, the QUAO analysis has been applied to the RHF-SCF wave functions. QUAOs can be thought of as minimal basis set orbitals, deformed from the respective free atoms due to the bonding structure in the molecule. The QUAOs are embedded in the actual molecular wave function. A series of singular value decompositions of the overlap matrix between the molecular orbitals and a highly accurate atomic minimal basis set (AAMBS) allow for the extraction of the QUAOs.37,38,58The QUAOs are “adapted” to the chemical environment of the molecule (i.e. “oriented”) by minimizing the number of interatomic first order density matrix elements with large absolute values.59 In this way, these “oriented QUAOs” capture the essence of covalent bonding in a system.38 Throughout the present work, the terms QUAO and oriented QUAO will be used interchangeably. QUAOs can be thought of as the ab initio counterpart of the early qualitative concept of hybrid orbitals. The s- and p-characters of each QUAO are determined by directly computing the s- and p-fractions of each QUAO.38
2.2 Kinetic bond orders
The first order density matrix ρ(1,2) is expressed in terms of the QUAOs as | (1) |
 | (2) |
2.3 Isodesmic reactions
In an isodesmic reaction, the number of bonds of each formal type (e.g. C–C single bond, C–C double bond, C–H bond) is conserved from reactants to products.65 The advantage of isodesmic reactions is that since each bond type is conserved, relative errors in electron correlation from reactants to products are minimized.66,67 Consequently, even Hartree–Fock can provide reasonable energy differences and second order perturbation theory is generally reliable. The following discussion considers a specific type of isodesmic reaction in which the reactant is a strained ring, and the product is an unstrained analog, such that all bond types are conserved. Pople et al. called such isodesmic reactions bond separation reactions.66 Bond separation reactions have also been used to characterize aromatic and anti-aromatic molecules.50,68,69 The isodesmic reactions for the molecules of interest here are shown in Fig. 1.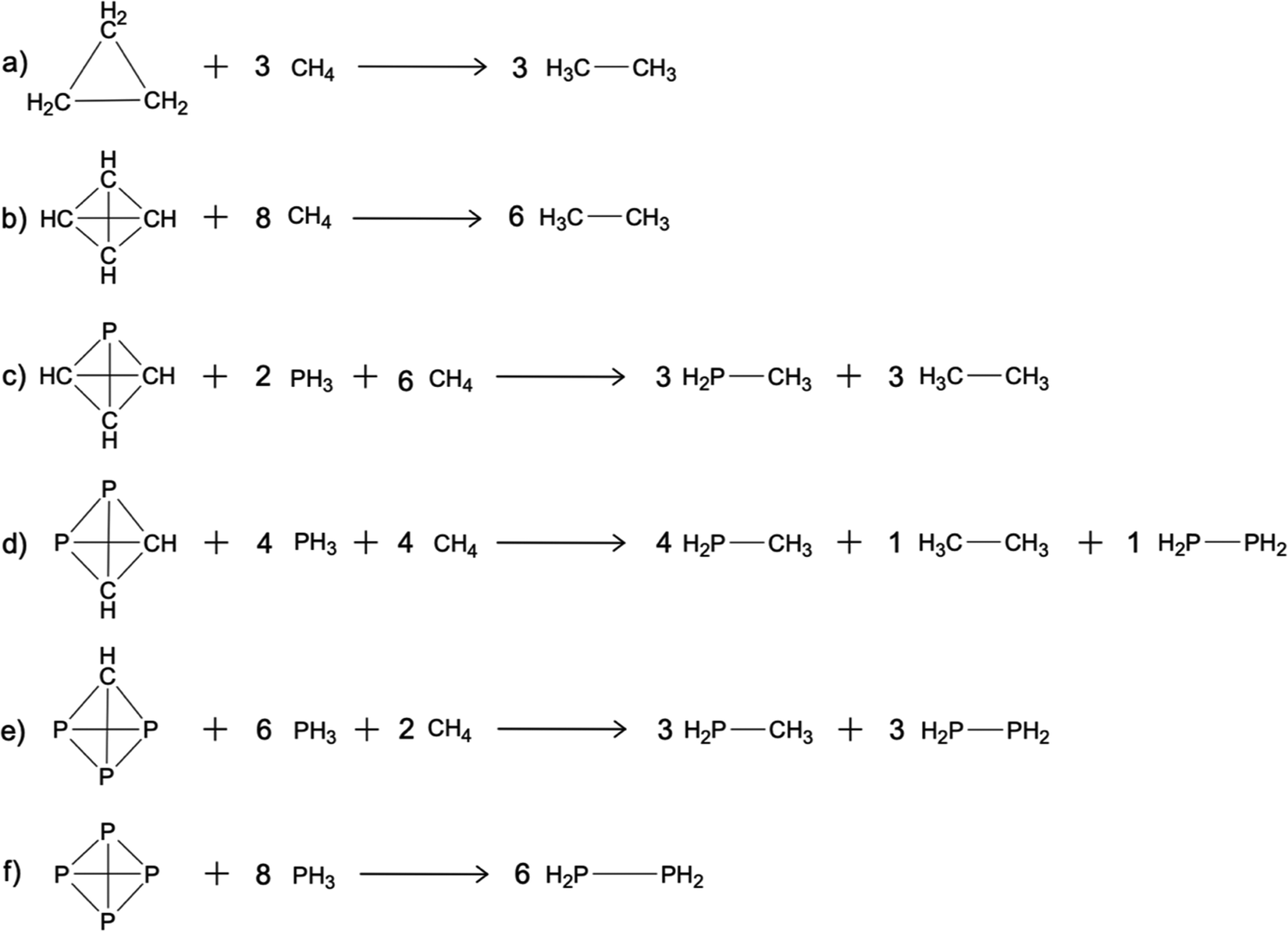 | ||
| Fig. 1 Isodesmic reactions for (a) cyclopropane, (b) td, (c) ptd, (d) p2td, (e) p3td, and (f) P4. | ||
The reaction for cyclopropane (Fig. 1a) has been previously employed by Hariharan and Pople,70 and has a reported reaction energy of −26.2 kcal mol−1 at the RHF/6-31G(d)//RHF/STO-3G level of theory. The negative reaction energy is a measure of the amount of strain in the parent ring compound. The isodesmic reaction for td (Fig. 1b) has previously been used by Glukhovstev and collaborators.47 Their results were obtained with the G2 level of theory71 with a computed reaction energy of −105.5 kcal mol−1 with zero point vibrational energy (ZPE) corrections and −100.2 kcal mol−1, without ZPE corrections. The reactions for ptd (Fig. 1c), p2td (Fig. 1d), and p3td (Fig. 1e) are designed to be as similar as possible to that in Fig. 1b. The reaction for the P4 molecule (Fig. 1f) was previously employed by Schoeller and collaborators,10 using both the HF method, and the coupled electron pair approximation72 (CEPA). Their basis sets consisted of specially designed Gaussian lobe functions. The computed reaction energies, excluding ZPE corrections are −5.3, −3.2 (HF) and 14.4 (CEPA) kcal mol−1, using two different basis sets for the HF calculations and a double zeta basis set for the CEPA calculations. The change from negative (HF) to positive (CEPA) isodesmic reaction energies is attributed to the inclusion of electron correlation, which, according to the authors, favors tighter, more stable ring structures.
3. Computational details
The geometries of all molecules described here were optimized both at the RHF and MP2 levels of theory using the 6-31G(d) basis set. Only singlet states were considered. Hessians were computed at the respective geometries to ascertain their stationary point nature, as well as to compute the respective zero-point energies. RHF zero-point energies have been scaled by 0.9135 in all cases, while MP2 zero-point energies have been scaled by 0.9646.73 QUAOs were obtained from the RHF/6-31G(d) wave functions of td, ptd, p2td, and p3td, calculated at the RHF/6-31G(d) and MP2/6-31G(d) optimized geometries. Only the results obtained at the RHF/6-31G(d) geometries are reported since the RHF/6-31G(d)//MP2/6-31G(d) results were essentially the same. Coupled cluster calculations within the resolution of the identity approximation, RI-CCSD(T)74/cc-pVTZ/cc-pVTZ-RI, were then performed at the MP2/6-31G(d) geometries to compute more accurate strain energies. Analogous calculations were done for ethane, cyclopropane, and P4, which will be used as reference systems. All calculations were performed using the GAMESS software51–53 and QUAOs were plotted using the MacMolPlt75 visualization software.4. Results and discussion
4.1 Geometries
The equilibrium structures of ethane, cyclopropane, td, ptd, p2td, p3td and P4 are displayed in Fig. 2a–g, respectively. The RHF/6-31G(d) and MP2/6-31G(d) geometries are qualitatively similar, and thus only the RHF geometries are displayed in Fig. 2. The geometric parameters of both the RHF and MP2 structures are listed in Table 1.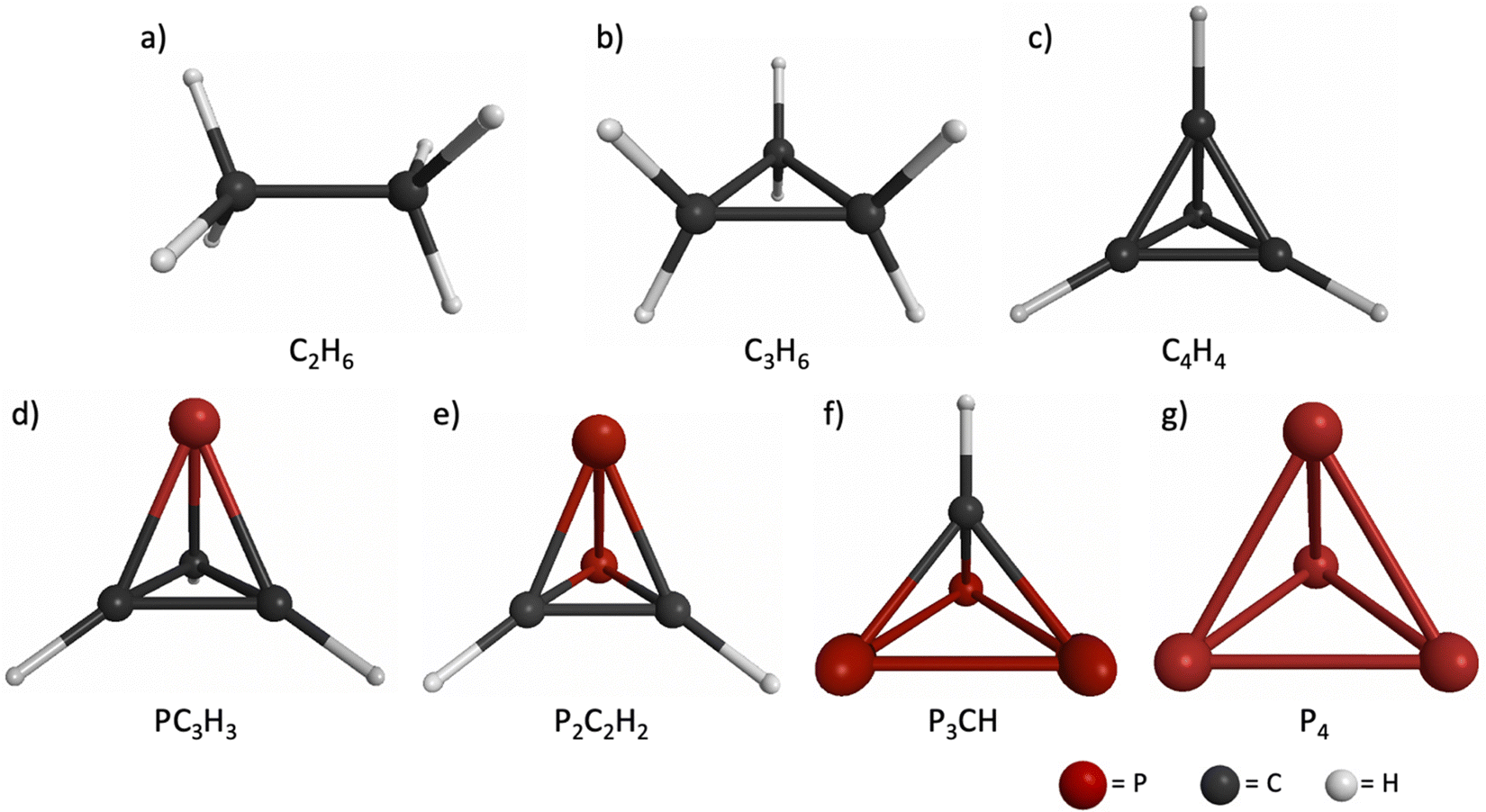 | ||
| Fig. 2 Equilibrium structures of (a) ethane, (b) cyclopropane, (c) td, (d) ptd, (e) p2t, (f) p3td, and (g) P4, optimized at the RHF/6-31G(d) level of theory. Carbon is shown in black, hydrogen is shown in white and phosphorous is shown in red. | ||
| Parameter | C2H6 | C3H6 | C4H4 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Calc. | Expt.76 | Theo.77 | Calc. | Expt.78 | Theo.78 | Calc. | Expt.29 | Theo.15 | |
| a Bond distances are given in angstroms and angles are given in degrees. | |||||||||
| r(C–C) | 1.527 (1.526) | 1.524 | 1.526 | 1.497 (1.503) | 1.501 | 1.502 | 1.463 (1.477) | 1.485 | 1.467 |
| r(C–H) | 1.086 (1.093) | 1.089 | 1.091 | 1.076 (1.085) | 1.083 | 1.078 | 1.063 (1.073) | — | 1.068 |
| ∠(C–C–C) | 60 (60) | — | — | 60 (60) | — | 60 | |||
| ∠(C–C–H) | 111.2 (111.2) | — | 111.2 | 118.1 (118.1) | — | — | 144.7 (144.7) | — | — |
| ∠(H–C–H) | 107.7 (107.7) | 106.9 | — | 114.0 (114.2) | — | 114.8 | |||
| PC3H3 | P2C2H2 | P3CH | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Calc. | Expt.31 | Theo.31 | Calc. | Expt.30 | Theo30 | Calc. | Expt.32 | Theo.32 | |
| r(C–C) | 1.453 (1.460) | 1.474 | 1.46 | 1.452 (1.450) | 1.462 | 1.460 | |||
| r(C–H) | 1.065 (1.076) | — | 1.073 | 1.067 (1.079) | — | — | 1.069 (1.081) | — | 1.076 |
| r(C–P) | 1.826 (1.862) | 1.845 | 1.838 | 1.814 (1.837) | 1.820–1.836 | 1.846 | 1.814 (1.827) | 1.788–1.841 | 1.831 |
| r(P–P) | 2.187 (2.245) | 2.308 | 2.213 | 2.172 (2.213) | — | 2.211 | |||
| ∠(C–C–C) | 60 (60) | — | 60 | ||||||
| ∠(C–C–H) | 140.5 (141.1) | — | 140.8 | 137.0 (137.3) | — | — | |||
| ∠(C–P–C) | 46.9 (46.9) | — | 46.8 | 47.2 (46.5) | 49.6–47.4 | 46.6 | |||
| ∠(C–C–P) | 66.6 (66.9) | — | 66.6 | 66.4 (66.8) | 66.3–66.6 | 66.7 | |||
| ∠(P–C–H) | 144.3 (143.0) | — | 143.9 | 139.7 (139.1) | — | — | 136.3 (135.6) | — | 135.8 |
| ∠(P–P–P) | 60 (60) | — | 60 | ||||||
| ∠(P–P–C) | 52.9 (52.3) | 50.5–51.2 | 53.2 | 53.2 (52.7) | — | 52.8 | |||
| ∠(P–C–P) | 74.2 (75.3) | 78.3 | 73.6 | 73.5 (74.6) | — | 74.3 | |||
The geometries presented in Fig. 2 and Table 1 are all local minima on the respective potential energy surfaces; both td and P4 have tetrahedral structures (Td symmetry). Ptd and p3td have a closed trigonal pyramidal geometry (C3v symmetry), both with the three equivalent atoms arranged in an equilateral triangle, while p2td has C2v symmetry. The bond angles in the ptd cage increase from 60° (C–C–C) in td to 66.6° (C–C–P) due to the substitution of a C–H group by a P atom. This increase in the angles centered about C is expected to relieve some strain in the ring. The C–H bond distances remain almost the same in ptd relative to td; however, the hydrogen atoms are pushed further down from the C–C–C plane in ptd relative to td. This is reflected in the similarity of the C–C–H angle in td and the P–C–H angle in ptd. In p2td, the P–C–P angle is the largest carbon-centered angle of any in the cages analyzed here (74.2° at the RHF/6-31G(d) geometry). Nonetheless, the C–C–P angles remain on the order of 66°. In p3td, the P–C–P angle is only slightly smaller than that in p2td (73.5° at the RHF/6-31G(d) geometry); however, all three C-centered angles in p3td are 73.5°. Thus, it can be initially deduced that p3td subjects C to the lowest geometric strain of all the cages analyzed here, because P can accommodate smaller angles. In general, the larger angles between the bonds containing C in P-containing cages can be thought of as a consequence of the longer C–P and P–P interatomic distances, relative to the C–C distances and the observation that P can accommodate smaller angles than C.
Since neither the parent td nor ptd and p2td have been isolated experimentally, the calculated geometric parameters in Table 1 are compared with experimental data that were determined for analogous compounds that contain bulky substituents, namely tetra-tert-butyltetrahedrane, tri-tert-butylphosphatetrahedrane, and di-tert-butyldiphosphatetrahedrane. For the latter, the experimental geometric parameters were obtained from a silver complex in which di-tert-butyldiphosphatetrahedrane acts as a ligand. It was reported that single crystals suitable for X-ray crystallography could not be grown.30 Similarly, there is currently no experimental data about the isolated structure of p3td. Thus, the data for a system in which P3CH is a ligand on an Fe center is used for comparison.32 Note that, when acting as ligands, the geometries of di-tert-butyldiphosphatetrahedrane and triphosphatetrahedrane are observed to experience distortions that lead to a loss of symmetry. Therefore, the ranges for the reported interatomic distances are shown in Table 1.
Despite the differences between the model and experimental systems, the agreement between the calculations and experiment is reasonable. As one might expect, the MP2 bond distances are in better agreement with experiment than those obtained using HF. Theoretical bond lengths for ethane were determined at the CCSD(T)/cc-pV5Z level of theory by Puzzarini and Taylor.77 As shown in Table 1, the results obtained at both the HF and MP2 levels of theory are in good agreement with the previous geometry predictions. For cyclopropane, the calculated bond distances in the present work are in good agreement with previous computational results reported by Gauss and collaborators.78 Their results were obtained at the CCSD(T)/cc-pVQZ level of theory. The values obtained at the MP2 level of theory are closer to these results than those calculated at the HF level of theory.
The calculated td bond distances presented here are also in good agreement with the previous computational results obtained at the M06-2X81/def2-TZVPP level of theory.15 Interestingly, td has a shorter C–C distance than that of cyclopropane, which is also shorter than the C–C distance in ethane. It is possible to identify a pattern for shorter interatomic distances as the molecular geometry becomes more strained.82 It has been previously shown that in highly strained rings, the bond path defined by the electron density does not necessarily follow the atom-to-atom direct line path.21,22,83 Rather, bond paths in highly strained rings like cyclopropane and td are curved, and as has been previously shown by Bader and collaborators,18 and by Boatz and Gordon.21 So, in molecules like these, a shorter distance does not necessarily imply a stronger bond. It may just indicate a more strained ring. The bonding nature of td will be further discussed below employing quasi-atomic orbitals.
The calculated ptd geometric parameters are in good agreement with those that have been reported by Riu and collaborators,31 both experimentally for the substituted derivative of ptd, and computationally for ptd itself. The geometry reported by Riu and collaborators was obtained at the M06-2X81/6-31G(d,p) level of theory. MP2 is in very good agreement with the M06-2X81/6-31G(d,p) results; MP2 also yields a C–C bond distance that is in closer agreement with experiment than HF. Interestingly, the C–C bond distance in ptd is even shorter than that of td, at both the HF and MP2 levels of theory. For the P–C distance, the HF value is too low by about the same amount as the MP2 distance is too long.
The theoretical geometry parameters shown in Table 1 were reported by Hierlmeier30 and collaborators for the methyl-substituted derivative of p2td, dimethyldiphosphatetrahedrane. The reported geometry was obtained at the TPSS-D3BJ/def2-TZVP level of theory. As can be seen in Table 1, there is overall reasonable agreement between the MP2 and the reported DFT results. Perfect agreement is not expected since the DFT results were obtained from a derivative of p2td. Relative to the experimental data, the P–P interatomic distance appears to be too short both at the RHF and MP2 levels of theory. However, this longer experimental P–P interatomic distance may be a consequence of di-tert-butyldiphosphatetrahedrane acting as a ligand.
For p3td, the calculated internuclear distances are in good agreement with the B3LYP-D3(BJ)/6-311G(d,p) results reported by Riu and collaborators.31 In this case, the MP2/6-31G(d) results appear to be in better agreement than RHF with the DFT results. As for the experimental data, both the RHF and MP2 calculated C–P distances are within the range reported for this interaction. Interestingly, the C–C interatomic distances in p2td and p3td are even shorter than those for td or cyclopropane. The strengths of these interactions will be further discussed below in terms of quasi-atomic orbitals.
For P4 the MP2 P–P internuclear distance is in good agreement with the experimental gas phase value reported by Cossairt and collaborators.79 The CCSD(T)/cc-pVTZ P–P distance reported by Persson and collaborators80 is in excellent agreement with the experimental value.
4.2 Quasi atomic orbitals, populations and bonding analysis
The QUAOs shown below are labeled according to their role in the bonding of the molecule. QUAOs are labeled as follows: the atomic symbol of the atom on which the QUAO is centered is listed first in a capital letter. If the orbital participates in bonding, the atomic symbol of the complementary atom (bonding partner) is listed second in lower case. Primes (′) are employed to differentiate between symmetrically equivalent atoms of a same element in the molecule. The third component indicates the type of bonding in which the QUAO participates or if it is a lone pair. The symbols σ, π, sl and pl indicate that the orbital is a σ-bonding orbital, a π-bonding orbital, an s-type lone pair or a p-type lone pair, respectively. For example, the label Cc′σ indicates a σ QUAO located on a carbon atom and oriented toward a different symmetrically equivalent C atom, c′.The oriented QUAOs obtained for ethane, cyclopropane, td, ptd, p2td, p3td and P4 are displayed in Fig. 3–9 respectively. The orbital labels and occupations are given below the respective orbital, bond orders are shown in bold between the interacting orbitals. KBOs (in kcal mol−1) are given next to the respective bond order. Only interactions with bond order magnitudes greater than 0.50 and kinetic bond orders with magnitude greater than 5.0 kcal mol−1 are shown (all other bond orders are 0.20 or lower).
 | ||
| Fig. 3 Oriented QUAOs of the C2H6 (ethane) RHF/6-31G(d) wave function at the RHF optimized geometry. Orbital labels are shown below the corresponding orbital; orbital populations are displayed next to each label. The bond order is shown in bold between interacting orbitals. Kinetic bond orders (in kcal mol−1) are shown to the right of the respective bond order. Only symmetry unique QUAOs are shown. Two equivalent Cc′σ orbitals are shown for clarity. | ||
 | ||
| Fig. 4 Oriented QUAOs of the C3H6 (cyclopropane) RHF/6-31G(d) wave function at the RHF optimized geometry. Orbital labels are shown below the corresponding orbital; orbital populations are displayed next to each label. The bond order is shown in bold between interacting orbitals. Kinetic bond orders (in kcal mol−1) are shown to the right of the respective bond order. Only symmetry unique QUAOs are shown. Two equivalent Cc′σ orbitals are shown for clarity. | ||
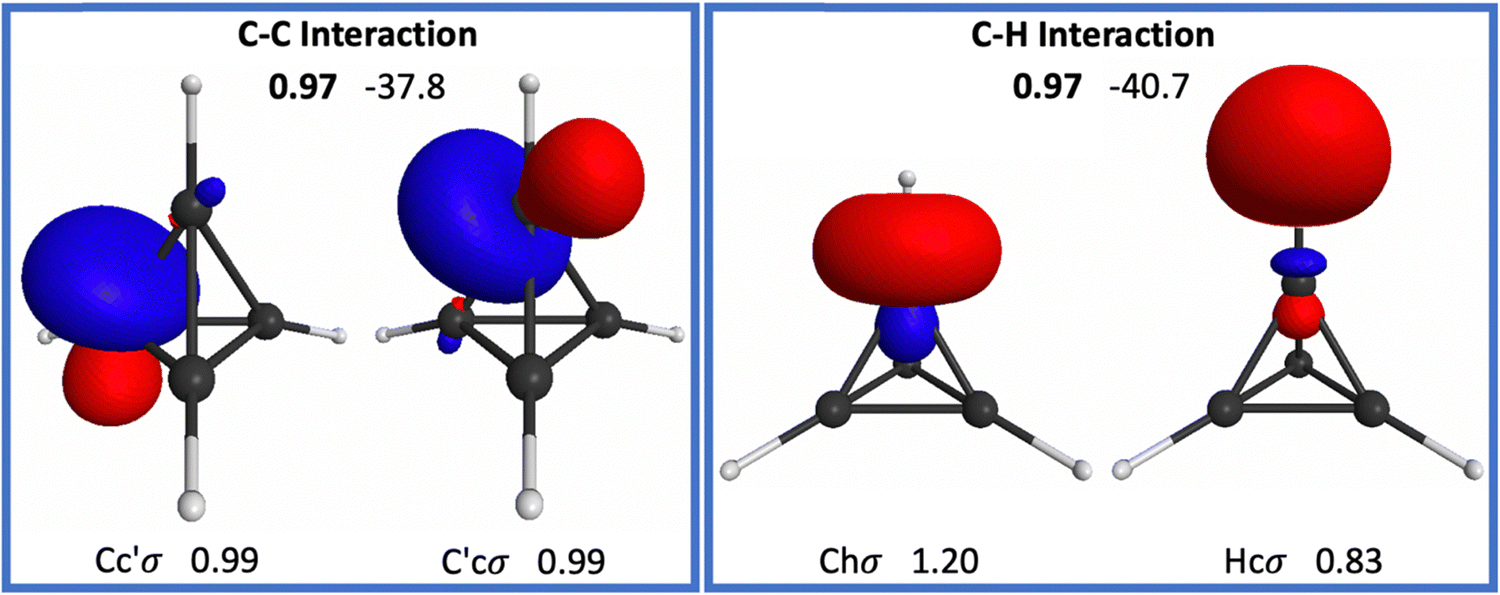 | ||
| Fig. 5 Oriented QUAOs of the C4H4 (td) RHF/6-31G(d) wave function at the RHF optimized geometry. Orbital labels are shown below the corresponding orbital; orbital populations are displayed next to each label. The bond order is shown in bold between interacting orbitals. Kinetic bond orders (in kcal mol−1) are shown to the right of the respective bond order. Only symmetry unique QUAOs are shown. Two equivalent Cc′σ orbitals are shown for clarity. | ||
 | ||
| Fig. 6 RHF/6-31G(d) oriented PC3H3 (ptd) QUAOs. Orbital labels are shown below the corresponding orbital; orbital populations are displayed next to each label. The bond order is shown in bold between interacting orbitals. Kinetic bond orders (in kcal mol−1) are shown to the right of the respective bond order. Only symmetry unique QUAOs are shown. Two equivalent Cc′σ orbitals are shown for clarity. | ||
 | ||
| Fig. 7 Oriented QUAOs of the P2C2H2 (p2td) RHF/6-31G(d) wave function at the RHF optimized geometry. Orbital labels are shown below the corresponding orbital; orbital populations are displayed next to each label. The bond order is shown in bold between interacting orbitals. Kinetic bond orders (in kcal mol−1) are shown to the right of the respective bond order. Only symmetry unique QUAOs are shown. Two equivalent Pp′σ and Cc′σ orbitals are shown for clarity. | ||
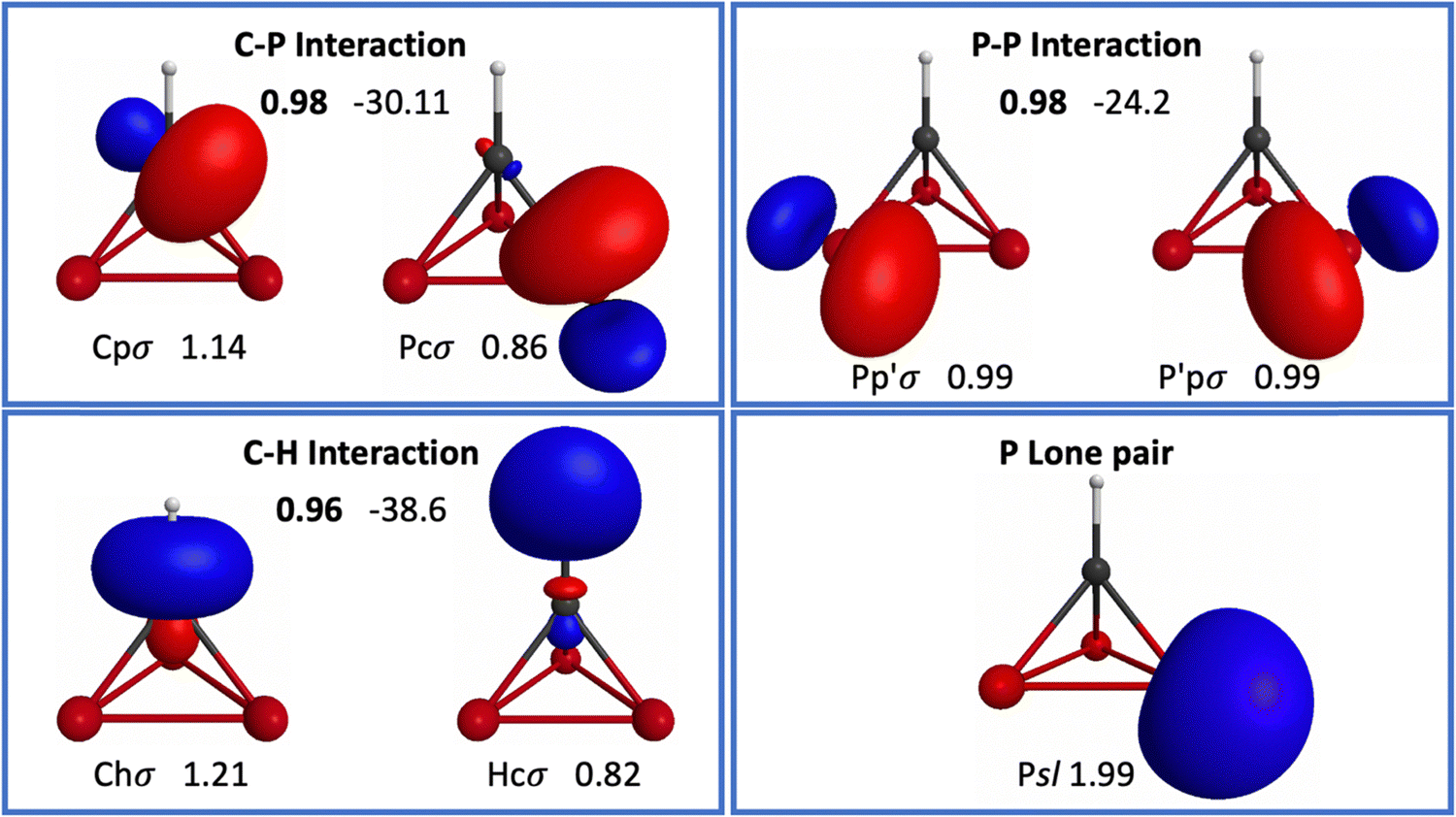 | ||
| Fig. 8 Oriented QUAOs of the P3CH (p3td) RHF/6-31G(d) wave function at the RHF optimized geometry. Orbital labels are shown below the corresponding orbital; orbital populations are displayed next to each label. The bond order is shown in bold between interacting orbitals. Kinetic bond orders (in kcal mol−1) are shown to the right of the respective bond order. Two equivalent Pp′σ orbitals are shown for clarity. | ||
 | ||
| Fig. 9 Oriented QUAOs of the P4 RHF/6-31G(d) wave function at the RHF optimized geometry. Orbital labels are shown below the corresponding orbital; orbital populations are displayed next to each label. The bond order is shown in bold between interacting orbitals. Kinetic bond orders (in kcal mol−1) are shown to the right of the respective bond order. Two equivalent Pp′σ orbitals are shown for clarity. | ||
The s- and p-fractions of the QUAOs of the molecules of interest in this work are summarized in Table 2. The BOs and KBOs of all relevant bonding interactions for the seven molecules are summarized in Table 3. Orbital populations are presented in Table 4. The RHF QUAO analysis was applied to the RHF/6-31G(d) and MP2/6-31G(d) geometries; the results were essentially the same for the two geometries for every molecule, thus only those for the RHF geometries are reported.
| Orbital | C2H6 | C3H6 | C4H4 | PC3H3 | P2C2H2 | P3CH | P4 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| s | p | s | p | s | p | s | p | s | p | s | p | s | p | |
| Chσ | 0.2 | 0.8 | 0.25 | 0.75 | 0.39 | 0.61 | 0.38 | 0.62 | 0.38 | 0.62 | 0.37 | 0.63 | ||
| Hcσ | 1.00 | 0 | 1.00 | 0 | 1.00 | 0 | 1.00 | 0 | 1.00 | 0 | 1.00 | 0 | ||
| Cc′σ | 0.22 | 0.78 | 0.16 | 0.84 | 0.15 | 0.85 | 0.17 | 0.83 | 0.19 | 0.81 | ||||
| Pcσ | 0.07 | 0.93 | 0.08 | 0.92 | 0.09 | 0.91 | ||||||||
| Cpσ | 0.13 | 0.87 | 0.15 | 0.85 | 0.17 | 0.83 | ||||||||
| Pp′σ | 0.05 | 0.95 | 0.06 | 0.94 | 0.07 | 0.93 | ||||||||
| Psl | 0.70 | 0.30 | 0.70 | 0.30 | 0.70 | 0.30 | 0.71 | 0.29 | ||||||
| Bond | C2H6 | C3H6 | C4H4 | |||
|---|---|---|---|---|---|---|
| BO | KBO | BO | KBO | KBO | KBO | |
| CHσ | 0.98 | −36.2 | 0.98 | −38.0 | 0.97 | −40.7 |
| CCσ | 0.99 | −44.7 | 0.97 | −38.8 | 0.97 | −37.8 |
| PC3H3 | P2C2H2 | P3CH | ||||
|---|---|---|---|---|---|---|
| BO | KBO | BO | KBO | BO | KBO | |
| CHσ | 0.97 | −39.9 | 0.96 | −39.2 | 0.96 | −38.6 |
| CCσ | 0.97 | −40.2 | 0.99 | −43.6 | ||
| CPσ | 0.96 | −26.2 | 0.97 | −27.9 | 0.98 | −30.1 |
| PPσ | 0.98 | −22.5 | 0.98 | −24.2 | ||
| P4 | ||
|---|---|---|
| BO | KBO | |
| PPσ | 0.99 | −26.1 |
| Orbital | C2H6 | C3H6 | C4H4 | PC3H3 | P2C2H2 | P3CH | P4 |
|---|---|---|---|---|---|---|---|
| Chσ | 1.12 | 1.14 | 1.20 | 1.20 | 1.20 | 1.21 | |
| Hcσ | 0.88 | 0.87 | 0.83 | 0.83 | 0.82 | 0.82 | |
| Cc'σ | 1.00 | 0.99 | 0.99 | 0.99 | 0.99 | ||
| Pcσ | 0.90 | 0.88 | 0.86 | ||||
| Cpσ | 1.09 | 1.11 | 1.14 | ||||
| Ppσ | 0.99 | 0.99 | 1.00 | ||||
| Psl | 1.99 | 1.99 | 1.99 | 2.00 |
QUAO populations (Table 4) show that, as one would expect, a small charge transfer occurs from H to C, in accordance with the difference in electronegativity between C and H. The Chσ orbitals have a population of 1.12, paired with a Hcσ orbital population of 0.88. The orbital population for the Cc′σ orbitals is exactly 1. As expected for a molecule like ethane, all orbitals centered on C atoms have very similar s- and p-characters; the s- and p-characters of the Chσ orbitals are 0.2 and 0.8, respectively, while the s- and p-characters of the Cc′σ orbitals are 0.22 and 0.78. This shows that the orbital structure of C is homogeneous across all of its bonding interactions. As for the bond orders and KBOs (Table 3), the CCσ interaction has a bond order of 0.99 and a KBO of −44.7 kcal mol−1. For the CHσ bonding interaction, the BO is 0.98 and the KBO is −36.2 kcal mol−1. As mentioned above, KBOs cannot be quantitatively translated into bonding energies, however, the results obtained for ethane provide a reference point for the KBOs of the CCσ and CHσ interactions in the molecules discussed below.
Orbital populations (Table 4) show that a slightly larger charge transfer from H to C occurs in cyclopropane than in ethane. The Chσ and Hcσ orbital populations are 1.14, and 0.87, respectively. There is also a higher s-character in the Chσ orbital, 0.25 in cyclopropane vs. 0.2 in ethane. There is concomitantly also greater p-character in the cyclopropane Cc′σ orbital. As discussed below, this behavior has been related to strain in molecules. Qualitatively, in cyclopropane the Cc′σ orbitals do not display the cylindrical symmetry around the interatomic axis that is observed for ethane. In fact, it can be seen in Fig. 4 that the orbitals are “tilted”, with lobes pointing outwards from the center of the molecule. This is a consequence of the small angles, and overall strain, in the molecule, as pointed out by Coulson and Moffit.20 This “tilted” structure can be related to the idea of a curved or bent bond19,20 in cyclopropane.18,21
The CHσ bonding interaction in cyclopropane has a KBO of −38.0 kcal mol−1, indicating a 1.8 kcal mol−1 stronger bond than that for the same interaction in ethane. The opposite is true for the CCσ bonding interaction, which has a KBO of −38.8 kcal mol−1, 5.9 kcal mol−1 weaker than that for the same interaction in ethane. This is consistent with the earlier comment that despite having a shorter interatomic distance than in ethane, the CCσ bonding interaction is weaker in cyclopropane than in ethane. The change from ethane to cyclopropane strengthens the CHσ bonding interactions, while weakening the CCσ ones.
The QUAO populations (Table 4) show that, as in ethane and cyclopropane, a charge transfer occurs from H to C atoms. However, the charge transfer is larger than in the aforementioned molecules; the Chσ orbitals and Hcσ orbitals have occupations of 1.20 and 0.83, respectively. Wiberg, Bader and Lau (WBL) have shown that this electron density shift from H to C increases as carbon is subjected to higher geometric strain.84 It has been suggested that small carbon-centered angles in molecular structures result in an increased p-character of the Cc′σ orbitals and thus an increase in the s-character of the Chσ orbitals.20,84 As shown in Table 2, the observations by WBL apply to td, in which the Cc′σ orbitals have a p-character of 0.85. The overall trend of the electron density shift from H to C, as well as the elevated p-character in Cc′σ orbitals is evident upon transitioning from ethane to cyclopropane, to td. The results also show that the higher the molecular strain, the greater the difference in the s- and p-characters between Cc′σ and Chσ orbitals. In fact, the higher the p-character in the Cc′σ orbitals, the higher the s-character in the Chσ orbitals. These results support the correlation between small bond angles and the changes in s- and p-character in orbitals, as observed by WBL. Despite the similarities in the s- and p-character of the Cc′σ orbitals in td and cyclopropane, there is a significant difference in their Chσ orbitals; the s-character of the Chσ orbitals is larger in td (0.39) than in cyclopropane (0.25). Although the CCC bond angles in the two molecules are similar, this significant difference in s-character might be related to the fact that there is one more Cc′σ orbital (which tends to maximize p-character in strained rings) and therefore one less Chσ orbital (which tends to maximize s-character) in td than in cyclopropane.
The CHσ bonding interaction in td has a BO and KBO of 0.97 and −40.7 kcal mol−1, respectively. For the CCσ bonds each carbon atom uses 3 orbitals labeled Cc′σ, each oriented toward a different C atom. The spatial arrangement of these orbitals is slightly bent away from the center of the molecule, similar to the bending present in cyclopropane.19 Consequently, internuclear distances do not correspond directly to bond lengths, as noted earlier. Bader and collaborators have reported that the ratio of the bond path length, defined by the electron density, to the equilibrium interatomic distance is larger than 1 in both cyclopropane and td, with td having the larger ratio. This was also related by the authors to a weakening of the CC bond.18
The CCσ bond has a BO and KBO of 0.97 and −37.8 kcal mol−1, respectively. This KBO is the most positive (weakest) for a CCσ interaction among ethane, cyclopropane, and td. Thus, strain in molecules weakens the structure of the ring or cage, despite the shorter internuclear distances, in agreement with the previous observations by Bader et al.18 and by Boatz and Gordon.21 The opposite is true for the CHσ bonds, which appear to become stronger as strain increases in the molecule.
The s- and p-characters of each orbital in ptd are shown in Table 2. All relevant bonding interactions in the RHF geometry are summarized in Table 3. Similar to td, the QUAO populations (Table 4) show that a charge transfer occurs from H to C in ptd. Populations in the Chσ and Hcσ orbitals are 1.20 and 0.83. The BO and KBO for the CHσ bond are 0.97 and −39.9 kcal mol−1, respectively. The latter is less than one kcal mol−1 weaker than the same interaction in td.
Carbon uses two Cc′σ orbitals to bind to the other C atoms in the structure. These orbitals have populations of 0.99, reflecting the symmetric, nonpolar nature of the CCσ bond in this molecule. The BO and KBO for the CCσ bonding interaction are 0.97 and −40.2 kcal mol−1, respectively. This KBO is 2.4 kcal mol−1 more negative than that for the CCσ bond in td, reflecting a slightly stronger CCσ bond in ptd than in td. As shown in Table 2, the s- and p-characters of the Cc′σ orbitals are 0.17 and 0.83, similar to the Cc′σ orbitals in td.
The occupations of the orbitals involved in the PCσ bond reflect a small charge transfer from P to C, as might be expected given the difference in their electronegativities. The inclusion of an electropositive atom, relative to C, in the structure results in a slightly higher electron population on the C–C–C ring of ptd (4.28e in the QUAOs of each C atom), than in td (4.17e in the QUAOs of each C atom).
The BO and KBO for the PCσ interaction are 0.96 and −26.2 kcal mol−1. The s- and p-characters of the Cpσ orbitals are 0.13 and 0.87, respectively, similar to the values found in the Cc′σ orbitals. The Pcσ orbitals have s- and p-characters of 0.07 and 0.93, respectively, reflecting the limited ability of phosphorous atoms to mix s- and p-type functions compared to C. As noted by Kutzelnigg,85 this lack of 3s–3p mixing is due to the rather different spatial extents of 3s and 3p orbitals, compared with 2s and 2p orbitals. The only lone pair in the structure is an orbital centered on P with an occupation of 1.99. This orbital has an s-character of 0.70, so it has been labeled Psl.
The s- and p-characters of each orbital in p2td are displayed in Table 2. The BOs and KBOs of all relevant bonding interactions at the RHF geometry are summarized in Table 3. The orbital populations (Table 4) of the Hcσ and Chσ orbitals show that a charge transfer from H to C occurs in p2td, similar to td and ptd. The Chσ orbital has s- and p-characters of 0.38 and 0.62, the same as for the Chσ orbital in ptd. The BO and KBO for the CHσ bond are 0.96 and −39.2 kcal mol−1, respectively. This KBO suggests a slightly weaker bond than for the same interactions in ptd and td.
The two C atoms in p2td interact via two symmetrically equivalent Cc′σ orbitals. The s- and p-characters of this orbital are 0.19 and 0.81, respectively. Interestingly, the Ccσ orbital in p2td has the highest s-character of all the Cc′σ orbitals participating in the ring/cage structures analyzed here. The s- and p-characters of the Cc′σ orbital in p2td are in between those of the Cc′σ orbitals in cyclopropane and ethane. This is not surprising as the CCP and PCP angles in the cage structure of p2td are both larger (Table 1) than the 60° C–C–C angles in the cyclopropane ring structure. Thus, the larger the angle between the bonds containing C, the higher the s-character (and the lower the p-character) of the orbitals it employs for bonding in the ring/cage structure, in accordance with previous observations.20,84 The BO and KBO of 0.99 and −43.6 kcal mol−1, respectively, imply that the CCσ bond in p2td is the strongest of its type in all of the ring/cage structures analyzed here. This KBO is 1.1 kcal mol−1 more positive (suggesting a weaker bond) than the KBO for the CCσ bond in ethane, and 4.8 kcal mol−1 more negative (suggesting a stronger bond) than the KBO for the same bond in cyclopropane. Hence, the strength of this bond in p2td is expected to be closer to that of the CCσ bond in ethane than to the one in cyclopropane. As for orbital populations, the occupation of 0.99 of the Ccσ orbital reflects the non-polar nature of the interaction between the two C atoms in p2td, as expected from the symmetry of the molecule.
Each C atom employs two symmetrically equivalent Cpσ orbitals to bond to each of the P atoms in p2td. Accordingly, every P atom employs two symmetrically equivalent Pcσ orbitals to bond to each of the C atoms in the molecule. The orbital populations of the Cpσ and Pcσ orbitals are 1.11 and 0.88, respectively. Thus, the PCσ bond is observed to be slightly more polarized toward the C atom than the PCσ bond in ptd. There is an increase in the s-character of both the Cpσ and Pcσ orbitals in p2td relative to ptd, with the increase being larger for the Cpσ orbital than for the Pcσ orbital. As for the Cc′σ orbitals, this increase in s-character can be interpreted in terms of the larger angles in the cage structure of p2td. Moreover, the increase in the s-character of the Cpσ and Cc′σ orbitals suggests that C is closer to the so-called sp3 hybridization in p2td than in ptd or td.
The BO and KBO for the CPσ bond in p2td are 0.97 and −27.9 kcal mol−1, respectively. This KBO is 1.7 kcal mol−1 more negative than the KBO for the CPσ interaction in ptd, reflecting a slightly stronger CPσ bond in p2td.
The two P atoms in p2td interact via two symmetrically equivalent Pp′σ orbitals. The s- and p-fractions of this orbital are 0.05 and 0.95, respectively. This reflects the lack of 3s–3p mixing in phosphorous discussed above. The BO and KBO for the PPσ bond are 0.98 and −22.5 kcal mol−1, respectively. As expected from the symmetry of p2td, the orbital occupation of 0.99 for the Pp′σ orbital reflects a non-polar PPσ bond. As in ptd, each P atom has a lone pair orbital, Psl, with an occupation of 1.99 and s- and p-characters of 0.70 and 0.30, respectively.
The s- and p-characters of each orbital in p3td are shown in Table 2. All relevant bonding interactions at the RHF geometry are summarized in Table 3. As expected from the orbital populations (Table 4) observed in td, ptd and p2td, a charge transfer occurs from H to C in p3td; the occupations of the Chσ and Hcσ orbitals are 1.21 and 0.82 (Table 4). Interestingly, this is the largest transfer of charge from H to C among all of the molecules in the present study. The Chσ orbital has s- and p-characters of 0.37 and 0.63, respectively. The Chσ orbital in p3td has the lowest s-character among the Chσ orbitals in the molecules with cage-like structures discussed here. In fact, when going from td to ptd to p2td to p3td, the s-character of the Chσ orbital decreases (and the p-character increases) as CH groups are substituted by P atoms. The BO and KBO for the CHσ bond are 0.96 and −38.6 kcal mol−1, respectively.
The C atom in p3td employs three symmetrically equivalent Cpσ orbitals to bond to the three P atoms in the system. As for ptd and p2td, orbital populations show that each CPσ bond is polarized toward the C atom. The Cpσ orbital has a population of 1.14, while the Pcσ orbital has a population of 0.86. Thus, the charge transfer from P to C appears to grow as one goes from ptd to p2td to p3td. This charge transfer is also accompanied by a higher s-character in the Cpσ orbital in p3td (0.17) relative to p2td (0.15) and ptd (0.13). Hence, a correlation between the population of an orbital and its s-character is observed, in a similar fashion as discussed above for CHσ bonds. This increase in the s-character can also be related to the larger angles between the CPσ bonds than the P–C–P angle suggests (Table 1), which is expected to decrease the geometric strain relative to td, ptd and p2td.
Interestingly, the s- and p-fractions in the Cpσ orbitals in p3td are the same as those of the Cc′σ orbitals in ptd. Nonetheless, in p3td, all of the C-centered orbitals participating in the bonding of the cage structure, namely the three Cpσ orbitals, have the same s- and p-fractions. On the other hand, in ptd, each C employs only two orbitals with this s/p ratio, and a highly p-type Cpσ orbital for the CPσ bond. As discussed below, this difference in the orbital structure of C can be correlated with a difference in the strain energies of these molecules.
Another difference regarding the Pcσ orbitals in p3td relative to p2td and ptd is that the p-character of this orbital is the lowest (0.91), and its s-character is the highest (0.09) in p3td. In addition, the s-character of the Pcσ orbitals grows as one goes from ptd to p2td to p3td. This trend can be related to the larger P–P–C and P–P–P angles (53.2° and 60°, respectively) in p3td compared to the C–P–C and P–P–C angles in p2td (47.2° and 52.9°, respectively), and to the C–P–C angle in ptd (46.9°).
The CPσ bond has a BO and KBO of 0.98 and −30.1 kcal mol−1, respectively. This KBO is 3.9 and 2.2 kcal mol−1 more negative than the KBOs of the same interaction in ptd and p2td, respectively. This suggests stronger CPσ bonds as one goes from ptd to p2td to p3td.
The PPσ bond has a BO and KBO of 0.98 and -24.2 kcal mol−1, respectively. This KBO is 1.7 kcal mol−1 more negative than the KBO for the same interaction in p2td, suggesting a stronger PPσ bond in p3td. The occupation of the Ppσ orbitals involved in this interaction is 0.99, reflecting the non-polar nature of this bond. The s- and p-characters of the Ppσ orbital are 0.06 and 0.94, respectively. These orbitals have lower s-character than their Pcσ counterparts. Both the Pcσ and Cpσ orbitals have higher s-characters in p3td than in p2td and ptd. Finally, every P atom has a lone pair orbital Psl with occupation of 1.99, and s- and p-fractions of 0.7 and 0.3, respectively, the same as in ptd and p2td.
Each P atom uses three orbitals, labeled Pp′σ, to bond to the other atoms in the molecule. These orbitals are bent away from the center of the molecule, in a similar fashion to the orbitals in cyclopropane, tetrahedrane, and the P-substituted tertrahedranes. The BO and KBO of the PPσ bonds are 0.99 and −26.1 kcal mol−1, respectively. This KBO is 3.6 and 1.9 kcal mol−1 more negative than the KBOs for the same interaction in p2td and p3td, respectively. Thus, a trend of stronger PPσ bonds is observed upon going from p2td to p3td to P4.
Each P atom has a lone pair in an orbital oriented away from the center of the molecule, similar to ptd, p2td and p3td. The s- and p-fractions of this orbital are 0.71 and 0.29, respectively. These fractions represent only a slight deviation from the s- and p-characters observed for the Psl orbital in the other P-containing cages. The s- and p-fractions of the Pp′σ orbitals are 0.07 and 0.93. As previously mentioned, small angles in molecular geometries increase the p-character of bonding orbitals.20,84 Phosphorus is more amenable to such an arrangement than is carbon, since the 3s and 3p orbitals do not like to mix as much as the 2s and 2p do.85 This is evident in all of the bonding QUAOs centered on P across all the systems analyzed here.
4.3 Isodesmic reactions
The RHF, MP2 and RI-CCSD(T) strain energies calculated with the isodesmic reactions displayed in Fig. 1 are shown in Table 5. The RHF and MP2 energy differences have been corrected with the respective scaled zero-point vibrational energies, and the RI-CCSD(T) strain energies have been corrected using the MP2 zero-point vibrational energy. Although the absolute values differ, the overall trend in strain energies is the same for the three levels of theory.| Molecule | Strain energy | ||
|---|---|---|---|
| RHF | MP2 | RI-CCSD(T) | |
| C3H6 | 21.3 | 18.6 | 19.1 |
| C4H4 | 115.7 | 106.4 | 103.9 |
| PC3H3 | 82.9 | 65.9 | 68.2 |
| P2C2H2 | 55.4 | 32.5 | 37.7 |
| P3CH | 31.2 | 4.5 | 10.1 |
| P4 | 8.6 | −20.2 | −15.22 |
The isodesmic reaction used for cyclopropane has been previously reported by Hariharan and Pople.70 Their RHF/6-31G(d)//RHF/STO-3G strain energy is 26.2 kcal mol−1. The isodesmic reaction for td has been previously used by Glukhovstev and collaborators.47 Their reported strain enthalpy, obtained from G2 calculations, is 100.29 kcal mol−1, in reasonable agreement with the MP2/6-31G(d) and RI-CCSD(T)/cc-pVTZ/cc-pVTZ-RI strain energies in Table 5. Strain in td and the P-substituted tetrahedranes has been previously assessed by Riu and collaborators,32 and their results display the same trend, namely, that strain decreases as CH groups are successively substituted by P atoms. The isodesmic reaction for the P4 molecule was used previously by Schoeller and collaborators.10 Their reported strain energies are 5.3 (SCF) and −14.4 (CEPA), in reasonable agreement with the strain energies reported in Table 4. In general, when correlation is added to the calculation via MP2 or RI-CCSD(T), the strain energy decreases.
Tetrahedrane is the most strained molecule of the five cage molecules considered here, with a computed strain energy greater than 100 kcal mol−1 compared to the cyclopropane strain energy of ∼20 kcal mol−1. This is not surprising since one may think of td as three fused cyclopropane rings.
MP2 and RI-CCSD(T) show that the substitution of a CH group by a P atom, upon transitioning from td to ptd, lowers the strain energy of the molecule by about 35 to 40 kcal mol−1. The substitution of a second CH group by one P atom, going from ptd to p2td, lowers the strain energy of the molecule by another ∼30 kcal mol−1. Substituting a third CH group by a P atom, going from p2td to p3td, lowers the strain energy by another ∼28 kcal mol−1. This trend is consistent with the observation that the angles centered on carbon atoms in the cage structures increase from ptd to p2td to p3td. The larger angles are expected to decrease some of the strain on the C atoms. Furthermore, these results reflect the greater ability of phosphorus to accommodate small bond angles on the order of 60° or less compared to carbon. Consistent with this observation, the strain in P4 is essentially non-existent; the much lower strain in P4 than in td has been noted previously.10,32
The results displayed in Table 5 show that RHF tends to overestimate strain energies; that is, MP2 and RI-CCSD(T) are observed to yield lower strain energies than RHF does. In all but one case (the td strain energy), MP2 is observed to overcorrect the RHF strain energy, with respect to RI-CCSD(T). Nonetheless, the trend of decreasing strain energy when going from td, to p2td, to p3td, to P4 is observed at all three levels of theory.
The insight provided by the QUAO analysis and the computed strain energies allow one to understand the different chemical behaviors of P4 and td: given the elevated strain energy of td, it will likely tend to isomerize (i.e. open the tetrahedral cage) to a more stable system, as has been described for its derivative: tetra-tert-butyltetrahedrane.25 Based on the latter experiments, Maier, Pfriem, Schäfer and Matusch report quantitative isomerization to tetra-tert-butylcyclobutadiene upon heating, breaking only 2 CCσ bonds and substantially reducing geometric strain. On the other hand, the low strain energy of P4 allows for its structure to remain unaltered in the absence of external disruptions. An example can be found in the experiments reported by Mal and collaborators,86 in which tetrahedral P4 was encapsulated in a metal–organic cage.87 According to the authors, P4 was stabilized in this system through van der Waals interactions with the phenylene groups in the body of the cage. The encapsulated P4 was reported to remain unaltered after 4 months of exposure to the atmosphere, and the observed stability was attributed to the spatial constraint that the cage exerted on P4. This constraint prevented it from reacting with the oxygen in the atmosphere, as P4 is known to easily undergo oxidation upon reaction with O2.86
The PPσ bonds in P4 have an experimental bond energy of 47.5 kcal mol−1,88 while the P–O bonds in phosphorous oxides have energies ranging from 80 to 156 kcal mol−1.89 This difference in bond strengths is reported to lead to a low activation barrier to oxidation, thereby explaining the pyrophoric nature of P4.86 In cyclopropane, calculations have estimated the CCσ bonds to have a bond energy of 71 kcal mol−1 at the RHF/6-31G(d,p) level of theory.90 KBOs suggest that the CCσ bonds in td are slightly weaker than the same bonds in cyclopropane. Thus, the bond energy of the CCσ bonds in td is expected to be slightly lower than the 71 kcal mol−1 theoretical estimate, but well above the 47.5 kcal mol−1 bond energy of the PPσ bonds in P4. Therefore, the bonds in the cage structure of td can be expected to be stronger than those in P4. Despite their geometric similarity, it appears that while both td and P4 tend to “break” their tetrahedral structures, the driving force for this process is different in the two molecules. On the one hand, the limited ability of C to employ orbitals with high p-character for Sigma-type bonding interactions leads to highly strained unstable geometries, as evidenced by the computed strain energies. On the other, the weak PPσ bonding interactions can lead to easily broken cages.
5. Conclusions
The bonding in tetrahedrane, phosphatetrahedrane, diphosphatetrahedrane and triphosphatetrahedrane has been analyzed in terms of quasi-atomic orbitals (QUAOs) using ethane, cyclopropane and P4 as reference systems. The analysis in terms of QUAOs was complemented with the computation of strain energies via isodesmic reactions. This analysis leads to the identification of multiple relationships between the trends in the bonding of the molecules analyzed here, and the trend of the computed strained energies.With the increase of strain in the analyzed molecules; that is, upon going from ethane to cyclopropane to tetrahedrane, the C orbitals used for bonding in the ring/cage structures have an increased p-character and are bent away from the center of the respective molecules. Despite the substantial structural difference between cyclopropane and tetrahedrane, the s- and p-characters of their Cc′σ orbitals are observed to be similar, unlike their strain energies. This suggests that, as geometric strain grows, it becomes more difficult for C to increase the p-character of the orbitals that it uses to bond in small-angle arrangements. The difficulty to increase the p-character of the orbitals of C can be interpreted as a consequence of the propensity of C to mix s and p functions, which contrasts with the limited ability of P to do so. The propensity to hybridize, or the lack thereof, has implications for the stability of molecules with strained geometries, as the computed strain energies make evident. The increase in strain is accompanied by an increase in the s-character of the Chσ orbitals, an increased transfer of charge from H to C, a weakening of the CCσ bonds and a strengthening of the CHσ bonds.
The substitution of a P atom for a CH group in tetrahedrane leads to larger angles in the cage structure of phosphatetrahedrane, a lower strain energy, slightly stronger CCσ bonds, and a lower p-character in the orbitals involved in the CCσ interaction, all relative to tetrahedrane. The substitution of one more P atom for a CH group in phosphatetrahedrane leads to larger C-centered angles in the cage structure of diphosphatetrahedrane, a lower strain energy, stronger CCσ and PCσ bonds, and a further lowering in the p-character of the orbitals involved in the CCσ and PCσ bonds, all relative to phosphatetrahedrane. The substitution of a third P atom for a CH group; that is, going from diphosphatetrahedrane to triphosphatetrahedrane, maximizes the angles between the bonds containing C in the cage-like structures analyzed here. A further decrease in the strain energy, stronger CCσ, PCσ and PPσ bonds, as well as lower p-characters in the orbitals involved in these interactions, relative to diphosphatetrahedrane, are observed. The overall trend of increasingly stronger CCσ bonds in the P-substituted tetrahedranes is accompanied by increasingly shorter C–C interatomic distances. Thus, while the generally short C–C distances in the cage-like structures do not imply stronger CCσ bonds relative to those in unstrained systems (e.g., the CCσ bond in ethane), shorter C–C interatomic distances within the cage structures analyzed here do appear to correlate with stronger CCσ bonds. The trend of decreasing strain energy in the P-substituted tetrahedranes shows that orbital structures with elevated p-characters are favorable for P atoms, leading to an essentially unstrained molecule in P4. The comparison of the present results with literature bond energies suggests that, despite the geometric similarity of tetrahedrane and P4, and their tendency to “break” the tetrahedral cages, the driving force of this process is different for the two molecules. For tetrahedrane, the limited ability of C to employ orbitals with high p-character for Sigma-type bonding interactions leads to a highly strained unstable molecule. For P4, the weak PPσ bonds can lead to an easily broken cage.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Prof. Klaus Ruedenberg for his insightful comments and suggestions on this work. The authors also thank Dr Dipayan Datta for his helpful guidance on the use of the RI-CCSD(T) code. Daniel Del Angel Cruz thanks Dr Adriana Palacios Rosas and Dr Miguel Ángel Ocaña Bribiesca for their valuable support to carry out this work. This work was supported by a Department of Energy Exascale Computing Project to the Ames Laboratory, project no. 17-SC-20-SC. Ames Laboratory is operated by Iowa State University under Contract No. DE-AC02-07CH11338.References
- S. Nagase, M. Nakano and T. Kudo, Strain and structures in the silicon analogues of tetrahedrane, prismane, and cubane. A theoretical study, J. Chem. Soc., Chem. Commun., 1987, 60 RSC.
- E. Kochanski and J. M. Lehn, The electronic structure of cyclopropane, cyclopropene and diazirine an ab initio SCF-LCAO-MO study, Theor. Chim. Acta, 1969, 14, 281–304 CrossRef CAS.
- A. Komornicki, F. Pauzat and Y. Ellinger, Ab initio prediction of structures, force constants and vibrational frequencies. Saturated three-membered rings cyclopropane, ethylene oxide, and ethyleneimine, J. Phys. Chem., 1983, 87, 3847–3857 CrossRef CAS.
- I. F. Tupitsyn, Yu. V. Puzanov and A. Yu, Shibaev, Quantum-chemical study of the force constants of CH bonds in molecules with three-membered rings, Theor. Exp. Chem., 1990, 25, 372–378 CrossRef.
- J. M. Schulman and T. J. Venanzi, Theoretical study of the tetrahedrane molecule, J. Am. Chem. Soc., 1974, 96, 4739–4746 CrossRef CAS.
- B. A. Hess and L. J. Schaad, Ab initio second-order Moeller-Plesset calculation of the vibrational spectrum of tetrahedrane, J. Am. Chem. Soc., 1985, 107, 865–866 CrossRef CAS.
- H. Kollmar, An MO theoretical study on the stability of tetrahedrane, J. Am. Chem. Soc., 1980, 102, 2617–2621 CrossRef CAS.
- M. Balci, M. L. McKee and P. R. Schleyer, Theoretical Study of Tetramethyl- and Tetra- tert -butyl-Substituted Cyclobutadiene and Tetrahedrane, J. Phys. Chem. A, 2000, 104, 1246–1255 CrossRef CAS.
- A. F. Sax and J. Kalcher, MC-SCF and CI calculations on the Si4H4 system, J. Comput. Chem., 1989, 10, 309–328 CrossRef CAS.
- W. W. Schoeller, V. Staemmler, P. Rademacher and E. Niecke, Theoretical studies on inorganic ring systems. Tetraphosphabicyclobutane, cyclotriphosphane, and white phosphorus: ring strain and hybridization, Inorg. Chem., 1986, 25, 4382–4385 CrossRef CAS.
- B. M. Gimarc and M. Zhao, Strain Energies in Homoatomic Nitrogen Clusters N4, N6, and N8, Inorg. Chem., 1996, 35, 3289–3297 CrossRef CAS.
- D. Allen. Clabo and H. F. Schaefer, Tetrasilatetrahedrane, J. Am. Chem. Soc., 1986, 108, 4344–4346 CrossRef.
- A. F. Sax and J. Kalcher, Quantum chemical calculations on Si4H4, J. Chem. Soc., Chem. Commun., 1987, 809 RSC.
- B. F. Yates, D. A. Clabo and H. F. Schaefer, Cyclic isomers of singlet Si4H4 related to tetrasilacyclobutadiene, Chem. Phys. Lett., 1988, 143, 421–427 CrossRef CAS.
- C. A. Bayse and M. Jaffar, Bonding analysis of the effect of strain on trigger bonds in organic-cage energetic materials, Theor. Chem. Acc., 2020, 139, 95 Search PubMed.
- T. M. Klapötke, Chemistry of High-Energy Materials, De Gruyter, 2019 Search PubMed.
- G. Zhou, J.-L. Zhang, N.-B. Wong and A. Tian, Computational studies on a kind of novel energetic materials tetrahedrane and nitro derivatives, J. Mol. Struct. THEOCHEM, 2004, 668, 189–195 CrossRef CAS.
- R. F. W. Bader, T. H. Tang, Y. Tal and F. W. Biegler-Koenig, Properties of atoms and bonds in hydrocarbon molecules, J. Am. Chem. Soc., 1982, 104, 946–952 CrossRef CAS.
- K. B. Wiberg, Bent Bonds in Organic Compounds, Acc. Chem. Res., 1996, 29, 229–234 CrossRef CAS.
- C. A. Coulson and W. E. Moffitt, I. The properties of certain strained hydrocarbons, Lond. Edinb. Dublin Philos. Mag. J. Sci., 1949, 40, 1–35 CrossRef CAS.
- J. A. Boatz and M. S. Gordon, Theoretical studies of three-membered ring compounds Y2H4X (Y = C, Si; X = CH2, NH, O, SiH2, PH, S), J. Phys. Chem., 1989, 93, 3025–3029 CrossRef CAS.
- J. A. Boatz, M. S. Gordon and R. L. Hilderbrandt, Structure and bonding in cycloalkanes and monosilacycloalkanes, J. Am. Chem. Soc., 1988, 110, 352–358 CrossRef CAS.
- P. Chakrabarti, P. Seiler, J. D. Dunitz, A. D. Schluter and G. Szeimies, Experimental evidence for the absence of bonding electron density between inverted carbon atoms, J. Am. Chem. Soc., 1981, 103, 7378–7380 CrossRef CAS.
- S. Tsutsui, K. Sakamoto, C. Kabuto and M. Kira, X-ray Crystallographic Analysis of a 3-Silacyclopropene with Electronegative Substituents on Silicon, Organometallics, 1998, 17, 3819–3821 CrossRef CAS.
- G. Maier, S. Pfriem, U. Schäfer and R. Matusch, Tetra-tert-butyltetrahedrane, Angew. Chem., Int. Ed. Engl., 1978, 17, 520–521 CrossRef.
- G. Maier, J. Neudert, O. Wolf, D. Pappusch, A. Sekiguchi, M. Tanaka and T. Matsuo, Tetrakis(trimethylsilyl)tetrahedrane+, J. Am. Chem. Soc., 2002, 124, 13819–13826 CrossRef CAS PubMed.
- G. Maier and D. Born, Tri-tert-butyl(trimethylsilyl)tricyclo[1.1.0.02, 4]-butane-a Second Tetrahedrane Derivative, Angew. Chem., Int. Ed. Engl., 1989, 28, 1050–1052 CrossRef.
- G. Maier, J. Neudert and O. Wolf, Tetrakis(trimethylsilyl)cyclobutadiene and Tetrakis(trimethylsilyl)tetrahedrane, Angew. Chem., Int. Ed., 2001, 40, 1674–1675 CrossRef CAS.
- H. Irngartinger, A. Goldmann, R. Jahn, M. Nixdorf, H. Rodewald, G. Maier, K.-D. Malsch and R. Emrich, Tetra-tert-butyltetrahedrane—Crystal and Molecular Structure, Angew. Chem., Int. Ed. Engl., 1984, 23, 993–994 CrossRef.
- G. Hierlmeier, P. Coburger, M. Bodensteiner and R. Wolf, Di-tert-butyldiphosphatetrahedrane: Catalytic Synthesis of the Elusive Phosphaalkyne Dimer, Angew. Chem., Int. Ed., 2019, 58, 16918–16922 CrossRef CAS PubMed.
- M.-L. Y. Riu, R. L. Jones, W. J. Transue, P. Müller and C. C. Cummins, Isolation of an elusive phosphatetrahedrane, Sci. Adv., 2020, 6, eaaz3168 CrossRef CAS PubMed.
- M.-L. Y. Riu, M. Ye and C. C. Cummins, Alleviating Strain in Organic Molecules by Incorporation of Phosphorus: Synthesis of Triphosphatetrahedrane, J. Am. Chem. Soc., 2021, 143, 16354–16357 CrossRef CAS PubMed.
- M. E. Wolf, E. A. Doty, J. M. Turney and H. F. Schaefer Iii, Highly Strained Pn(CH) 3 (Pn = N, P, As, Sb, Bi) Tetrahedranes: Theoretical Characterization, J. Phys. Chem. A, 2021, 125, 2612–2621 CrossRef CAS PubMed.
- E. D. Glendening, C. R. Landis and F. Weinhold, Natural bond orbital methods, WIREs Comput. Mol. Sci., 2012, 2, 1–42 CrossRef CAS.
- W. J. Hehre, R. Ditchfield, L. Radom and J. A. Pople, Molecular orbital theory of the electronic structure of organic compounds. V. Molecular theory of bond separation, J. Am. Chem. Soc., 1970, 92, 4796–4801 CrossRef CAS.
- J. J. Duchimaza Heredia, K. Ruedenberg and M. S. Gordon, Quasi-Atomic Bonding Analysis of Xe-Containing Compounds, J. Phys. Chem. A, 2018, 122, 3442–3454 CrossRef CAS PubMed.
- A. C. West, M. W. Schmidt, M. S. Gordon and K. Ruedenberg, A Comprehensive Analysis in Terms of Molecule-Intrinsic, Quasi-Atomic Orbitals. II. Strongly Correlated MCSCF Wave Functions, J. Phys. Chem. A, 2015, 119, 10360–10367 CrossRef CAS PubMed.
- A. C. West, M. W. Schmidt, M. S. Gordon and K. Ruedenberg, A comprehensive analysis of molecule-intrinsic quasi-atomic, bonding, and correlating orbitals. I. Hartree-Fock wave functions, J. Chem. Phys., 2013, 139, 234107 CrossRef PubMed.
- A. C. West, M. W. Schmidt, M. S. Gordon and K. Ruedenberg, A Comprehensive Analysis in Terms of Molecule-Intrinsic, Quasi-Atomic Orbitals. III. The Covalent Bonding Structure of Urea, J. Phys. Chem. A, 2015, 119, 10368–10375 CrossRef CAS PubMed.
- A. C. West, M. W. Schmidt, M. S. Gordon and K. Ruedenberg, A Comprehensive Analysis in Terms of Molecule-Intrinsic Quasi-Atomic Orbitals. IV. Bond Breaking and Bond Forming along the Dissociative Reaction Path of Dioxetane, J. Phys. Chem. A, 2015, 119, 10376–10389 CrossRef CAS PubMed.
- A. C. West, J. J. Duchimaza-Heredia, M. S. Gordon and K. Ruedenberg, Identification and Characterization of Molecular Bonding Structures by ab initio Quasi-Atomic Orbital Analyses, J. Phys. Chem. A, 2017, 121, 8884–8898 CrossRef CAS PubMed.
- J. J. Duchimaza Heredia, A. D. Sadow and M. S. Gordon, A Quasi-Atomic Analysis of Three-Center Two-Electron Zr–H–Si Interactions, J. Phys. Chem. A, 2018, 122, 9653–9669 CrossRef CAS PubMed.
- E. B. Guidez, M. S. Gordon and K. Ruedenberg, Why is Si 2 H 2 Not Linear? An Intrinsic Quasi-Atomic Bonding Analysis, J. Am. Chem. Soc., 2020, 142, 13729–13742 CrossRef CAS PubMed.
- S. M. Bachrach, Theoretical studies of phosphirane and phosphetane, J. Phys. Chem., 1989, 93, 7780–7784 CrossRef CAS.
- P. George, M. Trachtman, C. W. Bock and A. M. Brett, An alternative approach to the problem of assessing destabilization energies (strain energies) in cyclic hydrocarbons, Tetrahedron, 1976, 32, 317–323 CrossRef CAS.
- M. S. Gordon, Ring strain in cyclopropane, cyclopropene, silacyclopropane, and silacyclopropene, J. Am. Chem. Soc., 1980, 102, 7419–7422 CrossRef CAS.
- M. N. Glukhovtsev, S. Laiter and A. Pross, Thermochemistry of Cyclobutadiene and Tetrahedrane: A High-Level Computational Study, J. Phys. Chem., 1995, 99, 6828–6831 CrossRef CAS.
- K. K. Baldridge and M. S. Gordon, Theoretical studies of disilabenzenes, J. Organomet. Chem., 1984, 271, 369–379 CrossRef CAS.
- K. K. Baldridge and M. S. Gordon, Substituted silabenzenes, Organometallics, 1988, 7, 144–155 CrossRef CAS.
- K. K. Baldridge and M. S. Gordon, Potentially aromatic metallocycles, J. Am. Chem. Soc., 1988, 110, 4204–4208 CrossRef CAS.
- G. M. J. Barca, C. Bertoni, L. Carrington, D. Datta, N. De Silva, J. E. Deustua, D. G. Fedorov, J. R. Gour, A. O. Gunina, E. Guidez, T. Harville, S. Irle, J. Ivanic, K. Kowalski, S. S. Leang, H. Li, W. Li, J. J. Lutz, I. Magoulas, J. Mato, V. Mironov, H. Nakata, B. Q. Pham, P. Piecuch, D. Poole, S. R. Pruitt, A. P. Rendell, L. B. Roskop, K. Ruedenberg, T. Sattasathuchana, M. W. Schmidt, J. Shen, L. Slipchenko, M. Sosonkina, V. Sundriyal, A. Tiwari, J. L. Galvez Vallejo, B. Westheimer, M. Włoch, P. Xu, F. Zahariev and M. S. Gordon, Recent developments in the general atomic and molecular electronic structure system, J. Chem. Phys., 2020, 152, 154102 CrossRef CAS PubMed.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. Su, T. L. Windus, M. Dupuis and J. A. Montgomery, General atomic and molecular electronic structure system, J. Comput. Chem., 1993, 14, 1347–1363 CrossRef CAS.
- M. S. Gordon and M. W. Schmidt, Theory and Applications of Computational Chemistry, Elsevier, 2005, pp. 1167–1189 Search PubMed.
- G. Schoendorff, M. W. Schmidt, K. Ruedenberg and M. S. Gordon, Quasi-Atomic Bond Analyses in the Sixth Period: II. Bond Analyses of Cerium Oxides, J. Phys. Chem. A, 2019, 123, 5249–5256 CrossRef CAS PubMed.
- A. C. West, M. W. Schmidt, M. S. Gordon and K. Ruedenberg, Intrinsic Resolution of Molecular Electronic Wave Functions and Energies in Terms of Quasi-atoms and Their Interactions, J. Phys. Chem. A, 2017, 121, 1086–1105 CrossRef CAS PubMed.
- G. Schoendorff, K. Ruedenberg and M. S. Gordon, Multiple Bonding in Rhodium Monoboride. Quasi-atomic Analyses of the Ground and Low-Lying Excited States, J. Phys. Chem. A, 2021, 125, 4836–4846 CrossRef CAS PubMed.
- J. L. Galvez Vallejo, J. D. Heredia and M. S. Gordon, Bonding analysis of water clusters using quasi-atomic orbitals, Phys. Chem. Chem. Phys., 2021, 23, 18734–18743 RSC.
- M. W. Schmidt, E. A. Hull and T. L. Windus, Valence Virtual Orbitals: An Unambiguous ab Initio Quantification of the LUMO Concept, J. Phys. Chem. A, 2015, 119, 10408–10427 CrossRef CAS PubMed.
- J. Ivanic, G. J. Atchity and K. Ruedenberg, Intrinsic local constituents of molecular electronic wave functions. I. Exact representation of the density matrix in terms of chemically deformed and oriented atomic minimal basis set orbitals, Theor. Chem. Acc., 2008, 120, 281–294 Search PubMed.
- K. Ruedenberg, The Physical Nature of the Chemical Bond, Rev. Mod. Phys., 1962, 34, 326–376 CrossRef CAS.
- G. B. Bacskay, S. Nordholm and K. Ruedenberg, The Virial Theorem and Covalent Bonding, J. Phys. Chem. A, 2018, 122, 7880–7893 CrossRef CAS PubMed.
- M. W. Schmidt, J. Ivanic and K. Ruedenberg, Covalent bonds are created by the drive of electron waves to lower their kinetic energy through expansion, J. Chem. Phys., 2014, 140, 204104 CrossRef PubMed.
- C. A. Coulson and H. C. Longuet-Higgins, The electronic structure of conjugated systems I. General theory, Proc. R. Soc. Lond. Ser. Math. Phys. Sci., 1947, 191, 39–60 CAS.
- B. H. Chirgwin, Summation Convention and the Density Matrix in Quantum Theory, Phys. Rev., 1957, 107, 1013–1025 CrossRef CAS.
- T. Dudev and C. Lim, Ring Strain Energies from ab Initio Calculations, J. Am. Chem. Soc., 1998, 120, 4450–4458 CrossRef CAS.
- R. Ditchfield, W. J. Hehre, J. A. Pople and L. Radom, Molecular orbital theory of bond separation, Chem. Phys. Lett., 1970, 5, 13–14 CrossRef CAS.
- L. C. Snyder and H. Basch, Heats of reaction from self-consistent-field energies of closed-shell molecules, J. Am. Chem. Soc., 1969, 91, 2189–2198 CrossRef CAS.
- Y. Mo and P. von R. Schleyer, An Energetic Measure of Aromaticity and Antiaromaticity Based on the Pauling–Wheland Resonance Energies, Chem. – Eur. J., 2006, 12, 2009–2020 CrossRef CAS PubMed.
- M. S. Gordon, P. Boudjouk and F. Anwari, Are the silacyclopentadienyl anion and the silacyclopropenyl cation aromatic?, J. Am. Chem. Soc., 1983, 105, 4972–4976 CrossRef CAS.
- P. C. Hariharan and J. A. Pople, The effect of d-functions on molecular orbital energies for hydrocarbons, Chem. Phys. Lett., 1972, 16, 217–219 CrossRef CAS.
- A. Nicolaides, A. Rauk, M. N. Glukhovtsev and L. Radom, Heats of Formation from G2, G2(MP2), and G2(MP2,SVP) Total Energies, J. Phys. Chem., 1996, 100, 17460–17464 CrossRef CAS.
- R. Ahlrichs and P. Scharf, in Advances in Chemical Physics, ed. K. P. Lawley, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2007, pp. 501–537 Search PubMed.
- J. A. Pople, A. P. Scott, M. W. Wong and L. Radom, Scaling Factors for Obtaining Fundamental Vibrational Frequencies and Zero-Point Energies from HF/6-31G* and MP2/6-31G* Harmonic Frequencies, Isr. J. Chem., 1993, 33, 345–350 CrossRef CAS.
- D. Datta and M. S. Gordon, A Massively Parallel Implementation of the CCSD(T) Method Using the Resolution-of-the-Identity Approximation and a Hybrid Distributed/Shared Memory Parallelization Model, J. Chem. Theory Comput., 2021, 17, 4799–4822 CrossRef CAS PubMed.
- B. M. Bode and M. S. Gordon, Macmolplt: a graphical user interface for GAMESS, J. Mol. Graph. Model., 1998, 16, 133–138 CrossRef CAS PubMed.
- J. L. Duncan, D. C. McKean and A. J. Bruce, Infrared spectroscopic studies of partially deuterated ethanes and the r0, rz, and re structures, J. Mol. Spectrosc., 1979, 74, 361–374 CrossRef CAS.
- C. Puzzarini and P. R. Taylor, An ab initio study of the structure, torsional potential energy function, and electric properties of disilane, ethane, and their deuterated isotopomers, J. Chem. Phys., 2005, 122, 054315 CrossRef PubMed.
- J. Gauss, D. Cremer and J. F. Stanton, The re Structure of Cyclopropane, J. Phys. Chem. A, 2000, 104, 1319–1324 CrossRef CAS.
- B. M. Cossairt, C. C. Cummins, A. R. Head, D. L. Lichtenberger, R. J. F. Berger, S. A. Hayes, N. W. Mitzel and G. Wu, On the Molecular and Electronic Structures of AsP3 and P4, J. Am. Chem. Soc., 2010, 132, 8459–8465 CrossRef CAS PubMed.
- B. Joakim Persson, P. R. Taylor and T. J. Lee, Ab initio geometry, quartic force field, and vibrational frequencies for P4, J. Chem. Phys., 1997, 107, 5051–5057 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Density Functionals with Broad Applicability in Chemistry, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS PubMed.
- F. H. Allen, The geometry of small rings. I. Substituentinduced bond-length asymmetry in cyclopropane, Acta Crystallogr. B, 1980, 36, 81–96 CrossRef CAS.
- R. F. W. Bader, T. S. Slee, D. Cremer and E. Kraka, Description of conjugation and hyperconjugation in terms of electron distributions, J. Am. Chem. Soc., 1983, 105, 5061–5068 CrossRef CAS.
- K. B. Wiberg, R. F. W. Bader and C. D. H. Lau, Theoretical analysis of hydrocarbon properties. 2. Additivity of group properties and the origin of strain energy, J. Am. Chem. Soc., 1987, 109, 1001–1012 CrossRef CAS.
- W. Kutzelnigg, Orthogonal and non-orthogonal hybrids, THEOCHEM, 1988, 169, 403–419 CrossRef.
- P. Mal, B. Breiner, K. Rissanen and J. R. Nitschke, White Phosphorus Is Air-Stable Within a Self-Assembled Tetrahedral Capsule, Science, 2009, 324, 1697–1699 CrossRef CAS PubMed.
- P. Mal, D. Schultz, K. Beyeh, K. Rissanen and J. R. Nitschke, An Unlockable-Relockable Iron Cage by Subcomponent Self-Assembly, Angew. Chem., 2008, 120, 8421–8425 CrossRef.
- F. S. Dainton, X—X and X—O bond energies of phosphorus, arsenic and antimony and their importance in the kinetics of the oxidation of these elements, Trans. Faraday Soc., 1947, 43, 244–256 RSC.
- Y.-R. Luo, Comprehensive Handbook of Chemical Bond Energies, CRC Press, 1st edn, 2007 Search PubMed.
- D. Cremer and J. Gauss, Theoretical determination of molecular structure and conformation. 20. Reevaluation of the strain energies of cyclopropane and cyclobutane carbon-carbon and carbon-hydrogen bond energies, 1,3 interactions, and Sigma-aromaticity, J. Am. Chem. Soc., 1986, 108, 7467–7477 CrossRef CAS PubMed.
| This journal is © the Owner Societies 2023 |