
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHow a few help all: cooperative crossing of lipid membranes by COSAN anions†
David C.
Malaspina
 ,
Francesc
Teixidor
,
Clara
Viñas
* and
Jordi
Faraudo
*
,
Francesc
Teixidor
,
Clara
Viñas
* and
Jordi
Faraudo
*
Institut de Ciencia de Materials de Barcelona, ICMAB-CSIC, Campus de la UAB, E- 08193, Bellaterra, Spain. E-mail: clara@icmab.es; jfaraudo@icmab.es
First published on 6th October 2023
Abstract
Experimental results show that the presence of a concentration gradient of certain nano-ions (most notably cobaltabisdicarbollide ([o-COSAN]− anions), induce a current across intact artificial phospholipid bilayers in spite of the high Born free energy estimated for these ions. The mechanism underlying this observed translocation of nano-anions across membranes has yet to be determined. Here we show, using molecular dynamics simulations, that the permeation of [o-COSAN]− anions across a lipid bilayer proceeds in a cooperative manner. Single nano-ions can enter the bilayer but permeation is hampered by a free energy barrier of about 8kBT. The interaction between these nano-ions inside a leaflet induces a flip-flop translocation mechanism with the formation of transient, elongated structure inside the membrane. This cooperative flip-flop allows an efficient distribution of [o-COSAN]− anions in both leaflets of the bilayer. These results suggest the existence of a new mechanism for permeation of nano-ions across lipid membranes, relevant for those that have the appropriate self-assembly character.
Molecules able to passively permeate lipid membranes are of great interest for many applications in medicine and in biological research. These applications include the important case of drug delivery, for example. In general, the design of molecules able to cross lipid membranes in a passive, nonfacilitated way, is a difficult task.1,2 The only class of molecules that are able to rapidly cross lipid membranes are small, nonpolar gases1 (for example, permeability of O2 is between ∼ 20–100 cm s−1 depending on the lipid membrane composition3). Membrane permeation is more difficult -but still possible- for a small polar molecule.1,4 The water molecule is the paradigmatic example with a permeability coefficient of the order of 10−3 cm s−1 in artificial lipid membranes.4 Also, its mechanism of permeation has been studied in detail with large scale molecular dynamics simulations.5 In the case of more complex molecules such as drugs, only molecules with a delicate balance between size, polarity and lipophilicity are able to spontaneously permeate this barrier1,2,6 (typical permeabilities are of the order of 10−6 cm s−1 in this case).
Lipid bilayers are virtually impermeable against charged molecules, including small ions.1,7 Typical permeability of Cl− is between 10−12–10−8 cm s−1 depending on the particular lipids.8,9 Cations such as Na+ and K+ have even lower permeabilities,1,9 of the order of 10−14–10−12 cm s−1.
In the classical theory, permeation across the membrane involves first the partition inside the hydrophobic phase and then diffusion across the membrane (the solubility-diffusion mechanism). In this framework, the extremely low permeabilities of ions were attributed to the low dielectric constant (εr ∼ 2) of the hydrophobic region (hydrocarbon tails) of a lipid membrane.7,10 A low dielectric constant implies that the transfer of a charge from water (εr ∼ 80) to the membrane core has a very large Born energy cost, making the solubility-diffusion mechanism highly inefficient. There are other mechanisms beyond this solubility-diffusion mechanism that are also responsible for the slow, nonfacilitated permeation of ions.11 One alternative mechanism is due to the fact that ions tend to associate with phospholipids and that phospholipids are able to translocate from one leaflet of the bilayer to another (flip-flop mechanism). The flip-flop of a phospholipid-ion complex can transfer charge from one side of the bilayer to the other11,12 (for example, the permeability coefficient estimated from this mechanism for Cl− in phosphatidylserine bilayers is ∼ 10−11 cm s−112).
Another possible transport mechanism is related to the fact that membrane interfaces are soft and ions can induce local membrane deformations.9,11,16,17 In the case of thin lipid bilayers, made of lipids of short tails, these deformations are able to induce the spontaneous formation of transient pores in the lipid membrane which allow the transport of ions with a certain amount of solvation water.8,18 In thicker membranes, the deformation can propagate towards the membrane interior translocating the ion and an amount of accompanying water molecules without forming a complete hydrophilic pore.9,16,17 This mechanism is also expected to be responsible for the permeation of certain charged peptides across lipid bilayers.17,19 This process avoids the exposure of a charge to the low dielectric constant environment of the membrane interior but it is highly inefficient since the required fluctuations have substantial energy barriers and consequently they have low probability.
In this context, the experimental results obtained for certain anions of nanometric size are particularly relevant.20,21 These studies considered the ortho-cobaltabis(dicarbollide) [3,3′-Co(1,2-C2B9H11)2]− ion, known as [o-COSAN]− and some of its derivatives, which are elongated anions with a size ≈ 1 nm and 3D global aromaticity.22 When a 0.1 mM concentration of Na[o-COSAN] is applied to one side of a phospholipid planar bilayer, an steady anionic electric current of ∼ 0.2 nA is established across the membrane due to the concentration gradient.20 It has to be recalled that the lipid bilayer remained intact during the experiment and no pores were formed. Further experiments with a voltage-jump across the lipid bilayer also show fast translocation of these anions.21 [o-COSAN]− also shows translocation across natural cell membranes, as demonstrated recently for the case of mammalian cells23,24 and of Gram-positive bacterial membranes.25 Also, it has been shown that [o-COSAN]− is able to act as a carrier, transporting cationic peptides across membranes.26
These experiments demonstrate that lipid membranes are not acting as barriers to the [o-COSAN]− anion and its derivatives. This behaviour is remarkable in view of the high free energy cost associated to travel directly through a lipid bilayer due to its high Born energy. In ref. 20 this free energy cost was estimated as ∼ 80 kJ mol−1, or more than 30kBT, for an ideal COSAN anion (dimensions 1.1 nm × 0.6 nm).
It has been suggested20 that this remarkable membrane crossing ability of [o-COSAN]− could be related to the unusual dual hydrophobic and hydrophilic properties of [o-COSAN]−. The [o-COSAN]− anion has neither a hydrocarbon chain as a hydrophobic tail nor a polar group as a headgroup like in a conventional surfactant, but it is amphiphilic since it has a well defined hydrophilic polar region and a hydrophobic apolar region,13 as shown in Fig. 1. It exhibits a rich self-assembly behavior in solution which includes micelles and vesicles typical of surfactants.27–30 It is also able to bind to proteins31 and glucose units32 and self-assembles with polypyrrole33 and poly(ethylene oxide) (PEO) to give hybrid nanocomposites.34 Thus, the behaviour of [o-COSAN]−, with its rich spectrum of molecular interactions, is more like that of an anionic small molecule rather than that of an anion.
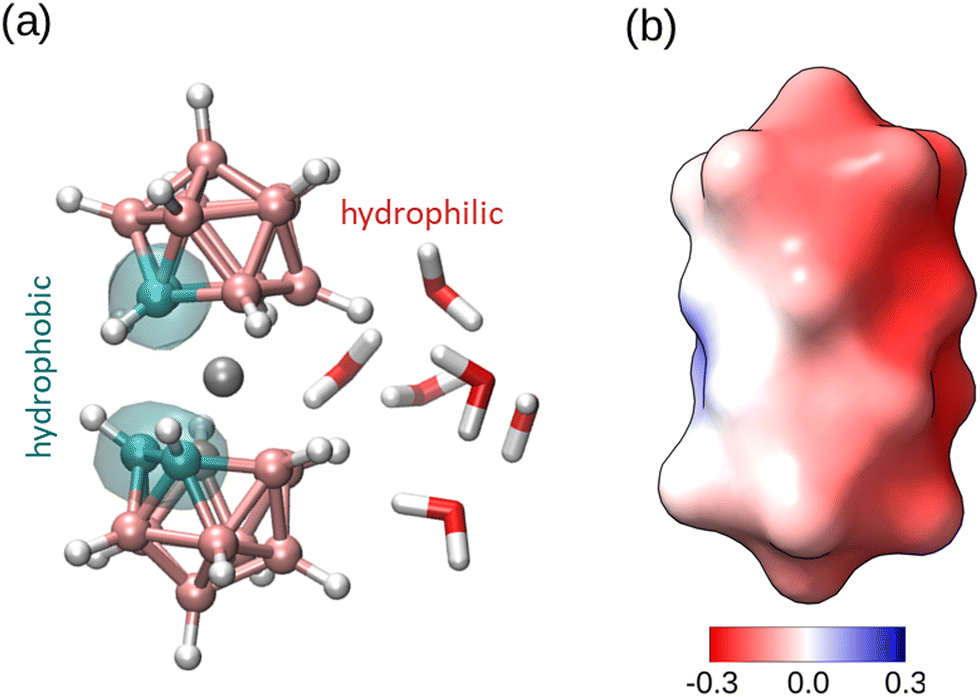 | ||
| Fig. 1 Structure of [o-COSAN]− in its cis rotameric conformation which is the most stable conformation in water.13 (a) Ball and stick representation with indication of the hydrophobic and hydrophilic regions, as obtained in previous MD simulations (see ref. 13). The color code is C: cyan, B: pink, Co: grey, H: white. The structure of the water solvation near the hydrophilic region is also indicated. (b) Surface representation colored according to the surface electrostatic potential (color scale in units of (kcal mol−1e, the highest positive potential is 0.14 and the most negative −0.29). The hydrophobic region around C atoms is mostly apolar and the negative charge of the anion is concentrated in the hydrophobic region (B atoms opposite to the C atoms). Image (a) made with VMD.14 Electrostatic potential in (b) calculated with Chimera.15 | ||
Elucidation of the detailed mechanism of [o-COSAN]− anion permeation in lipid bilayers is not only important from a fundamental perspective but it has also implications in biomedicine applications. We recall that [o-COSAN]− has shown potential as anticancer drug35 and as a antibacterial agent.25
In this work, we identify this lipid bilayer crossing mechanism by using all-atomic Molecular Dynamics (MD) simulations. Our results show that the permeation of [o-COSAN]−, in its cisoid rotamer, proceeds in a cooperative manner. Our simulations show that a single [o-COSAN]− anion is not able to cross the hydrophobic core of a lipid membrane due to the presence of a free energy barrier. Instead, a single [o-COSAN]− remains incorporated into a membrane leaflet as a membrane component due to its amphiphilic nature. However, the interaction between these nano-ions inside the membrane induces the formation of elongated structures and a flip-flop transport mechanism that allows translocation across the hydrophobic core and an efficient distribution of [o-COSAN]− anions in both bilayers of the membrane.
In our simulations, the phospholipid bilayer was composed by dipalmitoylphosphatidylcholine (DPPC) phospholipid (which is widely used experimentally in liposomes and in bilayers mimicking mammalian cells) and cholesterol (which helps to restrict the passage of molecules by increasing the packing of phospholipids). We considered a 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 DPPC:Chol ratio as in our previous work.36 Concerning the [o-COSAN]− nano-ion, it should be noted that it has three rotameric forms, designed as trans-, cis- and gauche. In this work, we will consider only the cis[o-COSAN]− rotamer, since it is the most stable one in water, as we have shown recently by DFT calculations.13 This cisoid rotamer of [o-COSAN]− has a charge distribution resembling that of a peculiar anionic surfactant, with a small apolar region defined by its four carbon atoms (which are neighbors only in the particular case of cis[o-COSAN]−) and an hydrophilic polar region that concentrates most of the anion charge (with boron atoms that strongly interact with water molecules).13
:3 DPPC:Chol ratio as in our previous work.36 Concerning the [o-COSAN]− nano-ion, it should be noted that it has three rotameric forms, designed as trans-, cis- and gauche. In this work, we will consider only the cis[o-COSAN]− rotamer, since it is the most stable one in water, as we have shown recently by DFT calculations.13 This cisoid rotamer of [o-COSAN]− has a charge distribution resembling that of a peculiar anionic surfactant, with a small apolar region defined by its four carbon atoms (which are neighbors only in the particular case of cis[o-COSAN]−) and an hydrophilic polar region that concentrates most of the anion charge (with boron atoms that strongly interact with water molecules).13
The forcefield employed for [o-COSAN]− in our simulations is also the same employed in our previous work,13 which is compatible with the CHARMM36 forcefield employed for the membrane. In all simulations water molecules were included explicitly. Full technical details of the simulations are given in the Methods section in the ESI.†
We first considered a simulation of a single [o-COSAN]− anion (with its sodium counterion) in water phase in a system containing a planar bilayer membrane (of size ∼ 73 nm2), as shown in Fig. 2. We observe spontaneous incorporation of the [o-COSAN]− nano-ion into the membrane. As shown in Fig. 2, the incorporation of [o-COSAN]− inside the bilayer followed several steps, highlighted in Fig. 2. First, the [o-COSAN]− initially in the water phase adsorbs on the headgroup region, with its long axis perpendicular to the phospholipid molecules and the hydrophobic carbon region of [o-COSAN]− oriented towards the lipid membrane. After remaining adsorbed over the bilayer (for about ∼ 20 ns), the [o-COSAN]− ion changes its orientation, aligning its long axis parallel to the hydrocarbon tails of the lipids. During this change in orientation, the ion moves from adsorption over the membrane towards the hydrophobic region of the lipid membrane. Once inside the hydrocarbon region of the lipid membrane, it remains in this orientation for the remainder of the simulation (∼ 200 ns) with the central Co atom of [o-COSAN]− located at a distance between 7 Å–11 Å from the center of the bilayer, without crossing the center of the membrane towards the other leaflet.
 | ||
| Fig. 2 Snapshots of a MD simulation showing the incorporation of a single [o-COSAN]− nano-ion at a lipid membrane. Left-panel: initial adsorption at lipid headgroups. Middle-panel: permeation of [o-COSAN]− into hydrophobic region (hydrocarbon tails). Right-panel: equilibrated system with [o-COSAN]− located in the hydrophobic region (hydrocarbon tails). In this particular snapshot, the distance between the central Co atom and the center of the membrane is 7.5 Å. The lipids are shown as lines, Na+ (in yellow) and [o-COSAN]− are shown with its van der Waals size. Water molecules are not shown for clarity. | ||
In order to verify whether a single [o-COSAN]− is able or not to spontaneously cross a lipid bilayer, we have calculated the free energy profile associated to [o-COSAN]− permeation using adaptive bias force (ABF) method.37 The results (Fig. 3) show the existence of a free energy minima (∼−32.35 kJ mol−1) inside the hydrocarbon region of the membrane, corresponding to the range of Co-bilayer center separations spontaneously found by the [o-COSAN]− ion in our previous non-biased MD simulation. The results also show a free energy difference of ∼ 21.7 kJ mol−1 (∼ 8kBT) between the center of the membrane and the equilibrium position of [o-COSAN]− inside the membrane.
 | ||
| Fig. 3 Free Energy profile (kJ mol−1) for the permeation of a single [o-COSAN]− across a lipid bilayer from MD-ABF simulations. As reaction coordinate we consider the distance between the central Co atom of [o-COSAN]− and the center of the bilayer (the bilayer is fully symmetric). The origin for the free energy was taken at the water phase. | ||
This implies that passive translocation of [o-COSAN]− from its equilibrium position at one leaflet of the bilayer to the equivalent position at the other leaflet is hampered by a free energy barrier of ∼ 8kBT. Interestingly, this barrier is smaller than the free energy cost (∼ 30kBT) estimated previously for [o-COSAN]− in ref. 20 using a simple Born energy calculation.
Let us now consider a simulation with a micelle made of 15 [o-COSAN]−, instead of considering a single [o-COSAN]− and maintaining same lipid bilayer (see Methods in the ESI† for details). Let us remark here that this was the typical size of a micelle obtained in our previous simulations,13 in agreement with experiments.27
The results are shown in Fig. 4 (snapshots) and Fig. 5 (time evolution of the density profile).
 | ||
| Fig. 4 Snapshots of a MD simulation corresponding to a system composed by a micelle made by 15 [o-COSAN]− ions interacting with a lipid membrane. (a) Initially, there is an aggregate of [o-COSAN]− molecules near the membrane and a single [o-COSAN]− from the aggregate lays horizontally over the membrane. (b) The [o-COSAN]− that was previously laying horizontally now penetrates the membrane vertically. (c) As the simulation proceeds [o-COSAN]− penetrate the membrane forming elongated structures. (d) Final configuration of the MD simulation showing the rearrangement of the [o-COSAN]− ions. [o-COSAN]− near the membrane and Na+ counterions are shown as van der Waals spheres. All other molecules are shown by lines. Water is not shown for easier visualization. | ||
 | ||
| Fig. 5 Average intrinsic density profile (atoms nm−3) of Co atoms from [o-COSAN]− inside a lipid bilayer for different time windows. Distances are calculated in reference to the P atoms in DPPC molecules (we took the origin of coordinates in the upper leaflet of the membrane). Different horizontal color lines indicate average location of different DPPC atoms, to show the relative position of the membrane. On the right scheme, we represented the [o-COSAN]− molecule and the lipid membrane. In the lipid membrane scheme, P, N and terminal C atoms of DPPC molecule are represented with VDW beads (their positions are indicated as horizontal lines in the left plot). | ||
Initially, the micelle is adsorbed over the bilayer and individual [o-COSAN]− anions enter to the membrane following the mechanism described before for a single [o-COSAN]− anion, as shown in Fig. 4. As time advances and several [o-COSAN]− are incorporated inside the membrane, they are able to interact and form dynamic, elongated structures inside the bilayer, as shown in the snapshots in Fig. 4. In these structures it is possible to find [o-COSAN]− anions at locations such as the center of the bilayer which were not possible for a single [o-COSAN]−, as seen in the snapshots of Fig. 4 and in the intrinsic density profile in Fig. 5. Initially, the peak of the [o-COSAN]− concentration is located at the hydrocarbon region, as expected from our analysis for a single [o-COSAN]− (Fig. 5). But as time advances, the distribution extends towards the central region of the membrane, showing that now [o-COSAN]− ions are allowed to explore the full bilayer, including the central region, and thus [o-COSAN]− ions are allowed to cross from one side to the other of the membrane. The interaction between [o-COSAN]− nano-ions and the formation of the elongated structures seen in Fig. 4 allows for a free energy gain that supersedes the free energy barrier experienced by single [o-COSAN]−.
A detailed description of the mechanism that allows [o-COSAN]− to cross the membrane is shown in Fig. 6. As seen in that figure, the mechanism involves the coordinated motion of several [o-COSAN]− inside the membrane. The sequence of events shown there is as follows. First, one [o-COSAN]− is spontaneously incorporated inside the membrane (Fig. 6a) at the equilibrium position expected from our previous free energy analysis (see Fig. 3). A second and a third [o-COSAN]− nano-ions enter the membrane (Fig. 6b and c, respectively). The three [o-COSAN]− associate into a transient elongated structure, shown in Fig. 6d and e. This transient structure finally ends up with two of the [o-COSAN]− at the membrane leaflet opposite to the one at which were originally incorporated (Fig. 6f). This correlated motion of [o-COSAN]− inside the membrane, that extends to time scales of about 300 ns can be clearly identified in the trajectory plot shown in Fig. 6. We designate this translocation mechanism as “cooperative flip-flop”, since it is a mechanism in which the “cooperation” between interacting [o-COSAN]− allows them to cross from one lipid layer to the other.
 | ||
| Fig. 6 Detail of [o-COSAN]− cooperative permeation mechanism. Each panel shows a snapshots corresponding to a different time: (a) 31.2 ns, (b) 99.2 ns, (c) 126.4 ns, (d) 146.0 ns, (e) 291.0 ns and (f) 330.1 ns. In order to showcase the mechanism, 3 specific [o-COSAN]− were emphasized (the membrane and other [o-COSAN]− were shown as lines or transparent). To facilitate distinguishing the interacting [o-COSAN]− we shown them in different color scheme (one in CPK colors, one in blue and one in orange). The bottom panel shows the time evolution of the position of two of these [o-COSAN]− nano-ions, indicating with arrows the events shown in the snapshots. The red line corresponds to the [o-COSAN]− incorporated to the membrane as shown in panel (a) and the black line to the one incorporated at the membrane as shown in panel (b) (the average position of the N atoms of DPPC lipids is also shown to indicate the location of the membrane). | ||
In conclusion, in this work, we have identified the mechanism of [o-COSAN]− membrane translocation by atomistic MD simulations, a mechanism that we designate as “cooperative flip-flop”, providing a mechanistic explanation for previous experimental observations.20,21
Our simulations show that a single [o-COSAN]− anion is spontaneously integrated into the hydrophobic region of a leaflet of a lipid membrane. However, a single [o-COSAN]− is not observed to spontaneously cross the lipid membrane in our simulations. Our calculations indicate a large Free Energy barrier (∼ 8kBT) at the center of the lipid bilayer. Spontaneous translocation of [o-COSAN]− is facilitated by the interaction between several [o-COSAN]− molecules inside the bilayer. Inside the bilayer, [o-COSAN]− anions self-assembly into highly dynamic elongated structures, that extend across the bilayer including the central region excluded to isolated [o-COSAN]−.
The motion of [o-COSAN]− molecules inside the bilayer is highly correlated. The [o-COSAN]− molecules are able to cross the central region of the bilayer helped by their mutual interactions by a cooperative mechanism that can be labelled as “cooperative flip-flop”. Therefore, the remarkable self-assembly ability of [o-COSAN]−, which allows them to form micelles and other structures in bulk solution,27–30 it is also involved in its ability to translocate across a membrane. It should be emphasized here that our computational study the particular case of a symmetric membrane with equal composition in both leaflets, as considered in previous experiments.20 Given the fact that real biological membranes have asymmetric composition, the next necessary step will be to consider simulations of asymmetric membranes of different compositions and investigate whether it has or not an impact on the mechanism observed in our simulations.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from MCIN/AEI/10.13039/501100011033 agency through Grants PID2021-124297NB-C33 and PID2019-106832RB-I00, and the “Severo Ochoa” Programme for Centres of Excellence in R&D (CEX2019-000917-S) awarded to ICMAB. We also thank the Government of Catalonia (AGAUR) for Grants 2021SGR01519 and 2021SGR442. We thank the Spanish national supercomputing network (BSC-RES) for the award of computer time at the Minotauro supercomputer. D. C. Malaspina is supported by the European Union Horizon 2020 research and innovation programme under Marie Sklodowska-Curie grant agreement No. 6655919.References
- N. J. Yang and M. J. Hinner, in Getting Across the Cell Membrane: An Overview for Small Molecules, Peptides, and Proteins, ed. A. Gautier and M. J. Hinner, Springer, New York, New York, NY, 2015, pp. 29–53 Search PubMed.
- S. M. Loverde, J. Phys. Chem. Lett., 2014, 5, 1659–1665 CrossRef CAS PubMed.
- W. K. Subczynski, J. S. Hyde and A. Kusumi, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 4474–4478 CrossRef CAS PubMed.
- A. Walter and J. Gutknecht, J. Membr. Biol., 1986, 90, 207–217 CrossRef CAS PubMed.
- C. Hong, D. P. Tieleman and Y. Wang, Langmuir, 2014, 30, 11993–12001 CrossRef CAS PubMed.
- C. R. W. Guimarães, A. M. Mathiowetz, M. Shalaeva, G. Goetz and S. Liras, J. Chem. Inf. Model., 2012, 52, 882–890 CrossRef PubMed.
- S. Paula and D. W. Deamer, Current Topics in Membranes and Transport, Academic Press, 1999, vol. 48, pp. 77–95 Search PubMed.
- S. Paula, A. Volkov and D. Deamer, Biophys. J., 1998, 74, 319–327 CrossRef CAS PubMed.
- I. Vorobyov, T. E. Olson, J. H. Kim, R. E. Koeppe, O. S. Andersen and T. W. Allen, Biophys. J., 2014, 106, 586–597 CrossRef CAS PubMed.
- A. Parsegian, Nature, 1969, 221, 844–846 CrossRef CAS PubMed.
- A. A. Gurtovenko, J. Anwar and I. Vattulainen, Chem. Rev., 2010, 110, 6077–6103 CrossRef CAS PubMed.
- Y. Toyoshima and T. E. Thompson, Biochemistry, 1975, 14, 1525–1531 CrossRef CAS PubMed.
- D. C. Malaspina, C. Viñas, F. Teixidor and J. Faraudo, Angew. Chem., Int. Ed., 2020, 59, 3088–3092 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- E. Awoonor-Williams and C. N. Rowley, Biochim. Biophys. Acta, Biomembr., 2016, 1858, 1672–1687 CrossRef CAS PubMed.
- W. Shinoda, Biochim. Biophys. Acta, Biomembr., 2016, 1858, 2254–2265 CrossRef CAS PubMed.
- A. Volkov, S. Paula and D. Deamer, Bioelectrochem. Bioenerg., 1997, 42, 153–160 CrossRef CAS.
- J. L. MacCallum, W. F. Bennett and D. P. Tieleman, Biophys. J., 2011, 101, 110–117 CrossRef CAS PubMed.
- C. Verdiá-Báguena, A. Alcaraz, V. M. Aguilella, A. M. Cioran, S. Tachikawa, H. Nakamura, F. Teixidor and C. Viñas, Chem. Commun., 2014, 50, 6700 RSC.
- T. I. Rokitskaya, I. D. Kosenko, I. B. Sivaev, Y. N. Antonenko and V. I. Bregadze, Phys. Chem. Chem. Phys., 2017, 19, 25122–25128 RSC.
- J. Poater, C. Viñas, I. Bennour, S. Escayola, M. Solà and F. Teixidor, J. Am. Chem. Soc., 2020, 142, 9396–9407 CrossRef CAS PubMed.
- M. Tarrés, E. Canetta, E. Paul, J. Forbes, K. Azzouni, C. Viñas, F. Teixidor and A. J. Harwood, Sci. Rep., 2015, 5, 7804 CrossRef PubMed.
- M. Tarrés, E. Canetta, C. Viñas, F. Teixidor and A. J. Harwood, Chem. Commun., 2014, 50, 3370–3372 RSC.
- I. Bennour, M. N. Ramos, M. Nuez-Martínez, J. A. M. Xavier, A. B. Buades, R. Sillanpää, F. Teixidor, D. Choquesillo-Lazarte, I. Romero, M. Martinez-Medina and C. Viñas, Dalton Trans., 2022, 51, 7188–7209 RSC.
- Y. Chen, A. Barba-Bon, B. Grüner, M. Winterhalter, M. A. Aksoyoglu, S. Pangeni, M. Ashjari, K. Brix, G. Salluce, Y. Folgar-Cameán, J. Montenegro and W. M. Nau, J. Am. Chem. Soc., 2023, 145, 13089–13098 CrossRef CAS PubMed.
- P. Bauduin, S. Prevost, P. Farràs, F. Teixidor, O. Diat and T. Zemb, Angew. Chem., Int. Ed., 2011, 50, 5298–5300 CrossRef CAS PubMed.
- C. Viñas, M. Tarrés, P. González-Cardoso, P. Farràs, P. Bauduin and F. Teixidor, Dalton Trans., 2014, 43, 5062–5068 RSC.
- M. Uchman, V. Ďordovič, Z. Tošner and P. Matějícek, Angew. Chem., 2015, 127, 14319–14323 CrossRef.
- R. Fernandez-Alvarez, Z. Medoš, Z. Tošner, A. Zhigunov, M. Uchman, S. Hervø-Hansen, M. Lund, M. Bešter-Rogač and P. Matějíček, Langmuir, 2018, 34, 14448–14457 CrossRef CAS PubMed.
- I. Fuentes, J. Pujols, C. Viñas, S. Ventura and F. Teixidor, Chem. – Eur. J., 2019, 25, 12820–12829 CrossRef CAS PubMed.
- T. Merhi, A. Jonchère, L. Girard, O. Diat, M. Nuez, C. Viñas and P. Bauduin, Chem. – Eur. J., 2020, 26, 13935–13947 CrossRef CAS PubMed.
- A. Errachid, D. Caballero, E. Crespo, F. Bessueille, M. Pla-Roca, C. A. Mills, F. Teixidor and J. Samitier, Nanotechnology, 2007, 18, 485301 CrossRef.
- D. Vrbata, V. Dord’ovic, J. Seitsonen, J. Ruokolainen, O. Janoušková, M. Uchman and P. Matejícek, Chem. Commun., 2019, 55, 2900–2903 RSC.
- I. Fuentes, T. García-Mendiola, S. Sato, M. Pita, H. Nakamura, E. Lorenzo, F. Teixidor, F. Marques and C. Viñas, Chem. – Eur. J., 2018, 24, 17239–17254 CrossRef CAS PubMed.
- I. Cabrera, I. Abasolo, J. L. Corchero, E. Elizondo, P. R. Gil, E. Moreno, J. Faraudo, S. Sala, D. Bueno, E. González-Mira, M. Rivas, M. Melgarejo, D. Pulido, F. Albericio, M. Royo, A. Villaverde, M. F. García-Parajo, S. Schwartz, N. Ventosa and J. Veciana, Adv. Healthcare Mater., 2016, 5, 829–840 CrossRef CAS PubMed.
- J. Comer, J. C. Gumbart, J. Hénin, T. Lelievre, A. Pohorille and C. Chipot, J. Phys. Chem. B, 2015, 119, 1129–1151 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Simulation Methods. See DOI: https://doi.org/10.1039/d3cp03614f |
| This journal is © the Owner Societies 2023 |