
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Gas phase H+, H3O+ and NH4+ affinities of oxygen-bearing volatile organic compounds; DFT calculations for soft chemical ionisation mass spectrometry†
Maroua
Omezzine Gnioua
 ab,
Anatolii
Spesyvyi
a and
Patrik
Španěl
*a
ab,
Anatolii
Spesyvyi
a and
Patrik
Španěl
*a
aJ. Heyrovský Institute of Physical Chemistry of the Czech Academy of Sciences, Dolejškova 2155/3, 18223 Prague 8, Czech Republic. E-mail: patrik.spanel@jh-inst.cas.cz
bFaculty of Mathematics and Physics, Charles University, V Holešovičkách 747/2, 18000 Prague 8, Czech Republic
First published on 31st October 2023
Abstract
Quantum chemistry calculations were performed using the density functional theory, DFT, to understand the structures and energetics of organic ions relevant to gas phase ion chemistry in soft chemical ionisation mass spectrometry analytical methods. Geometries of a range of neutral volatile organic compound molecules and ions resulting from protonation, the addition of H3O+ and the addition of NH4+ were optimised using the B3LYP hybrid DFT method. Then, the total energies and the normal mode vibrational frequencies were determined, and the total enthalpies of the neutral molecules and ions were calculated for the standard temperature and pressure. The calculations were performed for several feasible structures of each of the ions. The proton affinities of several benchmark molecules agree with the accepted values within ±4 kJ mol−1, indicating that B3LYP/6-311++G(d,p) provides chemical accuracy for oxygen-containing volatile organic compounds. It was also found that the binding energies of H3O+ and NH4+ to molecules correlate with their proton affinities. The results contribute to the understanding of ligand switching ion–molecule reactions important for secondary electrospray ionisation, SESI, and selected ion flow tube, SIFT, mass spectrometries.
1 Introduction
The ligand-switching ion–molecule reactions of hydrated H3O+ and NH4+ ions with oxygen-containing organic molecules are becoming increasingly important in soft chemical ionisation mass spectrometry techniques. Proton transfer from H3O+ was traditionally the dominant ionisation process1 in selected ion flow tube mass spectrometry (SIFT-MS).2–5 However, recently, with the transition from helium carrier gas to nitrogen, H2OH3O+ is just as important reagent ion.Another promising technique is secondary electrospray ionisation mass spectrometry, SESI-MS,6–10 where reactions occur between hydrated reagent ions H3O+(H2O)1–411,12 and the analyte molecules introduced into the surrounding gas for highly sensitive analyses.6–10 The complex ion chemistry occurring in the SESI ion source, largely involving gas-phase ligand switching, results in widely variable sensitivities for different classes of VOCs. The quantification sensitivity was observed to depend on the analyte molecule, M, dipole moment (D) and its proton affinity (PA). Additionally, sensitivity tends to decrease as the observed fraction of the H2OMH+ product relative to the MH+ product increases. Furthermore, it has been suggested that there may be other as-yet-unidentified factors that significantly impact sensitivity in this context.
The NH4+ reagent ions are used in various forms of chemical ionisation mass spectrometry (CI-MS),13–20 including proton transfer reaction mass spectrometry (PTR-MS),21–26 the PTR3 instruments,27 SIFT-MS,28 ion mobility spectrometry (IMS)29 and high kinetic energy ion mobility spectrometry (HiKE-IMS).30 Also, it was observed in SESI-MS that NH4+(H2O)1–3 become dominant ions even when only trace amounts of ammonia are present.31
The hydrated ions H2OH3O+ and H2ONH4+ are formed by three body association reactions of H3O+ and NH4+ ions with H2O molecules. It is well known that the proton transfer reactions of H3O+ and NH4+ with oxygen-containing organic compounds, M, proceed by forming the MH+ ions when the proton affinity of M is greater than that of H2O or NH3. On the other hand, the ligand-switching reactions are not as well characterised. Fig. 1 shows a scheme of the reaction system of H3O+, NH4+, M and H2O. The idea behind this study is thus to introduce the concepts of H3O+ and NH4+ affinities that will help to predict the energetics of the ligand switching reactivity of H2OH3O+ and H2ONH4+ ions with molecules to facilitate their use for quantitative mass spectrometric analyses.
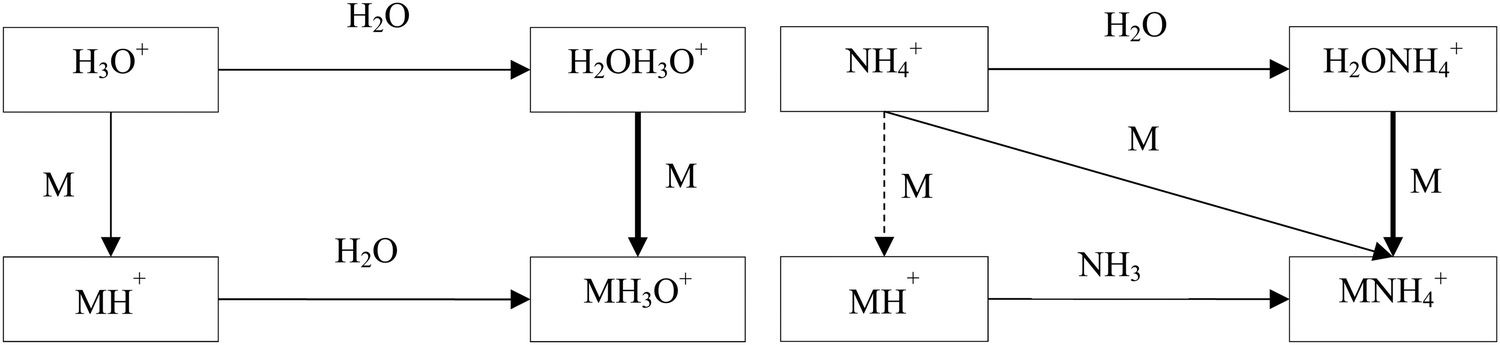 | ||
| Fig. 1 Ion chemistry is initiated by the H3O+ and NH4+ reactions with H2O and M molecules. | ||
Density functional theory (DFT) is one of the most widely used methods for quantum chemistry calculations of the structure of atoms, molecules, crystals, surfaces, and their interactions. It can predict a wide variety of molecular properties for unknown systems with low computational effort required. Over the past two decades, DFT has found widespread use in various branches of chemistry. It is now common practice to complement experimental studies by DFT calculations. Software and established approximations are now readily available, and DFT has emerged as an indispensable tool in scientific research because it serves two vital purposes: it can either validate experimental results or provide clarity when experiments yield ambiguous results. DFT is now routinely used to calculate various molecular properties, creating robust links between theoretical predictions and experimental observations. This approach often yields valuable insights into the structure, behaviour, and energy-related characteristics of molecules.32,33
DFT has been used to calculate PA of molecules since 1990.34,35 Several studies were recently carried out in an attempt to predict PA of organic molecules using ab initio or DFT computational approaches.36–38 Additionally, PA was calculated for indole39 as a benchmark using ab initio MP2 (Moller–Plesset with second-order energy correction) and DFT with two functionals B3LYP and M06-2X and five different basis sets to compare their agreement with available experimental values revealing that B3LYP is optimal in terms of computational cost and accuracy, even with a relatively restricted basis set 6-31+G(d,p). PA and IE were then calculated for several classes of molecules using this level of theory. Another recent study evaluated the PA of several hydrocarbons using several ab initio methods and different basis sets.40 On the basis of the previous work, it is evident that DFT using B3LYP is a suitable method, also because the performance of the current computers allows the calculations to be carried out typically in several hours.
The objective of this work is, therefore, to apply DFT as a reliable and facile computational method to determine proton affinities of organic molecules and enthalpy changes in association reactions of H3O+ and NH4+ with organic molecules. The results will be useful in predicting the reactivity of ions of the type MH+(H2O)n and MNH4+(H2O)n in soft chemical ionisation techniques like SIFT-MS and SESI-MS. The choice of molecules (acetone, propanol, 2-butenal, trans-2-heptanal, 2-heptanone, heptanal, 2,3-heptanedione and menthone) was guided by the results of the recent SESI-MS sensitivity study from our laboratory.41
2 Methods
All quantum chemistry calculations are carried out using ORCA 5.0.1 software.42 Molecular geometries of all neutral reactant molecules, their protonated forms and the adduct ion forms were first drawn using AVOGADRO43 software and then further optimised using ORCA with the B3LYP hybrid functional using 6-311++G(d,p) basis set (somewhat wider than 6-31+G(d,p) used previously39) and additionally with the D4 correction.44This level of theory was also used to calculate the normal mode vibrational frequencies and thermodynamic quantities of the neutral molecules and the ion structures. For this calculation to produce correct results, all vibrational frequencies must be calculated for the optimal geometry. If the geometry does not correspond to a local minimum on the energy surface, i.e. when it is a saddle point, then imaginary vibrational frequencies are listed in the calculation output. If that happens, it is necessary to modify the ion structure and repeat the calculation until all frequencies are real. The calculations were performed for several feasible structures of each of the ions, obtained by placing H3O+ or NH4+ moiety at several different sites of the organic molecule, and the lowest energy structure was chosen for inclusion in the results.
The total enthalpies of all neutral molecules and ions were thus calculated for the standard temperature and pressure (298.15 K, 1 atm). Enthalpies were calculated as the sum of the total electronic energy, Eele, the zero-point vibrational energy, EZPV, the temperature-dependent portion of the vibrational energy, Evib(T) and the thermal translational and rotational energies (5/2RT) of the molecule at 298 K.
| H = Eele + EZPV + Evib(T) + 5/2RT | (1) |
| H+ + M → MH+ | (2) |
| PA = H(MH+) − H(M) − 5/2RT = −ΔEele − ΔEZPV − Evib(T) + 5/2RT | (3) |
The energetics of the H3O+ and NH4+ adduct ions were calculated from the differences, ΔH, between the total enthalpies of their lowest energy structures and the enthalpies of their constituents, as will be discussed in Section 3.
3 Results and discussion
For neutral molecules, the structures are well established, and the B3LYP optimised structures were as expected. The DFT-optimised geometries of the neutral molecules agree well with the generally accepted structures.46 To find the lowest energy structure of MH+, MH3O+ and MNH4+ several initial geometries including configurations of H3O+ + M and MH+ + H2O were used as inputs for the geometry optimisation. The lowest enthalpy result was taken, and the corresponding geometries are illustrated in Fig. 2 and 3, and they are also listed as Cartesian coordinates in ESI.† Note that the structures of the adduct ions were identical when starting from the initial geometries composed of H3O+ and M moieties or from the MH+ and H2O moieties. The same applies to the NH3-containing structures in Fig. 3.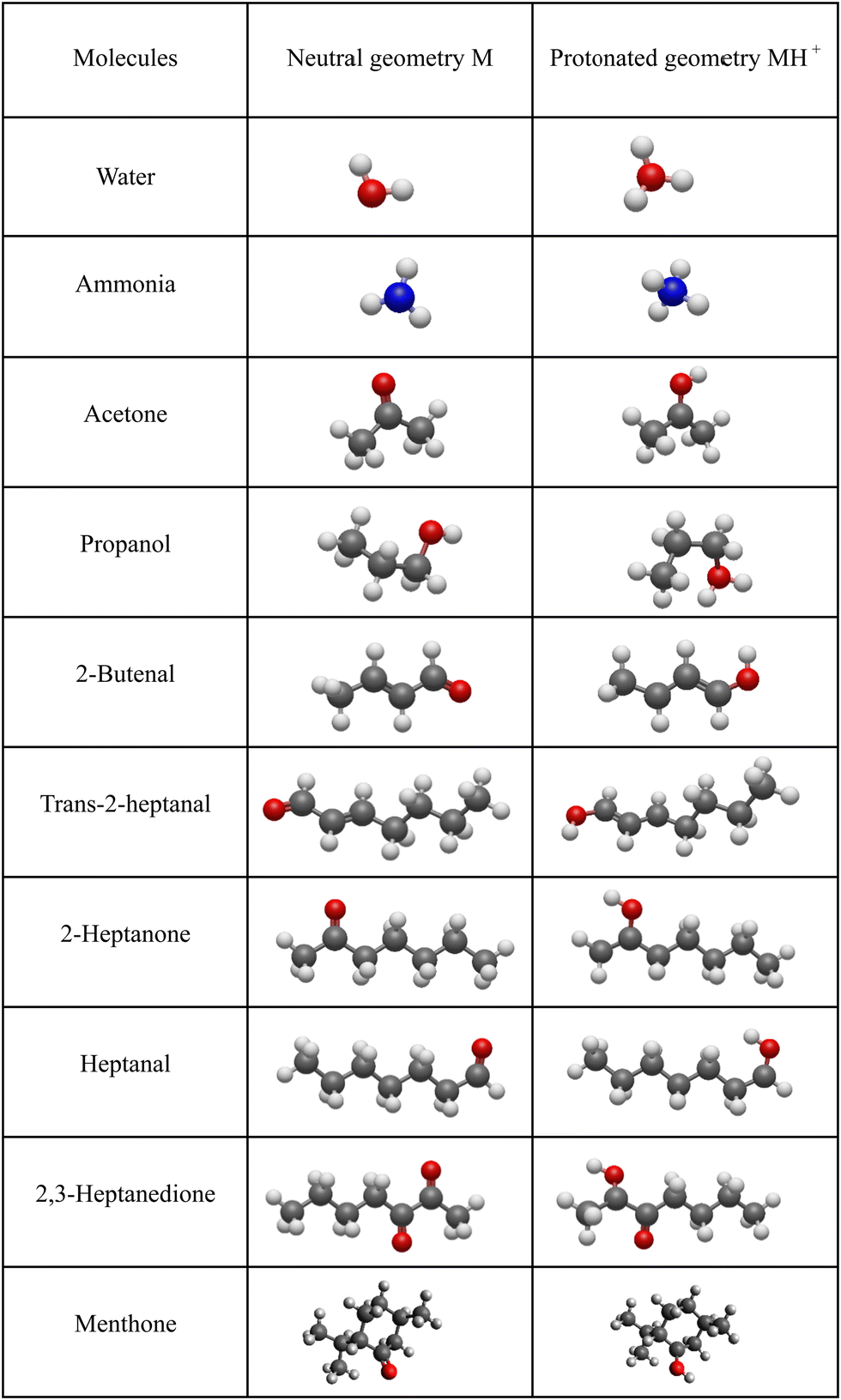 | ||
| Fig. 2 Optimised geometries of neutral and protonated molecules (C grey, O red, H white). | ||
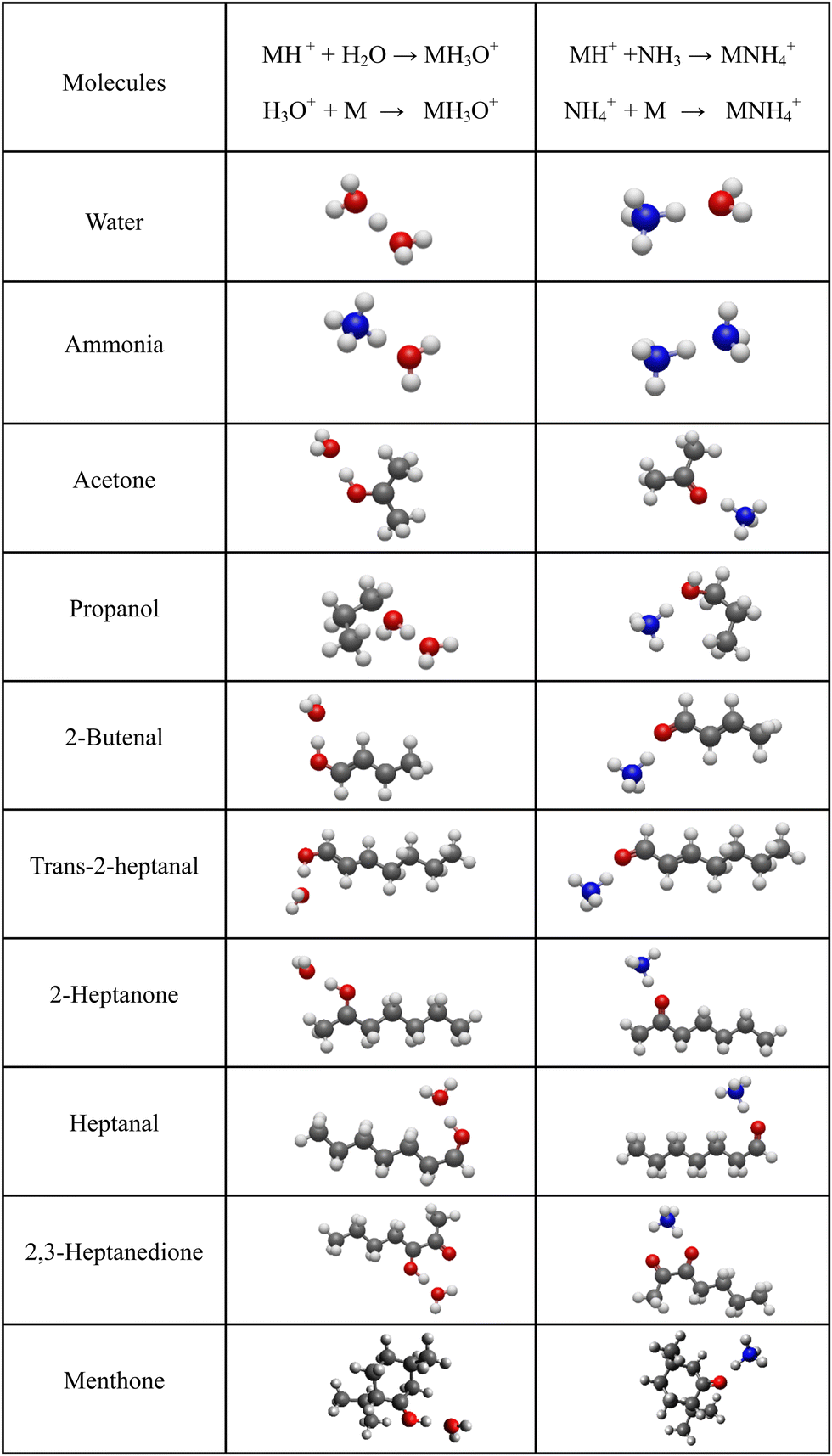 | ||
| Fig. 3 Optimised geometries of the H3O+ and NH4+ adducts. | ||
3.1 Proton affinity
Results of calculations of proton affinities using eqn (3) for selected volatile organic compounds are given in Table 1, where they can be compared with the values from the NIST Webbook database.47 Note that all values taken from the NIST database for the present study originated from the extensive tables of evaluated gas phase basicities and proton affinities of molecules as updated by Hunter and Lias 199848 which are based on experimental determinations of relative proton affinities from numerous previous studies. The values are not just copied, but they are adjusted on an internally consistent scale to match several absolute values based on the best data available at that time. The resulting proton affinity scale has been widely accepted and is generally considered to be reliable.49| Compound | Formula | PA (kJ mol−1) | |
|---|---|---|---|
| Theory | NIST | ||
| Water | H2O | 688 | 691 |
| Ammonia | NH3 | 852 | 853.6 |
| Acetone | C3H6O | 815 | 812 |
| Propanol | C3H8O | 785 | 786.5 |
| 2-Butenal | C4H6O | 839 | 830.8 |
| trans-2-Heptenal | C7H12O | 851 | |
| 2-Heptanone | C7H14O | 831 | |
| Heptanal | C7H14O | 797 | |
| 2,3-Heptanedione | C7H12O2 | 831 | |
| Menthone | C10H18O | 864 | |
Note that the PA calculated for H2O differs from the NIST value (691 kJ mol−1) only by 3 kJ mol−1, which is within the uncertainty given in the compilation by Hunter and Lias.48 The calculated PAs of other listed compounds are also within 3 kJ mol−1, NH3 (853.6 kJ mol−1) is 1.6 kJ mol−1 higher than our calculated value, acetone (812 kJ mol−1) is lower by 3 kJ mol−1, propanol (786.5 kJ mol−1) is higher by 1.5 kJ mol−1. All these are thus calculated within the so-called chemical accuracy (in traditional units quoted as ±1 kcal mol−1 corresponding to 4 kJ mol−1). This indicates that our choice of the B3LYP functional, D4 dispersion correction and 6-311++G(d,p) basis set is suitable for accurate PA calculations, considering that these NIST values, taken in general from Hunter and Lias,48 are well-established and confirmed from multiple sources.
However, there are no proton affinity values listed in NIST for aldehydes in Table 1, except for 2 butenal. For this molecule, the PA was determined only from a single experimental study of gas-phase proton transfer equilibria using ion trapping technique50 involving butenal in binary mixtures with acetone, methyl acetate and ethyl acetate (in the original traditional units) as an average value of 197 kcal mol−1 (824 kJ mol−1). This PA was later evaluated by using an updated set of reference values by Hunter and Lias as 830.8 kJ mol−1.48 Thus, the departure of the present calculated PA of 8.2 kJ mol−1 may be due to the uncertainty of the NIST value.
In summary, the PA of all VOCs included are greater than the PA of H2O and only trans-2-heptenal comes close to NH3, which has the highest PA in Table 1. This agrees with the experimental evidence that H3O+ reacts by proton transfer to all molecules in Table 1 and that NH4+ ions tend to form adduct ions with molecules, as we observed in recent experimental studies.28
3.2 Enthalpy changes
To evaluate the binding energies of H2O and NH3 to MH+ and of H3O+ and NH4+ to M, we have considered the following four association reactions:| MH+ + H2O → MH3O+ | (4) |
| MH+ + NH3 → MNH4+ | (5) |
| H3O+ + M → MH3O+ | (6) |
| NH4+ + M → MNH4+ | (7) |
| Compound | ΔH (kJ mol−1) | |||
|---|---|---|---|---|
| MH+ + H2O → MH3O+ | H3O+ + M → MH3O+ | MH+ + NH3 → MNH4+ | NH4+ + M → MNH4+ | |
| H2O | −157 | −157 | −255 | −90 |
| NH3 | −90 | −255 | −121 | −121 |
| C3H6O | −99 | −226 | −155 | −118 |
| C3H8O | −110 | −206 | −176 | −107 |
| C4H6O | −91 | −242 | −139 | −126 |
| C7H12O | −87 | −250 | −131 | −134 |
| C7H14O | −93 | −237 | −143 | −127 |
| C7H14O | −94 | −203 | −167 | −111 |
| C7H12O2 | −104 | −209 | −202 | −100 |
| C10H18O | −69 | −324 | −88 | −178 |
In SESI and SIFT ion chemistry, products of reactions (4)–(7) would further undergo ligand-switching reactions. To predict the energetics of such reactions, we propose that it is helpful to consider concepts of H3O+ affinity as the binding energy in reaction (6):
| ΔH = H(MH3O+) − H(M) − H(H3O+) | (8) |
| ΔH = H(MNH4+) − H(M) − H(NH4+) | (9) |
| H3O+M1 + M2 → H3O+M2 + M1 | (10) |
3.3 Overall trends
It is interesting to assess how the calculated affinities of molecules to H3O+ and NH4+ relate to their proton affinities. The values of ΔH corresponding to eqn (6) and (7) are plotted as functions of the proton affinities in Fig. 4. The most obvious observation is that all H3O+ affinities are greater than the NH4+ affinities. This can be only partly explained by the difference in proton affinities between H2O and NH3, which is 165 kJ mol−1. For this set of molecules, the ΔH values strongly correlate with their proton affinities but with slopes significantly smaller than 1. Note that some values are outside the general trend; for example, the binding energy of 2,3-heptanedione to NH4+ is somewhat below the trendline. This departure can be explained by the structure of this adduct ion (see Fig. 3), in which NH4+ binds to both O atoms in the dione molecule. Note that this molecule has two oxygen sites, and thus, the proton affinity was taken for the structure with the lowest enthalpy, as illustrated in Fig. 2.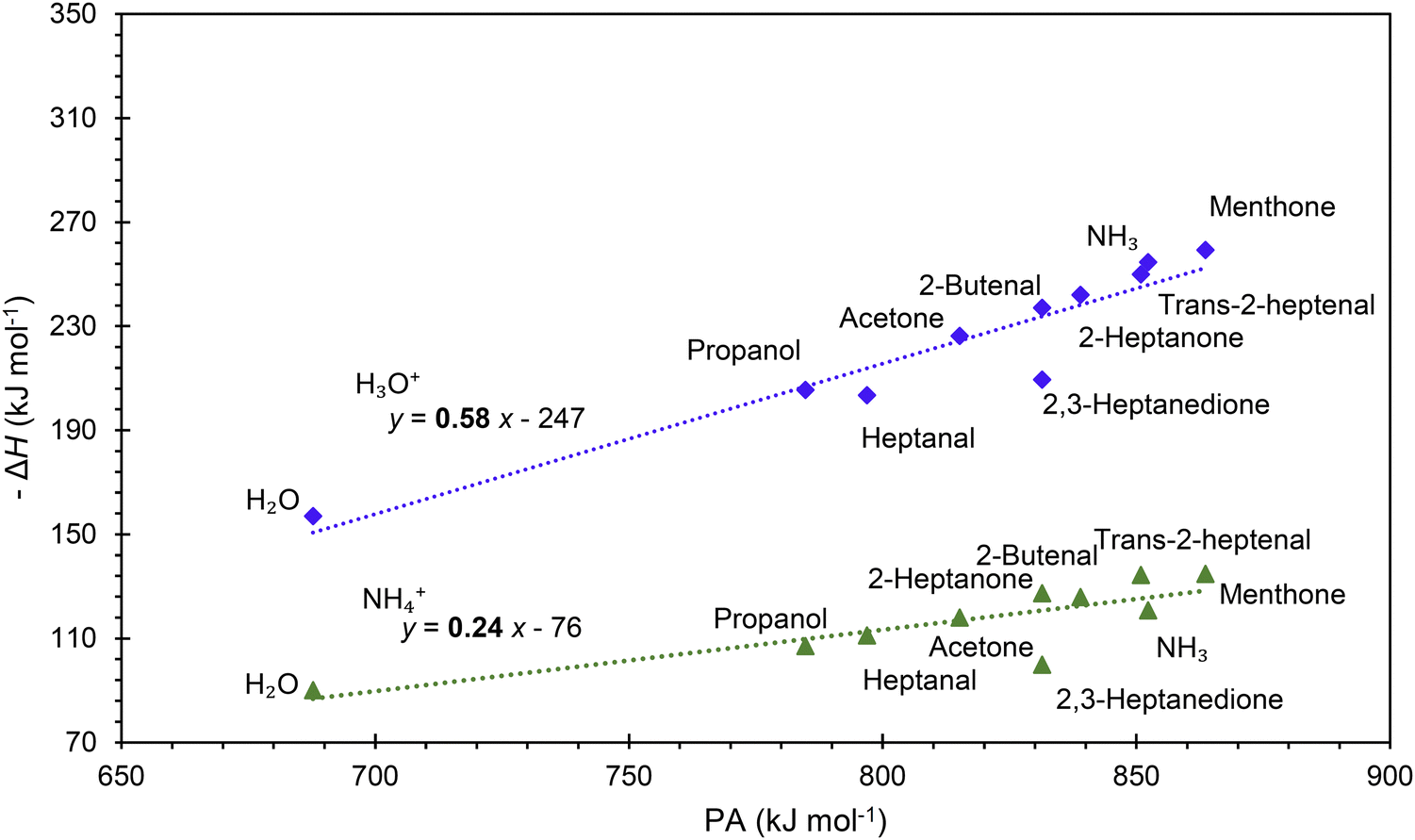 | ||
| Fig. 4 Correlation of H3O+ and NH4+ affinities (binding energies to M) with proton affinities. | ||
This observation is interesting and, to some degree, logical. It can be explained by the idea that a higher ability of molecule M to bind a proton will also likely result in an increased ability of MH2O or MNH3 neutral to bind a proton. However, the relationship between PA and H3O+ affinities is not straightforward and is also affected by the binding energy of neutral M to neutral H2O or NH3, which is independent of PA.
Conclusions
The results from the present computational study are promising as they facilitate predicting the energetics of the reactions of hydrated hydronium ions, H3O+ and ammonium ions, NH4+, with volatile organic compounds proceeding via ligand switching reactions. For example, the H3O+ affinity of all organic molecules in this study is greater than that of H2O and thus, H2OH3O+ will react exothermically with all of them. Such data are essential for SIFT-MS and SESI-MS to interpret the mass spectra and to provide reliable quantification methods. The computational method presented in this article can now be routinely used to calculate the proton affinities of the VOC molecules and their affinities to H3O+ and NH4+ ions that govern the energetics of hydrated ions of type MH+(H2O) and adducts of the type MNH4+. These values will have to be validated experimentally, for example, by studying ligand-switching reactions.51 The observed strong correlations between the proton affinities and H3O+ and NH4+ affinities are interesting and should be explored further, along with the computed thermochemical properties for identifying and quantifying gas phase reactions.Note, however, that the reaction rate coefficients can be influenced by factors other than energetics. Thus, while the H3O+ and NH4+ affinities indicate the energetics of ligand-switching reactions, the actual reaction kinetics may be influenced by additional factors. That said, the reaction equilibria that may take place in the atmospheric pressure region of ion sources like SESI are defined by Gibbs free energy changes, which can be readily calculated using the present method only by adding the entropy terms. This will be subject to further work that will compare DFT calculations of equilibrium rate constants with original experimental results.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
GAČR project: “Selected ion flow drift tube mass spectrometry with negative ions and nitrogen carrier gas” 21-25486S. Computational resources were in part provided by the e-INFRA CZ project (ID:90254), supported by the Ministry of Education, Youth and Sports of the Czech Republic.References
- D. Pagonis, K. Sekimoto and J. de Gouw, J. Am. Soc. Mass Spectrom., 2019, 30, 1330–1335 CrossRef CAS PubMed.
- P. Španěl, K. Dryahina and D. Smith, Int. J. Mass Spectrom., 2006, 249, 230–239 CrossRef.
- P. Španěl and D. Smith, Mass Spectrom. Rev., 2011, 30, 236–267 CrossRef PubMed.
- D. Smith and P. Španěl, Analyst, 2011, 136, 2009–2032 RSC.
- P. Španěl and D. Smith, Curr. Anal. Chem., 2013, 9, 525–539 CrossRef.
- C. Wu, W. F. Siems and H. H. Hill, Anal. Chem., 2000, 72, 396–403 CrossRef CAS PubMed.
- T. Bruderer, T. Gaisl, M. T. Gaugg, N. Nowak, B. Streckenbach, S. Muller, A. Moeller, M. Kohler and R. Zenobi, Chem. Rev., 2019, 119, 10803–10828 CrossRef CAS PubMed.
- A. T. Rioseras, K. D. Singh, N. Nowak, M. T. Gaugg, T. Bruderer, R. Zenobi and P. Martinez-Lozano Sinues, Anal. Chem., 2018, 90, 6453–6460 CrossRef PubMed.
- L. Bregy, A. R. Muggler, P. Martinez-Lozano Sinues, D. Garcia-Gomez, Y. Suter, G. N. Belibasakis, M. Kohler, P. R. Schmidlin and R. Zenobi, Sci. Rep., 2015, 5, 15163 CrossRef CAS PubMed.
- L. Bregy, Y. Nussbaumer-Ochsner, P. Martinez-Lozano Sinues, D. Garcia-Gomez, Y. Suter, T. Gaisl, N. Stebler, M. T. Gaugg, M. Kohler and R. Zenobi, Clin. Mass Spectrom., 2018, 7, 29–35 CrossRef.
- J. Lan, F. B. Parte, G. Vidal-de-Miguel and R. Zenobi, in Breathborne Biomarkers and the Human Volatilome, ed. J. Beauchamp, C. Davis and J. Pleil, Elsevier, Boston, 2nd edn, 2020, pp. 185–199 Search PubMed.
- A. T. Rioseras, M. T. Gaugg and P. Martinez-Lozano Sinues, Anal. Methods, 2017, 9, 5052–5057 RSC.
- A. Maquestiau, R. Flammang and L. Nielsen, Org. Mass Spectrom, 1980, 15, 376–379 CrossRef CAS.
- T. Keough and A. J. Destefano, Org. Mass Spectrom, 1981, 16, 527–533 CrossRef CAS.
- R. S. Blake, K. P. Wyche, A. M. Ellis and P. S. Monks, Int. J. Mass Spectrom., 2006, 254, 85–93 CrossRef CAS.
- A. Hansel, W. Scholz, B. Mentler, L. Fischer and T. Berndt, Atmos. Environ., 2018, 186, 248–255 CrossRef CAS.
- A. Zaytsev, M. Breitenlechner, A. R. Koss, C. Y. Lim, J. C. Rowe, J. H. Kroll and F. N. Keutsch, Atmos. Meas. Tech., 2019, 12, 1861–1870 CrossRef CAS PubMed.
- T. Berndt, W. Scholz, B. Mentler, L. Fischer, H. Herrmann, M. Kulmala and A. Hansel, Angew. Chem., Int. Ed., 2018, 57, 3820–3824 CrossRef CAS PubMed.
- S. Zhou, J. C. Rivera-Rios, F. N. Keutsch and J. P. D. Abbatt, Atmos. Meas. Tech., 2018, 11, 3081–3089 CrossRef CAS.
- J. B. Westmore and M. M. Alauddin, Mass Spectrom. Rev., 1986, 5, 381–465 CrossRef CAS.
- C. Shen, J. Li, H. Han, H. Wang, H. Jiang and Y. Chu, Int. J. Mass Spectrom., 2009, 285, 100–103 CrossRef CAS.
- Q. Zhang, X. Zou, Q. Liang, H. Wang, C. Huang, C. Shen and Y. Chu, J. Am. Soc. Mass Spectrom., 2019, 30, 501–508 CrossRef CAS PubMed.
- E. Canaval, N. Hyttinen, B. Schmidbauer, L. Fischer and A. Hansel, Front. Chem., 2019, 7, 191 CrossRef PubMed.
- M. Müller, F. Piel, R. Gutmann, P. Sulzer, E. Hartungen and A. Wisthaler, Int. J. Mass Spectrom., 2020, 447, 116254 CrossRef.
- L. Zhu, T. Mikoviny, A. Kolstad Morken, W. Tan and A. Wisthaler, Int. J. Greenhouse Gas Control, 2018, 78, 349–353 CrossRef CAS.
- B. Yuan, A. R. Koss, C. Warneke, M. Coggon, K. Sekimoto and J. A. de Gouw, Chem. Rev., 2017, 117, 13187–13229 CrossRef CAS PubMed.
- M. Breitenlechner, L. Fischer, M. Hainer, M. Heinritzi, J. Curtius and A. Hansel, Anal. Chem., 2017, 89, 5824–5831 CrossRef CAS PubMed.
- S. J. Swift, D. Smith, K. Dryahina, M. Omezzine Gnioua and P. Španěl, Rapid Commun. Mass Spectrom., 2022, 36, e9328 CrossRef CAS PubMed.
- A. Abedi, L. Sattar, M. Gharibi and L. A. Viehland, Int. J. Mass Spectrom., 2014, 370, 101–106 CrossRef CAS.
- M. Allers, A. T. Kirk, C. Schaefer, D. Erdogdu, W. Wissdorf, T. Benter and S. Zimmermann, J. Am. Soc. Mass Spectrom., 2020, 31, 2191–2201 CrossRef CAS PubMed.
- K. Dryahina, S. Som, D. Smith and P. Španěl, Rapid Commun. Mass Spectrom., 2021, e9047 CrossRef CAS PubMed.
- W. T. Yang, Phys. Rev. Lett., 1991, 66, 1438–1441 CrossRef CAS PubMed.
- C. D. Sherrill, J. Chem. Phys., 2010, 132, 110902 CrossRef PubMed.
- M. Toscano, N. Russo and J. Rubio, J. Chem. Soc., Faraday Trans., 1996, 92, 2681–2684 RSC.
- A. K. Chandra and A. Goursot, J. Phys. Chem., 1996, 100, 11596–11599 CrossRef CAS.
- Z. S. Safi and N. Wazzan, J. Comput. Chem., 2021, 42, 1106–1117 CrossRef CAS PubMed.
- A. V. Lebedev, J. Anal. Chem., 2020, 75, 1719–1730 CrossRef CAS.
- Y. Valadbeigi, H. Farrokhpour and M. Tabrizchi, J. Chem. Sci., 2014, 126, 1209–1215 CrossRef CAS.
- M. Bhatia, Comput. Theor. Chem., 2023, 1223, 114101 CrossRef CAS.
- E. E. Etim, O. E. Godwin and S. A. Olagboye, Int. J. Adv. Res. Chem. Sci., 2020, 7, 11–22 Search PubMed.
- K. Dryahina, M. Polášek, D. Smith and P. Španěl, Rapid Commun. Mass Spectrom., 2021, 35, e9187 CrossRef CAS PubMed.
- F. Neese, WIREs Comput Mol Sci., 2022, 12, e1606, DOI:10.1002/wcms.1606.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Cheminform., 2012, 4, 17 CrossRef CAS PubMed.
- E. Caldeweyher, C. Bannwarth and S. Grimme, J. Chem. Phys., 2017, 147, 034112 CrossRef PubMed.
- Z. Safi and W. Fares, in Conceptual Density Functional Theory and Its Application in the Chemical Domain, ed. N. Islam and S. Kaya, Apple Academic Press, New York, 2018, pp. 297–323 Search PubMed.
- S. Kim, J. Chen, T. Cheng, A. Gindulyte, J. He, S. He, Q. Li, B. A. Shoemaker, P. A. Thiessen, B. Yu, L. Zaslavsky, J. Zhang and E. E. Bolton, Nucleic Acids Res., 2022, 51, D1373–D1380 CrossRef PubMed.
- S. G. Lias, R. D. Levin and S. A. Kafafi, National Institute of Standards and Technology, Gaithersburg, 2016 Search PubMed.
- E. Hunter and S. Lias, J. Phys. Chem. Ref. Data, 1998, 27, 413 CrossRef CAS.
- B. J. Smith and L. Radom, J. Am. Chem. Soc., 1993, 115, 4885–4888 CrossRef CAS.
- J. H. Vajda and A. G. Harrison, Int. J. Mass Spectrom. Ion Processes, 1979, 30, 293–306 CrossRef.
- A. Spesyvyi, M. Lacko, K. Dryahina, D. Smith and P. Španěl, J. Am. Soc. Mass Spectrom., 2021, 32, 2251–2260 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp03604a |
| This journal is © the Owner Societies 2023 |