
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFull-dimensional automated potential energy surface development and detailed dynamics for the CH2OO + NH3 reaction
Cangtao
Yin
 * and
Gábor
Czakó
*
* and
Gábor
Czakó
*
MTA-SZTE Lendület Computational Reaction Dynamics Research Group, Interdisciplinary Excellence Centre and Department of Physical Chemistry and Materials Science, Institute of Chemistry, University of Szeged, Rerrich Béla tér 1, Szeged H-6720, Hungary. E-mail: cangtaoyin@foxmail.com; gczako@chem.u-szeged.hu
First published on 25th September 2023
Abstract
With the help of the ROBOSURFER program package, a global full-dimensional potential energy surface (PES) for the reaction of the Criegee intermediate, CH2OO, with the NH3 molecule is developed iteratively using different ab initio methods and the monomial symmetrization fitting approach. The final permutationally-invariant analytical PES is constructed based on 23447 geometries and the corresponding ManyHF-based CCSD(T)-F12b/cc-pVTZ-F12 energies. The accuracy of the PES is confirmed by the excellent agreement of its stationary-point properties and one-dimensional potential energy curves compared with the corresponding ab initio data. The reaction probabilities and integral cross sections are calculated for the ground-state and several vibrationally excited-state reactions by quasi-classical trajectory simulations. Remarkable is that the maximum impact parameter b where reactivity vanishes is almost independent of collision energy ranging from 1 to 40 kcal mol−1, and the reaction probability increases with increasing collision energy for this negative-barrier reaction. At the same time, a slight mode-specificity effect is observed. In addition, the deuterium effect is investigated and the sudden vector projection is discussed.
I. Introduction
Carbonyl oxides, also called Criegee intermediates, are found to play a significant role in the global atmospheric processes. Due to having C![[double bond, length half m-dash]](https://www.rsc.org/images/entities/char_e006.gif) O–O functional groups, Criegee intermediates are very reactive and they decompose to yield OH radicals1–5 or react with many atmospheric species such as water,6,7 sulfur dioxide,8–11 nitrogen dioxide,10,11 ammonia,12–18 methanol,19–21 organic and inorganic acids.22–28
O–O functional groups, Criegee intermediates are very reactive and they decompose to yield OH radicals1–5 or react with many atmospheric species such as water,6,7 sulfur dioxide,8–11 nitrogen dioxide,10,11 ammonia,12–18 methanol,19–21 organic and inorganic acids.22–28
As the most abundant alkaline atmospheric compound, ammonia is believed to play an important role in the formation of inorganic aerosols29,30 and secondary organic aerosols.31,32 In regard to the reaction of the simplest Criegee intermediate CH2OO with NH3, Misiewicz et al.12 calculated a rate coefficient of 5.36 × 10−14 cm3 molecule−1 s−1. The product of the CH2OO + NH3 reaction is predicted to be the NH2CH2OOH adduct, which may further undergo photolytic breaking of the fragile O–O bond or react with OH radicals. Later Liu et al.13 measured the rate coefficient of the CH2OO + NH3 reaction with two different experimental methods, the OH laser-induced fluorescence method and the UV transient absorption method. Weak temperature dependence was observed, consistent with the theoretical activation energy of −0.53 kcal mol−1 at the QCISD(T)/CBS//B3LYP/6-311+G(2d,2p) level. The rate coefficient was 5.64 × 10−14 cm3 molecule−1 s−1 and 8.1 × 10−14 cm3 molecule−1 s−1 by both methods at 298 K, within the error bars of the theoretical results. Chhantyal-Pun et al.14 also investigated the CH2OO + NH3 reaction both theoretically and experimentally. The average rate coefficient was 8.4 × 10−14 cm3 molecule−1 s−1 deduced at 293 K. Photoionization mass spectra indicated production of NH2CH2OOH functionalized organic hydroperoxide adduct from the reaction. Ab initio calculations performed at the CCSD(T)(F12*)/cc-pVQZ-F12//CCSD(T)(F12*)/cc-pVDZ-F12 level of theory predicted pre-reactive complex formation, consistent with previous studies. Master equation simulations of the experimental data using the ab initio computed structures identified submerged barrier heights of −0.50 kcal mol−1, similar with the results from Liu et al.12 Adjieufack et al.17 analyzed the flow of the electron density along the CH2OO + NH3 reaction from the perspective of bonding evolution theory. In addition to the initial adduct NH2CH2OOH, four reaction pathways including CH2(O)NH2˙ + OH˙, H2NC(O)H + H2O, H2CNH + HOOH and H2CNH + H2OO as afterward products are determined by Nazari and Saheb as possible product channels.18
First-principles dynamics investigations of the CH2OO + NH3 → NH2CH2OOH reaction have been hindered, because no potential energy surface (PES) is available in the literature. The only full-dimensional PES related to Criegee intermediates we can find is for the CH2OO + H2O reaction, reported by Wu et al.33 very recently, which was developed based on high-level CCSD(T)-F12a/aug-cc-pVTZ data. Extensive quasi-classical trajectory (QCT) calculations were performed on that PES providing detailed kinetics and dynamics results. In this research, we develop a full-dimensional ab initio PES for the CH2OO + NH3 → NH2CH2OOH reaction. Next, the QCT simulations are carried out on this PES. In addition, the normal-mode excitations and the deuterium effect are also investigated.
II. PES
A. Initial PES
Following our previous approach for the HBr/HI + C2H5 reactions,34,35 first we randomly displace the Cartesian coordinates13 of the vdW complex, the transition state, and the product of the CH2OO + NH3 → NH2CH2OOH reaction in the 0 Å–0.4 Å interval, resulting in 1000 geometries for each stationary point. For the entrance region, the distance between CH2OO and NH3 is randomly set between 3 and 8 Å, which generated 2000 geometries. Next the energies of these geometries at the ManyHF-based36 MP237/aug-cc-pVDZ38 level of theory are calculated using the MOLPRO program package39 as our initial candidate of geometry set. It should be noticed that for the geometries far from equilibrium, the Hartree–Fock (HF) method offers no guarantee to find the lowest-energy solution even if convergence is achieved. Therefore, in this work we use the ManyHF method recently developed in our group,36 which finds better HF solutions. Since geometries with too high energy are not needed in the global PES and could even damage the analytical PES, we exclude those geometries with energies higher than 400 kcal mol−1 relative to the global minimum of the set, which is the stationary point of the product in this case. Now we have a dataset consisting of 4557 geometries and the corresponding energies in hand.To get an initial full-dimensional analytical PES function the Monomial Symmetrization Approach (MSA)40 is used with the above data set. This function is an expansion of polynomials of the yij = exp(−rij/a) variables, where rij are the distances between two atoms and the non-linear parameter a is set to 2.0 bohr as we chose before in other systems.34,35 The method of weighted least squares is used for the fitting, with a weighting factor of E0/(E + E0), where E is the potential energy relative to the global minimum, and E0 is set to 0.1 (in hartree). There are 2121/11241 fitting coefficients in the fourth/fifth-order expansion. Since we have 4557 initial data points, we employed the fourth-order expansion at first.
B. PES development
 | ||
| Fig. 1 Distribution of the data points for the final PES. The energy zero point is set at the asymptote of reactants. The bin size is 1 kcal mol−1. | ||
C. PES evaluation and test
Fig. 2 shows the reactants, vdW complex, transition state, and the product of the CH2OO + NH3 reaction. Both the geometry and energy indicate a reactant-like TS. The energies obtained by three different methods (see the caption of Fig. 2) are in good agreement with each other, indicating low fitting errors of the analytical PES function and the fact that reliable results can be deduced from this PES.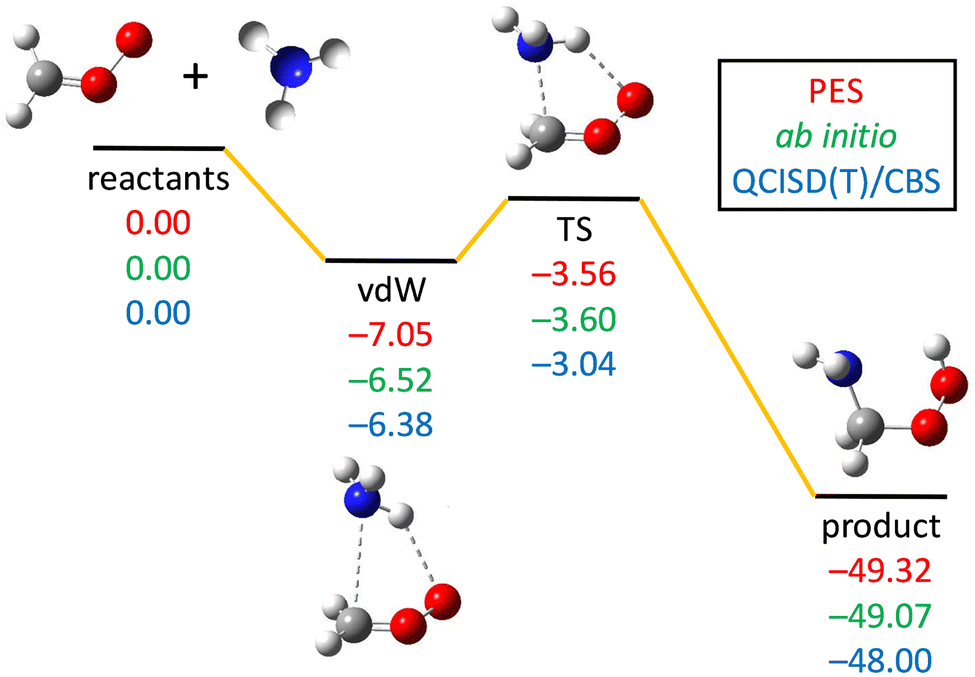 | ||
| Fig. 2 PES schematic of the CH2OO + NH3 → NH2CH2OOH reaction. All energies are in kcal mol−1 and relative to the CH2OO + NH3 asymptote at three different methods: obtained on the PES (red), the ManyHF-CCSD(T)-F12b/cc-pVTZ-F12 ab initio energies at geometries optimized on the PES (green), and the QCISD(T)/CBS relative energies taken from ref. 13 (blue). | ||
The imaginary frequency of the TS is 288i cm−1 on the PES, which is consistent with the ab initio calculations of 302i cm−1 (Misiewicz et al.12), 239i cm−1 (Liu et at.13), 268i cm−1 (Chhantyal-Pun et al.14), and 355i cm−1 (Nazari and Saheb18). The zero-point energies (ZPEs) of CH2OO, NH3, vdW complex, transition state, and product obtained on the present PES are 19.32 (19.54), 21.99 (21.54), 43.49 (42.83), 44.49 (43.92), and 46.70 (45.82) kcal mol−1, respectively (the ab initio values13 are listed in parentheses).
As shown in Fig. 3, three different potential energy curves are plotted: (1) the optimized reactants, CH2OO and NH3, are separated at the distance of 1 Å to 10 Å, in terms of C–N distance; (2) the C–N bond of optimized vdW complex geometry is elongated and shortened, while other degrees of the two parts are frozen; (3) the C–N bond of optimized transition state geometry is elongated and shortened, while other degrees of the two parts are frozen. Note that each curve uses different asymptote in Fig. 3.
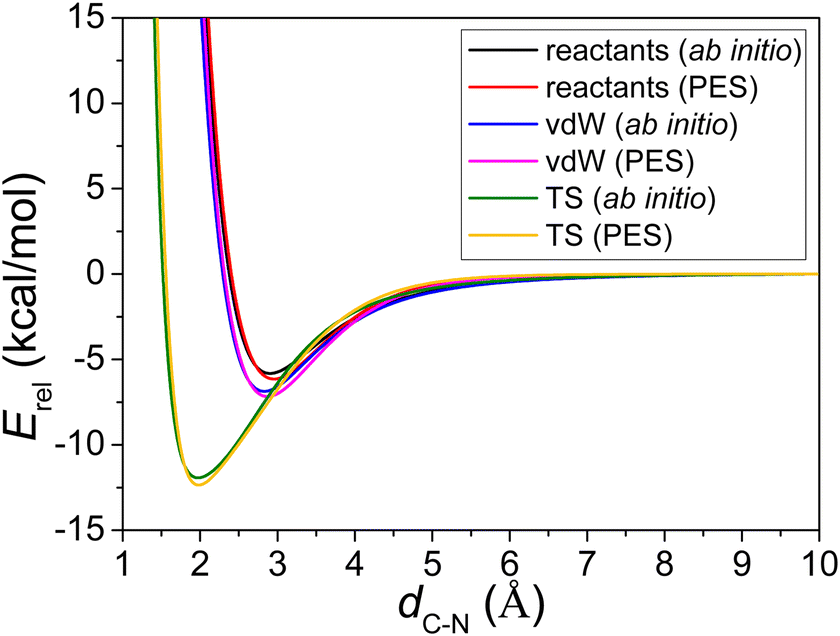 | ||
| Fig. 3 Comparisons of potential energies as a function of dC–N (distance between the C atom of CH2OO and the N atom of NH3) obtained from the PES and from ManyHF-CCSD(T)-F12b/cc-pVTZ-F12 ab initio calculations, while fixing all the other internal coordinates for the three situations. | ||
The energies of the wells with respect to the corresponding asymptote for the three situations described above are shown in Table 1. The comparison between the PES minima and ab initio minima shows a perfect chemical accuracy for this full-dimensional PES and its fitting errors are merely about 0.4 kcal mol−1. Table 1 also offers the positions of the minima located at the C–N distance for PES and ab initio (with less than 0.05 Å differences), and they show a good behavior of the analytical PES in the entrance channel.
| Reactants | vdW complex | Transition state | |
|---|---|---|---|
| Energy (kcal mol−1) | −6.1 (−5.8) | −7.2 (−6.9) | −12.3 (−11.9) |
| C–N distance (Å) | 2.95 (2.90) | 2.87 (2.83) | 1.98 (1.97) |
III. QCT simulations
A common approach used to study the dynamics of chemical reactions is the QCT method, which is computationally easier and faster than quantum methods. In the QCT method, the motion of atoms during the collision is described by classical mechanics. The QCT method has another advantage that it provides the possibility of visualization of the motion of atoms. Thus, one can get insight into the dynamics of the reaction by analyzing the motion of atoms during collisions.In this paper, the quasi-classical dynamics is carried out based on standard QCT simulations. Seven different collision energies Ecoll = 1, 2, 5, 10, 20, 30, and 40 kcal mol−1 are adopted as one of the initial conditions of the QCT simulations. Other initial conditions include: (1) the ZPEs of the reactants CH2OO and NH3 are set by standard normal-mode sampling;42 (2) the rotational angular momentum of each reactant is set to zero; (3) the spatial orientations of both reactants are randomly sampled; (4) the X/Y/Z-direction distance between the center of mass of the two reactants CH2OO and NH3 is set to 16/b/0 bohr, where the center of mass velocities are parallel with the X axis and the impact parameter b takes a value from 0 to bmax (no reaction occurs), increasing by 0.5 bohr each time. Two thousand trajectories for each b value are performed to get statistically accurate results. During the simulations, we choose 0.0726 fs as the time step for trajectory propagation. In this cycloaddition reaction, the two reactants collide after around 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 time steps. The product is formed around 20000 time steps if reaction occurs. We set 100000 time steps for each trajectory and we also stop the propagation when the largest interatomic distance becomes larger than the largest initial one by 1 bohr. In this case reactive trajectories can stay in the product region long enough and the non-reactive trajectories are halted much earlier to save time. There are of course other ways of defining the reactive event and stopping condition, for example, in the N2O + C2H2 reaction, the trajectories were halted when the distances of the two forming bonds C–N and C–O arrived at 1.6 Å.44 At the end less than 0.1% of the trajectories failed and gave unphysical results, which also indicates a good behavior of the newly-developed analytical PES.
000 time steps. The product is formed around 20000 time steps if reaction occurs. We set 100000 time steps for each trajectory and we also stop the propagation when the largest interatomic distance becomes larger than the largest initial one by 1 bohr. In this case reactive trajectories can stay in the product region long enough and the non-reactive trajectories are halted much earlier to save time. There are of course other ways of defining the reactive event and stopping condition, for example, in the N2O + C2H2 reaction, the trajectories were halted when the distances of the two forming bonds C–N and C–O arrived at 1.6 Å.44 At the end less than 0.1% of the trajectories failed and gave unphysical results, which also indicates a good behavior of the newly-developed analytical PES.
A. Reaction probabilities
Reaction probabilities of the ground-state reactants corresponding to seven different collision energies are plotted as a function of the impact parameter b in Fig. 4. Remarkable is that the reaction probability increases with increasing Ecoll for this negative barrier reaction. The reaction probabilities for collision energy as small as 1 kcal mol−1 are not zero which means no threshold energy for the CH2OO + NH3 → NH2CH2OOH reaction. By checking the non-reactive trajectories, we found that sometimes the two reactants rotate with each other, after a few rounds they separate and no reaction occurs. This often happens at low Ecoll. The maximum b where reactivity vanishes is around 5 bohr and almost independent of Ecoll. | ||
| Fig. 4 Reaction probabilities calculated with the QCT method at varying collision energies (given in kcal mol−1). | ||
B. The role of the vibrational excitations and deuterium effect
Comparing how efficiently translational and vibrational energy promote a reaction is popular. By checking the geometries of the reactants and product, we see that the CH2 is bent down after the reaction, the O–O bond and C–O bond are elongated around 0.11 Å and 0.14 Å, respectively, and also the NH3 becomes flatter. In addition, the reaction involves N–H bond breaking, as well as C–N bond and O–H bond forming. Clearly the NH-stretching excitation is expected to promote the reaction, so does the translational energy (which has already been proved by Fig. 4). Therefore, we pick the CH2 wagging, OO-stretching, CO-stretching, NH3-umbrella-wagging, NH-symmetric-stretching, and NH-asymmetric-stretching normal modes and investigate their efficiency for the reactivity. The motions corresponding to above vibrations are represented in Fig. 5. | ||
| Fig. 5 Six normal-mode vibrations studied in the present work. The normal-mode analysis is performed on the PES. | ||
In Fig. 6, following the same strategy as before,45,46 we present the reaction cross sections (σ) obtained with the QCT method for the reaction with both reactants in vibrational ground state as well as for six excited vibrational states described above. The results reflect a slight increment for vOO-str = 1 and vNH-str = 1 with both symmetric and asymmetric case, but a slight inhibition effect for vCO-str = 1 and vCH2-wag = 1. However, the excitation of the umbrella mode of NH3 has the negative effect to the reactivity at low Ecoll while enhances the reactivity at high Ecoll.
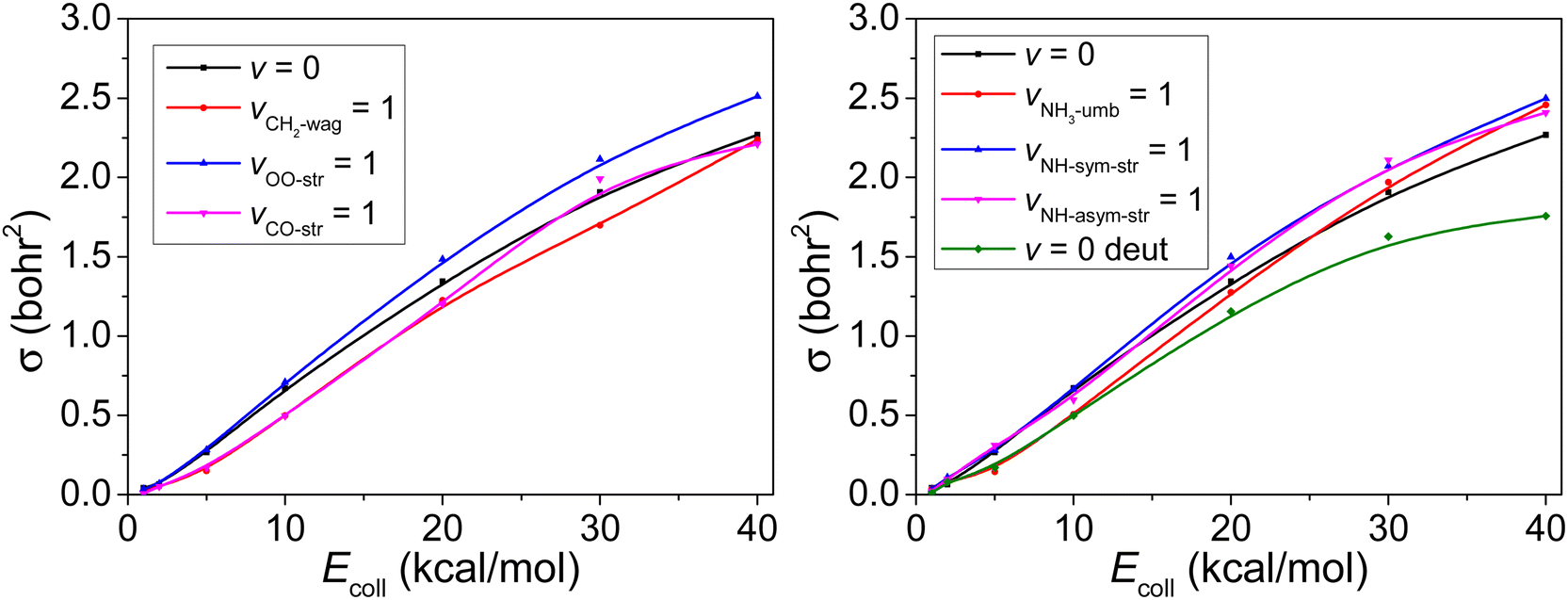 | ||
| Fig. 6 Integral cross sections as a function of the collision energy for the CH2OO (vx = 1) [x = CH2-wag, OO-str, CO-str] + NH3 (v = 0) (left panel) and CH2OO (v = 0) + NH3 (vy = 1) [y = NH3-umb, NH-sym-str, NH-asym-str] (right panel) reactions. The ground-state reaction case (v = 0) is shown in both panels for comparison. In addition, cross sections of the CH2OO (v = 0) + ND3 (v = 0) reaction are shown in the right panel, denoted as v = 0 deut. | ||
The deuterium effect by changing NH3 to ND3 is also shown in Fig. 6. Despite its usefulness, due to its classical nature, the QCT method does not treat tunneling effect and only the mass is different for H and D, as well as the corresponding ZPEs. As expected the reactivity of ND3 is weaker (about 20%) compared to NH3.
Additional information is provided by the sudden vector projection (SVP) model developed by Jiang and Guo.47,48 The idea beyond this model is the evaluation of the overlaps of the vibrational modes of the reactants with the reaction coordinate (imaginary mode) at TS structure. This is accomplished by calculating SVP values using the CCSD(T)/cc-pVDZ optimized geometries and the corresponding normal-mode vectors of the TS while the two reactants are placed far. The results are listed in Table 2.
| Normal mode | Energy (kcal mol−1) | SVP value | QCT |
|---|---|---|---|
| CH2 wag | 2.17 | 0.247 | −10% |
| OO-str | 2.77 | 0.084 | +10% |
| CO-str | 3.62 | 0.007 | −7% |
| NH3-umb | 3.39 | 0.113 | −1% |
| NH-sym-str | 9.81 | 0.016 | +9% |
| NH-asym-str | 10.17 | 0.031 | +6% |
| Translation | 0.818 |
As expect only the translational enhancement is consistent with the prediction of the SVP model, since the imaginary frequency of the TS in the CH2OO + NH3 → NH2CH2OOH reaction is only 288i cm−1, which could not represent the whole process of reaction very well. Comparing the SVP values and the QCT results we can see that indeed the SVP model failed to predict the vibrational enhancement in this reaction. And only a minor mode-specificity (within 10% based on the QCT results) is seen for the selected vibrational mode excitations.
IV. Conclusions
A full-dimensional PES for the CH2OO + NH3 → NH2CH2OOH reaction under the framework of the ROBOSURFER program package is developed and its dynamics are presented in detail by performing QCT simulations. The vdW complex, the TS, and the three 1D scans obtained on the PES agree well with the ab initio results. QCT simulations on this PES in a wide range of Ecoll are performed and no threshold energy is found for the CH2OO + NH3 → NH2CH2OOH reaction, which is expected due to the submerged-barrier, exothermic nature of the system. What unexpected is that the reaction probability increases with increasing collision energy for this negative-barrier reaction. As for the vibrational mode-specificity, a slight enhancement occurs for vOO-str = 1 and vNH-str = 1 with both the symmetric and asymmetric case, but a slight inhibition effect for vCO-str = 1 and vCH2-wag = 1 is observed. Interestingly, the excitation of the umbrella mode of NH3 has a negative effect to the reactivity at low Ecoll while enhances the reactivity at high Ecoll. The deuterium effect is also investigated by changing NH3 to ND3 and, as expected, the reactivity of ND3 is weaker compared to NH3. Only the translational enhancement is consistent with the prediction of the SVP model. The QCT calculations provide useful information on the microscopic mechanism of the reaction. At very low collision energies sometimes the two reactants rotate with each other, and after a few rounds they separate and no reaction occurs.Data availability
The data that support the findings of this study are available from the corresponding authors upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Tibor Győri and Viktor Tajti for the tips and discussions. This work was supported by the National Research, Development and Innovation Office–NKFIH, K-125317; Project no. TKP2021-NVA-19, provided by the Ministry of Innovation and Technology of Hungary from the National Research, Development and Innovation Fund, financed under the TKP2021-NVA funding scheme; and the Momentum (Lendület) Program of the Hungarian Academy of Sciences.References
- A. M. Green, V. P. Barber, Y. Fang, S. J. Klippenstein and M. I. Lester, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 12372 CrossRef CAS PubMed.
- C. Yin and K. Takahashi, Phys. Chem. Chem. Phys., 2017, 19, 12075 RSC.
- B. Long, J. L. Bao and D. G. Truhlar, Nat. Commun., 2019, 10, 2003 CrossRef PubMed.
- X. Zhou, Y. Liu, W. Dong and X. Yang, J. Phys. Chem. Lett., 2019, 10, 4817 CrossRef CAS PubMed.
- C. Robinson, L. Onel, J. Newman, R. Lade, K. Au, L. Sheps, D. E. Heard, P. W. Seakins, M. A. Blitz and D. Stone, J. Phys. Chem. A, 2022, 126, 6984 CrossRef CAS PubMed.
- C. Yin and K. Takahashi, Phys. Chem. Chem. Phys., 2018, 20, 20217 RSC.
- Y. H. Lin, C. Yin, K. Takahashi and J. J.-M. Lin, Commun. Chem., 2021, 4, 12 CrossRef CAS PubMed.
- C. M. Stangl, J. M. Krasnomowitz, M. J. Apsokardu, L. Tiszenkel, Q. Ouyang, S. Lee and M. V. Johnston, J. Geophys. Res.: Atmos., 2019, 124, 4800 CrossRef CAS.
- M. Yang, S. S. Ma, H. Ashraf, S. F. Pang and Y. H. Zhang, Atmos. Environ., 2020, 231, 117560 CrossRef CAS.
- O. Welz, J. D. Savee, D. L. Osborn, S. S. Vasu, C. J. Percival, D. E. Shallcross and C. A. Taatjes, Science, 2012, 335, 204 CrossRef CAS PubMed.
- C. A. Taatjes, O. Welz, A. J. Eskola, J. D. Savee, A. M. Scheer, D. E. Shallcross, B. Rotavera, E. P. F. Lee, J. M. Dyke, D. K. W. Mok, D. L. Osborn and C. J. Percival, Science, 2013, 340, 177 CrossRef CAS PubMed.
- J. P. Misiewicz, S. N. Elliott, K. B. Moore and H. F. Schaefer, Phys. Chem. Chem. Phys., 2018, 20, 7479 RSC.
- Y. Liu, C. Yin, M. C. Smith, S. Liu, M. Chen, X. Zhou, C. Xiao, D. Dai, J. J.-M. Lin, K. Takahashi, W. Dong and X. Yang, Phys. Chem. Chem. Phys., 2018, 20, 29669 RSC.
- R. Chhantyal-Pun, R. J. Shannon, D. P. Tew, R. L. Caravan, M. Duchi, C. Wong, A. Ingham, C. Feldman, M. R. McGillen and M. A. H. Khan, et al. , Phys. Chem. Chem. Phys., 2019, 21, 14042 RSC.
- W. Chao, C. Yin, K. Takahashi and J. J.-M. Lin, Phys. Chem. Chem. Phys., 2019, 21, 22589 RSC.
- W. Chao, C. Yin, Y. L. Li, K. Takahashi and J. J.-M. Lin, J. Phys. Chem. A, 2019, 123, 1337 CrossRef CAS PubMed.
- A. I. Adjieufack, M. M. Bake, C. N. Nguimkeu, J. Pilmé and I. M. Ndassa, Chem. Phys. Chem., 2021, 22, 1792 CrossRef CAS PubMed.
- A. Nazari and V. Saheb, Theor. Chem. Acc., 2022, 141, 66 Search PubMed.
- Y.-H. Lin, C. Yin, W.-H. Lin, Y.-L. Li, K. Takahashi and J. J.-M. Lin, J. Phys. Chem. Lett., 2018, 9, 7040 Search PubMed.
- W. Chao, Y.-H. Lin, C. Yin, W.-H. Lin, K. Takahashi and J. J.-M. Lin, Phys. Chem. Chem. Phys., 2019, 21, 13633 RSC.
- B. Tang and Z. Li, J. Phys. Chem. A, 2020, 124, 8585 CrossRef CAS PubMed.
- O. Welz, A. J. Eskola, L. Sheps, B. Rotavera, J. D. Savee, A. M. Scheer, D. L. Osborn, D. Lowe, A. M. Booth and P. Xiao, et al. , Angew. Chem., Int. Ed., 2014, 53, 4547 CrossRef CAS PubMed.
- E. S. Foreman, K. M. Kapnas and C. Murray, Angew. Chem., Int. Ed., 2016, 55, 10419 CrossRef CAS PubMed.
- A. Kumar, S. Mallick and P. Kumar, Phys. Chem. Chem. Phys., 2022, 24, 7458 RSC.
- V. J. Esposito, T. A. McHenry and M. I. Lester, Chem. Phys. Lett., 2022, 809, 140179 CrossRef CAS.
- C. A. Chung, C. W. Hsu and Y. P. Lee, J. Phys. Chem. A, 2022, 126, 5738 CrossRef CAS PubMed.
- R. Reynolds, M. Ahmed and K. R. Wilson, ACS Earth Space Chem., 2023, 7, 901 CrossRef CAS.
- P. L. Luo, Phys. Chem. Chem. Phys., 2023, 25, 4062 RSC.
- B. H. Baek, V. P. Aneja and Q. Tong, Environ. Pollut., 2004, 129, 89 CrossRef CAS PubMed.
- J. L. Hand, B. A. Schichtel, M. Pitchford, W. C. Malm and N. H. Frank, J. Geophys. Res.: Atmos., 2012, 117, D05209 Search PubMed.
- S. M. Murphy, A. Sorooshian, J. H. Kroll, N. L. Ng, P. Chhabra, C. Tong, J. D. Surratt, E. Knipping, R. C. Flagan and J. H. Seinfeld, Atmos. Chem. Phys., 2007, 7, 2313 CrossRef CAS.
- X. Ge, A. S. Wexler and S. L. Clegg, Atmos. Environ., 2011, 45, 524 CrossRef CAS.
- H. Wu, Y. Fu, W. Dong, B. Fu and D. H. Zhang, RSC Adv., 2023, 13, 13397 RSC.
- C. Yin, V. Tajti and G. Czakó, Phys. Chem. Chem. Phys., 2022, 24, 24784 RSC.
- C. Yin and G. Czakó, Phys. Chem. Chem. Phys., 2022, 24, 29084 RSC.
- T. Győri and G. Czakó, J. Chem. Phys., 2022, 156, 071101 CrossRef PubMed.
- C. Møller and M. S. Plesset, Phys. Rev., 1934, 46, 618 CrossRef.
- T. H. Dunning Jr., J. Chem. Phys., 1989, 90, 1007 CrossRef.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby, M. Schütz, et al., Molpro, version 2015.1, a package of ab initio programs, see https://www.molpro.net Search PubMed.
- Z. Xie and J. M. Bowman, J. Chem. Theory Comput., 2010, 6, 26 CrossRef CAS PubMed.
- T. Győri and G. Czakó, J. Chem. Theory Comput., 2020, 16, 51 CrossRef PubMed.
- W. L. Hase, Encyclopedia of Computational Chemistry, Wiley, New York, 1998, pp. 399–407 Search PubMed.
- T. J. Lee and P. R. Taylor, Int. J. Quantum Chem., 1989, 36, 199 CrossRef.
- Y. Liu and J. Li, ACS Omega, 2020, 5, 23343 CrossRef CAS PubMed.
- C. Yin and G. Czakó, Phys. Chem. Chem. Phys., 2023, 25, 3083 RSC.
- C. Yin and G. Czakó, Phys. Chem. Chem. Phys., 2023, 25, 9944 RSC.
- B. Jiang and H. Guo, J. Chem. Phys., 2013, 138, 234104 CrossRef PubMed.
- H. Guo and B. Jiang, Acc. Chem. Res., 2014, 47, 3679 CrossRef CAS PubMed.
| This journal is © the Owner Societies 2023 |