
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bounding [AnO2]2+ (An = U, Np) covalency by simulated O K-edge and An M-edge X-ray absorption near-edge spectroscopy†
Kurtis
Stanistreet-Welsh
 and
Andrew
Kerridge
*
and
Andrew
Kerridge
*
Department of Chemistry, Lancaster University, Lancaster, LA1 4YB, UK. E-mail: a.kerridge@lancaster.ac.uk
First published on 14th August 2023
Abstract
Restricted active space simulations are shown to accurately reproduce and characterise both O K-edge and U M4,5-edge spectra of uranyl in excellent agreement with experimental peak positions and are extended to the Np analogue. Analysis of bonding orbital composition in the ground and O K-edge core-excited states demonstrates that metal contribution is underestimated in the latter. In contrast, An M4/5-edge core-excited states produce bonding orbital compositions significantly more representative of those in the ground state. Quantum Theory of Atoms in Molecules analysis is employed to explain the discrepancy between K- and M-edge data and demonstrates that the location of the core–hole impacts the pattern of electron localisation in core-excited states. An apparent contradiction to this behaviour in neptunyl is rationalised in terms interelectronic repulsion between the unpaired 5f electron and the excited core–electron.
Introduction
Understanding actinide–ligand covalency is critical to a range of applications in, for example, the separation of spent nuclear fuels,1,2 catalysis,3,4 and magnetic materials.5,6 In particular, the design of new ligands used in the separation of minor actinides (An) from lanthanides (Ln) in high level nuclear waste takes advantage of relatively enhanced covalency in An–ligand bonding compared to Ln.2 While actinide covalency has been observed in a variety of complexes, the relative contribution from the actinide 6d- and 5f-orbitals is an active area of investigation, while the 6p, 7s and 7p shells may also play a significant role.7–17 From a theoretical perspective, covalency can be considered in terms of perturbation theory as a deviation from the ionic bonding limit.8,14,18 In a molecular orbital picture of electronic structure, the mixing of metal ϕM and ligand ϕL orbitals in a bonding molecular orbital φ can be expressed as: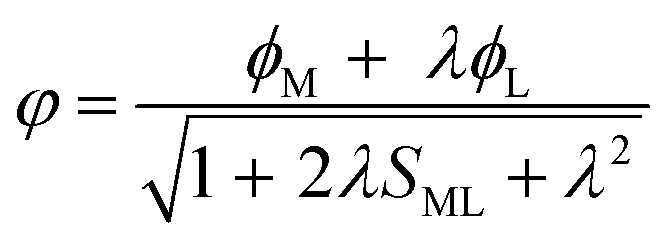 | (1) |
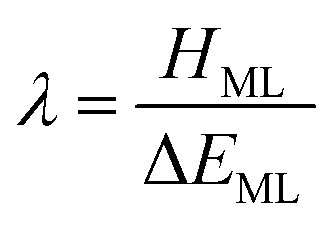 | (2) |
X-ray absorption near-edge spectroscopy (XANES) has, when combined with theoretical approaches, emerged as a powerful tool for probing metal–ligand covalency in a variety of f-element complexes. For example, in 2018, Su et al.,9 utilized Cl K-edge XANES in combination with density functional theory (DFT) simulations to determine the relative roles of An 5f- and 6d-orbital contributions to bonding in actinide(IV) hexachlorides, finding greater 6d-orbital contributions overall but increasing 5f-orbital participation when moving across the series due to increased energy-degeneracy driven covalency from Th to Pu. This study and others highlight the versatility of this technique for probing ground-state covalency.7,9–11,13,15,20–26
The oxygen K-edge XANES spectrum of uranyl was reported in 2002,15 and more recently, U and Np M4/5-edge XANES have been reported.11 In O K-edge XANES, pre-edge peaks result from dipole-allowed core-excitations from O 1s orbitals into valence anti-bonding orbitals. Intensity is driven by the degree of O 2p contribution within the latter, which by orthogonality arguments allows determination of the O 2p contribution to the bonding orbitals.15,23,24,26,27 The same principle applies to actinide M4/5-edge XANES, except here core-excitations occur from An 3d orbitals into empty valence orbitals with 5f character.11,22,23,25 Interpretation is based on the assumption that bonding orbital compositions probed in XANES core-excited states (CESs) are representative of those in the ground-state (GS). This may not be the case: GS and CES bonding orbitals could differ due to orbital relaxation in response to the core–hole and varying degrees of multiconfigurational character between the states, reflecting the differing electron correlation.
XANES simulations are challenging due to the nature of f-element systems: with strong electron correlation, pronounced relativistic effects and substantial multiconfigurational character, there is a need for robust theoretical approaches. Recently, Autschbach and co-workers have established the use of multiconfigurational restricted active space (RAS) methods in the successful simulation of XANES for a variety of f-element systems.20–24 Polly et al., has also shown RASSCF simulations to be successful in simulating uranyl 3d4f RIXS, further demonstrating the versatility of RAS approaches.25 A key advantage of RAS simulations is that they facilitate the treatment of multiconfigurational states and, critically, allow for orbital relaxation in the presence of the core–hole. Including orbital relaxation allows the key assumption in the application of XANES data to quantitatively determine ground state bonding properties, namely that the GS and CES electronic structures are sufficiently similar, to be assessed. Autschbach and co-workers have addressed this assumption using a RAS approach to simulate An M4/5-edge XANES for actinyl systems.22 This study showed metal contributions to π-bonds in CESs of actinyls are comparable to those of the GS. However, the need for some caution was indicated, as the metal contribution to the σ-bonds of neptunyl and plutonyl were found to be underestimated in comparison to the GS.
To date, a comprehensive RASSCF analysis of uranyl O K-edge XANES has not been reported, surprising given uranyl's ubiquity in hexavalent uranium chemistry. In this contribution, RASSCF XANES simulations for O K-edge and An M4/5-edge for both [UO2]2+ and [NpO2]2+ are presented and characterised. To the authors’ best knowledge, the O K-edge XANES spectrum for [NpO2]2+ is yet to be reported experimentally. We aim to determine the degree to which uranyl and neptunyl CES bonding orbitals are comparable to the GS when probed through these two complimentary XANES techniques. Interpretation is further supported by the application of Quantum Theory of Atoms in Molecules (QTAIM) analysis,28,29 which our group has previously used to examine both ground8,30,31 and valence excited states of uranyl.32 In this study, we make use of QTAIM to examine how covalency differs between the GS and CESs for both systems when probed through the two XANES techniques, providing a rationalisation of the reported orbital compositions.
Computational details
Theoretical XANES was simulated using spin–orbit coupled (SOC) restricted active-space self-consistent field (RASSCF) theory calculations in version 21.02 of OpenMolcas.33–36 Free actinyl systems have been shown in previous studies to be an appropriate model for simulating An M4/5-edge XANES, and more recently Misael et al., has shown that free uranyl is appropriate for simulating O K-edge XANES using equation of motion coupled cluster simulations.37 Free actinyl systems were therefore used here, exploiting the symmetry of the highest abelian point-group of D2h with the molecular axis orientation in the z-direction. For O K-edge XANES, [UO2]2+ and [NpO2]2+ bond lengths were set to experimental values at the greatest number of reported digits of 1.770 Å and 1.775 Å respectively.15,38,39 Experimental O K-edge XANES was performed on a Cs2UO2Cl4 crystal by Denning et al., and so an analogous crystal structure of Cs2NpO2Cl4 was chosen from the literature to inform the bond length for our neptunyl O K-edge simulation. Experimental An M4/5-edge XANES reported by Vitova et al.,11 was performed on [AnO2(H2O)5]2+ systems. For our simulations, the bond lengths were set to those obtained from extended X-ray absorption fine structure (EXAFS) measurements at 1.76 Å and 1.75 Å for U-O and Np-O respectively.40 Scalar relativistic effects were modelled using the second order Douglas–Kroll–Hess Hamiltonian, and Cholesky decomposition was utilized throughout in the calculation of two-electron integrals.41–45 All simulations employed the all-electron ANO-RCC basis sets of Roos, et al., at the TZP level of quality but with higher angular momentum An h-functions removed; U (9s8p6d4f2g), Np (9s8p6d4f2g), O (4s3p2d1f).46–48 RASSCF calculations were used to obtain the required number of spin-free ground-states and core-excited states (see Tables S1–S7 of (ESI†)). In RASSCF simulations, the active space is partitioned into three subspaces labelled RAS1/2/3 as shown in Fig. 1. In our XANES simulations, RAS1 comprised the core-orbitals, RAS2 the occupied bonding orbitals, and RAS3 contained both the formally non-bonding 5f orbitals and anti-bonding orbitals. Non-bonding 5f orbitals were placed within the RAS3 space to manage the number of possible configurations generated, making calculations more tractable. CESs were generated by allowing up to 1 or 2 additional electrons to occupy RAS3, and enforcing a single core–hole across RAS1: hereafter, we refer to these simulations, RASSCF(16,1,1;2,6,10) as RAS(S), and RASSCF(16,1,2;2,6,10) as RAS(SD), respectively. For O K-edge XANES, RAS1 comprised the linear combination of oxygen 1s orbitals, and for An M4/5-edge XANES comprised the set of five 3d metal orbitals. Uranyl singlet and triplet CES RAS(SD) wavefunctions were generated by allowing up to 2 electrons to populate RAS3, while neptunyl doublet and quartet RAS(SD) wavefunctions were generated by allowing up to 3 electrons, which accounts for the single unpaired 5f-electron already populating RAS3. Supersymmetry designations ensured that core-orbitals do not rotate out of RAS1 during the SCF procedure. RAS(S) and RAS(SD) GSs were obtained by removing the RAS1 core–hole constraint, which led to wavefunctions that are compatible for state-interaction with CESs, but with a fully occupied RAS1 subspace and RAS3 populations that reflect the expected GSs for these systems. RASSCF calculations produced a closed-shell singlet GS for [UO2]2+ while for [NpO2]2+ a set of four doublet states, spanning the odd-parity irreps, were generated due to the single unpaired electron that distributes across the non-bonding 5f orbitals. Scalar relativistic states were then obtained by taking the appropriate level of state-averaging (Tables S1–S7, ESI†) for both RAS(S) and RAS(SD) wavefunctions of different spin-multiplicities and irreducible representations. RAS(S) simulations were used to rationalise the peak assignments, results of which are included in the ESI,† while RAS(SD) results are presented within the main text as these are expected to be of higher quality. RAS(SD) wavefunctions capture the possible core-excitations between RAS1 and RAS3, but also a degree of correlation between the bonding and antibonding orbitals spanning RAS2 and RAS3. Dynamical correlation was included via multiconfigurational 2nd order perturbation theory (RASPT2),49–51 with an IPEA shift52 of 0.25 a.u. and imaginary shift53 0.5 a.u., which was found to offer a reasonable balance between converging intruder free solutions without significantly shifting the resulting RASPT2 state energies. These values are in line with related studies.21,22,24,25 The appropriate GS and CESs for each system were spin–orbit coupled post hoc via state interaction of the scalar relativistic states with a mean-field spin–orbit operator, making use of atomic mean-field integrals (AMFI), in the Restricted Active Space State Interaction (RASSI) formalism.45,54 Diagonal energies of the Hamiltonian matrix computed by RASSI were replaced by the calculated RASPT2 energies. Application of spin–orbit coupling to neptunyl, produced a degenerate ground state comprised of a linear combination of the four spin-free doublets that span the odd-parity irreps (ESI,† Section S1.2), while the spin–orbit coupled uranyl GS was comprised of only the closed shell 1Ag state. RASSI calculations provided spin–orbit coupled state energies and GS → CES electric-dipole transition oscillator strengths enabling transition sticks to be plotted, which were broadened using Lorentzian functions to generate the overall theoretical XANES curves. RASSI calculations also provided spin–orbit natural orbital (SONO) coefficients along with their natural populations and converted to MOLDEN format using a custom Molcas2Molden package which has been made freely available (see ref. 58).22,55–57 SONOs were utilized to perform peak assignments by inspection of the corresponding natural populations. Subsequent analysis was enabled using Molden2AIM for file conversion.58 Using SONOs to represent the density, Quantum Theory of Atoms in Molecules (QTAIM) analysis,28,29 along with AIM orbital composition determination were performed using version 19.02.13 of AIMALL59 and version 3.8 of Multiwfn60 respectively. Additional details are provided in the ESI.†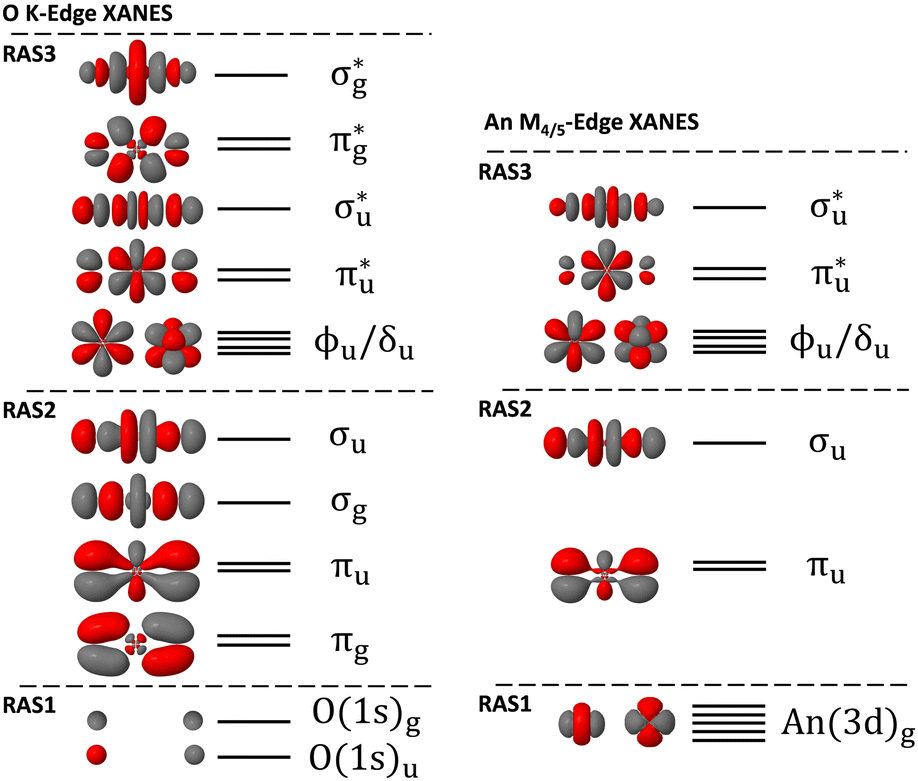 | ||
| Fig. 1 Diagram showing the active-spaces used for XANES RAS(SD) simulations of O K-edge (left) and An M4/5-edge (right). Orbitals are labelled in terms of the higher D∞h symmetry. Pseudonatural orbitals taken from GS uranyl RAS(SD) simulations and energy levels are qualitative in nature. | ||
Results and discussion
[AnO2]2+ O K-edge XANES
Three main peaks positioned at 531.4 eV, 534.1 eV, and 536.5 eV are reported in the experimental O K-edge XANES of uranyl by Denning et al.,15 assigned as core-excitations into the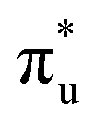 ,
, 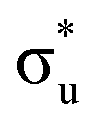 , and
, and 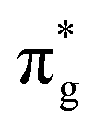 orbitals, respectively. Our simulations (Fig. 1) predict these peaks with exceptional accuracy, at unshifted values of 531.7 eV (Δ = +0.3 eV), 535.6 eV (Δ = +1.5 eV), and 537.1 eV (Δ = +0.6 eV), and can be assigned to the same core-excitations as those proposed by Denning. Analogous peaks are predicted for neptunyl at 531.5 eV, 534.8 eV, and 538.2 eV. The experimental XANES of uranyl also shows a possible shoulder on the first peak at 530.0 eV and a feature at 551.0 eV. These features are also present in our RAS(SD) simulations (Table S8, ESI†), occurring at 529.7 eV (Δ = −0.3 eV) and 551.7 eV (Δ = +0.7 eV), and assigned to core-excitations into the non-bonding 5f orbitals and
orbitals, respectively. Our simulations (Fig. 1) predict these peaks with exceptional accuracy, at unshifted values of 531.7 eV (Δ = +0.3 eV), 535.6 eV (Δ = +1.5 eV), and 537.1 eV (Δ = +0.6 eV), and can be assigned to the same core-excitations as those proposed by Denning. Analogous peaks are predicted for neptunyl at 531.5 eV, 534.8 eV, and 538.2 eV. The experimental XANES of uranyl also shows a possible shoulder on the first peak at 530.0 eV and a feature at 551.0 eV. These features are also present in our RAS(SD) simulations (Table S8, ESI†), occurring at 529.7 eV (Δ = −0.3 eV) and 551.7 eV (Δ = +0.7 eV), and assigned to core-excitations into the non-bonding 5f orbitals and 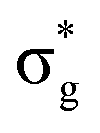 orbital, respectively. The predicted neptunyl O K-edge spectrum also contains a low energy shoulder at 529.5 eV, again attributed to excitation into non-bonding 5f orbitals. Fig. 2 presents the actinyl RAS(SD) simulated O K-edge spectra, shifted by −0.3 eV to align both spectra with the first experimental uranyl peak. Given that the excited electron originates from the core 1s-orbitals, and RAS2 bonding orbitals remain strongly occupied, these peak assignments suggest an energetic ordering for the anti-bonding orbitals as 5f <
orbital, respectively. The predicted neptunyl O K-edge spectrum also contains a low energy shoulder at 529.5 eV, again attributed to excitation into non-bonding 5f orbitals. Fig. 2 presents the actinyl RAS(SD) simulated O K-edge spectra, shifted by −0.3 eV to align both spectra with the first experimental uranyl peak. Given that the excited electron originates from the core 1s-orbitals, and RAS2 bonding orbitals remain strongly occupied, these peak assignments suggest an energetic ordering for the anti-bonding orbitals as 5f < 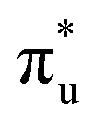 <
< 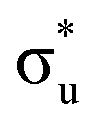 <
< 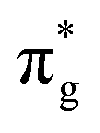 <
< 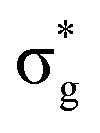 , directly aligning with that reported by Denning.12,15
, directly aligning with that reported by Denning.12,15
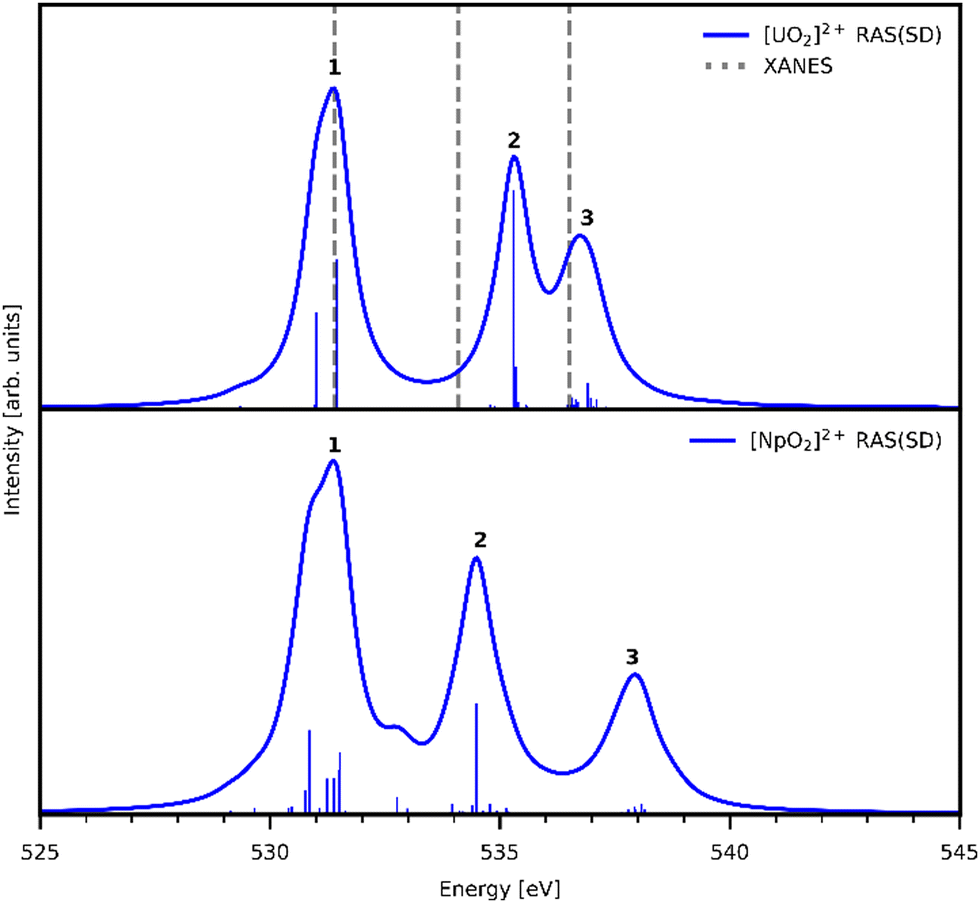 | ||
| Fig. 2 Simulated O K-edge XANES spectra for [UO2]2+ (top) and [NpO2]2+ (bottom). [UO2]2+ spectrum is compared to experimental peak positions (dashed lines15). Both theoretical actinyl spectra are shifted −0.3 eV to align with the first experimental peak position of [UO2]2+. Calculated spectra obtained with a 0.8 eV Lorentzian broadening of individual transitions. | ||
Table 1 reports the natural occupations of the RAS3 orbitals for key CESs used to characterise peaks 1–3 in Fig. 2. In the GS, the RAS3 orbitals can be considered unoccupied for [UO2]2+, whereas the single unpaired electron in [NpO2]2+ is represented by occupation across non-bonding 5f orbitals. For both O K-edge XANES simulations, CESs show increased multiconfigurational character (as indicated by fractional occupations of RAS3 orbitals) as the state energies increase from peaks 1 to 3 (Tables S20 and S21, ESI†). In the [UO2]2+ simulation, for example, peak 1 comprises a set of transitions (Fig. S9, ESI†) that correspond to clear core-excitations into the 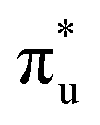 orbitals with limited multi-configurational character, of which the most intense transition has the natural orbital populations reported in Table 1. Peak 2 is comprised of a number of low intensity transitions with varied occupation of the non-bonding 5f orbitals along with
orbitals with limited multi-configurational character, of which the most intense transition has the natural orbital populations reported in Table 1. Peak 2 is comprised of a number of low intensity transitions with varied occupation of the non-bonding 5f orbitals along with 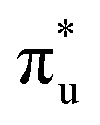 . It contains a dominant highly intense core-excitation into the
. It contains a dominant highly intense core-excitation into the 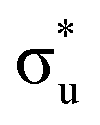 orbital with a natural population of 0.68, meaning that it can be attributed to this core-excitation alone. However, the set of four non-bonding 5f-orbitals, as well as the
orbital with a natural population of 0.68, meaning that it can be attributed to this core-excitation alone. However, the set of four non-bonding 5f-orbitals, as well as the 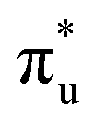 ,
, 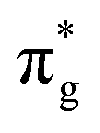 and
and 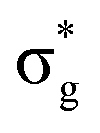 orbitals are also populated at 0.64, 0.16, 0.08 and 0.01 respectively, which alongside the 0.68 population of the
orbitals are also populated at 0.64, 0.16, 0.08 and 0.01 respectively, which alongside the 0.68 population of the 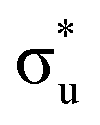 orbital, results in a total of 1.57 electrons occupying the RAS3 space in this CES. Of which, 0.57 can be attributed to electron redistribution from RAS2 bonding orbitals into the RAS3 orbitals to recover static correlation. The large density of transitions attributed to peak 3 continues this trend, with these multiconfigurational CESs further utilizing lower energy orbitals to optimise the electronic structure. Similar redistribution has been found in related studies.20,23,24 As such, despite the low population level of the
orbital, results in a total of 1.57 electrons occupying the RAS3 space in this CES. Of which, 0.57 can be attributed to electron redistribution from RAS2 bonding orbitals into the RAS3 orbitals to recover static correlation. The large density of transitions attributed to peak 3 continues this trend, with these multiconfigurational CESs further utilizing lower energy orbitals to optimise the electronic structure. Similar redistribution has been found in related studies.20,23,24 As such, despite the low population level of the 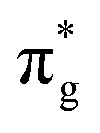 orbitals in CESs associated with peak 3, values are above those of all other states allowing characterisation as a
orbitals in CESs associated with peak 3, values are above those of all other states allowing characterisation as a 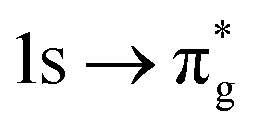 excitation. RAS(S) simulations reported in the ESI† re-enforce this interpretation, demonstrating their utility in enabling intuitive assignment. The [NpO2]2+ simulation exhibits the same characteristics, with a greater density of states across all peaks 1–3 due to the combined presence of the single 5f electron in RAS3 and configurations reflecting core-excitation. This high density of states renders ascribing the intensity of a peak to a single CES difficult. The issue is further compounded by the fact that, for peaks which can be attributed to a small number of intense core-excitations (e.g. peak 2 in [UO2]2+) the CESs themselves, as shown in Table 1, exhibit substantial partial occupation of all anti-bonding orbitals, obscuring the relationship between peak intensity and the degree of oxygen 2p orbital character in a specific anti-bonding orbital.
excitation. RAS(S) simulations reported in the ESI† re-enforce this interpretation, demonstrating their utility in enabling intuitive assignment. The [NpO2]2+ simulation exhibits the same characteristics, with a greater density of states across all peaks 1–3 due to the combined presence of the single 5f electron in RAS3 and configurations reflecting core-excitation. This high density of states renders ascribing the intensity of a peak to a single CES difficult. The issue is further compounded by the fact that, for peaks which can be attributed to a small number of intense core-excitations (e.g. peak 2 in [UO2]2+) the CESs themselves, as shown in Table 1, exhibit substantial partial occupation of all anti-bonding orbitals, obscuring the relationship between peak intensity and the degree of oxygen 2p orbital character in a specific anti-bonding orbital.
| System | Peak | 5f |
|
|
|
|
|---|---|---|---|---|---|---|
| [UO2]2+ O K-edge | 1 | 0.04 | 0.99 | 0.08 | 0.16 | 0.00 |
| 2 | 0.64 | 0.16 | 0.68 | 0.08 | 0.01 | |
| 3 | 1.02 | 0.54 | 0.05 | 0.26 | 0.00 | |
| [NpO2]2+ O K-edge | 1 | 1.48 | 0.62 | 0.08 | 0.10 | 0.02 |
| 2 | 1.32 | 0.40 | 0.72 | 0.06 | 0.03 | |
| 3 | 1.63 | 0.79 | 0.24 | 0.24 | 0.01 | |
| [UO2]2+ U M4-edge | 1 | 1.00 | 0.04 | 0.01 | — | — |
| 2 | 0.82 | 0.65 | 0.02 | — | — | |
| 3 | 1.11 | 0.74 | 0.10 | — | — | |
| [NpO2]2+ Np M5-edge | 1 | 1.94 | 0.42 | 0.02 | — | — |
| 2 | 1.79 | 1.01 | 0.12 | — | — | |
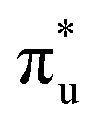
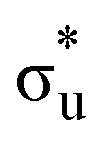
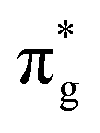
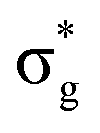
[AnO2]2+ M4/5-edge XANES
Uranyl metal M4-edge and neptunyl M5-edge XANES simulations, Fig. 3 and 4 respectively, are shown to be in good agreement with the experimental work of Vitova et al.,11 albeit after systematic shifting of 21.9 eV and 21.7 eV for uranyl and neptunyl, respectively. These shifts are consistent with two previous related studies.22,25 Simulations predict both the M4- and M5-edges with splittings of 172 eV and 179 eV for uranyl and neptunyl, respectively (see Fig. S11, ESI†), but discussion is restricted to the M4-edge of uranyl and M5-edge of neptunyl for which experimental data is reported. Fig. 3 shows that the U M4-edge simulation reflects the experimental peak separation with high accuracy, as well as the spectral profile. Our simulated spectrum shows relative intensity decreasing from peak 1 to 3 in line with observation. Three [UO2]2+ peaks were experimentally reported, positioned at 3726.7 eV, 3728.8 eV, and 3732.9 eV and assigned to core-excitations into the non-bonding 5f,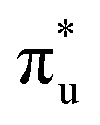 and
and 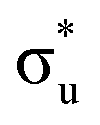 orbitals, respectively. The same peak assignments are also deduced from our simulations. Peaks 1 and 2 comprise several intense transitions with clear non-bonding 5f and
orbitals, respectively. The same peak assignments are also deduced from our simulations. Peaks 1 and 2 comprise several intense transitions with clear non-bonding 5f and 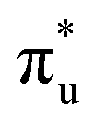 orbital occupations, respectively, while peak 3 has a larger density of low intensity transitions presenting
orbital occupations, respectively, while peak 3 has a larger density of low intensity transitions presenting 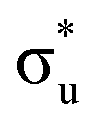 occupancy. This density of states also broadens the simulated peak 3 compared to experiment. As found in our O K-edge simulations, significant multiconfigurational character is apparent with partial occupation of lower energy orbitals utilized to optimize higher energy states. CESs attributed to peak 3 are a particularly apparent example of this, containing low levels of
occupancy. This density of states also broadens the simulated peak 3 compared to experiment. As found in our O K-edge simulations, significant multiconfigurational character is apparent with partial occupation of lower energy orbitals utilized to optimize higher energy states. CESs attributed to peak 3 are a particularly apparent example of this, containing low levels of 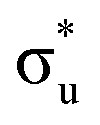 population but high levels of
population but high levels of 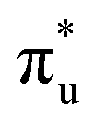 and non-bonding 5f orbital occupancy. The CES reported in Table 1 has a population of 0.10. However, this value is relatively high compared to background level occupancy of
and non-bonding 5f orbital occupancy. The CES reported in Table 1 has a population of 0.10. However, this value is relatively high compared to background level occupancy of 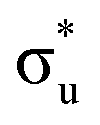 across all reported CESs in Table S22 (ESI†). This is in contrast to the RAS(S) simulation (Fig. S19, ESI†) in which peak 3 is comprised of three intense transitions with CESs having
across all reported CESs in Table S22 (ESI†). This is in contrast to the RAS(S) simulation (Fig. S19, ESI†) in which peak 3 is comprised of three intense transitions with CESs having 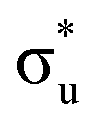 occupancies of above 0.99, and we note here how reducing the constraints on RASSCF wavefunctions in RAS(SD) simulations, while capturing increased static correlation through the inclusion of a greater range of electronic configurations, has an impact on the ability to intuitively characterize core-excitations in terms of orbitals transitions.
occupancies of above 0.99, and we note here how reducing the constraints on RASSCF wavefunctions in RAS(SD) simulations, while capturing increased static correlation through the inclusion of a greater range of electronic configurations, has an impact on the ability to intuitively characterize core-excitations in terms of orbitals transitions.
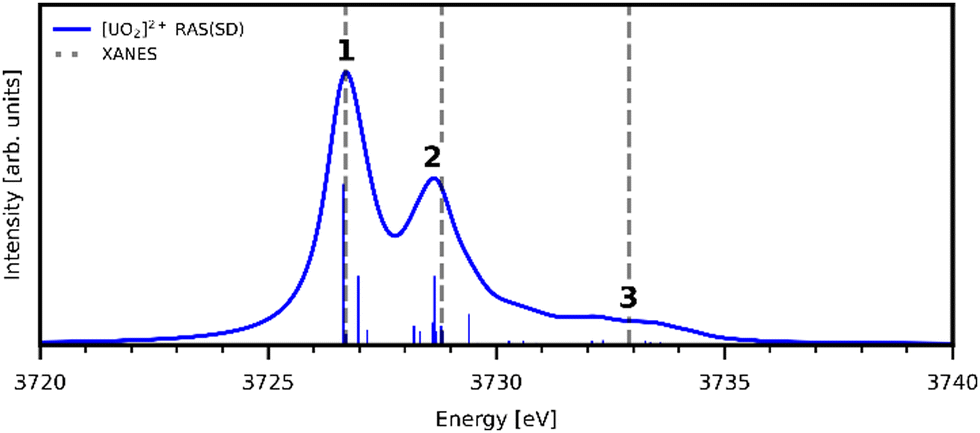 | ||
| Fig. 3 Simulated U M4-edge XANES for [UO2]2+ and experimental peak positions (dashed lines11). Theoretical spectrum is shifted to align with the first experimental peak position. Calculated spectra obtained with a 1.0 eV Lorentzian broadening of individual transitions. | ||
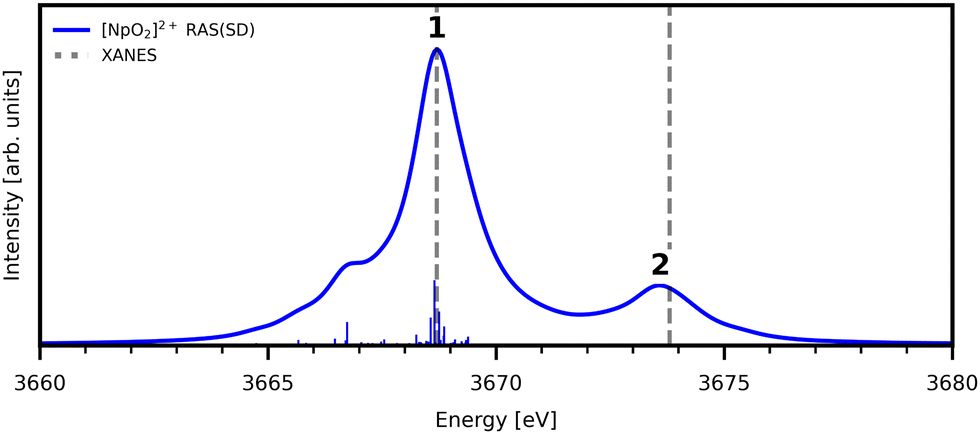 | ||
| Fig. 4 Simulated Np M5-edge XANES for [NpO2]2+ and experimental peak positions (dashed lines11). Theoretical spectrum is shifted to align with the first experimental peak position. Calculated spectra obtained with a 1.0 eV Lorentzian broadening of individual transitions. | ||
The experimental [NpO2]2+ M5-edge XANES presents a two-peak structure, with the first peak incorporating core-excitations into both the non-bonding 5f and 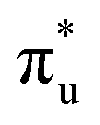 orbitals, and a resolved second peak corresponding to core-excitation into
orbitals, and a resolved second peak corresponding to core-excitation into 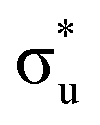 . A similar two peak profile is predicted by our simulations, Fig. 4, reflecting the experimental peak separations and the greater relative intensity of peak 1 as compared to peak 2. Peaks 1 and 2 are assigned to core-excitation into the
. A similar two peak profile is predicted by our simulations, Fig. 4, reflecting the experimental peak separations and the greater relative intensity of peak 1 as compared to peak 2. Peaks 1 and 2 are assigned to core-excitation into the 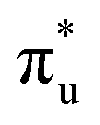 and
and 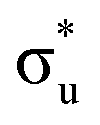 orbitals and a predicted shoulder at 3666.5 eV is assigned to core-excitations into the non-bonding 5f orbitals. The most intense transition responsible for the shoulder on peak 1 contains
orbitals and a predicted shoulder at 3666.5 eV is assigned to core-excitations into the non-bonding 5f orbitals. The most intense transition responsible for the shoulder on peak 1 contains 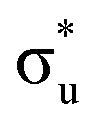 ,
, 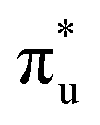 and non-bonding 5f orbital populations of 0.01, 0.22, and 1.86, respectively, showing clear excitation into the non-bonding 5f orbitals. The density of CESs increases from peak 1 to 2 along with the degree of multiconfigurational character. Peak 2 results from the broadening of a significantly large number of very low intensity core-excitations (see Fig. S16, ESI†) which are not visible on the scale presented in Fig. 4. These CESs are highly multiconfigurational but present levels of
and non-bonding 5f orbital populations of 0.01, 0.22, and 1.86, respectively, showing clear excitation into the non-bonding 5f orbitals. The density of CESs increases from peak 1 to 2 along with the degree of multiconfigurational character. Peak 2 results from the broadening of a significantly large number of very low intensity core-excitations (see Fig. S16, ESI†) which are not visible on the scale presented in Fig. 4. These CESs are highly multiconfigurational but present levels of 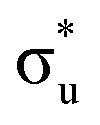 occupancy above those of other CESs, aligning with experimental assignment for this peak as well as RAS(S) assignments, with a population of 0.12 for the most intense transition out of a selection shown in Fig. S16 (ESI†).
occupancy above those of other CESs, aligning with experimental assignment for this peak as well as RAS(S) assignments, with a population of 0.12 for the most intense transition out of a selection shown in Fig. S16 (ESI†).
Bond characterisation
Peak intensities in XANES are related to the degree of oxygen-2p and actinide-5f character in anti-bonding orbitals for O K-edge and An M-edge respectively, which via orthogonality constraints, also allows bonding orbital contributions to be probed. For XANES to be a valid probe of GS covalency, it is a requirement that the GS and CES electronic structures do not differ significantly in their compositions. Here, we explore their similarity via QTAIM and SONO composition. Two QTAIM metrics have been extensively used to quantify covalency, the delocalisation index (δ) and the magnitude of the electron density at the bond critical point, ρBCP.1,8,31,32,61 The former quantifies the number of electrons shared between two atomic centres, while the latter quantifies charge accumulation in the bonding region through σ-interactions. For both O K-edge and An M-edge XANES simulations, ρBCP values for both [UO2]2+ and [NpO2]2+ range between 0.30 a.u. and 0.34 a.u. in the GS, and upon core-excitation these values remain relatively unchanged (Table S11, ESI†). This is unsurprising given that core-excitations do not involve depletion of electrons from the bonding orbitals, in contrast to valence excitations. This limited change in ρBCP between GS and CESs suggests electron density accumulation in the bonding region along the molecular axis is not significantly affected by core-excitation. However, the δ-values decrease (Table 2) for all CESs as expected when promoting core–electrons into anti-bonding orbitals, which acts to decrease the overall bond orders.| Simulation | Excitation | Δδ(An,O) | Δλ(An) | Δλ(O) |
|---|---|---|---|---|
| [UO2]2+ O K-edge |
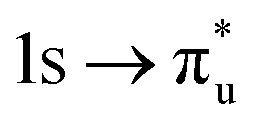
|
−0.55 | +0.71 | +0.20 |
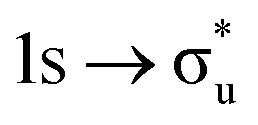
|
−0.62 | +0.83 | +0.22 | |
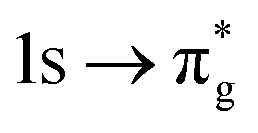
|
−0.68 | +0.99 | +0.21 | |
| [NpO2]2+ O K-edge |
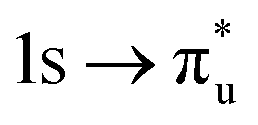
|
−0.47 | +0.60 | +0.17 |
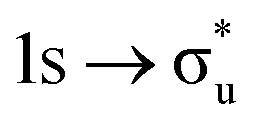
|
−0.58 | +0.77 | +0.21 | |
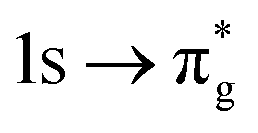
|
−0.72 | +0.96 | +0.26 | |
| [UO2]2+ M4-edge |
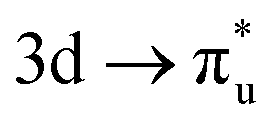
|
−0.19 | +0.37 | −0.01 |
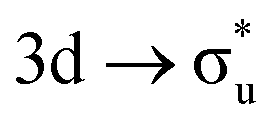
|
−0.37 | +0.61 | +0.07 | |
| [NpO2]2+ M5-edge |
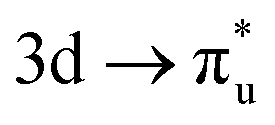
|
−0.14 | +0.38 | −0.08 |
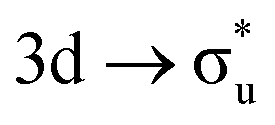
|
−0.42 | +0.54 | +0.16 | |
SONO compositions were determined to assess whether bonding orbitals differ in the GS and CESs, enabling the validity of XANES as a GS covalency probe to be examined. By including the bonding orbitals within the RAS2 space of RASSCF wavefunctions we ensure both orbital relaxation and correlation effects are accounted for. Table 3 quantifies the change in metal centre contributions to the GS and CES bonding SONOs for both O K-edge and An M-edge simulations. This analysis reveals that An contributions to O K-edge CES bonding SONOs are consistently underestimated, by between 7–10% and 1–13% for uranium and neptunium, respectively. For these CESs, increased electron localisation (Δλ) is identified on all centres (Table 2), ranging between +0.60 and +0.99 on the actinide and between +0.17 and +0.26 on the individual oxygen centres. Localisation onto oxygen centres can be attributed to the increased effective nuclear charge on oxygen due to the presence of the ligand core–hole. While increased electron localisation onto the metal centre can be attributed to the occupation of anti-bonding orbitals of predominantly metal character (Tables S16 and S18, ESI†) as well as significant partial occupation of non-bonding An 5f-orbitals. This is supported by the observation that for states which exhibit greater population of these orbitals, larger An electron localisation is found. For example, the O K-edge CESs assigned to the 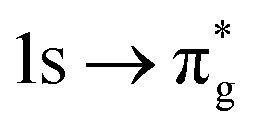 excitation (peak 3) for both actinyl systems, manifest the greatest multiconfigurational character and thus presents the largest Δλ(An) values of +0.99 and +0.96 for [UO2]2+ and [NpO2]2+, respectively. These multiconfigurational states also present the most significant decreases in the δ-values. Alongside the ligand core–hole, (which acts to withdraw electrons once shared in GS covalent bonds onto the ligand centre in CESs), significant redistribution of electrons from RAS2 bonding orbitals into RAS3 anti-bonding orbitals acts to reduce the number of electrons shared in bonding interactions, hence lowering δ-values.
excitation (peak 3) for both actinyl systems, manifest the greatest multiconfigurational character and thus presents the largest Δλ(An) values of +0.99 and +0.96 for [UO2]2+ and [NpO2]2+, respectively. These multiconfigurational states also present the most significant decreases in the δ-values. Alongside the ligand core–hole, (which acts to withdraw electrons once shared in GS covalent bonds onto the ligand centre in CESs), significant redistribution of electrons from RAS2 bonding orbitals into RAS3 anti-bonding orbitals acts to reduce the number of electrons shared in bonding interactions, hence lowering δ-values.
| Core-excitation | Bonding SONO | U | Np |
|---|---|---|---|
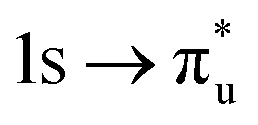
|
πu | −10% | −13% |
(25%  15%) 15%) |
(28%  15%) 15%) |
||
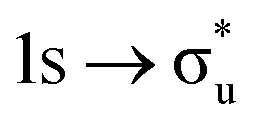
|
σu | −7% | −1% |
(53%  46%) 46%) |
(55%  54%) 54%) |
||
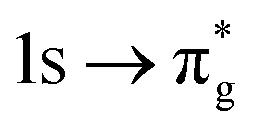
|
πg | −8% | −6% |
(17%  9%) 9%) |
(16%  10%) 10%) |
||
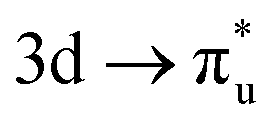
|
πu | +0% | +1% |
(26%  26%) 26%) |
(27%  28%) 28%) |
||
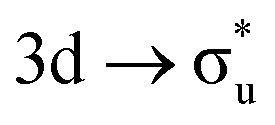
|
σu | +1% | −2% |
(56%  57%) 57%) |
(62%  60%) 60%) |
In contrast to O K-edge, SONO analysis (Table 3) shows An M-edge CESs comprise bonding orbital compositions significantly more representative of those in the GS. QTAIM analysis (Table 2) shows An M-edge CESs have increased electron localisation on the An centre only, with the exception of the 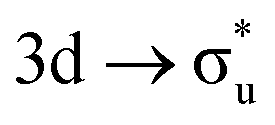 excitation in neptunyl, which we address separately below. In An M-edge CESs, the core–hole resides on the An centre and consequently the effective charge of the oxygen remains unchanged with oxygen localisation largely absent. Alongside the localising effect of the metal core–hole, increased electron localisation on An centres is also driven by the occupation of anti-bonding orbitals of predominantly metal character (Tables S17 and S19, ESI†) as well as significant partial occupation of non-bonding An 5f-orbitals. Analogous to the case in O K-edge, CESs manifesting greater multiconfigurational character coincide with greater decreases in δ-values compared to other M-edge states. This is highlighted by the U M-edge
excitation in neptunyl, which we address separately below. In An M-edge CESs, the core–hole resides on the An centre and consequently the effective charge of the oxygen remains unchanged with oxygen localisation largely absent. Alongside the localising effect of the metal core–hole, increased electron localisation on An centres is also driven by the occupation of anti-bonding orbitals of predominantly metal character (Tables S17 and S19, ESI†) as well as significant partial occupation of non-bonding An 5f-orbitals. Analogous to the case in O K-edge, CESs manifesting greater multiconfigurational character coincide with greater decreases in δ-values compared to other M-edge states. This is highlighted by the U M-edge 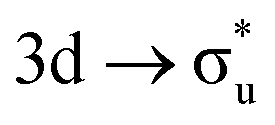 excitation, with the corresponding CES having a greater number of electrons distributed across RAS3 compared to the
excitation, with the corresponding CES having a greater number of electrons distributed across RAS3 compared to the 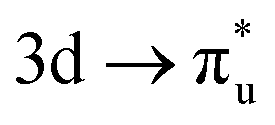 CES (peaks 2 and 3 in Table 1), which consequently have Δλ(An) and Δδ(An,O) values of +0.61 and −0.37 respectively, compared to +0.37 and −0.19 in the
CES (peaks 2 and 3 in Table 1), which consequently have Δλ(An) and Δδ(An,O) values of +0.61 and −0.37 respectively, compared to +0.37 and −0.19 in the 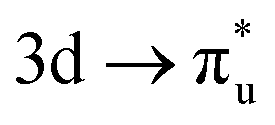 CES. For Np M-edge, the
CES. For Np M-edge, the 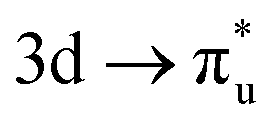 CES presents a localisation pattern similar to that of the U M-edge states, and the corresponding πu bonding SONO composition is comparable to the GS (Table 3). However, the Np M-edge
CES presents a localisation pattern similar to that of the U M-edge states, and the corresponding πu bonding SONO composition is comparable to the GS (Table 3). However, the Np M-edge 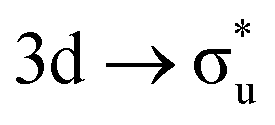 excitation is found to deviate from the localisation pattern of all other M-edge states. This CES reflects a localization pattern more akin to O K-edge states, and the SONO bonding orbital composition reflect this, with a 2% underestimation of the Np contribution to the σu bonding SONO compared to the GS.
excitation is found to deviate from the localisation pattern of all other M-edge states. This CES reflects a localization pattern more akin to O K-edge states, and the SONO bonding orbital composition reflect this, with a 2% underestimation of the Np contribution to the σu bonding SONO compared to the GS.
Overall, the QTAIM results suggest that the decrease in An contributions to bonding orbitals in O K-edge CESs as compared to the GS (Table 3) can be attributed to two contributing effects. First, the increased charge density on the An centre through occupation of an anti-bonding orbital of predominantly metal character, drives electron density in bonding orbitals away from the An centre. This effect is enhanced by the increased effective charge of the oxygen ligands due to the presence of the O 1s core–hole. These effects combine to produce the reported decrease in An contribution to bonding SONOs in the CESs as compared to the GS. In contrast, SONO analysis shows uranyl M-edge CESs comprise bonding orbital compositions significantly more representative of those in the GS. Here, any potential decrease in An contribution due to the occupation of an anti-bonding orbital, which again acts to drive electron density off the An centre in bonding orbitals, is counteracted by the core–hole being located on the An centre, increasing effective An charge and acting to retain electron density. These effects appear to largely cancel, leaving the bonding orbital compositions comparable to those in the GS. A previous study by Autschbach and coworkers also found the bonding orbital compositions for these U M-edge CESs to be comparable to those of the GS.22
For the 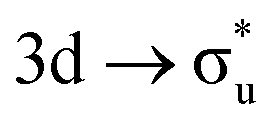 excitation in our neptunyl M-edge simulation, we find a decrease of 2% in the Np contribution which, although small, represents a deviation from the trend found for all other M-edge CESs. Whilst the same effects discussed for uranyl also manifest here, there is additional electrostatic repulsion between the unpaired 5f electron and the excited core–electron in
excitation in our neptunyl M-edge simulation, we find a decrease of 2% in the Np contribution which, although small, represents a deviation from the trend found for all other M-edge CESs. Whilst the same effects discussed for uranyl also manifest here, there is additional electrostatic repulsion between the unpaired 5f electron and the excited core–electron in 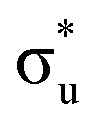 . This acts to drive electron density in the σu SONO off the metal centre and is substantial enough to alter the localisation pattern as seen in Table 2, with increased localisation onto the oxygen centres, in contrast to all other M-edge states. However, analogous repulsion does not appear to impact the localisation pattern of the
. This acts to drive electron density in the σu SONO off the metal centre and is substantial enough to alter the localisation pattern as seen in Table 2, with increased localisation onto the oxygen centres, in contrast to all other M-edge states. However, analogous repulsion does not appear to impact the localisation pattern of the 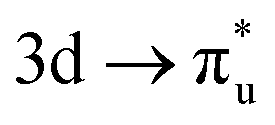 CES in neptunyl. This apparent contradiction can be understood by examination of the σu and πu SONO compositions. The σu SONO has a covalent composition in the GS, with dominant (62%) Np. The πu SONO, in contrast, has more ionic character, and is dominated by the (73%) oxygen contribution (Table S19, ESI†). This high oxygen contribution means the transfer of charge onto the oxygen centres due to the electrostatic interelectronic repulsion is energetically unfavorable, leaving the πu bonding SONO compositions comparable between the CES and GS, again with net localisation of electrons onto the An centre. Similar composition characteristics have also been reported by Autschbach and coworkers, who found a similar decrease in Np contribution to the σu bonding orbital in the CES.22
CES in neptunyl. This apparent contradiction can be understood by examination of the σu and πu SONO compositions. The σu SONO has a covalent composition in the GS, with dominant (62%) Np. The πu SONO, in contrast, has more ionic character, and is dominated by the (73%) oxygen contribution (Table S19, ESI†). This high oxygen contribution means the transfer of charge onto the oxygen centres due to the electrostatic interelectronic repulsion is energetically unfavorable, leaving the πu bonding SONO compositions comparable between the CES and GS, again with net localisation of electrons onto the An centre. Similar composition characteristics have also been reported by Autschbach and coworkers, who found a similar decrease in Np contribution to the σu bonding orbital in the CES.22
Overall, the SONO compositions suggest that the experimentally probed covalency of uranyl and neptunyl can be bounded with respect to the two complementary XANES techniques. Our simulations suggest that oxygen K-edge spectra serves to provide a lower bound for the metal contribution to the bonding orbitals, with uranium and neptunium contributions to the bonding SONOs being underestimated by up to 10% and 13% respectively. SONO compositions of An M-edge CESs suggest that the experimentally probed An M-edge XANEs states offer comparable orbital compositions between GS and CESs.
Conclusions
In summary, O K-edge XANES for [UO2]2+ and [NpO2]2+ has been simulated using RASSCF approaches for the first time. Simulated [UO2]2+ O K-edge XANES agree well with peak positions reported by Denning, confirming the same peak assignments.15 U M4- and Np M5-edge XANES have also been simulated and give good agreement with the experimental work of Vitova et al.,11 in terms of relative peak separations, intensities and assignments, suggesting that the predicted O K-edge spectrum of [NpO2]2+ is likely to be representative of the as-yet unreported experimental spectrum. RASSCF simulations reveal experimentally observed peaks to be comprised of multiple excitations, therefore ascribing the intensity to a single CES is not valid. For peaks which can be attributed to a small number of intense core-excitations, the CESs exhibit multiconfigurational character, obscuring the relationship between peak intensity and the degree of ligand character in a specific anti-bonding orbital. This work demonstrates the importance of considering the complementary data obtained from ligand K-edge and An M-edge XANES spectra in concert. XANES is regularly used to infer ground state bonding character and our analysis indicates that O K-edge XANES may underestimate the An contribution to GS bonding by up to 13%, serving as a lower bound. In contrast, An M-edge XANES provides electronic structures more consistent with the ground state, and theoretical considerations suggest that this may represent an upper bound. A qualitative rationalization for the reported SONO compositions is aided by QTAIM analysis of the core-states which reveal a reduction in δ-values across all CESs as expected when occupying anti-bonding orbitals, but different electron localisation patterns for O K-edge and An M-edge states. Two effects are thought to impact the reported orbital compositions: first, the increase of effective nuclear charge on the center in which the core–hole is localised acts to draw electron density in bonding orbitals to that center, and secondly, occupation of anti-bonding orbitals with predominant metal character acts to drive electron density away from the metal center in bonding orbitals. When the core–hole is localised on the ligand in O K-edge CESs, the two effects combine to produce the reported decrease in metal contribution. In An M-edge CES, the two contributing effects appear to largely cancel leaving bonding orbital compositions comparable to the GS. An exception to this trend is found in the apparently anomalous [NpO2]2+ M5-edge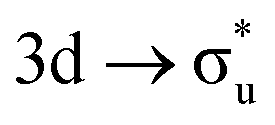 data, which can nonetheless be rationalised by consideration of interactions with the unpaired 5f electron, which results in an electron localisation pattern more comparable with the O K-edge CESs. The important role of the unpaired 5f electron in neptunyl indicates that future work should focus on further exploration of open-shell species as found throughout the actinide series.
data, which can nonetheless be rationalised by consideration of interactions with the unpaired 5f electron, which results in an electron localisation pattern more comparable with the O K-edge CESs. The important role of the unpaired 5f electron in neptunyl indicates that future work should focus on further exploration of open-shell species as found throughout the actinide series.
Conflicts of interest
There are no conflicts to declare.References
- N. Kaltsoyannis, Inorg. Chem., 2013, 52, 3407–3413 CrossRef CAS PubMed.
- H. H. Dam, D. N. Reinhoudt and W. Verboom, Chem. Soc. Rev., 2007, 36, 367–377 RSC.
- P. L. Arnold, M. W. McMullon, J. Rieb and F. E. Kühn, Angew. Chem., Int. Ed., 2015, 54, 82–100 CrossRef CAS PubMed.
- R. J. Batrice and M. S. Eisen, Chem. Sci., 2016, 7, 939–944 RSC.
- N. Magnani and R. Caciuffo, Inorganics, 2018, 6, 26, DOI:10.3390/inorganics6010026.
- K. R. Meihaus and J. R. Long, Dalton Trans., 2015, 44, 2517–2528 RSC.
- S. G. Minasian, J. M. Keith, E. R. Batista, K. S. Boland, D. L. Clark, S. A. Kozimor, R. L. Martin, D. K. Shuh and T. Tyliszczak, Chem. Sci., 2014, 5, 351–359 RSC.
- A. Kerridge, Chem. Commun., 2017, 53, 6685–6695 RSC.
- J. Su, E. R. Batista, K. S. Boland, S. E. Bone, J. A. Bradley, S. K. Cary, D. L. Clark, S. D. Conradson, A. S. Ditter, N. Kaltsoyannis, J. M. Keith, A. Kerridge, S. A. Kozimor, M. W. Löble, R. L. Martin, S. G. Minasian, V. Mocko, H. S. la Pierre, G. T. Seidler, D. K. Shuh, M. P. Wilkerson, L. E. Wolfsberg and P. Yang, J. Am. Chem. Soc., 2018, 140, 17977–17984 CrossRef CAS PubMed.
- S. A. Kozimor, P. Yang, E. R. Batista, K. S. Boland, C. J. Burns, D. L. Clark, S. D. Conradson, R. L. Martin, M. P. Wilkerson and L. E. Wolfsberg, J. Am. Chem. Soc., 2009, 131, 12125–12136 CrossRef CAS PubMed.
- T. Vitova, I. Pidchenko, D. Fellhauer, P. S. Bagus, Y. Joly, T. Pruessmann, S. Bahl, E. Gonzalez-Robles, J. Rothe, M. Altmaier, M. A. Denecke and H. Geckeis, Nat. Commun., 2017, 8, 16053 CrossRef CAS PubMed.
- R. G. Denning, J. Phys. Chem. A, 2007, 111, 4125–4143 CrossRef CAS PubMed.
- S. G. Minasian, J. M. Keith, E. R. Batista, K. S. Boland, D. L. Clark, S. D. Conradson, S. A. Kozimor, R. L. Martin, D. E. Schwarz, D. K. Shuh, G. L. Wagner, M. P. Wilkerson, L. E. Wolfsberg and P. Yang, J. Am. Chem. Soc., 2012, 134, 5586–5597 CrossRef CAS PubMed.
- M. L. Neidig, D. L. Clark and R. L. Martin, Coord. Chem. Rev., 2013, 257, 394–406 CrossRef CAS.
- R. G. Denning, J. C. Green, T. E. Hutchings, C. Dallera, A. Tagliaferri, K. Giarda, N. B. Brookes and L. Braicovich, J. Chem. Phys., 2002, 117, 8008–8020 CrossRef CAS.
- S. K. Cary, M. Vasiliu, R. E. Baumbach, J. T. Stritzinger, T. D. Green, K. Diefenbach, J. N. Cross, K. L. Knappenberger, G. Liu, M. A. Silver, A. E. DePrince, M. J. Polinski, S. M. Van Cleve, J. H. House, N. Kikugawa, A. Gallagher, A. A. Arico, D. A. Dixon and T. E. Albrecht-Schmitt, Nat. Commun., 2015, 6, 6827 CrossRef CAS PubMed.
- M. J. Polinski, E. B. Garner, R. Maurice, N. Planas, J. T. Stritzinger, T. G. Parker, J. N. Cross, T. D. Green, E. V. Alekseev, S. M. Van Cleve, W. Depmeier, L. Gagliardi, M. Shatruk, K. L. Knappenberger, G. Liu, S. Skanthakumar, L. Soderholm, D. A. Dixon and T. E. Albrecht-Schmitt, Nat. Chem., 2014, 6, 387–392 CrossRef CAS PubMed.
- W. Heitler and F. London, Z. Phys., 1927, 44, 455–472 CrossRef CAS.
- R. Hoffmann, J. Chem. Phys., 2004, 39, 1397–1412 CrossRef.
- Y. Qiao, G. Ganguly, C. H. Booth, J. A. Branson, A. S. Ditter, D. J. Lussier, L. M. Moreau, D. R. Russo, D.-C. Sergentu, D. K. Shuh, T. Sun, J. Autschbach and S. G. Minasian, Chem. Commun., 2021, 57, 9562–9565 RSC.
- G. Ganguly, D.-C. Sergentu and J. Autschbach, Chem. – Eur. J., 2020, 26, 1776–1788 CrossRef CAS PubMed.
- D.-C. Sergentu, T. J. Duignan and J. Autschbach, J. Phys. Chem. Lett., 2018, 9, 5583–5591 CrossRef CAS PubMed.
- D.-C. Sergentu and J. Autschbach, Dalton Trans., 2022, 51, 1754–1764 RSC.
- D.-C. Sergentu and J. Autschbach, Chem. Sci., 2022, 13, 3194–3207 RSC.
- R. Polly, B. Schacherl, J. Rothe and T. Vitova, Inorg. Chem., 2021, 60, 18764–18776 CrossRef CAS PubMed.
- E. I. Solomon, B. Hedman, K. O. Hodgson, A. Dey and R. K. Szilagyi, Coord. Chem. Rev., 2005, 249, 97–129 CrossRef CAS.
- F. Frati, M. O. J. Y. Hunault and F. M. F. de Groot, Chem. Rev., 2020, 120, 4056–4110 CrossRef CAS PubMed.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, Oxford, 1990 Search PubMed.
- I. Fryer-Kanssen and A. Kerridge, Chem. Commun., 2018, 54, 9761–9764 RSC.
- P. Di Pietro and A. Kerridge, Inorg. Chem., 2016, 55, 573–583 CrossRef CAS PubMed.
- S. Armstrong, T. Malcomson and A. Kerridge, Phys. Chem. Chem. Phys., 2022, 24, 26692–26700 RSC.
- J. Olsen, B. O. Roos, P. Jorgensen and H. J. A. Jensen, J. Chem. Phys., 1988, 89, 2185–2192 CrossRef CAS.
- F. Aquilante, J. Autschbach, A. Baiardi, S. Battaglia, V. A. Borin, L. F. Chibotaru, I. Conti, L. de Vico, M. Delcey, I. F. Galván, N. Ferré, L. Freitag, M. Garavelli, X. Gong, S. Knecht, E. D. Larsson, R. Lindh, M. Lundberg, P. Å. Malmqvist, A. Nenov, J. Norell, M. Odelius, M. Olivucci, T. B. Pedersen, L. Pedraza-González, Q. M. Phung, K. Pierloot, M. Reiher, I. Schapiro, J. Segarra-Martí, F. Segatta, L. Seijo, S. Sen, D.-C. Sergentu, C. J. Stein, L. Ungur, M. Vacher, A. Valentini and V. Veryazov, J. Chem. Phys., 2020, 152, 214117 CrossRef CAS PubMed.
- F. Aquilante, L. De Vico, N. Ferré, G. Ghigo, P. Malmqvist, P. Neogrády, T. B. Pedersen, M. Pitoňák, M. Reiher, B. O. Roos, L. Serrano-Andrés, M. Urban, V. Veryazov and R. Lindh, J. Comput. Chem., 2010, 31, 224–247 CrossRef CAS PubMed.
- F. Aquilante, J. Autschbach, R. K. Carlson, L. F. Chibotaru, M. G. Delcey, L. De Vico, I. Fdez. Galván, N. Ferré, L. M. Frutos, L. Gagliardi, M. Garavelli, A. Giussani, C. E. Hoyer, G. Li Manni, H. Lischka, D. Ma, P. Å. Malmqvist, T. Müller, A. Nenov, M. Olivucci, T. B. Pedersen, D. Peng, F. Plasser, B. Pritchard, M. Reiher, I. Rivalta, I. Schapiro, J. Segarra-Martí, M. Stenrup, D. G. Truhlar, L. Ungur, A. Valentini, S. Vancoillie, V. Veryazov, V. P. Vysotskiy, O. Weingart, F. Zapata and R. Lindh, J. Comput. Chem., 2016, 37, 506–541 CrossRef CAS PubMed.
- W. A. Misael and A. S. P. Gomes, Core excitations and ionizations of uranyl in Cs2UO2Cl4 from relativistic embedded damped response time-dependent density functional theory and equation of motion coupled cluster calculations, https://arxiv.org/abs/2302.07223, (accessed 19 June 2023).
- S. M. Cornet, M. P. Redmond, D. Collison, C. A. Sharrad, M. Helliwell and J. Warren, C. R. Chim, 2010, 13, 832–838 CrossRef CAS.
- M. P. Wilkerson, C. A. Arrington, J. M. Berg and B. L. Scott, J. Alloys Compd., 2007, 444–445, 634–639 CrossRef CAS.
- P. J. Hay, R. L. Martin and G. Schreckenbach, J. Phys. Chem. A, 2000, 104, 6259–6270 CrossRef CAS.
- A. Wolf, M. Reiher and B. A. Hess, J. Chem. Phys., 2002, 117, 9215–9226 CrossRef CAS.
- B. A. Hess, Phys Rev A, 1986, 33, 3742–3748 CrossRef CAS PubMed.
- B. A. Hess, Phys Rev A, 1985, 32, 756–763 CrossRef CAS PubMed.
- M. Douglas and N. M. Kroll, Ann Phys (N Y), 1974, 82, 89–155 CrossRef CAS.
- B. A. Heß, C. M. Marian, U. Wahlgren and O. Gropen, Chem. Phys. Lett., 1996, 251, 365–371 CrossRef.
- B. O. Roos, R. Lindh, P.-Å. Malmqvist, V. Veryazov and P.-O. Widmark, Chem. Phys. Lett., 2005, 409, 295–299 CrossRef CAS.
- B. O. Roos, R. Lindh, P.-Å. Malmqvist, V. Veryazov and P.-O. Widmark, J. Phys. Chem. A, 2004, 108, 2851–2858 CrossRef CAS.
- B. O. Roos, R. Lindh, P.-Å. Malmqvist, V. Veryazov and P.-O. Widmark, J. Phys. Chem. A, 2005, 109, 6575–6579 CrossRef CAS PubMed.
- S. Vancoillie, H. Zhao, V. T. Tran, M. F. A. Hendrickx and K. Pierloot, J. Chem. Theory Comput., 2011, 7, 3961–3977 CrossRef CAS PubMed.
- J. Finley, P.-Å. Malmqvist, B. O. Roos and L. Serrano-Andrés, Chem. Phys. Lett., 1998, 288, 299–306 CrossRef CAS.
- J. Finley, P.-Å. Malmqvist, B. O. Roos and L. Serrano-Andrés, Chem. Phys. Lett., 1998, 288, 299–306 CrossRef CAS.
- J. P. Zobel, J. J. Nogueira and L. González, Chem. Sci., 2017, 8, 1482–1499 RSC.
- N. Forsberg and P.-Å. Malmqvist, Chem. Phys. Lett., 1997, 274, 196–204 CrossRef CAS.
- P. Å. Malmqvist, B. O. Roos and B. Schimmelpfennig, Chem. Phys. Lett., 2002, 357, 230–240 CrossRef CAS.
- K. Stanistreet-Welsh, Molcas2Molden: Molcas INPORB to MOLDEN Convertor, https://github.com/k-stanistr-wel/Molcas2Molden, (accessed 24 March 2023).
- J. Autschbach, Comments Inorg. Chem., 2016, 36, 215–244 CrossRef CAS.
- F. Gendron, B. Pritchard, H. Bolvin and J. Autschbach, Inorg. Chem., 2014, 53, 8577–8592 CrossRef CAS PubMed.
- Zou Wenli, Molden2AIM: A utility program which can be used to create AIM-WFN, AIM-WFX, and NBO-47 files from a Molden file, https://github.com/zorkzou/Molden2AIM, (accessed 24 March 2023).
- T. A. Keith, AIMAll, version 19.02.13, TK Gristmill Software, Overland Park KS, USA, 2019.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- J. P. W. Wellington, A. Kerridge and N. Kaltsoyannis, Polyhedron, 2016, 116, 57–63 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp03149g |
| This journal is © the Owner Societies 2023 |