
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStarch/PVOH aqueous solutions: a chemical–physical characterization
Paolino
Caputo
 ab,
Valeria
Loise
*ab,
Maria Francesca
Colella
*ab,
Michele
Porto
ab,
Rosachiara A.
Salvino
ab,
Cesare
Oliviero Rossi
ab and
Giuseppina
De Luca
ab
ab,
Valeria
Loise
*ab,
Maria Francesca
Colella
*ab,
Michele
Porto
ab,
Rosachiara A.
Salvino
ab,
Cesare
Oliviero Rossi
ab and
Giuseppina
De Luca
ab
aDepartment of Chemistry and Chemical Technologies, University of Calabria, Via P. Bucci, Rende (CS), 87036, Italy. E-mail: valeria.loise@unical.it
bUdR INSTM of Calabria, Italy
First published on 22nd September 2023
Abstract
This work investigates the relationship between the structure and physicochemical properties of three different starches in starch/polyvinyl alcohol aqueous solutions. For this purpose, accurate nuclear magnetic resonance (NMR) analyses were performed to determine the role that the starch structure plays in the formation of binder solutions. Moreover, a dynamic shear rheometer (DSR) was used to investigate the mechanical properties of the solutions and correlate them with the structure of each starch. Complete characterization of the analysed starches and the starch/PVOH solutions was also carried out through light scattering measurements. Furthermore, by crossing the data coming from NMR and light scattering with those coming from rheology, the best solution was identified. Finally, to confirm the interaction mechanism between starch and PVOH, thermogravimetric analysis and an NMR study, using 1H and 13C NMR spectra, were carried out on the film obtained from the best solution. The analyses carried out showed that PVOH has a stabilizing effect on starch/PVOH solutions, and the starch with the greatest branching degree is the one that forms a more structured network.
1. Introduction
1.1 Starch: background
Starches are abundant biodegradable biopolymers produced and stored in the photosynthetic tissue of plants. They are made up of polysaccharides, i.e. a large number of glucose units linked together, and come essentially in the form of amylose and amylopectin. Amylose is a linear α-1-4 linked d-glucopyranose, its molecular weight varies according to the degree of polymerization from 500 in high-amylose maize starch to more than 6000 in potato starch.1 The amylose chains take a double helix conformation so as to have the hydrocarbon fractions of the anhydroglucose units inside the double helix itself.2 Moreover, the amylose content has a considerable influence on the characteristics of the starch. In fact, for example, as recently reported by Falua et al.,3 the amylose-rich starches have better thermal stability, and also the gel properties are strongly influenced by the amylose content. On the other hand, amylopectin is formed from linear α-1-4 linked D-glucopyranose and at the O-6 position of the glucose unit, the branches occur through the formation of α-1-6 bonds. Its degree of polymerization is around 10.000/100.000.4 In general, normal starches contain around 20–30% amylose and 70–80% amylopectin packed as granules of different sizes and shapes depending on different sources. The starch granule is formed by a succession of hollow, semi-crystalline and concentric spheres separated by a partially hydrated amorphous material. The arrangement of amylose and amylopectin within the starch granule shows semi-crystalline properties that make it water-insoluble at room temperature.5 However, the granules can be hydrated if heated at high temperatures in an aqueous environment. Under these conditions, the starch granules hydrate progressively and, by swelling, they lose their crystalline and ordered structure and move on to a disordered structure. Indeed, the structure of starch is stabilized by hydrogen bonds between the amylose double helix and the amylopectin side chains. High temperatures and the addition of water have the power to break these weak internal interactions and consequently amylopectin and amylose enter into the solution forming bonds with the water molecules. This ability to retain water is affected by the amylose content and by side chain lengths of the amylopectin but also by the characteristics of the granules, such as the size and presence of holes and channels. Therefore, free water decreases and a viscous suspension is obtained as a result of irreversible changes in the starch such as disruption of the granular and crystalline structure and loss of birefringence. This irreversible process is known as gelatinization.6–8On an industrial scale, starches are largely used in the food industry as thickening agents, in the paper industry as adhesives and wet strength agent,9 in the pharmaceutical industry as excipients, and in the polymer industry where their characteristic of plasticity is exploited to obtain biodegradable composite materials to be used as alternatives to non-biodegradable and petroleum-based plastic materials. Especially, in the field of polymers, the need to safeguard the environment and limit the use of petroleum-derived polymers has driven research and industries in the field of materials towards new biodegradable materials from renewable sources. In this context, starches, due to their high bio-renewable power, are particularly attractive.9 Precisely for these reasons, Tedeschi et al.10 have recently studied a new material for food packaging with antioxidant properties, obtained from corn starch and Tetradesmus obliquus microalga.
1.2 Alternative starch source: a brief review
According to the report released by the Agriculture Department of the province of Manitoba, the annual growth rate for the global market of starch has been estimated at 5.1%.11 This pushes researchers to find new sources for the production of starches, which obviously are not competitive with the food needs of the world population. Traditional crops (starch, i.e. potatoes, corn, wheat and cassava) have recently been joined by alternative sources for the extraction of starch, such as food waste from fruits and vegetables.12 In 2017 Hernadez-Carmona et al.13 conducted a study on the extraction of starch from green plantain peel waste produced by the food industry, obtaining an average 29% yield from the dry mass. In the same year Nakthong et al.14 published an article highlighting the thermopalstic properties of starch extracted from pineapple stems as agricultural waste. Also Rinju et al. extracted starch from pineapple stems as agricultural waste.15 Instead, Santana et al.16 have carried out the extraction of starch from turmeric with very low yields, about 3%, highlighting how the extraction yield depends on the cultivation conditions of the plant from which the starch is extracted. In 2018 Tesfaye et al.17 were able to obtain a 64% extraction yield, on a dry weight basis, of starch from avocado seed scraps from a fresh juice store. Moreover, Noor et al.18 performed a study on optimizing the yield of ultrasound-assisted extraction of starch from sago pith waste derived from the sago flour manufacturer. Instead, Choy et al.19 extracted starch from lime, papaya, banana, rambutan and jackfruit waste obtained from a local fruit vendor. They found that the starch content was less than 20% for the samples tested, except in the case of jackfruit seeds whose starch content was around 50%. Moreover, recently, researchers have begun to find alternative sources for the extraction of starches, such as algae, both marine and freshwater. In 2017, for example, Gifuni et al.20 found that starch from microalgae Chlorella sorokiniana was crystalline, and its molecular weight and thermal properties were very close to starch from other sources. Subsequently, in 2020, Ramli et al.21 have carried out the extraction of starch from marine microalgae (Klebsormidium flaccidum), finding that the characteristics of this starch are similar to those of commercial corn starch. Moreover, the structural properties of starch extracted from microalgae exceed those of corn starch. In 2021, Di Caprio et al.22 performed the extraction of starch from a strain of the microalga Tetradesmus obliquus (generally known as Scenedesmus obliquus), finding that the gelatinization temperature and the average size of the starch granules are lower than in traditional starch sources.1.3 How to improve the mechanical performance
Although starch is cheap, renewable and biodegradable unfortunately, in its pure form, it lacks those mechanical properties such as strength, impermeability, workability and heat stability, typical of petroleum-derived polymers.23However, in order to improve these issues, starch can be blended with other polymers that may be more or less biodegradable, such as poly(ε-caprolactone) (PCL), poly(lactic acid) (PLA), and polyvinyl alcohol (PVOH). In particular, PVOH is a linear synthetic polymer produced by partial or total hydrolysis of polyvinyl acetate to remove acetate groups. The amount of hydroxylation determines its physical, chemical and mechanical properties. PVOH is successfully used in a wide range of industrial fields for its excellent optical and physical properties. However, it has a high cost, low biodegradation rate and poor moisture barrier properties.24
To improve its eco-friendly properties and reduce costs, PVOH is often mixed with other polymers, including starches, in order to obtain biodegradable plastics. Indeed, both PVOH and starch are polar polymers; therefore, the preparation of PVOH and starch composites can result in stable eco-friendly materials with excellent mechanical properties and barrier behaviour. In most of the studies in the literature on PVOH and starch blends, the authors' interest focuses on the possible applications of these materials in different fields and depending on the possible uses, they try to improve the material's properties using some physical and chemical methods such as crosslinking.25 In contrast, there is very little information about the possible interactions between starch and PVOH molecules as such and the influence that different starches, native or modified, have on the final composite properties.26 Therefore, in order to get insight into this field, we have undertaken in this work, the study of the structural and physicochemical properties of aqueous solutions consisting of PVOH and three different native starches. The aim of the research was two-fold: (a) the investigation, at a molecular level, of the correlation between the structures of the starches and the possible interactions that these create in solution with the PVOH molecules by using experimental NMR data coming from the different magnetically active nuclei and (b) the macroscopic characterization in terms of physicochemical properties of starch/PVOH aqueous solutions using rheological experiments and thermogravimetric measurements. It should be emphasized that the NMR technique is particularly suitable for studies in isotropic solution since it is commonly used for the characterization at the molecular level of synthetic or natural compounds and the qualitative and quantitative study of complex mixtures.27 Beyond this, the technique is also used in anisotropic media to obtain structural and conformational information on even large molecules through residual dipolar couplings (RDCs).28,29
The interest in the study at the molecular level of these particular starch/PVOH solutions is primarily industrial.
Indeed, at present, for example, the composite market, i.e. particle board and nonwoven, uses binders30 based on toxic or potentially toxic substances.31 These same binders are widely used in kitchen utensils, paints, and paper treatment agents.32 Therefore, it is evident, that a potential replacement of these binders would have a huge impact on the health of workers in the sector.33 Furthermore, their production is not free from the emission of pollutants. Consequently, finding a valid alternative to these compounds would also meet the increasingly necessary measures for sustainable growth.34,35 Consequently, the use of green, biodegradable and eco-friendly compounds is highly desirable.
Hence, understanding what role the starch structure plays in the PVOH binder is crucial for choosing the one with the best characteristics.
2. Experimental
2.1 Chemicals and materials
Three different commercial wheat corn starches labelled as S1, S2 and S3, and polyvinyl alcohol (PVOH with MW = 61.00 approx. and degree of hydrolysis between 98.0 and 98.8 mol%) purchased from Sigma Aldrich Company, were analysed. Furthermore, a viscosity standard supplied by VWR Chemicals, with a viscosity of 8.7 cSt at 25 °C was used for the rheological analysis.2.2 Sample preparation
The aqueous starch solutions with 20% dry matter were prepared by mixing starch powder with distilled water in a flask equipped with a magnetic stirrer and reflux condenser. After the addition, the stirring was continued for about 15 minutes by increasing the temperature up to 100 °C to induce starch gelatinization.The PVOH solution was obtained by dissolving PVOH in distilled water and stirring continuously under reflux for about 1 hour.
Finally, starting from the appropriate quantities of the previous starch and PVOH solutions, the final binder solution with a starch/PVOH ratio of 70/30 and with a total dry matter of about 15% was prepared. After mixing the solution, it was mechanically stirred for 10 minutes at 50 °C.
2.3 Rheological characterization
Rheological measurements on binder solutions were carried out using a strain-controlled rheometer (RFS III, Rheometric Scientific, USA) equipped with a parallel plate geometry (gap 1 mm, φ = 50 mm within the temperature range 25–80 °C) and a Peltier system (±0.1 °C) for temperature control.To prevent errors due to evaporation, measuring geometries were surrounded by a viscosity standard at low viscosity. Three different kinds of experiments were carried out as follows: (a) dynamic shear experiments were performed in a frequency range between 0.100 and 15.9 Hz (all dynamic experiments were conducted within the visco-elastic region), hereafter frequency sweep and (b) steady experiments where the viscosity was measured as a function of the shear rate.
2.4 Thermogravimetric analysis
Thermogravimetric analysis was performed on a PerkinElmer TGA-6 instrument. Each analysis was performed from 100 °C to 300 °C at a temperature scan rate of 2 °C min−1. This analysis makes it possible to monitor the weight loss of the sample examined during a linear increase in temperature.2.5 NMR spectroscopy
The NMR samples of starches were prepared as follows: 25 mg of starch was placed in a small vial and 0.75 mL of DMSO-d6 was added. The vials were closed with a glass stopper, wrapped with a parafilm and heated at 353 K until the starch dissolved, then the whole mixture was transferred to a 5 mm NMR tube using a Pasteur glass pipet. The tube was sealed and wrapped with the parafilm and 1H, 13C–{1H} and HMQC NMR spectra were recorded for each sample. Afterwards, to improve the integration of anomeric protons, a few drops of deuterated trifluoroacetic acid (TFA-d1) were added directly to each sample and the 1H NMR spectra were recorded again.27 Each of the latter experiments was repeated three times to check for repeatability.The NMR sample of PVOH was prepared by dissolving 20 mg of PVOH in a solution of D2O/DMSO-d6 directly in the NMR tube. From this sample, 1H and 13C–{1H} NMR spectra were recorded.
From the starch/PVOH mixture, two NMR samples were prepared as follows: (a) sample A containing the freshly prepared mixture using DMSO-d6 as the deuterated solvent; (b) sample B of a film obtained starting from an aliquot of the mixture dried in the oven for 24h and dissolved in DMSO-d6. From these samples, the 1H, 13C–{1H} spectra were recorded.
All NMR spectra were recorded at 315 K on a high-resolution Bruker Avance 500 MHz spectrometer (11.74 T) (Fällanden, Switzerland), operating at a Larmor frequency of 500.13 MHz for 1H, equipped with a 5 mm TBO probe (Triple Resonance Broadband Observe) and a standard variable-temperature unit BVT-3000. Data were processed using Bruker TopSpin 3.6 software.35
1H and 13C NMR chemical shifts (δ) are reported in parts per million (ppm) and referenced to tetramethylsilane (TMS), using the solvent residual peak as an internal reference, 2.549 ppm for 1H and 39.5 ppm for 13C. DMSO-d6 (99.5% D atom) and TFA-d1 (99% D atom) were purchased from Sigma Aldrich (Italy) and used as received.
3. Results and discussion
3.1 Rheological analysis
In Fig. 1, the graphs of frequency sweep tests of the starch solutions S1, S2 and S3 at different temperatures are reported. Frequency sweep is performed at varying frequencies during a constant applied strain. Thanks to these measures it is possible to determine the time-dependent behaviour of a sample. In fact, high frequencies correspond to short time scales, while low frequencies correspond to longer time scales. Here a trend of a complex modulus, i.e. the resistance of the sample to deform, as defined in eqn (1), as a function of frequency is presented. | (1) |
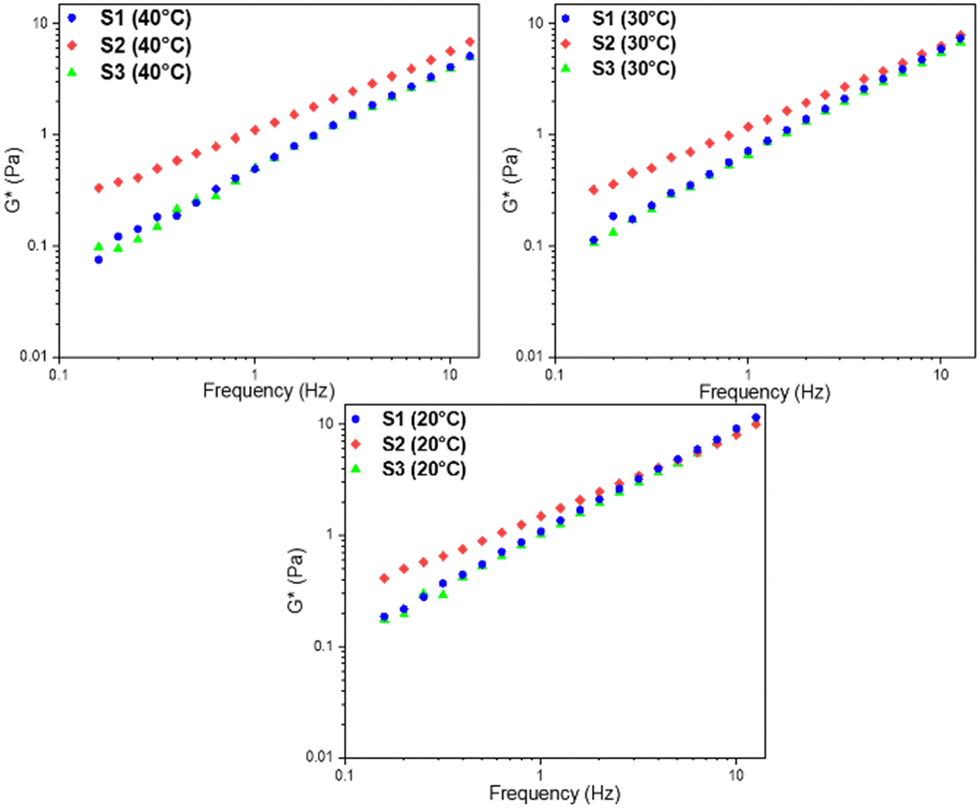 | ||
| Fig. 1 Comparison between the frequency sweep tests of the starch solutions S1, S2 and S3 at different temperatures (error < 1%). | ||
As can be noted from Fig. 1, S1 and S3 solutions have similar mechanical behaviour in the investigated temperature range, while the S2 solution has a higher complex modulus. In general, a high complex modulus value describes the formation of a more structured colloidal network, i.e. a bio-polymeric network.36 In the case of S1 and S3, the two networks are similar in terms of link points and interaction force; in contrast, S2 has a higher number of link points and higher interaction force.
By making a fitting of the type 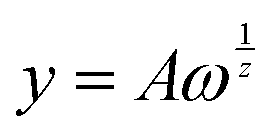 on the frequency sweep measurements of the solutions, it is possible to obtain two coefficients, which are the slope and the intercept of the least square line in a log–log plot. These two coefficients, specifically called A and z, are related to the strength of the structure and to the extension of the three-dimensional lattice.37 From Fig. 2a, it can be observed that the value of A decreases for all starch solutions with increasing temperature, thus showing a reduction in the interaction force, probably due to purely kinetic effects. Moreover, the trend of z (Fig. 2b) is almost flat with respect to temperature for all solutions. The z behaviour provides evidence for how the formed network maintains its tridimensional structure under heating conditions. It is worth noting that the values of A and z are always higher for S2 solutions.
on the frequency sweep measurements of the solutions, it is possible to obtain two coefficients, which are the slope and the intercept of the least square line in a log–log plot. These two coefficients, specifically called A and z, are related to the strength of the structure and to the extension of the three-dimensional lattice.37 From Fig. 2a, it can be observed that the value of A decreases for all starch solutions with increasing temperature, thus showing a reduction in the interaction force, probably due to purely kinetic effects. Moreover, the trend of z (Fig. 2b) is almost flat with respect to temperature for all solutions. The z behaviour provides evidence for how the formed network maintains its tridimensional structure under heating conditions. It is worth noting that the values of A and z are always higher for S2 solutions.
 | ||
| Fig. 2 Weak-gel parameters for the starch solutions S1, S2 and S3 at different temperatures (error ± 0.1). | ||
With regards to the binder, the trend of A decreases with increasing temperature again. Both coefficients, A and z, are greater for the binder containing starch S2 (Fig. 3). The analysis of parameters A and z shows that starch S2 forms a more structured and stronger network. This observation could be related to the branching degree of the S2 starch, in fact, the presence of the branch could allow the formation of more point links and a more coordinated system. These results could also be correlated with the dynamic light scattering (DLS) analysis. Each starch was dissolved in distilled water and their particle size was analysed using a Zetasizer Nano ZS (Malvern Instruments Inc., Malvern, U.K). 1 ml of diluted starch nanoparticle dispersions (10 time in DI water) was added to plastic cuvettes and placed in the DLS instrument for measurements. The DLS measurements showed that the S3 and S1 starches form aggregates of similar sizes both in the starch solutions and in the binder solutions. Moreover, the solutions containing these two starches are characterized by much smaller aggregates than those obtained in the case of S2 starch. For example, the dimensions of the aggregates that are formed in the binder solutions containing starch S3 and starch S1 are about 100 and about 80 nm, respectively, while for the binder solution containing S2, the dimensions of the aggregates are of few microns.
 | ||
| Fig. 3 Weak-gel parameters for the binder solutions at different temperatures (error ± 0.1). | ||
From Fig. 4, S1 and S3 have very similar flow curves, especially at low temperatures (20 and 40 °C), in contrast at high temperatures (60 and 80 °C) they are slightly different. Probably this behaviour is due to the evaporation control of the measurement system. Shear flow can be depicted as layers of fluid sliding over one another with each layer moving faster than the one beneath it. The uppermost layer has the maximum velocity while the bottom layer is stationary. For shear flow to take place, a shear force must act on the fluid. The coefficient of proportionality between the shear stress and shear rate is defined as the shear viscosity or dynamic viscosity (η), which is a quantitative measure of the internal fluid friction, and is associated with damping or loss of kinetic energy in the system.
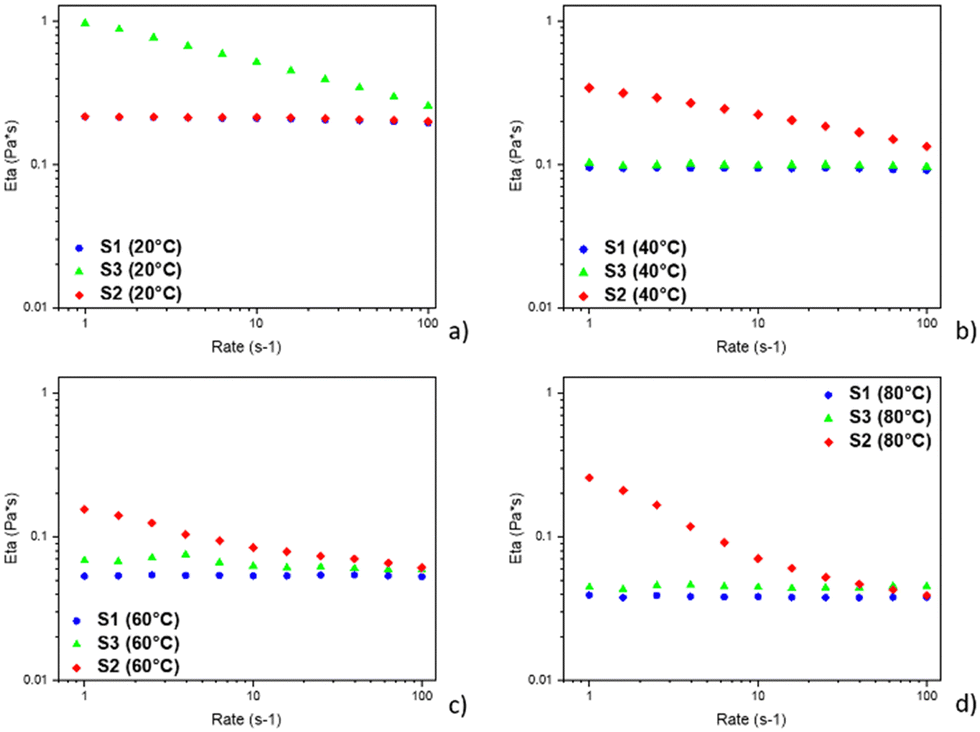 | ||
| Fig. 4 Comparison between flow curves of the starch solutions at different temperatures. | ||
According to the shear rate, the viscosity of S1 and S3 remains constant. This indicates that the two samples are Newtonian systems therefore colloidal systems formed by aggregates modelled as non-interacting spheres.38 On the other hand, S2 exhibits a shear thinning behaviour, having a higher viscosity value compared to S1 and S3. Its profile is typical for large aggregate, and it is modelled as elongated spheres that are oriented along the flow.39
In Fig. 5, it is worth noting that the three binders have very similar flow curves (shear thinning). Moreover, PVOH seems to have a stabilizing effect. In fact, as can be seen from Fig. 5, the viscosity of the binder solutions is scarcely affected by the temperature. This behaviour could depend on the fact that the solutions containing PVOH are stabilized by the formation of hydrogen bonds between the PVOH and starch.
 | ||
| Fig. 5 Comparison between flow curves of the binder solutions at different temperatures (error < 1%). | ||
3.2 NMR characterization
The molecular characterization of the three starches was carried out on the basis of the 1D, 1H and 13C–{1H} NMR experiments, 2D heteronuclear correlation NMR spectroscopy (1H–13C HMQC) and by comparison with the published data.40,41 In Fig. 6, the 1H and 13C–{1H} NMR spectra of KP starch with the relevant chemical shifts of both 1H and 13C nuclei associated with each group of lines are reported. As can be seen from the spectra in the figure, there are no additional peaks compared to those typical of the basic starch structure and this means that the KP sample is an unmodified starch. This also occurs for the other two starches whose spectral profiles are similar to that of KP. From the 1H NMR spectra, it is possible to trace the degree of branching (DB), which represents the percentage of branches (α-1,6 links) with respect to the total number of bonds present in starch, for the three different starches. DB can be calculated from the area of the anomeric protons using the following equation:40where I(α-1,4) represents the area of the anomeric proton signals at 5.11 ppm involved in the 1–4 bond of the linear chain, denoted as H1, and the area of the anomeric proton signals originated from terminal non-reducing ends (from 5.03 to 5.4 ppm), while I(α-1,6) represents the area of anomeric proton signals at 4.75 ppm involved in a branch point, denoted as H1′.

 | ||
| Fig. 6 Chemical structure, nuclei numbering, 1H and 13C–{1H} NMR spectra of S2 starch in DMSO-d6 at 315 K and signal assignment. | ||
From the observation of the proton spectrum in Fig. 7, it is evident how the anomeric protons indicated with H1 and H1′ are poorly defined due to the presence of other resonances from starch hydroxyl groups (OH) in the same region (4.10–5.61 ppm) and from the signal of the residual water peak at 3.14 ppm. In order to carry out a good integration of the anomeric protons and hence reduce the errors in the measurement, it is convenient to have well isolated signals for these protons. An efficient way to obtain clearly integrable signals of anomeric protons is the one proposed by Tizzotti et al.40 based on the addition of a small amount of TFA-d1. Indeed, the addition of TFA-d1 to the starch sample dissolved in DMSO-d6 causes a rapid exchange between the TFA-d1 deuterium and the protons of the starch OH groups and those of the residual water, giving rise to a single peak strongly displaced from the anomeric zone (δ >8 ppm). Moreover, the chemical shift of the final signal of the exchangeable protons can be tuned to an appropriate value by adjusting the quantity of TFA-d1 added. Note that the addition of a small amount of TFA-d1 does not affect the chemical shifts of the other proton signals of the starch as shown in Fig. 7, where the comparison between the 1H NMR spectrum of S2 starch in DMSO-d6 prior to and after the addition of TFA-d1 is reported.
 | ||
| Fig. 7 1H NMR spectrum of S2 starch in DMSO-d6 at 315 K prior (lower panel in continue blue line) and after (upper panel in dashed red line) addition of TFA-d1. | ||
The well resolved signals of anomeric protons H1 and H1′ after the addition of TFA-d1 allows a clear integration and reproducible calculation of DB. The DB calculated for the S2 starch was 2.83 ± 0.02%, which agrees with the data reported in the literature for native starches whose DB values are between 1 and 5%.42 A similar situation also took place for the other two starch samples and by operating in the same way, after the addition of TFA-d1, their DB% values were calculated from the area of the anomeric signals and their values are reported in Table 1. As can be seen from the table, the S2 starch is the one with the highest DB value.
| Starch type | DB% |
|---|---|
| S1 | 2.56 ± 0.02 |
| S2 | 2.83 ± 0.02 |
| S3 | 2.02 ± 0.02 |
The characterization of the PVOH molecule was carried out in an aqueous solution by adding a small amount of DMSO-d6. In Fig. 8, the 1H and 13C–{1H} NMR spectra of PVOH with the relevant chemical shifts of both 1H and 13C nuclei associated with each group of lines assigned are reported.
 | ||
| Fig. 8 (a) General structure of PVOH which highlights the stereochemistry of the three possible polymeric triads that generate slightly different 1H and 13C NMR signals: isotactic triad (mm), heterotactic triad (rm) and syndiotactic triad (rr).43,44 (b) 1H and (c) 13C–{1H} NMR spectra recorded for the PVOH sample at 315 K. | ||
On the basis of the rheological data, it was hypothesized that the three starch/PVOH binder solutions were stabilized by the establishment of interactions, possibly hydrogen bonds, between the two components in solution. Moreover, the binder solution consisting of S2 which has the highest value of DB, and PVOH shows the highest viscosity which probably induces particular macroscopic characteristics in the molecular aggregates. In order to understand the structure and nature of the interactions existing between starch and PVOH molecules, the binder solution, and a film of these two constituents, S2/PVOH, was also studied via NMR.
Fig. 9 shows the 1H NMR spectrum of the fresh binder mixture of S2 and PVOH (sample A in dashed and purple line) using DMSO-d6 as the deuterated solvent, and the 1H NMR spectrum of a film, obtained by drying the mixture, dissolved in DMSO-d6 (sample B spectral profile as a continuous blue line) are reported. The same figure also shows the 1H spectra of PVOH (dashed green) and S2 starch (dashed orange) in DMSO-d6 as a comparison. As can be seen from Fig. 9, in the case of the fresh mixture (sample A), the water peak is so intense as to hide the weak signals relating to the two components of the binder. In contrast, in the case of the film (sample B), the water signal, although present, does not cover the other signals, and, very interestingly, this peak resonates at a very different frequency compared to the previous sample and even slightly shifted compared to what is reported in the literature.45
 | ||
| Fig. 9 Comparison between 1H NMR spectra of sample B (continue blue line), sample A (dashed purple line, - - -), PVOH (dashed green line, -•-•-) and S2 starch (dashed orange line, ---) in DMSO-d6 at 315 K. | ||
This is because the water in the case of the film does not act as a solvent as it is present in less quantities than DMSO. Furthermore, the peak of water resonating at a slightly different frequency from that reported in the literature could be explained by the presence of a possible interaction of the water molecules with the two components of the binder. If, on the other hand, the spectrum obtained from the film is compared with that of the individual substances (respectively highlighted in the dashed green and purple curve), there are no other signals but only those of the two initial solutions, except for the OH signals whose chemical shifts are slightly shifted compared to the single components. This means that in the formation of the binder, there is no breaking and formation of new covalent bonds, but only weak interactions are established between the two components, such as the hypothesized hydrogen bond. This is also confirmed by the 13C spectra shown in Fig. 10.
 | ||
| Fig. 10 Comparison between 13C NMR spectra of sample B (continue blue line), sample A (dashed purple line, - - -), PVOH (dashed green line, -•-•-) and S2 starch (dashed orange line, ---) in DMSO-d6 at 315 K. | ||
In this case, the profile of the film (in dashed blue curve) is given by the superposition of PVOH and starch spectra (dashed green and orange spectra), with a slight difference in terms of chemical shift as in the case of the proton spectrum. Unlike the proton spectrum in which the water signal hid the other signals, a sufficiently resolved 13C spectrum was obtained in the case of the mixture just prepared. This spectrum (in dashed purple line) is similar in profile to the film spectrum but with some differences: (a) the signals show a more marked variation in chemical shift probably due to the presence of numerous interactions with the solvation water, (b) the signal related to the triad mm is the CH signal of PVOH which varies the most. This is probably due to a situation in which the configuration of three consecutive residues allows better interaction with water and thus this triad could represent a key fragment in understanding the interactions existing within the binder and that gives the material its macroscopic properties.
3.3 Thermogravimetric analysis
A certain quantity of binder was poured into a Petri dish and left to dry in an oven at 60/70 °C for 24 h in order to obtain a film. Thermograms of S2 starch, PVOH and film from binder are shown in Fig. 11. | ||
| Fig. 11 TGA of starch, PVOH and film from the binder. | ||
As can be observed, in the case of starch, the first decomposition occurs at around 100 °C and is due to the loss of water, due to the hydrophilicity of the starch.46 Subsequently, the major degradation takes place, which finishes at around 370 °C, and corresponds to a weight loss of about 80%. This loss is due to the breaking of the chains as well as to the oxidation of the rings, as reported in the literature.47 With regards to PVOH, according to the literature data,47,48 the most important decomposition leads to a weight loss of about 70%, which occurs between 300 and 400 °C. This thermal event is caused both by the formation of the polyene due to the decomposition of the side-groups, and by the breaking of the C–C bonds of the polymer chain. Moreover, from Fig. 11, the thermogram of the film obviously does not show the stage due to loss of water. The major thermal event begins at around 270 °C like that of PVOH and ends at around 370 °C as for starch. In general, the thermogram of the film is the sum of the thermograms of starch and PVOH. This is in line with the results obtained by NMR which show the formation of only hydrogen bonds between starch and PVOH.
4. Conclusions
In conclusion, the analyses carried out make it possible to hypothesize the establishment of hydrogen bonds between starch and PVOH in the formation of the binder. Although through NMR analysis of the binder solution, it is not possible to appreciate the formation of weak interactions due to the presence of excessive water whose signals mask the signals of starch and PVOH, it is reasonable to assume that the formation of such interactions does not depend on the evaporation effect of the solvent. In fact, flow curves show that PVOH has a stabilizing effect on binder solutions. Finally, it was important to note that both the S2 starch solution and the PVOH+S2 binder solution showed better mechanical properties. S2 starch, being the one with the highest degree of branching, is capable of giving rise to more structured networks.Author contributions
Paolino Caputo: writing – review & editing. Valeria Loise: conceptualization and writing – original draft. Maria Francesca Colella: investigation and data curation. Michele Porto: investigation and data curation. Rosachiara Salvino: investigation and data curation. Cesare Oliviero Rossi: methodology and validation. Giuseppina De Luca: methodology, validation, and supervision.Conflicts of interest
There are no conflicts to declare.References
- Y. Ai and J. l Jane, in Encyclopedia of Food and Health, ed. B. Caballero, P. M. Finglas and F. Toldrá, Academic Press, Oxford, 2016, pp. 165–174 Search PubMed.
- J. l Jane, in Chemical and Fuctional Properties of Food Saccharides, ed. P. Tomasik, CRC Press LLC, 2004 Search PubMed.
- K. J. Falua, A. Pokharel, A. Babaei-Ghazvini, Y. Ai and B. Acharya, Valorization of Starch to Biobased Materials: A Review, Polymers, 2022, 14(11), 2215 CrossRef CAS PubMed.
- W. Bergthaller, J. Hollmann and P. Johannis, J. Chem. Educ., 2008, 85, 579–612 Search PubMed.
- K. Villwock and J. N. BeMiller, The Architecture, Nature, and Mystery of Starch Granules. Part 1: A Concise History of Early Investigations and Certain Granule Parts, Starch – Stärke, 2022, 74, 2100183 CrossRef CAS.
- Y. I. Cornejo-Ramírez, O. Martínez-Cruz, C. L. Del, F. J. Wong-Corral, J. Borboa-Flores, J. Cinco-Moroyoqui, Y. Isbeth, O. Martínez-Cruz, C. L. Del, F. J. Wong-Corral, J. Borboa-Flores, F. J. Cinco-Moroyoqui, Y. I. Cornejo-Ramírez, O. Martínez-Cruz, C. L. Del Toro-Sánchez, F. J. Wong-Corral, J. Borboa-Flores, F. J. Cinco-Moroyoqui, D. De Investigación, U. De Sonora and C. P. México, The structural characteristics of starches and their functional properties The structural characteristics of starches and their functional properties, CyTA – J. Food, 2018, 1003–1017 CrossRef.
- L. Copeland, J. Blazek, H. Salman and M. C. Tang, Form and functionality of starch, Food Hydrocolloids, 2009, 23, 1527–1534 CrossRef CAS.
- Y. Kulshreshtha, E. Schlangen, H. M. Jonkers, P. J. Vardon and L. A. van Paassen, CoRncrete: A corn starch based building material, Constr. Build. Mater., 2017, 154, 411–423 CrossRef CAS.
- I. Francolini, L. Galantini, F. Rea, C. Di Cosimo and P. Di Cosimo, Polymeric Wet-Strength Agents in the Paper Industry: An Overview of Mechanisms and Current Challenges, Int. J. Mol. Sci., 2023, 24(11), 9268 CrossRef CAS PubMed.
- A. M. Tedeschi, F. Di Caprio, A. Piozzi, F. Pagnanelli and I. Francolini, Sustainable Bioactive Packaging Based on Thermoplastic Starch and Microalgae, Int. J. Mol. Sci., 2023, 23(1), 178 CrossRef PubMed.
- Global Market for Starch and Starch Products, https://www.gov.mb.ca/agriculture/markets-and-statistics/trade-statistics/pubs/starch-world-market.pdf, (accessed 5 September 2023).
- D. H. Kringel, A. R. G. Dias, E. da, R. Zavareze and E. A. Gandra, Fruit Wastes as Promising Sources of Starch: Extraction, Properties, and Applications, Starch – Stärke, 2020, 72, 1900200 CrossRef CAS.
- F. Hernández-Carmona, Y. Morales-Matos, H. Lambis-Miranda and J. Pasqualino, Starch extraction potential from plantain peel wastes, J. Environ. Chem. Eng., 2017, 5, 4980–4985 CrossRef.
- N. Nakthong, R. Wongsagonsup and T. Amornsakchai, Characteristics and potential utilizations of starch from pineapple stem waste, Ind. Crops Prod., 2017, 105, 74–82 CrossRef CAS.
- R. Rinju and B.-S. Harikumaran-Thampi, Characteristics of Starch Extracted from the Stem of Pineapple Plant (Ananas comosus) – an Agro Waste from Pineapple Farms, Braz. Arch. Biol. Technol., 2021, 64, e21190276 CrossRef CAS.
- Á. L. Santana, G. L. Zabot, J. F. Osorio-Tobón, J. C. F. Johner, A. S. Coelho, M. Schmiele, C. J. Steel and M. A. A. Meireles, Starch recovery from turmeric wastes using supercritical technology, J. Food Eng., 2017, 214, 266–276 CrossRef.
- T. Tesfaye, M. Gibril, B. Sithole, D. Ramjugernath, R. Chavan, V. Chunilall and N. Gounden, Valorisation of avocado seeds: extraction and characterisation of starch for textile applications, Clean Technol. Environ. Policy, 2018, 20, 2135–2154 CrossRef CAS.
- M. H. Mohamed Noor, N. Ngadi, A. N. Suhaidi, I. Mohammed Inuwa and L. Anako Opotu, Response Surface Optimization of Ultrasound-Assisted Extraction of Sago Starch from Sago Pith Waste, Starch – Stärke, 2022, 74, 2100012 CrossRef CAS.
- S. Y. Choy, K. M. N. Prasad, T. Y. Wu, M. E. Raghunandan, B. Yang, S.-M. Phang and R. N. Ramanan, Isolation, characterization and the potential use of starch from jackfruit seed wastes as a coagulant aid for treatment of turbid water, Environ. Sci. Pollut. Res., 2017, 24, 2876–2889 CrossRef CAS PubMed.
- I. Gifuni, G. Olivieri, I. R. Krauss, G. D’Errico, A. Pollio and A. Marzocchella, Microalgae as New Sources of Starch: Isolation and Characterization of Microalgal Starch Granules, Chem. Eng. Trans., 2017, 57, 1423–1428 Search PubMed.
- R. N. Ramli, C. K. Lee and M. A. Kassim, Extraction and Characterization of Starch from Microalgae and Comparison with Commercial Corn Starch, IOP Conf. Ser. Mater. Sci. Eng., 2020, 716, 12012 CrossRef CAS.
- F. Di Caprio, R. Chelucci, I. Francolini, P. Altimari and F. Pagnanelli, Extraction of microalgal starch and pigments by using different cell disruption methods and aqueous two-phase system, J. Chem. Technol. Biotechnol., 2022, 97, 67–78 CrossRef CAS.
- N. Tudorachi, C. N. Cascaval, M. Rusu and M. Pruteanu, Testing of polyvinyl alcohol and starch mixtures as biodegradable polymeric materials, Polym. Test., 2000, 19, 785–799 CrossRef CAS.
- X. Tang and S. Alavi, Recent advances in starch, polyvinyl alcohol based polymer blends, nanocomposites and their biodegradability, Carbohydr. Polym., 2011, 85, 7–16 CrossRef CAS.
- S. K. Vineeth, R. Gadhave and P. Gadekar, Investigation of crosslinking ability of sodium metabisulphite with polyvinyl alcohol–corn starch blend and its applicability as wood adhesive, Indian Chem. Eng., 2021, 64, 1–11 Search PubMed.
- S. Hatna, B. Raj and S. Rudrappa, Structure–property relation in polyvinyl alcohol/starch composites, J. Appl. Polym. Sci., 2004, 91, 630–635 CrossRef.
- R. A. Salvino, M. F. Colella and G. De Luca, NMR-based metabolomics analysis of Calabrian citrus fruit juices and its application to industrial process quality control, Food Control, 2021, 121, 107619 CrossRef CAS.
- M. E. Di Pietro, C. Aroulanda, G. Celebre, D. Merlet and G. De Luca, The conformational behaviour of naproxen and flurbiprofen in solution by NMR spectroscopy, New J. Chem., 2015, 39, 9086–9097 RSC.
- M. E. Di Pietro, T. Margola, G. Celebre, G. De Luca and G. Saielli, A combined LX-NMR and molecular dynamics investigation of the bulk and local structure of ionic liquid crystals, Soft Matter, 2019, 15, 4486–4497 RSC.
- M. J. Denton and P. N. Daniels, Textile terms and definitions, The Textile Institute, Manchester, UK, 11th edn, 2002 Search PubMed.
- A. K. Behera and S. Manna, Evaluation of conductivity and mechanical properties of natural fiber reinforced soy-melamine formaldehyde based green composite, Polym. Compos., 2022, 43, 1546–1556 CrossRef CAS.
- S. W. Kariuki, J. Wachira, M. Kawira and G. Murithi, Formaldehyde Use and Alternative Biobased Binders for Particleboard Formulation: A Review, J. Chem., 2019, 2019, 5256897 Search PubMed.
- A. Arias, S. González-García, G. Feijoo and M. T. Moreira, Tannin-based bio-adhesives for the wood panel industry as sustainable alternatives to petrochemical resins, J. Ind. Ecol., 2022, 26, 627–642 CrossRef CAS.
- A. Vujanović, J. Puhar, D. Krajnc, P. Awad and L. Čuček, Reducing the environmental impacts of the production of melamine etherified resin fibre, Sustainable Prod. Consum., 2022, 29, 479–494 CrossRef.
- Bruker, TopSpin, https://www.bruker.com/en/products-and-solutions/mr/nmr-software/topspin.html, (accessed 9 May 2022).
- L. Coppola, R. Gianferri, I. Nicotera, C. Oliviero and G. Antonio Ranieri, Structural changes in CTAB/H2O mixtures using a rheological approach, Phys. Chem. Chem. Phys., 2004, 6, 2364–2372 RSC.
- L. Bohlin, A theory of flow as a cooperative phenomenon, J. Colloid Interface Sci., 1980, 74, 423–434 CrossRef CAS.
- H. Barnes, J. F. Hutton and K. Walters, An introduction to Rheology, Elsevier, 1989 Search PubMed.
- G. R. Del Sorbo, S. Prévost, E. Schneck, M. Gradzielski and I. Hoffmann, On the Mechanism of Shear-Thinning in Viscous Oppositely Charged Polyelectrolyte Surfactant Complexes (PESCs), J. Phys. Chem. B, 2020, 124, 909–913 CrossRef CAS PubMed.
- M. J. Tizzotti, M. C. Sweedman, D. Tang, C. Schaefer and R. G. Gilbert, New (1)h NMR procedure for the characterization of native and modified food-grade starches, J. Agric. Food Chem., 2011, 59, 6913–6919 CrossRef CAS PubMed.
- P. Cuenca, S. Ferrero and O. Albani, Preparation and characterization of cassava starch acetate with high substitution degree, Food Hydrocolloids, 2020, 100, 105430 CrossRef CAS.
- G. S. Nilsson, L. Gorton, K.-E. Bergquist and U. Nilsson, Determination of the Degree of Branching in Normal and Amylopectin Type Potato Starch with 1H-NMR Spectroscopy Improved resolution and two-dimensional spectroscopy, Starch – Stärke, 1996, 48, 352–357 CrossRef CAS.
- O. Olabisi and K. Adewale, Handbook of Thermoplastics, Taylor & Francis, 2nd edn, 2016, p. 36 Search PubMed.
- X. Hong, L. Zou, J. Zhang and L. Wang, Preparation of high molecular weight polyvinyl alcohol by emulsifier-free emulsion polymerization, E3S Web Conf., 2020, 206, 2023 CrossRef CAS.
- G. R. Fulmer, A. J. M. Miller, N. H. Sherden, H. E. Gottlieb, A. Nudelman, B. M. Stoltz, J. E. Bercaw and K. I. Goldberg, NMR Chemical Shifts of Trace Impurities: Common Laboratory Solvents, Organics, and Gases in Deuterated Solvents Relevant to the Organometallic Chemist, Organometallics, 2010, 29, 2176–2179 CrossRef CAS.
- Y. Liu, L. Yang, C. Ma and Y. Zhang, Thermal Behavior of Sweet Potato Starch by Non-Isothermal Thermogravimetric Analysis, Materials, 2019, 12(5), 699 CrossRef CAS PubMed.
- S. Ismail, N. Mansor and Z. Man, A Study on Thermal Behaviour of Thermoplastic Starch Plasticized by [Emim] Ac and by [Emim] Cl, Proc. Eng., 2017, 184, 567–572 CrossRef CAS.
- N. Azeez, Thermogravimetric Analysis on PVA/PVP Blend Under Air Atmosphere, Engineering and Technology Journal, 2016, 34(13), 2433–2442 CrossRef.
| This journal is © the Owner Societies 2023 |