
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Two phases of trans-stilbene in a polystyrene matrix
Renata
Karpicz
 a,
Gabriele
Kareivaite
a,
Mindaugas
Macernis
*b,
Darius
Abramavicius
b and
Leonas
Valkunas
ab
a,
Gabriele
Kareivaite
a,
Mindaugas
Macernis
*b,
Darius
Abramavicius
b and
Leonas
Valkunas
ab
aCenter for Physical Sciences and Technology, Saulėtekio av. 3, Vilnius, Lithuania
bInstitute of Chemical Physics, Faculty of Physics, Vilnius University, Saulėtekio av. 3, Vilnius, Lithuania. E-mail: mindaugas.macernis@ff.vu.lt
First published on 17th July 2023
Abstract
Variability in the spectral properties of solid conformations of stilbene under various external conditions still remains obscure. The photophysical properties of trans-stilbene solution in solid polystyrene glass have been studied by absorption and time-resolved fluorescence. Concentration-induced quenching has been observed for small concentrations of stilbene. At large concentrations, the spectroscopic characteristics become split between the two phases of the sample: single-molecule properties are responsible for absorption, while the micro-crystalline phase dominates in fluorescence. Ab initio and molecular dynamics analyses suggest permanent twisting of the stilbene molecular structure upon crystallization, which supports spectroscopic phase separation.
Introduction
Technologically promising organic molecules display various specific photoinduced intra- and inter-molecular transformations, such as, for instance, trans–cis isomerization or variability of fluorescence lifetimes depending on environmental conditions. Stilbene (Scheme 1) is a pronounced example of such a type of molecule.1–4 Stilbene molecules have been extensively studied from the mid-60s of the last century under various conditions such as in solutions and supersonic jets, at different pressures and at various temperatures.2,5–14 They can be found in two forms, namely, cis and trans forms. The process of cis–trans isomerization upon photoexcitation of stilbene is well established and is characterized by means of fluorescence decay lifetime in different solvents.2,4,14,15 These molecules are usually considered model molecules for studies of photoinduced intramolecular isomerization processes. By means of time-resolved Raman spectroscopy and high-quality quantum chemistry calculations it has been recently confirmed that the isomerization process reflects a smooth gradual evolution along a reaction coordinate.16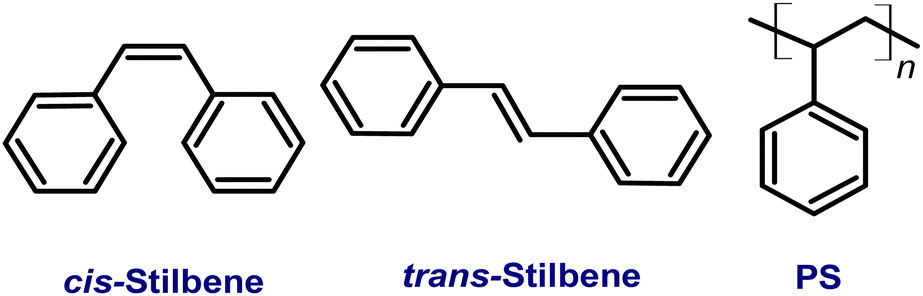 | ||
| Scheme 1 Structures of cis-stilbene, trans-stilbene and polystyrene (PS). | ||
Solid stilbene compounds are usually of trans-stilbene (Tstilbene) form and have been used in engineering scintillating devices and possess the ability to distinguish between the effects caused by different illumination.17 However, knowledge of their internal structure is still lacking. Possible technological applications require extensive knowledge of photophysical processes of various complex heterostructured materials based on stilbene. The variability in the spectral properties of solid conformations of Tstilbene with temperature18–21 in aggregated form22–24 has been investigated for many decades, but still remains obscure. From our recent studies it has been found that the temperature dependence of the steady-state and time-resolved fluorescence of solid-state systems and films of Tstilbene is attributed to cooperative exciton state formation in condensed phases, when the molecular isomerization process is suppressed.25 For comparison, time-resolved fluorescence measurements have also been performed for stilbene solutions in chloroform and for stilbene in a polystyrene matrix, which specifically behaves as a highly viscous solution. Sensitivity of the fluorescence decay lifetimes for different samples at various temperatures suggests the emergence of aggregation-induced exciton diffusion in solid forms of Tstilbene, which specifically shapes the fluorescence spectrum. It is demonstrated that this type of excitonic dynamics dominates over different single-molecular excitation decay pathways in the solid phase.25
However, detailed studies of aggregate formation of Tstilbene molecules is still lacking. Here, we present comparative studies of the absorption and fluorescence spectra as well as the fluorescence kinetics of Tstilbene solutions in a polystyrene (PS) matrix as a function of the solute concentration. Our results reveal the formation of nano-crystals in concentrations above 20%. The experimental data are analysed and interpreted together with ab initio and molecular dynamics (MD) computational analysis. Our results demonstrated that at high concentrations Tstilbene properties could be attributed to two phases, which are observed experimentally. Calculations reveal molecular packing upon crystallization and suggest that absorption occurs in the single-molecule phase, while fluorescence is dominated in the micro-crystalline phase. Such separation could possibly be employed in optical quantum technologies.
1. Materials and methods
1.1. Experiments
Tstilbene molecules (molecular structure is shown in Scheme 1) were purchased from Sigma-Aldrich. For film preparation we used polystyrene (PS, glass transition point Tg = 112 °C, Mw = 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) as the host material doped with stilbene molecules at different concentrations. A polystyrene matrix (Scheme 1) is used as a solid solvent because it is transparent in the spectral region to the red from 280 nm and has minimal effects on both absorption and fluorescence in the case of a single, non-aggregated sample.25 Films were prepared in a UV-isolated room using chloroform solutions (initial PS concentration for all solutions was the same, 8 mg mL−1) by spin-coating (1000 rpm for 40 s, with KW-4A apparatus) onto clean 15–24 mm quartz substrates. The average film thickness ranged from 120 to 160 nm. The concentration (weight percentage) of Tstilbene varied from 0.5% to 80%.
000) as the host material doped with stilbene molecules at different concentrations. A polystyrene matrix (Scheme 1) is used as a solid solvent because it is transparent in the spectral region to the red from 280 nm and has minimal effects on both absorption and fluorescence in the case of a single, non-aggregated sample.25 Films were prepared in a UV-isolated room using chloroform solutions (initial PS concentration for all solutions was the same, 8 mg mL−1) by spin-coating (1000 rpm for 40 s, with KW-4A apparatus) onto clean 15–24 mm quartz substrates. The average film thickness ranged from 120 to 160 nm. The concentration (weight percentage) of Tstilbene varied from 0.5% to 80%.
Absorption spectra were recorded using a Jasco V670 spectrometer. Steady-state fluorescence spectra and fluorescence decay kinetics were recorded using a time-correlated single photon counting spectrophotometer (Edinburgh Instruments F920) with a time resolution of about 100 ps. A picosecond pulsed diode PLED-300 emitting about 850 ps duration pulses in sub-nanoseconds was used for excitation at 300 nm with an average power of 0.08 mW m−2 and with a 10 MHz pulse repetition rate. All fluorescence spectra were corrected for instrument sensitivity.
The optical density at the excitation wavelength was in the 0.01–0.9 range. The measurement error did not exceed 1–2%. The fluorescence decay kinetics were approximated using one-component or two-component exponentials taking into account the instrumental function with different characteristic decay times.
Note that stilbene could be found in two molecular forms, cis- and trans-stilbene, and these two forms can be interchanged when irradiated with UV light in liquid solutions. This is highly unlikely in our preparations because our samples are solid films. However, even if cis-stilbene existed in the films, spectroscopically it exhibits blue-shifted absorption (peaked at 270 nm) compared to Tstilbene (it has multiple absorption bands at 267, 288, 300, 312, and 327 nm). So cis-stilbene could be only weakly excited in our fluorescence experiment. Additionally, cis-stilbene fluorescence spectra are characterized by one broad fluorescence band with a maximum at 430–440 nm and a very low fluorescence quantum yield, which could be easily filtered out.26
1.2. Computational details
Quantum chemical calculations have been used in our previous studies on stilbene20,25 and similar molecules,27 where the details of the theoretical analysis are described. Density functional theory (DFT) calculations on Tstilbene clusters have been performed using the Gaussian 16 package.28 The B3LYP and CAM-B3LYP functionals in combination with the cc-pVDZ basis set have been used. Initially equilibrated clusters were generated using the classical molecular dynamics (MD) approach with AMBER 21 software29,30 (together with the Packmol package31 for structure generation). MD force fields are not optimized for Tstilbene, hence, the force field was reoptimized for the molecular models described below. Two types of models were used (i) when a single entity (“molecule”) in MD is a single Tstilbene and (ii) when the single “molecule” in MD is a Tstilbene dimer. The reference Tstilbene molecules and dimers are optimized by DFT at the B3LYP/cc-pVDZ level. Partial atomic charges were determined using the RESP procedure as it is required in the antechamber program from AMBER.29 An MD simulation box (size 350 × 350 × 350 Å3), filled with 100, 250, 500 and 1000 Tstilbenes, was used for MD simulations. MD simulations were performed at room temperature for 140 ps.2. Experimental results
Detailed analysis of the fluorescence and absorption spectra of the films containing different concentrations of the Tstilbene molecules was carried out. The absorption spectra of the films presented in Fig. 1A contain three main peaks at 300 nm, 312 nm and 327 nm. By increasing the Tstilbene concentration, it is possible to distinguish the additional peaks at 267 nm and 288 nm. Absorption at 260–270 nm, which clearly manifests itself at the lowest concentrations (0.5–2%), is mainly determined by the PS matrix. The absorption intensity at the stilbene region (290–340 nm) linearly increases with the increase of the concentration of Tstilbene, while this linear dependence disappears when the concentration exceeds 5% (see the inset of Fig. 1A). At larger concentrations the background of white light (presumably due to light scattering) becomes visible and it grows with the concentration.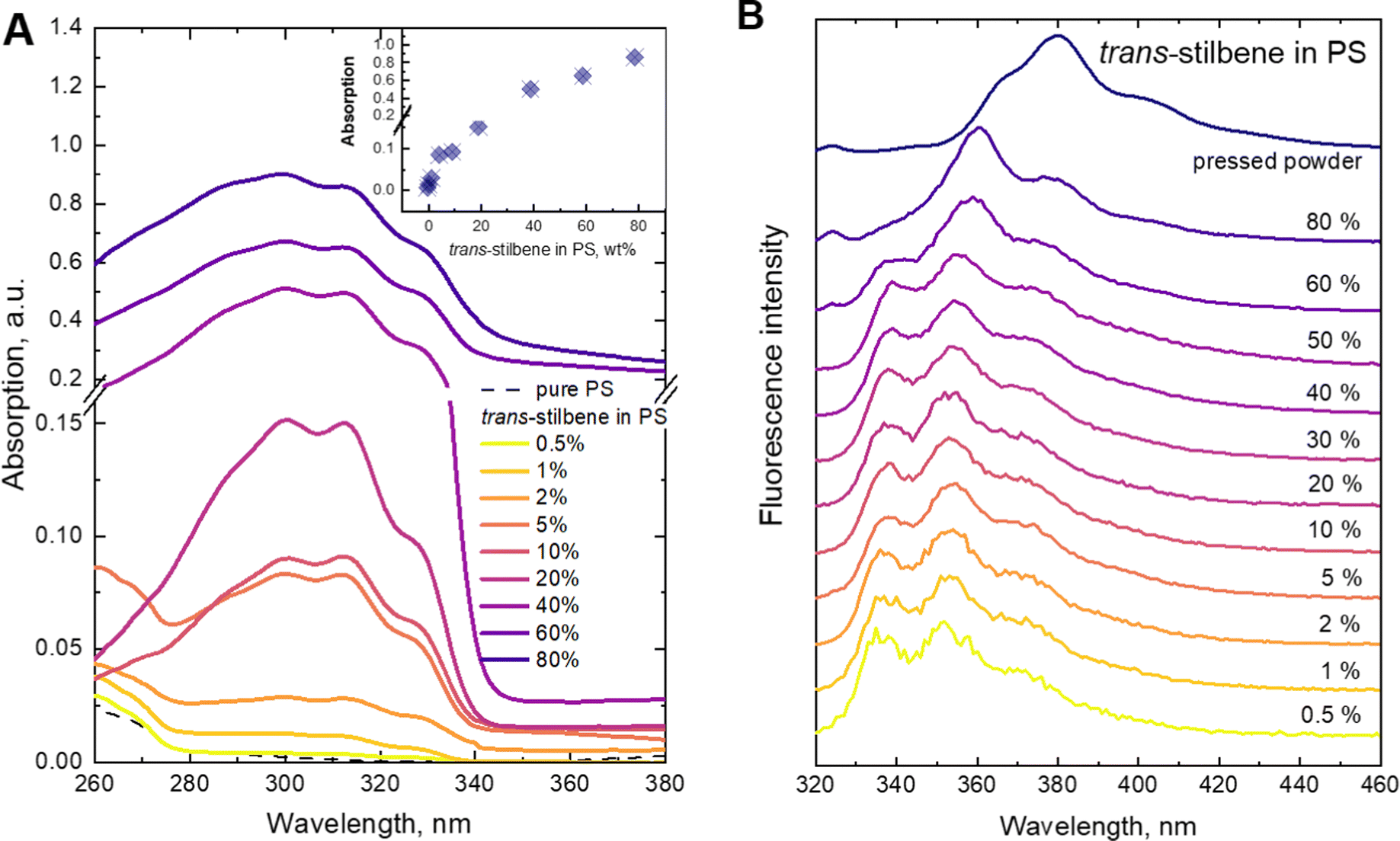 | ||
| Fig. 1 (A) Normalized absorption spectra of trans-stilbene in the PS matrix; in the inset, the dependence of the absorption at 312 nm on the trans-stilbene concentration in the PS matrix is shown. (B) Normalized fluorescence spectra of trans-stilbene in the PS matrix under 300 nm excitation. | ||
Fluorescence spectra were recorded for the same set of samples. For this purpose, the excitation wavelength of the samples was tuned to 300 nm, which corresponds to the absorption peak of the Tstilbene molecules. As shown in Fig. 1B, the fluorescence spectra do not demonstrate considerable variation with concentration at its lower values (less than 50%). At higher concentrations the fluorescence spectrum is largely red-shifted with changes in the peak structure and redistribution of intensities. The integrated fluorescence intensity also depends on the concentration of stilbene, as shown in Fig. 2. The initial linear increase of the fluorescence intensity with the increase of the stilbene concentration (up to 10%) should evidently support the attribution of the fluorescence signal to the molecular emission at these concentrations. A subsequent decrease of the fluorescence intensity at higher concentrations (>10%) is accompanied by variation of the fluorescence spectrum. Such intensity and spectral changes with the concentration might be caused by gradual aggregation of the stilbene molecules.
 | ||
| Fig. 2 Integrated fluorescence intensity of trans-stilbene dependence on concentration in the PS matrix under 300 nm excitation. Molecular structure of trans-stilbene is also shown. | ||
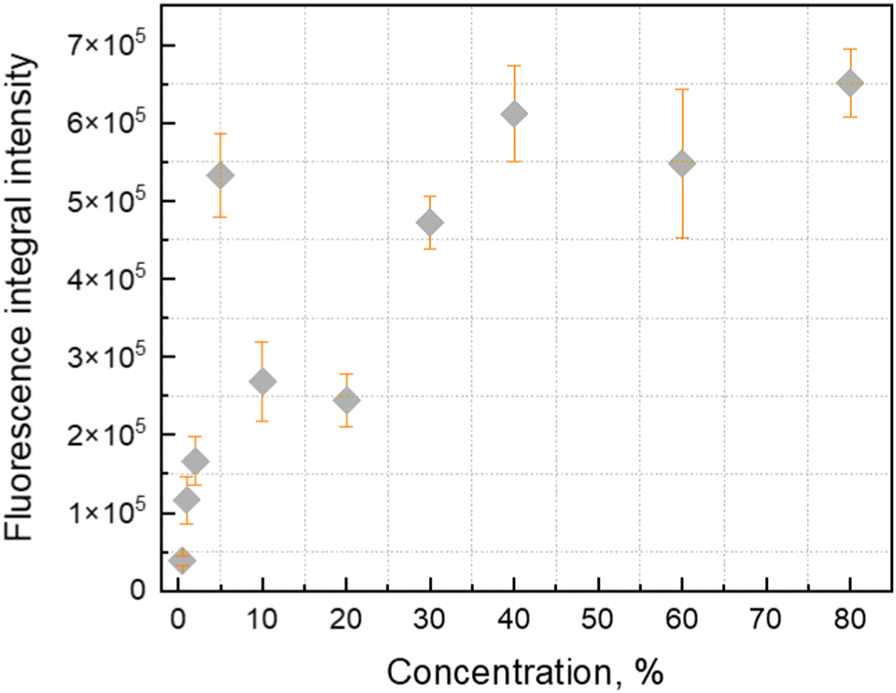
Fluorescence kinetics are more sensitive to the concentration of the stilbene molecules in the sample, as shown in Fig. 3. An interplay between three distinct processes depending on the concentration is evidently distinguished. The fast exponential decay component is dominating at lower concentrations. The corresponding decay time gradually reduces from 0.75 ns at the lowest concentrations to 0.17 ns at 40%. Starting from 60% the kinetics become nonexponential demonstrating the emergence of the second type—slow—ns timescale kinetics. We found that the decay becomes slow and again exponential in the pressed sample (third type kinetics), when the polystyrene matrix is absent.
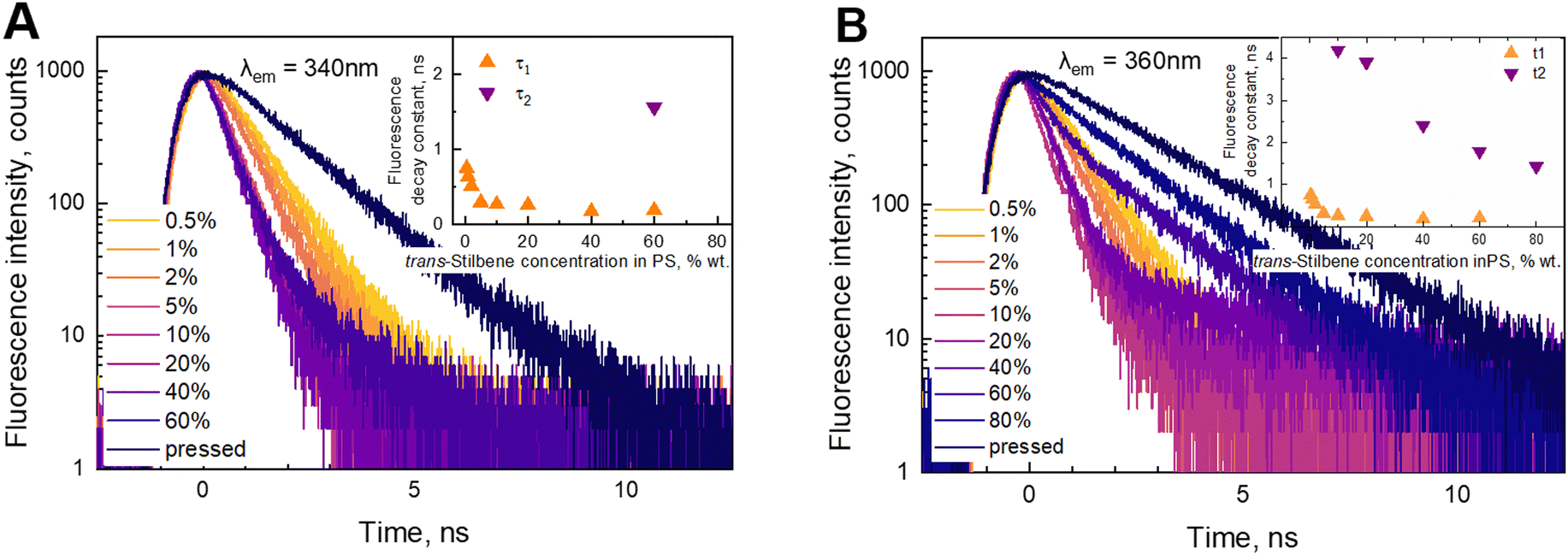 | ||
| Fig. 3 Fluorescence decay kinetics of trans-stilbene in the PS matrix at 340 nm (A) and 360 nm (B) under 300 nm excitation. Dependences of the fluorescence decay constants (τ1 and τ2) on the stilbene concentration in the PS matrix are presented in the insets. | ||
The data of the analysis of the fluorescence kinetics corresponding to different fluorescence bands are presented in Table 1. Two fluorescence bands were studied at 340 and 360 nm. Our experimental data showed that up to 5% concentration, the decay behaves identically—increase of the concentration leads to faster decay. At higher concentrations (10–40%), the 360 nm band shows an additional slow component, while the 340 nm band still retains the mono-exponential character. The decay time of the fast component at the two wavelengths is the same. According to our experimental data, we found that the 340 nm band still shows properties of a separate molecule, while at 360 nm, there is an overlap with the fluorescence from the aggregated species. Complete aggregation is achieved at the pressed sample. In this case we find a new slow mono-exponential decay. We concluded that this result correlates with the fluorescence spectrum (Fig. 1B). The fluorescence spectrum of the pressed sample has no intensity at 340 nm.
| Trans-stilbene concentration in PS, % wt | Absorption band positions, nm | Fluorescence band positions, nm | τ 1 340, ns | τ 2 340, ns | τ 1 360, ns | τ 2 360, ns |
|---|---|---|---|---|---|---|
| 0.5 | 300, 312, 327 | 336, 352, 372 | 0.75 (100%) | — | 0.75 (100%) | — |
| 1 | 300, 312, 327 | 337, 353, 372 | 0.64 (100%) | — | 0.65 (100%) | — |
| 2 | 300, 313, 328 | 337, 353, 372 | 0.52 (100%) | — | 0.53 (100%) | — |
| 5 | 286, 300, 312, 327 | 338, 354, 372 | 0.3 (100%) | — | 0.31 (100%) | — |
| 10 | 286, 300, 312, 327 | 338, 353, 373 | 0.27 (100%) | — | 0.26 (93%) | 4.2 (7%) |
| 20 | 287, 300, 312, 327 | 338, 353, 373 | 0.27 (100%) | — | 0.25 (85%) | 3.9 (15%) |
| 40 | 267, 288, 300, 312, 327 | 338.5, 355, 374 | 0.17 (100%) | — | 0.18 (98%) | 2.4 (2%) |
| 60 | 267, 288, 300, 313, 328 | 339, 359, 377, 400 | 0.19 (91%) | 1.6b (9%) | 0.21 (47%) | 1.8 (53%) |
| 80 | 268, 288, 300, 313, 329 | 340, 360, 379, 400 | — | — | — | 1.4 (100%) |
| Pressed | 345, 365, 380, 403 | 1.6 (100%) | 2.2 (100%) |
In order to check the validity of the results, we have decomposed the fluorescence spectra into two components: the molecular spectrum, corresponding to the 0.5% concentration sample, and the spectrum of the aggregated species, corresponding to the spectrum obtained at 80% concentration. Fluorescence at intermediate concentrations is then assumed to be given by the weighted superpositions of these two components. The reconstructed spectrum is presented in Fig. 4A. The recorded experimental spectra are shown as well. This result nicely demonstrates that by varying stilbene concentrations, the stilbene molecules aggregate into complexes (or nanocrystals) and the resulting sample consists of two phases—the molecular part and the aggregated part. The two phases have different fluorescence spectra and different fluorescence kinetics. This is confirmed by the images shown in Fig. 4B obtained by transmission electron microscopy (TEM), which demonstrate the emergence of crystallized formations in the solution as the concentration is increased. At low concentrations the sample is homogeneous. At 10% concentration 20 nm nanostructures can be observed. They combine into larger aggregates at 60–80% concentrations. It should be noted that TEM images suggest the presence of a molecular phase which is not crystallized, but it shows almost no fluorescence compared to the crystallized part. Hence, the fluorescence is mostly from 100–300 nm crystalline forms. The crystallization is confirmed by diffraction patterns.
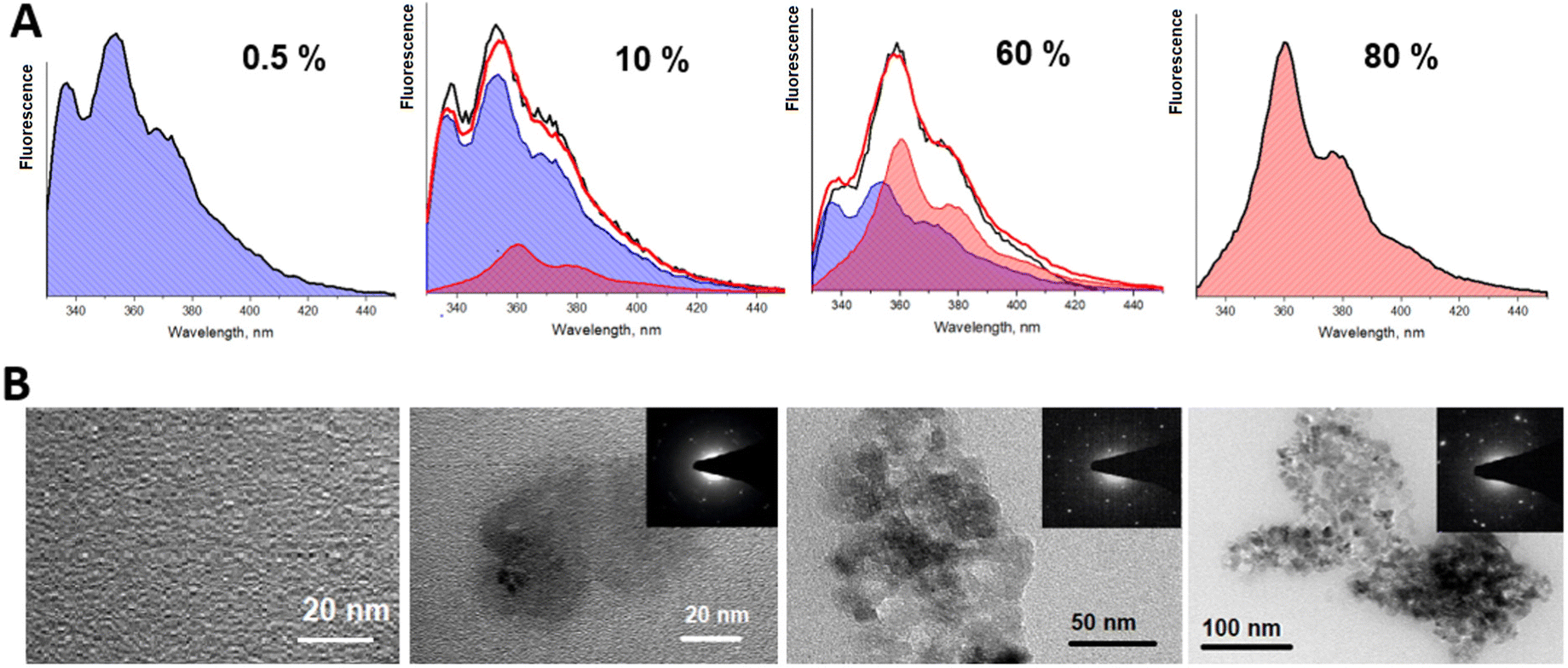 | ||
| Fig. 4 (A) The normalized fluorescence spectra obtained (black line) and reconstructed (red line) at different concentrations of trans-stilbene in the PS matrix. Fluorescence spectra filled in blue demonstrate a molecular part of trans-stilbene fluorescence and those of aggregates (or nanocrystals) in pink. Note that there is still a small contribution of molecular fluorescence at 80% concentration. Amplitudes of decomposition are presented in Fig. 6. (B). TEM images of the same films. | ||
Our experimental data showed that Tstilbene molecules demonstrate aggregation-induced quenching of the excited state for the single-molecular form in low concentration solutions. In this case the shape of the absorption spectrum does not change with concentration. A completely different picture follows from fluorescence at high concentrations in a polycrystal state where the fluorescence becomes red-shifted and the fluorescence decay kinetics are slow. It is important to relate the spectral properties to the microscopic structure. Two microscopic levels of structural reorganization are possible upon crystallization: (1) reorganization of the molecular structure within a single molecule and (2) emergence of intermolecular interactions upon packing. In order to study these properties we performed a hybrid computational study by using density functional theory (DFT) and molecular dynamics (MD) simulations.
3. Calculations and analysis
In our previous study the quantum chemical analysis of Tstilbene dimers was carried out25 and it was shown that dimerization leads to a slight twisting of dihedral angles starting from the planar Tstilbene geometry20,25 Additional quantum dynamics (QD) calculations over an 80 fs time scale demonstrated the persistence of the ±15° twisting at room temperature. This indicates that the twisted configuration remains stable, even when considering the fluctuations. However, this did not lead to a conclusive picture regarding Tstilbene aggregation on a large scale.To consider Tstilbene packing in large clusters we employed MD simulations. As the first attempt we performed the “traditional” MD analysis by considering Tstilbene molecules as independent entities. The obtained MD trajectory showed no indication of Tstilbene clusterization (Fig. 5A). The possible reason for this is the absence of dipole moments in single Tstilbenes; hence, the electrostatic intermolecular interactions are weak. In this case, only π-stacking interactions,32–34 which typical MD simulations do not take into account, may be necessary for clusterization. To check this hypothesis we performed MD simulations by taking the whole DFT-optimized Tstilbene dimer (where π-stacking interactions are in place) as a single “supermolecule”. This type of MD simulation resulted in various types of clusters within 1.5 ps with 4–6 Tstilbenes in the clusters, which remained stable for at least 1 ps (Fig. 5B).
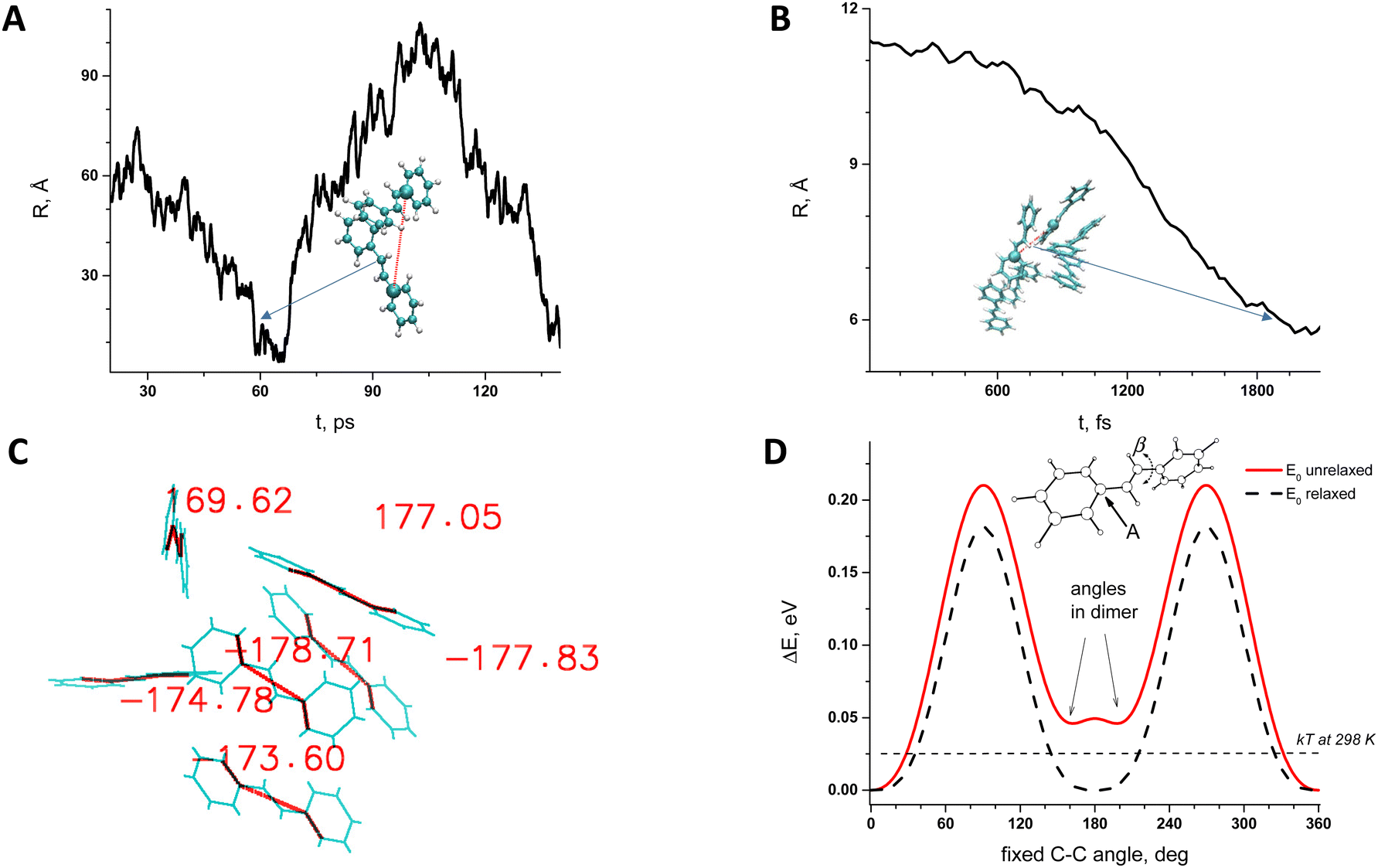 | ||
| Fig. 5 Molecular dynamics simulations and global minima according to DFT studies: (A) – trans-stilbene parametrized as monomers can create temporary dimers even in 100 ps simulations time line; (B) – trans-stilbene parametrized as dimers can create many complexes even in its first 2 ps of the simulations; (C) – dihedral angles after optimisations with the B3LYP computational level for chosen complexes of the 6 trans-stilbenes from MD simulations (figure B); (D) – potential energy surface of a single trans-stilbene molecule with respect to twisting along the single-bond (β angle). In figures (A) and (B), the distance R is chosen between dimers and two atoms labelled as (A) in figure (D). | ||
The obtained clusters from MD simulations were taken as initial structures for further quantum chemistry refinement using DFT. DFT geometry optimization was performed for one specific six-Tstilbene cluster using the CAM-B3LYP functional (Fig. 5C) (and B3LYP for comparison). We note that all Tstilbene molecules in the refined cluster turned into a permanently twisted (by 10–40 degrees) form. This result correlates with our previous study where we showed that dimerization of stilbene results in molecular twisting.20,27Fig. 5D indicates that such twisting is not accidental. Twisting Tstilbene along the single bond using unrelaxed conditions (when hydrogens are fixed) yields the double-well potential energy surface with a minimum exactly at 30 degrees away from the ideal planar trans configuration. Such an unrelaxed configuration is available even for a single Tstilbene molecule via thermal activation at room temperature.
To relate the structural information to that from spectroscopy, we have also calculated the excitation energies of single Tstilbene and of the Tstilbene cluster. We find that the cluster has six nearby excitations with energies shifted by −0.118 (0.0), −0.078 (1.44), −0.048 (0.27), 0 (1.22), +0.032 (0.44), and +0.092 (1.04) eV from the single-stilbene optical transition (oscillator strength in D2 is given in parenthesis; the single stilbene oscillator strength is 0.97 D2). Notice that the lowest energy excitation has zero oscillator strength (is optically dark). This is the same result as for the Tstilbene dimer.20,27 The cluster thus should act as the excitation quencher, because an energetically favourable state is not fluorescing. The next state is also red-shifted, and it is optically brighter than that of the single molecule. Neither of these states has the CT character; hence, they are purely excitonic.
The computational results correlate with our spectroscopy data. First of all, the molecular packing in clusters is not compact, so molecules do not form extended molecular crystals. As a result, the disordered molecular clustering results in excitation energies scattered around the molecular transition. The fine restructuring due to aggregation is not observable due to the broad absorption bands. However, the cluster shows red-shifted low energy excitations, which correlates with the red-shifted fluorescence spectrum in aggregated stilbene. More importantly this result correlates with the slow fluorescence decay in the aggregate form due to the vanishing oscillator strength of the lowest energy excited state of the stilbene cluster.
Discussion and conclusions
Detailed studies of the variability of the Tstilbene absorption and fluorescence spectra in the PS matrix depending on the concentration are carried out experimentally and by means of relevant hybrid MD simulations and DFT modelling. As follows from our experimental data the absorption spectra do not vary much with concentration. This implies that (i) either stilbene molecules remain weakly perturbed by each other in the course of aggregation and, thus, molecular features are not affected in the electronic ground state even at high concentrations or (ii) only non-crystalline regions of the solvent efficiently absorb the light. However, fluorescence shows a completely different picture: the spectrum barely depends on the concentration up to 60%, while a substantial shift of the fluorescence spectra emerges at higher concentrations. Naturally, this spectral shift is attributed to the formation of aggregates in the PS matrix which is supported by observations from the TEM measurements. Indeed, these data demonstrate that some evidence of stilbene crystallization can be observed even at low concentrations (starting from 10%) with the growth of microcrystals in size at higher concentrations. The modelling data support these observations.The inconsistency of the sensitivity between absorption and fluorescence spectra depending on concentrations can be understood in terms of the origin of molecular orbitals. Due to molecular symmetry, the dipole moment of the molecule in the electronic ground state is absent and, thus, the intermolecular interactions are weak even at relatively short distances. A different picture emerges when considering fluorescence. The excited state potential surface is known to be completely flat in trans configurations.1,2,5,6,12,20,23 The molecule thus easily experiences different configurations in the excited state. However, such molecular twisting is restricted in the crystalline state of our samples.
The integrated fluorescence intensity presented in Fig. 2 is yet another complicated dependence that is difficult to explain in this context. At low concentrations <5% separate molecules should certainly dominate in the fluorescence spectra and, thus, the intensity should linearly increase with the concentration. At 5% the only thing that can happen is related to the formation of small nanoparticles, most probably dimeric structures. Apparently, such small nanoparticles act as fluorescence quenchers (which follows from calculations of the clusters and dimers25). The relative content of dimers grows up to 20% of the stilbene concentrations, resulting in a decrease of the fluorescence yield. These species could not be observed in the fluorescence spectrum, so they do not display any contribution in the spectra shown in Fig. 1B. For concentrations from 20 to 40%, a different kind of aggregate (nano-crystals) starts to dominate. These nanocrystals are fluorescent and the fluorescence yield increases as their concentration increases. At concentrations >40% the excitation migration becomes efficient resulting in equilibration among fluorescent and quenching species, so that the fluorescence intensity stays almost constant. Notice that at these concentrations light scattering is also very efficient and so this is an additional factor that may restrict the growth of fluorescence intensity.
It should be noted that stilbene crystals were present with polystyrene in the investigated materials. From a theoretical point of view the formation of stilbene crystals in polystyrene is puzzling and the influence of polystyrene is still not clear. Styrene monomers are structurally similar to stilbene. So presumably stilbene should interact with styrene very similarly to with another stilbene. In this case the sample would remain homogneneous at all concentrations of stilbene. As shown in Fig. 4B at high concentrations, there are strict boundaries of the crystalline phase. This implies that stilbene molecules combine with other stilbenes and “pushes” styrene out so that stilbene-only crystals are formed. It turns out that inter-stilbene interaction must be higher than that of stilbene–styrene. This is supported by Fig. 6, which indirectly shows the fractions of single-molecular phase stilbene and of crystallized stilbene. The result is highly non-linear, thus showing that the single-molecule phase stays appreciable until very high concentrations (up to 80%). As a result, the single-molecule phase is maintained from a moderate local stilbene concentration up to very high concentrations (up to 80%) in the whole sample. The single-molecule phase of Tstilbene corresponds to the flat molecular structure and is responsible for the concentration-independent absorption spectrum. Notice that we were unable to obtain the absorption spectrum of the pressed sample. As molecules crystallize, they become twisted and their absorption diminishes. As the crystal contains excitations lower than those in the single-molecule phase, the crystal becomes an excitation acceptor, thus being responsible for fluorescence.
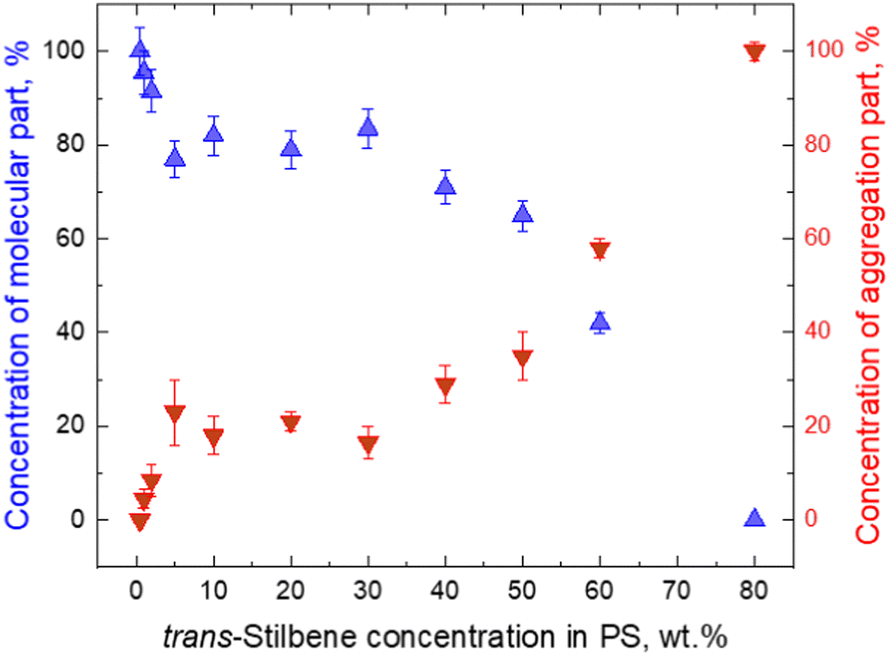 | ||
| Fig. 6 Molecular (blue triangles) and aggregate (red triangles) parts of fluorescence trans-stilbene dependence on concentration in the PS matrix. The points present amplitudes in decomposition of Fig. 4A. | ||
Phase separation is also observed in the fluorescence spectra. Most notably they are different at two wavelengths. Both at 340 and 360 emission wavelengths fluorescence exponentially decays at small concentrations (0.5–5%), while the decay rate increases with concentration. The increase of the decay rate with concentration is not easy to explain. Phenomenologically the effect is called concentration quenching. On a microscopic level, this behaviour can be explained by assuming excitation energy transfer from fluorescent monomers to quenching sites (dimers or similar aggregates). The increasing concentration effectively leads to shortening of the distance between monomers and dimers so the transfer rate to the quencher increases and the decay time becomes smaller. The exponential regime suggests that the process is one-step transport limited by transfer time.
Increasing the concentration further makes the fluorescence kinetics become non-exponential indicating an excitation diffusion process, i.e., multistep energy transport to excitation quenchers. This continues until larger crystals are formed (at concentrations above 80%); these crystals demonstrate their own decay kinetics which is exponential again, while much slower. At this regime the diffusion to the quenchers (possibly on the surface of microcrystals) is faster than the decay time at the quencher sites. These quencher sites, corresponding to nanocrystals, are different from quenchers at low concentrations of stilbene aggregates since their lifetime is different.
Concluding our study suggests the following picture: stilbene solid solution demonstrates formation of two phases as the concentration is increased. The first phase corresponds to the ideal solution of stilbene molecules in a PS matrix. The molecules are in the trans state, and spectroscopically they behave as isolated from each other. This phase demonstrates concentration-induced quenching. This effect may be due to dimerization: the dimers are disordered and the molecules twist away from the pure trans configuration, as suggested by calculations—correspondingly they become fluorescence centers. Excitations easily diffuse among molecules in the aggregates. At even larger concentrations the second nano-crystalline phase becomes dominating. The regularity of the crystal supports extremely fast excitation diffusion. Fluorescent sites at this phase are not known; however, they are all the same—the fluorescence decay kinetics is purely single-exponential. The two phases are distinct not only by their physical state but at long times after excitation can be associated with specific optical processes: nanocrystals are responsible for fluorescence, while the single-molecule phase is responsible for absorption. Such properties may be important for optical quantum technologies where excitation becomes separable from de-excitation.
Author contributions
All authors agreed to the final version of the manuscript. Renata Karpicz: methodology, data curation, formal analysis, and funding acquisition. Gabriele Kareivaite: data curation and visualization. Mindaugas Macernis: methodology, data curation, formal analysis, and writing – editing. Darius Abramavicius: conceptualization, writing – review and editing, and funding acquisition. Leonas Valkunas: supervision, conceptualization, project administration, and writing – original draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by Horizon Europe FLORIN (Project No. 101086142) and the Research Council of Lithuania (Grant No. S-MIP-23-48). The authors would like to thank Dr. Martynas Skapas for TEM nanostructure characterization. Computations were performed on resources at the supercomputer “VU HPC” Saulėtekis of Vilnius University at Faculty of Physics.Notes and references
- D. H. Waldeck, Chem. Rev., 1991, 91, 415–436 CrossRef CAS.
- G. I. Likhtenshtein, Stilbenes: applications in chemistry life sciences and materials science, Wiley-VCH, 2010 Search PubMed.
- T. Teka, L. Zhang, X. Ge, Y. Li, L. Han and X. Yan, Phytochemistry, 2022, 197, 113128 CrossRef CAS PubMed.
- C. Wang, M. D. J. Waters, P. Zhang, J. Suchan, V. Svoboda, T. T. Luu, C. Perry, Z. Yin, P. Slavíček and H. J. Wörner, Nat. Chem., 2022, 14, 1126–1132 CrossRef CAS PubMed.
- G. Orlandi and W. Siebrand, Chem. Phys. Lett., 1975, 30, 352–354 CrossRef CAS.
- D. J. S. Birch and J. B. Birks, Chem. Phys. Lett., 1976, 38, 432–436 CrossRef CAS.
- J. A. Syage, W. R. Lambert, P. M. Felker, A. H. Zewail and R. M. Hochstrasser, Chem. Phys. Lett., 1982, 88, 266–270 CrossRef CAS.
- J. A. Syage, P. M. Felker and A. H. Zewail, J. Chem. Phys., 1984, 81, 4706–4723 CrossRef CAS.
- S. A. Kovalenko and A. L. Dobryakov, Chem. Phys. Lett., 2013, 570, 56–60 CrossRef CAS.
- A. A. Heikal, J. S. Baskin, L. Bañares and A. H. Zewail, J. Phys. Chem. A, 1997, 101, 572–590 CrossRef CAS.
- R. Improta and F. Santoro, J. Phys. Chem. A, 2005, 109, 10058–10067 CrossRef CAS PubMed.
- O. Ünsalan, N. Kuş, S. Jarmelo and R. Fausto, Spectrochim. Acta, Part A, 2015, 136, 81–94 CrossRef PubMed.
- M. Sumitani, N. Nakashima, K. Yoshihara and S. Nagakura, Chem. Phys. Lett., 1977, 51, 183–185 CrossRef CAS.
- G. R. Fleming, S. H. Courtney and M. W. Balk, J. Stat. Phys., 1986, 42, 83–104 CrossRef.
- B. Carmeli and A. Nitzan, Phys. Rev. Lett., 1983, 51, 233–236 CrossRef.
- S. Takeuchi, S. Ruhman, T. Tsuneda, M. Chiba, T. Taketsugu and T. Tahara, Science, 2008, 322, 1073–1077 CrossRef CAS PubMed.
- S. K. Lee, Y. H. Cho, B. H. Kang, W. G. Lee, J. K. Kim, Y. K. Kim, G. D. Kim and N. Z. Galunov, Prog. Nucl. Sci. Technol., 2011, 292–295 CrossRef.
- R. M. Hochstrasser, J. Mol. Spectrosc., 1962, 8, 485–506 CrossRef CAS.
- M. K. Chaudhuri and S. C. Ganguly, J. Phys. C, 1969, 2, 1560 CrossRef CAS.
- J. Catalán, Chem. Phys. Lett., 2006, 421, 134–137 CrossRef.
- N. Ostapenko, M. Ilchenko, Y. Ostapenko, O. Kerita, V. Melnik, E. Klishevich, N. Galunov, I. Lazarev and M. Chursanova, Mol. Crys. Liq. Crys., 2018, 671, 104–112 CrossRef CAS.
- M. Aguiar, L. Akcelrud, M. Pinto, T. D. Z. Atvars, F. Karasz and J. Saltiel, J. Photosci., 2003, 10, 149–155 CAS.
- D. Riedel, M. Cranney, M. Martin, R. Guillory, G. Dujardin, M. Dubois and P. Sonnet, J. Am. Chem. Soc., 2009, 131, 5414–5423 CrossRef CAS PubMed.
- D. Tzeli, G. Theodorakopoulos, I. D. Petsalakis, D. Ajami and J. Rebek, J. Am. Chem. Soc., 2011, 133, 16977–16985 CrossRef CAS PubMed.
- R. Karpicz, N. Ostapenko, Y. Ostapenko, Y. Polupan, I. Lazarev, N. Galunov, M. Macernis, D. Abramavicius and L. Valkunas, Phys. Chem. Chem. Phys., 2021, 23, 3447–3454 RSC.
- M. Sajadi, A. L. Dobryakov, E. Garbin, N. P. Ernsting and S. A. Kovalenko, Chem. Phys. Lett., 2010, 489, 44–47 CrossRef CAS.
- M. Macernis, B. P. Kietis, J. Sulskus, S. H. Lin, M. Hayashi and L. Valkunas, Chem. Phys. Lett., 2008, 466, 223–226 CrossRef CAS.
- G. W. T. M. J. Frisch, H. B. Schlegel, G. E. Scuseria, J. R. C. M. A. Robb, G. Scalmani, V. Barone, H. N. G. A. Petersson, X. Li, M. Caricato, A. V. Marenich, B. G. J. J. Bloino, R. Gomperts, B. Mennucci, H. P. Hratchian, A. F. I. J. V. Ortiz, J. L. Sonnenberg, D. Williams-Young, F. L. F. Ding, F. Egidi, J. Goings, B. Peng, A. Petrone, D. R. T. Henderson, V. G. Zakrzewski, J. Gao, N. Rega, W. L. G. Zheng, M. Hada, M. Ehara, K. Toyota, R. Fukuda, M. I. J. Hasegawa, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, K. T. T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, M. J. B. F. Ogliaro, J. J. Heyd, E. N. Brothers, K. N. Kudin, T. A. K. V. N. Staroverov, R. Kobayashi, J. Normand, A. P. R. K. Raghavachari, J. C. Burant, S. S. Iyengar, M. C. J. Tomasi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, R. L. M. J. W. Ochterski, K. Morokuma, O. Farkas and D. J. F. J. B. Foresman, Gaussian, Gaussian, Inc., Wallingford CT, 2019 Search PubMed.
- H. M. A. D. A. Case, K. Belfon, I. Y. Ben-Shalom, S. R. Brozell, D. S. Cerutti, T. E. Cheatham III, V. W. D. Cruzeiro, T. A. Darden, R. E. Duke, G. Giambasu, M. K. Gilson, H. Gohlke, A. W. Goetz, R. Harris, S. Izadi, S. A. Izmailov, C. Jin, K. Kasavajhala, M. C. Kaymak, E. King, A. Kovalenko, T. Kurtzman, T. S. Lee, S. LeGrand, P. Li, C. Lin, J. Liu, T. Luchko, R. Luo, M. Machado, V. Man, M. Manathunga, K. M. Merz, Y. Miao, O. Mikhailovskii, G. Monard, H. Nguyen, K. A. O’Hearn, A. Onufriev, F. Pan, S. Pantano, R. Qi, A. Rahnamoun, D. R. Roe, A. Roitberg, C. Sagui, S. Schott-Verdugo, J. Shen, C. L. Simmerling, N. R. Skrynnikov, J. Smith, J. Swails, R. C. Walker, J. Wang, H. Wei, R. M. Wolf, X. Wu, Y. Xue, D. M. York, S. Zhao and P. A. Kollman, AMBER, University of California, San Francisco, 2021 Search PubMed.
- M. Macernis, V. Mickus, J. Ahonen, L. Diska, J. Franukevicius and J. Sulskus, arXiv, 2022, preprint, arXiv:2210.00934, DOI:10.48550/arXiv.2210.00934.
- L. Martínez, R. Andrade, E. G. Birgin and J. M. Martínez, J. Comput. Chem., 2009, 30, 2157–2164 CrossRef PubMed.
- H. Tachikawa, R. Iura and H. Kawabata, Sci. Rep., 2019, 9, 2377 CrossRef PubMed.
- X. Ye, Z.-H. Li, W. Wang, K. Fan, W. Xu and Z. Hua, Chem. Phys. Lett., 2004, 397, 56–61 CrossRef CAS.
- J. M. Živković, I. M. Stanković, D. B. Ninković and S. D. Zarić, Cryst. Growth Des., 2020, 20, 1025–1034 CrossRef.
| This journal is © the Owner Societies 2023 |