
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynergistic theoretical and experimental study on the ion dynamics of bis(trifluoromethanesulfonyl)imide-based alkali metal salts for solid polymer electrolytes†
Brigette Althea
Fortuin
 abc,
Jon
Otegi
b,
Juan Miguel
López del Amo
a,
Sergio Rodriguez
Peña
ab,
Leire
Meabe
a,
Hegoi
Manzano
*b,
María
Martínez-Ibañez
*a and
Javier
Carrasco
*ad
abc,
Jon
Otegi
b,
Juan Miguel
López del Amo
a,
Sergio Rodriguez
Peña
ab,
Leire
Meabe
a,
Hegoi
Manzano
*b,
María
Martínez-Ibañez
*a and
Javier
Carrasco
*ad
aCentre for Cooperative Research on Alternative Energies (CIC energiGUNE), Basque Research and Technology Alliance (BRTA), Alava Technology Park, Albert Einstein 48, 01510 Vitoria-Gasteiz, Spain. E-mail: jcarrasco@cicenergigune.com; mmartinez@cicenergigune.com
bDepartment of Physics, University of the Basque Country (UPV/EHU), 48940 Leioa, Spain. E-mail: hegoi.manzano@ehu.eus
cALISTORE-European Research Institute, CNRS FR 3104, Hub de l’Energie, Rue Baudelocque, 80039 Amiens Cedex, France
dIKERBASQUE, Basque Foundation for Science, Plaza Euskadi 5, 48009 Bilbao, Spain
First published on 12th September 2023
Abstract
Model validation of a well-known class of solid polymer electrolyte (SPE) is utilized to predict the ionic structure and ion dynamics of alternative alkali metal ions, leading to advancements in Na-, K-, and Cs-based SPEs for solid-state alkali metal batteries. A comprehensive study based on molecular dynamics (MD) is conducted to simulate ion coordination and the ion transport properties of poly(ethylene oxide) (PEO) with lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) salt across various LiTFSI concentrations. Through validation of the MD simulation results with experimental techniques, we gain a deeper understanding of the ionic structure and dynamics in the PEO/LiTFSI system. This computational approach is then extended to predict ion coordination and transport properties of alternative alkali metal ions. The ionic structure in PEO/LiTFSI is significantly influenced by the LiTFSI concentration, resulting in different lithium-ion transport mechanisms for highly concentrated or diluted systems. Substituting lithium with sodium, potassium, and cesium reveals a weaker cation-PEO coordination for the larger cesium-ion. However, sodium-ion based SPEs exhibit the highest cation transport number, indicating the crucial interplay between salt dissociation and cation-PEO coordination for achieving optimal performance in alkali metal SPEs.
1. Introduction
The ever-increasing global energy demand tied with increasing environmental concerns1 regarding climate change has progressively led towards the incorporation of renewable energy sources, such as wind and solar energy, within national grids. However, considering the intermittent nature of natural energy sources, energy storage is vital towards the realization of a sustainable and efficient energy producing sector. The current market leader for energy storage systems, the lithium-ion battery (LIB), shows a high potential towards mitigating energy fluctuations from renewable energy sources within the grid.2 Considering the rapid expansion of the energy storage market in addition to the rising cost and scarcity of lithium,3,4 the need for developing alternative battery technologies to LIBs, is essential. Furthermore, demand for higher energy density, increased sustainability, abundant and economically viable energy storage systems have led to a surge in the exploration for alternative battery technologies beyond LIBs in recent years.5–7 Post-lithium-ion batteries (post-LIBs), such as lithium–metal batteries (LMBs),8–11 sodium-ion batteries (NIBs),4,12,13 potassium-ion batteries (KIBs),14–17 and cesium-ion batteries (CIBs)18–20 are gaining momentum. Numerous advantages of these post-LIB technologies include higher redox potentials; Li+/Li (−3.04 V vs. SHE), Na+/Na (−2.71 V vs. SHE), K+/K (−2.93 V vs. SHE), and Cs+/Cs (−3.03 V vs. SHE), along with superior theoretical specific capacities,21 an abundance of both sodium and potassium in the Earth's crust posing an attractively low cost alternative to lithium, and the higher diffusion coefficients (low diffusion barrier) of cesium-based electrodes resulting in hindered dendrite formation for cesium-ion batteries.20,22 However, despite its promise, alkali metal batteries face high reactivity, with electrode and electrolyte components spontaneously reacting with most polar aprotic electrolyte solvents, resulting in unstable solid electrolyte interphases, which may affect cycle performance.21,23 The limited stability and lifetime challenges are further reduced by failure mechanisms, such as dendritic growth on the anode and the current collector, internal short-circuits and electrolyte decomposition, resulting in electrolyte loss.21,23,24 Addressing these challenges, the successful development of alternative all-solid-state alkali metal battery technologies necessitates the advancement of alkali metal salts, considering their compatibility with various polymer matrices and alkali metal-based electrodes. Therefore, a fundamental understanding of ion coordination as well as ion transport mechanisms is crucial for assessing the overall performance of these SPEs.Atomistic modelling of polymer electrolytes is an effective approach to accelerate the search towards new alkali metal salts and alkali metal-based electrolyte components, especially considering the recent advances in computing power.8,25–29 The growing utilization of modelling techniques is driven by several factors, including reduced environmental impact compared to experimental processes, faster simulation times compared to traditional experimental methods, and the capability to acquire information that may not be readily attainable through experimental characterization techniques.8,30–34 Yet, a key element for the successful integration of computational-based methods in materials development is comprehensive model validation against well-defined, high-quality experimental data, which is often lacking but crucially needed in the field.
Herein, we present a comprehensive study of the well-known PEO/LiTFSI SPE as a function of LiTFSI concentration. Our approach involves predicting ion coordination and ion transport using atomistic molecular dynamics (MD) simulations. A key aspect of our study is the validation of the model, where we have taken particular care to combine three different techniques: Raman, magic angle spinning nuclear magnetic resonance (MAS-NMR), and electrochemical impedance spectroscopies. This combination of well-defined experiments is crucial for validating our theoretical models and simulations. Notably, such a synergistic model validation study has been lacking in the literature on the PEO/LiTFSI SPEs, and our work fills this important gap. Furthermore, our validated theoretical approach provides valuable insight that extends beyond the PEO/LiTFSI system. Specifically, it allows us to predict similar properties for alternative alkali metal-based systems, such as PEO/XTFSI (X = Na, K, and Cs), broadening the scope of our study. These insights offer valuable guidance for designing new salts for SPEs, thus contributing to the advancement of the field.
2. Theoretical and experimental methods
2.1. Theoretical methods and computational details
Classical MD simulations were conducted on PEOn/LiTFSI for four different EO/Li+ ratios (n = 6, 16, 20 and 32). To ensure the robustness of our results and eliminate any potential bias arising from the choice of MD software, we employed two of the most widely used codes in the literature: Gromacs35 and LAMMPS.36 Convergence tests were meticulously conducted to validate the consistency of results obtained from both codes. These tests, based on the total ionic conductivity as a function of temperature, enabled us to identify the optimal simulation time and box size for the studied systems (cf. Fig. S1–S3, ESI†). Based on these tests, a simulation time of 100 ns and a medium-sized box containing 40 ion pairs and 40 polymer chains were deemed sufficient to achieve accurate and comparable outcomes.Considering that LAMMPS generally exhibits slower simulation performance compared to Gromacs,37,38 we selected Gromacs to investigate the influence of salt concentration on Li-containing systems. This choice was particularly advantageous for highly diluted systems, where longer MD simulations are needed to obtain suitable statistical averages, making Gromacs the preferred option due to its higher speed. In contrast, LAMMPS was exclusively utilized to study one intermediate concentration (n = 20) while also exploring the role of different alkali metals (Li+, Na+, K+, and Cs+). By adopting this approach, we aimed to ensure the validity and reliability of our findings while investigating the diverse aspects of our research.
Specifically, we considered the following computational setups. For the simulations performed using Gromacs,35 the simulation boxes consisted of 40 PEO chains with 24 EO repeating units in each chain (Mw = ∼1056 g mol−1), and 160, 60, 48 and 30 LiTFSI ion pairs, for n = 6, 16, 20, and 32, respectively. Initial boxes were generated randomly as implemented in Gromacs. With LAMMPS36 we examined the ion coordination and transport properties for PEO20/XTFSI systems, where X = Li, Na, K, or Cs. In this case, the simulation boxes consisted of 40 PEO chains with 20 EO repeating units in each chain (Mw = ∼880 g mol−1), and 40 XTFSI ion pairs with initial configurations containing randomly positioned molecules, generated using Packmol.39
Prior to the MD production simulations, an efficient and multistep protocol was applied to prepare and equilibrate the systems. It is worth noting that due to the implementation of Gromacs programming, the initial simulation box needs to be relatively large, resulting in low densities, e.g., 1.649 × 10−1 g cm−3 for PEO32/LiTFSI. To ensure the appropriate geometry of the system, the first step in this case involves an energy minimization process where the positions of the molecules are adjusted to achieve a state of minimum energy. Accordingly, an initial NPT structural compression step was carried out using the Berendsen thermostat and the Parrinello–Rahman barostat (with a relaxation time of 5 ps for all cases). This compression was performed at −267 °C (10 K) under a pressure of 98.99 atm (100 bar) to obtain densities closer to experimental values, i.e., simulated (experimental): 1.352 (1.234) g cm−3 (PEO6/LiTFSI), 1.193 (1.188) g cm−3 (PEO16/LiTFSI), 1.159 (1.179) g cm−3 (PEO20/LiTFSI), and 1.167 (1.164) g cm−3 (PEO32/LiTFSI). Subsequently, a gradual heating process to 327 °C (600 K) at 1 atm and equilibration in an NVT ensemble were conducted to prevent the formation of possible metastable configurations. The temperature increase followed an exponential pattern based on the time constant (1 ps). Afterwards, the systems underwent a cooling NPT procedure to reach the simulation temperature of 70 °C (343 K) and pressure of 1 atm. The final configuration was further equilibrated for 1 ns under similar conditions. Finally, the production MD simulations were performed in the NVT ensemble for 200 ns (PEOn/LiTFSI) or 100 ns (PEO20/XTFSI) to ensure a fully diffusive regime.
All-atom optimized potentials for liquid simulations (OPLS-AA) force field34,40–44 were utilized to describe the energy potentials of PEO, Li+, Na+, K+, Cs+, and TFSI−, including the force field parameters (bond stretching, bond-angle, dihedral angle, and Lennard-Jones potential). The force field parameters used in this study are available in the ESI† (Tables S1, S2 and Fig. S4). The trajectories obtained by the MD simulations were analyzed using Travis analyzer.45,46
We utilized the default atomic charges from the OPLS-AA force field for the PEO atoms. However, to enhance the accuracy and suitable description of our systems, we conducted an optimization of the TFSI− anion's structure in the gas-phase and computed its corresponding atomic charges using density functional theory (DFT) calculations. Subsequently, we incorporated these recomputed charges to update the original OPLS-AA force field. The DFT calculations were performed with the Fritz Haber Institute ab initio molecular simulations (FHI-AIMS) software,47,48 incorporating the Becke's three parameters (B3) exchange functional with the Lee–Yang–Parr (LYP) nonlocal correlation functional (B3LYP)49,50 adopted with the “tier2” standard basis set in the FHI-AIMS code. The partial charges of TFSI− were calculated using the electrostatic potential (ESP) method26,27,30,34,51–54 and scaled to −0.7. The charges of Li+, Na+, K+, and Cs+ were also scaled to +0.7 each to maintain charge neutrality. This scaling factor is used to account for the effects of polarization, which are not considered in conventional classical MD simulations since the charges are treated as unchangeable point charges. The value of 0.7 is often used in the literature for simulating similar PEO-based polymer electrolytes.26,27,52–54 The cut-off for van der Waals forces and the real space of Ewald summation was selected to be 10 Å, with the fast smooth particle mesh Ewald (PME) electrostatics55,56 to treat Coulomb interactions in periodic systems.
Considering the computed MD trajectories, the diffusion coefficient of each species was deduced from its mean square displacement (MSD), using the Einstein relation:57
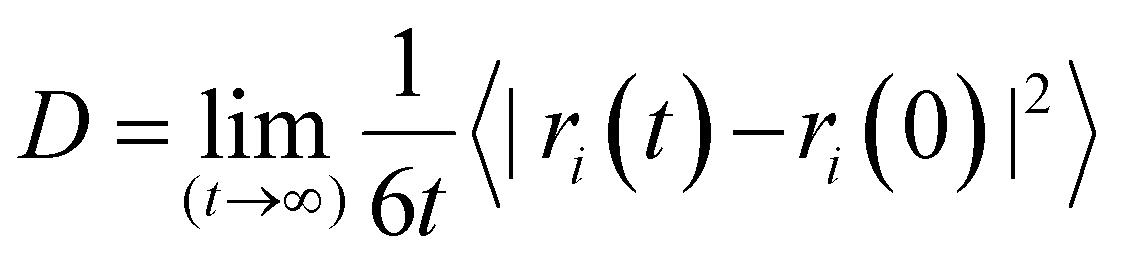 | (1) |
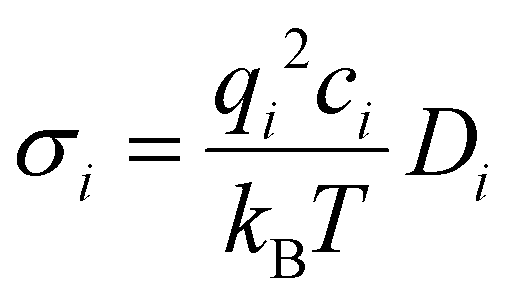 | (2) |
The cation transport numbers based on MD simulations were calculated using eqn (3):
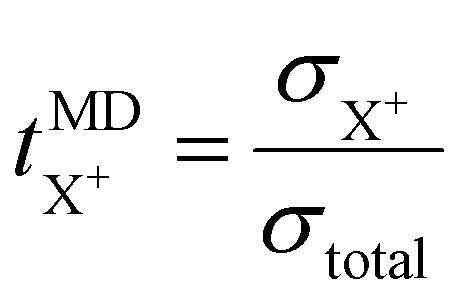 | (3) |
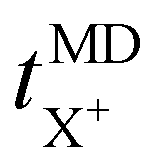 represents the cation transport number, σX+ the ionic conductivity, and σtotal the total ionic conductivity, determined from the sum of the cation and anion conductivities, for the respective cation species (X+ = Li+, Na+, K+, or Cs+) with the TFSI− anion.
represents the cation transport number, σX+ the ionic conductivity, and σtotal the total ionic conductivity, determined from the sum of the cation and anion conductivities, for the respective cation species (X+ = Li+, Na+, K+, or Cs+) with the TFSI− anion.
2.2. Materials
PEO (Mw = 5 × 106 g mol−1), LiTFSI (99.9%), and acetonitrile (ACN) were purchased from Sigma Aldrich. Prior to use, LiTFSI was dried overnight at 100 °C under vacuum. An argon-filled glovebox was utilized to conduct all the procedures related to the moisture or oxygen sensitive materials (MBraun, H2O and O2 < 0.5 ppm).2.3. Preparation of polymer electrolyte membranes
We used the solvent-casting preparation method to fabricate five different PEOn/LiTFSI formulations, with ethylene oxide (EO)/Li+ ratios of 6, 16, 20, 32, and 64. The PEO-systems were dissolved in ACN and subjected to a two-step drying process to remove any residual solvent: (i) the solution was left to dry under ventilation at 35 °C for 24 h, and (ii) it was further dried under dynamic vacuum at 50 °C for an additional 24 h. Subsequently, PEO-based SPEs were prepared using the hot pressing method at 60 °C and 3 tons, with varying processing times. For the measurement of ionic conductivity, SPEs with a target thickness of 300 μm and a diameter of 4 mm were used. Meanwhile, lithium symmetric cells were constructed with SPEs having a thickness of 100 μm and a diameter of 16 mm.2.4. Differential scanning calorimetry (DSC)
A DSC Discovery 2500 (TA Instrument) instrument was used to study the phase transition behaviour of the SPEs from −80 to 100 °C, with heating and cooling rates of 10 K min−1. Following this, the SPE samples (ca. 10 mg) were sealed in Al crucibles in an argon-filled glovebox. Thermal properties, including the values for the glass transition temperature (Tg, midpoint of the heat capacity change), melting temperature (Tm, maximum of the endothermic peak), and melting enthalpy (ΔHm, area below the endothermic peak), were deduced from the second heating scan. The calculation of the crystalline fraction (χC) of the polymer electrolytes was performed using eqn (4):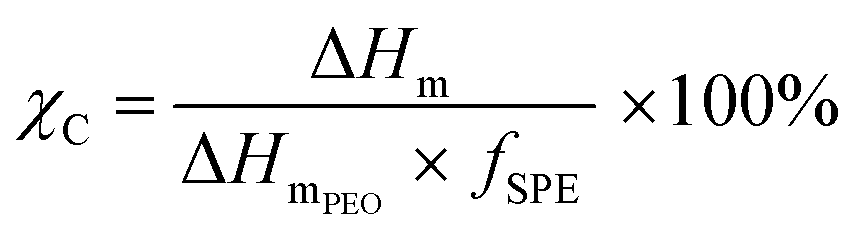 | (4) |
2.5. Ionic conductivity and lithium transference number
CR2032 type coin cells were assembled in an argon-filled glovebox to measure both the ionic conductivity and the lithium-ion transference number for the respective studied systems. The ionic conductivity was measured by using two stainless steel (SS) blocking electrodes, with the configuration SS|SPE|SS and a SPE area of 0.1257 cm2, based on electrochemical impedance spectroscopy (EIS), performed on a Multichannel VMP3 (Biologic, Claix France) with a signal amplitude of 10 mV over the frequency range 10−1–106 Hz, at 40 and 70 °C. A stabilization period of 8 h was employed for the measurement at 40 °C, and 2 h for the measurement at 70 °C. The following equation was employed to calculate the ionic conductivity, eqn (5):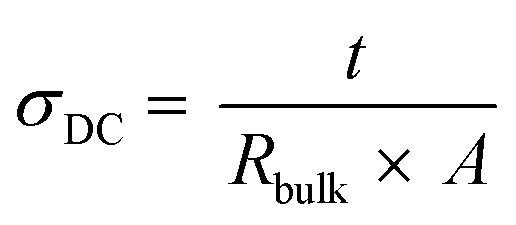 | (5) |
Lithium-ion symmetric cells were assembled using a Li|SPE|Li configuration. The lithium-ion transference number was measured following the procedures as described by Bruce and Vincent et al.,59 measured at 40 and 70 °C. CR2032 type coin cells were employed, sandwiching the SPEs between two lithium discs, with a lithium–metal area of 1.54 cm2. Prior to EIS measurements, the cells were left to stabilize at 40 °C for 8 h, and at 70 °C for 6 h. Eqn (6) was employed to calculate the lithium-ion transference number, 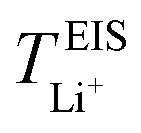 :
:
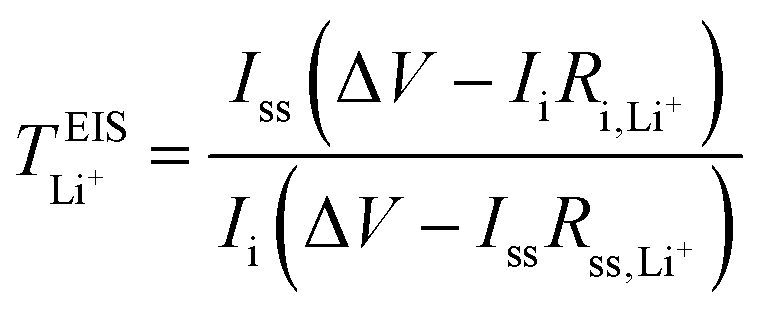 | (6) |
2.6. Raman spectroscopy
Raman spectra were collected using a Renishaw inVia confocal Raman spectrometer (serial number 16H981) with an incident laser with a wavelength of 532 nm (laser spot size: 0.8 m; spatial resolution: 0.4 m). The Raman spectra, collected in the range of 3000–300 cm−1, were recorded at 25, 40, and 70 °C with increasing temperature, and a stabilization time of 2 h (40 °C) and 1 h (70 °C). To increase the signal-to-noise ratio (S/N), each plotted spectrum is the average of 10 accumulations of approximately 3 minutes each. The spectra shown has been normalized from 0 to 1. The samples were placed in a sealed, air-tight cell built with a Raman-inactive glass window and assembled in an argon-filled glovebox to prevent contamination from ambient atmosphere (e.g., air, water).2.7. MAS-NMR spectroscopy
7Li MAS-NMR spectroscopy was employed to determine the chemical environments of lithium-ion upon concentration changes within the PEO matrix. The experiments were recorded using a WB 500 MHz Bruker Advance III spectrometer equipped with a 2.5 mm probe and was conducted on LiTFSI powder, concentrated PEO6/LiTFSI and diluted PEO32/LiTFSI SPEs. All samples were spun at a magic angle with a MAS frequency of 20 kHz. The 7Li MAS-NMR spectra were recorded by using a single pulse experiment (3 μs pulse), with its chemical shift referenced to a 0.1 M solution of LiCl. The temperature-variation NMR spectra were recorded by varying the temperature, from 40 to 80 °C, with a stabilization time of approximately 1 h in between measurements. DMFIT software was used to analyze the spectra.603. Results and discussion
Ion coordination, lithium-ion speciation, and ion transport properties were assessed through MD simulations, focusing on particle density changes around lithium cation or TFSI− anions within the PEO polymer matrix. The MD simulations were conducted at 70 °C, as classical MD simulations are limited to studying molecular kinetics and cannot properly simulate the thermodynamic properties of semicrystalline regions that might be present at lower temperatures. Moreover, the modelling of the PEO64/LiTFSI system was excluded since longer time and length scales are required to suitably account for the diffusivity of ionic species within PEO at such low concentrations.53,54,61,62The computational framework was validated at 70 °C using experimental techniques, including DSC to analyze polymer chain dynamics, Raman, and NMR spectroscopies for probing the ion coordination environments, and EIS for examining ion transport properties.
The validation of the computational framework enabled the prediction of various properties of alternative alkali metal-ions. MD simulations were performed on PEO20/XTFSI, investigating the effects of substituting lithium with sodium, potassium, or cesium cations on ionic transport mechanisms. The analysis included ion coordination, ion conductivity, and ion transport, which are critical factors influencing the performance of SPEs in all-solid-state batteries.
3.1. Model validation of PEOn/LiTFSI SPEs
Fig. 1(a) and (b) displays both the RDFs and CNs of interaction of the reference particle, lithium, with either PEO, denoted as Li+−O−(PEO), or TFSI− ions, denoted as Li+−O−(TFSI−), via their respective oxygen atoms. Comparing the RDFs of the lithium-polymer (Li+−O−(PEO)), and lithium-TFSI− (Li+−O−(TFSI−) interactions, Li+−O−(PEO) possesses a significantly higher RDF peak intensity and an average CN of 5 compared to the latter Li+−O−(TFSI−). At first glance, based on the CNs, nearly complete LiTFSI dissociation is observed for most of the investigated systems, attributed to the preferential solvation of the oxygen dense EO units from PEO.28,30,34,54,63
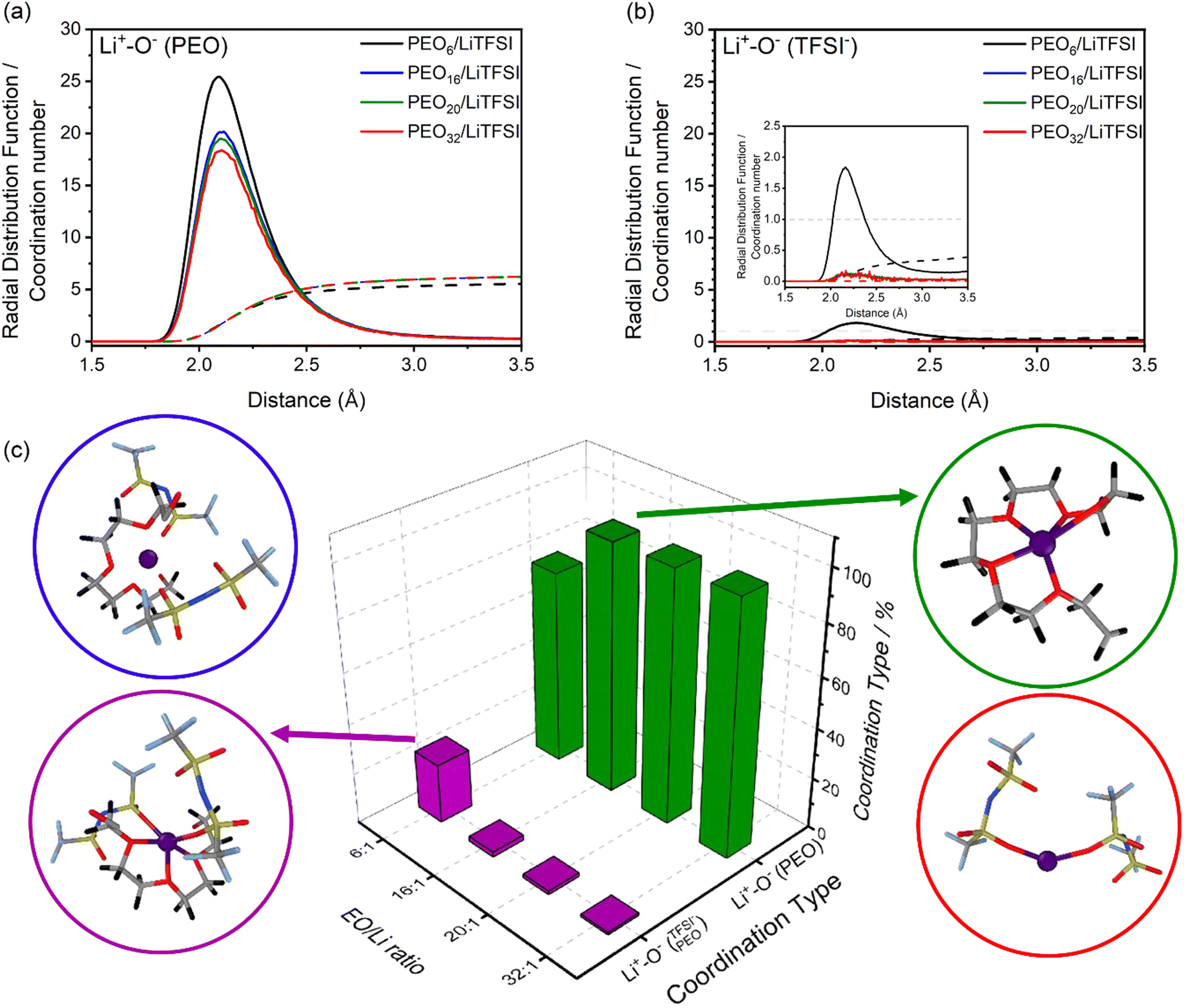 | ||
Fig. 1 RDFs (solid line) and CNs (dashed line) for PEOn/LiTFSI SPEs, indicating the Li+–O− interaction between lithium-ions and (a) PEO, or (b) TFSI− ions. (c) Lithium-ion molecular speciation analysis with lithium-ion as the reference species. The different types of lithium-ion speciation are represented by different colours: isolated Li+ (blue), Li+−O−(PEO) (green), 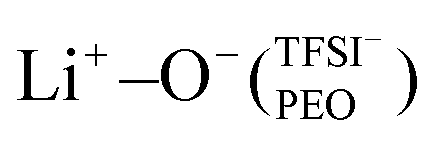 (purple), and Li+−O−(TFSI−) (red). RDFs and lithium-ion molecular speciation analyses were calculated from MD simulations conducted at 70 °C. (purple), and Li+−O−(TFSI−) (red). RDFs and lithium-ion molecular speciation analyses were calculated from MD simulations conducted at 70 °C. | ||
The incomplete LiTFSI dissociation observed at highly concentrated PEO6/LiTFSI strongly suggests the formation of ion pairs. This phenomenon can be attributed to two primary factors. Firstly, the increased probability of electrostatic interactions leads to a higher likelihood of ion cluster formation. Secondly, the reduced number of EO coordination sites available for lithium-ion solvation plays a significant role. This scenario becomes evident at such high salt concentration, where the EO/Li+ ratio is around 6, causing the coordination between ether oxygen atoms and Li ions to approach saturation. Consequently, it is natural for some Li ions to coordinate with TFSI− oxygen atoms instead.
Advancing the prediction to a more in-depth analysis regarding the lithium-ion coordination environment, lithium-ion molecular speciation analysis is performed as a function of LiTFSI salt concentration. Based on RDF analysis, the local minimum of the first RDF peak, for both Li+−O−(PEO) and Li+−O−(TFSI−) coordination types, is observed at 3 Å (cf.Fig. 1(a) and (b)).
This local minimum corresponds to the edge of the first solvation shell for Li+-ions, therefore, it is selected as the cut-off distance for the lithium-ion molecular speciation analysis. The effects of LiTFSI salt concentration upon the types of ionic species, such as polymer–Li+, polymer–polymer, and various ion–ion interaction interdependencies are plotted in Fig. 1(c) as percentage probability. The lithium-ion molecular speciation remains consistent regardless of the EO/Li+ ratio, in good agreement with RDF analyses and previous literature findings.26,52–54 Predominantly, lithium ions interact with PEO via its oxygen atoms, denoted in the following as Li+−O−(PEO). The occurrence of Li+−O−(PEO) speciation increases as the LiTFSI salt concentration decreases, observed in both PEO20/LiTFSI and PEO32/LiTFSI (99%), and similarly in PEO16/LiTFSI (98%). However, there is a significant decrease in this speciation for highly concentrated PEO6/LiTFSI (77%) (cf. Table S3, ESI†). Additionally, a second type of lithium-ion molecular speciation, where lithium ions interact with the oxygen atoms from both TFSI− anions and PEO chains, is exclusively present in PEO6/LiTFSI (23%), denoted as 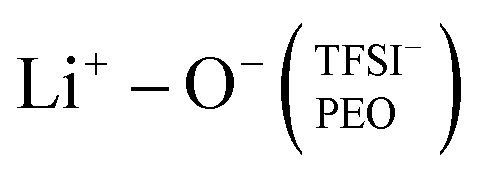 . The relatively higher LiTFSI salt concentration and lower PEO concentration in PEO6/LiTFSI limit the available PEO oxygen solvation sites, which hinders LiTFSI dissociation and may promote ion cluster formation,26,52 since lithium ions are preferentially solvated by the oxygen atoms from PEO,64–67 there is a higher probability of
. The relatively higher LiTFSI salt concentration and lower PEO concentration in PEO6/LiTFSI limit the available PEO oxygen solvation sites, which hinders LiTFSI dissociation and may promote ion cluster formation,26,52 since lithium ions are preferentially solvated by the oxygen atoms from PEO,64–67 there is a higher probability of 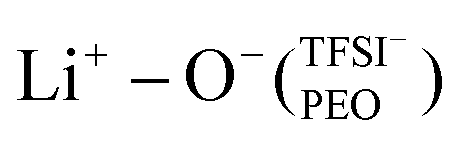 speciation in this case.
speciation in this case.
The study of ion transport incorporates MSD analysis, a useful tool for examining the displacement of particles over time. MSD analysis allows computation of diffusion coefficients (eqn (1)) and lithium-ion transport numbers (eqn (3)). As shown in Fig. 2(a), it is evident that TFSI− anions exhibit greater displacement than lithium ions regardless of the EO/Li+ ratio of the SPEs.
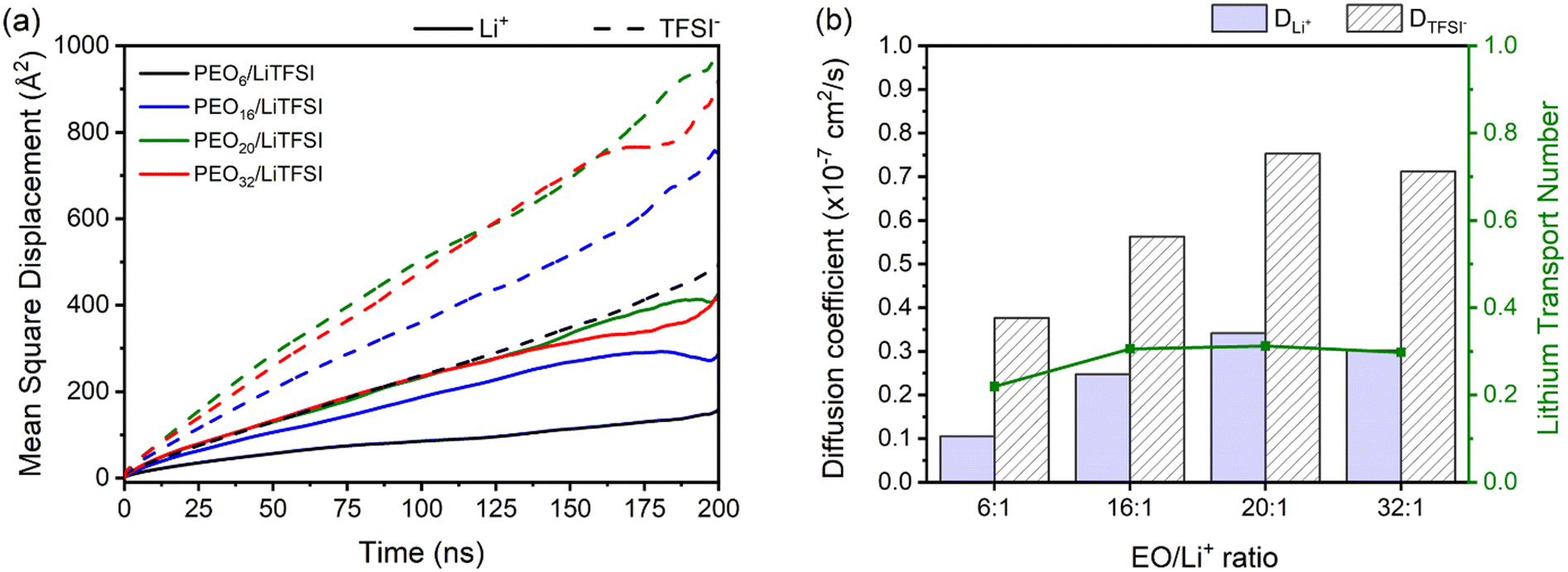 | ||
Fig. 2 (a) MSD functions for lithium- (solid line) and TFSI− ions (dashed line) as a function of LiTFSI salt concentration. (b) Self-diffusion coefficients for the studied EO/Li+ ratios, for Li+ (solid column) and TFSI− (striped column) as well as lithium-ion transport numbers, 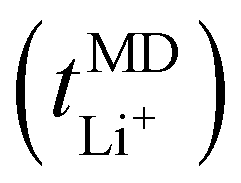 (solid line). MSDs, self-diffusion coefficients, and lithium-ion transport numbers were calculated from MD simulations conducted at 70 °C. (solid line). MSDs, self-diffusion coefficients, and lithium-ion transport numbers were calculated from MD simulations conducted at 70 °C. | ||
Upon closer inspection, it is evident that higher ion mobilities are observed for PEO20/LiTFSI, closely followed by PEO16/LiTFSI. In contrast, for PEO6/LiTFSI, the effects of ion pair formation become evident through notably lower Li+ and TFSI− ion mobilities in this highly concentrated system, as revealed by RDF analyses. When comparing the MSD of lithium ions to the oxygen atoms from PEO, as depicted in Fig. S5 (ESI†), it becomes apparent that lithium-ion mobility is primarily limited by polymer chain movement, as the MSD peaks for the respective atoms consistently correlate. In moderately and highly diluted PEOn/LiTFSI systems, the MSD curves for lithium-ion mobility are lower compared to the displacement of oxygen atoms from PEO. Remarkably, for the highly concentrated PEO6/LiTFSI, the lithium-ion transport appears to be nearly equivalent to PEO motion, suggesting the presence of a distinct lithium-ion transport mechanism in this case.
Self-diffusion coefficients as a function of LiTFSI concentration, plotted in Fig. 2(b), tend to be higher for TFSI− compared to Li+ ions, irrespective of the LiTFSI concentration. Discernably, more dilute EO/Li+ ratios of 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 32:1 display higher Li+ and TFSI− ion self-diffusion coefficients, substantiated by RDF analyses which indicate nearly complete LiTFSI dissociation for these ratios. Fig. 2(b) additionally displays the lithium-ion transport number
:1 and 32:1 display higher Li+ and TFSI− ion self-diffusion coefficients, substantiated by RDF analyses which indicate nearly complete LiTFSI dissociation for these ratios. Fig. 2(b) additionally displays the lithium-ion transport number 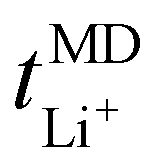 . The
. The 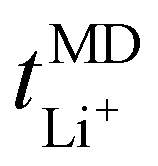 trend remains relatively consistent, except for PEO6/LiTFSI, which shows the lowest value of 0.21. For the other studied PEOn/LiTFSI systems,
trend remains relatively consistent, except for PEO6/LiTFSI, which shows the lowest value of 0.21. For the other studied PEOn/LiTFSI systems, 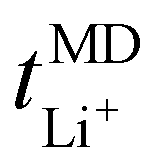 only slightly increases, with PEO16/LiTFSI (0.31), PEO20/LiTFSI (0.31), and PEO32/LiTFSI (0.30) displaying comparable values (cf. Table S4, ESI†). Interestingly, the modelled ion pair effects predicted for PEO6/LiTFSI do not seem to significantly alter the ion transport effects.
only slightly increases, with PEO16/LiTFSI (0.31), PEO20/LiTFSI (0.31), and PEO32/LiTFSI (0.30) displaying comparable values (cf. Table S4, ESI†). Interestingly, the modelled ion pair effects predicted for PEO6/LiTFSI do not seem to significantly alter the ion transport effects.
The lithium-ion transport mechanism can be deduced from the analysis shown in Fig. S6(a) and (b) (ESI†), where a reference lithium-ion is selected to demonstrate lithium-ion diffusion concerning the oxygen atoms from PEO, referred to as the O index, or the oxygen atoms from the TFSI− anions, denoted as the TFSI index. The MD trajectory involved 24 EO monomers to represent 40 PEO chains in the system, which included either 160 (PEO6/LiTFSI) or 30 (PEO32/LiTFSI) additional Li-TFSI ion pairs. Each single PEO chain is visualized separated by delineated dash lines in Fig. S6 (ESI†), and similarly, each TFSI− ion is separated accordingly. It is important to emphasize that while we focus on a single Li+ ion in the analysis displayed in Fig. S6 (ESI†) to illustrate the diffusion mechanism, the same mechanism applies when considering other Li+ ions as reference. Essentially, the lithium-ion transport mechanism at high concentrations is inferred from the collective data presented in Fig. 1(c) and Table S3 (ESI†), and the data used in Fig. S6 (ESI†) serves as a representative example. It is observed that the lithium-ion dissociates from TFSI− ions at the beginning of the simulation, moves towards a PEO chain, and interacts with oxygen atoms from both the TFSI− ions and PEO. Eventually, the lithium-ion diffuses along a single polymer chain while remaining coordinated with TFSI− ions for the remainder of the simulation time. This verifies that lithium-ion diffusion for concentrated PEO6/LiTFSI occurs via diffusion along a single polymer chain. In the case of PEO32/LiTFSI, the lithium-ion barely interacts with TFSI− ions, clearly jumping from one PEO chain to another, confirming that the lithium-ion transport mechanism for moderately dilute PEOn/LiTFSI systems occurs via an ion-hopping mechanism.
In the case of highly diluted PEO64/LiTFSI, the absence of a Tg can be ascribed to several factors: the exceptionally low LiTFSI content and significantly high PEO content result in a highly crystalline SPE (56%); additionally, the Tg dependency on the temperature sweep rate70 may also contribute to the absence of a Tg signal.
The studied SPEs with EO/Li+ ratios of 16 and 20 display comparable Tg values of −35 °C, however, the slightly more diluted PEO32/LiTFSI exhibits the lowest Tg (−43 °C), indicating improved polymer chain flexibility. Lower temperatures are required to expedite polymer motion in this case, leading to enhanced polymer mobility and, consequently, faster ion dynamics and Li+-ion mobility, as supported by the MSD analysis from the MD simulations.
The melting transition temperature, Tm, supplies information regarding the temperature range at which the SPE is molten and its long-range structure transitions to a disordered amorphous system (cf. Fig. S7 and Table S5, ESI†). Contrary to the trend for the Tg, the Tm increases slightly as LiTFSI content decreases, from Tm = 49 to 62 °C, for PEO16/LiTFSI and PEO64/LiTFSI, respectively, which indicates that the lithium salt promotes the amorphization of the crystalline domains resulting in lower melting temperatures. Additionally, the enthalpy of the melting transition, ΔHm, has been used to estimate the crystallinity degree, χc, of the studied PEO-based SPEs, which represents the fraction of the polymer that is in a relatively ordered state. Based on the data presented in Table S5 (ESI†), a clear trend emerges, showing an increase in χc with decreasing LiTFSI concentration. This observation can be attributed to the increase in the proportion of semi-crystalline PEO and the corresponding decrease in the amount of LiTFSI salt. This trend confirms the amorphization effect of polar LiTFSI within the PEO matrix.
Proceeding with the validation of the model, Raman and MAS-NMR spectroscopies were utilized to evaluate the accuracy of the computational predictions concerning the impact of salt concentration on the lithium-ion coordination environment. Raman spectra were obtained for various EO/Li+ ratios at temperatures of 25 °C (cf. Fig. S8(a), ESI†), 40 °C (cf. Fig. S8(b), ESI†), and 70 °C (Fig. 3(a)). Peak deconvolution was then performed to quantify the extent of LiTFSI association as a function of LiTFSI concentration and temperature, with results shown for PEO6/LiTFSI in Fig. 3(b) (cf. Table S6, ESI†). For further insight, we conducted MAS-NMR measurements on two distinct SPE systems: a highly concentrated PEO6/LiTFSI and a highly diluted PEO32/LiTFSI (see Fig. 4 and Table S7, ESI†). These measurements allowed us to discern notable dissimilarities in ion dynamics as a function of concentration and temperature.
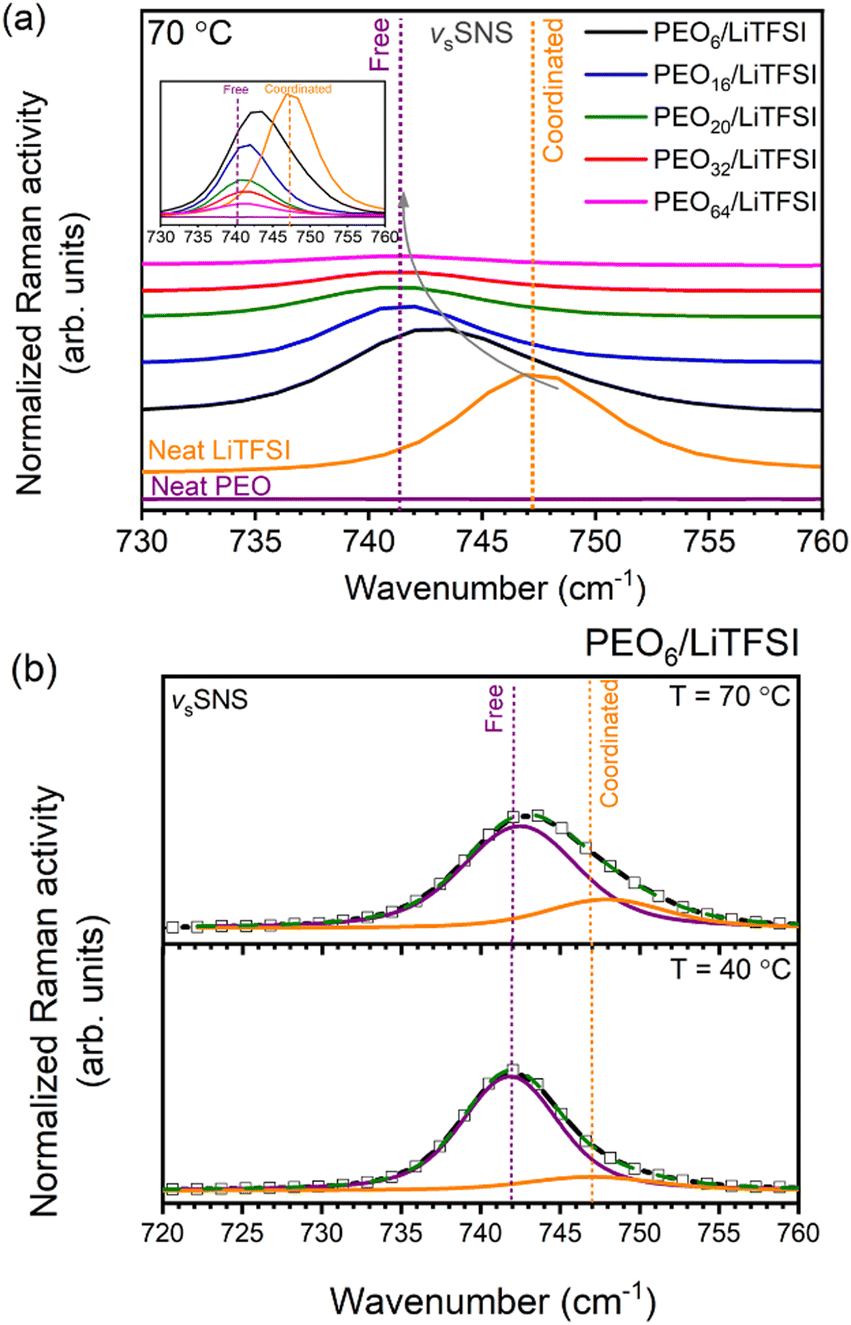 | ||
| Fig. 3 Raman spectra for the 730 (720)–760 cm−1 vibrational window illustrating the (a) S–N–S vibration for PEOn/LiTFSI SPEs at 70 °C, with spectra of neat LiTFSI and neat PEO at 25 °C. The grey arrow represents the downshift observed upon decreased LiTFSI concentration. Raman spectra of (b) PEO6/LiTFSI at 40 (bottom) and 70 °C (top). The solid black line represents recorded spectra, the solid green line with symbols corresponds to the cumulative fit of Voigt character performed by peak deconvolution analysis, with the contribution from the coordinated character (orange) and the free TFSI− ion character (purple). The two vertical dotted lines correspond to the coordinated contact ion pair position at 747 cm−1 (orange) and the free ion pair position at 740 cm−1 (purple), respectively. | ||
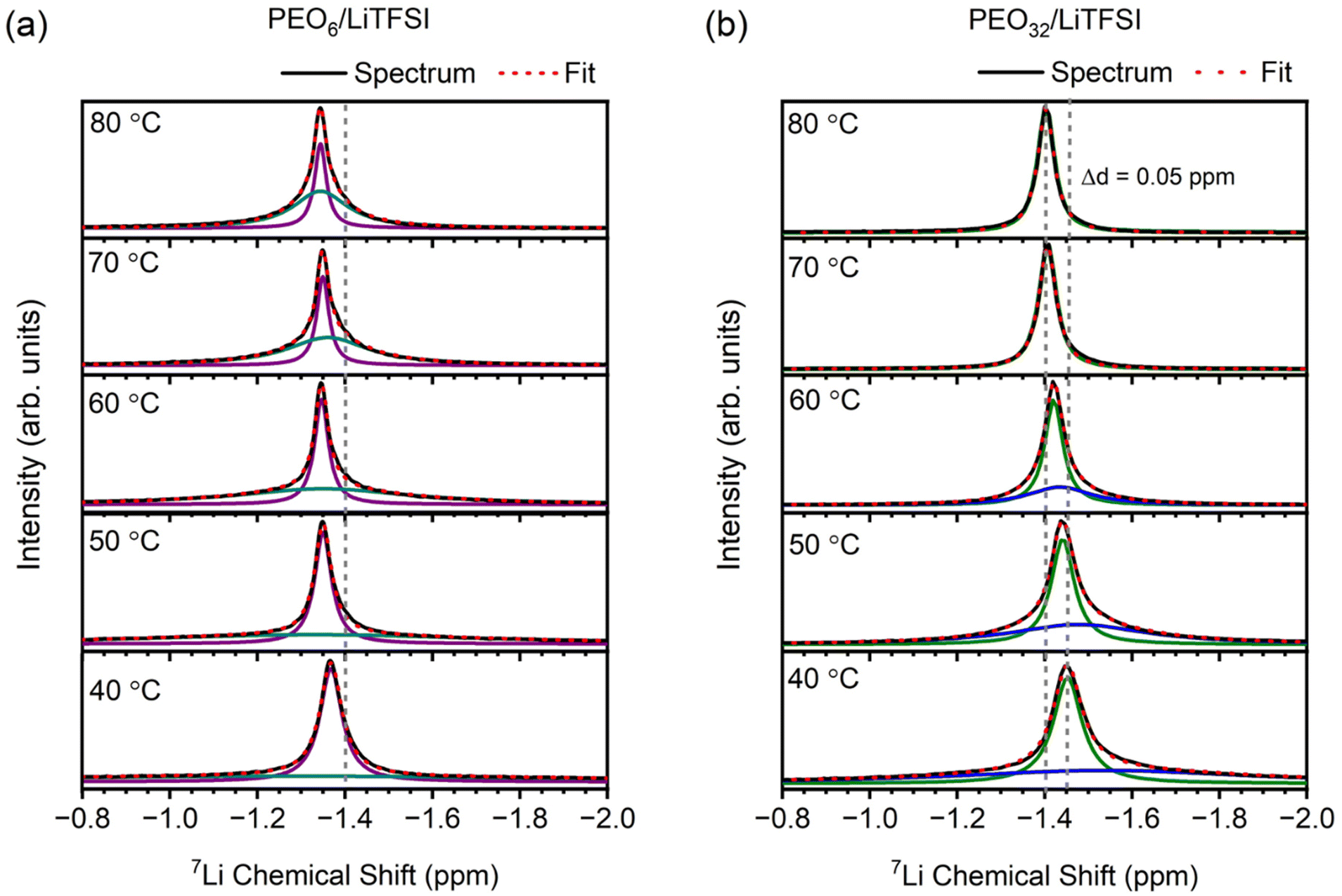 | ||
| Fig. 4 7Li MAS-NMR spectra for (a) PEO6/LiTFSI and (b) PEO32/LiTFSI at 40 °C, 50 °C, 60 °C, 70 °C, and 80 °C. The solid black line represents the recorded spectra, the dashed red lines represent the peak fitting, whilst the solid purple and cyan lines in (a), and the solid green and blue lines in (b), both respectively represent two distinct lithium-ion dynamics. | ||
Analysis of the Raman spectra reveals distinct vibrational peaks for the S–N–S vibration at 70 °C. Coordinated LiTFSI ion pairs form, as evidenced by the signal at 747 cm−1, while free ion pairs appear at 740 cm−1, consistent with previous studies.30,34,71–74 Upon a decrease in LiTFSI salt concentration, from highly concentrated PEO6/LiTFSI to highly diluted PEO64/LiTFSI, a clear downshift is observed from 743 to 741 cm−1, corresponding to coordination that changes from partially coordinated Li+–TFSI− to the free TFSI− form. These experimental results align with the predictions from the MD simulations, confirming the accuracy of the computational model. Essentially, the increased dissociation of LiTFSI at lower LiTFSI salt concentrations is evident from the Raman trends, further validating the simulated coordination environment.
The quantification of the effect of both LiTFSI concentration and temperature changes upon the lithium-ion coordination environment was attained by performing peak deconvolution of the studied PEOn/LiTFSI SPEs at various temperatures of 25 °C, 40 °C and 70 °C (cf. Table S6 and Fig. S8, ESI†). The temperature effect proves to produce significant changes in the quantification of the two different coordination environments principally detected for LiTFSI.71,72 It is important to highlight that the Raman spectra collected at 25 °C in this study was performed prior to a preheating treatment step,71 for which non-preheated and preheated SPEs’ spectra may show considerable differences in the S–N–S vibration window for more concentrated PEO6/LiTFSI, assigned to the small amount of ion pairs present in the amorphous phase.71 Examining the effect of concentration changes, mainly PEO6/LiTFSI and PEO16/LiTFSI display notable differences, with the remaining SPEs showing comparable results, i.e., complete LiTFSI dissociation. A moderate contribution from coordinated LiTFSI is seen for PEO6/LiTFSI at 40 °C (15%), significantly increased at 70 °C (23%). Clearly, the local ionic structure of highly concentrated PEO6/LiTFSI is affected by the melting transition of the SPE.71,72 Conversely, in the case of PEO16/LiTFSI, a negligible decrease in the coordinated LiTFSI form is observed, transitioning from 7% at 40 °C to 4% at 70 °C. These results suggest that more dilute PEOn/LiTFSI systems (n > 6) tend to retain their ionic structure despite changes in temperature.
MAS-NMR spectroscopy is a well-established technique to probe both ion and polymer dynamics at short-range order length scales (1–2 Å) for a given nucleus. This technique offers detailed information about chemical environments and their temporal fluctuations. Thus, we used 7Li solid-state NMR to gain a deeper understanding of the changes in the local lithium-ion structure and dynamics75,76 as a function of lithium salt concentration at various temperatures. The line broadening observed in the 7Li MAS-NMR resonances can be correlated to the rates and amplitudes of local fluctuations in the Li+ environments, arising from dynamic averaging of local dipolar and quadrupolar interactions. Slower polymer and lithium-ion dynamics result in broader NMR signals, whilst faster dynamics lead to narrower signals.75,77 In Fig. 4, the variable temperature 7Li MAS-NMR spectra of PEO32/LiTFSI exhibit a transition from a two- to a one-component system, with both signals becoming narrower as the temperature increases. The broader component eventually disappears in the temperature range of 60–70 °C. In contrast, the variable temperature 7Li MAS-NMR spectrum of PEO6/LiTFSI remains consistent with a predominantly two-component system, where the broader component becomes narrower and increases in quantity with rising temperature.
The deconvolution of the 7Li MAS-NMR spectra from Fig. 4 was performed to quantify the respective contributions from each component (cf. Table S7, ESI†). A two-component system is observed for PEO32/LiTFSI, comprising a minor contribution of faster lithium-ion dynamics as a narrower component at 40 °C (46%), increasing consistently up to 60 °C (63%), and subsequently increasing significantly as the temperature is increased to 70 °C (100%), notably transpiring at the melting phase transition temperature (Tm = 56 °C). Considering that lithium-ions are preferentially solvated by PEO, as predicted by the RDF and MSD analyses from MD simulations, lithium-ion mobility is interlinked with PEO segmental motion, hence, two modes of lithium-ion transport are regarded: (1) lithium-ion transport via ion hopping between the coordination sites between different PEO chains or segments, or (2) lithium-ion transport without a change of coordination site and via segmental motion of the polymer.30,54,78,79 Since PEO32/LiTFSI is a relatively dilute SPE system, and LiTFSI is entirely dissociated within the PEO matrix at 70 °C, the faster lithium-ion dynamics (narrower 7Li MAS-NMR signals in Fig. 4) are evidently attributed to the Li+–PEO coordination, as detected by MD simulations and confirmed by Raman spectroscopy. Reviewing the Raman peak deconvolution results at 40 °C, LiTFSI is once again determined to be completely dissociated, inferring that the two-component lithium-ion dynamics deduced by NMR are solely attributed to lithium-ions coordinated to PEO, occurring by either one of the two lithium-ion transport modes. Based on MD simulations (cf. Fig. S6(b), ESI†) and similar MD simulations performed on PEO/LiTFSI systems from literature,54,78 lithium-ion mobility between different coordination sites between different PEO chains is faster than lithium-ion mobility arising by the lithium-ion coordination via PEO segmental motion. Therefore, the narrower component representing faster lithium-ion dynamics, occurs distinctly via lithium-ion transport between different PEO coordination sites and different polymer chains for PEO32/LiTFSI at 70 °C.
Continuing with this reasoning, it is interesting to note an opposite trend for PEO6/LiTFSI, with the minor contribution of faster lithium-ion dynamics from the two-component system proceeding to decrease with increasing temperature. However, considering the significantly lower number of PEO coordination sites present due to the high LiTFSI salt concentration, additional lithium-ion transport modes may prevail. Based on the lithium-ion transport mechanism deduced in Fig. S6(a) (ESI†) and a similar study performed for PEO/LiTFSI systems,54 at high LiTFSI concentrations, lithium-ions preferentially coordinate with PEO due to the presence of ion pairs and ion clusters. However, despite this preference, the coordination of lithium ions to TFSI− anions enhances lithium-ion transport. Notably, ion pairs and ion clusters offer the most stable structure for transporting lithium cations, resulting in higher lithium-ion mobilities compared to lithium ions solely coordinated to PEO.54 Consequently, the limited polymer segmental motion induced by the high LiTFSI concentration leads to the faster lithium-ion dynamics observed as a minority. At 40 °C, this minority constitutes 47%, decreasing to 32% at 70 °C. These faster lithium-ion dynamics are noticeably distinguished as lithium ions in the form of ion pairs and ion clusters. This observation finds further supported from MD simulations and Raman spectroscopy, where the minority of the lithium ions is found to be associated with TFSI− anions.
Considering that temperatures higher than the Tm of the PEO6/LiTFSI SPE lead to a greater presence of amorphous regions, facilitating predominantly favoured lithium-PEO coordination, there is an observed increase in slower lithium-ion dynamics (reaching 68% at 70 °C). This observation is further confirmed by Raman spectroscopy, which indicates the presence of dissociated LiTFSI species, and MD simulations, which show Li+–PEO coordination.
Ascertaining the accuracy of the model's ability to predict the effects of concentration on the lithium-ion transport, studying the temperature dependence of the ionic conductivity may convey information about the primary charge carriers and their transport properties within the studied systems. The Arrhenius plot in Fig. 5(a) displays the ionic conductivity of the studied PEOn/LiTFSI systems at 40 °C and 70 °C. The semicrystalline nature of PEO is apparent since the ionic conductivities at 40 °C, a lower temperature than the melting phase transition temperatures (cf. Table S5, ESI†), are significantly lower compared to the ionic conductivities at 70 °C, at which completely amorphous behaviour of the SPEs are observed. Observing the effect of LiTFSI concentration, at temperatures lower than the melting phase transition, i.e., 40 °C, both the crystallinity degree and the extent of LiTFSI concentration are factors which may affect the ionic conductivity. An increased crystallinity degree signifies an increased fraction of ordered polymer chains, decreasing ionic conductivity, whilst an upsurge in LiTFSI concentration may produce increased ion–ion interdependent interactions, generally restricting ion mobility to a greater magnitude. This rationale follows for more concentrated SPEs, where PEO6/LiTFSI possesses lower ionic conductivity (4.2 × 10−6 ± 7.7 × 10−7 S cm−1) compared to highly diluted PEO64/LiTFSI (1.6 × 10−5 ± 26 × 10−6 S cm−1). Regarding moderately dilute systems, a trend of increasing ionic conductivity with increasing LiTFSI content is apparent, with PEO32/LiTFSI (2.0 × 10−5 ± 6.2 × 10−6 S cm−1) displaying slightly lower ionic conductivity compared to PEO20/LiTFSI (4.0 × 10−5 ± 8.9 × 10−6 S cm−1), while PEO16/LiTFSI exhibits the highest ionic conductivity (1.7 × 10−4 ± 4.0 × 10−5 S cm−1). At a higher temperature of 70 °C, the ionic conductivities of the studied SPE systems are relatively comparable and fall within a range of 4.0 × 10−4− 1.6 × 10−5 S cm−1, for PEO6/LiTFSI and PEO16/LiTFSI, respectively. The comparative ionic conductivity is attributed to enhanced ion transport in the amorphous regions of the SPEs. This improvement arises from increased polymer free volume and improved segmental motion of the polymer chains in the amorphous phase.
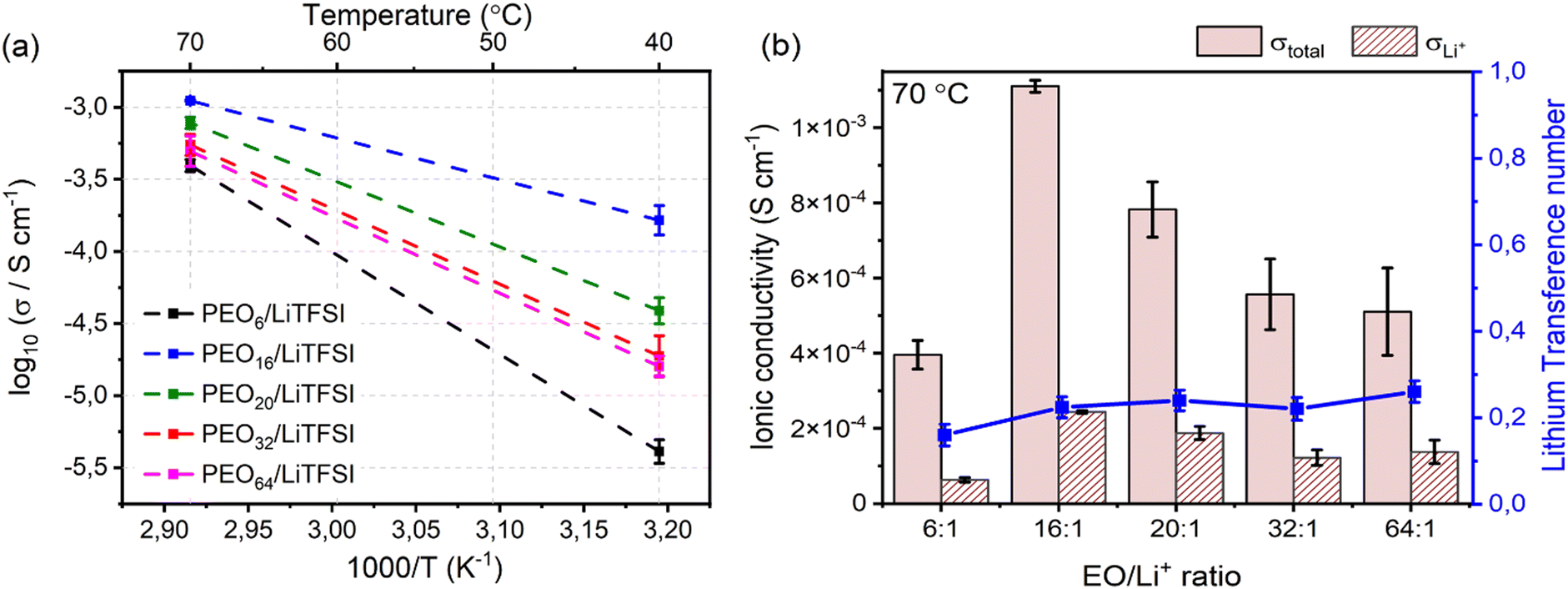 | ||
| Fig. 5 (a) Temperature dependence of the ionic conductivity for PEOn/LiTFSI SPEs with varying LiTFSI salt concentration. (b) Histogram illustrating the total (solid) and Li+ (stripe) ionic conductivity with error bars, and lithium-ion transference number (blue) of PEOn/LiTFSI SPEs, where n = 6, 16, 20, 32, or 64, at 70 °C. | ||
Delving further into the ion dynamics, the total ionic conductivity, σtotal, and lithium-ion conductivity, σLi+, is provided in Fig. 5(b). By comparing the contribution of the lithium-ion conductivity to the total ionic conductivity, the effect of LiTFSI concentration on the ion transport is apparent. PEO16/LiTFSI possesses both the highest total and lithium-ion conductivity, with PEO20/LiTFSI following closely behind. It may be interesting to highlight that PEO32/LiTFSI and PEO64/LiTFSI display comparable σtotal and σLi+, albeit the latter is almost twice as dilute as the former. Based on the MD simulations, MSD and self-diffusion coefficient analyses predicted comparable ion transport properties for PEO20/LiTFSI and PEO32/LiTFSI, although slightly lower for PEO16/LiTFSI. This discrepancy may be due to several factors: (1) the polymer chain length utilized in the MD simulations (40 PEO chains with 24 EO units per chain, translated into a Mw of approximately 1,056 g mol−1) may not accurately represent the performance of experimentally measured SPEs (PEO chains with a of Mw of 5 × 106 g mol−1); (2) a higher solvent polarity may result in a system consisting of primarily free ions solvated by PEO, which may yield higher self-diffusion coefficients within the polymer matrix; (3) partial charges and charge density may differ depending on the partial charge calculation method; and (4) time and length scales of the simulation adapted to meticulously describe the experimental system may induce additional computational cost.
Now, drawing attention to the lithium-ion transference number. Firstly, the lithium-ion transference number determined experimentally via EIS, 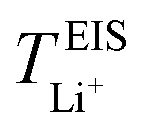 , is different than the lithium-ion transport number,
, is different than the lithium-ion transport number, 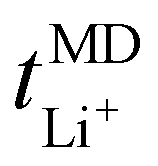 , calculated previously from MD simulations. The former is based on chronoamperometry, deducing the number of moles of lithium-ions migrating per Faraday of charge, which may include contributions from free Li+ ions, ion pairs, ion triplets or ion clusters.27,54,70,78 The calculation of the lithium-ion transport number based on MD simulations,
, calculated previously from MD simulations. The former is based on chronoamperometry, deducing the number of moles of lithium-ions migrating per Faraday of charge, which may include contributions from free Li+ ions, ion pairs, ion triplets or ion clusters.27,54,70,78 The calculation of the lithium-ion transport number based on MD simulations, 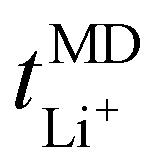 , is conducted by analyzing the averaged displacements and their self-diffusion coefficients, or proportion of current carried by the ionic species within the polymer matrix, i.e., free Li+ or TFSI− ions. Based on Fig. 5(b), the overall trend of the lithium-ion transference number follows a higher
, is conducted by analyzing the averaged displacements and their self-diffusion coefficients, or proportion of current carried by the ionic species within the polymer matrix, i.e., free Li+ or TFSI− ions. Based on Fig. 5(b), the overall trend of the lithium-ion transference number follows a higher 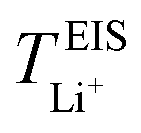 with decreasing LiTFSI salt concentration, similar to values deduced from MD simulations (cf. Table S4, ESI†), albeit slightly higher. The slightly higher predicted
with decreasing LiTFSI salt concentration, similar to values deduced from MD simulations (cf. Table S4, ESI†), albeit slightly higher. The slightly higher predicted 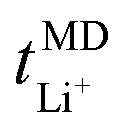 values may be due to the deviation of the force field based on various factors, as discussed earlier. PEO16/LiTFSI, PEO20/LiTFSI, and PEO32/LiTFSI display similar
values may be due to the deviation of the force field based on various factors, as discussed earlier. PEO16/LiTFSI, PEO20/LiTFSI, and PEO32/LiTFSI display similar 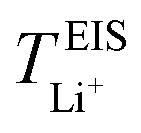 values. A lower
values. A lower 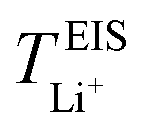 for highly concentrated PEO6/LiTFSI compared to the aforementioned SPEs is attributed to the overall restricted lithium-ion mobility related to the lower lithium-ion ionic conductivity, which introduces more charge carriers and decreases ionic conductivity,68,69 whereas highly diluted PEO64/LiTFSI exhibits a higher
for highly concentrated PEO6/LiTFSI compared to the aforementioned SPEs is attributed to the overall restricted lithium-ion mobility related to the lower lithium-ion ionic conductivity, which introduces more charge carriers and decreases ionic conductivity,68,69 whereas highly diluted PEO64/LiTFSI exhibits a higher 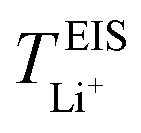 due to higher polymer free volume, facilitating faster ion mobility.
due to higher polymer free volume, facilitating faster ion mobility.
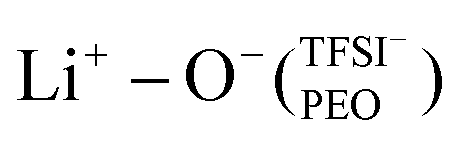 coordination type (1%). Experimental techniques, such as Raman spectroscopy, validated the extent of LiTFSI salt association as a function of LiTFSI salt concentration by inspecting the S–N–S vibration, where solely dissociated LiTFSI was found, further examined by MAS-NMR spectroscopy to possess faster lithium-ion dynamics. Therefore, for diluted SPEs, the lithium-ions coordinated to PEO in the form of free Li+ ion follows faster lithium-ion transport, diffusing via an ion-hopping mechanism between different PEO chains and segments.
coordination type (1%). Experimental techniques, such as Raman spectroscopy, validated the extent of LiTFSI salt association as a function of LiTFSI salt concentration by inspecting the S–N–S vibration, where solely dissociated LiTFSI was found, further examined by MAS-NMR spectroscopy to possess faster lithium-ion dynamics. Therefore, for diluted SPEs, the lithium-ions coordinated to PEO in the form of free Li+ ion follows faster lithium-ion transport, diffusing via an ion-hopping mechanism between different PEO chains and segments.
| EO/Li+ ratio 70 °C | MD simulations | Raman spectroscopy | 7Li MAS-NMR spectroscopy | |||
|---|---|---|---|---|---|---|
| Li+–PEO coordination (%) | Li+–TFSI−–PEO coordination (%) | Dissociated LiTFSI (%) | Associated LiTFSI (%) | Narrower component (faster Li+-ion mobility) (%) | Broader component (slower Li+-ion mobility) (%) | |
| 6:1 |
77 | 23 | 77 ± 2 | 23 ± 7 | 32 | 68 |
| 32:1 |
99 | 1 | 100 ± 3 | 0 | 100 | 0 |
In the case of PEO6/LiTFSI, MD simulations predicted two distinct lithium-ion coordination environments, predominantly Li+−O−(PEO) (77%), and a minor 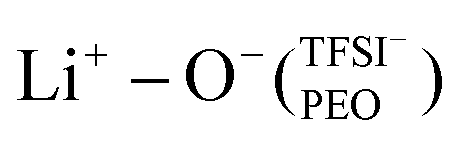 coordination (23%). Raman spectroscopy validated the findings from MD simulations, as a majority of free TFSI− ions (77 ± 2%) and a minority of contact ion pair form (23 ± 7%). Further analysis of the lithium-ion dynamics by MAS-NMR lead to the determination of two distinct components as the temperature rose from 40 °C to 80 °C; faster lithium-ion dynamics (32%), and slower lithium-ion dynamics (68%) at 70 °C. Since lithium-ions are preferentially solvated by PEO, Li+−O−(PEO) remains the dominant speciation, however, these lithium-ions possess slower lithium-ion dynamics. Although a minor contribution,
coordination (23%). Raman spectroscopy validated the findings from MD simulations, as a majority of free TFSI− ions (77 ± 2%) and a minority of contact ion pair form (23 ± 7%). Further analysis of the lithium-ion dynamics by MAS-NMR lead to the determination of two distinct components as the temperature rose from 40 °C to 80 °C; faster lithium-ion dynamics (32%), and slower lithium-ion dynamics (68%) at 70 °C. Since lithium-ions are preferentially solvated by PEO, Li+−O−(PEO) remains the dominant speciation, however, these lithium-ions possess slower lithium-ion dynamics. Although a minor contribution, 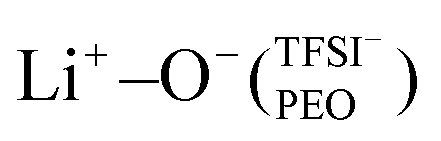 speciation facilitates faster lithium-ion dynamics, since ion pairs and ion clusters are more stable structures for lithium-ion transport compared to PEO.54 Notably, the crucial difference in analyzing how ion coordination affects ion transport and dynamics requires consideration of the salt concentration.
speciation facilitates faster lithium-ion dynamics, since ion pairs and ion clusters are more stable structures for lithium-ion transport compared to PEO.54 Notably, the crucial difference in analyzing how ion coordination affects ion transport and dynamics requires consideration of the salt concentration.
The prediction of both ion coordination and transport based on MD simulations was successfully validated by utilizing experimental techniques such as Raman, MAS-NMR, and EIS. This model may aid the examination of cation effects on the ion coordination and transport properties of alternative alkali metal solid polymer electrolytes, as we examine in the following section.
3.2. Beyond lithium-based batteries: sodium, potassium, and cesium
The validation of the PEOn/LiTFSI computational framework allows for its extension to other cations beyond lithium-ion batteries, by substituting lithium with sodium, potassium, or cesium cations. Alternative solid-state electrolytes based on extensively studied PEO may be modelled, such as PEO20/NaTFSI, PEO20/KTFSI, and PEO20/CsTFSI, towards the realization of alkali metal batteries.Addressing ion coordination, RDFs and respective CNs of the studied PEO20/XTFSI systems were computed by analyzing the coordination between the alkali metal cations and the oxygen atoms from both PEO and TFSI− ions, presented in Fig. 6(a) and (b), respectively. The first peak of the RDFs in Fig. 6(a) indicates that the coordination distance between the cation and the EO units increases with cation size, from 2.1 Å for lithium-based PEO20/LiTFSI, to 3.1 Å for cesium-based PEO20/CsTFSI. Additionally, the number of coordinated oxygen atoms in the first coordination shell, indicated by the width of the first peak in the RDF, increases from 6 for PEO20/LiTFSI and 7 for PEO20/NaTFSI, to 8 for both PEO20/KTFSI and PEO20/CsTFSI.
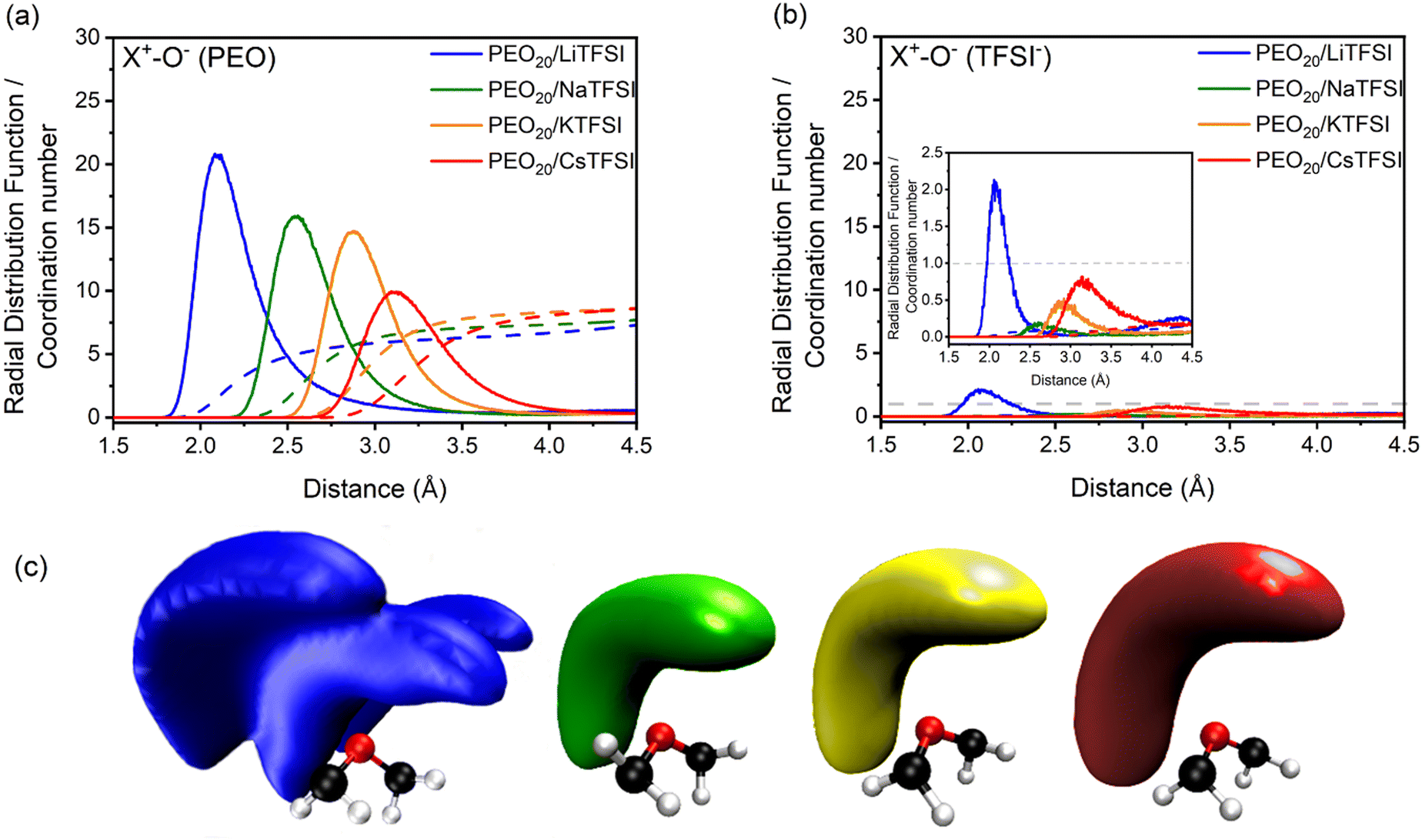 | ||
| Fig. 6 Lithium-ion coordination for PEO20/XTFSI systems, where X = Li, Na, K, and Cs. RDFs (solid lines) and CNs (dashed lines) between (a) different cations and oxygen atoms of the EO units from PEO, and (b) oxygen atoms from TFSI− ions, at 70 °C. (c) SDFs representing the coordination between the EO units from PEO (ball and stick) and cations (surface volume), from left to right: Li+ (blue), Na+ (green), K+ (yellow), and Cs+ (red) cations. | ||
In the case of cation–anion coordination, the RDFs representing the cation coordination with TFSI− ions, plotted in Fig. 6(b), display CN values close to zero for the first coordination shell, which indicates nearly complete dissociation, irrespective of the cation nature. This observation aligns well with the decrease in dissociation energies observed in calculations of X-TFSI clusters in the gas-phase, as the cationic size increases.80
Moreover, the computed spatial distribution functions (SDFs) in Fig. 6(c) indicates stronger coordination between the lithium cation and the PEO chains, due to the presence of three lobes as the surface volume. The remaining cations, however, display a different surface volume, observed as single lobes at larger distances from the EO monomers the larger the cation size. These findings verify the complete dissociation predicted by RDF analyses. The preferential cation-PEO coordination observed for all cations demonstrates a stronger cation-PEO coordination for smaller cations, attributed to the shorter distance between the respective cation and the oxygen atoms from PEO, which may hinder cation transport due to increased intermolecular forces.
Fig. 7(a) displays the ionic conductivities of the modelled PEO20/XTFSI systems. Additionally, Fig. 7(b)–(d) illustrates experimentally obtained values for PEO20/LiTFSI, PEO20/NaTFSI, and PEO20/KTFSI, respectively.80
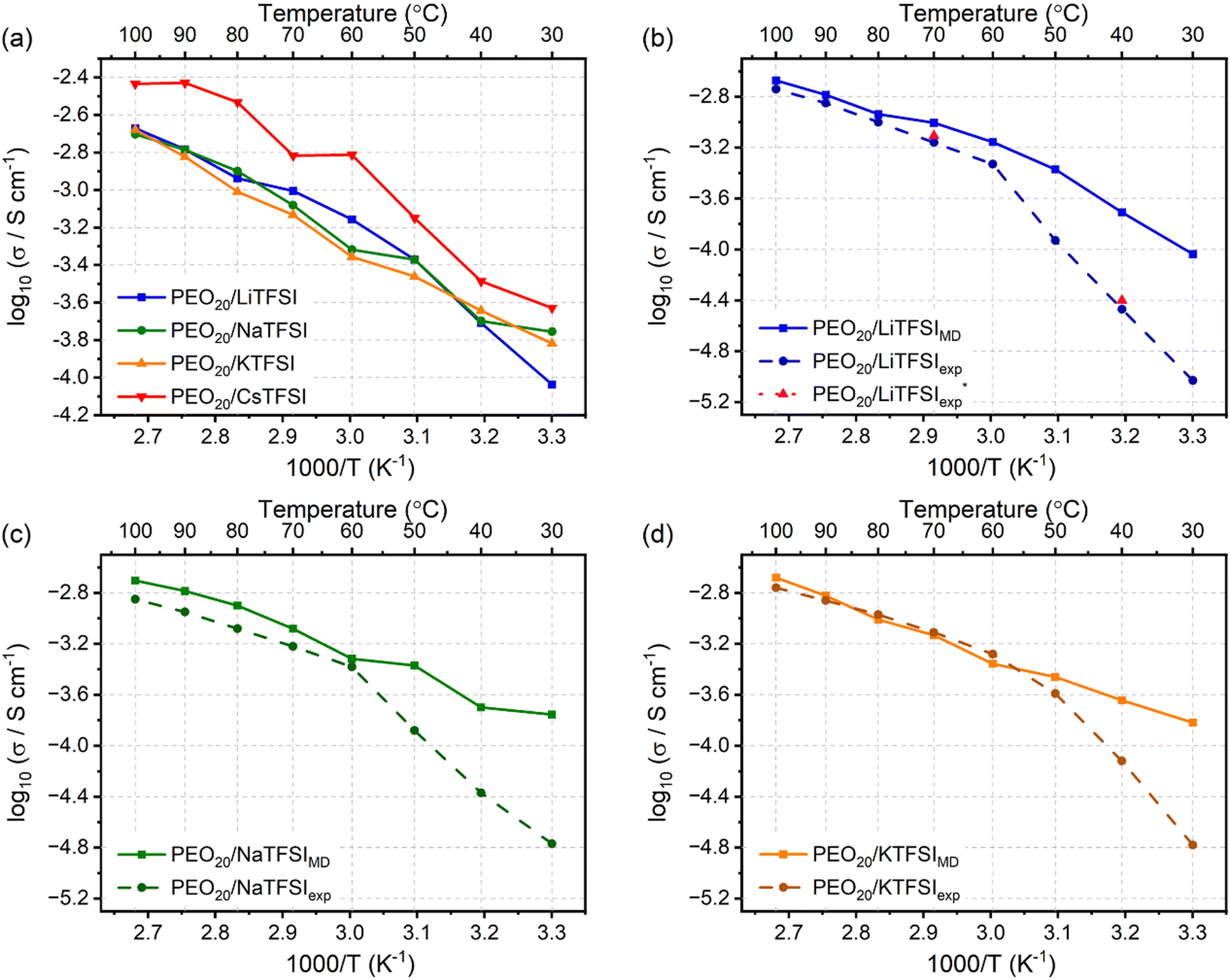 | ||
| Fig. 7 Ionic conductivity of the (a) PEO20/XTFSI systems, where X = Li, Na, K, or Cs, at different temperatures. Comparison of MD simulated (solid line) and experimentally obtained (dashed line)80 ionic conductivities, including this study (pink) (PEO20/LiTFSI), for (b) PEO20/LiTFSI (blue), (c) PEO20/NaTFSI (green) and (d) PEO20/KTFSI (orange) SPE systems. | ||
The ionic conductivities of the four alkali metal based SPEs show similar trends, as inferred from Fig. 7(a). Notably, PEO20/LiTFSI, PEO20/NaTFSI, and PEO20/KTFSI possess similar ionic conductivity values, while PEO20/CsTFSI displays the highest ionic conductivity. This result may be surprising, as similar ion transport for cations of different sizes and masses seem contradictory. However, this contradiction can be solved by referring to the weaker cation coordination to the PEO chains, as discussed earlier: larger cations coordinate to the EO monomers at greater distances, favouring weaker cation coordination and faster cation transport.
At temperatures higher than the Tm of PEO, the ionic conductivities for PEO20/LiTFSI, PEO20/NaTFSI, and PEO20/KTFSI predicted by the simulations and those determined by experiments80 are in good agreement, proving the accuracy of the model at temperatures above the melting point of PEO. However, at temperatures lower than 50 °C, the theoretical ionic conductivities differ to experimental values. This is attributed to the semicrystalline nature of PEO present at temperatures below its phase transition temperature, which impedes ion transport due to the lower degree of amorphous regions, resulting in lower ionic conductivity. Classical MD simulations model molecular kinetics, with limited ability to simulate morphological changes, such as semi-crystallinity, hence, the discrepancy with experimental values, observed for all three alkali metal based SPEs.
Fig. 8 displays the cation transport numbers for the investigated PEO20/XTFSI systems. While no distinct trend can be readily inferred from these values, PEO20/NaTFSI exhibits the highest cation transport number across most of the studied temperatures. This finding aligns well with previous experimental reports in the literature, where a cation transport number as high as 0.66 for Na-ions was observed in a PEO/NaTFSI system.80 At temperatures higher than 60 °C, the 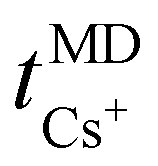 numbers are generally higher than the cation transport numbers observed for either lithium or potassium over the studied temperature range. This implies greater cationic conductivity for both Na- and Cs-based SPEs. It is interesting to note that the
numbers are generally higher than the cation transport numbers observed for either lithium or potassium over the studied temperature range. This implies greater cationic conductivity for both Na- and Cs-based SPEs. It is interesting to note that the 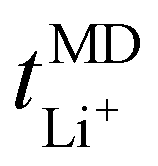 is found to be slightly higher for PEO20/LiTFSI using LAMMPS compared to Gromacs, (cf.Fig. 8 and 2a, respectively).
is found to be slightly higher for PEO20/LiTFSI using LAMMPS compared to Gromacs, (cf.Fig. 8 and 2a, respectively).
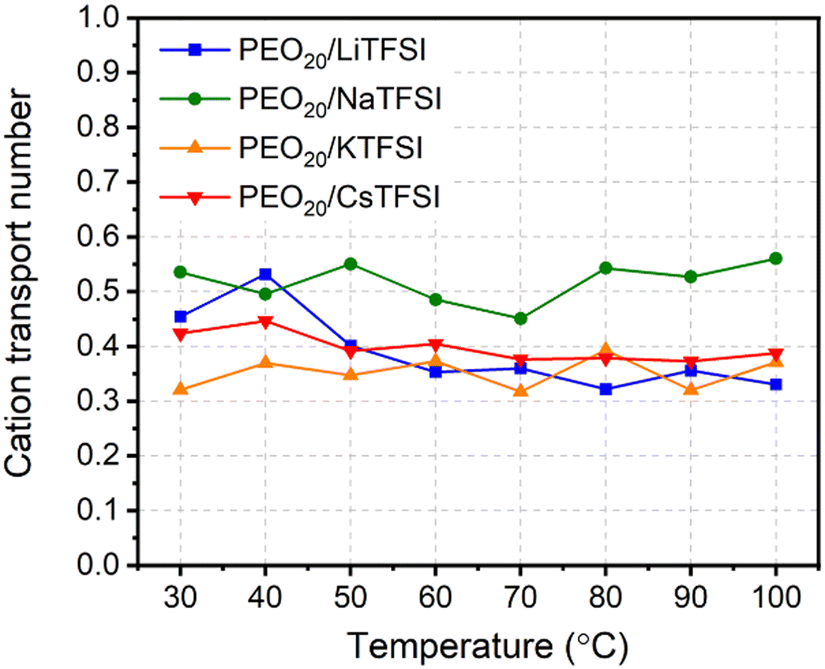 | ||
| Fig. 8 Cation transport numbers of the PEO20/XTFSI systems as a function of temperature, where X = Li, Na, K, and Cs. | ||
The activation energies of the transport mechanisms were calculated by fitting the ionic conductivity data shown in Fig. 7(a) to the Arrhenius equation (cf. eqn (S2) and Fig. S9, ESI†). Similar activation energy values were obtained for the four investigated cations, suggesting that the ion transport mechanism is independent of cation size.
In summary, the ion conductive properties of the studied alkali metal salts in the PEO matrix show no significant differences in order of magnitude, ascribed to the structural properties of the system, whereby larger cation sizes result in weaker coordination with the polymer chains from PEO. As a result, the greater mass of these cations is compensated for by the weaker cation coordination, making the studied sodium, potassium, and cesium systems equally suitable as polymer electrolytes compared to their lithium counterpart. These findings suggest that the use of sodium, potassium, or cesium SPEs used in alkali metal or alkali-ion batteries, utilizing sodium, potassium, or cesium as their primary charge carriers, may exhibit similar bulk electrolyte performance compared to lithium-based SPEs.
4. Conclusion
In moderately to highly diluted PEOn/LiTFSI systems (n ≥ 16), fully dissociated LiTFSI was found using MD simulations, while PEO6/LiTFSI exhibited some extent of ion pairing, as inferred by RDF analysis. Lithium-ion speciation in PEO revealed two coordination types: Li+−O−(PEO) and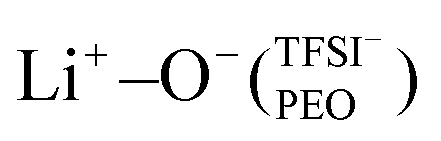 , with the former in more dilute PEOn/LiTFSI systems and the latter exclusively in concentrated PEO6/LiTFSI. Raman spectroscopy confirmed the MD simulations’ predictions, showing either free TFSI− ions (dissociated LiTFSI) or contact ion pairs (associated LiTFSI). MAS-NMR was further utilized to study the lithium-ion dynamics at varying temperatures for PEO32/LiTFSI and PEO6/LiTFSI, which consistently exhibited two distinct lithium-ion dynamics, continuously present for PEO6/LiTFSI regardless of the temperature, while one component disappeared at temperatures above 60 °C for PEO32/LiTFSI. Different ion transport mechanisms were observed depending on the LiTFSI salt content in the SPE.
, with the former in more dilute PEOn/LiTFSI systems and the latter exclusively in concentrated PEO6/LiTFSI. Raman spectroscopy confirmed the MD simulations’ predictions, showing either free TFSI− ions (dissociated LiTFSI) or contact ion pairs (associated LiTFSI). MAS-NMR was further utilized to study the lithium-ion dynamics at varying temperatures for PEO32/LiTFSI and PEO6/LiTFSI, which consistently exhibited two distinct lithium-ion dynamics, continuously present for PEO6/LiTFSI regardless of the temperature, while one component disappeared at temperatures above 60 °C for PEO32/LiTFSI. Different ion transport mechanisms were observed depending on the LiTFSI salt content in the SPE.
Moderately and highly dilute systems (n ≥ 16) had free Li+ ions diffusing via ion-hopping between coordination sites of different PEO chains. Concentrated PEO6/LiTFSI showed associated LiTFSI species, limiting lithium-ion mobility due to the lack of available PEO coordination sites. However, despite lower lithium-ion mobility in PEO6/LiTFSI than SPEs with n ≥ 16, we found that concentrated PEO6/LiTFSI exhibited faster ion dynamics through ion pairs and clusters compared to lithium-ion transport via PEO coordination sites.
When considering ion coordination and transport mechanism for alternative alkali metal-ions (sodium, potassium, and cesium) at a fixed EO/X+ ratio of 20:1, we found that the cation coordination distance increased with larger cations due to their increased volume. However, all studied alkali metal-salts consistently experienced complete dissociation, attributed to PEO's strong solvating ability. Substituting cations in the PEO matrix showed comparable coordination numbers (ranging from 5 to 8) for lithium, potassium, and cesium ions. PEO's solvating ability seemed to reach its limit for larger cations like potassium (with an atomic radius of 2.31 Å), resulting in similar CNs for potassium and cesium (with an atomic radius of 2.62 Å).
MD simulation results for ionic conductivity agreed well with experimental data for PEO20/LiTFSI, PEO20/NaTFSI, and PEO20/KTFSI at temperatures of 50 °C and above. PEO20/CsTFSI exhibited higher ionic conductivity due to weaker coordination between Cs ions and PEO, enhancing cationic mobility. Additionally, cationic transport number analysis showed sodium-ion having the highest cation transport number among the studied cations over the entire temperature range, indicating an optimal balance between cation coordination with PEO and NaTFSI salt dissociation.
In conclusion, the design of novel SPEs should consider not only the extent of salt dissociation but also the type of coordination structures present, as these factors influence charge carrier mobility, allowing tuning of ionic conductivity and cation transport numbers. Our MD simulation-based framework accurately predicted LiTFSI salt association with concentration and facilitated the investigation of alternative alkali metal ions, revealing the impact of cation size on ion dynamics and transport. Finding a balance between cation size and salt dissociation is crucial for advancing the design of novel polymer electrolytes for all-solid-state batteries.
Author contributions
The manuscript includes computational and experimental experiments performed by B. A. F. (Section 3.1), and computational experiments by J. O. (Section 3.2). The manuscript was primarily written by B. A. F., with additions by J. O. (Section 3.2). H. M., M. M.-I. and J. C. provided conceptualization and supervision of the work. Reviewing and editing was performed by all the authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research was supported by funding as a part of the DESTINY PhD program, funded by the European Union's Horizon2020 research and innovation program under the Marie Skłodowska-Curie Actions COFUND (Grant No. 945357), and funding through the Basque Government PhD Grant. The authors also acknowledge funding from ‘Departamento de Educación, Política Lingüística y Cultura del Gobierno Vasco’ (Grant No. IT1358-22), the Basque Government (PRE_2022_1_0034), and thank SGI/IZO-SGIker UPV/EHU for providing supercomputing resources.References
- A. Rahman, O. Farrok and M. M. Haque, Environmental impact of renewable energy source based electrical power plants: Solar, wind, hydroelectric, biomass, geothermal, tidal, ocean, and osmotic, Renewable Sustainable Energy Rev., 2022, 161, 112279 CrossRef.
- H. A. Behabtu, M. Messagie, T. Coosemans, M. Berecibar, K. Anlay Fante, A. A. Kebede and J. V. Mierlo, A Review of Energy Storage Technologies’ Application Potentials in Renewable Energy Sources Grid Integration, Sustainability, 2020, 12, 24 CrossRef.
- L. V. Garcia, Y.-C. Ho, M. M. Myo Thant, D. S. Han and J. W. Lim, Lithium in a Sustainable Circular Economy: A Comprehensive Review, Processes, 2023, 11, 2 CrossRef.
- Y. Ji, J. Li and J. Li, Recent Development of Electrolyte Engineering for Sodium Metal Batteries, Batteries, 2022, 8, 10 CrossRef.
- S. Kaskel, J.-Q. Huang and H. Sakaebe, Lithium-Sulfur Batteries: Current Achievements and Further Development, Batteries Supercaps, 2022, 5, 12 Search PubMed.
- D. Wu, X. Li, X. Liu, J. Yi, P. Acevedo-Peña, E. Reguera, K. Zhu, D. Bin, N. Melzack, R. G. A. Wills, J. Huang, X. Wang, X. Lin, D. Yu and J. Ma, 2022 Roadmap on aqueous batteries, J. Phys.: Energy, 2022, 4, 041501 Search PubMed.
- G. Lu, Z. Wang, S. Zhang, J. Ding, J. Luo and X. Liu, Cathode materials for halide-based aqueous redox flow batteries: recent progress and future perspectives, Nanoscale, 2023, 15, 4250–4260 RSC.
- H. Zhang, F. Chen and J. Carrasco, Energy Storage Mater., 2021, 36, 77–90 CrossRef.
- J. Jiao, G. Lai, L. Zhao, J. Lu, Q. Li, X. Xu, Y. Jiang, Y.-B. He, C. Ouyang, F. Pan, H. Li and J. Zheng, Self-Healing Mechanism of Lithium in Lithium Metal, Adv. Sci., 2022, 9(12), 2105574 CrossRef CAS PubMed.
- L. Dong, C. Zhang and W. Liu, All-in-One Structured Lithium-Metal Battery, Adv. Sci., 2022, 9(17), 2200547 CrossRef CAS PubMed.
- S. Kim, G. Park, S. J. Lee, S. Seo, K. Ryu, C. H. Kim and J. W. Choi, Lithium-Metal Batteries: From Fundamental Research to Industrialization, Adv. Mater., 2022, 2206625 Search PubMed.
- S. Wang, B. Peng, J. Lu, Y. Jie, X. Li, Y. Pan, Y. Han, R. Cao, D. Xu and S. Jiao, Recent Progress in Rechargeable Sodium Metal Batteries: A Review, Chem. – Eur. J., 2023, 29, e202202380 CrossRef CAS PubMed.
- L. Zhang, Y. Xia, H. Yang, S. Xiao, J. Zhou, Y. Cao and T. Qian, The current status of sodium metal anodes for improved sodium batteries and its future perspectives, APL Mater., 2022, 10, 070901 CrossRef CAS.
- T. A. Pham, K. E. Kweon, A. Samanta, V. Lordi and J. E. Pask, Solvation and Dynamics of Sodium and Potassium in Ethylene Carbonate from ab Initio Molecular Dynamics Simulations, J. Phys. Chem. C, 2017, 121, 21913–21920 CrossRef CAS.
- H. Ding, Y. Feng, J. Zhou, X. Yu, L. Fan and B. Lu, Superstable potassium metal batteries with a controllable internal electric field, Fundam. Res., 2022, 16–27 Search PubMed.
- J. Park, J. Lee, M. H. Alfaruqi, W.-J. Kwak, J. Kim and J.-Y. Hwang, Initial investigation and evaluation of potassium metal as an anode for rechargeable potassium batteries, J. Mater. Chem. A, 2020, 8, 16718–16737 RSC.
- A. D. Khudyshkina, P. A. Morozova, A. J. Butzelaar, M. Hoffmann, M. Wilhelm, P. Theato, S. S. Fedotov and F. Jeschull, Poly(ethylene oxide)-Based Electrolytes for Solid-State Potassium Metal Batteries with a Prussian Blue Positive Electrode, ACS Appl. Polym. Mater., 2022, 4, 2734–2746 CrossRef CAS.
- I. R. de Larramendi, I. Lozano, M. Enterría, R. Cid, M. Echeverría, S. R. Peña, J. Carrasco, H. Manzano, G. Beobide, I. Landa-Medrano, T. Rojo and N. Ortiz-Vitoriano, Unveiling the Role of Tetrabutylammonium and Cesium Bulky Cations in Enhancing Na-O2 Battery Performance, Adv. Energy Mater., 2022, 12, 2102834 CrossRef CAS.
- F. Ding, W. Xu, X. Chen, J. Zhang, Y. Shao, M. H. Engelhard, Y. Zhang, T. A. Blake, G. L. Graff, X. Liu and J.-G. Zhang, Effects of Cesium Cations in Lithium Deposition via Self-Healing Electrostatic Shield Mechanism, J. Phys. Chem. C, 2014, 118, 4043–4049 CrossRef CAS.
- B. Lu, X. Liu, J. Qu and Z. Li, Monolayer H-MoS2 with high ion mobility as a promising anode for rubidium (cesium)-ion batteries, Nanoscale Adv., 2022, 4, 3756–3763 RSC.
- H. Wang, D. Yu, C. Kuang, L. Cheng, W. Li, X. Feng, Z. Zhang, X. Zhang and Y. Zhang, Alkali Metal Anodes for Rechargeable Batteries, Chem, 2019, 5, 313–338 CAS.
- Y. Zhao, X. Zhang, J. Xiao, H. Fan, J. Zhang, H. Liu, Y. Liu, H. Yuan, S. Fan and Y. Zhang, Effect of Mg Cation Diffusion Coefficient on Mg Dendrite Formation, ACS Appl. Mater. Interfaces, 2022, 14, 6499–6506 CrossRef CAS PubMed.
- H. Liu, X.-B. Cheng, Z. Jin, R. Zhang, G. Wang, L.-Q. Chen, Q.-B. Liu, J.-Q. Huang and Q. Zhang, Recent advances in understanding dendrite growth on alkali metal anodes, EnergyChem, 2019, 1, 100003 CrossRef.
- A. Stoddart, Alkali metal batteries: Preventing failure, Nat. Rev. Mater., 2018, 3, 18021 CrossRef.
- C. Ling, A review of the recent progress in battery informatics, NPJ Comput. Mater., 2022, 8, 33 CrossRef.
- T. Mabuchi, K. Nakajima and T. Tokumasu, Molecular dynamics study of ion transport in polymer electrolytes of all-solid-state li-ion batteries, Micromachines, 2021, 12, 9 CrossRef PubMed.
- H. Gudla, C. Zhang and D. Brandell, Effects of Solvent Polarity on Li-ion Diffusion in Polymer Electrolytes: An All-Atom Molecular Dynamics Study with Charge Scaling, J. Phys. Chem. B, 2020, 124, 8124–8131 CrossRef CAS PubMed.
- M. Ebadi, L. T. Costa, C. M. Araujo and D. Brandell, Modelling the Polymer Electrolyte/Li-Metal Interface by Molecular Dynamics simulations, Electrochim. Acta, 2017, 234, 43–51 CrossRef CAS.
- T. Xie, A. France-Lanord, Y. Wang, J. Lopez, M. A. Stolberg, M. Hill, G. M. Leverick, R. Gomez-Bombarelli, J. A. Johnson, Y. Shao-Horn and J. C. Grossman, Accelerating amorphous polymer electrolyte screening by learning to reduce errors in molecular dynamics simulated properties, Nat. Commun., 2022, 13, 3415 CrossRef CAS PubMed.
- B. A. Fortuin, L. Meabe, S. R. Peña, Y. Zhang, L. Qiao, J. Etxabe, L. Garcia, H. Manzano, M. Armand, M. Martínez-Ibañez and J. Carrasco, Molecular-Level Insight into Charge Carrier Transport and Speciation in Solid Polymer Electrolytes by Chemically Tuning Both Polymer and Lithium Salt, J. Phys. Chem. C, 2023, 127, 1955–1964 CrossRef CAS PubMed.
- M. Andersson, M. Streb, J. Y. Ko, V. Löfqvist Klass, M. Klett, H. Ekström, M. Johansson and G. Lindbergh, Parametrization of physics-based battery models from input–output data: A review of methodology and current research, J. Power Sources, 2022, 521, 230859 CrossRef CAS.
- A. A. Franco, A. Rucci, D. Brandell, C. Frayret, M. Gaberscek, P. Jankowski and P. Johansson, Boosting Rechargeable Batteries R&D by Multiscale Modeling: Myth or Reality?, Chem. Rev., 2019, 119, 4569–4627 CrossRef CAS PubMed.
- E. Redondo-Iglesias, P. Venet and S. Pelissier, Modelling Lithium-Ion Battery Ageing in Electric Vehicle Applications—Calendar and Cycling Ageing Combination Effects, Batteries, 2020, 6, 1 CrossRef.
- L. Meabe, S. R. Peña, M. Martinez-Ibañez, Y. Zhang, E. Lobato, H. Manzano, M. Armand, J. Carrasco and H. Zhang, Insight into the ionic transport of solid polymer electrolytes in polyether and polyester blends, J. Phys. Chem. C, 2020, 124, 17981–17991 CrossRef CAS.
- S. Pronk, S. Páll, R. Schulz, P. Larsson, P. Bjelkmar, R. Apostolov, M. R. Shirts, J. C. Smith, P. M. Kasson, D. Van Der Spoel, B. Hess and E. Lindahl, GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit, Bioinformatics, 2013, 29, 845–854 CrossRef CAS PubMed.
- S. Plimpton, Fast Parallel Algorithms for Short-Range Molecular Dynamics, J. Comput. Phys., 1995, 117, 1–19 CrossRef CAS.
- N. Kondratyuk, V. Nikolskiy, D. Pavlov and V. Stegailov, GPU-accelerated molecular dynamics: State-of-art software performance and porting from Nvidia CUDA to AMD HIP, Int. J. High Perform. Comput. Appl., 2021, 35, 312–324 CrossRef.
- C. Kutzner, S. Páll, M. Fechner, A. Esztermann, B. L. de Groot and H. Grubmüller, More bang for your buck: Improved use of GPU nodes for GROMACS 2018, J. Comput. Chem., 2019, 40, 2418–2431 CrossRef CAS PubMed.
- L. Martínez, R. Andrade, E. G. Birgin and J. M. Martínez, PACKMOL: A package for building initial configurations for molecular dynamics simulations, J. Comput. Chem., 2009, 30, 2157–2164 CrossRef PubMed.
- A. S. L. Gouveia, C. E. S. Bernardes, L. C. Tomé, E. I. Lozinskaya, Y. S. Vygodskii, A. S. Shaplov, J. N. C. Lopes and I. M. Marrucho, Ionic liquids with anions based on fluorosulfonyl derivatives: from asymmetrical substitutions to a consistent force field model, Phys. Chem. Chem. Phys., 2017, 19, 29617–29624 RSC.
- W. Jorgensen, D. Maxwell and J. Tirado-Rives, Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids, J. Am. Chem. Soc., 1996, 118(45), 11225–11236 CrossRef CAS.
- R. C. Rizzo and W. L. Jorgensen, OPLS all-atom model for amines: Resolution of the amine hydration problem, J. Am. Chem. Soc., 1999, 121, 4827–4836 CrossRef CAS.
- E. K. Watkins and W. L. Jorgensen, Perfluoroalkanes: Conformational Analysis and Liquid-State Properties from ab Initio and Monte Carlo Calculations, J. Phys. Chem. A, 2001, 105, 4118–4125 CrossRef CAS.
- W. L. Jorgensen and N. A. Mcdonald, Development of an all-atom force field for heterocycles. Properties of liquid pyridine and diazenes, THEOCHEM, 1998, 424(1–2), 145–155 CrossRef CAS.
- M. Brehm, M. Thomas, S. Gehrke and B. Kirchner, TRAVIS—A free analyzer for trajectories from molecular simulation, J. Chem. Phys., 2020, 152, 164105 CrossRef CAS PubMed.
- M. Brehm and B. Kirchner, TRAVIS - A Free Analyzer and Visualizer for Monte Carlo and Molecular Dynamics Trajectories, J. Chem. Inf. Model., 2011, 51, 2007–2023 CrossRef CAS PubMed.
- V. Blum, R. Gehrke, F. Hanke, P. Havu, V. Havu, X. Ren, K. Reuter and M. Scheffler, Ab initio molecular simulations with numeric atom-centered orbitals, Comput. Phys. Commun., 2009, 180, 2175–2196 CrossRef CAS.
- V. Havu, V. Blum, P. Havu and M. Scheffler, Efficient O (N) integration for all-electron electronic structure calculation using numeric basis functions, J. Comput. Phys., 2009, 228, 8367–8379 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Development of the Colic-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 37, 785 CrossRef PubMed.
- A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- L. Qiao, U. Oteo, Y. Zhang, S. R. Peña, M. Martínez-Ibañez, A. Santiago, R. Cid, L. Meabe, H. Manzano, J. Carrasco, H. Zhang and M. Armand, Trifluoromethyl-free anion for highly stable lithium metal polymer batteries, Energy Storage Mater., 2020, 32, 225–233 CrossRef.
- N. Molinari, J. P. Mailoa and B. Kozinsky, Effect of High Salt Concentration on Ion Clustering and Transport in Polymer Solid Electrolytes: a Molecular Dynamics Study of PEO-LiTFSI, J. Chem. Mater., 2018, 30(18), 6298–6306 CrossRef CAS.
- D. J. Brooks, B. V. Merinov, W. A. Goddard, B. Kozinsky and J. Mailoa, Atomistic Description of Ionic Diffusion in PEO-LiTFSI: Effect of Temperature, Molecular Weight, and Ionic Concentration, Macromolecules, 2018, 51, 8987–8995 CrossRef CAS.
- P. Kang, L. Wu, D. Chen, Y. Su, Y. Zhu, J. Lan, X. Yang and G. Sui, Dynamical Ion Association and Transport Properties in PEO-LiTFSI Electrolytes: Effect of Salt Concentration, J. Phys. Chem. B, 2022, 126, 4531–4542 CrossRef CAS PubMed.
- T. Darden, D. York and L. Pedersen, Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, A smooth particle mesh Ewald method, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS.
- H. Mehrer, Diffusion in Solids. Fundamentals, Methods, Materials, Diffusion-Controlled Processes, Springer-Verlag Berlin Heidelberg, 2007 Search PubMed.
- G. D’Angelo, G. Tripodo, G. Carini, A. Bartolotta, G. Di Marco and G. Salvato, Low-temperature excess specific heat and fragility in polymers: Crystallinity dependence, J. Chem. Phys., 1998, 109, 7625–7631 CrossRef.
- J. Evans, C. A. Vincent and P. G. Bruce, Electrochemical measurement of transference numbers in polymer electrolytes, Polymer, 1987, 28, 2324–2328 CrossRef CAS.
- D. Massiot, F. Fayon, M. Capron, I. King, S. le Calvé, B. Alonso, J.-O. Durand, B. Bujoli, Z. Gan and G. Hoatson, Modelling one- and two-dimensional solid-state NMR spectra, Magn. Reson. Chem., 2002, 40, 70–76 CrossRef CAS.
- H. Lee, A. H. de Vries, S.-J. Marrink and R. W. Pastor, A Coarse-Grained Model for Polyethylene Oxide and Polyethylene Glycol: Conformation and Hydrodynamics, J. Phys. Chem. B, 2009, 113, 13186–13194 CrossRef CAS PubMed.
- D. N. Theodorou and U. W. Suter, Detailed molecular structure of a vinyl polymer glass, Macromolecules, 1985, 18, 1467–1478 CrossRef CAS.
- J. Atik, D. Diddens, J. H. Thienenkamp, G. Brunklaus, M. Winter and E. Paillard, Cation-Assisted Lithium-Ion Transport for High-Performance PEO-based Ternary Solid Polymer Electrolytes, Angew. Chem., Int. Ed., 2021, 60, 11919–11927 CrossRef CAS PubMed.
- C. Labrèche and J. Prud’homme, Preferential solvation and free volume as interrelated features governing ion conduction in plasticised polyether electrolytes, J. Power Sources, 1999, 81–82, 130–136 CrossRef.
- Y. Song, L. Yang, J. Li, M. Zhang, Y. Wang, S. Li, S. Chen, K. Yang, K. Xu and F. Pan, Synergistic Dissociation-and-Trapping Effect to Promote Li-Ion Conduction in Polymer Electrolytes via Oxygen Vacancies, Small, 2021, 17, 2102039 CrossRef CAS PubMed.
- B. M. Savoie, M. A. Webb and T. F. I. I. I. Miller, Enhancing Cation Diffusion and Suppressing Anion Diffusion via Lewis-Acidic Polymer Electrolytes, J. Phys. Chem. Lett., 2017, 8, 641–646 CrossRef CAS PubMed.
- W. Gorecki, M. Jeannin, E. Belorizky, C. Roux and M. Armand, Physical properties of solid polymer electrolyte PEO(LiTFSI) complexes, J. Phys.: Condens. Matter, 1995, 7, 6823 CrossRef CAS.
- S. Hong, Y. Wang, N. Kim and S. B. Lee, Polymer-based electrolytes for all-solid-state lithium–sulfur batteries: from fundamental research to performance improvement, J. Mater. Sci., 2021, 56, 8358–8382 CrossRef CAS.
- N. A. Stolwijk, C. Heddier, M. Reschke, M. Wiencierz, J. Bokeloh and G. Wilde, Salt-Concentration Dependence of the Glass Transition Temperature in PEO–NaI and PEO–LiTFSI Polymer Electrolytes, Macromolecules, 2013, 46, 8580–8588 CrossRef CAS.
- D. Brandell, J. Mindemark and G. Hernández, Polymer-based Solid State Batteries, De Gruyter, 2021 Search PubMed.
- L. Edman, Ion association and ion solvation effects at the crystalline-amorphous phase transition in PEO-LiTFSI, J. Phys. Chem. B, 2000, 104, 7254–7258 CrossRef CAS.
- I. Rey, J. C. Lassègues, J. Grondin and L. Servant, Infrared and Raman study of the PEO-LiTFSI polymer electrolyte, Electrochim. Acta, 1998, 43, 1505–1510 CrossRef CAS.
- L. Suo, D. Oh, Y. Lin, Z. Zhuo, O. Borodin, T. Gao, F. Wang, A. Kushima, Z. Wang, H.-C. Kim, Y. Qi, W. Yang, F. Pan, J. Li, K. Xu and C. Wang, How Solid-Electrolyte Interphase Forms in Aqueous Electrolytes, J. Am. Chem. Soc., 2017, 139, 18670–18680 CrossRef CAS PubMed.
- J. C. Lassègues, J. Grondin, C. Aupetit and P. Johansson, Spectroscopic identification of the lithium ion transporting species in LiTFSI-doped ionic liquids, J. Phys. Chem. A, 2009, 113, 305–314 CrossRef PubMed.
- L. van Wüllen, T. Echelmeyer, N. Voigt, T. K.-J. Köster and G. Schiffmann, Local Li Cation Coordination and Dynamics in Novel Solid Electrolytes, Oldenbourg Wissenschaftsverlag, 2010, 224, 1735–1769 Search PubMed.
- B.-H. Wang, T. Xia, Q. Chen and Y.-F. Yao, Probing the Dynamics of Li+ Ions on the Crystal Surface: A Solid-State NMR Study, Polymers, 2020, 12(2), 391 CrossRef CAS PubMed.
- D. J. Morales and S. Greenbaum, NMR Investigations of Crystalline and Glassy Solid Electrolytes for Lithium Batteries: A Brief Review, Int. J. Mol. Sci., 2020, 21, 9 Search PubMed.
- O. Borodin and G. D. Smith, Mechanism of Ion Transport in Amorphous Poly(ethylene oxide)/LiTFSI from Molecular Dynamics Simulations, Macromolecules, 2006, 39, 1620–1629 CrossRef CAS.
- Z. Xue, D. He and X. Xie, Poly(Ethylene Oxide)-Based Electrolytes for Lithium-Ion Batteries, J. Mater. Chem. A, 2015, 3, 19218–19253 RSC.
- U. Oteo, M. Martinez-Ibañez, I. Aldalur, E. Sanchez-Diez, J. Carrasco, M. Armand and H. Zhang, Improvement of the Cationic Transport in Polymer Electrolytes with (Difluoromethanesulfonyl)(trifluoromethanesulfonyl)imide Salts, ChemElectroChem, 2019, 6, 1019–1022 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supporting tables and figures including force field parameters, chemical structures, lithium-ion speciation analysis, MD-based ionic conductivity convergence tests, MSD, DSC, EIS, Raman spectroscopy, MAS-NMR spectroscopy, and activation energies of the transport mechanisms for the different investigated systems. See DOI: https://doi.org/10.1039/d3cp02989a |
| This journal is © the Owner Societies 2023 |