
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective modulation of alkali metal ions on acetylcholinesterase†
Xia
Mu‡
 a,
Shengwei
Yuan‡
ab,
Dinglin
Zhang‡
a,
Rui
Lai
a,
Chenyi
Liao
*a and
Guohui
Li
*a
a,
Shengwei
Yuan‡
ab,
Dinglin
Zhang‡
a,
Rui
Lai
a,
Chenyi
Liao
*a and
Guohui
Li
*a
aState Key Laboratory of Molecular Reaction Dynamics, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian, China. E-mail: cliao2@dicp.ac.cn; ghli@dicp.ac.cn
bUniversity of Chinese Academy of Sciences, Beijing, China
First published on 28th October 2023
Abstract
Acetylcholinesterase (AChE) is an important hydrolase in cholinergic synapses and a candidate target in the treatment of Alzheimer's disease. The lithium treatment widely used in neurological disorders can alter the AChE activity, yet the underlying mechanism of how the ion species regulate the enzymatic activity remains unclear. In this work, we performed combined quantum mechanics/molecular mechanics (QM/MM) and molecular dynamics (MD) simulations and well-tempered metadynamics to understand the modulation of human AChE (hAChE) activity using three alkali metal ions (Li+, Na+, and K+) in different concentrations. Our simulations show that the binding affinity and catalytic activity are affected by different ion species through allosteric ion coordination geometries on the hAChE complex and distant electrostatic screening effect. A Li+ cluster involving D330, E393, and D397 residues and three Li+ ions was found to be highly conserved and can be critical to the enzyme activity. Binding energy calculations indicate that the electrostatic screening from allosterically bound cations can affect the key residues at the catalytic site and active-site gorge, including E199. Furthermore, an increase in ion concentration can lead to lower reactivity, especially for Li+ ions, which exhibit more cation-hAChE contacts than Na+ and K+. The selective ion binding and their preferred modulation on hAChE are highly related to ion species. This work provides a molecular perspective on selective modulation by different ion species of the enzyme catalytic processes.
1. Introduction
Acetylcholinesterase (AChE) is a serine hydrolase responsible for the termination of nervous signaling by hydrolyzing the neurotransmitter acetylcholine (ACh) in cholinergic synapses.1,2 Its non-hydrolytic functions are also found as morphometric processes along the nervous system.3,4 The inhibition of AChE leads to the accumulation of ACh in the synaptic space and causes the malfunction of central and peripheral nervous systems. Many AChE inhibitors like organophosphates are widely used as insecticides in agriculture.5 However, some organophosphates are nerve agents, which can cause irreversible damage to the human nervous system, even death.6,7 In recent years, AChE has gained increasing interest as a target for Alzheimer's disease (AD) treatment due to the connection between cholinergic activity and AD.3,8–10 Several clinical drugs used for the treatment of AD, like donepezil, are inhibitors of AChE.11–14 Modulation of AChE using multiple factors including ions and small molecules is gaining increasing interest to reveal its various activities in the nervous system and clinical treatments.The catalytic site of AChE consists of three parts: a long gorge around 20 Å from the surface to in-deep active sites, Ser–His–Glu as the catalytic triad for hydrolysis, and the oxyanion hole for transition stabilization. A natural allosteric site at the entrance of the gorge was identified and called the peripheral anionic site (PAS).15,16 Modulators binding on the PAS may alter the enzyme activity by steric effects from a narrowed gorge or by changing the conformation of active sites. The PAS contains three polar residues, including Y72, Y124 and D74, on the enzyme surface and two aromatic residues, W286 and W86, in the substrate gorge as a bottleneck for substrate diffusion. Experiment suggests that the two residues participate in reaction via a cation–π interaction between the aromatic ring and the choline part of the substrate.15,17 A few allosteric sites were also studied. For example, Chierrito et al. used STD-NMR experiments and molecular modeling and showed that their synthesized AChE inhibitor molecule binds to the allosteric site B, which contains residues R247, Q291, and R296.18 Bondžić et al. used experiments and molecular docking and found an allosteric site for binding voluminous negatively charged molecules and named it β-allosteric site, which contains residues R21, K23, and Y105.19 Two new possible inhibition allosteric sites were identified recently by Roca which include the E81-E452-R463 group and R296-H405-W532-Q413-N533.20 In general, the AChE exhibits high performance in hydrolyzing ACh with kcat/KM ≈ 2 × 108 M−1 S−1.1 The diffusion of the substrate is the rate-limiting step for the whole catalysis which is electrostatically controlled. A large dipole along the gorge, about 505 Debye, is thought to be the force that pulls positively-charged ACh from the enzyme surface to the active sites.21 Indeed, some studies have suggested that the cationic ions can modulate the catalytic efficiency of an enzyme, especially those with a charged substrate like ACh.17,22,23 Due to the vast presence of lithium, sodium and potassium ions in biological systems, it is of interest to elucidate the effects of alkali metal ions on the allosteric modulation of AChE.
Lithium has received increasing attention for its industrial applications and pollution of the environment.24 But its protective impact on neurotransmitters makes it more appealing for clinical treatment. For example, lithium is widely used in the therapy of bipolar disorder because it may alter the concentration of ACh in the rat brain.25 Experiment shows that lithium treatment in vivo significantly decreased the AChE activity but made no changes in vitro.26,27 Although the mechanism of lithium action remains unclear, there is growing knowledge that Li+ may exert its physiological function by affecting the signal transduction proteins or the post-transition process of AChE.28 These proteins include G-protein-coupled pathways especially the sodium and potassium channels for a competitive effect.29 This raises the question of what is the difference between the modulation mechanisms of Li+ ions on physiological activities compared to those of Na+ and K+ ions.
Vizi et al. for the first time found a difference between the effects of Na+ and Li+ ions that ACh release was unaffected at 25 mM Na+ concentration but reduced the ACh output while Li+ increased to 117.9 mM.30 Tsakiris and co-workers suggested that, at 115 mM concentration of Na+ and Li+, Li+ has a greater activation impact on soluble AChE.31 A decreased activity was found with increasing ionic strength from a concentration range of 1–600 mM.17,23 A different negative effect from Li+, Na+ and K+ on AChE has also been studied.22 Rodrigo found that 100 mg L−1 Li+ increases the activity of AChE in zebrafish.32 Irina et al. observed a decrease in the activity of the reactivation of phosphorylated hAChE with Li+ concentration from 50–1000 mM.33 Thainara et al. demonstrated that higher Li+ exposure would decrease AChE activity with concentration in the range of 100–750 μg L−1 after 28 day in Mtilus galloprovincialis system.34 Although experiment research produced conflicting findings, there is a certain relationship between ion concentration and enzymatic activity. How alkali metal ions affect the enzyme structure and activity is not clear.
In this work, combined quantum mechanics/molecular mechanics (QM/MM) and molecular dynamics (MD) simulations are performed to study the modulation of three alkali metal ions, Li+, Na+, and K+, on AChE activity at different concentrations. In general, Li+ is mildly toxic, but its concentration over 2.86 mM in blood may lead to death.35 The Li+ ion concentration used in clinical treatment is usually in the range of 0.6–1.2 mM in neurons; serum concentration >2 mM is considered toxic.29,36 Fundamental understandings of the regulation mechanism by environmental factors on AChE activity are essential to the rational design of AChE inhibitors. Here, we study systems at 15 mM and 150 mM for the normal and high concentrations, respectively. Large time-scale MD simulations were performed to reveal the potential binding sites for ions and the conformational changes of the enzyme. The QM/MM MD method was applied to study the catalytic efficiency of the enzyme under different conditions. The selective ion binding and their preferred modulation are highly related to ion species. This work helps to understand the ion modulation on AChE in catalytic processes.
2. Computational methods
AChE with three alkali metal ions (Li+, Na+ and K+) at normal (15 mM) and high (150 mM) concentrations are studied, totaling six systems. We first applied MM-MD to study the conformational changes induced by different ion species and concentrations. Next, we applied QM/MM-MD to examine the enzyme catalysis activity changes at different ion concentrations in the ACh hydrolysis process.The initial structure of hAChE was extracted from the Protein Data Bank (PDB ID 6O4W).37 This structure presents a dimeric hAChE–donepezil complex, but only chain A was used in this work. The three missing initial residues in the crystal structure are ignored. The sequence of enzyme was renumbered with the active site as S200, E331 and H444 in this work. Based on the well-characterized reaction mechanism (as shown in Scheme 1) of AChE catalysis, we docked the substrate ACh to the active site and adjusted its position to the Michaelis complex based on early experimental and theoretical studies.38–42
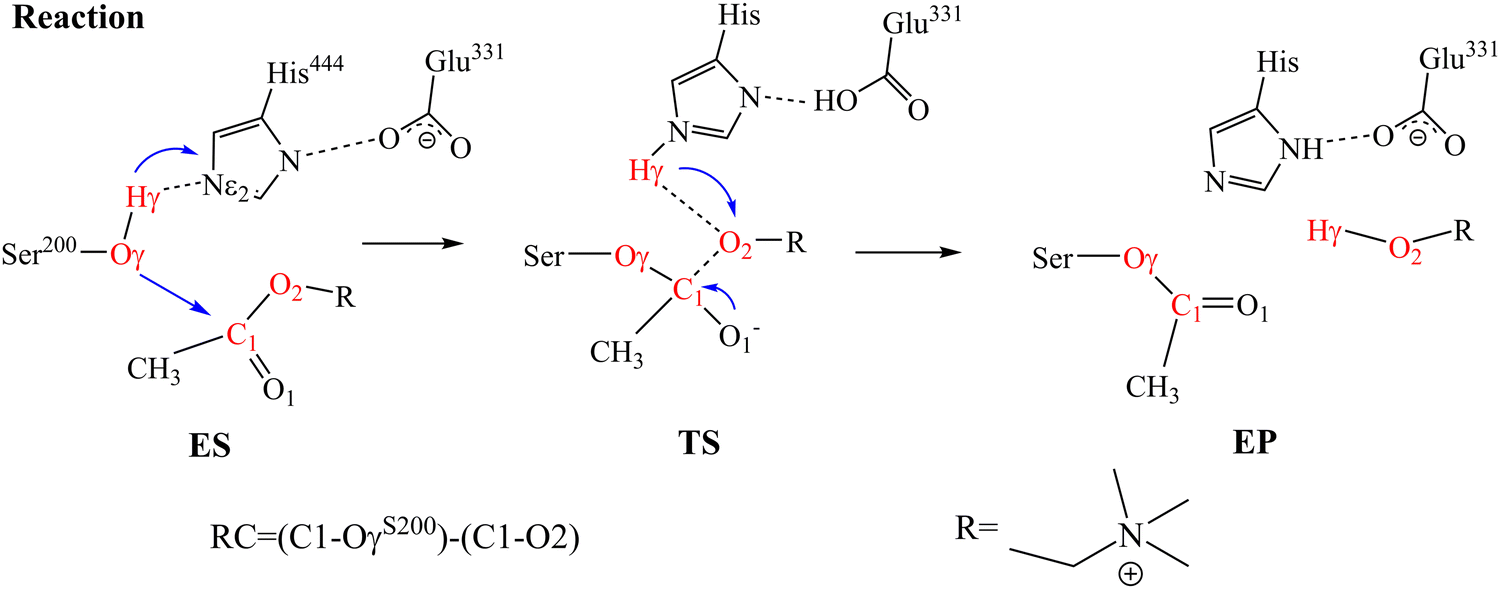 | ||
| Scheme 1 Proposed mechanism of ACh hydrolysis catalyzed by AChE at the active site consisting of S200, E331, and H444. Atoms involved in the reaction coordinate (RC) are colored in red. | ||
For all systems, the hAChE and substrate complex was solvated with 24055 pre-equilibrated TIP3P water molecules43 in a 85 × 107 × 105 Å3 periodic boundary condition44 rectangular box with at least 12 Å from the protein to the nearest box edge. With a system volume of around 100 × 10−23 L, we added 9 ions to the 15 mM system and 90 ions to the 150 mM system according to the normal saline concentration of 0.1539 mol L−1, and chloride ions were used to neutralize the system. At the pre-equilibrium stage, the system was equilibrated for 1 ns with an integration time step of 1 fs at a temperature of 300 K using the Nosé–Hoover scheme,45,46 and pressure at 1 bar. For the non-bonded interactions, a 10.0 Å cut-off was used for the van der Waals interactions and short-range electrostatics, with the long-range electronics evaluated using the particle-Mesh Ewald summation scheme.47,48 The bonds involving hydrogen were constrained with SHAKE algorithm.49 At the production stage, we employed a 4 fs time step with hydrogen mass repartitioning (HMR) approach to accelerate MD simulations. All MD simulations were carried out with the ff14SB force field using the AMBER 18 package.50 Finally, we carried out five parallel simulations for each system, 1000 ns for each independent simulation, totaling about 5 μs for all MM-MD simulations.
To study the enzyme activity with different ion species and concentrations, the QM/MM models were built to simulate the ACh hydrolysis reaction. In this work, a semi-empirical method, the third-order density functional tight-binding (DFTB3) theory,51 was employed to describe the electronic structure for the atoms in the QM region with 3OB-3-1 parameters.52–55 The atoms in the MM region were treated with the ff14SB AMBER force field.56 After constructing the reaction model, QM/MM-MD simulations were performed to equilibrate the system. Each system was studied with five parallel simulations, and each independent simulation lasted for 1 ns. For data analysis, we only use the final 1000 structures from each simulation. The proposed mechanism for ACh hydrolysis catalyzed by AChE is illustrated in Scheme 1. In the active site, the S200 of AChE transfers its hydrogen to the general base H444 and results in the formation of nucleophile O−. The O− forms bond with the C1 atom on the substrate and leads to bond breaking in the C1–O2. To explore the free energy surface (FES) involving these residues, we applied well-tempered metadynamics (WT-MetaD)57,58 with multiple walker58 using AMBER 18 interfaced with Plumed 2.6.0.59,60 The collective variables (CVs) for the QM/MM WT-MetaD simulation were defined as CV1 = (Oγ–C1)–(C1–O2) for the distance difference between C1–OγS200 and C1–O2 and CV2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Hγ-Nε2 for the distance difference between the Hγ atom of Ser200 and the Nε of H444. CV1 involves the nucleophilic attack and elimination on substrate. CV2 reflects the proton transfer process during substrate hydrolysis. For each system, a total of five walkers were applied with each simulation lasting at least 500 ps until the FES converged. We set up with a Gaussian height of 0.2 kcal mol−1, a width of 0.2 Å, and a bias factor of 10.0. We deposit a Gaussian on the landscape in every 100 MD steps (100 fs) to improve the sampling efficiency of the conformation space.
Hγ-Nε2 for the distance difference between the Hγ atom of Ser200 and the Nε of H444. CV1 involves the nucleophilic attack and elimination on substrate. CV2 reflects the proton transfer process during substrate hydrolysis. For each system, a total of five walkers were applied with each simulation lasting at least 500 ps until the FES converged. We set up with a Gaussian height of 0.2 kcal mol−1, a width of 0.2 Å, and a bias factor of 10.0. We deposit a Gaussian on the landscape in every 100 MD steps (100 fs) to improve the sampling efficiency of the conformation space.
3. Results and discussion
3.1 Selective ion binding on the hAChE complex
We carried out five parallel 1000 ns MD simulations for each of the six systems. Generally, with the crystal structure as reference, hAChE reached equilibrium at around 400 ns with RMSD around 2.0–2.5 Å in 15 mM (Fig. S1, ESI†), little fluctuation for the catalysis-related residues (Fig. S3, ESI†) and DSSP secondary structure results of hAChE fragments (Fig. S5, ESI†) with a large fluctuation value. The RMSD in 150 mM are smaller, 1.3–1.9 Å. The radius of gyrations (Rg) of hAChE in Li+, Na+, and K+ systems (Fig. S4, ESI†) shows some structural perturbation from high salt, compared with 15 mM. Considering different radii of Li+, Na+ and K+, we chose 3.5 Å, 3.7 Å and 4.0 Å as the cut-off distances to count the strong ion–hAChE contacts as displayed in Fig. 1. With the smallest ion radius, Li+ forms more contacts with hAChE than Na+ and K+. In the Li+ system of 15 mM, strong ion–hAChE contacts are found between Li+ and D71/E289(Group-1), P165/D301/D303(Group-2), and D330/E393/D397(Group-3). As illustrated in Fig. 2, the negatively charged aspartic and glutamic residues bind with Li+via their carboxyl groups, whereas the proline binds with Li+ with the backbone oxygen atom. When the concentration increased to 150 mM, more interactions were found between Li+ and the hAChE surface. For example, binding residues consisting of E78 and E81 were found to coordinate together with Li+, while D92 or D131 interacts with Li+ separately. Also, E289 interacts with Li+ together with E282 in the 150 mM system, while with D71 in the 15 mM system. Group 3 is the most stable coordination cluster, accounting for 60% trajectory in 15 mM Li+, Na+ and K+ simulations. This group is thought to be a common motif in more than 31 AChE sequences and considered to be helpful in enzyme stabilization and activity.61 As the concentration increases in Na+ and K+ systems, the binding strength decreased slightly. Overall, no additional ion binding sites were observed as the ion concentration increased over 9-fold from 15 mM to 150 mM. | ||
| Fig. 1 Fractions of frames where the ion–hAChE contact are present during the MD simulation in Li+, Na+, and K+ systems of 15 mM (A), (C) and (E) and 150 mM (B), (D) and (F), respectively. Residues that coordinated with the same ion(s) are marked with the same color, except black colored residue that interacts with ions solely by itself. | ||
 | ||
| Fig. 2 Identified coordination geometry of bound Li+ on hAChE complex. (A) An overall view of the ion-binding sites on the hAChE's surface with (B)–(D) displaying the strong hAChE–ion interacting sites. The ACh binding site is in cyan, while Li+-bound residues are highlighted in purple. | ||
We further examine the Li+ binding mode in the 15 mM system (Fig. 2). The active site of AChE lies next to the PAS at the base of the narrow 20 Å deep gorge.62–65 Group-1 is formed by D71, E289 and two Li+ by electrostatic interactions (view 1 in Fig. 2). It locates at the entrance of the active site gorge, and may hinder a substrate's entry. Thus, the Li+ cluster with D71 and E289 plays as a gorge lock decreasing enzyme activity. In contrast, group-2 and one Li+ that locates far away from the active site entrance may have little effect on the enzyme activity (view 2 in Fig. 2). The group-3 and three Li+ was found conserved in all Li+ systems as the most stable Li+ coordination geometry (view 3 in Fig. 2). While group-1 and group-2 only involve residues in loop regions, group-3 contains two residues in helices and one in loop region. These three residues exhibit small dynamic fluctuations according to their RMSF values (Fig. S3, ESI†), and are also found to bind with a nitrate ion in the crystal structure (PDB ID 6O4W) in position overlap of the three bound Li+ in our study. Moreover, the Li+ coordination residue D330 is next to E331, a key residue in enzyme catalysis. Thus, further detailed analysis was carried out on the allosteric effect of those Li+ binding sites and hAChE activity.
3.2 Allosteric path of ion binding sites
To demonstrate the allosteric effect of Li ion binding on enzyme active sites, we carried out the dynamics network analysis using WISP procedure66 with VMD and plotted the optimal path, shown in Fig. 3. The optimal pathway between two groups is considered as the potential allosteric pathway, and the shortest path in WISP representing the most possible situation. Each Li+ binding group generates a total of 10 optimal paths, with path colors ranging from blue to yellow and a decrease in the radius of the path-pipe. When all three ion binding groups are used as the starting point for analyzing, all 10 paths are located between group-3 and active sites (S200, E331, and H444), indicating that the group consists of D330, E393 and D397 is the most important effect site among them. This result is consistent with that only this group is common for Na+ and K+ systems. The shortest optimal pathway in group-3 is D330-V328-H444 with a length of 1.3; only one residue V328 is involved indicating a stronger allosteric relationship between D330 and H444. As for group-2, there is a much longer path which consists of P165-G166-N167-P298-F120-A201-S200 with a length of 2.8. Totally, five residues insert into P165 and S200. Different from group-3, the ending point is S200 rather than H444. For group-1, the shortest optimal path is found to be E289-S290-V291-F335-H444, about 3.1 for this path length. Overall, D330 and V328 play as communication hubs in those pathways, indicating their critical roles in the activity of AChE. Li+ modulation on AChE is implemented through the allosteric network of these Li+ binding groups.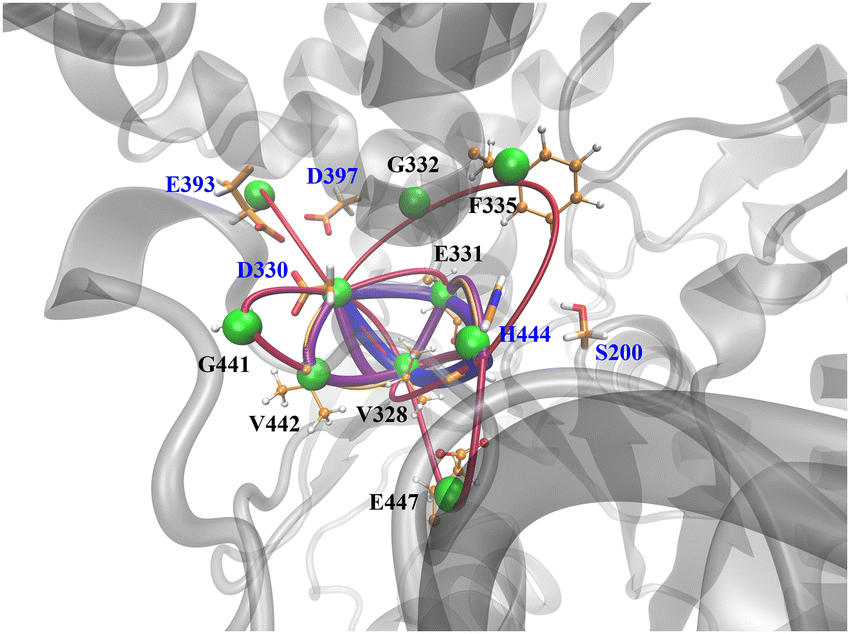 | ||
| Fig. 3 Optimal paths from ion-binding sites (group-3) to active sites. The green ball represents the node on the allosteric path. The shorter path exhibits a larger pipe radius, vice versa. The optimal path of ion-binding sites (group-1 and group-2) can be found in Fig. S7 (ESI†). | ||
3.3 Ion effect on the binding affinity of the hAChE complex
Since it is suggested that the Li+ clusters formed at the entrance of the active site gorge have potential for regulating the activity of hAChE complex, we further look into the binding affinity of the hAChE complex in different ion environments. The distributions of ACh–AChE distance measured by Oγ(S200) and C1(ACh) for different systems are shown in Fig. 4. In 15 mM, while the ACh–AChE distance in the Li+ system is calculated to be around 7.0 Å, it exhibits multiple distance peaks such as 6.0, 8.0, and 10–11 Å in K+ and Na+ systems. In 150 mM, the ACh–AChE distance in the Li+ system becomes more disturbed with multiple distance peaks around 7.0, 10–11, and 12 Å, while the distance in K+ and Na+ systems decreased. The populated ACh–AChE distance in the K+ system of 150 mM becomes similar (∼7 Å) to that of the Li+ system of 15 mM. Thus, the ACh molecule shifts its preferred binding position as the salt concentration increased, particularly for the Li+ system, indicating a decrease tendency in the binding affinity in the hAChE complex. Previous research demonstrated that the “dipole moment” of the active-site gorge has promoted a substrate to enter the enzyme.17,67 The electrostatic screening by high salt concentration may disrupt the “dipole” guidance along the active-site gorge. | ||
| Fig. 4 Distribution of nucleophilic distance between ACh and hAChE by calculating the distance between Oγ (S200) and C1(ACh) in Li+, Na+, and K+ systems of 15 mM and 150 mM in MD simulations. | ||
To quantify the binding affinity of the hAChE complex, we employed the generalized Born and surface area continuum solvation (MM/GBSA) method68 to compute the ACh binding energy in different ion environments. As shown in Table 1, the ACh binding energy decreases in 15 mM Li+, Na+, and K+ systems as −24.2, −26.6, and −29.3 kcal mol−1, respectively, which is consistent with the ACh–AChE distance distribution tendency, shown in Fig. 4. The ACh binding energy decreases in all ion systems as the salt concentration increases to 150 mM with a similar tendency to those in the 15 mM systems. However, the binding affinity in the Li+ system is more sensitive to salt concentration with a decrease of 5 kcal mol−1, while it is affected the least in the Na+ system with a decrease of 1.4 kcal mol−1.
| Ion species | 15 mM | 150 mM |
|---|---|---|
| Li+ | −24.2 ± 3.6 | −19.3 ± 5.6 |
| Na+ | −26.6 ± 3.8 | −25.2 ± 4.8 |
| K+ | −29.3 ± 3.0 | −26.8 ± 3.0 |
To examine how much the salt concentration affects local substrate–residue interactions near the active-site gorge,17,69 we carried out the per-residue free energy decomposition to identify the key ligand–protein interactions. As presented in Table 2, residues with significant impact on ACh binding primarily locate at the active site (catalytic site), active-site gorge, and ion-binding sites. For example, favorable residues near the catalytic site include G118, G119, S122, E199, E331, H444, and Y446. Important residues along the active-site gorge include D71 and W83, whereas D71 also belongs to the ion-binding site. In other studies, W83, Y121 and Y446 are found to stabilize ACh via the π–cation interactions.70,71 The importance of W83 and Y446 to ligand binding is also supported in our study. Primarily, W83 is found to contribute to ligand binding in all ion systems with more than −3.0 kcal mol−1. The E199 near the catalytic site S200 is thought to be critical for substrate orientation and displays a higher positive energy of 3.8 kcal mol−1 in the Li+ system, compared with lower positive energies in Na+ and K+ systems of 15 mM. As the salt concentration increases to 150 mM, global and local electrostatic screening causes an increase in the variety of positive-impact (negative energies) and negative-impact (positive energies) residues (Table 2). Overall, different ion species lead to different influences on the binding affinity of the hAChE complex. The hAChE complex in the Li+ system with more Li+–hAChE contacts (Fig. 1) displays less binding affinity compared with that in the K+ system with fewer K+–hAChE contacts.
| Residue | Li+ (15 mM) | Na+ (15 mM) | K+ (15 mM) | Li+ (150 mM) | Na+ (150 mM) | K+ (150 mM) |
|---|---|---|---|---|---|---|
| D71 | — | — | — | — | 0.87 | — |
| W83 | −4.1 | −4.1 | −3.0 | −3.3 | −4.8 | −4.8 |
| G118 | — | — | — | −1.1 | — | −0.8 |
| G119 | — | — | — | −0.4 | — | — |
| S122 | — | −1.3 | — | — | — | — |
| E199 | 3.8 | 1.5 | 2.1 | 5.3 | 3.7 | 3.2 |
| E331 | — | — | — | 0.8 | 0.5 | 0.9 |
| Y334 | −1.0 | — | −1.7 | — | −0.7 | −1.9 |
| H444 | — | — | −1.0 | — | — | — |
| Y446 | −1.2 | — | −2.5 | — | −1.0 | −1.2 |
| E447 | — | — | — | 0.9 | 0.5 | 0.9 |
3.4 Allosteric modulation of alkali metal ions on AChE
Our MD demonstrates that the salt environment disrupted the catalytic triad S200-H444-Glu331, causing the substrate molecule to lose its reactive conformation (Fig. S6, ESI†). Other theoretical studies have revealed a nucleophilic distance of roughly 2.2–3.0 Å in the ACh hydrolysis catalyzed by AChE.40,72,73 In principle, a stable binding structure is essential for studying the reaction mechanism (the RMSD of five QM/MM-MD for all systems can be found in Fig. S2, ESI†). We have summarized some key parameters of the ACh–AChE interactions in QM/MM-MD in Table 3 (data for the 150 mM system is shown in Table S1, ESI†). As mentioned above, the S200–H444 and S200–ACh distances are critical for ACh hydrolysis, and these distances are shorter and more stable from the Li+, Na+ to K+ systems. The Hγ(S200)–Nε2(H444) is 4.3 Å in the Li+ system, 3.1 Å in the Na+ system, and 2.0 Å in the K+ system, respectively. The nucleophilic distance of the S200-ACh in Li+ system is around 4.5 Å, whereas in the K+ system it is just 2.7 Å and much easier for a nucleophilic attack. Furthermore, as the ion radius increases from Li+ to K+, the interactions between the carbonyl oxygen and the “oxyanion–hole” would become more stable. The W83 in the active-site gorge kept the cationic N atom on ACh at a constant distance by the positive ion–π interaction. With all the key geometry parameters in the reasonable range, the binding structure has reached a relatively stable equilibrium for use to study the ACh hydrolysis reaction. In the following, using QM/MM MD simulation, we will focus on the enzyme acetylation reaction and examine the Michaelis complex to gain better understanding of the catalytic activity of AChE in different solutions.| Distance (Å) | 15 mM Li+ | 15 mM Na+ | 15 mM K+ |
|---|---|---|---|
| Hγ(S200)–Nε2(H444) | 4.3 ± 1.0 | 3.1 ± 0.9 | 2.0 ± 0.2 |
| Oγ(S200)–C1(ACh) | 4.5 ± 1.5 | 3.7 ± 1.6 | 2.7 ± 0.3 |
| Hδ1(H444)–Oε2(E331) | 2.0 ± 0.7 | 2.0 ± 0.3 | 3.5 ± 2.2 |
| O1–H(G118) | 3.8 ± 0.8 | 2.6 ± 1.3 | 2.0 ± 1.2 |
| O1–H(G119) | 2.7 ± 0.9 | 2.4 ± 1.3 | 1.8 ± 0.1 |
| O1–H(A201) | 3.7 ± 2.0 | 3.1 ± 2.2 | 2.1 ± 0.2 |
| N1–W83-ring | 4.4 ± 0.3 | 4.3 ± 0.2 | 4.2 ± 0.2 |
| N1–Y334-ring | 10.4 ± 0.6 | 11.0 ± 1.0 | 10.9 ± 1.0 |
| N1–Y446-ring | 5.8 ± 0.6 | 7.9 ± 1.2 | 6.7 ± 0.6 |
All FES results from the WT-MetaD for acetylation step of ACh hydrolysis are shown in Fig. 5. The representative structures of the species involved in the 15 mM Li+ system are shown in Fig. 6 (structures of 15 mM Na+ and 15 mM K+ in Fig. S8 and S9, ESI†). The key geometric data are listed in Table 5. For a clearly display, we summarize the energies of the stationary states in Table 4. There are two transition states involved in the acetylation step. The first step is the nucleophilic attack of S200-Oγ to C1 of ACh as well as the proton transfer of HγS200 to the general base H444, which leads to the formation of a tetrahedral intermediate (EI). Then, the orientation of H444 turns to ACh's leaving group leading to the donation of the proton to the substrate. Simultaneously, the ester link C1–O2 bond breaks, producing an acetylated enzyme (EP). The reaction mechanism is consistent with those reported in the literature.40,72,73 Interestingly, the activation energies are rather different with the presence of different metal ions in solution. In the first step (from ES to EI), the activation energies for TS1 are 3.44 kcal mol−1, 6.81 kcal mol−1 and 6.76 kcal mol−1, respectively, for Li+, Na+ and K+ ions, suggesting that the protonation of H444 requires less activation free energy in the presence of Li+ in solution. The free energies for the EI state are −3.93 kcal mol−1, 1.29 kcal mol−1 and 1.02 kcal mol−1, respectively, for the systems with Li+, Na+ and K+, implicating that a more stable EI state is involved in the system with Li+. Similarly, the Li+-system has lower TS2 energy compared to those with Na+ and K+ ions. In addition, all the free energies for the K+ system are slightly higher than the Li+ and Na+ systems, as shown in Fig. 5. The free energy of TS2 is relatively lower than that of TS1, suggesting that the first step is the rate-limiting step. Therefore, with Li+ in the solution, hAChE has the highest catalytic activity among the three studied alkali metal ions.
 | ||
| Fig. 5 Free energy surface (FES) for the ACh hydrolysis reaction in different ion environments. | ||
 | ||
| Fig. 6 Representative structures of species involved in the reaction obtained from the WT-MetaD of hydrolysis reaction catalyzed by hAChE in 15 mM Li+ solution. The snapshot structure of stationary points in hAChE hydrolysis ACh reaction with 15 mM Na+ and K+ systems can be found in Fig. S8 and S9 (ESI†). | ||
| ES | TS1 | EI | TS2 | EP | |
|---|---|---|---|---|---|
| 15 mM Li+ | 0.00 | 3.44 | −3.93 | 1.29 | −3.55 |
| 15 mM Na+ | 0.00 | 6.81 | 1.29 | 2.36 | −0.02 |
| 15 mM K+ | 0.00 | 6.76 | 1.02 | 3.84 | −2.19 |
| 150 mM Li+ | 0.00 | 6.81 | −2.26 | 2.23 | −2.20 |
| 150 mM Na+ | 0.00 | 2.94 | 0.67 | 1.96 | −3.00 |
| 150 mM K+ | 0.00 | 3.82 | −4.55 | −1.02 | −5.65 |
Two transition states are also located in the 150 mM systems. The stationary structures of the 150 mM Li+ system are shown in Fig. 7, and the representative structures for Na+ and K+ systems are shown in Fig. S10 and S11 (ESI†). The activation energies for TS1 are 2.94 kcal mol−1 for Na+ ions and 3.82 kcal mol−1 for K+ ions, which are ∼3 kcal mol−1 lower than that in the 15 system. A similar tendency is also observed in the EI, TS2, and EP of 150 mM Na+ and K+ systems compared with the 15 mM system. This implies that an increase in salt concentration promotes ACh hydrolysis in Na+ and K+ systems. On the contrary, an increase in Li+ concentration significantly exhibits a higher activation energy for TS1 (6.81 kcal mol−1) and TS2 (2.23 kcal mol−1), implying an inhibitory effect for hydrolysis. Therefore, the enzyme activity is inhibited by an increase in Li+ but enhanced when the concentrations of Na+ and K+ increase.
 | ||
| Fig. 7 Representative structures for the species involved in the ACh hydrolysis catalyzed by hAChE in 150 mM Li+ solution obtained from WT-MetaD simulation. | ||
By comparing the reaction processes of 15 mM and 150 mM Li+ in Tables 5 and 6, the main difference takes place in the formation of TS1 from ES. In the initial ES state, ACh remains closer to S200 with a distance at 2.4 Å in the 15 mM system, but 2.8 Å in the 150 mM system. Furthermore, the oxyanion–hole residues (G118, G119 and A201) also form more stable interactions than those in the high concentration system. This result is consistent with the results for nucleophilic distance distribution shown in Fig. 4 and the binding energies shown in Table 1. During the formation of TS1, C1 is obviously activated in 15 mM, as evidenced by d(Oγ–C1) = 1.9 Å, while in the 150 mM model this distance is 2.2 Å. This indicates that proton transfer and nucleophilic attack happen in weak correlation in high concentration systems. The increased salt concentration reduced the substrate binding affinity and caused it to shift away from the near nucleophilic attacking conformation towards a higher transition state. Therefore, the increase in ion concentration leads to lower reaction activity, especially for Li+ ions, in agreement with more Li+–hAChE contacts identified in classical MD simulation.
| Distance (Å) | ES | TS1 | EI | TS2 | EP |
|---|---|---|---|---|---|
| Oγ(S200)–C1 | 2.4 | 1.9 | 1.6 | 1.4 | 1.3 |
| C1–O2 | 1.3 | 1.4 | 1.6 | 1.9 | 3.3 |
| Hγ(S200)–Nε2(H444) | 1.8 | 1.2 | 1.1 | 1.8 | 2.4 |
| Hγ(S200)–O2 | 2.8 | 2.6 | 1.7 | 1.0 | 1.0 |
| Hδ1(H444)–Oε2(E331) | 1.9 | 1.6 | 1.9 | 2.4 | 1.9 |
| O1–H(G118) | 1.8 | 1.7 | 1.8 | 1.9 | 1.9 |
| O1–H(G119) | 2.0 | 1.8 | 1.8 | 1.9 | 1.8 |
| O1–H(A201) | 1.9 | 1.8 | 1.7 | 1.8 | 2.1 |
| Distance (Å) | ES | TS1 | EI | TS2 | EP |
|---|---|---|---|---|---|
| Oγ(S200)–C1 | 2.8 | 2.2 | 1.4 | 1.3 | 1.3 |
| C1–O2 | 1.4 | 1.4 | 1.7 | 2.5 | 3.3 |
| Hγ(S200)–Nε2(H444) | 1.8 | 1.3 | 1.0 | 1.4 | 2.3 |
| Hγ(S200)–O2 | 3.4 | 2.0 | 1.9 | 1.2 | 0.9 |
| Hδ1(H444)–Oε2(E331) | 1.7 | 1.7 | 1.8 | 1.8 | 1.8 |
| O1–H(G118) | 3.3 | 3.5 | 1.8 | 1.9 | 1.8 |
| O1–H(G119) | 2.1 | 1.8 | 2.0 | 1.9 | 1.9 |
| O1–H(A201) | 2.4 | 2.1 | 1.7 | 2.0 | 1.9 |
4. Conclusions
We applied QM/MM and MD simulations and well-tempered metadynamics to investigate the effect of Li+, Na+, and K+ on the ACh binding affinity and hAChE catalytic activity at different salt concentrations. Large time-scale classical MD simulations were performed to reveal the allosteric modulation by different ion species and the conformational changes in hAChE. QM/MM MD simulations with the umbrella sampling method was used to understand the allosteric effects of alkali metal ions on the catalytic efficiency of hAChE. Our simulations show that the binding affinity and catalytic activity are affected by different ion species through allosteric ion coordination geometries on the hAChE complex and distant electrostatic screening effect. A Li+ cluster consisting of D330, E393, D397 and three Li+ was found conserved in all Li+ systems and could be critical to the enzyme activity. Binding energy calculations indicate that the electrostatic screening from allosterically bound cations affects the key residues at the catalytic site and active-site gorge, including E199. Furthermore, the increase of ion concentration leads to lower reaction activity, especially for Li+ ions, consistent with more identified Li+–hAChE contacts. This work provides a molecular perspective on ion modulation in enzyme catalytic process.Author declarations
Guohui Li proposed the concept; Xia Mu, Chenyi Liao, Dinglin Zhang and Shengwei Yuan performed molecular simulation, data analysis and manuscript writing. Chenyi Liao and Rui Lai edited the manuscript.Data availability
The data that support the findings of this study are available within the article and its ESI.†Conflicts of interest
The authors have no conflicts to disclose.Acknowledgements
This work was supported by the National Key R & D Program of China (2019YFA0709400), the Strategic Priority Research Program of Chinese Academy of Sciences (XDB 37000000), and the National Natural Science Foundation of China (21933010, 22207108, 22303102).References
- T. L. Rosenberry, Adv. Enzymol. Relat. Areas Mol. Biol., 1975, 43, 103–218 CAS.
- D. M. Quinn, Chem. Rev., 1987, 87, 955–979 CrossRef CAS.
- I. Silman and J. L. Sussman, Curr. Opin. Pharmacol., 2005, 5, 293–302 CrossRef CAS PubMed.
- T. Lazarevic-Pasti, A. Leskovac, T. Momic, S. Petrovic and V. Vasic, Curr. Med. Chem., 2017, 24, 3283–3309 CrossRef CAS PubMed.
- M. B. Colovic, D. Z. Krstic, T. D. Lazarevic-Pasti, A. M. Bondzic and V. M. Vasic, Curr. Neuropharmacol., 2013, 11, 315–335 CrossRef CAS PubMed.
- J. Bajgar, in Advances in Clinical Chemistry, ed. G. S. Makowski, 2004, vol. 38, pp. 151–216 Search PubMed.
- L. G. Costa, Clinica Chimica Acta, 2006, 366, 1–13 CrossRef CAS PubMed.
- N. C. Inestrosa, A. Alvarez, C. A. Perez, R. D. Moreno, M. Vicente, C. Linker, O. I. Casanueva, C. Soto and J. Garrido, Neuron, 1996, 16, 881–891 CrossRef CAS PubMed.
- P. Anand, B. Singh and N. Singh, Bioorg. Med. Chem., 2012, 20, 1175–1180 CrossRef CAS PubMed.
- T. H. Ferreira-Vieira, I. M. Guimaraes, F. R. Silva and F. M. Ribeiro, Curr. Neuropharmacol., 2016, 14, 101–115 CrossRef CAS PubMed.
- A. Takeda, E. Loveman, A. Clegg, J. Kirby, J. Picot, E. Payne and C. Green, Int. J. Geriatr Psych., 2006, 21, 17–28 CrossRef CAS PubMed.
- R. A. Hansen, G. Gartlehner, A. P. Webb, L. C. Morgan, C. G. Moore and D. E. Jonas, Clin. Interv. Aging., 2008, 3, 211–225 CAS.
- C. C. Tan, J. T. Yu, H. F. Wang, M. S. Tan, X. F. Meng, C. Wang, T. Jiang, X. C. Zhu and L. Tan, J. Alzheimers Dis., 2014, 41, 615–631 CAS.
- J. Korabecny, K. Spilovska, E. Mezeiova, O. Benek, R. Juza, D. Kaping and O. Soukup, Curr. Med. Chem., 2019, 26, 5625–5648 CrossRef CAS PubMed.
- Y. Bourne, P. Taylor, Z. Radic and P. Marchot, EMBO J., 2003, 22, 1–12 CrossRef CAS PubMed.
- F. Tong, R. M. Islam, P. R. Carlier, M. Ma, F. Ekstrom and J. R. Bloomquist, Pestic. Biochem. Physiol., 2013, 106, 79–84 CrossRef CAS PubMed.
- S. A. Botti, C. E. Felder, S. Lifson, J. L. Sussman and I. Silman, Biophys. J., 1999, 77, 2430–2450 CrossRef CAS PubMed.
- T. P. C. Chierrito, S. Pedersoli-Mantoani, C. Roca, C. Requena, V. Sebastian-Perez, W. O. Castillo, N. C. S. Moreira, C. Perez, E. T. Sakamoto-Hojo, C. S. Takahashi, J. Jimenez-Barbero, F. J. Canada, N. E. Campillo, A. Martinez and I. Carvalho, Eur. J. Med. Chem., 2017, 139, 773–791 CrossRef CAS PubMed.
- A. M. Bondzic, T. D. Lazarevic-Pasti, A. R. Leskovac, S. Z. Petrovic, M. B. Colovic, T. N. Parac-Vogt and G. V. Janjic, Eur. J. Pharm. Sci., 2020, 151, 105376 CrossRef PubMed.
- C. Roca, C. Requena, V. Sebastian-Perez, S. Malhotra, C. Radoux, C. Perez, A. Martinez, J. Antonio Paez, T. L. Blundell and N. E. Campillo, J. Enzym Inhib Med. Ch., 2018, 33, 1034–1047 CrossRef CAS.
- A. J. Franjesevic, S. B. Sillart, J. M. Beck, S. Vyas, C. S. Callam and C. M. Hadad, Chem. – Eur. J., 2019, 25, 5337–5371 CrossRef CAS PubMed.
- G. Petraglio, M. Bartolini, D. Branduardi, V. Andrisano, M. Recanatini, F. L. Gervasio, A. Cavalli and M. Parrinello, Proteins: Struct., Funct., Bioinf., 2008, 70, 779–785 CrossRef CAS PubMed.
- B. Lu and J. A. McCammon, PMC biophysics, 2010, 3, 1 CrossRef PubMed.
- H. Barbosa, A. M. V. M. Soares, E. Pereira and R. Freitas, Sci. Total Environ, 2023, 857, 159374 CrossRef CAS PubMed.
- A. Z. Ronai and S. E. Vizi, Biochem. Pharmacol., 1975, 24, 1819–1820 CrossRef CAS PubMed.
- R. D. Oliveira, K. J. Seibt, E. P. Rico, M. R. Bogo and C. D. Bonan, Neurotoxicol. Teratol., 2011, 33, 651–657 CrossRef CAS PubMed.
- M. H. Hillert, I. Imran, M. Zimmermann, H. Lau, S. Weinfurter and J. Klein, J. Neurochem., 2014, 131, 42–52 CrossRef CAS PubMed.
- B. Rahinizadeh-Robuyi, B. Afsharimani, L. Moezi, T. Ebrahimi, S. E. Mehr, T. Mombeini, M. H. Ghahremani and A. R. Dehpour, J. Cardiovasc. Pharmacol., 2007, 50, 641–646 CrossRef PubMed.
- I. F. Tselnicker, V. Tsemakhovich, I. Rishal, U. Kahanovitch, C. W. Dessauer and N. Dascal, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 5018–5023 CrossRef CAS PubMed.
- E. S. Vizi, A. Ronai, J. Knoll and P. Illes, Neuropharmacology, 1972, 11, 521–530 CrossRef CAS PubMed.
- S. Tsakiris and P. Kouniniotoukrontiri, Biochem. Cell Biol., 1988, 66, 382–388 CrossRef CAS PubMed.
- R. Zanandrea, M. S. Abreu, A. Piato, L. J. G. Barcellos and A. C. V. V. Giacomini, Neurosci. Lett., 2018, 664, 34–37 CrossRef CAS PubMed.
- I. V. Zueva, S. V. Lushchekina and P. Masson, Bioscience Rep., 2018, 38, BSR20180609 CrossRef CAS PubMed.
- T. Viana, N. Ferreira, B. Henriques, C. Leite, L. De Marchi, J. Amaral, R. Freitas and E. Pereira, Environ. Pollut., 2020, 267, 115458 CrossRef CAS PubMed.
- H. Aral and A. Vecchio-Sadus, Ecotoxicol. Environ. Saf., 2008, 70, 349–356 CrossRef CAS PubMed.
- W. Young, Cell Transplant., 2009, 18, 951–975 Search PubMed.
- O. Gerlits, K.-Y. Ho, X. Cheng, D. Blumenthal, P. Taylor, A. Kovalevsky and Z. Radic, Chem.-Biol. Interact., 2019, 309, 108698 CrossRef CAS PubMed.
- Y. K. Zhang, J. Kua and J. A. McCammon, J. Am. Chem. Soc., 2002, 124, 10572–10577 CrossRef CAS PubMed.
- A. V. Nemukhin, S. V. Lushchekina, A. V. Bochenkova, A. A. Golubeva and S. D. Varfolomeev, J. Mol. Model., 2008, 14, 409–416 CrossRef CAS PubMed.
- Y. Z. Zhou, S. L. Wang and Y. K. Zhang, J. Phys. Chem. B, 2010, 114, 8817–8825 CrossRef CAS PubMed.
- S. V. Lushchekina, I. A. Kaliman, B. L. Grigorenko, A. V. Nemukhin and S. D. Varfolomeev, Russ. Chem. Bull., 2011, 60, 2196–2204 CrossRef CAS.
- A. V. Nemukhin, B. L. Grigorenko, D. I. Morozov, M. S. Kochetov, S. V. Lushchekina and S. D. Varfolomeev, Chem.-Biol. Interact., 2013, 203, 51–56 CrossRef CAS PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- G. Makov and M. C. Payne, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 4014–4022 CrossRef CAS PubMed.
- S. Nose, J. Chem. Phys., 1984, 81, 511–519 CrossRef CAS.
- W. G. Hoover, Phys Rev A, 1985, 31, 1695–1697 CrossRef PubMed.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS.
- J. P. Ryckaert, G. Ciccotti and H. J. C. Berendsen, J. Comput. Phys., 1977, 23, 327–341 CrossRef CAS.
- I. Y. B.-S. D. A. Case, S. R. Brozell, D. S. Cerutti, T. E. Cheatham, III, V. W. D. Cruzeiro, T. A. Darden, D. G. R. E. Duke, G. Giambasu, T. Giese, M. K. Gilson, H. Gohlke, A. W. Goetz, D. Greene, R. Harris, Y. H. N. Homeyer, S. Izadi, A. Kovalenko, R. Krasny, T. Kurtzman, T. S. Lee, S. LeGrand, P. Li, C. Lin, T. L. J. Liu, R. Luo, V. Man, D. J. Mermelstein, K. M. Merz, Y. Miao, G. Monard, C. Nguyen, A. O. Nguyen, F. Pan, R. Qi, D. R. Roe, A. Roitberg, C. Sagui, S. Schott-Verdugo, J. Shen, C. L. Simmerling, J. Swails, R. C. Walker, J. Wang, H. Wei, L. Wilson, R. M. Wolf, X. Wu, L. Xiao, D. M. York, P. A. Kollaman and Y. Xiong, AMBER 18 package, 2019 Search PubMed.
- Y. Yang, H. Yu, D. York, Q. Cui and M. Elstner, J. Phys. Chem. A, 2007, 111, 10861–10873 CrossRef CAS PubMed.
- M. Gaus, A. Goez and M. Elstner, J. Chem. Theory Comput., 2013, 9, 338–354 CrossRef CAS PubMed.
- M. Gaus, X. Y. Lu, M. Elstner and Q. Cui, J. Chem. Theory Comput., 2014, 10, 1518–1537 CrossRef CAS PubMed.
- M. Kubillus, T. Kubar, M. Gaus, J. Rezac and M. Elstner, J. Chem. Theory Comput., 2015, 11, 332–342 CrossRef CAS PubMed.
- I. R. Bothwell, D. P. Cogan, T. Kim, C. J. Reinhardt, W. A. van der Donk and S. K. Nair, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 17245–17250 CrossRef CAS PubMed.
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, J. Chem. Theory Comput., 2015, 11, 3696–3713 CrossRef CAS PubMed.
- A. Laio and M. Parrinello, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 12562–12566 CrossRef CAS PubMed.
- P. Raiteri, A. Laio, F. L. Gervasio, C. Micheletti and M. Parrinello, J. Phys. Chem. B, 2006, 110, 3533–3539 CrossRef CAS PubMed.
- M. Bonomi, D. Branduardi, G. Bussi, C. Camilloni, D. Provasi, P. Raiteri, D. Donadio, F. Marinelli, F. Pietrucci, R. A. Broglia and M. Parrinello, Comput. Phys. Commun., 2009, 180, 1961–1972 CrossRef CAS.
- G. A. Tribello, M. Bonomi, D. Branduardi, C. Camilloni and G. Bussi, Comput. Phys. Commun., 2014, 185, 604–613 CrossRef CAS.
- I. Silman, V. L. Shnyrov, Y. Ashani, E. Roth, A. Nicolas, J. L. Sussman and L. Weiner, Protein Sci., 2021, 30, 966–981 CrossRef CAS PubMed.
- M. Harel, D. M. Quinn, H. K. Nair, I. Silman and J. L. Sussman, J. Am. Chem. Soc., 1996, 118, 2340–2346 CrossRef CAS.
- Y. C. Xu, J. P. Colletier, M. Weik, H. L. Jiang, J. Moult, I. Silman and J. L. Sussman, Biophys J., 2008, 95, 2500–2511 CrossRef CAS PubMed.
- B. Sanson, J. P. Colletier, Y. C. Xu, P. T. Lang, H. L. Jiang, I. Silman, J. L. Sussman and M. Weik, Protein Sci., 2011, 20, 1114–1118 CrossRef CAS PubMed.
- S. M. Cheng, W. L. Song, X. J. Yuan and Y. C. Xu, Sci. Rep., 2017, 7, 3219 CrossRef PubMed.
- A. T. Van Wart, J. Durrant, L. Votapka and R. E. Amaro, J. Chem. Theory Comput., 2014, 10, 511–517 CrossRef CAS PubMed.
- A. J. Franjesevic, S. B. Sillart, J. M. Beck, S. Vyas, C. S. Callam and C. M. Hadad, Chemistry, 2019, 25, 5337–5371 CrossRef CAS PubMed.
- S. Genheden and U. Ryde, Expert Opin. Drug Discovery, 2015, 10, 449–461 CrossRef CAS PubMed.
- R. R. Chapleau, C. A. McElroy, C. D. Ruark, E. J. Fleming, A. B. Ghering, J. J. Schlager, L. D. Poeppelman and J. M. Gearhart, J. Biomol. Screen, 2015, 20, 1142–1149 CrossRef CAS PubMed.
- J. P. Colletier, A. Royant, A. Specht, B. Sanson, F. Nachon, P. Masson, G. Zaccai, J. L. Sussman, M. Goeldner, I. Silman, D. Bourgeois and M. Weik, Acta Crystallogr., Sect. D: Struct. Biol., 2007, 63, 1115–1128 CrossRef CAS PubMed.
- Y. C. Xu, J. P. Colletier, M. Weik, G. R. Qin, H. L. Jiang, I. Silman and J. L. Sussman, Biophys J., 2010, 99, 4003–4011 CrossRef CAS PubMed.
- H. Tachikawa, M. Igarashi, J. Nishihira and T. Ishibashi, J. Photochem. Photobiol., B, 2005, 79, 11–23 CrossRef CAS PubMed.
- J. L. Fattebert, E. Y. Lau, B. J. Bennion, P. Huang and F. C. Lightstone, J. Chem. Theory Comput., 2015, 11, 5688–5695 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: More details on RMSDs, structures of ACh hydrolysis, and optimized geometric distances for ACh hydrolysis. See DOI: https://doi.org/10.1039/d3cp02887a |
| ‡ These authors contributed equally to this work. |
| This journal is © the Owner Societies 2023 |