
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Towards a realistic prediction of catalyst durability from liquid half-cell tests†
Timo
Imhof
 *a,
Roberta K. F.
Della Bella
b,
Björn M.
Stühmeier
b,
Hubert A.
Gasteiger
b and
Marc
Ledendecker
*ac
*a,
Roberta K. F.
Della Bella
b,
Björn M.
Stühmeier
b,
Hubert A.
Gasteiger
b and
Marc
Ledendecker
*ac
aTechnical University of Darmstadt, Peter-Grünberg-Strasse 10, 64287 Darmstadt, Germany. E-mail: timo.imhof@tu-darmstadt.de
bTechnical University of Munich, Lichtenbergstrasse 4, 85748 Garching, Germany
cTechnical University of Munich, Schulgasse 22, 94315 Straubing, Germany. E-mail: marc.ledendecker@tum.de
First published on 7th July 2023
Abstract
Liquid half-cell measurements provide a convenient laboratory method for determining relevant parameters of electro-catalysts applied in e.g. polymer electrolyte membrane fuel cells. While these measurements may be effective in certain contexts, their applicability to real-world systems, such as single-cells in a membrane electrode assembly (MEA) configuration, is not always clear. This is particularly true when assessing the stability of these systems through accelerated stress tests (ASTs). Due to different electrode compositions and operating conditions, nanoscale degradation proceeds differently. Nevertheless, given the high demands of MEA measurements in terms of time, testing equipment complexity, and amount of catalyst material, application-relevant predictions of catalyst durability from liquid half-cell tests are highly desirable. This study combines electrochemical and nanoparticle analysis based on transmission electron microscopy to conduct a typical voltage cycling AST for rotating disc electrode (RDE) measurements, showing that the loss of the electrochemically active surface area (ECSA) of the used Pt/Vulcan catalyst is strongly enhanced at 80 °C compared to room temperature, which goes along with increased nanoparticle coarsening. Additionally, a high ionomer/carbon mass ratio (I/C = 0.7) accelerates the ECSA loss, and further investigations of its influence suggest a combination of several factors, including the high local proton concentration and the presence of adsorbing anions. At the same temperature (80 °C) and I/C ratio (0.7), the ECSA loss vs. AST cycle number of the Pt/Vulcan catalyst is essentially identical for a voltage cycling AST conducted in either an RDE half-cell or an MEA configuration, suggesting that liquid electrolyte half-cell based ASTs can provide application-relevant results. Thus, our study points out a way for predicting the stability of electro-catalysts in MEAs based on RDE experiments that require less specialized equipment and only μg-quantities of catalysts.
Introduction
The transition from fossil fuels to renewable energy sources is a necessary step to fight carbon dioxide-related climate change, and it has the potential to mitigate geopolitical risks and conflicts.1,2 The sustainable production of hydrogen, e.g. through water electrolysis, and its use as a substitute in industrial processes and transportation offers a promising route to achieve independence from fossil energy sources.3,4 Polymer electrolyte membrane fuel cells (PEMFCs) fuelled by green hydrogen are considered to play an important role in this transformation to alternative power sources for stationary and automotive applications.5–7 Besides costs, mainly arising from the required precious metal catalysts, and the performance, the long-term stability remains a major subject of optimization on the way to a broad commercialization of PEMFCs.8–14 It strongly depends on the durability of the active catalyst layer, which undergoes significant restructuring and degradation on the nano- and microscale over operation time. During normal operation of a PEMFC stack, transient variations in the load drawn from the stack lead to cathode potential variations between the open circuit voltage (under H2/air ∼ 0.95 V vs. the reversible hydrogen reference electrode scale (VRHE)) and lower potentials of ∼ 0.6 VRHE (at high current densities). This induces losses of the electrochemically active surface area (ECSA) of the carbon-supported platinum (Pt/C) or platinum alloy catalysts at the cathode, which is commonly mimicked by either load cycling (current-controlled, typically done with PEMFC (short-)stacks) or voltage cycling (potential-controlled, typically done with single- or half-cells) accelerated stress tests (ASTs).15 The associated degradation process has been identified as a major contributor to the overall aging-related performance loss of a PEMFC and has not yet been fully overcome.16–21 Although the underlying degradation mechanisms during voltage cycling have been studied extensively over the last decade, there is still an increasing need to fully understand the exact mechanisms and the respective factors that affect the platinum surface area loss under normal operating conditions. Predictions about the durability of a given catalyst material are commonly made on the basis of voltage cycling ASTs, with the loss/retention of the ECSA as the main reference parameter.22–25 Depending on the upper potential limit (UPL), loading conditions targeting metallic nanoparticle degradation only (≤1.0 VRHE) and/or start/stop conditions that additionally include carbon support corrosion (up to 1.6 VRHE) can be simulated. The ECSA loss observed in a fuel cell under operating conditions with potentials below 1.0 VRHE is predominantly ascribed to two mechanisms that occur upon the voltage cycling-induced formation of soluble platinum species such as Pt2+, as illustrated in Fig. 1:26 (1) μm-scale diffusion of the dissolved Pt species across the electrode and either formation of electrically isolated Pt particles in the ionomer phase near/in the membrane by cross-over hydrogen in MEA-based ASTs or loss of Pt species into the bulk of the electrolyte phase in liquid electrolyte half-cell rotating disc electrode (RDE) based ASTs (platinum loss); (2) nm-scale diffusion of dissolved Pt species and their re-deposition during periods of low potential (i.e. in a load cycling AST during high-load phases) onto Pt particles on the carbon support, leading to growth of the active Pt nanoparticles by Ostwald ripening (particle growth).17,27 In the liquid half-cell configuration, dissolved Pt species may accumulate in the electrolyte, deposit on the walls of the cell, or be reduced and deposited on the counter electrode. It was also found that especially Pt/C catalysts with a high amount of small, ∼2 nm diameter, Pt-nanoparticles suffer from Pt dissolution into the electrolyte phase already at potentials above 0.65 VRHE.28 Previous studies by Ferreira et al. estimated that both processes contribute roughly equally to the overall ECSA loss of a typical Pt/Vulcan catalyst in the cathode catalyst layer of an MEA when degraded via a voltage cycling AST under H2/N2 (anode/cathode).17 In the same study, a dependency of the degradation mechanisms on the location within the catalyst layer of the MEA was found by performing a bright-field TEM analysis on cross-sections of 100 nm thick slices of degraded MEA samples, cut along different distances from the electrode/membrane interface. Pt loss into the ionomer phase was more pronounced for catalyst particles located closer to the membrane interface and Ostwald ripening was prevalent for catalyst particles located closer to the gas diffusion layer (GDL). So far, such a variation of degradation mechanisms across the thickness of the catalyst film has not been studied for the much thinner RDE catalyst films, but it is reasonable to assume that such a variation also occurs, depending on whether the catalyst particles are located near the interface between the catalyst layer and the RDE disk substrate or near the interface between the catalyst layer and the liquid electrolyte. | ||
| Fig. 1 Scheme of the two dominating degradation mechanisms of carbon-supported Pt catalysts during voltage cycling ASTs with an upper potential limit up to 1.0 VRHE, namely Pt particle growth via Ostwald Ripening and Pt loss into the electrolyte phase. In the MEA configuration, platinum ions such as Pt2+ that have diffused away from the catalyst layer are typically reduced by crossover hydrogen coming from the anode side to form isolated Pt deposits in the ionomer phase, whereas in RDE configuration, Pt ions dissolve into the liquid electrolyte. | ||
Single-cell measurements with MEAs are typically conducted for PEMFC performance and stability studies but require large quantities of catalyst to prepare MEAs (gram scale), many hours of in-cell MEA conditioning, and complex testing equipment. Hence, simplified half-cell configurations are more convenient, especially in the early stages of research, e.g., for the characterization of new catalysts. Due to their simplicity and low demand of catalyst material on the milligram scale, classical hydrodynamic electroanalytical techniques such as RDE measurements in a half-cell configuration with a liquid electrolyte are popular tools to determine relevant parameters of electro-catalysts such as the ECSA and kinetic and mass-specific activities.29,30 The control over the hydrodynamic boundary layer via the rotation speed facilitates the determination of kinetic rate constants using the RDE setup in comparison to the MEA.30,31 On the other hand, mass transport and the interplay of gaseous, liquid, and solid phases at the triple phase boundary which cannot be simulated in the RDE configuration make the interpolation from RDE to MEA results challenging. Recent reviews by Ehelebe et al. and Larazidis et al. particularly emphasize the challenges associated with the interpretation of catalyst stability data derived from degradation studies in liquid electrolyte half-cell configurations.32,33
The differences in catalyst degradation between liquid and solid electrolytes have been discussed in several studies. For instance, Sharma et al. conducted ASTs in a liquid electrolyte (1 M H2SO4) and half-MEA setups and observed more severe ECSA degradation in the liquid electrolyte for a voltage cycling AST conducted at room temperature.34 Ahluwalia et al. and Martens et al., on the other hand, observed more severe degradation (ECSA loss and particle growth) in the MEA at 60 °C and 80 °C compared to aqueous half-cell tests conducted at room temperature.26,35 Gilbert et al. and Kim et al. suggested using a flowing or frequently exchanged electrolyte instead of a stagnant electrolyte at room temperature to achieve application-relevant catalyst degradation, whereas Yu et al. claimed to have better recreated MEA degradation by increasing the Pt-ion concentration in the electrolyte while conducting the experiments at 80 °C.36–38 Riese et al. provided a qualitative comparison of ASTs conducted in an RDE setup with results obtained from MEA tests and suggested the application of elevated temperatures in the RDE for better comparability of ECSA degradation in the RDE with that in an MEA.39 The importance of temperature has also been considered in studies by Polymeros et al. and Urchaga et al., who measured the stability of Pt/C catalysts in an RDE setup at 60 °C or 70 °C.40,41 A drawback of the existing literature on PEMFC catalyst degradation derived from liquid electrolyte half-cell measurements is a general lack of comparability due to varying parameters such as electrolyte, temperature, pH, and degradation protocols. ASTs traditionally used by the US Department of Energy (DOE) or the Fuel Cell Commercialization Conference of Japan (FCCJ) in the RDE configuration are conducted at room temperature and the voltage cycling profiles differ from those applied in the MEA configuration.42–44 Since different catalyst materials may respond differently to different potential limits, scanning profiles, and treatment time, a variation of the degradation protocol does not appear productive in the search for a generalized approach. To our knowledge, systematic work providing parameters that can be applied in e.g. an RDE experiment to achieve an application-relevant prediction of catalyst durability in an MEA is still missing. Although various attempts have been made to achieve similar ECSA degradation in an RDE setup compared to an MEA setup, there is still a lack of understanding of which experimental conditions are critical to overcome the poor comparability of ECSA degradation observed in RDE and MEA setups.
Therefore, the objective of this work is to achieve an application-relevant liquid electrolyte half-cell-based AST to predict the voltage cycling durability of Pt/C catalysts and to elaborate on the parameters that most strongly affect the extent of catalyst degradation and its mechanism as well as to discuss the associated challenges. To this end, we conducted a detailed comparative examination of the same Pt/C catalyst and the same degradation protocol in MEA and RDE configurations. For liquid electrolyte half-cell measurements, degradation was systematically investigated as a function of temperature, ionomer content of the electrode coating on the RDE substrate, pH, and type of electrolyte. The applied aging protocol is based on a slight modification of the specifications of the US Department of Energy that is established as part of standard routine MEA testing.44 ECSA determination by CO stripping and TEM analysis of degraded catalysts with determination and analysis of Pt particle size distributions were performed to follow the course of aging, to correlate changes in specific Pt surface area with the structure of individual particles, and to correlate MEA and RDE half-cell measurements. In addition to a comprehensive overview of the examined testing conditions, the developed RDE-based AST protocol yields comparable ECSA loss rate profiles for an MEA- and RDE-based voltage cycling AST.
Results and discussion
To obtain a detailed picture of the degradation mechanisms and the effects of temperature, ionomer content, forced convection, and electrolyte, the catalyst stability was probed by an AST with a square-wave potential profile with a lower potential limit of 0.6 VRHE (LPL) and an upper potential limit of 1.0 VRHE (UPL), with a hold time of 1 second at each potential. The tests were performed up to 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles using a 20 wt% Pt/Vulcan catalyst with a particle size of 2.7 ± 0.8 nm (TEM). The progression in the ECSA loss was determined by CO-stripping experiments which were conducted after various aging cycles within the accelerated stress tests. Background-corrected CO stripping peaks and the corresponding cyclic voltammograms (CV) recorded at 80 °C in N2-saturated 0.1 M HClO4 in an RDE configuration are exemplarily shown in Fig. 2(a) and (b).
000 cycles using a 20 wt% Pt/Vulcan catalyst with a particle size of 2.7 ± 0.8 nm (TEM). The progression in the ECSA loss was determined by CO-stripping experiments which were conducted after various aging cycles within the accelerated stress tests. Background-corrected CO stripping peaks and the corresponding cyclic voltammograms (CV) recorded at 80 °C in N2-saturated 0.1 M HClO4 in an RDE configuration are exemplarily shown in Fig. 2(a) and (b).
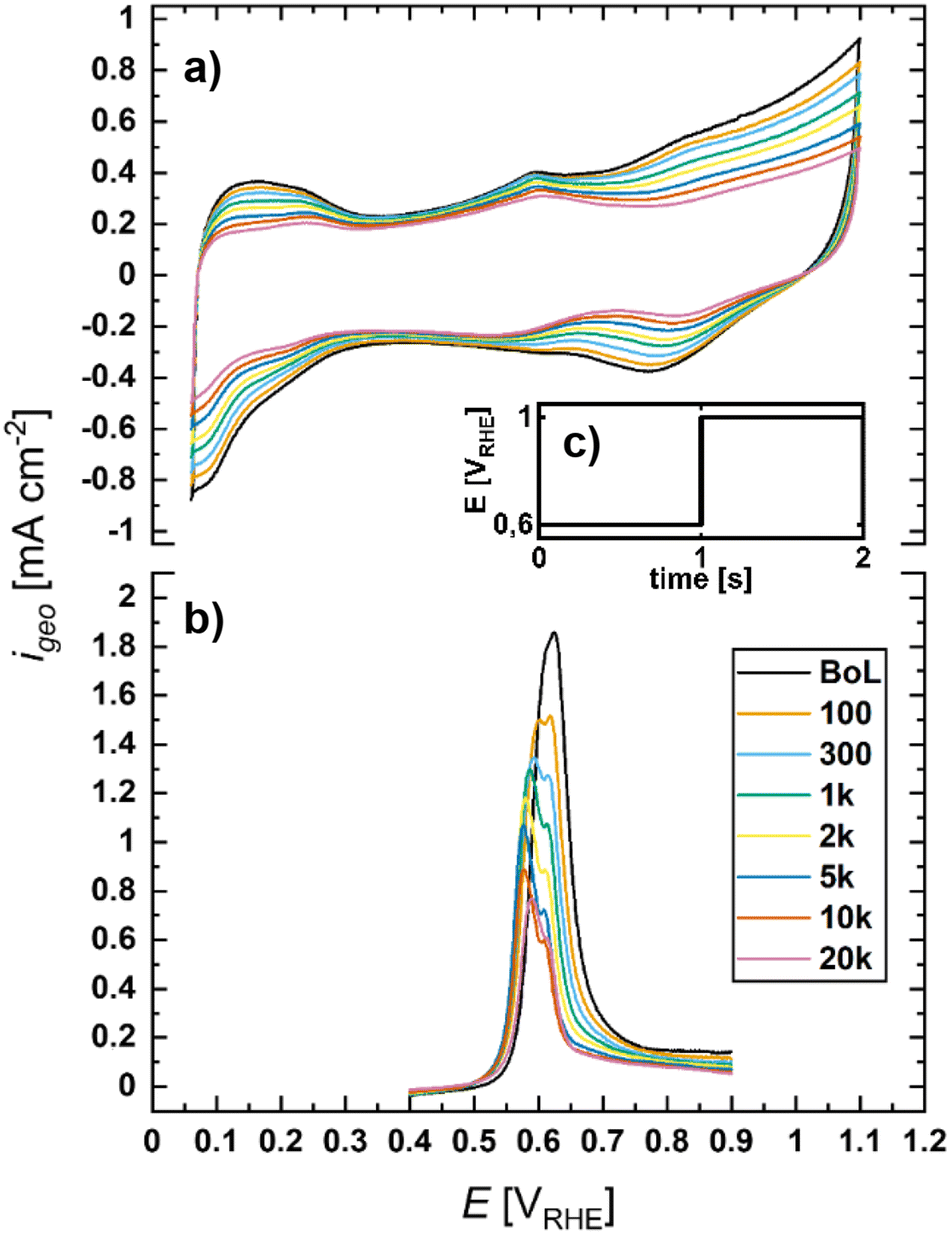 | ||
| Fig. 2 RDE-based voltage cycling AST (square-wave profiles between 0.6–1.0 VRHE with 1 s holds at LPL and UPL at 0 rpm) of a Pt/Vu catalyst (coated without ionomer) in 0.1 M HClO4 conducted at 80 °C: (a) Cyclic voltammograms (0.05 V s−1) in the N2-saturated electrolyte recorded at 80 °C at different points during the AST conducted at the same temperature; (b) corresponding background-corrected CO-stripping voltammograms (0.05 V s−1) recorded at 80 °C; (c) voltage profile of one AST cycle. | ||
Influence of temperature
To determine the effect of temperature on electrochemical stability, RDE measurements were conducted in a jacketed glass cell coupled to a thermostat at 80 °C; for measurements at 22 °C, a Teflon cell was used. Non-adsorbing 0.1 M HClO4 was used as the electrolyte and the catalyst films were prepared without ionomers. The development of the ECSA normalized to the beginning-of-life (BoL) ECSA (determined from CO-stripping voltammetry at 22 °C) over the course of aging is plotted against the number of aging cycles (CN) in a semi-logarithmic plot as shown in Fig. 3. The initial active surface area of the catalyst was 83 m2 g−1 and 68 m2 g−1 when measured at room temperature and 80 °C, respectively. The discrepancy between these values is most likely caused by a change in the equilibrium of CO adsorption and desorption reactions as described by Garrick et al., who observed a 20% decrease in CO stripping-based ECSA values when going from 30 °C to 90 °C.45 For this reason, only relative values normalized to the BoL ECSA determined at the same temperature as the respective AST are given in this work. At room temperature, ca. 67% of the initial ECSA was retained after 20k cycles whereas increasing the temperature to 80 °C decreases the ECSA to roughly 35% of the initially measured value. This is in accordance with the results of Urchaga et al., who observed increased catalyst degradation at elevated temperatures in RDE measurements.41 It is furthermore plausible, as the initial step of the dominating degradation mechanisms, i.e. the formation of soluble platinum species, is expected to be a temperature-activated process.26 Interestingly, at 80 °C the ECSA loss follows a linear trend in the semi-logarithmic plot, which is similar to what has been observed in several studies, in which voltage cycling ASTs were conducted with MEAs.21,24,46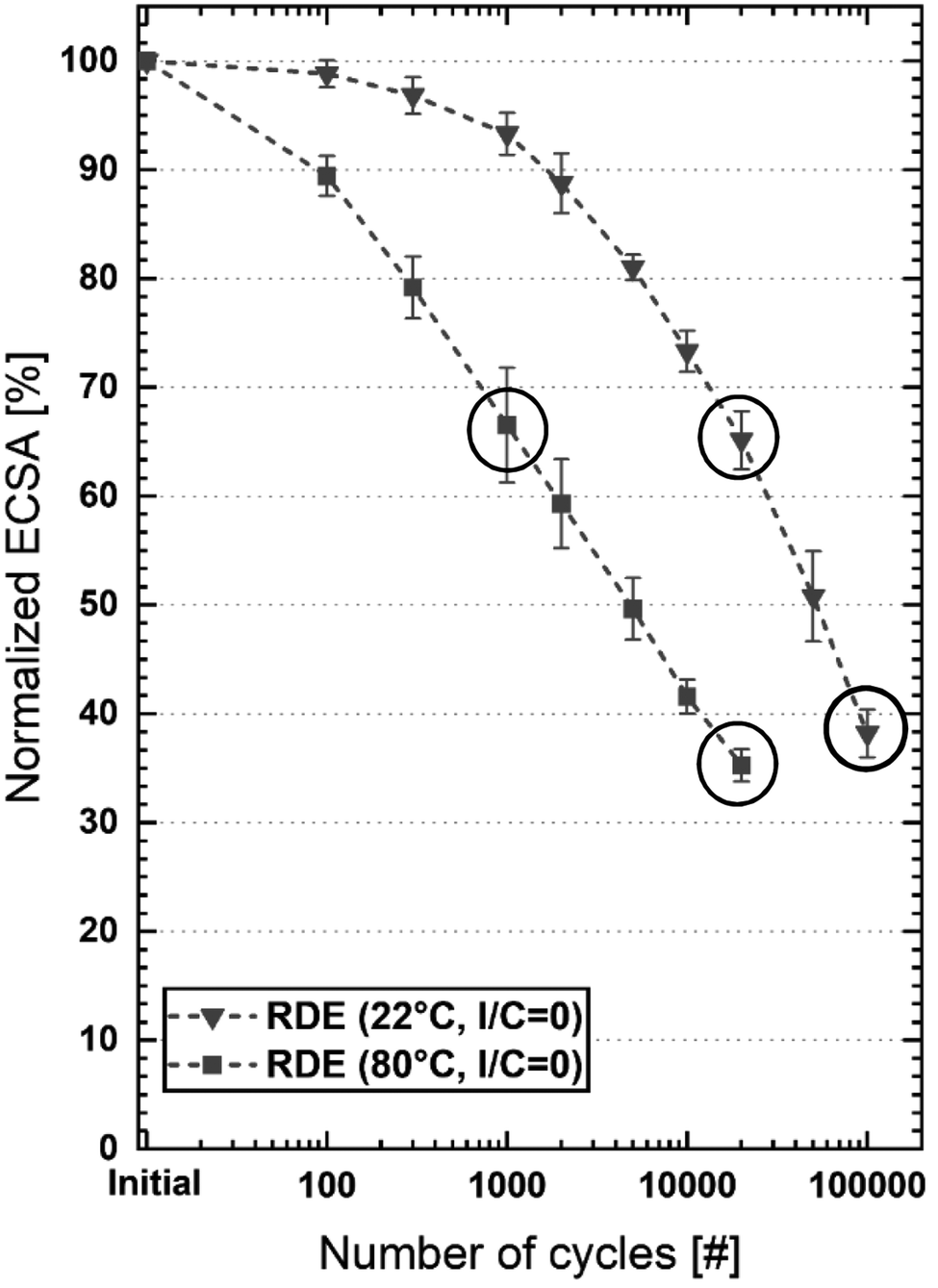 | ||
| Fig. 3 ECSA normalized to the BoL ECSA measured before the AST (initial) at the same temperature as the respective AST, plotted against the number of aging cycles. ECSAs were calculated from the individual CO oxidation charges recorded at the respective AST temperature (the error bars depict the standard deviation of 3 repeat experiments). | ||
In order to compare the changes in catalyst morphology after a similar extent of ECSA degradation for the voltage cycling AST conducted at 22 and 80 °C (see Fig. 2(d)), TEM images were recorded. For this purpose, samples were chosen at different degradation states, i.e. at the beginning-of-life, at ∼65% ECSA retention (marked by the uppermost circles in Fig. 2(d)), and at ∼35% ECSA retention (lowermost circles in Fig. 2(d)). The resulting TEM images for each case are shown in Fig. 4(a), (b), (d), and (e), and the results of the particle size distribution analysis for each case are depicted in Fig. 4(c) and (e). In order to provide statistical relevance for the TEM analysis of each sample, multiple single TEM images with a sampling size between 100 and 200 particles were analysed, which resulted in a minimum count of ca. 1000 particles on a minimum of 6 individual Vulcan carbon support particle aggregates. Additionally, we determined and correlated individual particle size distributions (PSDs) of 34 separate Vulcan particle aggregates from two different samples degraded over 100k AST cycles (0.6/1.0 VRHE square waves with 1 s holds) at room temperature. The results are given in Fig. S5 (ESI†). The mean Pt particle diameters obtained from these individual PSDs lie between 3.5 nm and 5.5 nm and we assume that this variation may have resulted from a variation of degradation mechanisms across the catalyst film thickness (like typically observed for degraded catalyst layers from MEAs, but less pronounced due to the much thinner catalyst layer used for the RDE experiment).17 Individual mean and surface area normalized (Sauter) diameters plotted in Fig. S6 (ESI†) do not show outliers, which indicates that our TEM sampling can be considered representative for the aging of the catalyst film.
 | ||
| Fig. 4 TEM images of Pt/Vu catalyst samples after 20k (22 °C, a) and 1k (80 °C, b) degradation cycles of the voltage cycling test shown in Fig. 3 (marked by the uppermost circles), with approximately 65% retained ECSA;. (c) Pt particle size distributions of these two degraded catalysts in comparison to the pristine catalyst. TEM images of catalyst samples after 100k (22 °C, d) and 20k (80 °C, e) degradation cycles of the same AST with roughly 35% retained ECSA each (marked by the lowermost circles in Fig. 3), (f) Pt particle size distributions of these two samples in comparison to the pristine catalyst. | ||
When the ECSA retention is 65%, the particles age similarly at 80 °C and room temperature (Fig. 4(a) and (b)), whereas in the case of 35%, marked differences exist between both temperatures (Fig. 4(d) and (e)). This indicates that the degradation leading to the first ∼35% of the ECSA loss is similar at both temperatures (Fig. 4(c)), even though it requires more cycles at room temperature to lead to the same ECSA loss. This might for instance be due to the complete dissolution of the nanoparticles with sizes <2 nm, which are supposed to be the least stable and almost disappeared after cycling at both temperatures (see Fig. 4(c)). With a ∼65% ECSA loss obtained after more extended cycling (Fig. 4(d) and (e)), a comparison of the particle size distribution (PSD) in Fig. 4(f) shows that the particles grew less at 22 °C as indicated by the mean Pt diameter (d = 4.3 ± 1.4 nm), whereas at 80 °C, a higher frequency of significantly grown nanoparticles results in a broader distribution and a larger mean particle diameter (d = 5.4 ± 2.4 nm). The PSD of the pristine catalyst (dark grey bars in Fig. 4(c)) can be fitted very accurately with a Gaussian function (R = 0.998, see Fig. S7, ESI†). In the following, we used the R values of the Gaussian fits to gauge the skewness of the PSDs of the degraded catalysts and to draw conclusions regarding the underlying degradation mechanisms. The PSDs of the samples degraded at 22 °C (red bars in Fig. 4(c) and (f)) are highly symmetrical and can be fitted with R2 values of ≥ 0.99 (see also Fig. S8 and S9, ESI†). This result is an indication that the particle growth is homogeneous across the catalyst layer at room temperature due to Ostwald ripening.47 On the other hand, the PSD after 20k cycles at 80 °C is more skewed (R2 = 0.976; see Fig. S11, ESI†), which points towards a wider distribution of particle sizes across the catalyst layer thickness. This may result for instance from variations in the ripening mechanism (e.g. due to different diffusion rates of Pt2+ ions into the electrolyte and re-deposition) and/or additional contributions from other aging mechanisms such as coalescence.
Although carbon corrosion, which enables particle migration and consequent particle agglomeration, is kinetically hindered and does not contribute significantly to platinum ECSA loss, e.g. through particle detachment at potentials lower than 1.0 VRHE,48 it cannot be excluded completely in ASTs with UPLs ≤ 1.0 V, as demonstrated by Pizzutilo et al.49 Catalysed especially by non-oxidized platinum nanoparticles, it is measurable at potentials as low as 0.6 VRHE and may lead to particle aggregation/agglomeration to some extent.50,51 Cremers and co-workers found that the carbon corrosion rate strongly increases with temperature, which gives a possible explanation for the herein observed differences in particle growth between 80 °C and 22 °C.52 The different mechanisms of agglomeration of Pt nanoparticles in MEAs during the AST have been discussed thoroughly by Ferreira and co-workers, who also observed a significant fraction of coalesced platinum nanoparticles after conducting ASTs with an UPL of 1.0 VRHE.28 The authors state that particles located in close proximity to each other do not necessarily have to possess significant mobility on the carbon support to form agglomerates. In this case, particle coalescence occurs via Ostwald ripening: either by diffusion of Pt atoms on the carbon support (2D) or as ions through the electrolyte phase (3D). Either way, after the RDE-based AST at 80 °C, a certain fraction of the particles with non-spherical shapes in the TEM images appear to have originated from a coalescence of previously closely neighbouring particles (see also Fig. S17, ESI†). Given these results and the much longer treatment time required for the measurements at room temperature (60 h vs. 12 h), we assume that the contribution of the different degradation mechanisms to the overall ECSA loss depends on the temperature, especially in the regime of ≤70% ECSA retention. Hence, ASTs conducted in liquid half-cell configurations at room temperature are not suitable to deliver application-relevant results.
Lastly, we correlated the electrochemically quantified retained ECSAs with the observed particle growth from the TEM via the surface area normalized (Sauter) diameter, calculated viaeqn (1), from the individual particles counted for each sample. Assuming a spherical shape of the particles, the ECSA values can be calculated from the TEM-based Sauter diameters (further on referred to as ECSASauter) by eqn (2).
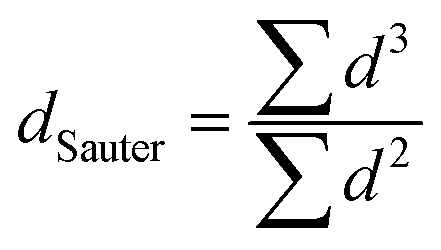 | (1) |
 | (2) |
The Sauter diameter calculated from the TEM-derived particle diameters of the pristine catalyst is 3.2 nm (see Fig. S4, ESI†). According to eqn (2), this gives an initial ECSASauter of 87 m2 g−1, which is in good agreement with the CO-stripping-based value of 83 m2 g−1 measured at 22 °C. For the comparison with the measured (relative) retained ECSA values (further referred to as ECSAmeasured), all ECSASauter values given in this paper are normalized to the ECSASauter of the pristine catalyst. The normalized ECSASauter values for the samples degraded at 22 °C/100k cycles and at 80 °C/20k cycles, using the Sauter diameters listed in Fig. 4(f), are 60% and 39% respectively. The ECSAmeasured values are lower at 39% and 35% (marked by the lowermost circles in Fig. 3). This is plausible, since the TEM-based calculation can only capture Ostwald ripening (producing spherical particles) and excludes the other potentially occurring degradation mechanisms such as detachment and coalescence of Pt particles or loss of platinum ions in the electrolytes. Assuming that ECSASauter accurately covers the ECSA loss caused by this mechanism, the difference between ECSASauter and ECSAmeasured must be attributed to other degradation mechanisms. Given the absence of potentials above 1.0 VRHE that would lead to significant carbon corrosion and of other factors such as bubbles formation and forced convection, we exclude detachment of Pt particles from the carbon support and catalyst detachment from the electrode as possible degradation mechanisms.41 Hence, we consider Pt (-ion) loss into the electrolyte phase as well as the formation of Pt aggregates and non-spherical particles on the carbon support, which are not covered by the TEM particle diameter analysis, as the other significant degradation mechanisms in the potential range of the applied AST.17
At room temperature, a larger gap between ECSASauter and ECSAmeasured is observed, which means that a higher fraction of the total ECSA loss cannot be explained by Ostwald ripening only compared to 80 °C. Studies on the electrochemical platinum dissolution by Urchaga et al., Xing et al., and Cherevko et al. reported similar Pt-ion concentrations in the electrolyte after identical voltage cycling treatments at room temperature in comparison to elevated temperatures.41,53,54 This suggests that the mechanism of platinum loss into the electrolyte phase does not seem to be increased significantly at higher temperatures. Given the longer treatment time in the room temperature AST (60 h vs. 12 h), we assume that a higher amount of platinum was lost in the electrolyte phase, thereby explaining the larger difference between ECSAmeasured and ECSASauter.
In addition to temperature, we investigated the effect of RDE rotation rate during the AST. It was shown by Arenz et al. and by Nagai et al. that forced convection induced by rotation increases the ECSA loss upon voltage cycling.55,56 At 1600 rpm, the ECSA retention was slightly lower compared to 0 rpm at both 22 °C and 80 °C (Fig. S1, ESI†). The effect was not as pronounced as reported in the literature, which might be due to the higher upper potential limits applied in the cited studies that additionally lead to carbon support corrosion and particle detachment.
Influence of the ionomer content
As another parameter, the ionomer content in the catalyst ink was altered. The characterization of newly developed catalyst materials by RDE commonly includes activity measurement protocols that are run before a voltage cycling AST. To access specific activities without possible poisoning by e.g. sulfonic acid groups, the use of ionomers is usually kept to a minimum.57 In cases where it is used, e.g. when it is necessary to prepare stable and reproducible catalyst films, the amount is typically below what is applied in an MEA (i.e. I/C ratios near 0.1 rather than ∼0.7–1 in MEAs). Since the main goal in this study was to achieve a better comparability of the ECSA degradation observed in the RDE configuration with the results from single-cell MEA measurements, we aimed for a similar ionomer content as conventionally used for MEA measurements and prepared catalyst films with an I/C ratio of 0.7 using Nafion®. Voltage cycling ASTs with the higher ionomer content were performed at both 22 °C and 80 °C. The catalyst ink recipe used here with a high water content (15%vol isopropanol) is expected to lead to significant ionomer–Pt interactions based on the report by van Cleve et al.58 The ECSA loss curves shown in Fig. 5 demonstrate that the severity of degradation increases when high ionomer content is incorporated into the catalyst film: after 20k cycles at 80 °C less than 20% of the ECSA remains at an I/C ratio of 0.7 compared to 35% for the film without Nafion®. A similar trend applies to the data obtained at 22 °C. These findings are in agreement with what has been mentioned but not further specified by Shinozaki et al. in their work on RDE activity measurements with I/C ratios ranging between 0 and 1.35.57 | ||
| Fig. 5 Normalized ECSA loss over the course of voltage cycling ASTs (0.6/1.0 VRHE square-waves with 1 s holds in the RDE configuration with 0.1 M HClO4 at 0 rpm) at 22 and 80 °C for Pt/Vu catalyst films with and without ionomers (the error bars depict the standard deviation of 3 repeat experiments). | ||
In order to obtain a detailed picture of the morphology and structure of the catalyst after aging, particle size distributions were determined from TEM images for the samples with and without ionomers degraded at 80 °C (Fig. 6). In the case of an ionomer-free electrode, the PSD is narrower but has a similar mean particle diameter of d = 5.4 ± 2.4 nm in comparison to 5.7 ± 3.2 nm for the case with ionomers. Gaussian fits shown in Fig. S11 and S12 (ESI†) reveal that the PSD of the degraded catalyst film with ionomers is less accurately describable by a Gaussian fit than for the catalyst film without ionomers (R2 = 0.940 vs. R2 = 0.976). This indicates that the variation in degradation mechanisms across the catalyst film thickness is increased through the incorporation of ionomers. Different rates of Pt ion loss in the electrolyte and re-deposition, for instance depending on the position of the catalyst particle or its exposure to the ionomer may result in the observed variations in particle growth. The difference between ECSASauter and ECSAmeasured values (Table 1) is larger in the case of the catalyst layer with a high I/C ratio, suggesting that the mechanisms of coalescence and/or overall Pt loss in the electrolytes are enhanced too.
 | ||
| Fig. 6 Representative TEM images of degraded catalyst samples without Nafion® (a) and with Nafion® (b) after 20k degradation cycles (marked by circles in Fig. 5) and Pt particle size distribution of the respective samples (c). | ||
The variation in the degradation mechanisms imposed by the ionomers might be caused by altered conditions in the catalyst layer compared to those present in the perchloric acid electrolyte, such as pH, which may ultimately result in different dissolution rates of platinum. The high density of negatively charged sulfonate groups in the ionomer requires a high concentration of protons as counter ions to guarantee charge neutrality. In combination with being a solid super-acid with a pKa of −6, the pH value is thus expected to be lower at the liquid/solid interface compared to that of 0.1 M HClO4.59,60 A concentration of approximately 1 M is expected to be present in wet Nafion® according to the calculation found in Table S2 (ESI†). Studies by Furuya et al. and Topalov and co-workers indeed demonstrate that lowering the pH of the electrolyte increases the dissolution of platinum.61,62 At the same time, it was shown that the presence of adsorbing ions in the form of sulphate/bisulphate in the sulphuric acid electrolyte does not significantly influence the platinum dissolution rate.61 Nevertheless, nanoparticle degradation may proceed differently in ClO4− and –SO3− containing electrolytes due to different adsorption energies, Pt solubilities, and ionic strengths. Other adverse effects could arise from degradation of the ionomer. Studies by Helmly et al. found that degradation of ionomers releases fluoride which could accumulate over time and affect the degradation rate.63 HF is known to dissolve and remove oxide layers from platinum surfaces and was already identified by Guilminot et al. as a possible factor for accelerated Pt nanoparticle degradation alongside sulfonate groups or sulfates.64 Martens et al. observed thin films of damaged ionomers close to the Pt surface upon electrochemical oxidation driven by formed hydrogen peroxide.65 Although they applied potentials above 1.0 VRHE, ionomer degradation may also have taken place to a minor extent in our experiments. Yano et al. observed increased H2O2 formation on Pt/C catalysts after ASTs which could eventually cause ionomer degradation towards the end of ASTs.66
Influence of the electrolyte
In order to identify the cause of the increased degradation when using an ionomer-containing catalyst film, ASTs were conducted on the RDE setup varying the electrolyte type to mimic the properties of the ionomer. To study the effect of lower pH (higher acidity) on the Pt surface area loss, 1.0 M HClO4 was used. The characteristic of adsorbing anions in the ionomer is simulated by using a 0.05 M H2SO4 electrolyte.30 The catalyst films did not contain ionomers in order to solely study the effect of the liquid electrolyte properties.The course of the ECSA loss at 80 °C is given in Fig. 7a in comparison to the previous data obtained in 0.1 M HClO4 with and without Nafion®. The ECSA loss without ionomers is accelerated by both H2SO4 (green) and low pH (1 M HClO4, red) compared to 0.1 M perchloric acid electrolyte (black). The values are between those of the catalyst films with and without ionomers cycled in 0.1 M HClO4. The same trends were observed for measurements at room temperature (see also Fig. S2, ESI†). TEM images and particle size distributions of the degraded catalysts in 1 M HClO4vs. 0.05 M H2SO4, both having a similar relative ECSA of ca. 26% after the voltage cycling AST, are shown in Fig. 7(b)–(d). In the case of the 0.05 M H2SO4 electrolyte, most of the smaller particles with the original particle size of 2–3 nm have disappeared. This results in larger mean and Sauter diameters compared to 1 M perchloric acid. As exemplarily highlighted by red and green circles in Fig. 6(b) and (c), we observed more coalescence of neighbouring particles (via 2D/3D Ostwald ripening) to form larger, non-spherical particles and aggregates in the 1 M HClO4 sample (Fig. 7(b)). In contrast, in the case of 0.05 M H2SO4, TEM analysis revealed that most of the particles retained their spherical shape and just increased in size (cf.Fig. 7(c)). Gaussian fits of the PSDs (Fig. S13 and S14, ESI†) show similar R2 values of 0.961 (1 M HClO4) and 0.959 (0.05 M H2SO4). The mean particle diameters and EoL ECSA values of all measurements shown in Fig. 7(a) are summarized in Table 1.
 | ||
| Fig. 7 (a) Normalized ECSA loss over the course of voltage cycling ASTs at 80 °C (0.6/1.0 VRHE square-waves with 1 s holds) in the RDE configuration (at 0 rpm) with 0.05 M sulphuric acid and 1.0 M perchloric acid in comparison to 0.1 M HClO4 with and without Nafion® for Pt/Vu catalyst films (the error bars depict the standard deviation of 3 repeat experiments). Representative TEM images of degraded catalyst samples after 20k cycles in 1 M HClO4(b) and in 0.05 M H2SO4(c) and Pt particle size distribution of the respective samples (d). | ||
The difference between ECSASauter and ECSAmeasured is similar when comparing 0.1 M HClO4 with 0.05 M H2SO4 (ca. 10–15%), but is larger when compared with 1 M HClO4 (ca. 25%) and largest for catalyst films with an I/C ratio of 0.7 (ca. 40%, measured in 0.1 M HClO4). We assume that Pt loss as dissolved ions in the electrolyte depends on the acid strength, thus is more severe in the ionomeric electrolyte and in 1 M HClO4, which is consistent with the literature reporting higher platinum dissolution at lower pH.61,62 The same literature reports no significant influence of the type of ion (perchlorate vs. sulfate electrolyte) which goes along with our observations. The measured pH values of the electrolytes used are given in Table S1 (ESI†).
RDE vs. MEA comparison
Finally, the presented results were compared to MEA measurements with the same catalyst and under the same AST conditions. As shown in Fig. 8, the ECSA loss curve of the RDE measurement with a catalyst film with an I/C ratio of 0.7 at 80 °C (blue squares) shows an almost identical course as the one obtained in the MEA (black squares) at the same temperature and for a catalyst layer with a quasi-identical I/C ratio (0.65 vs. 0.7). A major part of the remaining discrepancy arises during the first 100 cycles, which could be explained by the different catalyst activation protocols ran before the measurements. Either incomplete activation or the onset of the degradation already during activation in the RDE protocol is a plausible explanation of a lower ECSA loss in the first step of the RDE AST. Nevertheless, the absolute ECSA losses as well as the profiles of the ECSA loss versus cycle number are strikingly similar. On the other hand, the ECSA losses under conventionally used RDE conditions, i.e., with ionomer-free catalyst films conducted in 0.1 M HClO4 at room temperature (blue triangles) are vastly different, which can be ascribed primarily to the difference in temperature and, to a lesser extent, the absence/presence of ionomers in the catalyst layer. | ||
| Fig. 8 Comparison of normalized ECSA losses of Pt/Vu catalyst layers over the course of voltage cycling ASTs (0.6/1.0 VRHE square-waves with 1 s holds): (i) in the RDE configuration with 0.1 M perchloric acid either without ionomers in the catalyst film acquired at 22 °C or with an I/C ratio of 0.7 acquired at 80 °C (at Pt loadings of 10 μg cm−2); (ii) in an MEA operated at 80 °C using a catalyst layer with an I/C ratio of 0.65 (at a Pt loading of ca. 100 μg cm−2). The error bars depict the standard deviation of 3 repeat experiments in the case of the RDE measurements and 2 repeat measurements in the case of the MEA tests. | ||
In a recent publication, Della Bella et al. found that by using different square-wave (SW) based voltage cycling ASTs with upper potentials between 0.85–1.0 V, a linear trend of the ECSA loss (normalized to the initial ECSA value) vs. the logarithm of the number of cycles for ECSA losses between ∼20–80% was obtained for different MEAs subjected to the various ASTs.46 It has been explained that this linear correlation is caused by having the same underlying ECSA loss mechanisms independent of the UPL and the hold time, namely by the voltage cycling induced platinum dissolution followed by both (i) the diffusion of platinum ions into the ionomer phase where metallic Pt would precipitate due to the reaction with crossover hydrogen and (ii) the re-deposition of platinum ions onto the catalyst leading to Ostwald ripening.17 The analogous mechanisms prevail during aging tests in liquid half-cells34,41,67 and, apparently, lead to a similar linear trend of the ECSA loss versus the logarithm of the number of cycles for the RDE measurements conducted at 80 °C (see Fig. 8). Mechanistically, the loss of platinum into the electrolyte phase is different in the RDE configuration, where it remains as a dissolved platinum species in very low concentrations due to the extremely high electrolyte volume to catalyst mass ratio (if 100% of the platinum attached to the disc electrode were to be dissolved, the resulting platinum concentration would be only ∼0.1 μM). Since these concentrations are too low to allow for a significant back-transport of platinum dissolved in the electrolyte to the catalyst film on the disc, the loss of platinum into the electrolyte phase can also be considered as quasi-permanent, as in the case of platinum particles precipitated in the ionomer phase of the MEA.
A linear regression line fit of the semi-logarithmic plots of normalized ECSA between ∼20–80% ECSA loss versus the logarithm of cycle number was performed for voltage cycling ASTs conducted at 80 °C for the RDE data acquired shown in Fig. 7(a) as well as for the MEA data shown in Fig. 8.
The slopes obtained from these fits are listed in Table 2, showing that the value for the RDE measurements is closest to the MEA measurement when a similar I/C ratio is used for both the catalyst film on the RDE (I/C = 0.7) and for the MEA (I/C = 0.65), yielding 29.9 ± 0.8%/dec (RDE; experiment Nr. 2 in Table 2) vs. 32.4 ± 1.2%/dec (MEA; experiment Nr. 5). The values of the slopes of the RDE catalyst films without ionomers were smaller, indicating slower degradation of the catalyst. It is also important to note that the starting point of the linear regression, i.e. the first ∼20% of the ECSA loss at a cycle number of 100, was also closest in the former case with ionomers, indicating that the onset degradation mechanisms are most similar in RDE and MEA.
| Exp. Nr. | Setup | Electrolyte and electrode I/C ratio | Slope [%/dec] |
|---|---|---|---|
| 1 | RDE | 0.1 M HClO4, I/C = 0 | 23.8 ± 0.2 |
| 2 | RDE | 0.1 M HClO4, I/C = 0.7 | 29.9 ± 0.8 |
| 3 | RDE | 0.05 M H2SO4, I/C = 0 | 27.3 ± 0.6 |
| 4 | RDE | 1 M HClO4, I/C = 0 | 26.6 ± 0.3 |
| 5 | MEA | Nafion®; I/C = 0.65 | 32.4 ± 1.2 |
Representative sampling for post-AST TEM analysis is difficult for an MEA due to the aforementioned dependency of the degradation mechanisms on the location within the catalyst layer.17 In order to fully understand the origin of the similar results in ECSA degradation over the course of voltage cycling ASTs carried out under similar operating conditions (i.e. T = 80 °C, similar ionomer content), a more profound analysis of the contributions of the underlying degradation mechanisms to the overall ECSA loss should be done for both configurations in the future.
Conclusions
In summary, our full parametric study could demonstrate how application-relevant stability predictions of electrocatalysts can be achieved by mimicking MEA operating conditions and catalyst layer composition in half-cell measurements in a liquid electrolyte such as an RDE setup. Electrochemical surface area and electron microscopy image analysis revealed the importance of temperature and ionomer/carbon ratio for the realization of realistic catalyst degradation. Notably, MEA and RDE measurements conducted at the same temperature of 80 °C and a quasi-identical I/C ratio in the catalyst layer (0.65–0.7) resulted in comparable ECSA loss rates over a voltage cycling AST. Our results further provide detailed insights into the discrepancies in degradation mechanisms that affect the aging of Pt/C catalysts in liquid electrolyte half-cell configurations when different pH values, electrolyte types, ionomer contents, and temperatures are applied. Herein, temperature was found to have the most significant impact on degradation, accelerating it by a factor of five when transitioning from room temperature to 80 °C. Subsequently, the exposure to ionomers also contributed to the degradation process. Our results further highlight the challenges regarding the comparison of existing ex situ degradation studies between each other as well as their interpolation to real application. Our findings suggest that in order to obtain comparable data, the protocols traditionally used by the Department of Energy (DOE) and the Fuel Cell Commercialization Conference of Japan (FCCJ) should be extended to higher temperatures up to 80 °C and relevant I/C ratio should be used. Finally, our optimized RDE-AST protocol opens the way for measuring the stability of novel catalysts within hours, with little prerequisites for instrumentation, and with only μg-quantities of catalysts, while at the same time determining ECSA losses over cycling that are in very good quantitative agreement with analogous measurements conducted in the MEA configuration.Experimental section
RDE Measurements
Catalyst films for ASTs in liquid half cells were prepared by dispersing the catalyst (19.8 wt% Pt/V (TEC10V20E, Tanaka, Japan)) in a 15 vol% isopropanol/water (ultrapure-grade) mixture with an ultrasound vial-treater and subsequent drop-casting of 20 μl of the dispersion onto a ∅ 5 mm glassy carbon electrode of an RDE Tip. The loading was 10 μgPt cm−2 and in cases where ionomer was used, the respective amount of a 20% Nafion® solution was added to the ink before ultrasonication. With a respective loading of 40 μg cm−2 of the Vulcan carbon support, this results in catalyst films with an estimated thickness of ca. 0.8 μm. All measurements at 22 °C were conducted in a Teflon cell with three separate compartments for the working electrode (RDE), the reference electrode (Ag/AgCl, 3M KCl, Metrohm, which was separated from the working electrode compartment by a Luggin capillary and a Nafion® membrane), and the counter electrode (graphite rod); the electrolyte volume in the working electrode compartment was 100 ml. Measurements at 80 °C were conducted in a jacketed glass cell heated by a thermostat in a water bath. The Ag/AgCl reference electrode was replaced by a dynamic hydrogen reference electrode (HydroFlex® from Gaskatel). To prevent electrolyte evaporation, a vigreux condenser was placed at one outlet of the cell and the whole setup was sealed tightly with Parafilm®. In this way, electrolyte evaporation was mitigated sufficiently, so that the level of liquid (100 ml) in the cell remained constant during an overnight (ca.14 h) measurement. The setups were equipped with a Gamry Reference 600 potentiostat and either a Radiometer Analytical rotation controller or an Origalys Origatrod radiometer. The measurement protocol included catalyst activation via cyclovoltammetry, measurement of the reference electrode potential, CO-stripping before AST and the AST in the given order. More precisely, each catalyst film was subjected to 50 CVs (0.05–1.0 VRHE, 200 mV s−1) for surface cleaning. Next, the Pt/C working electrode was converted into a RHE by introducing hydrogen gas for the measurement of the potential of the reference electrode via CV (−0.02 VRHE to 0.03 VRHE, 1 mV s−1). The intercept at a current of 0 A was taken as the potential of the reference electrode. This step was followed by a CO-stripping. All ECSA values given in this work are derived from the CO oxidation charge obtained by the integration of the corresponding peak in the CO stripping CV (0.05–1.10 VRHE, 0.05 Vs−1) after subtracting a CV recorded in N2-saturated electrolyte to correct for non-faradaic contributions. For the calculation, a surface charge density of 195 μC cm−2Pt was used. In this way, the ECSA was measured several times throughout the AST (initially and after 100, 300, 1k, 2k, 5k, 10k and 20k cycles) which consisted of 20k potential cycles with a square-wave profile of 1 second hold each at 0.6 and 1.0 VRHE. The electrolytes were prepared from concentrated ultrapure-grade acids (70% HClO4 Carl Roth, 96% H2SO4 Merck) and ultrapure water (VWR). In order to evaluate whether instability of the perchloric acid electrolyte at 80 °C68 and eventually formed chloride or chlorine, which may adsorb and be reduced to chloride on the Pt/C electrode during AST, influences the ECSA loss in our experiments, additional experiments in sulphuric acid electrolyte were conducted. On the one hand, the concentration of the electrolyte and thus the pH value was varied, and on the other hand, different concentrations of chloride in the 0.05 M H2SO4 electrolyte were studied by adding respective amounts of a 1 M HCl solution. The results are shown in Fig. S22 and S23 and discussed in the ESI.†MEA preparation and measurement
In order to compare the ECSA degradation during voltage cycling-based ASTs for Pt/Vulcan in liquid half cells with that obtained from single cell PEM fuel cell measurements, MEAs were prepared by the decal transfer method according to Della Bella et al. using the same catalyst as for the RDE measurements, using an I/C ratio of 0.65 (800EW ionomer, 3M).46 The respective loadings for the anode and cathode were determined as 0.105 ± 0.003 mgPt cm−2MEA and 0.096 ± 0.001 mgPt cm−2MEA. The thickness of the cathode catalyst layer is ca. 7.5 μm.MEA aging and characterization tests were performed on a customized G60 test station (Greenlight innovation Corp., USA) equipped with a potentiostat (Reference3000, Gamry, UK) using a 5 cm2 active area single-cell hardware with graphite flow fields (7 parallel channels, one serpentine, 0.5 mm lands/channels, manufactured by Poco Graphite, Entegris GmbH, USA, according to our design). Gas diffusion layers (GDLs, H14C10, Freudenberg KG, Germany) with a thickness of 170 ± 5 μm were compressed by 12 ± 0.2% by means of quasi-incompressible PTFE-coated fiberglass gaskets (Fiberflon, Fiberflon GmbH & Co. KG, Germany). The cells were assembled with a torque of 12 Nm resulting in a gas diffusion layer compression of ≈1.5 MPa on the 5 cm2 active area (for details see Simon et al.).69
The MEA characterization and aging tests were done according to the measurement procedure of Della Bella et al.46 For the reader's convenience, the relevant experimental methods for this work are summarized in the following. Prior to each measurement, 8 cycles voltage-controlled ramp-in procedure using 1390 nccm H2 (anode) and 3320 nccm air (cathode) at 80 °C, 100% RH and 150 kPaabs was performed: 0.6 V (45 min), open circuit voltage (OCV, 5 min), and 0.85 V (10 min). The cathode ECSA was determined from CO-stripping measurements by adsorbing dry CO (100 nccm flow of 10% CO in N2) for 3 min at 40 °C and 150 kPaabs at a constant cathode potential of 0.1 VRHE. In order to remove residual CO from the gas lines and the cell, a 1 h purge was done using high purity N2 (5.0). Oxidation of the adsorbed CO was acquired by recording a cathode CV from the holding potential up to 1.1 VRHE at a scan rate of 100 mV s−1. Afterward, two subsequent scans were recorded in order to fully oxidize the CO from the catalyst surface. During the CO stripping procedure, the anode was fed with 200 nccm of 5% H2′/Ar. A voltage cycling-based accelerated stress test was performed under H2/N2 (200 nccm/75 nccm) configuration on the anode/cathode at 80 °C, 95% RH and ambient pressure. A Reference3000 potentiostat was used in order to control the voltage during square-wave (SW) based voltage cycling, whereby the potential step was completed in less than 1 s. The same aging protocol was used for the RDE experiments, namely voltage cycling between 0.6 V and 1.0 V with a vertex potential hold of 1 s. For the MEA aging tests, two individual MEAs with similar cathode and anode loading were prepared and evaluated, where the determined normalized ECSA values refer to the average of these two measurements and the error bars represent the min/max values.
TEM analysis
Samples for TEM analysis were prepared by re-dispersing the catalyst film from the RDE tip in isopropanol and drop-casting the dispersion on a TEM grid (Au) with Lacey carbon film. The images were recorded using a Philips CM20 transmission electron microscope and particle size distributions were determined by measuring a count of ≥1000 particles from at least 6 different carbon support particle agglomerates for each sample using ImageJ.70Author contributions
The initial conceptualization of this study was done by T. I., R. K. F. D. B., and B. M. S. and was further developed by all authors. T. I. carried out and analysed the RDE and TEM measurements. R. K. F. D. B. conducted and analysed the MEA measurements. The manuscript was written by T. I. T. I., R. K. F. D. B., B. M. S, H. G., and M. L. contributed to the interpretation of the results and reviewed and edited the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. L. and T. I. acknowledge Paul Roumeliotis for assistance with the RDE stability measurements and fruitful discussions. Financial support in the framework of the POREForm project funded by the German Federal Ministry for Economic Affairs and Energy (BMWK funding number 03ETB027C) is gratefully acknowledged. ML acknowledges the Federal Ministry of Education and Research (BMBF) in the framework of NanoMatFutur (SynKat, FK: 03XP0265) for financial support.Notes and references
- B. San-Akca, S. D. Sever and S. Yilmaz, Energy Res. Soc. Sci., 2020, 70, 101690 CrossRef PubMed.
- C. W. Su, K. Khan, M. Umar and W. Zhang, Energy Policy, 2021, 158, 112566 CrossRef.
- A. Sartbaeva, V. L. Kuznetsov, S. A. Wells and P. P. Edwards, Energy Environ. Sci., 2008, 1, 79–85 RSC.
- M. Pagliaro, A. G. Konstandopoulos, R. Ciriminna and G. Palmisano, Energy Environ. Sci., 2010, 3, 253 RSC.
- A. Buttler and H. Spliethoff, Renewable Sustainable Energy Rev., 2018, 82, 2440–2454 CrossRef CAS.
- S. Ardo, D. Fernandez Rivas, M. A. Modestino, V. Schulze Greiving, F. F. Abdi, E. Alarcon Llado, V. Artero, K. Ayers, C. Battaglia, J. P. Becker, D. Bederak, A. Berger, F. Buda, E. Chinello, B. Dam, V. Di Palma, T. Edvinsson, K. Fujii, H. Gardeniers, H. Geerlings, S. M. Hashemi, S. Haussener, F. Houle, J. Huskens, B. D. James, K. Konrad, A. Kudo, P. P. Kunturu, D. Lohse, B. Mei, E. L. Miller, G. F. Moore, J. Muller, K. L. Orchard, T. E. Rosser, F. H. Saadi, J. W. Schüttauf, B. Seger, S. W. Sheehan, W. A. Smith, J. Spurgeon, M. H. Tang, R. Van De Krol, P. C. K. Vesborg and P. Westerik, Energy Environ. Sci., 2018, 11, 2768–2783 RSC.
- I. Staffell, D. Scamman, A. Velazquez Abad, P. Balcombe, P. E. Dodds, P. Ekins, N. Shah and K. R. Ward, Energy Environ. Sci., 2019, 12, 463–491 RSC.
- B. G. Pollet, S. S. Kocha and I. Staffell, Curr. Opin. Electrochem., 2019, 16, 90–95 CrossRef CAS.
- S. Mezzavilla, C. Baldizzone, A. C. Swertz, N. Hodnik, E. Pizzutilo, G. Polymeros, G. P. Keeley, J. Knossalla, M. Heggen, K. J. J. Mayrhofer and F. Schüth, ACS Catal., 2016, 6, 8058–8068 CrossRef CAS.
- J. Knossalla, J. Mielby, D. Göhl, F. R. Wang, D. Jalalpoor, A. Hopf, K. J. J. Mayrhofer, M. Ledendecker and F. Schüth, ACS Appl. Energy Mater., 2021, 4, 5840–5847 CrossRef CAS.
- J. Knossalla, P. Paciok, D. Göhl, D. Jalalpoor, E. Pizzutilo, A. M. Mingers, M. Heggen, R. E. Dunin-Borkowski, K. J. J. Mayrhofer, F. Schüth and M. Ledendecker, J. Am. Chem. Soc., 2018, 140, 15684–15689 CrossRef CAS PubMed.
- R. A. Armengol, J. Lim, M. Ledendecker, K. Hengge and C. Scheu, Nanoscale Adv., 2021, 3, 5075–5082 RSC.
- S. Geiger, O. Kasian, M. Ledendecker, E. Pizzutilo, A. M. Mingers, W. T. Fu, O. Diaz-Morales, Z. Li, T. Oellers, L. Fruchter, A. Ludwig, K. J. J. Mayrhofer, M. T. M. Koper and S. Cherevko, Nat. Catal., 2018, 1, 508–515 CrossRef CAS.
- E. Pizzutilo, J. Knossalla, S. Geiger, J.-P. Grote, G. Polymeros, C. Baldizzone, S. Mezzavilla, M. Ledendecker, A. Mingers, S. Cherevko, F. Schüth and K. J. J. Mayrhofer, Adv. Energy Mater., 2017, 7, 1700835 CrossRef.
- M. S. Wilson, F. H. Garzon, K. E. Sickafus and S. Gottesfeld, J. Electrochem. Soc., 1993, 140, 2872–2877 CrossRef CAS.
- Y. Shao-Horn, W. C. Sheng, S. Chen, P. J. Ferreira, E. F. Holby and D. Morgan, Top. Catal., 2007, 46, 285–305 CrossRef CAS.
- P. J. Ferreira, G. J. la O’, Y. Shao-Horn, D. Morgan, R. Makharia, S. Kocha and H. A. Gasteiger, J. Electrochem. Soc., 2005, 152, A2256 CrossRef.
- R. M. Darling and J. P. Meyers, J. Electrochem. Soc., 2003, 150, A1523 CrossRef CAS.
- R. L. Borup, A. Kusoglu, K. C. Neyerlin, R. Mukundan, R. K. Ahluwalia, D. A. Cullen, K. L. More, A. Z. Weber and D. J. Myers, Curr. Opin. Electrochem., 2020, 21, 192–200 CrossRef CAS.
- X. Wang, R. Kumar and D. J. Myers, Electrochem. Solid-State Lett., 2006, 9, 225–228 CrossRef.
- R. Makharia, S. Kocha, P. Yu, M. A. Sweikart, W. Gu, F. Wagner and H. A. Gasteiger, ECS Trans., 2006, 1, 3–18 CrossRef CAS.
- C. H. Paik, G. S. Saloka and G. W. Graham, Electrochem. Solid-State Lett., 2007, 10, 39–42 CrossRef.
- R. L. Borup, J. R. Davey, F. H. Garzon, D. L. Wood and M. A. Inbody, J. Power Sources, 2006, 163, 76–81 CrossRef CAS.
- G. S. Harzer, J. N. Schwämmlein, A. M. Damjanović, S. Ghosh and H. A. Gasteiger, J. Electrochem. Soc., 2018, 165, F3118–F3131 CrossRef CAS.
- P. Zihrul, I. Hartung, S. Kirsch, G. Huebner, F. Hasché and H. A. Gasteiger, J. Electrochem. Soc., 2016, 163, F492–F498 CrossRef CAS.
- R. K. Ahluwalia, S. Arisetty, J.-K. Peng, R. Subbaraman, X. Wang, N. Kariuki, D. J. Myers, R. Mukundan, R. Borup and O. Polevaya, J. Electrochem. Soc., 2014, 161, F291–F304 CrossRef CAS.
- E. F. Holby, W. Sheng, Y. Shao-Horn and D. Morgan, Energy Environ. Sci., 2009, 2, 865–871 RSC.
- K. Yu, D. J. Groom, X. Wang, Z. Yang, M. Gummalla, S. C. Ball, D. Myers and P. J. Ferreira, Microsc. Microanal., 2014, 20, 482–483 CrossRef.
- T. J. Schmidt, H. A. Gasteiger, G. D. Stäb, P. M. Urban, D. M. Kolb and R. J. Behm, J. Electrochem. Soc., 1998, 145, 2354–2358 CrossRef CAS.
- U. A. Paulus, T. J. Schmidt, H. A. Gasteiger and R. J. Behm, J. Electroanal. Chem., 2001, 495, 134–145 CrossRef CAS.
- T. J. Schmidt, J. Electrochem. Soc., 1998, 145, 2354 CrossRef CAS.
- T. Lazaridis, B. M. Stühmeier, H. A. Gasteiger and H. A. El-sayed, Nat. Catal., 2022, 5, 363–373 CrossRef CAS.
- K. Ehelebe, D. Escalera-López and S. Cherevko, Curr. Opin. Electrochem., 2021, 29, 100832 CrossRef CAS.
- R. Sharma and S. M. Andersen, ACS Catal., 2018, 8, 3424–3434 CrossRef CAS.
- S. Martens, L. Asen, G. Ercolano, F. Dionigi, C. Zalitis, A. Hawkins, A. Martinez Bonastre, L. Seidl, A. C. Knoll, J. Sharman, P. Strasser, D. Jones and O. Schneider, J. Power Sources, 2018, 392, 274–284 CrossRef CAS.
- J. A. Gilbert, N. N. Kariuki, X. Wang, A. J. Kropf, K. Yu, D. J. Groom, P. J. Ferreira, D. Morgan and D. J. Myers, Electrochim. Acta, 2015, 173, 223–234 CrossRef CAS.
- H. Y. Kim, D. S. Im, U. H. Son, H. R. Lee and H. I. Joh, Carbon Lett., 2021, 32, 313–319 CrossRef.
- H. Yu, M. J. Zachman, C. Li, L. Hu, N. N. Kariuki, R. Mukundan, J. Xie, K. C. Neyerlin, D. J. Myers and D. A. Cullen, ACS Appl. Mater. Interfaces, 2022, 14, 20418–20429 CrossRef CAS PubMed.
- A. Riese, D. Banham, S. Ye and X. Sun, J. Electrochem. Soc., 2015, 162, F783–F788 CrossRef CAS.
- G. Polymeros, C. Baldizzone, S. Geiger, J. P. Grote, J. Knossalla, S. Mezzavilla, G. P. Keeley, S. Cherevko, A. R. Zeradjanin, F. Schüth and K. J. J. Mayrhofer, Electrochim. Acta, 2016, 211, 744–753 CrossRef CAS.
- P. Urchaga, T. Kadyk, S. G. Rinaldo, A. O. Pistono, J. Hu, W. Lee, C. Richards, M. H. Eikerling and C. A. Rice, Electrochim. Acta, 2015, 176, 1500–1510 CrossRef CAS.
- DOE Durability Working Group, Rotating Disk-Electrode Aqueous Electrolyte Accelerated Stress Tests for PGM Electrocatalyst/Support Durability Evaluation, 2011, URL: https://www.energy.gov/eere/fuelcells/articles/rotating-disk-electrode-aqueous-electrolyte-accelerated-stress-tests-pgm, access date: 17/03/2023.
- A. Ohma, K. Shinohara, A. Iiyama, T. Yoshida and A. Daimaru, ECS Trans., 2011, 41, 775–784 CrossRef CAS.
- U.S. Department of Energy, Fuel Cell 2016 Multi-Year Research, Development, and Demonstration Plan, 2016, URL: https://www.energy.gov/sites/default/files/2017/05/f34/fcto_myrdd_fuel_cells.pdf, access date: 17/03/2023.
- T. R. Garrick, T. E. Moylan, M. K. Carpenter and A. Kongkanand, J. Electrochem. Soc., 2017, 164, F55–F59 CrossRef CAS.
- R. K. F. Della Bella, B. M. Stühmeier and H. A. Gasteiger, J. Electrochem. Soc., 2022, 169, 044528 CrossRef CAS.
- A. Kregar and T. Katrašnik, Open Phys., 2019, 17, 779–789 CrossRef CAS.
- P. T. Yu, W. Gu, R. Makharia, F. T. Wagner and H. A. Gasteiger, ECS Trans., 2006, 3, 797–809 CrossRef CAS.
- E. Pizzutilo, S. Geiger, J.-P. Grote, A. Mingers, K. J. J. Mayrhofer, M. Arenz and S. Cherevko, J. Electrochem. Soc., 2016, 163, F1510–F1514 CrossRef CAS.
- N. Linse, L. Gubler, G. G. Scherer and A. Wokaun, Electrochim. Acta, 2011, 56, 7541–7549 CrossRef CAS.
- F. Liu, Z. Gao, J. Su and L. Guo, ChemSusChem, 2022, 15, 1–13 Search PubMed.
- C. Cremers, T. Jurzinsky, J. Meier, A. Schade, M. Branghofer, K. Pinkwart and J. Tübke, J. Electrochem. Soc., 2018, 165, F3307–F3315 CrossRef CAS.
- L. Xing, M. A. Hossain, M. Tian, D. Beauchemin, K. T. Adjemian and G. Jerkiewicz, Electrocatalysis, 2014, 5, 96–112 CrossRef CAS.
- S. Cherevko, A. A. Topalov, A. R. Zeradjanin, G. P. Keeley and K. J. J. Mayrhofer, Electrocatalysis, 2014, 5, 235–240 CrossRef CAS.
- M. Arenz and A. Zana, Nano Energy, 2016, 29, 299–313 CrossRef CAS.
- T. Nagai, H. Murata and Y. Morimoto, J. Electrochem. Soc., 2014, 161, F789–F794 CrossRef CAS.
- K. Shinozaki, J. W. Zack, S. Pylypenko, B. S. Pivovar and S. S. Kocha, J. Electrochem. Soc., 2015, 162, F1384–F1396 CrossRef CAS.
- T. Van Cleve, S. Khandavalli, A. Chowdhury, S. Medina, S. Pylypenko, M. Wang, K. L. More, N. Kariuki, D. J. Myers, A. Z. Weber, S. A. Mauger, M. Ulsh and K. C. Neyerlin, ACS Appl. Mater. Interfaces, 2019, 11, 46953–46964 CrossRef CAS PubMed.
- S. J. Sondheimer, N. J. Bunce, M. E. Lemke and C. A. Fyfe, Macromolecules, 1986, 19, 339–343 CrossRef CAS.
- A. Kusoglu and A. Z. Weber, Chem. Rev., 2017, 117, 987–1104 CrossRef CAS PubMed.
- A. A. Topalov, S. Cherevko, A. R. Zeradjanin, J. C. Meier, I. Katsounaros and K. J. J. Mayrhofer, Chem. Sci., 2014, 5, 631–638 RSC.
- Y. Furuya, T. Mashio, A. Ohma, M. Tian, F. Kaveh, D. Beauchemin and G. Jerkiewicz, ACS Catal., 2015, 5, 2605–2614 CrossRef CAS.
- S. Helmly, M. J. Eslamibidgoli, K. A. Friedrich and M. H. Eikerling, Electrocatalysis, 2017, 8, 501–508 CrossRef CAS.
- E. Guilminot, A. Corcella, M. Chatenet, F. Maillard, F. Charlot, G. Berthomé, C. Iojoiu, J.-Y. Sanchez, E. Rossinot and E. Claude, J. Electrochem. Soc., 2007, 154, B1106 CrossRef CAS.
- I. Martens, L. G. A. Melo, M. M. West, D. P. Wilkinson, D. Bizzotto and A. P. Hitchcock, ACS Catal., 2020, 10, 8285–8292 CrossRef CAS.
- H. Yano, M. Watanabe, A. Iiyama and H. Uchida, Nano Energy, 2016, 29, 323–333 CrossRef CAS.
- Y. Shao-Horn, W. C. Sheng, S. Chen, P. J. Ferreira, E. F. Holby and D. Morgan, Top. Catal., 2007, 46, 285–305 CrossRef CAS.
- F. Solymosi, Acta Phys. Chem., 1977, 23, 317–354 CAS.
- C. Simon, F. Hasché and H. A. Gasteiger, J. Electrochem. Soc., 2017, 164, F591–F599 CrossRef CAS.
- J. Speder, A. Zana, I. Spanos, J. J. K. Kirkensgaard, K. Mortensen, M. Hanzlik and M. Arenz, J. Power Sources, 2014, 261, 14–22 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional characterization including ECSA loss data plots, TEM images and fits of the particle size distributions. See DOI: https://doi.org/10.1039/d3cp02847j |
| This journal is © the Owner Societies 2023 |