
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
How inversion relates to disordering tendencies in complex oxides
Vancho
Kocevski†
,
Ghanshyam
Pilania‡
 and
Blas P.
Uberuaga
*
and
Blas P.
Uberuaga
*
Materials Science and Technology Division, Los Alamos National Laboratory, Los Alamos, NM 87545, USA. E-mail: blas@lanl.gov
First published on 27th September 2023
Abstract
Complex oxides exhibit great functionality due to their varied chemistry and structures. They are quite flexible in terms of the ordering of cations, which can also impact their functional properties to a large extent. Thus, the propensity for a complex oxide to disorder is a key factor in optimizing and discovering new materials. Here, we show that the propensity to disorder cations in perovskites, pyrochlores, and spinels correlates with the energy to “invert” the structure – to directly swap the cations across the sublattices. This relatively simple metric, involving only two energetic calculations per compound, qualitatively captures disordering trends amongst compounds across these three families of materials and is quantitative in several cases. This provides a fast and robust metric to determine those complex oxides that are easy or hard to disorder, providing new avenues for quick screening of compounds for cation-ordering-dependent functionalities.
1 Introduction
Chemical disorder in complex oxides, taking the form of cation antisites in the dilute limit, is one of the material parameters that is critical for understanding performance. For example, the ability of a complex oxide to accommodate disorder has been linked to its amorphization resistance.1–3 Further, many functional properties of complex oxides, including conductivity,4–7 mechanical properties,8 energy density,9 and magnetic properties,10 are sensitive to the level of cation disorder in the material. Thus, it is of general interest to understand which oxides will readily disorder and which will not.Computationally, one can estimate the energetic cost of disordering a material by using approaches such as special quasirandom structures (SQS)11 or small set of ordered structures (SSOS),12 which mimic the correlations of a random distribution of atoms in relatively compact simulation cells that can be easily handled in a computationally efficiently manner using density functional theory (DFT) based methods. These approaches have proven quite powerful in aiding the community in understanding how complex materials might disorder, with applications to spinels,13,14 pyrochlores,15 perovskites,16 and other complex oxides.17–19 However, despite this success, it would still be beneficial to have an even simpler metric for screening materials that might disorder more or less easily, a metric that is even more computationally efficient to determine.
In our previous work,20 we showed that, for inter-lanthanide perovskites where both cations are lanthanides, there is a clear relationship between the energy to disorder the material and the energy to invert it – to swap the A and B cations. This leads to a broader hypothesis that this relationship might hold for other compounds and crystal structures. The basic idea is that compounds which exhibit energetic degeneracy in cation orderings will be thus more easily disordered as they have little preference for the specific arrangement of cations within their structure. In this work, we expand on this hypothesis. We demonstrate that the concept of inversion, normally used in the context of spinels, can be used as a metric for determining the disordering tendencies of a given complex oxide. That is, the propensity of an oxide to disorder is directly correlated with the energy to invert the chemistry, to swap the cations in the ordered structure. While there are exceptions to this correlation, we find it holds broadly across a range of chemistries and crystal structures. This insight should provide new avenues for discovering oxides that can or cannot be easily disordered.
2. Methodology
DFT calculations were performed using the Vienna Ab-initio Simulation Package (VASP) code,21–24 with the Perdew, Burke and Ernzerhof (PBE)25 exchange correlation functional, and the projector augmented wave (PAW) method.26,27 For the lanthanides (Ln) we used the PAW potentials from the VASP distribution with formal valence of 3, that is “Ln_3'” pseudopotentials for all lanthanides except lanthanum, for which we used the “La” pseudopotential. We used a 520 eV cut-off energy for the plane wave basis set, and 10−6 eV energy and 10−2 eV Å−1 force convergence criteria, respectively. A Γ-centered k-point spacing of < 0.025 Å−1 was applied for sampling the Brillouin zone for each structure. The systems were considered to be spin-polarized (ISPIN = 2), with ferromagnetic ordering of the cations - all cation spins were set to up at the beginning of all calculations. During the structural relaxation, the atomic positions, cell shape and volume were allowed to change. Note that to make the relaxation process more efficient, we used the memory conserving symmetrization of the chanrge density as implemented in VASP, by setting the tag ISYM = 2. Our tests showed that removing the symmetry (ISYM = 0) gives negligible change in the total energy of the systems (< 0.001 eV per atom).3. Models
We examine the relationship between disordering and inversion in three crystal structures – perovskite, pyrochlore, and spinel, illustrated in Fig. 1 – with the associated space groups Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m, Fdm, and Fdm, respectively. In the case of perovskites, we build upon our prior work,20 focused on inter-lanthanide perovskites that are orthorombic, with a Pnma space group. The coordination environment of the cations in these structures is very different. In the Ã
m, Fdm, and Fdm, respectively. In the case of perovskites, we build upon our prior work,20 focused on inter-lanthanide perovskites that are orthorombic, with a Pnma space group. The coordination environment of the cations in these structures is very different. In the Ã![[B with combining tilde]](https://www.rsc.org/images/entities/char_0042_0303.gif) O3 perovskite, the à site is 8-fold cordinated while the site is 6-fold coordinated. (For clarity, here and in what follows, we use à to represent site A while A without a tilde represents the cation A.) In Ã22O7, the à sites are 8-fold coordinated and the are 6-fold. Finally, in spinel, à is tetrahedrally coordinated and is octahedrally coordinated. However, spinels can accommodate a natural level of so-called inversion, in which B cations occupy the tetrahedrally coordinated à site. The extent to which this occurs is called inversion and can vary from i = 0–1, where a value of 1 represents all of the à sites being occupibed by B cations. In principle, while not typically called inversion, similar behavior can occur in other oxides. In pyrochlore, the A and B cations can swap sites, leading to cation disorder. In this work, we will generically call the swapping of cations inversion, in analogy with the spinels.
O3 perovskite, the à site is 8-fold cordinated while the site is 6-fold coordinated. (For clarity, here and in what follows, we use à to represent site A while A without a tilde represents the cation A.) In Ã22O7, the à sites are 8-fold coordinated and the are 6-fold. Finally, in spinel, à is tetrahedrally coordinated and is octahedrally coordinated. However, spinels can accommodate a natural level of so-called inversion, in which B cations occupy the tetrahedrally coordinated à site. The extent to which this occurs is called inversion and can vary from i = 0–1, where a value of 1 represents all of the à sites being occupibed by B cations. In principle, while not typically called inversion, similar behavior can occur in other oxides. In pyrochlore, the A and B cations can swap sites, leading to cation disorder. In this work, we will generically call the swapping of cations inversion, in analogy with the spinels.
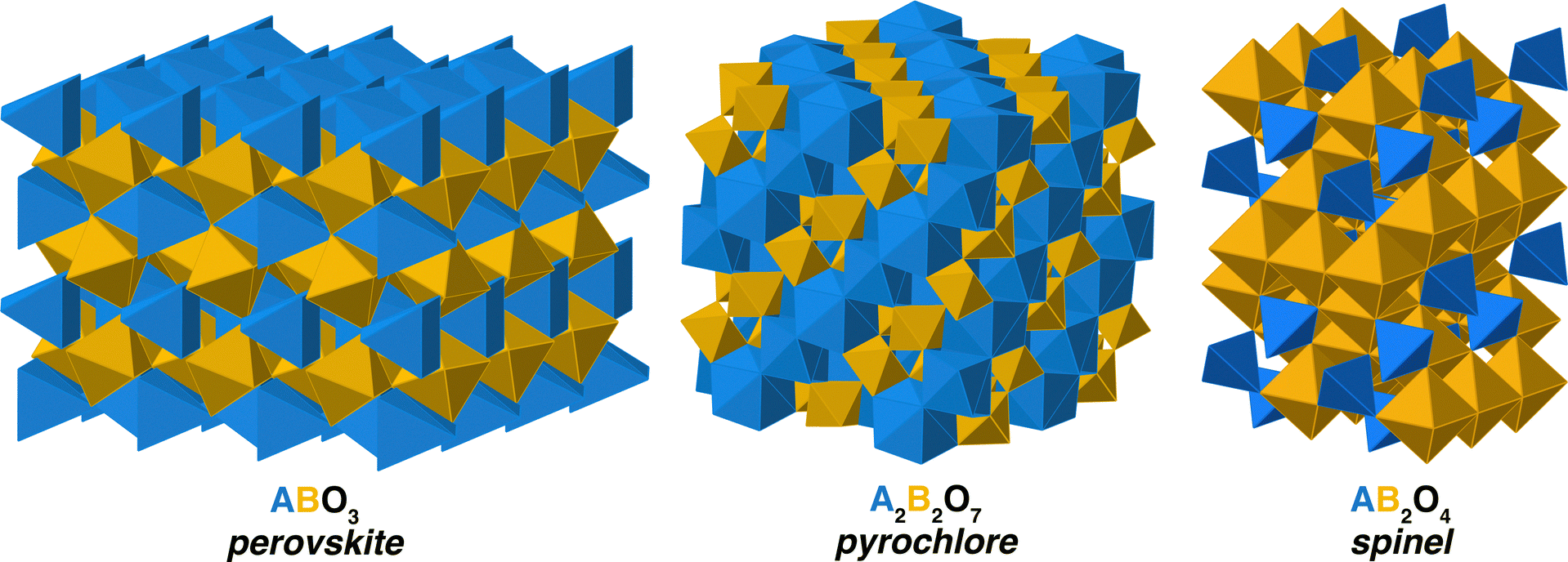 | ||
| Fig. 1 The crystal structures of the complex oxides considered in this work. The colors of the polyhedra represent the different sites for cation occupancy. Oxygen ions, sitting at the corners of the polyhedra, are not shown. | ||
Another distinguishing feature of these oxides is the fact that they are not all fully dense relative to a parent structure.28 In particular, as the two cations in pyrochlore are aliovalent, these compounds are oxygen deficient relative to the parent BO2 fluorite structure, and thus there are structural vacancies on the oxygen sublattice. Similarly, spinels are cation deficient relative to the parent AO rocksalt structure and there are cation vacancies in the structure. This will become important later.
The working hypothesis of this study is that the propensity for disorder is related to the degeneracy of various ordered structures that might be possible for the given compound. That is, if there are multiple ways of ordering the cations in the compound and those are all of similar energy, the compound has no great preference for how the cations are arranged and thus disordering might be relatively favorable. In contrast, if there is a strongly preferred ordering, then alternative cation arrangements, including those in which the cations are disordered, would be energetically costly. Thus, by comparing the relative energetics of different orderings, we can assess the propensity for disorder.
To that end, we compare the two extreme cases of cation ordering – the groundstate thermodynamically preferred ordered structure and the fully ‘inverse’ structure in which A and B cations are swapped. In the perovskite and pyrochlore structures, this procedure simply means that all A and B cation positions are swapped. In spinel, because the stoichiometry is not 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, this is a bit more complicted. In the inverse structure, all à sites are filled by B cations, and half of the sites are filled with A cations. The specific ordering we choose for the A and B cations on the site correspond to the ground state inverse structure we have used previously and which is illustrated in Fig. 1. Further, in some spinels, the inverse structure is actually the ground state and thus the normal structures is, in the context of the current work, the inverted structure. In all cases, when we refer to the energy of inversion ΔEinversion, we mean starting with the groundstate – whether it is normal or inverse in the conventional sense – and inverting it.
:1, this is a bit more complicted. In the inverse structure, all à sites are filled by B cations, and half of the sites are filled with A cations. The specific ordering we choose for the A and B cations on the site correspond to the ground state inverse structure we have used previously and which is illustrated in Fig. 1. Further, in some spinels, the inverse structure is actually the ground state and thus the normal structures is, in the context of the current work, the inverted structure. In all cases, when we refer to the energy of inversion ΔEinversion, we mean starting with the groundstate – whether it is normal or inverse in the conventional sense – and inverting it.
Finally, to compare inversion to disordering, we need to describe the disordered state. We use the SQS approach to mimic the random correlations of the random solid solution in a computational tractable supercell. SQSs for the three crystal structures were generated using the mcsqs tool,29 part of the ATAT distribution, and contain 160, 176, and 224 atoms for perovskites, pyrochlore, and spinels, respectively. In the case of spinel, we constructed a second set of SQSs by changing the run time of the mcsqs script. The results from that set are in good agreement with the first and so are not discussed further. We will refer to the energy of the SQS structure relative to the groundstate ordered structures as ΔErandom. A full list of all compounds considered and reported here is given in Table 1.
Our final procedure and concept are schematically illustrated in Fig. 2 for the case of pyrochlore. We simply compare the difference in energy between the ‘inverse’ and ‘normal’ structures with that of the fully random structure to determine if there is a correlation between the two energies.
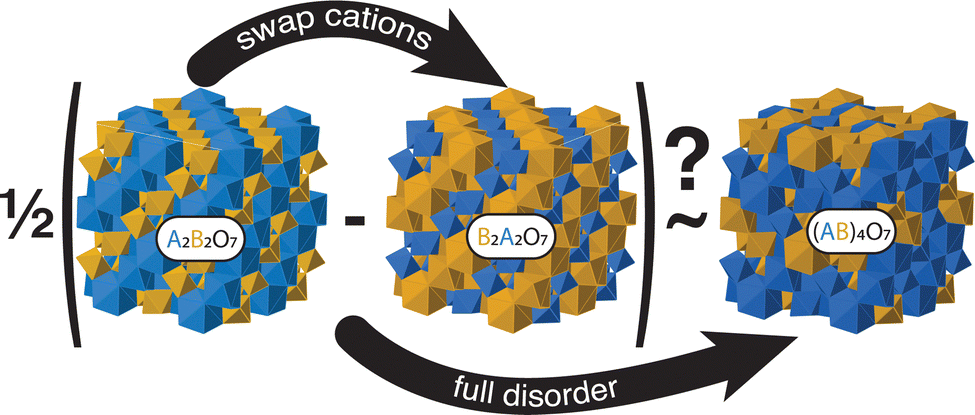 | ||
| Fig. 2 Illustrated for the example of pyrochlore, the basic concept behind this work is that the energy of the disordered state, represented here by the structure (AB)4O7, is an average of the two extreme states of ordering, the groundstate ‘normal’ A2B2O7 and the inverse B2A2O7 in which the A and B cations are simply swapped. | ||
4. Results and discussion
Fig. 3 shows the relationship between the energy to disorder a compound compared to the energy to invert the compound, as a function of crystal structure. The perovskite structured compounds all fall on or near a line described by Vegard's law, in which the energy is simply half of the energy difference between the normal and the inverted structure. The supposition here is that the disordered state, described by a 50/50 mixture of cations, is halfway between the normal and inverted state. For perovskite, this relationship holds quite well, with some possible deviation for compounds with lower energies of inversion.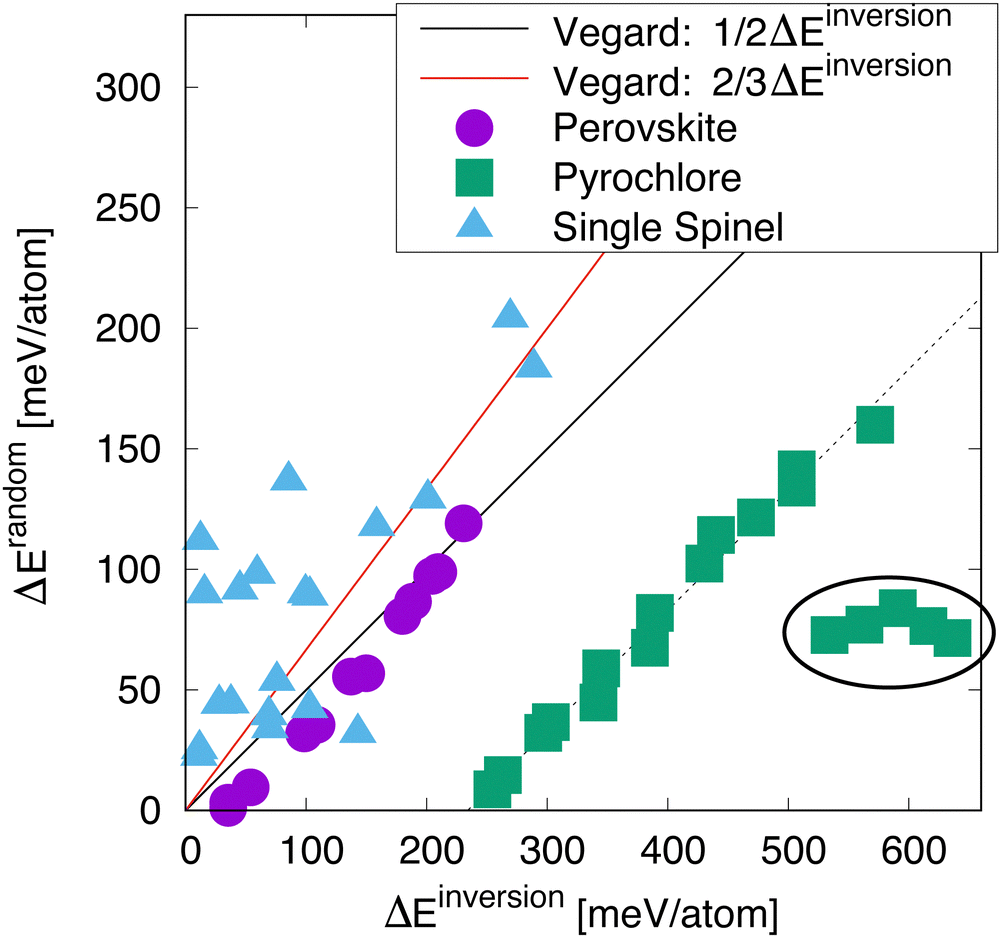 | ||
| Fig. 3 Energy to randomize the cation distribution versus inverting it for a selection of perovskites, spinels, and pyrochlores (the full list is given in Table 1). The lines are estimates from a 1/2 (black) and 2/3 (red) linear interpolation between the ground state and the inverted structure – referred to here as “Vegard”. The circled group of compounds are the titanate pyrochlores. | ||
For the most part, the energy to disorder the pyrochlores is also a linear function of the inversion energy, though offset from a perfect Vegard's law relationship. This deviation is due to the fact that, in the inverted structure, symmetry prevents the oxygen structural vacancies from relaxing while they are able to relax in the symmetry-broken disordered SQS. Thus, the inverted structure is in some sense an overestimation of the tendency for disordering. Looking at these structures in more detail, we indeed find that several oxygen ions relax significantly in the SQS as opposed to in the inverted structure. Some of the pyrochlores also exhibit some significant deviation from the rest, as highlighted by the circled region. These are pyrochlores in which B = Ti, a family of compounds known to exhibit non-monotonic behavior with A cation chemistry, including in the critical amorphization temperature.3 This complex behavior has been attributed to the varying nature of electronic hybridization as a function of the A cation in these compounds.30 This will be further discussed below. However, disregarding the known anomalous behavior of the titanates, the rest of the pyrochlores indeed exhibit a clear relationship between the disordering and inversion energies.
Finally, we turn our attention to the spinels. While the relationship between disordering and inversion is not nearly as straight-forward as for perovskites or pyrochlores, we still see an overall trend in which a higher energy for inversion correlates to a higher energy for disorder. Further, this relationship roughly tracks with a different Vegard relationship in which the inversion energy is scaled by two-thirds, as the random state corresponds to a structure with i = 2/3. However, it is very clear that the data for the spinels is much more scattered than for the other structures. While we have not identified a single reason for this, it seems that magnetism is at least partially responsible for this scatter, which is discussed later.
The results in Fig. 4 confirm the supposition that the deviation in behavior from ideal Vegard's law is due to relaxation, at least for the pyrochlores. The difference in the average relaxation of oxygen atoms in the random structures vs. the inverted structures (ΔΔd) is plotted against the deviation of the random energy from the Vegard's law interpolation between the end members (ΔΔE). (In these calculations, the reference is an ideal structure with the same cation distributions and the ion relaxation is calculated as the distance between the same ion in the relaxed and the ideal structures; the shape of the relaxed cell is scaled to that of the ideal cell to eliminate “displacements” due to changes in volume/shape.) The perovskites show very little relaxation from the ideal as-constructed structures and, correspondingly, small deviations in ΔΔE. The pyrochlores, in contrast, exhibit much higher values of ΔΔd and thus greater values of ΔΔE. This is, in part, a consequence of the higher symmetry of the inverted structure, which stabilizes the ordered structure. In particular, ΔΔE is negative, highlighting the enhanced stabilization that the relaxation of the oxygen ions provide in random structures. The spinels again exhibit more complex behavior. While ΔΔd is relatively small – on the order of that seen for the perovskites – ΔΔE is significantly larger. As we previously noted, we do not have a firm explanation for this larger scatter; we presume it is a consequence of the magnetic structure of these oxides. All calculations were initiated in a ferromagnetic state, which may not be the ground state for many of these compounds. If all three structures do not relax to a similar magnetic state, there will be an extra contribution to the energetics that we are not accounting for here.
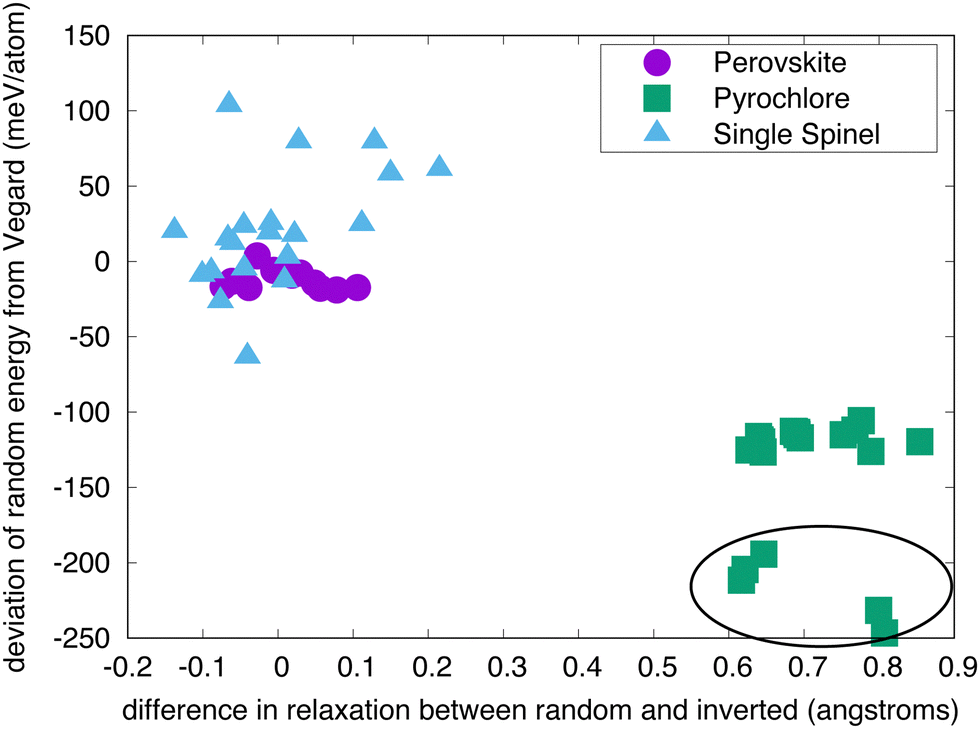 | ||
| Fig. 4 Deviation of the randomization energy from “Vegard” as a function of the difference in oxygen relaxation in the random vs. inverted structure for perovskites, pyrochlores, and spinels. The circled group of compounds are the titanate pyrochlores. | ||
To test the generality of this relationship, we also considered so-called double spinels, in which three cations order across the two sublattices in spinel.31 In the ground state, the structure looks very much like the inverse single spinel structure, except that the A cation on the site is replaced by a third C cation – this is illustrated in Fig. 5. In this ABCO4 structure, there are two scenarios to create a partially inverted structure: by swapping cations between the à and sites or the à and ![[C with combining tilde]](https://www.rsc.org/images/entities/char_0043_0303.gif) sites (swapping cations between the and sites does not change anything since these reside on the inversion-invariant octahedral sublattice). Each case leads to a potentially higher energy structure. Here, we correlate the SQS energy, in which all three cations are randomly distributed, with the energy of each of the inverted structures. The reasoning here is that if the next highest structure is much higher in energy, again the cations have a very strong preference for a specific ordering. On the other hand, there are scenarios where some orderings are energetically degenerate and others are not, so we consider both cases. The results are highlighted in Fig. 5. Again, we see a strong correlation between the disordering and inversion energies. This correlation is reasonably strong for both inverted structures. Interestingly, if we examine the lower energy inverted structure (labeled “Mid” in Fig. 5), the correlation is perhaps better but off of the Vegard's law trend line. In contrast, the higher energy inverted structure is closer to the trend line but exhibits more scatter, reminiscent of the single spinels. At this time, we do not have a reason for this behavior. While there are intuitive reasons for the data to be below the line, as it was for pyrochlore, there is no obvious reason for it to be higher, except that the mixing of all three cations in the double spinel structure does not correspond to any one inverse structure. That is, we cannot view the disorder state for a three component material as an interpolation between an inverted structure in which only two cations are swapped. Even so, we see there is a strong correlation between the two energies, indicating that the idea that disordering is related to structural degeneracy still holds.
sites (swapping cations between the and sites does not change anything since these reside on the inversion-invariant octahedral sublattice). Each case leads to a potentially higher energy structure. Here, we correlate the SQS energy, in which all three cations are randomly distributed, with the energy of each of the inverted structures. The reasoning here is that if the next highest structure is much higher in energy, again the cations have a very strong preference for a specific ordering. On the other hand, there are scenarios where some orderings are energetically degenerate and others are not, so we consider both cases. The results are highlighted in Fig. 5. Again, we see a strong correlation between the disordering and inversion energies. This correlation is reasonably strong for both inverted structures. Interestingly, if we examine the lower energy inverted structure (labeled “Mid” in Fig. 5), the correlation is perhaps better but off of the Vegard's law trend line. In contrast, the higher energy inverted structure is closer to the trend line but exhibits more scatter, reminiscent of the single spinels. At this time, we do not have a reason for this behavior. While there are intuitive reasons for the data to be below the line, as it was for pyrochlore, there is no obvious reason for it to be higher, except that the mixing of all three cations in the double spinel structure does not correspond to any one inverse structure. That is, we cannot view the disorder state for a three component material as an interpolation between an inverted structure in which only two cations are swapped. Even so, we see there is a strong correlation between the two energies, indicating that the idea that disordering is related to structural degeneracy still holds.
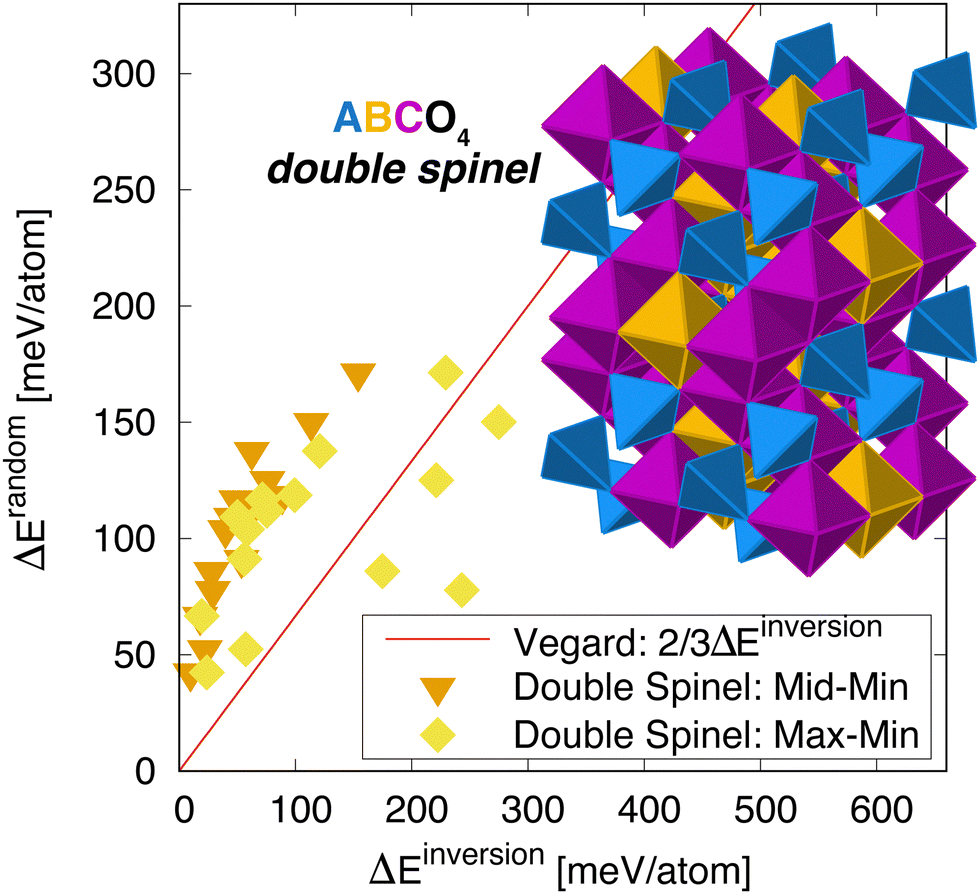 | ||
| Fig. 5 Energy to randomize the cation distribution versus inverting it for double spinels (the full list is given in Table 1). The line is an estimate from 2/3 linear interpolation between the ground state and the inverted structure – referred to here as “Vegard”. Two sets of data are shown, one in which the inversion energy is measured for the lower energy inverted structure relative to the ground state, labeled ‘Mid-Min’ and another in which the higher energy inverted structure is considered, labeled ‘Max–Min’. The inset shows the cation ordering within the double spinel structure. | ||
Thus, we find an overall strong correlation between the energy to disorder a complex oxide and the energy to invert the cations. This relationship holds across chemistries and for several different crystal structures, though there are clear exceptions to the rule as demonstrated by the titanate pyrochlores. However, the near-universality of this relationship suggests that the inversion energy can be used as a good proxy for the disordering energy and can be used as a metric to screen compounds that are more or less easily disordered.
The quest to find new multi-component oxides that do or do not disorder, including high entropy oxides,32,33 has led to the development of new descriptors to quickly assess that propensity. For example, the entropy forming ability (EFA) describes how the energy distribution of a range of random structures relates to likelihood of disordering the material.34 If the distribution is narrow – indicating that structures tend to have the same energy – the material is easy to disorder and conversely, if the distribution is broad, a disordered state is less likely to form. Our metric – the inversion energy – is in some sense a limit of this concept, where we identify one structure that describes the energetic degeneracy of different cation arrangements and thus the propensity for disorder. Of course, calculating the energy of one structure is more computationally efficient than doing the same for an ensemble of structures. The EFA has been quantitatively correlated with experimental synthesizability for multi-component materials and thus has been shown to be predictive and generalizable. Further work is needed to extend the inversion energy to multi-component systems, but the fact that it correlates with disordering for the three-component double spinels suggests this is possible.
The titanate pyrochlores are the clear exception to the trends discussed here. In the inverted pyrochlore structure, titanium cations are forced into 8-fold coordination environments. This is a particularly unstable configuration, as titanium cannot accommodate coordinations greater than 6.35,36 That fact is reflected in our observation that the titanate pyrochlores, in the inverted structure, see greater levels of oxygen relaxation than the other pyrochlore compounds. This highlights a limit to the use of inverted structures to understand disordering: if the inverted structure represents an extremely unstable configuration, it will not correlate with the random structure to any great extent. Further, the disordering energies for these compounds are not so high, a reflection of the greater cation relaxation in the random titanate pyrochlore structures than in the other pyrochlore chemistries. That is, these structures are so unstable that they rearrange to a higher degree. The cations do not move in the ordered pyrochlore structures (both normal and inverted), a consequence of the high symmetry of the cation arrangement.
This difference in behavior is further illustrated by comparing the radial distribution functions (RDF) for oxygen in two pyrochlores – Dy2Zr2O7 (DZO) and Dy2Ti2O7 (DTO). This comparison, shown in Fig. 6, reveals that, for DZO, the RDF for oxygen in the random structure spans the distances observed for the normal and inverse structures. This supports the premise that the random structure lies, in some sense, between these two extreme cation ordering cases. However, for DTO, this is not true. The random structure exhibits oxygen distances that lie outside of the range spanned by the normal and inverse structures, suggesting that there is much more relaxation in this case, in agreement with the results in Fig. 4. This is further demonstrated by comparing the structures, shown in the insets, in which both the oxygen and the cations exhibit significantly more relaxation for DTO than for DZO. Together, this suggests that, for some compounds, there are further considerations that are not captured by the two ordered endmembers.
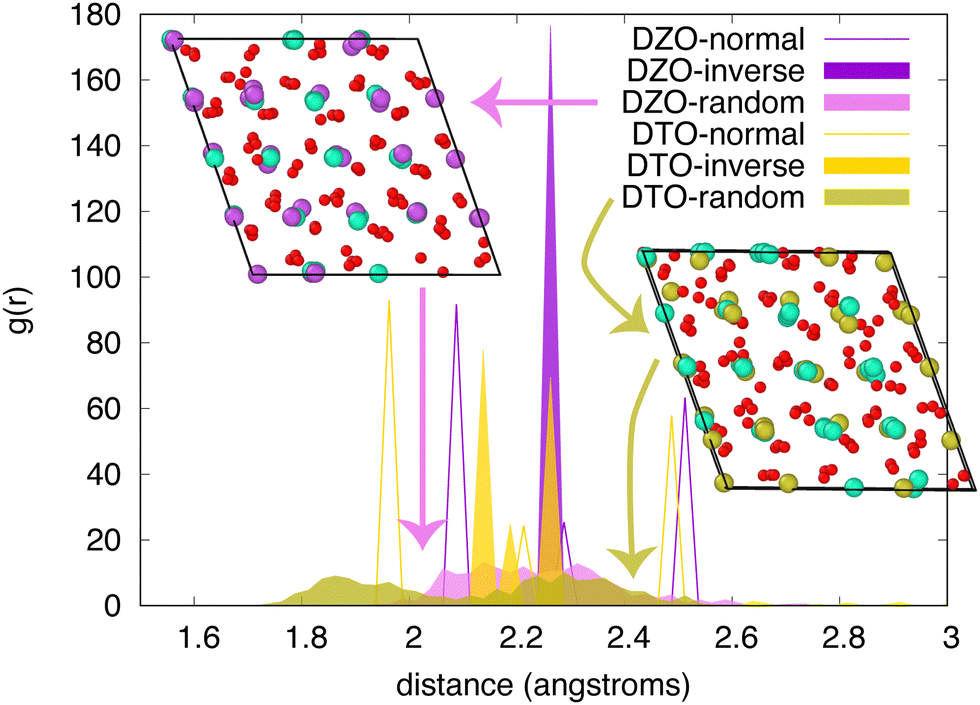 | ||
| Fig. 6 Radial distribution function of oxygen in Dy2Zr2O7 (DZO) and Dy2Ti2O7 (DTO) for the normal, inverse, and random structures. The insets highlight the atomic structure of the random case for both compounds. In these structures, red spheres represent oxygen, cyan spheres are Dy, purple spheres are Zr, and yellow spheres are Ti. | ||
| Structure | Compound | ΔEinversion (meV) | ΔErandom (meV) | Structure | Compound | ΔEinversion (meV) | ΔErandom (meV) |
|---|---|---|---|---|---|---|---|
| Pyrochlore | Nd2Ti2O7 | 636.218 | 71.509 | Single Spinel | (Cd)Al2O4 | 99.578 | 89.872 |
| Pyrochlore | Sm2Ti2O7 | 616.297 | 76.517 | Single spinel | (Al)AlNiO4 | 69.076 | 39.301 |
| Pyrochlore | Gd2Ti2O7 | 590.881 | 83.939 | Single spinel | (Cd)CdGeO4 | 45.058 | 91.395 |
| Pyrochlore | Dy2Ti2O7 | 563.114 | 77.045 | Single spinel | (Co)CoMnO4 | 142.862 | 31.909 |
| Pyrochlore | Er2Ti2O7 | 534.207 | 72.820 | Single spinel | (Mg)Fe2O4 | 37.610 | 44.152 |
| Pyrochlore | Nd2Hf2O7 | 430.417 | 102.402 | Single spinel | (Ga)GaMgO4 | 11.119 | 22.480 |
| Pyrochlore | Sm2Hf2O7 | 389.320 | 81.951 | Single spinel | (Ga)GaNiO4 | 102.938 | 42.237 |
| Pyrochlore | Gd2Hf2O7 | 344.801 | 58.909 | Single spinel | (In)InMgO4 | 11.580 | 25.185 |
| Pyrochlore | Dy2Hf2O7 | 303.076 | 36.601 | Single spinel | (Zn)ZnGeO4 | 59.471 | 97.927 |
| Pyrochlore | Er2Hf2O7 | 263.306 | 14.675 | Single spinel | (Mg)Al2O4 | 27.888 | 43.992 |
| Pyrochlore | Nd2Sn2O7 | 572.115 | 160.002 | Single spinel | (Co)CoGeO4 | 15.610 | 89.896 |
| Pyrochlore | Sm2Sn2O7 | 507.060 | 133.941 | Single spinel | (Cd)Cr2O4 | 269.338 | 204.265 |
| Pyrochlore | Gd2Sn2O7 | 506.935 | 141.496 | Single spinel | (Ni)Cr2O4 | 158.499 | 117.782 |
| Pyrochlore | Dy2Sn2O7 | 473.168 | 121.541 | Single spinel | (Cd)Fe2O4 | 103.084 | 88.853 |
| Pyrochlore | Er2Sn2O7 | 439.986 | 114.330 | Single spinel | (Ge)Mg2O4 | 12.458 | 111.995 |
| Pyrochlore | Nd2Zr2O7 | 385.260 | 67.506 | Single spinel | (Ge)Ni2O4 | 85.527 | 136.653 |
| Pyrochlore | Sm2Zr2O7 | 342.526 | 44.795 | Single spinel | (Cd)Rh2O4 | 288.803 | 183.534 |
| Pyrochlore | Gd2Zr2O7 | 296.789 | 32.212 | Single spinel | (Mn)Rh2O4 | 200.974 | 129.177 |
| Pyrochlore | Dy2Zr2O7 | 254.479 | 8.765 | Single spinel | (Mg)Ti2O4 | 69.928 | 33.880 |
| Pyrochlore | Er2Zr2O7 | 214.367 | -12.609 | Single spinel | (Mn)Ti2O4 | 75.765 | 53.570 |
| Perovskite | LaNdO3 | 35.542 | 3.331 | Double spinel | (Ti)MgFeO4 | 9.703 | 42.376 |
| Perovskite | LaEuO3 | 98.533 | 31.926 | Double spinel | (Mn)TiCoO4 | 17.959 | 66.702 |
| Perovskite | LaTbO3 | 137.351 | 55.429 | Double spinel | (Ga)MgAlO4 | 22.078 | 52.410 |
| Perovskite | LaErO3 | 188.698 | 86.556 | Double spinel | (Co)MnRhO4 | 28.870 | 77.806 |
| Perovskite | LaTmO3 | 204.272 | 97.279 | Double spinel | (In)MgTiO4 | 53.651 | 91.200 |
| Perovskite | LaLuO3 | 230.608 | 118.968 | Double spinel | (Zn)GeMgO4 | 44.652 | 109.233 |
| Perovskite | NdLuO3 | 209.607 | 98.914 | Double spinel | (Cd)GeMgO4 | 49.203 | 116.873 |
| Perovskite | EuLuO3 | 149.646 | 56.988 | Double spinel | (Zn)GeCoO4 | 39.702 | 103.955 |
| Perovskite | TbLuO3 | 108.802 | 35.528 | Double spinel | (Cd)GeCoO4 | 61.680 | 111.977 |
| Perovskite | ErLuO3 | 53.908 | 9.536 | Double spinel | (Zn)GeNiO4 | 62.399 | 137.645 |
| Perovskite | TmLuO3 | 35.443 | 1.047 | Double spinel | (Al)NiCrO4 | 27.720 | 86.021 |
| Perovskite | NdTmO3 | 179.743 | 80.703 | Double spinel | (Ga)NiCrO4 | 75.841 | 125.120 |
| Double spinel | (Cd)FeAlO4 | 88.568 | 118.636 | ||||
| Double spinel | (Cd)CrAlO4 | 113.474 | 150.112 | ||||
| Double spinel | (Cd)RhAlO4 | 154.046 | 171.383 |
Finally, cation disordering is also related to the amorphization resistance of a complex oxide under irradiation,2,37,38 though this relationship is sensitive to the crystal structure itself.39 Thus, having a metric to identify compounds which easily disorder will also provide insight into which compounds exhibit higher resistance to amorphization. At the same time, we also expect that this same metric can be used to identify compounds which can more easily be synthesized or grown in a thin film geometry, as compounds that are harder to amorphize might also be easier to grow in crystalline form, though they may be also harder to achieve in an ordered state. Thus, this proposed metric can be used to potentially screen for materials that are more or less susceptible to amorphization.
5. Conclusion
In conclusion, we have found that the energy to invert the cation structure in a complex oxide directly correlates to the energy to randomize the cations. That is, the inverted structure can be viewed as a limiting case with the random structure falling in between according to Vegard's law. Even in cases where other factors such as magnetism complicate the picture, there is still a trend that can be used to screen materials based on their propensity for disordering. This metric complements other approaches in the literature to identify compounds in which disordering cations is energetically favorable or not, providing new avenues for screening materials.Author contributions
Research was conceived by all authors. Calculations were performed by V. K. All contributors discussed and analyzed the results. All authors contributed to the writing of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the U.S. Department of Energy, Office of Science, Basic Energy Sciences, Materials Sciences and Engineering Division. Los Alamos National Laboratory is operated by Triad National Security, LLC, for the National Nuclear Security Administration of U.S. Department of Energy (Contract No. 89233218CNA000001).References
- L. Minervini, R. W. Grimes and K. E. Sickafus, J. Am. Ceram. Soc., 2000, 83, 1873–1878 CrossRef CAS.
- K. Sickafus, L. Minervini, R. Grimes, J. Valdez, M. Ishimaru, F. Li, K. McClellan and T. Hartmann, Science, 2000, 289, 748–751 CrossRef CAS.
- J. Lian, J. Chen, L. Wang, R. C. Ewing, J. M. Farmer, L. A. Boatner and K. Helean, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 134107 CrossRef.
- H. L. Tuller, J. Phys. Chem. Solids, 1994, 55, 1393–1404 CrossRef CAS.
- P. F. Ndione, Y. Shi, V. Stevanovic, S. Lany, A. Zakutayev, P. A. Parilla, J. D. Perkins, J. J. Berry, D. S. Ginley and M. F. Toney, Adv. Funct. Mater., 2014, 24, 610–618 CrossRef CAS.
- C. R. Kreller, J. A. Valdez, T. G. Holesinger, J. Morgan, Y. Wang, M. Tang, F. H. Garzon, R. Mukundan, E. L. Brosha and B. P. Uberuaga, J. Mater. Chem. A, 2019, 7, 3917–3923 RSC.
- C. R. Kreller and B. P. Uberuaga, Curr. Opin. Solid State Mater. Sci., 2021, 25, 100899 CrossRef CAS.
- C. Toher, C. Oses, M. Esters, D. Hicks, G. N. Kotsonis, C. M. Rost, D. W. Brenner, J.-P. Maria and S. Curtarolo, MRS Bull., 2022, 47, 194–202 CrossRef CAS.
- A. Abdellahi, A. Urban, S. Dacek and G. Ceder, Chem. Mater., 2016, 28, 3659–3665 CrossRef CAS.
- A. Harbi, H. Moutaabbid, Y. Li, C. Renero-Lecuna, M. Fialin, Y. Le Godec, S. Benmokhtar and M. Moutaabbid, J. Alloys Compd., 2019, 778, 105–114 CrossRef CAS.
- A. Zunger, S.-H. Wei, L. Ferreira and J. E. Bernard, Phys. Rev. Lett., 1990, 65, 353 CrossRef CAS PubMed.
- C. Jiang and B. P. Uberuaga, Phys. Rev. Lett., 2016, 116, 105501 CrossRef PubMed.
- S.-H. Wei and S. Zhang, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 045112 CrossRef.
- C. Jiang, K. E. Sickafus, C. R. Stanek, S. P. Rudin and B. P. Uberuaga, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 024203 CrossRef.
- C. Jiang, C. Stanek, K. Sickafus and B. Uberuaga, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 104203 CrossRef.
- W.-J. Yin, Y. Yan and S.-H. Wei, J. Phys. Chem. Lett., 2014, 5, 3625–3631 CrossRef CAS.
- V. Alexandrov, N. Grønbech-Jensen, A. Navrotsky and M. Asta, J. Nucl. Mater., 2014, 444, 292–297 CrossRef CAS.
- D. Music, S. Konstantinidis and J. M. Schneider, J. Phys.: Condens. Matter, 2009, 21, 175403 CrossRef PubMed.
- C. Stanek, C. Jiang, B. Uberuaga, K. Sickafus, A. Cleave and R. Grimes, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 174101 CrossRef.
- V. Kocevski, G. Pilania and B. P. Uberuaga, Mater. Des., 2023, 228, 111830 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49, 14251–14269 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- B. P. Uberuaga and K. E. Sickafus, Comput. Mater. Sci., 2015, 103, 216–223 CrossRef CAS.
- A. van de Walle, P. Tiwary, M. de Jong, D. Olmsted, M. Asta, A. Dick, D. Shin, Y. Wang, L.-Q. Chen and Z.-K. Liu, Calphad, 2013, 42, 13–18 CrossRef CAS.
- J. M. Farmer, L. A. Boatner, B. C. Chakoumakos, M.-H. Du, M. J. Lance, C. J. Rawn and J. C. Bryan, J. Alloys Compd., 2014, 605, 63–70 CrossRef CAS.
- G. Pilania, V. Kocevski, J. A. Valdez, C. R. Kreller and B. P. Uberuaga, Commun. Mater., 2020, 1, 1–11 CrossRef.
- C. M. Rost, E. Sachet, T. Borman, A. Moballegh, E. C. Dickey, D. Hou, J. L. Jones, S. Curtarolo and J.-P. Maria, Nat. Commun., 2015, 6, 8485 CrossRef CAS.
- B. Jiang, C. A. Bridges, R. R. Unocic, K. C. Pitike, V. R. Cooper, Y. Zhang, D.-Y. Lin and K. Page, J. Am. Chem. Soc., 2020, 143, 4193–4204 CrossRef PubMed.
- P. Sarker, T. Harrington, C. Toher, C. Oses, M. Samiee, J.-P. Maria, D. W. Brenner, K. S. Vecchio and S. Curtarolo, Nat. Commun., 2018, 9, 4980 CrossRef PubMed.
- G. Sattonnay and R. Tétot, J. Phys.: Condens. Matter, 2014, 26, 055403 CrossRef CAS PubMed.
- G. Pilania, B. Puchala and B. P. Uberuaga, npj Comput. Mater., 2019, 5, 7 CrossRef.
- K. E. Sickafus, R. W. Grimes, J. A. Valdez, A. Cleave, M. Tang, M. Ishimaru, S. M. Corish, C. R. Stanek and B. P. Uberuaga, Nat. Mater., 2007, 6, 217–223 CrossRef CAS PubMed.
- G. Pilania, K. R. Whittle, C. Jiang, R. W. Grimes, C. R. Stanek, K. E. Sickafus and B. P. Uberuaga, Chem. Mater., 2017, 29, 2574–2583 CrossRef CAS.
- B. P. Uberuaga, M. Tang, C. Jiang, J. A. Valdez, R. Smith, Y. Wang and K. E. Sickafus, Nat. Commun., 2015, 6, 8750 CrossRef CAS PubMed.
Footnotes |
| † Present address: Michigan Institute for Computational Discovery and Engineering (MICDE), University of Michigan, Ann Arbor, MI, USA. |
| ‡ Present address: GEProbabilistic Design and Materials Informatics Group, GE Research, Niskayuna, NY 12309, USA. |
| This journal is © the Owner Societies 2023 |