
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe hydration of an oxy-polycyclic aromatic compound: the case of naphthaldehyde†
Jordan A.
Claus
 a,
Celina
Bermúdez
ab,
Valérie
Vallet
a,
Laurent
Margulès
a and
Manuel
Goubet
*a
a,
Celina
Bermúdez
ab,
Valérie
Vallet
a,
Laurent
Margulès
a and
Manuel
Goubet
*a
aUniv. Lille, CNRS, UMR 8523 – PhLAM – Physique des Lasers, Atomes et Molécules, F-59000 Lille, France. E-mail: jordan.claus@univ-lille.fr; valerie.vallet@univ-lille.fr; laurent.margules@univ-lille.fr; manuel.goubet@univ-lille.fr
bDepartamento de Química Física y Química Inorgánica, Facultad de Ciencias – I.U. CINQUIMA, Universidad de Valladolid, Valladolid 47011, Spain. E-mail: celina.bermudez@uva.es
First published on 23rd August 2023
Abstract
The study of the intermolecular interactions of polycyclic aromatic compounds, considered as important pollutants of the Earth's atmosphere since they are emitted by the partial combustion of fuels, is essential to understand the formation and aging of their aerosols. In this study, the hydration of α-naphthaldehyde and β-naphthaldehyde isomers was investigated through a combination of Fourier transform microwave spectroscopy and quantum chemical calculations. Monohydrate structures were observed experimentally for both isomers, with two hydrate structures observed for β-naphthaldehyde and only one for α-naphthaldehyde, consistent with computational predictions. Analysis of the monohydrate structures indicated that the β-isomer exhibits higher hydrophilicity compared to the α-isomer, supported by electronic densities, hydration energies, and structural considerations. Further computational calculations were conducted to explore the planarity of the naphthaldehyde hydrates. Different levels of theory were employed, some of these revealing slight deviations from planarity in the hydrate structures. Low-frequency out-of-plane vibrational modes were examined, and the inertial defect was used to assess the planarity of the hydrates. The results suggested that the hydrates possess a predominantly planar structure, in agreement with the highest level of computational calculations and the absence of c-type transitions in the experimental spectra. Additionally, calculations were extended to dihydrate structures by attaching two water molecules to the naphthaldehyde isomers. The most stable dihydrate structures were predicted to be combinations of the observed monohydrate positions. However, experimental observation of the most stable dihydrate structures was challenging due to their very low vapour pressure, calling for complementary experiments using laser ablation nozzles.
1 Introduction
Volatile organic compounds (VOCs) are emitted in the atmosphere by biogenic and anthropogenic sources,1 and have a tremendous impact such as disrupting the chemical and physical properties of the atmosphere, as well as on human health. Among VOCs, polycyclic aromatic compounds (PACs) are considered as a major class of pollutants of the Earth's atmosphere,2 which are also known as carcinogenic agents.3,4 Within this class, polycyclic aromatic hydrocarbons (PAHs) have received the most attention with emissions estimated to be between 331 and 818 Gg per year.5 However, the importance of oxygenated PAH derivatives (oxy-PAHs) shall not be neglected as they may be almost as abundant as their parent PAHs.6,7 Both species are emitted by incomplete combustion processes such as residential heating and vehicle exhausts.8,9 In addition, oxy-PAHs are secondary products from atmospheric reactions on PAHs.6,7PACs are precursors of the so-called secondary organic aerosols (SOAs) which are an intricate mixture composed of molecular aggregates that play a major role in the atmosphere properties: scattering the solar radiation, influencing the Earth's radiative balance and acting as condensation nuclei. The formation SOAs in the atmosphere is a complicated process involving VOCs, ozonolysis, reactions with OH radicals and nitrogen oxides (NOx)10 as well as photolysis.11 Factors like humidity and the nature of the precursor influence in the generation of the SOAs. On one side, the relative humidity might alter the chemical composition of the SOAs12 and increase the aerosol production.13 On the other side, the effect of the precursor is mainly observed in the amount of SOAs formed; when the precursor is an oxygenated aromatic compound, the aerosol production yield is enhanced in comparison to other aromatic species such as reduced or heterocyclic aromatic species,14 most probably because oxy-PAHs vapour pressures are about 10 times lower than their corresponding parent PAH species.7,15
Since water is an abundant solvent in the atmosphere present under different states, investigating the hydration of precursors (e.g. oxy-PAHs) is a milestone for understanding the formation of SOAs and all processes connected with this phenomenon. Indeed, intermolecular interactions in SOAs are essential and need to be unveiled through molecular-level investigations. Hydrogen bonds (HB) are relevant for the stabilization of molecular complexes as may occur between precursors and water molecules. The interest in the hydration of PACs is very recent, so that the information remains scarce: investigations on the acenaphthene–water,16 anthracene–water17 and pyrene–water18 complexes conformational landscape has been made recently. Interestingly, there can be up to four water molecules in polar–π interactions (located above the aromatic rings) and furthermore, water complexes retain virtually the same structure as the free water cluster. However, in the case of a heterocyclic PAC (i.e. containing one or more atom other than a carbon in its structure), the possible anchoring sites are very varied depending on the solvent.19 When water is the solvent, it seems that the HB is preferentially formed with the electronegative heteroatom. This behaviour has been observed on a heterocyclic oxy-PAH (dibenzofuran),20 a partially aromatic oxy-PAH (naphthoquinone),21 a cyano-PAH22 and a heterocyclic nitro-PAH.23 Nonetheless, an interesting slightly different case deserves investigation: the hydration of a homocylic oxy-PAH which contains a functional group attached to fully aromatic rings. This raises a question about the interaction between the molecule and water: will the water be located above the aromatic rings, similarly to the pure PAH water complexes, or preferentially interacts with the functional group similarly to with hetero-PAH?
Besides its relevance to atmospheric sciences, the exponential growth in the number of publications dealing with hydrogen bonding shows at least that the importance of studying the hydrogen bond is now well established.24 The particular properties of this type of non-covalent interaction, in terms of structure (asymmetry, non-linearity) and dynamics (strong vibrational couplings, predissociation), impose an experimental and theoretical treatment different from “standard” molecules. Nowadays, spectroscopic studies combining experiment and theory have become a standard. In particular, the spectroscopic observation of a sample in jet-cooled conditions allows to obtain experimental data directly comparable to the same theoretical data thanks to the isolation of the complex in an environment with little or no external interaction and lowering its internal energy to a few degrees Kelvin. It makes it possible to “calibrate” the theoretical approach on the experimental results, giving it sufficient confidence to use/discuss the purely theoretical physico-chemical results (molecular structure, energies, vibrational modes). The interconnections between theory and experiment take place at all stages. (i) Upstream for predictive purposes: the first obstacle in high resolution spectroscopy is undoubtedly the analysis of the spectrum. Indeed, the identification of the observed signals can only be done by comparison with an a priori predictive spectrum, which can only be generated from molecular parameters calculated as accurately as possible in the absence of a prior experimental study. (ii) In parallel for a reciprocal support: higher-level calculations, which can be calibrated on available intermediate experimental results, are necessary in more complex cases to permit a thorough analysis, such as the study of excited states, the exploration of a conformational landscape or the presence of large amplitude motions involving the multiplication of signals by tunnel effect. (iii) Downstream in order to estimate the relevance of the theoretical molecular parameters by comparison with the experimental data: once the spectroscopic study has been finalized, the comparison between available theoretical and experimental data makes it possible to calibrate the calculations by pointing out the most appropriate approach. Then, the coherence between theory and experiment allows to use these calculations with enough confidence to interpret the observations.
In this paper, we report the study of naphthaldehyde–water complexes using rotational spectroscopy supported by quantum chemistry calculations. Naphthaldehyde (C11H8O), already studied in a previous paper,25 is a homocylic oxy-PAH containing two 6-membered rings with an aldehyde group attached to one of the rings. This results in two non-isoenergetic isomers (α and β) and two conformations (cis and trans) for each isomer, i.e. four structures are possible. Naphthaldehyde is of atmospheric interest given that it has been shown to be among the predominant substances of total oxy-PAHs measured on urban sites.2
2 Methods
2.1 Experimental setup
The pure rotational spectra of α- and β-naphthaldehyde–water complexes were recorded in the 2–20 GHz frequency range using supersonic jet cavity-based Fourier transform microwave (FP-FTMW) spectrometers already described elsewhere.26,27 The two samples, α- and β-naphthaldehyde, were purchased from Merck and TCI chemicals and used without further purification. The reservoir28 containing the sample was heated at 410 K to vaporize an optimal quantity of the naphthaldehyde. Liquid water was placed in a tube at room temperature before the reservoir. The carrier gas (neon or argon) was flown through at an absolute pressure of about 3 bar. Then, the mixture was injected at a frequency of 1.5 kHz through a Series 9 General Valve pinhole nozzle (1 mm diameter) connected to a vacuum Fabry–Perot cavity, creating a supersonic jet expansion. Molecules were polarized by 2 μs-duration microwave pulses. The Free Induction Decay (FID) signals were detected using a heterodyne detection at 30 MHz and digitized at a sampling rate of 120 MHz. A fast Fourier transform of the FID signal allowed to obtain the spectral domain signal, where transitions were observed as Doppler doublets due to the coaxial arrangement of the jet and the Fabry–Perot cavity. The frequency grid, linked to the number of data points, was set to 1.8 kHz representing the line accuracy.2.2 Theoretical calculations
First, a conformational search was made using molecular dynamic Monte-Carlo based-method (MCMM) with extended torsional sampling parameters, aiming to investigate all possible structures. In order to select the obtained structures, electronic structure calculations were performed using Gaussian16 rev C.01 software29 on the PhLAM laboratory high performance computing cluster. Equilibrium structures of each conformer were optimized using density functional theories (DFT), namely B3LYP30,31 and B9832, with respectively Pople split-valence triple-zeta basis set 6-311++G(d,p)33 and Dunning's correlation consistent basis set aug-cc-pVXZ (X = T, Q).34 Structures were also optimized ab initio using the second-order Møller–Plesset perturbation theory35 (MP2) with Dunning's correlation consistent triple zeta basis set aug-cc-pVTZ. DFT and MP2 geometry optimizations were done with the tight convergence option, the tight SCF convergence criterion and the ultrafine integral grid option. Afterward, harmonic frequencies calculations were performed to obtain the zero-point energy (ZPE) corrections, the harmonic vibrational frequencies and the quartic centrifugal distortion constants.Natural Bond Orbitals (NBO)36 analysis were performed with the NBO 7 program37 on the optimized geometries to give further information about the distribution of electronic density in atoms and bonds by transforming the delocalized electronic wave functions into optimized electron pairs, divided in Lewis- and non-Lewis-types. With the aim of unveiling intra- and intermolecular interactions such as HBs, Quantum Theory of Atoms in Molecules (QTAIM)38–40 were performed with AIMAll program41 together with Non-Covalent Interactions (NCI).42 Since a HB can be seen as a charge transfer between two atoms, the one acting as a proton donor and the other as an acceptor, its strength can be evaluated throughout the stabilization energy (i.e. E(2) in NBO calculations determined by second-order perturbation theory).
In order to quantify as accurately as possible the amount of energy involved in the intermolecular bonds (i.e. the hydration energies), basis set superposition error (BSSE) corrections were made using the counterpoise method43 on relevant optimized structures.
3 Results
3.1 Calculated conformational landscape
The results of the conformational search performed by the quantum chemical calculations indicate that water is attached to naphthaldehyde via HBs, acting either as proton donor or as proton acceptor, or by π interactions. All theoretical results from the calculations performed in this study agree qualitatively (see Table S1 of the ESI†). In order to evaluate the influence of long-range electron correlation effects on DFT values, tests calculations including London dispersion were performed at the B3LYP-D3/6-311++G(d,p) level on the two lowest energy conformers of the α-naphthaldehyde monohydrate. As a result, relative energy, structures and dipole moments get closer to the MP2 values than the ones from the “standard” B3LYP, what we interpret as a correction of DFT calculations without dispersion. Consequently, the following discussions are mostly based on the MP2/aug-cc-pVTZ results which are considered as the most reliable ones. All most favourable structures found have been classified into four groups as a function of the intermolecular bond types that stabilize the complexes (see Fig. 1). The first group denoted (I) corresponds to species where water is acting as a proton donor with the carbonyl oxygen and as a proton acceptor with the carbonyl hydrogen. The second group denoted (II) is similar to the first one since water both acts as proton donor and acceptor, but interacting with one hydrogen of the aromatic ring instead of the hydrogen of the aldehyde group. In the third group denoted (III), water only acts as proton acceptor, sharing one or its two lone electron pairs of the oxygen with hydrogen atoms of naphthaldehyde. The last group denoted (IV) contains all complexes where water is linked to naphthaldehyde via π interactions.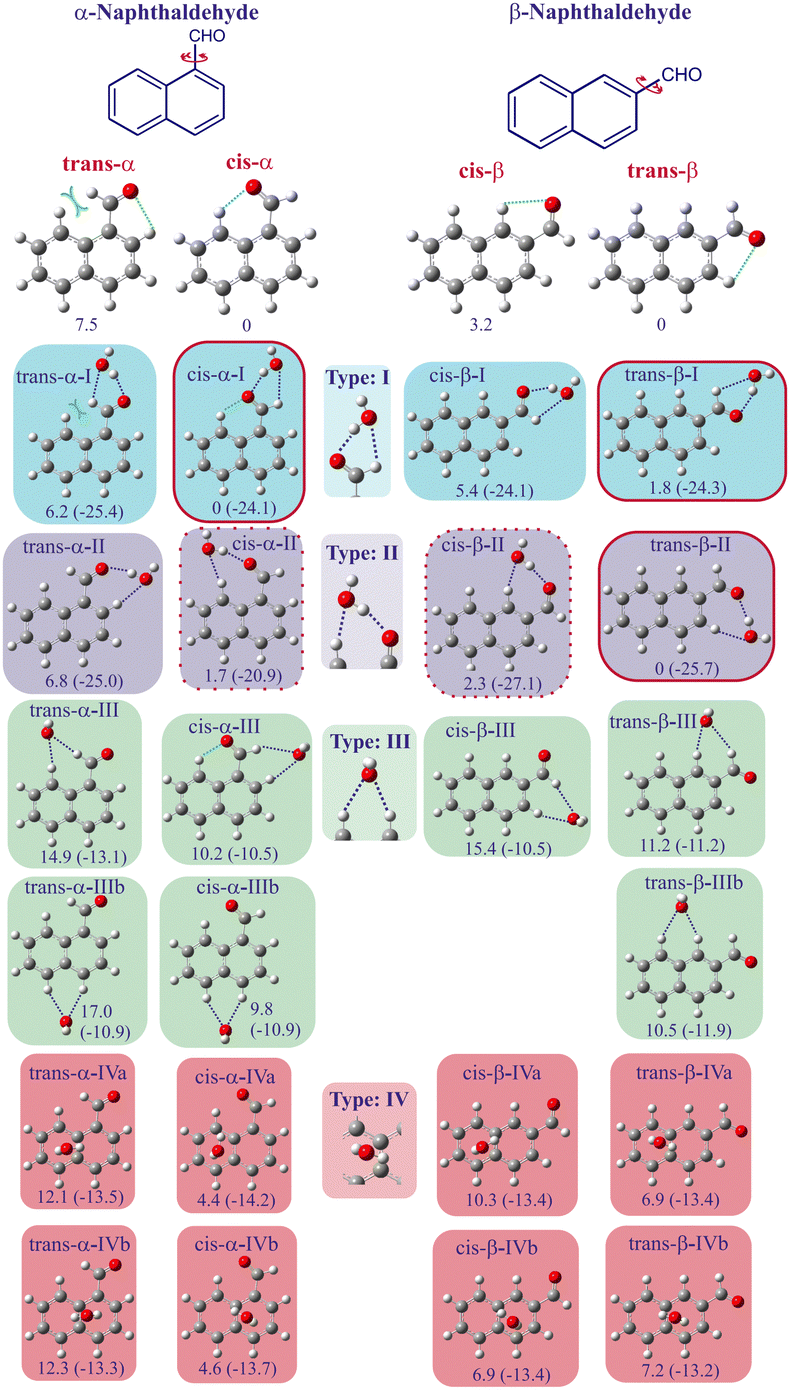 | ||
| Fig. 1 Conformational landscape of the naphthaldehyde–water complexes, classified by conformation and interaction types. Structures and energies are calculated at MP2/aug-cc-pVTZ level of theory, considering ZPE correction. Relative energies are calculated between conformations with the same isomer of the naphthaldehyde subunit (12 structures at the left for α and 11 structures at the right for β). Values are expressed in kJ mol−1. Values in parenthesis are the hydration energies (BSSE corrected). | ||
Concerning the α-naphthaldehyde monohydrate, the most stable structure, cis-α-I, belongs to the group I and is stabilized by two HBs between water and the carbonyl group. The next conformer in energy, cis-α-II, is around 1.73 kJ mol−1 higher in energy and is also a cis-α-naphthaldehyde conformer but linked to water in a type II interaction (water as a proton donor with the carbonyl oxygen and a proton acceptor with the closest hydrogen of the aromatic ring). All other conformers are too high in energy to be expected experimentally in our jet-cooled conditions.
For the β-naphthaldehyde monohydrate, calculations identified three possible structures within less than 2.5 kJ mol−1. The most stable structure, trans-β-II, belongs to the group II (whereas the most stable structure of the α-naphthaldehyde monohydrate belongs to the group I). The second structure, trans-β-I, 1.78 kJ mol−1 higher in energy, belongs to group I (thus sharing the same HBs as cis-α-I). The third structure, cis-β-II about 2.27 kJ mol−1 higher than trans-β-II, also belongs to the group II but its naphthaldehyde subunit is its less stable conformation cis.
The most probable anchoring sites for water on naphthaldehyde from these quantum chemistry calculations results will then serve to guide the experimental searches for hydrates signals. Based on the above energetic considerations, only type I and II complexes are expected to be observed under our jet-cooled conditions. In the following subsections, we describe the experimental results for each isomer of naphthaldehyde. Afterward, conformational preferences for the hydration of naphthaldehyde isomers will be discussed.
3.2 Monohydrate species of α-naphthaldehyde
The cis-α-I conformer is a nearly prolate (κ = −0.710) asymmetric rotor, with a strong dipole moment along the a-axis (|μa| = 3.2 D), a small dipole moment along the b-axis (|μb| = 0.7 D) and null along the c-axis, which is the axis perpendicular to the aromatic rings plane. The cis-α-II conformer is a slightly prolate (κ = −0.120) asymmetric rotor, with a strong dipole moment along the a-axis (|μa| = 3.0 D), a moderate dipole moment along the b-axis (|μb| = 1.4 D) and a small (|μc| = 0.5 D) along the c-axis, which is the axis perpendicular to the aromatic rings plane.Thus, scans around the predictions, based on the theoretical constants, for the a-type transitions of the R-branch were conducted to observe lines corresponding to the characteristic pattern of a nearly prolate species with groups of lines separated of about B+C. After optimizing the experimental conditions (carrier gas pressure, vaporization temperature and polarisation power), we obtained a set of low intense transitions that follows that pattern. Those lines were fitted to a semi-rigid rotor Watson Hamiltonian in the A-reduction in the Ir representation44 using the SPFIT/SPCAT software package.45 Then, an iterative process of predicting-assigning further lines was followed to include finally up to 112 a- and b-type transitions in the analysis (Jmax = 20, Ka,max = 5) reproduced at instrumental accuracy (RMS error of 1.6 kHz). Results of the fit are shown and compared to calculated values in Table 1 and the list of assigned transitions can be found in Table S8 of the ESI.† The good agreement between theoretical and experimental rotational constants clearly indicates that the observed monohydrate corresponds to the cis-α-I structure, for which coordinates are provided in Tables S2 and S3 of ESI.†
| cis-α-I | trans-β-II | trans-β-I | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Experiment | B98a | MP2a | Experiment | B98a | MP2a | Experiment | B98a | MP2a | |
| N is the number of transitions included into the fit.a With aug-cc-pVTZ basis set.b Fixed to MP2/aug-cc-pVTZ level of theory value. | |||||||||
| A/MHz | 1307.4471(12) | 1296.0 | 1317.5 | 1927.92684(68) | 1950.9 | 1946.1 | 2264.510(14) | 2369.6 | 2250.2 |
| B/MHz | 494.338636(70) | 489.7 | 501.9 | 403.000865(33) | 402.3 | 408.9 | 343.330363(75) | 334.7 | 349.3 |
| C/MHz | 358.918985(34) | 355.4 | 363.4 | 333.577804(18) | 333.6 | 337.9 | 298.269224(88) | 293.3 | 302.4 |
| Δ J/kHz | 0.02242(15) | 0.02396 | 0.01865 | 0.017218(43) | 0.015697 | 0.012197 | 0.02258(19) | 0.03516 | 0.01858 |
| Δ JK/kHz | 0.3014(18) | 0.5385 | 0.2170 | −0.04804(93) | −0.04238 | −0.01709 | −0.4773(81) | −0.8554 | −0.3486 |
| Δ K/kHz | −0.104(67) | −0.321 | −0.121 | 0.72(28) | 0.70 | 0.52 | [3.15]b | 7.50 | 3.15 |
| δ J/kHz | 0.005557(81) | 0.005352 | 0.004788 | 0.003776(28) | 0.003426 | 0.002673 | [0.004263]b | 0.007926 | 0.004263 |
| δ K/kHz | 0.1820(26) | 0.2794 | 0.1316 | 0.0659(46) | 0.0605 | 0.0500 | [0.05424]b | 0.10144 | 0.05424 |
| μ a/D | Observed | 4.2 | 3.2 | Observed | 3.8 | 3.1 | Observed | 4.6 | 3.7 |
| μ b/D | Observed | 0.8 | 0.7 | Observed | 1.4 | 1.4 | Not observed | 0.5 | 0.4 |
| μ c/D | Not observed | 0.0 | 0.0 | Not observed | 0.7 | 0.0 | Not observed | 0.0 | 0.0 |
| N | 112 | — | — | 100 | — | — | 36 | — | — |
| RMS/kHz | 1.6 | — | — | 0.7 | — | — | 2.3 | — | — |
Further explorations were conducted to find higher energy monohydrates, particularly cis-α-II, without success. In order to estimate our detection level, the signal-to-noise ratio (SNR) of the most intense line observed for cis-α-I  was measured. Then, the ratio between its calculated intensity and the one of the corresponding line for cis-α-II (both out of SPCAT program at a rotational temperature of 10 K) has been devided by the measured SNR. This gives an estimate of a factor by which the intensity of the strongest signal of cis-α-II must be multiplied in order not to come out of the noise (SNR = 1), i.e. it corresponds to an estimation of the maximum relative amount of cis-α-II in the jet which is estimated to 1% or less of cis-α-I. This result is discussed in Section 4.1.
was measured. Then, the ratio between its calculated intensity and the one of the corresponding line for cis-α-II (both out of SPCAT program at a rotational temperature of 10 K) has been devided by the measured SNR. This gives an estimate of a factor by which the intensity of the strongest signal of cis-α-II must be multiplied in order not to come out of the noise (SNR = 1), i.e. it corresponds to an estimation of the maximum relative amount of cis-α-II in the jet which is estimated to 1% or less of cis-α-I. This result is discussed in Section 4.1.
3.3 Monohydrate species of β-naphthaldehyde
The trans-β-II conformer is a nearly prolate (κ = −0.912) asymmetric rotor, with a strong dipole moment along the a-axis (|μa| = 3.1 D), a moderate dipole moment along the b-axis (|μb| = 1.4 D) and a null dipole moment along the c-axis, which is the axis perpendicular to the aromatic rings plane. The trans-β-I is also a nearly prolate (κ = −0.952) asymmetric rotor, with a strong dipole moment along the a-axis (|μa| = 3.7 D), a weak dipole moment along the b-axis (|μb| = 0.4 D) and a null dipole moment along the c-axis for the same reason. The cis-β-II conformer is a nearly prolate (κ = −0.685) asymmetric rotor, with a strong dipole moment along the a-axis (|μa| = 2.5 D), a medium-to-strong dipole moment along the b-axis (|μb| = 1.9 D) and a null dipole moment along the c-axis as well.Following the same steps as for the α-naphthaldehyde water complex, searches for the a-type transitions of the R-branch for trans-β-II structure, which is the most stable complex, were performed until the pattern of lines separated by B+C was found. Then, an iteration process of predicting-assigning further lines was conducted until the model reached the experimental accuracy. Up to 100 a- and b-type transitions were assigned (Jmax = 20, Ka,max = 4) with an RMS of 0.7 kHz. Results of the analysis are shown in Table 1 and the list of assigned transitions in Table S10 of the ESI.† The good agreement between theoretical and experimental rotational constants clearly indicates that the observed monohydrate corresponds to the trans-β-II structure, for which coordinates are provided in Tables S6 and S7 of ESI.†
Once the signals of the most stable trans-β-II have been assigned, searches for the two next monohydrates, namely trans-β-I and cis-β-II, were conducted following the same procedure. Lines corresponding to trans-β-I were found. The resulting simulation includes 36 a-type transitions (Jmax = 16, Ka,max = 3) with an RMS of 2.3 kHz. Results of the analysis are shown in Table 1 and the list of assigned transitions in Table S9 of the ESI.† Noticeably, a smaller number of transitions and only of a-type are included in the fit compared to the other complexes. Indeed, the combination of a lower levels population of this less stable conformation and the weaker dipole moment component along b-axis prevents the observation of more than the most intense a-type transitions of the R-branch. Coordinates of the corresponding structure are provided in Tables S4 and S5 of ESI.†
Further explorations were conducted to find the third possible monohydrate, cis-β-II, without success. Following the same procedure as for cis-α-II, the maximum relative amount of cis-β-II in the jet is estimated to 4% or less of trans-β-II.
4 Discussion
4.1 Hydration of naphthaldehyde
On the basis of these combined experimental and calculated results, the conformational preference of naphthaldehyde to be hydrated can be discussed.Looking at the panorama of monohydrate complexes found for naphthaldehyde (see Fig. 1), the relative energies between conformers depend on the precursor isomer (α, β) and on its conformation (cis, trans). Water will preferentially bind to naphthaldehyde via a combination of two intermolecular HBs, one where water acts as a proton acceptor and the other as a proton donor, corresponding to species of groups I and II. Relative stabilities within these two groups are discussed hereafter. Groups III (where water is doubly proton acceptor) and IV (π interactions) species are calculated too high in energy to be expected and are indeed not observed: below roughly 4 and 6.5 kJ mol−1 above the most stable structure depending on the isomer, only groups I and II structures are present. For all structures of the naphthaldehyde unit, group III monohydrates are by far the highest in energy (>10 kJ mol−1) so that water acting as a proton acceptor through only its oxygen atom is the less probable hydration process. It is confirmed by the fact that this group displays the lowest values of binding energies (<−12 kJ mol−1). Group IV monohydrates are more stable than Group III but are still less probable than group I and II when the naphthaldehyde unit is in its most stable conformation. In any case, all group IV structures are at least 4 kJ mol−1 higher in energy than the most stable ones. Here again, it is confirmed by the fact that this group displays binding energy values almost 2 times smaller than those of groups I and II. These results contrast with the observed species for the monohydrate complexes of pure PAHs,16 where the complexes are mainly stabilized by π interactions. Therefore, it seems obvious that the presence of an electronegative functional group alters the preference of the intermolecular interactions: the solvent molecule (water here) is not anymore preferentially placed above the aromatic plane but, instead, is placed within the aromatic plane around the functional group.
Within group I, the energy difference between cis-α-I and trans-α-I (6.23 kJ mol−1) is relatively close to that found between the two conformations of bare α-naphthaldehyde (7.54 kJ mol−1).25 This is even more evident for the β-structures of this group (3.59 kJ mol−1 compared to 3.23 kJ mol−1). It is in agreement with the roughly equivalent values of the binding energies within this group (around −24 kJ mol−1). This may suggest that the stabilization of these monohydrates is mostly due to the conformational landscape of the bare precursors. This is not the same for group II, where energy differences between hydrates are 5.11 kJ mol−1 and 2.27 kJ mol−1 for α and β isomer, respectively, suggesting that the stabilization process involved within hydration is affecting more the higher energy conformers than the most stable ones of the precursors, i.e. intermolecular HBs are stronger when water binds to the less stable conformation. It is clear for the both isomers where the binding energy in trans-α-II is 4 kJ mol−1 larger than in cis-α-II while it is 1.4 kJ mol−1 larger in cis-β-II than in trans-β-II. Such an effect can go up to a structural modification where the conformation of the flexible parent molecule is not the most stable one within the most stable hydrate.46 However, in the present cases, the naphthaldehyde subunits are rigid enough to keep their most stable conformations in the most favourable monohydrates.
Finally, the experimental results call for caution regarding the calculation methods.21 In the case of β-naphthaldehyde where two monohydrates are predicted within less than 2 kJ mol−1, namely trans-β-I and trans-β-II, experiments clearly point toward the latter being the most stable structure. This is in agreement with the prediction at the MP2/aug-cc-pVTZ level. Although the energy difference between these two conformers changes significantly from MP2 to B98 levels (1.79 kJ mol−1 and 0.24 kJ mol−1, respectively), the agreement is still qualitatively good with experiments. However, the stability order is reversed with an even more reduced energy difference (less than −0.12 kJ mol−1) at the B3LYP/aug-cc-pVTZ level. Therefore, the use of several quantum chemical methods, including ab initio if computational cost permits it, seems mandatory to check the coherence of the predictions.
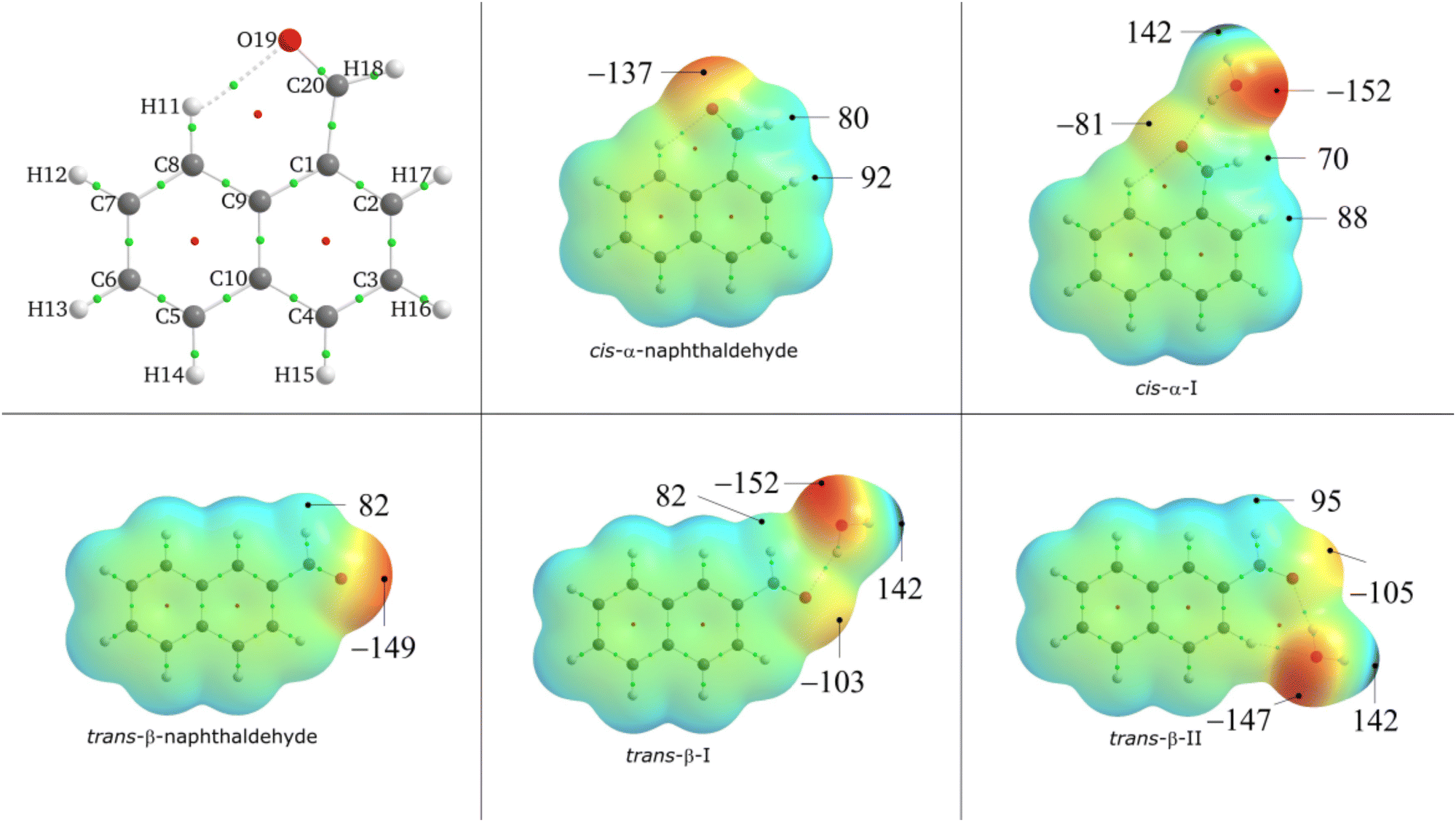 | ||
| Fig. 2 Molecular electrostatic potentials mapped on the 0.001Eh isodensity surface calculated at the MP2/aug-cc-pVQZ level of theory for cis-α-naphthaldehyde, trans-β-naphthaldehyde and their complexes. Values are expressed in kJ mol−1. | ||
The monohydration energy of cis-α-naphthaldehyde, corrected from BSSE, is estimated to −24.1 kJ mol−1 in cis-α-I. The monohydration energy in the next conformer cis-α-II is estimated to −20.9 kJ mol−1. This relatively large energy difference of 3.2 kJ mol−1 indicates that the second hydrate is less likely to be formed, which is in agreement with the experimental observation of the cis-α-I only.
Electronic densities, hydration energies and structural considerations all tend to suggest that β-naphthaldehyde would be more hydrophilic than α-naphthaldehyde.
4.2 Conformer selective expansion conditions
As already observed,47 the most favourable trans-β-II is observed with neon and argon as carrier gases whereas the slightly higher in energy trans-β-I is observed only with neon (see Fig. 3). In such a case, a relaxation effect in the supersonic expansion is considered at first. Indeed, the higher is Z of the carrier gas, the more efficient is the relaxation within the conformational landscape. However, relaxation occurs only if the interconversion barrier between conformers is low enough, typically a few hundreds of wavenumbers.48 Calculations were performed in order to find the transition state (TS) between the two conformers and determine the barrier height. Quadratic synchronous transit (QST)49 as implemented in Gaussian16 failed to converge to a TS structure, most probably because interconversion involves two or more internal motions, preventing the calculation of a fully relaxed barrier height. Several attempts on relaxed coordinate scans were made on different coordinates (C20–O19–O22 in-plane, for both structures; C2–C20–O19–O22 out-of-plane) and calculation methods (DFT, ab initio), in order to determine the barrier, also without success. Therefore, only an estimation of the barrier (unrelaxed) can be given, which oscillates between 5.4 and 7.9 kJ mol−1 for the first coordinates and between 4.8 and 6.5 kJ mol−1 for the second coordinates. However, those values need to be taken with caution since, as already mentioned, interconversion may involve two or more internal motions. These barrier values are in contradiction with a conformational relaxation within a supersonic expansion.48 Then, a more probable explanation is the better ability of Ne to form high energy HB complexes than Ar. This might be due to either a better energy transfer to Ne than Ar within the three-body collisions process stabilizing complexes at the nozzle exit or to the solvation of precursors by Ar50 preventing the formation of high energy complexes.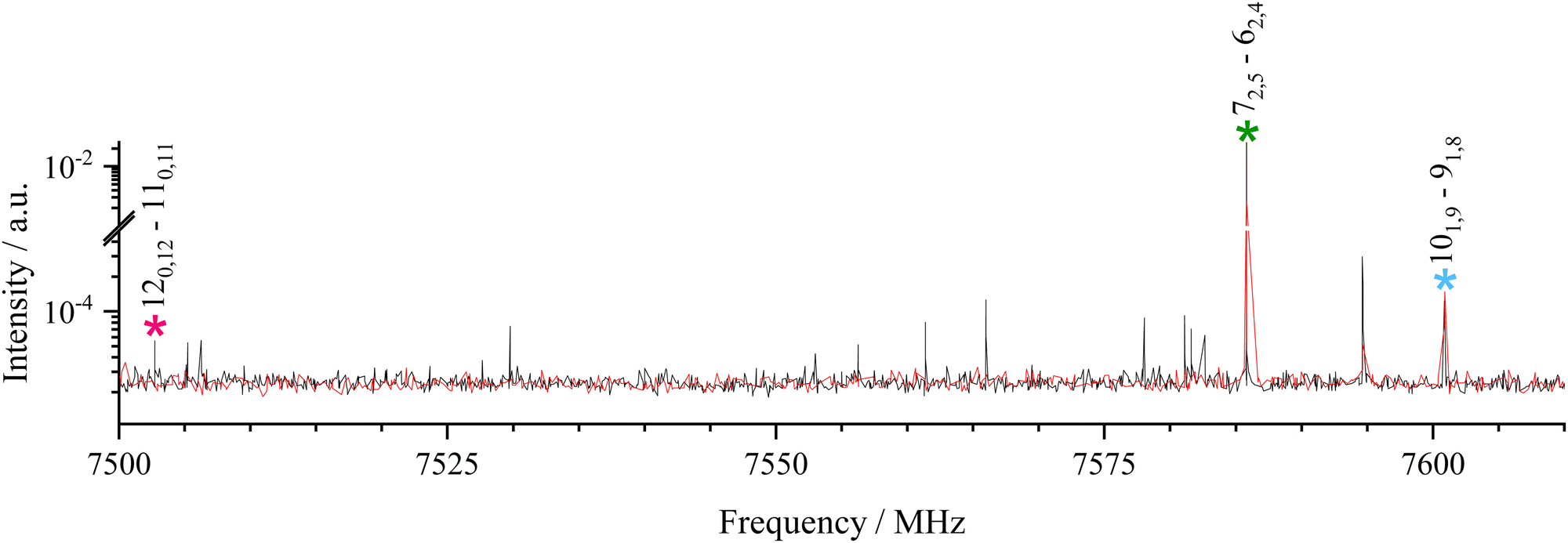 | ||
Fig. 3 Part of a low resolution scan of β-naphthaldehyde–water complexes, using neon (black curve) and argon (red curve): trans-β-naphthaldehyde is marked with a green asterisk, trans-β-II with a blue asterisk and trans-β-I with a pink asterisk. The assignment  is used. Unmarked signals are assigned to 13C isotopologues of β-naphthaldehyde. is used. Unmarked signals are assigned to 13C isotopologues of β-naphthaldehyde. | ||
In any case, the number of observed lines, their relative intensities and the carrier gas effect clearly indicate that the energetically most favourable conformer is trans-β-II, in agreement with ab initio calculations at the MP2 level.
4.3 Planarity of monohydrates
Calculations raised the question of planarity of naphthaldehyde hydrates. In the following, the case of the hydrated α isomer is discussed but the same trends were observed for the β isomer hydrates. The DFT calculations (B3LYP and B98) with aug-cc-pVTZ basis sets and MP2 ab initio calculations using the cc-pVTZ basis set showed that water is slightly out of plane with its free hydrogen forming a torsional angle that roughly ranges from 10° to 30° with the rings plane. In contrast, MP2 calculations with the aug-cc-pVTZ basis set indicate that the equilibrium structure is planar. Intermolecular vibrational modes thus need to be investigated. Calculations display three low-frequency modes below 100 cm−1. From the DFT/aug-cc-pVTZ and MP2/cc-pVTZ levels of theory at planarity, one of these frequencies has a negative value, corresponding to the aldehyde hydrogen out-of-plane motion. Consequently, coordinate scans along this motion using these levels of theory were calculated. MP2/aug-cc-pVTZ calculation results in one minimum which is planar, whereas all DFT calculations and MP2/cc-pVTZ give two symmetric and iso-energetic non-planar minima (see Fig. 4).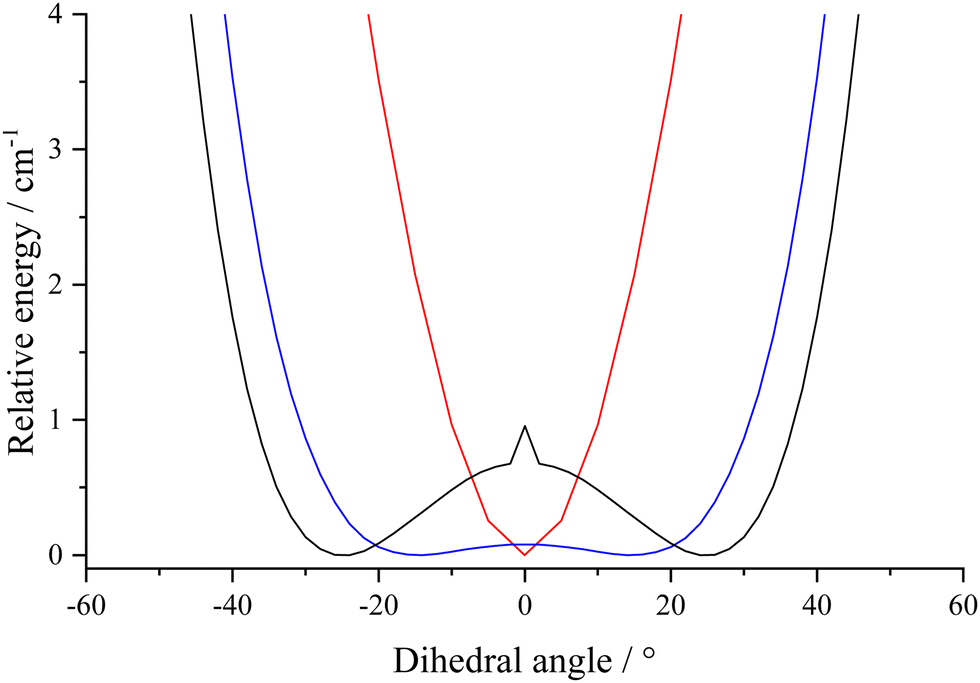 | ||
| Fig. 4 Scans from calculations along the dihedral angle C–O–O–H defining the position of the free H atom of water with respect to the symmetry plan of naphthaldehyde in cis-α-I structure: red = MP2/aug-cc-pVTZ, blue = B3LYP/aug-cc-pVTZ and black = B98/aug-cc-pVTZ. | ||
To split the difference, we used the inertial defect to probe the planarity. For a rigid planar molecule, its inertial defect is equal to zero but it can be negative at zero point in some cases by taking into account low frequency out-of-plane vibrational modes, such as here with a value of −0.81(63) amu Å2 cm−1. Thus, a value of the zero point inertial defect Δ0 can be computed using Oka's formula:51
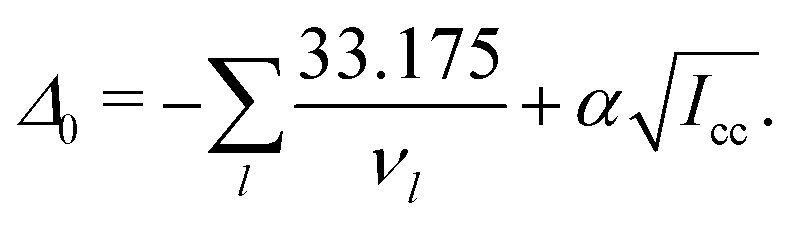 | (1) |
The first term takes into account out-of-plane vibrational modes whereas the second, through α, is empirical and depends on the type of molecule through the c-axis moment of inertia Icc.
According to the most recent α value for a PAC52 and the calculated frequencies of the two lowest out-of-plane modes, ν63 = 26.8 cm−1 and ν61 = 56.6 cm−1 (B98/aug-cc-pVTZ), eqn (1) gives an inertial defect of −0.850(12) amu Å2 cm−1 using only the lowest mode and −1.436(12) amu Å2 cm−1 if both modes are taken into account. The use of only the lowest frequency mode clearly offers better agreement with the experimental value but the use of a number of modes equal to the number of rings deserves to be considered. Uncertainties on the zero point inertial defect are difficult to estimate since there is no experimental frequency values in the literature (here we used the uncertainty of α52 and of our C rotational constant). However, a study showed, by comparing systematic errors on attributed vibrational modes, that DFT calculations give values closer to experimental ones than the MP2 level of theory in the harmonic approximation.53 To estimate uncertainties related to out-of-plane low frequency values, Δ0 using eqn (1) can be plotted as a function of the lowest out-of-plane frequency values around their B98/aug-cc-pVTZ values. Fig. 5 shows the error between the experimental inertial defect and its assumed value using α,52 and our values of Icc and the two lowest calculated frequencies. The black dashed curve represents all couples of (ν63,ν61) values that give a null error and the red cross stands for the couple (ν63,ν61) given by the B98/aug-cc-pVTZ calculation. Assuming that DFT calculations give the right value of ν63, there is no possible value for ν61 that retrieves to the black dashed curve in the range of 0–200 cm−1. Conversely, if ν61 is fixed to its DFT value, ν63 value would be equal to 54 cm−1 for a null error. By determining the minimum distance from calculated frequencies (red cross) to the null error curve (dash line), the closest point (red dot) coordinates are (ν63 = 47 cm−1, ν61 = 66.7 cm−1).
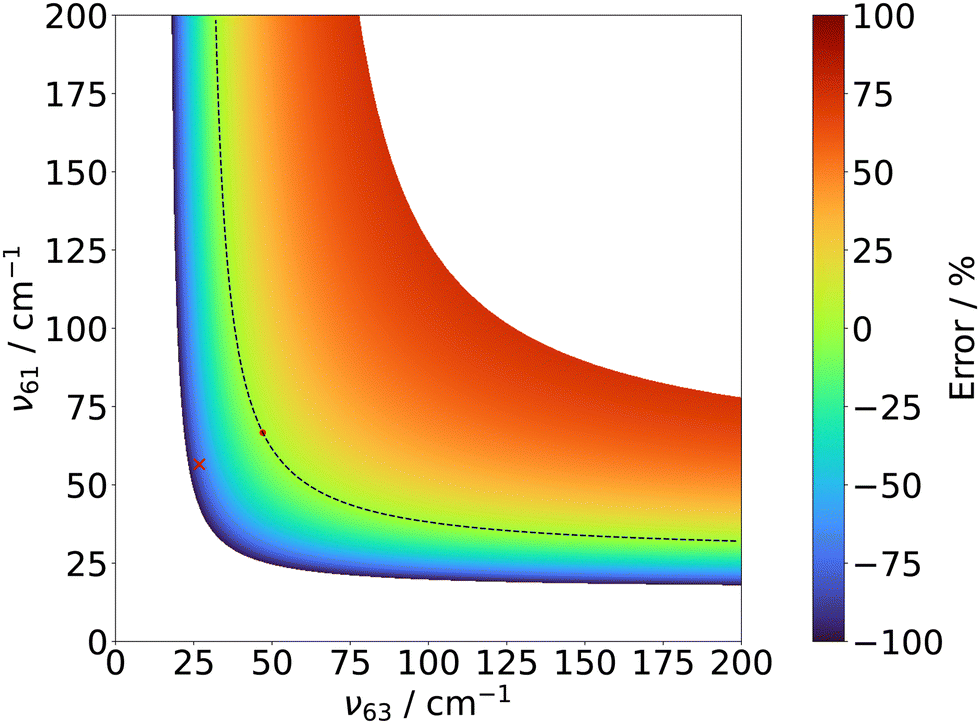 | ||
| Fig. 5 Simulation of inertial defect on cis-α-I structure based on Oka's formula (see eqn (1)). Colormap represents error between experimental and theoretical inertial defect. The red cross stands for the value given by the calculated values of (ν63, ν61) and the red dot stands for the couple of (ν63, ν61) which gives the minimal error, obtained via the closest “distance” from the calculated frequencies. | ||
Finally, a body of evidences tends to point toward a planar structure of the hydrates. The highest level of calculation (MP2/aug-cc-pVTZ) indicates planar hydrates. The use of only the lowest frequency to estimate Δ0, as initially stated by Oka, clearly agrees with planar structures. Using the two lowest frequencies, deviations from calculated values needed to correspond to planarity are reasonable enough to fall within expected deviations of few tens of cm−1 when dealing with harmonic calculations of very low frequency large amplitude motions.53 Experimentally, the non-observation of c-type transitions also suggests planar structures.
4.4 Formation of larger hydrates
Using the same method as for monohydrates, calculations were performed to predict the attachment of two or more water molecules to the naphthaldehyde isomers. The lowest energy structures correspond to a mix between the three first groups: resulting combinations are between I and II or between II and III but there are no structures combining I and III nor belonging to group IV (π interactions with the rings). The dihydrates structures calculated to be the most stable for both isomers are I and II combinations, so that the positions I and II are preferred as for monohydrates and in agreement with the case of naphthoquinone.21 The most favourable structures and their relative energies can be found in Tables S14 and S15 of the ESI.† Attempts to observe experimentally the most stable dihydrate for each isomer, namely cis-α-I-II and trans-β-I-II, were unsuccessful. Due to its very low vapour pressure (expected to be around 1 μbar at 300 K for both conformers), thermal vaporization of the sample is most probably not sufficient to extract dihydrates signals from the noise, calling for complementary experiments using a laser ablation nozzle.5 Conclusions
In this study, insights into the structural preferences and hydrophilic properties of α-naphthaldehyde and β-naphthaldehyde is provided from the comprehensive investigation of the hydration behavior of these polycyclic aromatic compounds using a combination of experimental and computational methods.The experimental observation of monohydrate structures for both isomers, along with their agreement with computational predictions, confirms the existence of stable hydrates. The presence of an electronegative functional group clearly alters the anchoring of water, which binds preferentially to the functional group rather than to the aromatic rings via a π interaction. The fact that two hydrate structures were observed for β-naphthaldehyde while only one was observed for α-naphthaldehyde suggests that the β-isomer exhibits higher hydrate formation propensity. This is supported by the analysis of electronic densities, hydration energies, and structural considerations, which all point towards a higher hydrophilicity of the β-isomer compared to the α-isomer.
In addition, computational calculations were employed to investigate the planarity of the naphthaldehyde monohydrates. Although slight deviations from planarity were observed in the low-level calculated structures, the analysis of low-frequency out-of-plane vibrational modes and the inertial defect indicated that the hydrates predominantly adopt a planar structure. This finding is consistent with the absence of c-type transitions in the experimental spectra, further supporting the planar nature of the hydrates.
The exploration of dihydrate structures by attaching two water molecules to the naphthaldehyde isomers revealed the most stable configurations, which were found to be combinations of the observed monohydrate positions. However, experimental detection of these dihydrates proved challenging due to their very low vapour pressure. Future experiments utilizing laser ablation nozzles may offer a promising way to overcome this challenge and facilitate the observation of dihydrate structures, enabling further investigation of their properties and interactions.
The present study, combining experimental and computational approaches at the molecular level, paves the way for further investigations into the hydration behavior of volatile organic compounds such as these oxygenated polycyclic aromatic hydrocarbons. For example, studies of isomer selective hygroscopic properties54 of their aerosols could deepen our understanding of the formation and aging of secondary organic aerosols.
Author contributions
Conceptualization: M. G.; data curation: all authors; formal analysis: J. A. C., C. B., V. V.; funding acquisition: L. M., M. G.; investigation: all authors; methodology: M. G, C. B., V. V.; project administration: L. M., M. G.; software: all authors; supervision: L. M., M. G.; validation: all authors; visualization: J. A. C., C. B.; writing – original draft: J. A. C., C. B.; writing – review & editing: all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the CaPPA project and by the CPER ECRIN funded by the French National Research Agency (ANR) through the PIA 11-LABX-0005-01, the I-SITE ULNE/ANR-16-IDEX-0004 ULNE, the Regional Council Hauts-de-France and the European Funds for Regional Economic Development (FEDER).Notes and references
- J. L. Jimenez, M. R. Canagaratna, N. M. Donahue, A. S. Prevot, Q. Zhang, J. H. Kroll, P. F. DeCarlo, J. D. Allan, H. Coe, N. L. Ng, A. C. Aiken, K. S. Docherty, I. M. Ulbrich, A. P. Grieshop, A. L. Robinson, J. Duplissy, J. D. Smith, K. R. Wilson, V. A. Lanz, C. Hueglin, Y. L. Sun, J. Tian, A. Laaksonen, T. Raatikainen, J. Rautiainen, P. Vaattovaara, M. Ehn, M. Kulmala, J. M. Tomlinson, D. R. Collins, M. J. Cubison, E. J. Dunlea, J. A. Huffman, T. B. Onasch, M. R. Alfarra, P. I. Williams, K. Bower, Y. Kondo, J. Schneider, F. Drewnick, S. Borrmann, S. Weimer, K. Demerjian, D. Salcedo, L. Cottrell, R. Griffin, A. Takami, T. Miyoshi, S. Hatakeyama, A. Shimono, J. Y. Sun, Y. M. Zhang, K. Dzepina, J. R. Kimmel, D. Sueper, J. T. Jayne, S. C. Herndon, A. M. Trimborn, L. R. Williams, E. C. Wood, A. M. Middlebrook, C. E. Kolb, U. Baltensperger and D. R. Worsnop, Science, 2009, 326, 1525–1529 CrossRef CAS PubMed.
- S. Tomaz, P. Shahpoury, J. L. Jaffrezo, G. Lammel, E. Perraudin, E. Villenave and A. Albinet, Sci. Total Environ., 2016, 565, 1071–1083 CrossRef CAS PubMed.
- H. C. A. Brandt and W. P. Watson, Ann. Occup. Hyg., 2003, 47, 349–378 CAS.
- R. G. Harvey, Polycyclic hydrocarbons and cancer, Elsevier, 1982, vol. 70, pp. 386–393 Search PubMed.
- H. Shen, Y. Huang, R. Wang, D. Zhu, W. Li, G. Shen, B. Wang, Y. Zhang, Y. Chen, Y. Lu, H. Chen, T. Li, K. Sun, B. Li, W. Liu, J. Liu and S. Tao, Environ. Sci. Technol., 2013, 47, 6415–6424 CrossRef CAS PubMed.
- M. Tsapakis and E. G. Stephanou, Environ. Sci. Technol., 2007, 41, 8011–8017 CrossRef CAS PubMed.
- I. J. Keyte, R. M. Harrison and G. Lammel, Chem. Soc. Rev., 2013, 42, 9333–9391 RSC.
- K. Ravindra, R. Sokhi and R. Van Grieken, Atmos. Environ., 2008, 42, 2895–2921 CrossRef CAS.
- G. Karavalakis, V. Boutsika, S. Stournas and E. Bakeas, Sci. Total Environ., 2011, 409, 738–747 CrossRef CAS PubMed.
- V. Perraud, E. A. Bruns, M. J. Ezell, S. N. Johnson, Y. Yu, M. L. Alexander, A. Zelenyuk, D. Imre, W. L. Chang, D. Dabdub, J. F. Pankow and B. J. Finlayson-Pitts, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 2836–2841 CrossRef CAS PubMed.
- A. Hodzic, S. Madronich, P. S. Kasibhatla, G. Tyndall, B. Aumont, J. L. Jimenez, J. Lee-Taylor and J. Orlando, Atmos. Chem. Phys., 2015, 15, 9253–9269 CrossRef CAS.
- L. Jia and Y. Xu, Aerosol Sci. Technol., 2014, 48, 1–12 CrossRef CAS.
- M. Tajuelo, A. Rodríguez, M. T. Baeza-Romero, A. Aranda, Y. Díaz-de Mera and D. Rodríguez, Atmos. Res., 2019, 230, 104631 CrossRef CAS.
- A. Akherati, Y. He, M. M. Coggon, A. R. Koss, A. L. Hodshire, K. Sekimoto, C. Warneke, J. De Gouw, L. Yee, J. H. Seinfeld, T. B. Onasch, S. C. Herndon, W. B. Knighton, C. D. Cappa, M. J. Kleeman, C. Y. Lim, J. H. Kroll, J. R. Pierce and S. H. Jathar, Environ. Sci. Technol., 2020, 54, 8568–8579 CrossRef CAS PubMed.
- B. Yuan, W. W. Hu, M. Shao, M. Wang, W. T. Chen, S. H. Lu, L. M. Zeng and M. Hu, Atmos. Chem. Phys., 2013, 13, 8815–8832 CrossRef.
- A. L. Steber, C. Pérez, B. Temelso, G. C. Shields, A. M. Rijs, B. H. Pate, Z. Kisiel and M. Schnell, J. Phys. Chem. Lett., 2017, 8, 5744–5750 CrossRef CAS PubMed.
- E. Rossich Molina, B. Xu, O. Kostko, M. Ahmed and T. Stein, Phys. Chem. Chem. Phys., 2022, 24, 23106–23118 RSC.
- H. Leboucher, A. Simon and M. Rapacioli, J. Chem. Phys., 2023, 158, 114308 CrossRef CAS PubMed.
- R. Knochenmuss, R. K. Sinha and S. Leutwyler, Annu. Rev. Phys. Chem., 2020, 71, 189–211 CrossRef CAS PubMed.
- D. Bernhard, M. Fatima, A. Poblotzki, A. L. Steber, C. Pérez, M. A. Suhm, M. Schnell and M. Gerhards, Phys. Chem. Chem. Phys., 2019, 21, 16032–16046 RSC.
- S. Baweja, S. Panchagnula, M. E. Sanz, L. Evangelisti, C. Pérez, C. West and B. H. Pate, J. Phys. Chem. Lett., 2022, 13, 9510–9516 CrossRef CAS PubMed.
- K. Egashira, Y. Ohshima and O. Kajimoto, J. Phys. Chem. A, 2001, 105, 1131–1139 CrossRef CAS.
- D. Loru, A. L. Steber, P. Pinacho, S. Gruet, B. Temelso, A. M. Rijs, C. Pérez and M. Schnell, Phys. Chem. Chem. Phys., 2021, 23, 9721–9732 RSC.
- E. Arunan, G. R. Desiraju, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R. H. Crabtree, J. J. Dannenberg, P. Hobza, H. G. Kjaergaard, A. C. Legon, B. Mennucci and D. J. Nesbitt, Pure Appl. Chem., 2011, 83, 1619–1636 CrossRef CAS.
- M. Goubet, M. A. Martin-Drumel, F. Réal, V. Vallet and O. Pirali, J. Phys. Chem. A, 2020, 124, 4484–4495 CrossRef CAS PubMed.
- S. Kassi, D. Petitprez and G. Wlodarczak, J. Mol. Struct., 2000, 517, 375–386 CrossRef.
- M. Tudorie, L. H. Coudert, T. R. Huet, D. Jegouso and G. Sedes, J. Chem. Phys., 2011, 134, 074314 CrossRef CAS PubMed.
- S. Kassi, D. Petitprez and G. Wlodarczak, J. Mol. Spectrosc., 2004, 228, 293–297 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian-16 Revision C.01, 2016 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5656 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- H. L. Schmider and A. D. Becke, J. Chem. Phys., 1998, 108, 9624–9631 CrossRef CAS.
- L. A. Curtiss, K. Raghavach Ari, P. C. Redfern, V. Rassolov and J. A. Pople, J. Chem. Phys., 1998, 109, 7764–7776 CrossRef CAS.
- T. H. Dunning, J. Chem. Phys., 1989, 90, 1007–1023 CrossRef CAS.
- C. Møller and M. S. Plesset, Phys. Rev., 1934, 46, 618–622 CrossRef.
- E. D. Glendening, C. R. Landis and F. Weinhold, J. Comput. Chem., 2013, 34, 1429–1437 CrossRef CAS PubMed.
- E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, P. Karafiloglou, C. R. Landis and F. Weinhold, NBO 7.0, 2018 Search PubMed.
- R. F. W. Bader, Acc. Chem. Res., 1985, 18, 9–15 CrossRef CAS.
- R. F. W. Bader, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- R. Bader, Atoms in Molecules: A Quantum Theory, Clarendon Press, 1994 Search PubMed.
- T. A. Keith, AIMAll (Version 19.10.12), TK Gristmill Software, 2019, http://aim.tkgristmill.com Search PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- S. F. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- J. K. G. Watson, in Vibrational spectra and structure, ed. J. R. Durig, Elsevier, Amsterdam, 1977, vol. 6, pp. 1–89 Search PubMed.
- H. M. Pickett, J. Mol. Spectrosc., 1991, 148, 371–377 CrossRef CAS.
- E. M. Neeman and T. R. Huet, Phys. Chem. Chem. Phys., 2021, 23, 2179–2185 RSC.
- M. Goubet, R. A. Motiyenko, F. Réal, L. Margulès, T. R. Huet, P. Asselin, P. Soulard, A. Krasnicki, Z. Kisiel and E. A. Alekseev, Phys. Chem. Chem. Phys., 2009, 11, 1719–1728 RSC.
- R. S. Ruoff, T. D. Klots, T. Emilsson and H. S. Gutowsky, J. Chem. Phys., 1990, 93, 3142–3150 CrossRef CAS.
- C. Peng and H. B. Schlegel, Isr. J. Chem., 1993, 33, 449–454 CrossRef CAS.
- A. Moudens, R. Georges, M. Goubet, J. Makarewicz, S. E. Lokshtanov and A. A. Vigasin, J. Chem. Phys., 2009, 131, 204312 CrossRef CAS PubMed.
- T. Oka, J. Mol. Struct., 1995, 352–353, 225–233 CrossRef.
- S. Chawananon, O. Pirali, M. Goubet and P. Asselin, J. Chem. Phys., 2022, 157, 064301 CrossRef CAS PubMed.
- A. Cuisset, G. Mouret, O. Pirali, P. Roy, F. Carier, H. Nouali and J. Demaison, J. Phys. Chem. B, 2008, 112, 12516–12525 CrossRef CAS PubMed.
- W. Ahmad, C. Coeur, A. Tomas, T. Fagniez, J.-B. Brubach and A. Cuisset, Appl. Opt., 2017, 56, E116–E122 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Enlarged version of Fig. 1; full set of calculated relative energies, rotational constants and dipole moment components; calculated Cartesian coordinates of observed monohydrate structures; lists of assigned rotational transitions, with (obs–calc), of observed monohydrates; calculated natural charges on atoms of observed monohydrates and corresponding isolated naphthaldehyde subunits; calculated structures, dipole moment components and relative energies of the most stables dihydrates. See DOI: https://doi.org/10.1039/d3cp02649c |
| This journal is © the Owner Societies 2023 |