
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
New insights into the protein stabilizing effects of trehalose by comparing with sucrose†
Kajsa
Ahlgren‡
 a,
Christoffer
Olsson‡
b,
Inna
Ermilova
a and
Jan
Swenson
*a
a,
Christoffer
Olsson‡
b,
Inna
Ermilova
a and
Jan
Swenson
*a
aDivision of Nano-Biophysics, Department of Physics, Chalmers University of Technology, Gothenburg SE-412 96, Sweden. E-mail: jan.swenson@chalmers.se
bDivision of Biomedical imaging, Department of Biomedical Engineering and Health Systems, KTH Royal Institute of Technology, Stockholm SE-114 28, Sweden
First published on 31st July 2023
Abstract
Disaccharides are well known to be efficient stabilizers of proteins, for example in the case of lyophilization or cryopreservation. However, although all disaccharides seem to exhibit bioprotective and stabilizing properties, it is clear that trehalose is generally superior compared to other disaccharides. The aim of this study was to understand this by comparing how the structural and dynamical properties of aqueous trehalose and sucrose solutions influence the protein myoglobin (Mb). The structural studies were based on neutron and X-ray diffraction in combination with empirical potential structure refinement (EPSR) modeling, whereas the dynamical studies were based on quasielastic neutron scattering (QENS) and molecular dynamics (MD) simulations. The results show that the overall differences in the structure and dynamics of the two systems are small, but nevertheless there are some important differences which may explain the superior stabilizing effects of trehalose. It was found that in both systems the protein is preferentially hydrated by water, but that this effect is more pronounced for trehalose, i.e. trehalose forms less hydrogen bonds to the protein surface than sucrose. Furthermore, the rotational motion around dihedrals between the two glucose rings of trehalose is slower than in the case of the dihedrals between the glucose and fructose rings of sucrose. This leads to a less perturbed protein structure in the case of trehalose. The observations indicate that an aqueous environment closest to the protein molecules is beneficial for an efficient bioprotective solution.
1 Introduction
Protein based drugs are becoming more and more predominant in the pharmaceutical market, such as Repatha, Empliciti and many others.1 However, the use of larger and more complex drug molecules presents new challenges related to maintaining these molecules intact and stable during an extended period of time, and in many different environments.2,3 For this purpose, different carbohydrates or polyols are often employed as stabilizing agents for proteins.4–6 These are used for protein stabilization in a large number of applications, such as during lyophilization, cryopreservation, or in vitro stabilization at room temperature.7,8 Specific carbohydrates often used are the disaccharide molecules trehalose and sucrose. Trehalose in particular is often considered as one of the most efficient and versatile stabilizing co-solutes, since it has shown a large ability to preserve the properties of proteins and many other biological molecules after different types of storages.9,10 However, the reason for why trehalose generally out-performs sucrose and other types of sugars for such applications is still widely debated. It might be expected that sugars stabilize proteins and other biomolecules by direct interaction with them, and that trehalose therefore is most efficient either because it interacts stronger with the biomolecules or simply because it exhibits a higher glass transition temperature, Tg, than other disaccharides, thereby forming a more immobile shell around the biomolecules. This hypothesis is, however, not supported by experimental and computational observations, which have shown that the sugar molecules are instead preferentially excluded from direct interaction with the biomolecular surface.11–13 Thus, the biomolecules are preferentially hydrated, by direct interaction with water, which requires another mechanism for explaining the stabilizing properties of disaccharides. In this case, experiments have shown14,15 that the hydration layer around protein molecules acts as a bridge between the sugar and the protein. The better stabilizing properties of trehalose are then assumed to be due to a stronger dynamic coupling of trehalose to water,16–18 which, in turn, leads to a stronger reduction of the protein dynamics,19–21 since the protein dynamics is “slaved” by the dynamics of the hydration layer.22,23An alternative explanation for why preferential hydration is beneficial for protein stability is that it also leads to a more pronounced excluded volume effect, which stabilizes the native state of the protein by a pure entropic effect.24–29 The reason for this is that a denaturation of the protein leads to a larger solvent-accessible surface area of the protein, which implies that the disaccharide molecules have to be excluded from a larger surface area causing a correspondingly larger Gibbs energy cost.28,29 The excluded volume effect increases also with an increasing density of the solution surrounding the protein, and since the density of the solvent becomes higher when a disaccharide is added to water, this leads also to a protein stabilizing effect of disaccharides.29 The main aim of this study is to investigate whether the hypothesis of a more pronounced preferential hydration effect for trehalose, compared to sucrose, and the associated excluded volume theory are experimentally supported or whether there are other explanations for the different bioprotective properties of the two disaccharides.
In a previous study13 we investigated the molecular structure and dynamics of a system of myoglobin in an aqueous trehalose solution by neutron diffraction combined with empirical potential structure refinement (EPSR) modeling, as well as quasielastic neutron scattering (QENS) and molecular dynamics (MD) simulations. In that study, we found strong experimental support for that the trehalose molecules are, indeed, preferentially excluded from the protein surface, but nevertheless were able to slow down the protein dynamics by interacting with, and thereby also slowing down, the protein hydration water, as described in ref. 13. However, we do not know whether these observations are particularly pronounced for trehalose or are similar for other disaccharides, such as sucrose. We elucidate this by comparing the structural and dynamical results we obtained in our previous study with the findings in the present study of myoglobin in an analogous sucrose solution. However, here we have also added X-ray diffraction measurement on both systems as well as new and longer MD simulations, and therefore we also present slightly updated results on the trehalose containing system, for accurate comparison. Thus, the EPSR produced structural model of the trehalose system, presented in Olsson et al.,13 was rerun with the added X-ray diffraction data. The resulting model did however show similar results as shown in Olsson et al.13
The present results show that the sucrose molecules are also preferentially excluded from interacting with the protein surface, but to a lesser extent than trehalose. Since Timasheff et al.11,12 have predicted that preferential exclusion from the protein surface is a key feature of co-solutes that are particularly suitable for stabilizing proteins, this implies that our finding can explain why trehalose stabilize proteins more efficiently than sucrose. The less preferential exclusion of sucrose from the protein surface is also in agreement with results from MD simulations by e.g. Lerbret,30 Corradini,21 as well as our own MD and free energy simulations. In further consistency with the stronger stabilizing effect of trehalose is our finding that trehalose slows down the protein dynamics more than sucrose, despite that trehalose exhibits less direct interactions with the protein surface.
2 Method
2.1 Sample preparation
We investigated six isotopically different samples for the two three-component (water–sugar–myoglobin) systems, with the same molar concentration of 1956![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :51:1 (corresponding to a weight ratio of 2:1:1 for the fully protonated samples). The isotope compositions of these six samples were the following:
:51:1 (corresponding to a weight ratio of 2:1:1 for the fully protonated samples). The isotope compositions of these six samples were the following:
1. D2O–D-sugar–myoglobin (with deuteration of all exchangeable OH-groups)
2. D2O–H-sugar–myoglobin (with deuteration of all exchangeable OH-groups)
3. H2O–D-sugar–myoglobin
4. H2O–H-sugar–myoglobin
5. 50–50 mol% mixture of samples 1 and 2
6. 50–50 mol% mixture of samples 1 and 4.
Three isotopically different samples (with H2O, D2O, and HDO) were also prepared for the two-component system water–myoglobin (1956:1 water:myoglobin molar ratio), as a reference.
Protonated α,α-trehalose (in dihydrated form) and protonated sucrose (in anhydrous form) were purchased from Sigma-Aldrich and used without any further purification. Samples with these sugars are denoted as H-sugar. The corresponding deuterated sugars (referred to as D-sugar) were purchased from Omicron Biochemicals in anhydrous form. In these sugars only their carbon-bound hydrogens were exchanged for deuterium. The hydrogen atoms in the hydroxyl groups of the sugars were the same as the hydrogen atoms in the water of the same sample. Thus, when the water was D2O these hydrogens were exchanged by repeatedly dissolving in D2O and drying the sugars under vacuum at 70 °C. Myoglobin was also purchased from Sigma-Aldrich and was freeze-dried in either H2O (for samples 3 and 4) or D2O (for samples 1 and 2) before use. This was done to remove residual water molecules, and to deuterate the exchangeable protein hydrogens. The pH of the different solutions was approximately 7.8 for the fully deuterated samples and 6.0 for the fully protonated samples.
2.2 Neutron diffraction experiments
All samples were measured inside 1 mm thick Ti0.676Zr0.324 containers, which were sealed with a PTFE O-ring and mounted onto an automatic sample changer. The neutron diffraction experiments were performed on the NIMROD diffractometer31 at the ISIS Pulsed Neutron and Muon Source, STFC Rutherford Appleton Laboratory, UK. All measurements were carried out at a temperature of 300 K. The structure factors, S(Q), were obtained from the raw diffraction data using the GUDRUN suite (2015 version).32 The GUDRUN software package correct the data for e.g. inelasticity effects, subtract background and empty can scattering, as well as normalize the scattering to a vanadium standard, as described in more detail in ref. 32.2.3 X-ray diffraction experiments
The fully protonated neutron diffraction samples (Mb in H-sugar and H2O) were used for the X-ray diffraction measurements. Samples were placed in 2 mm wide silica glass capillaries, and placed in an Ag anode X-ray diffractometer (Empyrean) giving a wavelength of 0.5609 Å. The data were corrected for background scattering, multiple scattering and attenuation and converted to an interference differential cross section scale by using GUDRUN-X,33 which is the X-ray diffraction version of the GUDRUN software package.2.4 EPSR modeling
The EPSR method34 is a Monte Carlo based method in which an atomistic model of the investigated system is built and assigned with a reference potential. An empirical potential – based on the difference between the simulated and experimental structure factors (S(Q)) is then added to the reference potential, followed by an equilibration of the system based on the new potential. This process is then iterated until the difference between the simulated and experimental S(Q)s becomes negligible. The method is extensively described in e.g. Ref. 34.In this study we only used EPSR modeling to obtain more detailed information about the short-range order structure in the sugar solution and at the protein–solvent interface. Structural correlations between different protein molecules were elucidated in a previous publication,35 where the small-angle scattering data of the present experiments were analyzed. In that study, it was found that the protein molecules were relatively evenly distributed in the water–sugar solution, without any indication of protein clusters. Therefore, to study the nearest environment of a protein molecule, it is sufficient to simulate systems containing only one myoglobin molecule, 51 disaccharide molecules, and 1956 water molecules, which correspond to the molar concentrations of the experimental samples. The number density of the three-component samples was determined to be 0.1094 atoms per Å3, which gives a cubic box size of 46 Å. The reference potential was set identical to that in ref. 36 and 37 for the water, where the SPC/E force field was used. For the two types of sugar molecules the OPLS-AA force field38 was used, with Lennard-Jones parameters as listed in Table S1 of the ESI.† The protein structure was obtained from the protein data bank (PDB ID: 1DWR). The reference potential for this protein was loosely based on the OPLS-AA force-field.39 The Lennard-Jones and Coulomb parameters for the different atom types in myoglobin are also presented in Table S1 of ESI.†
The EPSR simulations ran until the agreement between the measured and simulated S(Q) did not improve anymore. Thereafter, 3000 different configurations of the systems were produced to obtain sufficient statistics for calculations of e.g. pair-correlation functions, g(r). From the relatively good fits to the experimental S(Q)s, shown in Fig. 1 for (a) sucrose (suc) and for (b) trehalose (tre) (where the dashed lines display the experimental data while the solid lines represent the EPSR fits), it can be concluded that the structural models produced by EPSR can be regarded as representative models of the local structure around a protein molecule. The g(r) between specific atom types as well as the 3D images of the systems were produced using VMD 1.9.4.40
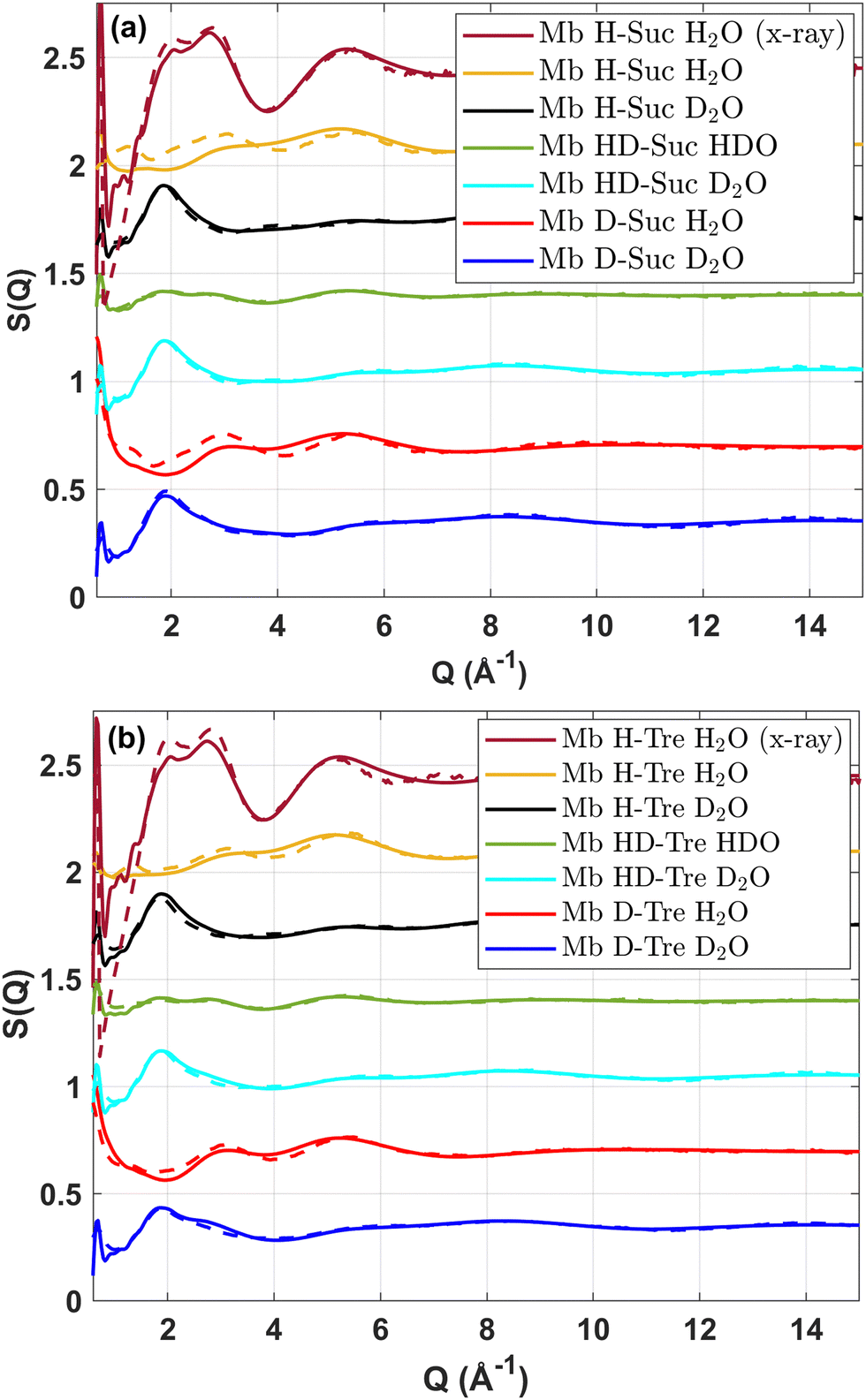 | ||
| Fig. 1 Structure factors of six isotopically different samples measured with neutron scattering or X-ray scattering for (a) sucrose and (b) trehalose. The dashed lines represent the experimental data while the solid lines represent the EPSR fits. | ||
It was also verified that the resulting models were not dependent on the starting configurations by (i) running the simulations with another randomized distribution of the solutes and (ii) starting the simulations with the sugar molecules preferentially bonded to the protein surface (by using an initial artificial protein–sugar attractive force, which was later turned off). In both cases, the final configurations gave similar fits to the experimental data and also similar partial pair correlation functions, gij(r).
2.5 QENS measurements
The QENS measurements were also performed at the neutron spallation source ISIS. In this case the IRIS spectrometer, as described in detail in ref. 41, was used. The measurements were carried out at 300 K on three isotopically different compositions of each three-component system: Mb in D-sugar and H2O, Mb in H-sugar and H2O, and Mb in H-sugar and D2O. As reference systems, we also measured Mb in D2O, Mb in H2O, H-sugar in D2O, and D-sugar in H2O. The samples were placed in annular aluminum alloy cans with a sample thickness of 0.1 or 0.25 mm, depending on the hydrogen content in the sample, to ensure a sample scattering of less than 10%. The PG002 analyzer configuration was used, which gave an experimental energy transfer window of ±0.4 meV and an energy resolution of 17.5 μeV FWHM. The spectrometer has 51 detectors, which were grouped into 10 groups with Q-values in the range 0.42–1.8 Å−1.The dynamic structure factor, S(Q,w), of each sample was obtained by de-convoluting the measured spectra with the resolution function, R(Q,w),42 which, in turn, was measured by taking the spectra of a vanadium standard. The S(Q,w)s were, however, not directly analyzed, but Fourier transformed to intermediate scattering functions, I(Q,t), which were used for further analysis. For all data corrections and analysis the Mantid software package42 was used.
2.6 QENS Fitting procedure
Since the time scales of the water, sugar and protein dynamics are expected to be rather different, the experimentally obtained I(Q,t)'s were fitted by three stretched exponential (KWW) functions. The total I(Q,t) for the sample of isotope composition (i) was thereby fitted by the following function: | (1) |
The fitting of the Ii(Q,t)'s for the two-component systems (either water and sugar, or water and protein) were performed correspondingly by using two isotope compositions and two stretched exponential relaxation processes. The average relaxation times were calculated from the obtained fit parameters, by the following equation:43
 | (2) |
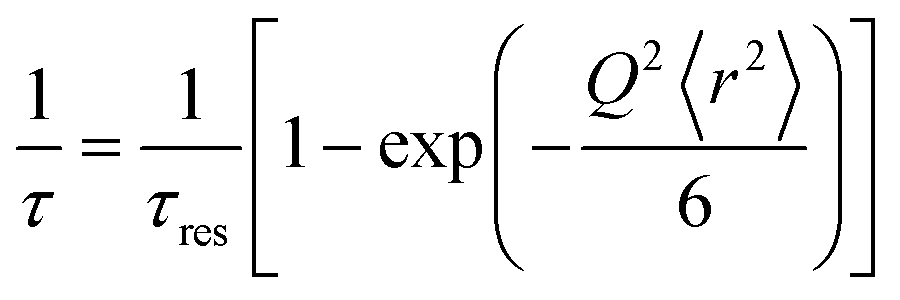 | (3) |
 | (4) |
2.7 Classical MD simulations
In order to perform a classical MD simulation for sucrose and trehalose, we implemented a force field derived from the general amber force field (GAFF).44 Specifically, partial atomic charges were computed using the Hartree–Fock method with the 6-31G(d) basis set, which is implemented in Gaussian 16 software,45 with applied restrained electrostatic potential (RESP)46 fitting method. The structures of sugars and their partial atomic charges can be observed in the archive uploaded in the Zenodo repository.For MD simulations the structure of myoglobin was the same as in the EPSR simulations (PDB ID: 1DWR47). The force field utilized for the protein was CHARMM36.48–50 Computational boxes were created in a following way: four molecules of myoglobin were placed in a large cubic box with sides of 12 nm each. The distance between molecules was at least of 8 Å in order to avoid any overlaps and clustering of compounds. Then 204 sugar molecules were randomly added keeping the distances between them to at least 3 Å. Water (7824 molecules) and ions were inserted at a distance of 2 Å from each other and other molecules in the system. System containing only counter ions had 8 Na, while systems containing ions for mimicking pH had 21 Na and 13 Cl ions. The water model was TIP3p.51,52
Every system was equilibrated for 200 ns and then simulated for additionally 800 ns with a time-step of 2 fs in NPT ensemble using Berendsen barostat.53 The compressibility was 0.000045 bar−1 and a coupling constant of 10 ps was used in order to keep the pressure of 1 atm. The temperature was set to 298 K. Long-range electrostatics was handled using the Particle Mesh Ewald (PME) algorithm54 with a Fourier spacing of 1.2 Å. The cut-off value was 9 Å with a scheme Verlet55 and van der Waals modifier Potential-shift. A LINCS algorithm56,57 with 12 iterations was utilized for constraining bonds. Output was recorded in the trajectory every 2 ps. The integrator was leap-frog.58 The MD engine utilized for all simulations was GROMACS-2019.59,60
2.8 Rotational free energy calculations
Rotational free energies were carried out using the well-tempered metadynamics technique.61–63 Rotations around the selected dihedral were investigated in 3 different environments for both sugars: a single molecule in water (1 sugar molecule was in the box with 1728 water molecules), a single sugar molecule selected in a box with myoglobin, sugar and counter ions (the boxes were taken from classical MD simulations), a single sugar molecule selected in a box with myoglobin, sugar and ions relevant for the pH level from experiments (the boxes were taken from classical MD simulations).For performing these simulations the software plumed-2.5.464 together with GROMACS-201959,60 were used. In order to guarantee a good sampling every simulation was carried out for 1 μs. As 2 collective variables (CVs), dihedrals φ and ψ of the sugar molecules (see Fig. 2) were chosen in these calculations with a grid spacing of 0.1 rad on the interval [−π;π], which was dumped every 100000 steps. In Fig. 2 one can see an isosurface for potential of mean force (PMF) which illustrates the output from well-tempered metadynamics simulations for 2 CVs. Later such an isosurface is integrated firstly into PMF for each variable and then into a rotational free energy for every dihedral.
 | ||
| Fig. 2 Illustration of PMF, selected dihedrals and integrated profile for cosine of angles. | ||
Settings for the plumed64 engine were the following. The bias factor (γ) was 50. The temperature in all simulations was 298 K. The width of the collective variable (σ) was 0.35 rad and the height of the Gaussian function was 1.2 kJ mol−1. Gaussian functions were deposited every 500 steps.
3 Results and discussion
3.1 Structural modeling from experimental data
Fig. 3 shows partial gij(r) between surface protein oxygens and water or sugar oxygens, as computed by the EPSR modeling. There it can be seen that close to the oxygens of the protein we find a strong peak of water at about 2.8 Å for both sugar solutions, and a much weaker correlation with the oxygen atoms of the sugars. This proportion between the two peaks were shown to be an indicator of preferential hydration in our previous study.13 From the presented data it can be concluded that the protein is surrounded by water rather than by sugars. In the system with trehalose the preferential hydration is more pronounced than in the system with sucrose. At the same time sucrose associates with myoglobin more than trehalose, although the preferential hydration effect is substantial also for sucrose. Corresponding partial gij(r) for nitrogen and carbon atoms at the protein surface are shown in Fig. S1 of ESI.† | ||
| Fig. 3 Partial pair correlation functions between surface protein oxygens (Op) and water oxygens (Ow) or sugar oxygens (Osuc/Otre). Dashed lines are from the trehalose model, whereas solid lines are from the sucrose model. The surface protein oxygens were defined as those oxygens which are within 2.5 Å of any atom in the solvent. | ||
The preferential hydration effect is further evident from the coordination numbers, given in Table 1, obtained by calculating the average number of the given atom–atom pairs within a distance of 3.4 Å. From this table it is evident that the probability for a protein oxygen to coordinate to a water oxygen (via hydrogen bonding) is about 20 and 40 times higher than the probability to coordinate to a sucrose oxygen and a trehalose oxygen, respectively. In consistency with the more pronounced preferential hydration effect for the trehalose system is also the observation that the coordination number between sugar oxygens and water oxygens is higher for the sucrose system. From the sugar–sugar coordination numbers it is also clear that there is a somewhat higher probability for sucrose to form smaller clusters, although there is no particular tendency for any of the two disaccharides to form clusters, as discussed in some more detail for the corresponding two-component systems without myoglobin.65
| N O–O (suc) | N O–O (tre) | |
|---|---|---|
| Protein–water | 338 | 451 |
| Protein–sugar | 15.5 | 9.63 |
| Sugar–water | 23.9 | 19.3 |
| Sugar–sugar | 2.85 | 2.15 |
| Water–water | 3.72 | 4.08 |
The pronounced preferential hydration effect and the finding that it is stronger for trehalose than sucrose can also visualized directly from the structural models. Fig. 4 shows 3D images of the EPSR produced structural models of the three-component systems containing sucrose (left) and trehalose (right), respectively. From this figure it is evident that a major part of the protein surface is hydrogen bonding to water (blue parts) and that a much smaller part of the protein surface is not directly interacting with water (red parts) in the case of trehalose. These parts of the protein surface are instead interacting with the disaccharide without any intermediate hydration layer (blue parts). However, it should here be noted that although the preferential hydration effect is clearly visible in Fig. 4 the effect seems to be less pronounced than indicated from the large difference between protein-water and protein–sugar coordination numbers, as shown in Table 1. The reason for this is that the sugar molecules are much larger than the water molecules, and therefore each sugar molecule occupies a much larger protein surface area than a single water molecule.
 | ||
| Fig. 4 Left panel: 3D image of the EPSR produced structural model of the three-component system with sucrose. Right panel: 3D image of the corresponding model of the three-component system with trehalose. Red parts are exposed protein surface without hydration water, blue parts are water molecules at the surface of the protein molecule and white stick figures are sucrose and trehalose molecules, respectively. | ||
These structural findings are not only supporting the preferential hydration model,11–13 but also the excluded volume theory24–29 since a more pronounced preferential hydration effect leads also to that the native state of the protein becomes more stable by entropic effects. Thus, more thermal energy is needed to denature the protein in the case of disaccharides which are preferentially excluded from the protein surface.
3.2 Dynamics from QENS data
The finding that the preferential hydration effect is more pronounced for trehalose than sucrose makes it interesting to elucidate how that affects the dynamics of the three components in the systems. Fig. 5(a) and (b) displays the intermediate scattering functions, I(Q,t), at Q = 1.06 Å−1 for the two- and three-component systems of both trehalose and sucrose. The solid lines are the fit while the markers display the experimental data. The incoherent scattering cross section of hydrogen is about 40 times larger than that of deuterium and other types of atoms in the systems. Therefore, the scattering due to non-hydrogen atoms can almost be neglected and samples H-Tre D2O and H-Suc D2O in Fig. 5(a) and Mb H-Tre D2O and Mb H-Suc D2O in Fig. 5(b) give an indication of the dynamics of the sugar (and protein in the three-component systems). In the two-component systems both the sugar and water dynamics of the sucrose and trehalose systems are quite similar, although slightly faster in the sucrose system. In the three-component system the protein dynamics is mostly represented by Mb H-Tre D2O and Mb H-Suc D2O in addition to the dynamics of the sugar. For both these samples it is possible to observe a stretched relaxation with a time span extending beyond the time window of the IRIS spectrometer (due to its limited energy resolution), as seen in Fig. 5(b). Hence, from the figure it is evident that a substantial fraction of the protein dynamics occurs on a longer time scale than the 100 ps reached by the IRIS spectrometer. However, for the sugar and protein dynamics observed in the experimental time-window, it is seen to be slightly faster for the sucrose system. The water dynamics can also be extracted from the I(Q,t) plots by determining how much I(Q,t) is changed by replacing D2O with H2O. From Fig. 5(a) it is clear that the water dynamics is considerably faster than the sugar dynamics, but also that there is not a large difference in the I(Q,t) for water between the two sugar solutions However, for the three-component systems it is evident that the water dynamics is faster for the sucrose containing system, as shown in Fig. 5(b). | ||
| Fig. 5 Intermediate scattering functions for the two-component systems (a) H-Tre D2O and H-Suc D2O (black lines with ring and star markers respectively) as well as D-Tre H2O and H-Suc H2O (red lines with cross and diamond markers respectively) and three-component systems (b) Mb H-Tre D2O and Mb H-Suc D2O (black lines with ring and star markers respectively), Mb D-Tre H2O and Mb H-Suc H2O (red lines with cross and diamond markers respectively) as well as Mb H-Tre H2O (green line with plus plus marker) at T = 300 K and at a momentum transfer of Q = 1.06 Å−1. | ||
To more accurately determine the average relaxation times of the different components, and how they vary with Q, the data presented in Fig. 5 were fitted by eqn (1), and the obtained relaxations times and stretching parameters were thereafter used to obtain average relaxation times by eqn (2). The inverse of these average relaxation times are shown as a function of Q2 in Fig. 6(a) for the two- and (b) for the three-component systems. As can be seen in the figure, the Q2-dependences are rather well described by the Gaussian jump-length diffusion model (eqn (3)). However, it should be noted that the observed “crossover” from a nearly Q2-dependence at the lowest Q-values to an almost Q-independent average relaxation time at the highest Q-values can also be, at least partly, due to that translational diffusion (with an expected Q2-dependence) dominates the scattering intensity at low Q-values, whereas rotational and other Q-independent motions may dominate the scattering intensity at high Q-values. Thus, the observed dependencies on Q may also be a result of a combination of translational diffusion and rotational (and other Q-independent) motions. Furthermore, in the case of protein dynamics, most of the motions are expected to be of local character, but over several Å (i.e. protein fluctuations), and therefore giving rise to a translational diffusion-like Q-dependence, although the slowest intramolecular fluctuations as well as the long range translation diffusion of the entire protein molecules are too slow for being observed on the experimental time scale, as shown in Fig. 5(b).
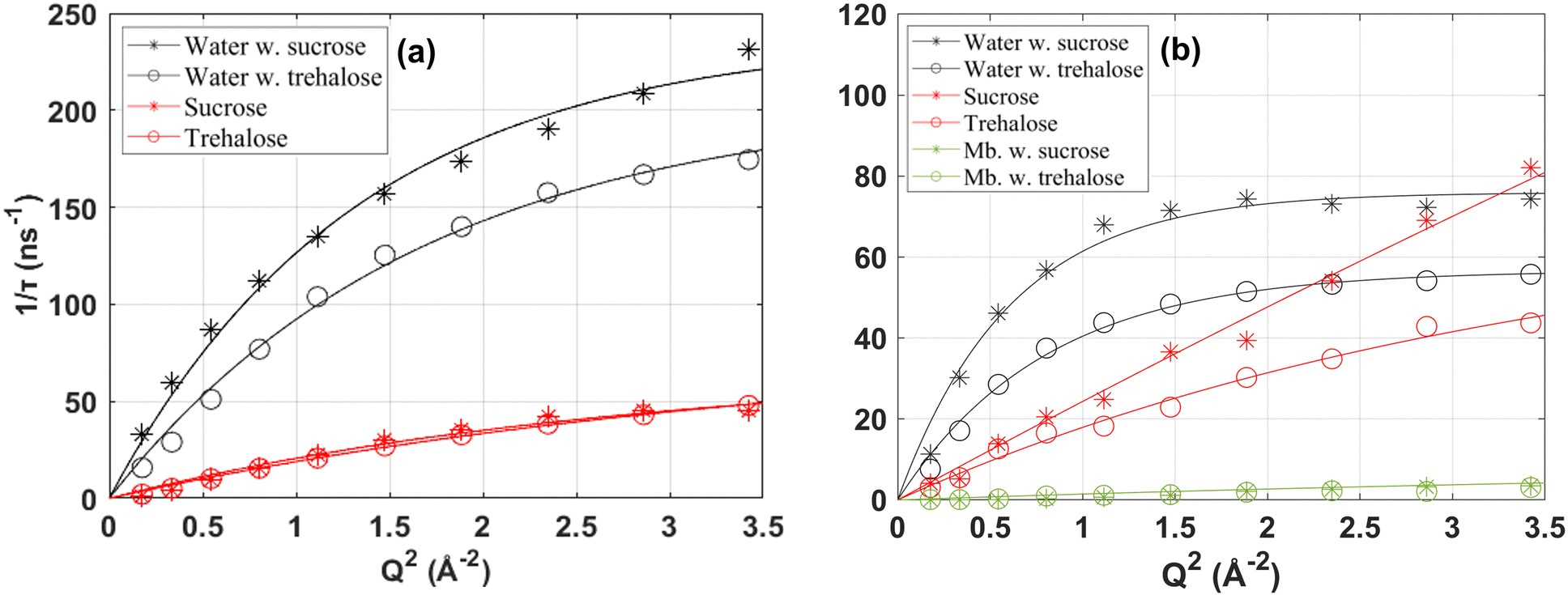 | ||
| Fig. 6 Inverse average relaxation times as a function of Q2 for the water and sugar in (a) the two-component systems as well as for the protein in (b) the three-component systems. Fits by the Gaussian jump-length diffusion model (eqn (3)) are shown by the solid lines. | ||
However, despite the difficulties mentioned above to determine the exact nature of the observed dynamics the Gaussian jump-length diffusion model is good to use for extracting the diffusion constant of the water and disaccharide, by combining it with eqn (4), since the deviation from a Q2-dependence at higher Q does not affect the obtained diffusion constant (only the values of the fit parameters in eqn (3)). In Table 2, the diffusion constants for both the sucrose and trehalose systems, as well as for water in the protein solution without any sugar13 are displayed. It should here be noted that the values are average values, and particularly for water the more bulk-like water should diffuse considerably faster than the hydration water which hydrogen bond to the protein surface and/or the sugar molecules, as shown in other studies.66,67 The diffusion constants given in Table 2 more or less confirm the qualitative discussion given above for the I(Q,t) plots, i.e. that both the water and sugar dynamics are faster in both the two- and three-component systems of sucrose, although the difference for the sugar dynamics is within the large error bars. Furthermore, the relative differences in the dynamics seem to be slightly larger for the three-component system than for the two-component system, likely due to that the more pronounced preferential hydration effect for trehalose gives rise to a sugar–water solution of a lower water concentration. The values presented in Table 2 cannot be directly compared with similar studies in the literature since different concentrations and/or temperatures have been used in previous studies. Nevertheless, a qualitative comparison can be made with e.g. Ref. 68 where they also used QENS to study aqueous solutions of trehalose and sucrose, respectively, at the concentration 20 water molecules per sugar molecule (we have 38 water molecules per sugar molecule in this study) and the temperature 323 K. They obtained diffusion constants of 15.5 × 10−10 m2 s−1 and 17.5 × 10−10 m2 s−1 for water in the solutions of trehalose and sucrose, respectively. For the sugar molecules the values were 1.63 × 10−10 m2 s−1 and 2.24 × 10−10 m2 s−1 for trehalose and sucrose, respectively. Thus, also in this study the diffusion of both the water and sugar molecules were faster for the sucrose solution. An even more quantitative comparison can be made with diffusion constants obtained from pulsed-gradient-spin–echo nuclear magnetic resonance (NMR),18 where the authors obtained the values 10.0 × 10−10 m2 s−1 and 10.2 × 10−10 m2 s−1 for water in the solutions of trehalose and sucrose, respectively, at 303 K and a concentration of 32 wt% sugar. For the sugar molecules the values were 1.27 × 10−10 m2 s−1 and 1.62 × 10−10 m2 s−1 for trehalose and sucrose, respectively. Thus, all diffusion constants from NMR are somewhat lower than from QENS, but it is expected since NMR probes the long-range macroscopic diffusion constant, whereas QENS probes the more local diffusion constant on a few nm length scale. Due to compositional inhomogeneities in a two- or three-component system the local diffusion tends to be faster than the macroscopic diffusion, particularly in the case of different diffusion constants of the different components. Another interesting finding in ref. 69 is that the difference in the diffusion constant betweeen trehalose and sucrose increases dramatically with increasing sugar concentration, indicating that trehalose forms a stronger and more immobile network structure than sucrose. This, in combination with the observation that the protein becomes more preferentially hydrated in the case of trehalose, may be a key factor for the generally superior stabilizing effect of trehalose.
| Component | D x (10−10 m2 s−1) |
|---|---|
| Water in three-comp | 12.5 ± 2.5 (7.09 ± 1.09) |
| Water in two-comp (sug) | 18.0 ± 2.8 (12.3 ± 2.0) |
| Sugar in three-comp | 2.49 ± 0.37 (2.06 ± 0.46) |
| Sugar in two-comp | 2.45 ± 0.59 (2.16 ± 0.36) |
| Water in two-comp (mb) | 18.9 ± 1.0 |
3.3 Classical molecular dynamics simulations
In order to verify that all simulations were well-equilibrated, properties like root mean square deviation (RMSD) and total energies were computed for simulated boxes during the whole simulation time.Fig. S2 in ESI,† demonstrates RMSD computed for each protein in every simulated system. After 200 ns of equilibration the RMSD of the atoms in all systems is less than 3 Å compared to the starting configuration, which is an acceptably small value.69,70
The protein backbone is known to be responsible for the structural stability of the protein, as well as for its overall shape, i.e. its tertiary structure.71,72 Therefore, the interactions with the protein backbone should be of relevance for the protein stability. Since sucrose has one difference from trehalose, which is its fructose ring, it can be expected that the rotational mobility is different for trehalose and sucrose. This can be investigated by computing rotational correlation functions, using the second order Legendre polynomial58,73 for the dihedral angle between fructose and glucose and between the two glucose rings, respectively. Fig. 7 shows such correlation functions, where it can be seen that the rotation around the dihedral between fructose and glucose rings is faster than the rotation around the dihedral between two glucose rings. Thus, this rotational mobility is higher for sucrose than for trehalose. Moreover, the presence of additional ions slows down the rotation for both sugars. Similar results have previously been found for diluted (4 wt% disaccharides in water) systems with the use of MD simulations.30,74 The present results thus show that trehalose exhibits a slower rotational dynamics than sucrose for the drier and more crowded system presented here. Interestingly however, there exists experimental indications that, for extremely dry systems75 and pure disaccharide melts,76 that sucrose exhibits a slower rotational dynamics. However, in the case of the disaccharide melts it was shown that both sucrose and trehalose exhibited the slowest rotational dynamics compared to other, similar, disaccharides, although with sucrose being slightly more rigid.
 | ||
| Fig. 7 Rotational correlation functions computed between 2 rings of sugars. (a) Systems with only counter ions. (b) Systems with additional ions for reaching the experimental pH. | ||
Since this rotational motion is likely to induce protein motions if the sugar interacts directly with the protein, a slower rotational motion and less direct sugar–protein interactions can be another reason why trehalose often stabilizes proteins more efficient than sucrose. However, in order to understand if this difference is due to the molecules themselves or due to that the molecules are located in different environments it is worth to study the rotation by free energy calculations.
3.4 Rotational free energy calculations
PMF profiles for rotations around selected dihedrals can give important information about free energy barriers which might hinder molecular motions, while computed free energies from one position to another one can explain if a certain rotation can happen spontaneously at the selected conditions.Fig. 8 demonstrates PMF profiles for single sugar molecules in 6 simulations. The highest value of rotational PMF for both dihedrals in sucrose and trehalose φ and ψ is observed for cosine equal to 1 (the least probable conformation), while the lowest one is for cosine equal to −1 (the most probable conformation). The sucrose molecule has a higher mobility around the dihedral φ than around the dihedral ψ, while trehalose appears to have a similar mobility around both dihedrals. From visual comparison of curves it can be concluded that values of rotational potential of mean force for simulations with trehalose are higher than for sucrose. Thus, sucrose is rotationally more mobile than trehalose. This statement can also be confirmed by the calculated rotational free energies given in Table 3.
 | ||
| Fig. 8 PMF for single sugar molecules separated into φ and ψ. (a) Dihedral φ of sucrose. (b) Dihedral ψ of sucrose. (c) Dihedral φ of trehalose. (d) Dihedral ψ of trehalose. The description of legend: “no protein” – for a single sugar molecule in water; “protein” – for a single sugar molecule in water with protein and counter ions; “protein + ions” – for a single sugar molecule in water with protein and ions for reaching the experimental pH. Convergence, sampling and total PMF profiles are shown in Fig. S3–S6 of ESI.† | ||
| System | φ | ψ |
|---|---|---|
| Sucrose | −14.89 | −9.28 |
| Sucrose + counter ions | −14.42 | −14.22 |
| Sucrose + ions for pH | −15.79 | −7.98 |
| Trehalose | −9.26 | −9.47 |
| Trehalose + counter ions | −8.72 | −10.35 |
| Trehalose + ions for pH | −9.69 | −10.81 |
These results indicate that binding to the protein backbone is a disadvantage of sucrose as a stabilizer, because this part of the protein is responsible for its structure and shape. The faster dihedral rotation of sucrose is likely to induce motions of the the protein backbone, which, in turn, leads to destabilization. In contrast, trehalose plays the role of a stable buffer around the protein, without almost any direct sugar–protein interactions which can affect the protein stability negatively. Thus, although sucrose and trehalose have the same chemical composition, they demonstrate different abilities to stabilize the globular protein myoglobin. Small structural differences of the sugars seem to play a crucial role for the interactions with the proteins and its related protein stability.
Finally, it should be noted that the present study was performed on a globular protein, and it is not clear whether similar results would be obtained for other types of proteins with other tertiary structures. Moreover, the different characteristics of trehalose and sucrose may affect different types of proteins differently, even if they interact similarly with the different proteins. This may cause sucrose to be a more efficient protein stabilizer than trehalose for some types of proteins. Similarly, it would be of interest to study how other types of biomolecules are preserved by trehalose and sucrose, respectively. For instance, how they affect the preservation of modern nucleic acids which are used as compounds of mRNA gene-therapies.2 Thus, from this study it is difficult to know how universal the observed behaviour of trehalose is for other types of proteins and biomolecules. However, we believe that the results obtained in this study should be representative for most globular proteins and provide insights into why trehalose is generally a more efficient protein stabilizer than sucrose.
4 Conclusion
In this work we have compared the structural and dynamical properties of sucrose and trehalose solutions and how the water and sugar molecules interact with the protein myoglobin. The neutron scattering experiments and advanced computer simulations provide a consistent picture, where relatively small differences can have important implications for the stability of globular proteins. The most important differences are:(a) There is a clear preferential hydration effect for both sugar solutions, but it is more pronounced for the trehalose solution.
(b) Trehalose slows down the water dynamics more than sucrose, and this, in turn, makes the protein dynamics slower, by the slaving mechanism.22,23
(c) The rotational motion around dihedrals between the two glucose rings of trehalose is slower than around the dihedrals between the glucose and fructose rings of sucrose. In addition, trehalose binds to fewer oxygens of amino-acid residues in myoglobin's backbone than sucrose.
Collectively, all these three key results suggests that trehalose perturbs less the preferred aqueous environment around the protein as well as alters the protein structure less than sucrose, thus, trehalose is able to stabilize the protein more efficient than sucrose without almost any direct interactions with the protein surface. The more pronounced preferential hydration of trehalose leads also to a stronger excluded volume effect, which stabilizes the native state of the protein by a pure entropic effect.
Author contributions
J. S. designed the project and supervised K. A., C. O. and I. E. C. O. prepared all the samples. C. O. and J. S. performed the experiments. K. A. and C. O. did the analysis of the neutron scattering data and EPSR modeling. I. E. performed the MD simulations and free-energy calculations. The manuscript was written by all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Tristan Youngs for his help with the NIMROD experiments and related analysis. We thank Tom Headen for his help with the X-ray diffraction measurements and data corrections. We also thank Victoria Garcia Sakai for her help with the IRIS experiments. Furthermore, we gratefully acknowledge the Science and Technology Facilities Council (STFC) for access to the neutron beam at ISIS, on the IRIS spectrometer (RB number 1600024) and NIMROD diffractometer (RB number 1500094). The computations were performed on resources provided by the Swedish National Infrastructure for Computing (SNIC). In National Supercomputer Center (NSC) Tetralith cluster was employed for calculations through projects SNIC2021/5-470, SNIC2019/3-553 and SNIC2019/7-36 and SNIC2022/5-511. In High Performance Computing Center North (HPC2N) Kebnekaise cluster was used for simulations with the project numbers SNIC2019/5-74, SNIC2020/5-45, SNIC2020/10-22, SNIC2020/6-53, SNIC2022/5-86, SNIC2022/5-43, SNIC2022/22-539, SNIC2022/22-1019. At PDC Center for High Performance Computing we would like to thank for letting us use cluster Dardel with the project SNIC2022/22-1030. Finally, this work was financially supported by the Swedish Research Council (Vetenskapsrådet), grant no. 2019-04020.Notes and references
- H. D. Lagassé, A. Alexaki, V. Simhadri, N. Katagiri, W. Jankowski, Z. Sauna and C. Kimchi-Sarfaty, F1000Research, 2017, 6, 113 Search PubMed.
- M. N. Uddin and M. A. Roni, Vaccines, 2021, 9, 1033 CrossRef CAS PubMed.
- S. Frokjaer and D. E. Otzen, Nat. Rev. Drug Discovery, 2005, 4, 298–306 CrossRef CAS PubMed.
- J. H. Crowe, L. M. Crowe and S. A. Jackson, Arch. Biochem. Biophys., 1983, 220, 477–484 CrossRef CAS PubMed.
- N. K. Jain and I. Roy, Protein Sci., 2009, 18, 24–36 CAS.
- K. Gekko and T. Morikawa, J. Biochem., 1981, 90, 51–60 CrossRef CAS PubMed.
- M. S. Marcial-Coba, T. Cieplak, T. B. Cahú, A. Blennow, S. Knøchel and D. S. Nielsen, Food Funct., 2018, 9, 5868–5879 RSC.
- Z. P. Tolstyka, H. Phillips, M. Cortez, Y. Wu, N. Ingle, J. B. Bell, P. B. Hackett and T. M. Reineke, ACS Biomater. Sci. Eng., 2016, 2, 43–55 CrossRef CAS PubMed.
- J. K. Kaushik and R. Bhat, J. Biol. Chem., 2003, 278, 26458–26465 CrossRef CAS PubMed.
- M. Sola-Penna and J. R. Meyer-Fernandes, Arch. Biochem. Biophys., 1998, 360, 10–14 CrossRef CAS PubMed.
- T. Arakawa and S. N. Timasheff, Biochem., 1982, 21, 6536–6544 CrossRef CAS PubMed.
- T. Arakawa and S. N. Timasheff, Biochem., 1985, 24, 6756–6762 CrossRef CAS PubMed.
- C. Olsson, S. Genheden, V. Garcia Sakai and J. Swenson, J. Phys. Chem. B, 2019, 123, 3679–3687 CrossRef CAS PubMed.
- P. Mason, G. Neilson, C. Dempsey, A. Barnes and J. Cruickshank, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 4557–4561 CrossRef CAS PubMed.
- S. Perticaroli, M. Nakanishi, E. Pashkovski and A. P. Sokolov, J. Phys. Chem. B, 2013, 117, 7729–7736 CrossRef CAS PubMed.
- S. Magazù, F. Migliardo and M. T. F. Telling, J. Phys. Chem. B, 2006, 110, 1020–1025 CrossRef PubMed.
- S. Magazù, F. Migliardo and M. T. F. Telling, Eur. Biophys. J., 2007, 36, 163–171 CrossRef PubMed.
- N. Ekdawi-Sever, J. J. de Pablo, E. Feick and E. von Meerwall, J. Phys. Chem. A, 2003, 107, 936–943 CrossRef CAS.
- G. Cottone, J. Phys. Chem. B, 2007, 111, 3563–3569 CrossRef CAS PubMed.
- R. D. Lins, C. S. Pereira and P. H. Hünenberger, Proteins: Struct., Funct., 2004, 55, 177–186 CrossRef CAS PubMed.
- D. Corradini, E. G. Strekalova, H. E. Stanley and P. Gallo, Sci. Rep., 2013, 3, 1–10 Search PubMed.
- P. W. Fenimore, H. Frauenfelder, B. H. McMahon and F. G. Parak, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 16047–16051 CrossRef CAS PubMed.
- H. Frauenfelder, G. Chen, J. Berendzen, P. W. Fenimore, H. Jansson, B. H. McMahon, I. R. Stroe, J. Swenson and R. D. Young, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 5129–5134 CrossRef CAS PubMed.
- A. J. Saunders, P. R. Davis-Searles, D. L. Allen, G. J. Pielak and D. A. Erie, Biopolymers, 2000, 53, 293–307 CrossRef CAS PubMed.
- J. A. Schellman, Biophys. J., 2003, 85, 108–125 CrossRef CAS PubMed.
- K. A. Parker and Y.-H. Lim, J. Am. Chem. Soc., 2004, 126, 15968–15969 CrossRef CAS PubMed.
- T. F. O'Connor, P. G. Debenedetti and J. D. Carbeck, Biophys. Chem., 2007, 127, 51–63 CrossRef PubMed.
- G. Graziano, Int. J. Biol. Macromol., 2012, 50, 230–235 CrossRef CAS PubMed.
- S. Cozzolino, A. Tortorella, P. Del Vecchio and G. Graziano, Life, 2021, 11, 652 CrossRef CAS PubMed.
- A. Lerbret, F. Affouard, A. Hedoux, S. Krenzlin, J. Siepmann, M.-C. Bellissent-Funel and M. Descamps, J. Phys. Chem. B, 2012, 116, 11103–11116 CrossRef CAS PubMed.
- D. T. Bowron, A. K. Soper, S. Jones, K. Ansell, S. Birch, J. Norris, L. Perrott, D. Riedel, N. J. Rhodes, S. R. Wakefield, A. Botti, M. A. Ricci, F. Grazzi and M. Zoppi, Rev. Sci. Instrum., 2010, 81, 033905 CrossRef CAS PubMed.
- Disordered Materials, Database, https://www.isis.stfc.ac.uk/groups/disordered-materials/database/, (Accessed: May 2023).
- A. K. Soper, N. Gudrun and X. Gudrun, Rutherford Appleton Laboratory Technical report: RAL-TR-2011-013 https://epubs.stfc.ac.uk/work/56240, 2011.
- A. K. Soper, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 104204 CrossRef.
- C. Olsson and J. Swenson, Mol. Phys., 2019, 117, 3408–3416 CrossRef CAS.
- S. E. Pagnotta, S. E. McLain, A. K. Soper, F. Bruni and M. A. Ricci, J. Phys. Chem. B, 2010, 114, 4904–4908 CrossRef CAS PubMed.
- C. Olsson, H. Jansson, T. Youngs and J. Swenson, J. Phys. Chem. B, 2016, 120, 12669–12678 CrossRef CAS PubMed.
- K. Chu, J. Vojtchovský, B. H. McMahon, R. M. Sweet, J. Berendzen and I. Schlichting, Nature, 2000, 403, 921–923 CrossRef CAS PubMed.
- W. Damm, A. Frontera, J. Tirado-Rives and W. L. Jorgensen, J. Comput. Chem., 1997, 18, 1955–1970 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- C. Carlile and M. Adams, Phys. B, 1992, 182, 431–440 CrossRef CAS.
- O. Arnold, J. Bilheux, J. Borreguero, A. Buts, S. Campbell, L. Chapon, M. Doucet, N. Draper, R. Ferraz Leal, M. Gigg, V. Lynch, A. Markvardsen, D. Mikkelson, R. Mikkelson, R. Miller, K. Palmen, P. Parker, G. Passos, T. Perring, P. Peterson, S. Ren, M. Reuter, A. Savici, J. Taylor, R. Taylor, R. Tolchenov, W. Zhou and J. Zikovsky, Nucl. Instrum. Methods Phys. Res., Sect. A, 2014, 764, 156–166 CrossRef CAS.
- M. Beé, Quasielastic Neutron Scattering, Adam Hilger, 1988 Search PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, 2016 Search PubMed.
- C. I. Bayly, P. Cieplak, W. Cornell and P. Kollman, J. Phys. Chem., 1993, 97, 10269–10280 CrossRef CAS.
- K. Chu, J. Vojtchovsky, B. H. McMahon, R. M. Sweet, J. Berendzen and I. Schlichting, Nature, 2000, 403, 921–923 CrossRef CAS PubMed.
- J. B. Lim, B. Rogaski and J. B. Klauda, J. Phys. Chem. B, 2012, 116, 203–210 CrossRef CAS PubMed.
- J. Huang and A. D. MacKerell Jr, J. Comput. Chem., 2013, 34, 2135–2145 CrossRef CAS PubMed.
- F.-Y. Lin and A. D. MacKerell Jr, J. Inf. Model., 2018, 59, 215–228 CrossRef PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- W. L. Jorgensen and C. Jenson, J. Comput. Chem., 1998, 19, 1179–1186 CrossRef CAS.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. D. Nola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- H. Grubmüller, H. Heller, A. Windemuth and K. Schulten, Mol. Simul., 1991, 6, 121–142 CrossRef.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- B. Hess, C. Kutzner, D. van der Spoel and E. Lindahl, J. Chem. Theory Comput., 2008, 4, 435–447 CrossRef CAS PubMed.
- W. F. Van Gunsteren and H. J. Berendsen, Mol. Simul., 1988, 1, 173–185 CrossRef.
- H. J. C. Berendsen, D. van der Spoel and R. Drunen, Comput. Phys. Commun., 1995, 91, 43–56 CrossRef CAS.
- B. Hess, J. Chem. Theory Comput., 2008, 4, 116–122 CrossRef CAS PubMed.
- A. Barducci, G. Bussi and M. Parrinello, Phys. Rev. Lett., 2008, 100, 020603 CrossRef PubMed.
- M. Bonomi, A. Barducci and M. Parrinello, J. Comput. Chem., 2009, 30, 1615–1621 CrossRef CAS PubMed.
- G. Bussi and A. Laio, Nat. Rev. Phys., 2020, 1–13 Search PubMed.
- G. A. Tribello, M. Bonomi, D. Branduardi, C. Camilloni and G. Bussi, Comput. Phys. Commun., 2014, 185, 604–613 CrossRef CAS.
- C. Olsson and J. Swenson, J. Phys. Chem. B, 2020, 124, 3074–3082 CrossRef CAS PubMed.
- J. D. Nickels, H. O'Neill, L. Hong, M. Tyagi, G. Ehlers, K. L. Weiss, Q. Zhang, Z. Yi, E. Mamontov, J. C. Smith and A. P. Sokolov, Biophys. J., 2012, 103, 1566–1575 CrossRef CAS PubMed.
- S. Perticaroli, G. Ehlers, C. B. Stanley, E. Mamontov, H. O'Neill, Q. Zhang, X. Cheng, D. A. Myles, J. Katsaras and J. D. Nickels, J. Am. Chem. Soc., 2017, 139, 1098–1105 CrossRef CAS PubMed.
- S. Magazù, V. Villari, P. Migliardo, G. Maisano and M. T. F. Telling, J. Phys. Chem. B, 2001, 105, 1851–1855 CrossRef.
- H. Geng, F. Chen, J. Ye and F. Jiang, Comput. Struct. Biotechnol. J., 2019, 17, 1162–1170 CrossRef CAS PubMed.
- L. Heo and M. Feig, Proteins: Struct., Funct., Genet., 2018, 86, 177–188 CrossRef CAS PubMed.
- Z. E. Reinert and W. S. Horne, OBC, 2014, 12, 8796–8802 CAS.
- S. A. Hollingsworth and P. A. Karplus, Biomol. Concepts, 2010, 1, 271–283 CAS.
- A. V. Komolkin, A. Laaksonen and A. Maliniak, J. Chem. Phys., 1994, 101, 4103–4116 CrossRef CAS.
- I. Choi, Y. S. Huh and D. Erickson, Microfluid. Nanofluid., 2012, 12, 663–669 CrossRef CAS.
- C. Olsson, R. Zangana and J. Swenson, Phys. Chem. Chem. Phys., 2020, 22, 21197 RSC.
- K. Kaminski, E. Kaminska, P. Wlodarczyk, S. Pawlus, D. Kimla, A. Kasprzycka, M. Paluch, J. Ziolo, W. Szeja and K. L. Ngai, J. Phys. Chem. B, 2008, 112, 12816–12823 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp02639f |
| ‡ These authors contributed equally to this work. |
| This journal is © the Owner Societies 2023 |