
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mapping the electronic transitions of protonation sites in peptides using soft X-ray action spectroscopy†
Juliette
Leroux
 *ab,
Amir
Kotobi
bc,
Konstantin
Hirsch
d,
Tobias
Lau
d,
Carlos
Ortiz-Mahecha
e,
Dmitrii
Maksimov
f,
Robert
Meißner
ce,
Bart
Oostenrijk
g,
Mariana
Rossi
f,
Kaja
Schubert
b,
Martin
Timm
d,
Florian
Trinter
hi,
Isaak
Unger
j,
Vicente
Zamudio-Bayer
d,
Lucas
Schwob
b and
Sadia
Bari
*bgk
*ab,
Amir
Kotobi
bc,
Konstantin
Hirsch
d,
Tobias
Lau
d,
Carlos
Ortiz-Mahecha
e,
Dmitrii
Maksimov
f,
Robert
Meißner
ce,
Bart
Oostenrijk
g,
Mariana
Rossi
f,
Kaja
Schubert
b,
Martin
Timm
d,
Florian
Trinter
hi,
Isaak
Unger
j,
Vicente
Zamudio-Bayer
d,
Lucas
Schwob
b and
Sadia
Bari
*bgk
aCIMAP, CEA/CNRS/ENSICAEN/Université de Caen Normandie, 14050 Caen, France
bDeutsches Elektronen-Synchrotron DESY, Germany. E-mail: juliette.leroux@desy.de; sadia.bari@desy.de
cHelmholtz-Zentrum Hereon, Institute of Surface Science, 21502 Geesthacht, Germany
dAbteilung Hochempfindliche Röntgenspektroskopie, Helmholtz-Zentrum Berlin für Materialien und Energie, 12489 Berlin, Germany
eHamburg University of Technology, Institute of Polymers and Composites, 21073 Hamburg, Germany
fMax Planck Institute for the Structure and Dynamics of Matter, 22761 Hamburg, Germany
gThe Hamburg Centre for Ultrafast Imaging, Hamburg, Germany
hInstitut für Kernphysik, Goethe-Universität Frankfurt am Main, 60438 Frankfurt am Main, Germany
iMolecular Physics, Fritz-Haber-Institut der Max-Planck-Gesellschaft, 14195 Berlin, Germany
jDepartment of Physics and Astronomy, Uppsala University, 75120 Uppsala, Sweden
kZernike Institute for Advanced Materials, University of Groningen, 9747 AG Groningen, The Netherlands
First published on 18th September 2023
Abstract
Near-edge X-ray absorption mass spectrometry (NEXAMS) around the nitrogen and oxygen K-edges was employed on gas-phase peptides to probe the electronic transitions related to their protonation sites, namely at basic side chains, the N-terminus and the amide oxygen. The experimental results are supported by replica exchange molecular dynamics and density-functional theory and restricted open-shell configuration with single calculations to attribute the transitions responsible for the experimentally observed resonances. We studied five tailor-made glycine-based pentapeptides, where we identified the signature of the protonation site of N-terminal proline, histidine, lysine and arginine, at 406 eV, corresponding to N 1s → σ*(NHx+) (x = 2 or 3) transitions, depending on the peptides. We compared the spectra of pentaglycine and triglycine to evaluate the sensitivity of NEXAMS to protomers. Separate resonances have been identified to distinguish two protomers in triglycine, the protonation site at the N-terminus at 406 eV and the protonation site at the amide oxygen characterized by a transition at 403.1 eV.
Introduction
In the biological medium, the charge states of molecules such as peptides, proteins, and nucleic acids play a significant role as they influence conformation and reactivity. Within the physiological pH range, an important mechanism altering the charge state is the protonation and deprotonation of specific chemical groups. To improve our understanding of biological processes and the structure–function relationship, we need to localize the (de)protonation sites in biomolecules. Experimentally, protonation sites in peptides have been studied using different techniques over the last few decades mainly by gas-phase basicity studies1–4 in order to evaluate proton affinities of chemical groups. According to the proton affinities of the different sites, protonation in amino acids and small peptides occurs preferably at the nitrogen site of the most basic side chains (arginine, histidine, and lysine with proton affinities of 250.2, 231.8, and 237.1 kcal mol−1, respectively5) and at the N-terminus primary amine –NH2.1 However, in certain cases, for example, when the number of protons exceeds the number of basic residues, protonation can also occur on the peptide backbone. Action spectroscopy techniques, and in particular infrared (IR) spectroscopy, are powerful tools for the determination of molecular structures in the gas phase that can be used to assess the protonation site of molecules as big as small peptides.6,7Protonated triglycine has been studied theoretically and experimentally by many groups mostly to understand its structure and sites of protonation that remains controversial. In her theoretical study on glycine oligomers, Chung-Phillips concluded that the most stable conformer of triglycine is protonated at an amide oxygen in the backbone.8 Wu and McMahon employed infrared multiphoton dissociation (IRMPD) to determine the structure and protonation site of triglycine. They showed that, under their experimental conditions, two distinct protomers of triglycine with different protonation sites were responsible for the recorded spectrum,9 one protonated at the N-terminus of the tripeptide and the other protonated on the first amine oxygen. The same conclusion was drawn by Grégoire and co-workers in a study in which they combined resonant IRMPD of protonated triglycine and Car–Parrinello molecular dynamics calculations.7 However, in a recent study, Li and co-workers found that the protonation site of triglycine is the N-terminal nitrogen rather than the amine oxygen, which they found to be at least 0.14 kcal mol−1 more stable.10 The authors also calculated the X-ray absorption spectra at the carbon, nitrogen, and oxygen K-edges of triglycine protomers with different conformations. They observed clear differences between the linear and folded conformers of triglycine, with resonances unique to some conformers and with the strongest effects being observed at the nitrogen and oxygen edges. This theoretically demonstrated that near-edge X-ray absorption fine structure (NEXAFS) spectroscopy can be sensitive to the different protonation sites of a peptide. Furthermore, this method has already been applied to determine the position of hydrogen atoms and the protonation state of biomolecules in the liquid phase and in crystals.11,12
Over the last few decades, soft X-ray spectroscopy has become an important technique for studying the electronic structure of biomolecular systems, both in the condensed13,14 and gas phases.15–17 In fact, soft X-ray excitation is element specific, so tuning the photon energy to a resonance allows site-selective excitation of a molecule to specific unoccupied molecular orbitals. Very recently, Wang and co-workers demonstrated using near-edge X-ray absorption mass spectrometry (NEXAMS) that the electronic structure of gas-phase oligonucleotides is influenced by the protonation site.18 In a previous study, Otero and Urquhart observed that peptides in the solid phase show differences in their N 1s NEXAFS spectra depending on the protonation state of the amine group.19 For amine-protonated species, they observed a feature at a high energy of around 406 eV, well above the resonance around 402 eV characterizing an unprotonated amine group.
To go further and map the different electronic transitions responsible for the protonation site in peptides, we studied tailor-made glycine-based pentapeptides. Glycine is the simplest amino acid with a single hydrogen as its side chain and has been studied extensively.2,20–26 Therefore, the study of peptides made from glycines and one basic amino acid at the C-terminus, or a proline at the N-terminus, allowed us to avoid probing effects from side chains’ atoms that are not at the actual protonation site, while keeping the system simple for comparison with calculations. To probe the electronic structures of the different protonation sites in peptides, we studied five different glycine-based protonated pentapeptides by soft X-ray action spectroscopy. As protonation occurs preferentially at nitrogen sites, we present here a study at the nitrogen K-edge of model peptides with known protonation sites: [GGGGG + H]+, [PGGGG + H]+, [GGGGH + H]+, [GGGGR + H]+, and [GGGGK + H]+ (noted G5, PG4, G4H, G4R, and G4K, respectively, in the following), where G stands for glycine, P for proline, H for histidine, R for arginine, and K for lysine, as well as protonated triglycine [GGG + H]+, denoted G3 in the following. The chemical structures of G5 and of the amino acids that make up the other pentapeptide sequences are shown in Fig. 1. G5 and PG4 are both N-terminal-protonated but with primary and secondary amines, respectively. Histidine, arginine, and lysine are basic residues, and the protonation occurs on their side chains. The study of triglycine allows us to test the theory of Li and co-workers.10
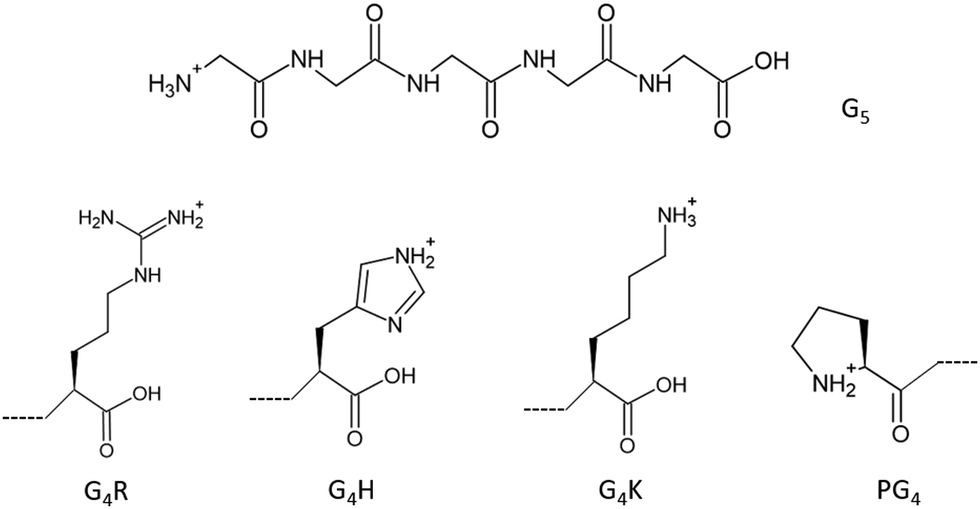 | ||
| Fig. 1 Chemical structure of protonated pentaglycine (top) and the chemical structures of the last amino acid of G4R, G4H, G4K, and the first amino acid of the sequence PG4, from left to right (bottom). | ||
Methods
All custom peptides were purchased from ProteoGenix (Schiltigheim, France), at >95% purity and used without further purification. The sample solutions were prepared at 30 μM concentration in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 water/methanol with 1% of formic acid to ensure the protonation of the peptides.
:1 water/methanol with 1% of formic acid to ensure the protonation of the peptides.
The experiments have been performed during two experimental campaigns, one at the BESSY II synchrotron (HZB, Berlin, Germany) and one at the PETRA III synchrotron (Deutsches Elektronen-Synchrotron DESY, Germany) on two different apparatus with a similar principle. The results for protonated G4H, G4R, and PG4 have been obtained by coupling our home-built tandem mass spectrometer to the P04 soft X-ray beamline of PETRA III. This setup relies on a high-fluence electrospray ionization (ESI) source to introduce the ions into the gas phase. The electrosprayed ions are transported through a heated capillary into the first pumping stage which hosts an ion funnel that collects and focuses the ions. They continue through a radio frequency (RF) octupole ion guide to a quadrupole mass filter where the ions of interest are selected according to their mass-to-charge (m/z) ratio. Then, a second octupole and a set of Einzel lenses guide and focus the ions into an RF 3D Paul ion trap where they are accumulated prior to irradiation with photons. To allow efficient trapping of the ions, their kinetic energy is cooled down to room temperature by collisions with helium buffer gas. All cationic products are eventually extracted from the trap and analyzed in a reflectron time-of-flight (Re-TOF) mass spectrometer (m/Δm = 1800). The duration of the synchrotron light exposure, typically 2 seconds, is controlled with an optomechanical shutter. At the N K-edge, the energy bandwidth was set to 250 meV. The photon energy scans were performed by steps of 200 meV across the N K-edge (397–412 eV). A photodiode placed downstream of the ion trap was used to record the photon flux. The NEXAMS spectra obtained during this beamtime were normalized to photon flux and precursor ion intensity to remove the contribution of the ESI source fluctuations. To account for all sources of background ions, the data acquisition was divided into repeated cycles of three mass scans. First, a mass spectrum of the ESI only was recorded to measure the precursor ion intensity and fragments originating from energetic collisions with the buffer gas. Second, the mass spectrum resulting from irradiation of the trapped molecules was recorded. For the third spectrum, the ESI is switched off and the mass spectrum resulting from the irradiation of the residual gas was recorded. The mass spectrum resulting from the ESI and the one resulting from the irradiation of the residual gas were subtracted from the mass spectrum resulting from the irradiation of the trapped molecules. To obtain the final mass spectrum, a series of a hundred cycles was accumulated to average over long-term fluctuations in the source. Therefore, in the final subtracted mass spectrum, the precursor ion peak of each molecule appears negative due to this procedure.
The results on protonated G3, G5, and G4K have been obtained at the UE52_PGM Ion Trap beamline of the BESSY II synchrotron, as described elsewhere.27,28 Briefly, the instrument is equipped, similar to the setup described above, with an ESI source, a quadrupole mass filter, a cryogenically cooled linear ion trap and a Re-TOF mass spectrometer. For G4K, the temperature of the trap was set at 290 K. Contrary to side-chain protonation, backbone protonation is more sensitive to a peptide's conformation. In an attempt to reduce the number of conformers in the trap and limit them to the lowest energy conformers, the temperature of the trap was set to 8 K for G3 and G5. The N K-edge spectra were recorded by steps of 100 meV and 210 meV bandwidth, across the range 397–410 eV (only to 408 eV for G4K). Measurements at the O K-edge for G5 and G3 were additionally performed to assess the effect of a potential amide oxygen protonation. Here, the photon–energy bandwidth was 200 meV and the photon energy was ramped in 100 meV steps between 528 and 545 eV. A photodiode located behind the interaction region was used to record the photon flux for data normalization. Note that the design of the ion trap instrument limits the detection of the irradiation products to a small m/z window. The extraction parameters were set to observe fragments over a mass range of about m/z 50–150 for G4K and over a mass range of m/z 25–70 for the measurements of G5 and G3. The NEXAMS spectra, or total ion yield (TIY) spectra, can be assumed to be good approximations of the X-ray absorption cross section, and thus comparable to the calculated DFT/ROCIS oscillator strengths. Indeed, the partial ion yields for individual fragments are the results of the convolution of the absorption cross section of the molecule with the fragments’ branching ratio. By summing up, the contribution of all fragments into a “total ion yield” spectrum, we approach the result of an absorption spectrum.29 Nevertheless, disagreement in spectral intensity could arise in the case of detecting multiple ions from the same photoabsorption event or due to discrepancies in the overall detection efficiency of ions. NEXAMS, or TIY NEXAFS, has been extensively used and compared with X-ray absorption calculations.17,18,30–35
Theoretical calculations
Structural candidates for G3 and the different pentapeptides were obtained with different structure search techniques. For G3, a random structure search was performed, where all dihedral angles of the molecule were allowed to rotate with random angles. The search was conducted using the GenSec package.36,37 We sampled 600 structures starting from the three different protonation states labelled GGGH1, GGGH2 and GGGH3 (see Fig. 5). These structures were fully optimized with the PBE038 exchange-correlation functional including the pairwise TS-vdW corrections39 with the FHI-aims program package.40 The final optimized structures were ranked by their total energy, and we selected conformers lying on the lowest 1 eV range for each protonation state. In total, we obtained 10 GGGH1, 10 GGGH2, and 5 GGGH3 conformers. This search strategy was targeted at finding low-temperature, enthalpically stabilized conformers. We subsequently calculated in silico X-ray absorption spectra using the density functional theory (DFT) combined with the restricted-open-shell configuration interaction with singles excitations (ROCIS) for the lowest energy conformer of each protonation state.41 The calculated X-ray absorption spectra of GGGH3 do not reproduce the experiment at all, the results for this conformer will not be discussed further in this paper. Apart from the fact that only a few conformations were found in the ab initio based GenSec structure search performed in this work, there is evidence in the literature that only a few conformers contribute to the spectroscopic signals of small glycine peptides, as shown in the study by Wu and Mc Mahon.9 In addition, the extensive conformer search performed by Hudgins and co-workers found essentially only one stable conformation in the case of glycine pentapeptides and smaller glycines.42A different strategy was adopted for the pentapeptides, targeted at finding higher temperature stable conformers. We conducted molecular dynamics simulations and DFT/ROCIS calculations for the pentapeptides using the GROMACS program (version 2018.8) and the ORCA electronic structure package,43 respectively. To efficiently sample the potential energy surface (PES), exponentially distributed replica exchange molecular dynamics (REMD) simulations with different temperatures ranging from 300 K to 648 K were performed based on the Amber ff14SB force field44 in GROMACS. The conformers generated by the REMD simulation were represented by the values of their dihedral angles for the backbone and side chains. Calculating these descriptors for each snapshot of the REMD simulation resulted in a feature dataset of the REMD trajectory. These features were used to cluster conformers with similar structures using the probabilistic analysis of molecular motifs (PAMM)45,46 such that we could identify recurring molecular conformations of different metastable states sampled during REMD. PAMM is specifically designed to work in relatively high-dimensional spaces and can partition the underlying probability distribution function into modes corresponding to different conformers. The lowest REMD energy conformation of each identified cluster was chosen as the representative conformer of that cluster and considered for the calculation of the absorption spectra, as discussed by Kotobi and co-workers.47 A Boltzmann weighting factor based on the REMD energy differences between the representative conformers and normalized by the cluster population was employed to estimate the statistical relevance of the various metastable states and to obtain weighted average spectra considering a canonical ensemble of the conformers. Conformers, from REMD, having a weighting factor less than 2% of the contribution were assumed to be negligible. All theoretical absorption spectra presented in this paper were obtained using the DFT/ROCIS approach, employing the TZVP Ahlrichs basis set48 in combination with the B3LYP exchange–correlation functional.49 The optimized conformers were used to calculate transition energies and dipole moments for carbon, nitrogen and oxygen K-edges. We calculated the average absorption spectra by weighting the contribution of different conformers, using the previously calculated weighting factors. The final absorption spectra were then broadened, using a pseudo-Voigt profile with a full width at half-maximum (FWHM) of 0.6 eV.50 The shift in the energy of the theoretical spectra is done by matching the first resonance of the theoretical spectra to the corresponding one in the experimental spectra, representing π*CONH electronic transitions in the peptide backbone, as explained later.51,52 This shift is due to the B3LYP functional underestimating the core excitation energies.53,54 Such a method has already been used successfully to compare DFT calculations with soft X-ray absorption spectra.15,17,23,55
Results and discussion
Photodissociation mass spectra analysis
Fig. 2(a)–(f) show the mass spectra measured for peptides G3 (m/z 190), G5 (m/z 304), PG4 (m/z 344), G4H (m/z 384), G4R (m/z 403), and G4K (m/z 375) at the photon energy of 401.6 eV (π*(CONH) resonance, as discussed later). The photodissociation mass spectra over the full mass range for the six peptides are shown in the ESI† (Fig. S1–S6). As can be seen in Fig. S1–S6 (ESI†), for all peptides, no intact photoionized precursor ions have been observed. Fragments are labelled according to the peptide fragment nomenclature established by Roepstorff, Fohlman, and Biemann.56,57 A table summarizing the different fragments is shown in ESI.†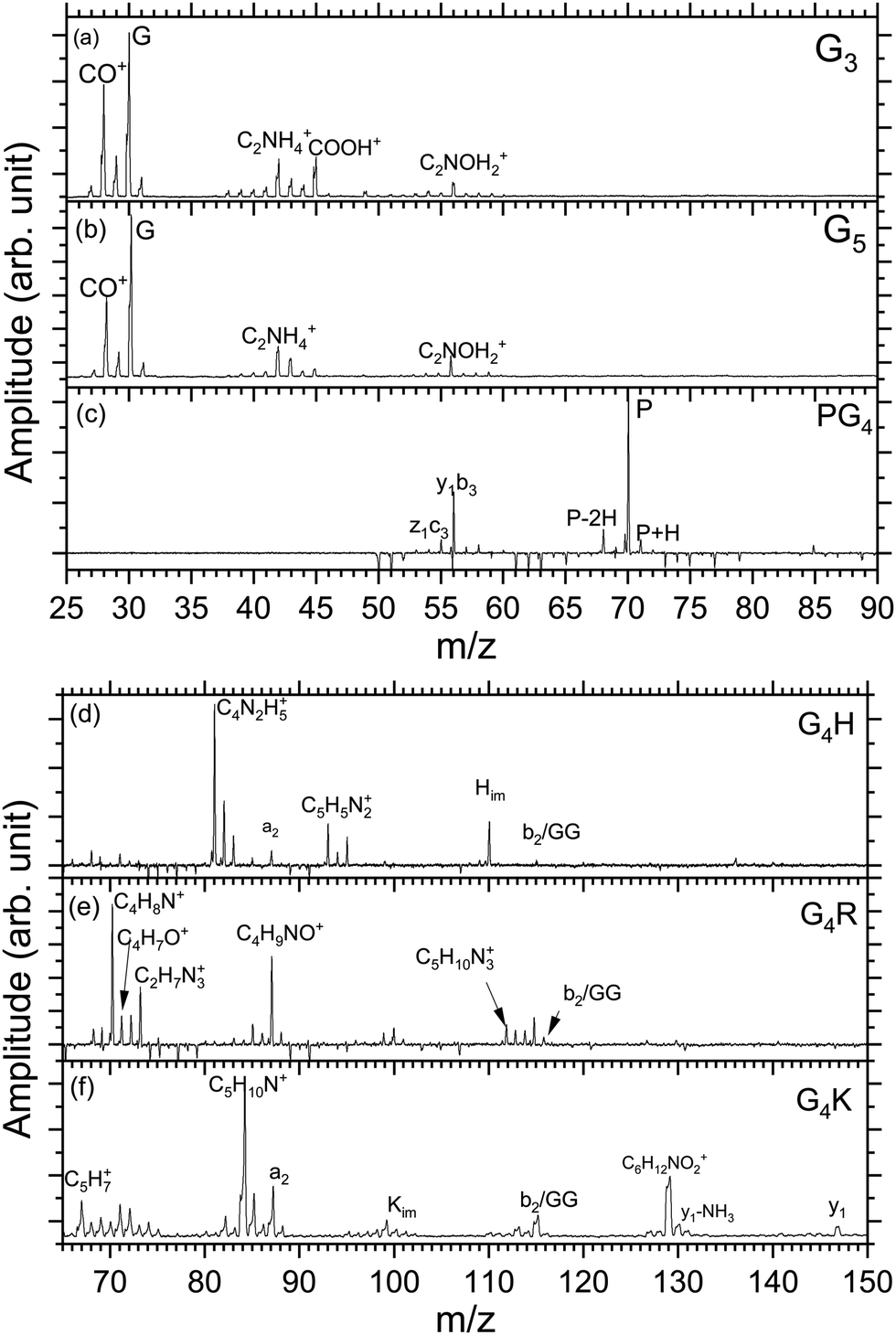 | ||
| Fig. 2 Low mass region of the photodissociation mass spectra of all investigated protonated peptides measured at 401.6 eV photon energy. The mass range changes between the two sets of mass spectra to show the main fragments produced by photodissociation. Peaks of negative intensity are due to the background subtraction. | ||
Triglycine and pentaglycine are composed of the same residues, which reflects in their similar mass spectra (Fig. 2(a) and (b)) with fragments stemming from the backbone. m/z 28 corresponds to a fragment that can be attributed to C2H4+ or CH2N+, m/z 29 is attributed to the CHO+ fragment. The immonium ion of glycine is observed at m/z 30 and at m/z 31 is the fragment CH3O+. m/z 42 corresponds to C2H4N+, m/z 43 to CHNO+, m/z 45 to COOH+, and m/z 56 to C2NOH2+. These fragments have also been reported for neutral glycine under VUV ionization at 21.21 eV by Lago and co-workers58 and by Majer and co-workers using electron-impact ionization.59
In the photodissociation mass spectrum of PG4 (Fig. 2(c)), fewer fragments are observed. The most prominent one is the fragment at m/z 70, which is the immonium ion of the proline amino-acid residue, that is also produced by collision-induced dissociation (CID) as observed by Zhang and co-workers.60 The other two fragments at m/z 68 and m/z 71 are related to proline as well. The two fragments at m/z 56 and m/z 55 are attributed to internal backbone fragments, C2NOH2+ and C2NOH+, respectively.
In the spectrum of G4H, shown in Fig. 2(d), most of the ions observed result from fragmentation taking place at the side chain of histidine. The spectrum shows at m/z 110 the immonium ion of histidine (C5H8N3+), as observed in the CID experiment,61 accompanied by an ammonia loss (–NH3) at m/z 93 and an additional loss of HCN at m/z 83. The fragments at m/z 82 and 81 result from further hydrogen losses, the latter being the dominant fragment. The peak at m/z 87 is attributed to the a2 fragment. The fragment at m/z 115 can be attributed to the internal backbone fragment GG or the b2 fragment which have the same elemental composition.
For the mass spectrum of G4R, presented in Fig. 2(e), some fragments remain unassigned and the others are mainly due to fragmentation at the protonated arginine side chain. We do not observe the immonium ion of arginine at m/z 129 but we do observe the related ions at m/z 112, 87, 70. Other fragments at m/z 71, 72, 73, 88 and 85 are attributed to the arginine residue, and the fragments' elemental compositions are given in Table S1 (ESI†). Only the fragments at m/z 70, 71, 72, 88, and 112 have been reported in CID experiments.45 Note that the intense peak at m/z 87 most likely also has a contribution from the a2 ion. GG/b2 fragments are also observed at m/z 115.
In the spectrum of G4K (Fig. 2(f)), we were not able to clearly identify either the fragments around m/z 70 or those around m/z 100. These fragments are not background and were therefore included in the analysis. We observed the y1 fragment at m/z 147 and an –NH2 loss from the y1 fragment at m/z 131. m/z 67 and m/z 84 attributed to C5H7+ and C5H10N+ ions from the lysine side chain have been also observed in the CID.60 Other lysine-related fragments are observed at m/z 129, m/z 112, and m/z 101, the latter being the immonium ion of lysine. m/z 87 is the a2 fragment from the peptide. The fragment at m/z 115 can be attributed to the internal backbone fragment GG or the b2 fragment for G4H and G4R. As the amino-acid sequences of G4K and G4H are the same for the first four residues, it is expected to observe a2 and b2 for all peptides under study except PG4. The internal fragment GG should also be a common fragment of all peptides. The fact that it is not observed in PG4 (see Fig. S2, ESI†) suggests the fragment m/z is most likely due to a b2 fragment rather than GG in G4H, G4R and G4K.
To summarize, in the mass spectra of G4K, G4H, and G4R, the majority of the ions are attributed to fragments from the side chain of the basic residues in the peptides, lysine, histidine, and arginine, respectively. PG4, G5, and G3 do not contain a basic side chain, thus the fragments observed arise only from backbone dissociation. Most of the observed fragments were also reported in collision-induced dissociation (CID) experiments60 suggesting that, in our case, internal conversion followed by intramolecular vibrational redistribution of internal energy is the main process following photoexcitation/photoionization and Auger decay, eventually leading to statistical dissociation of the peptides.
Different protonation sites on amino groups in peptides
The total ion yields (TIY) of each molecule were calculated by summing the areas under the peaks of all detected fragments in the photo-induced mass spectra, for each photon energy over the scanned energy range. No specific fragmentation channels could be identified over the scanned photon energies for the different studied peptides, as shown in the different partial ion yield spectra in the ESI† (Fig. S7–S12).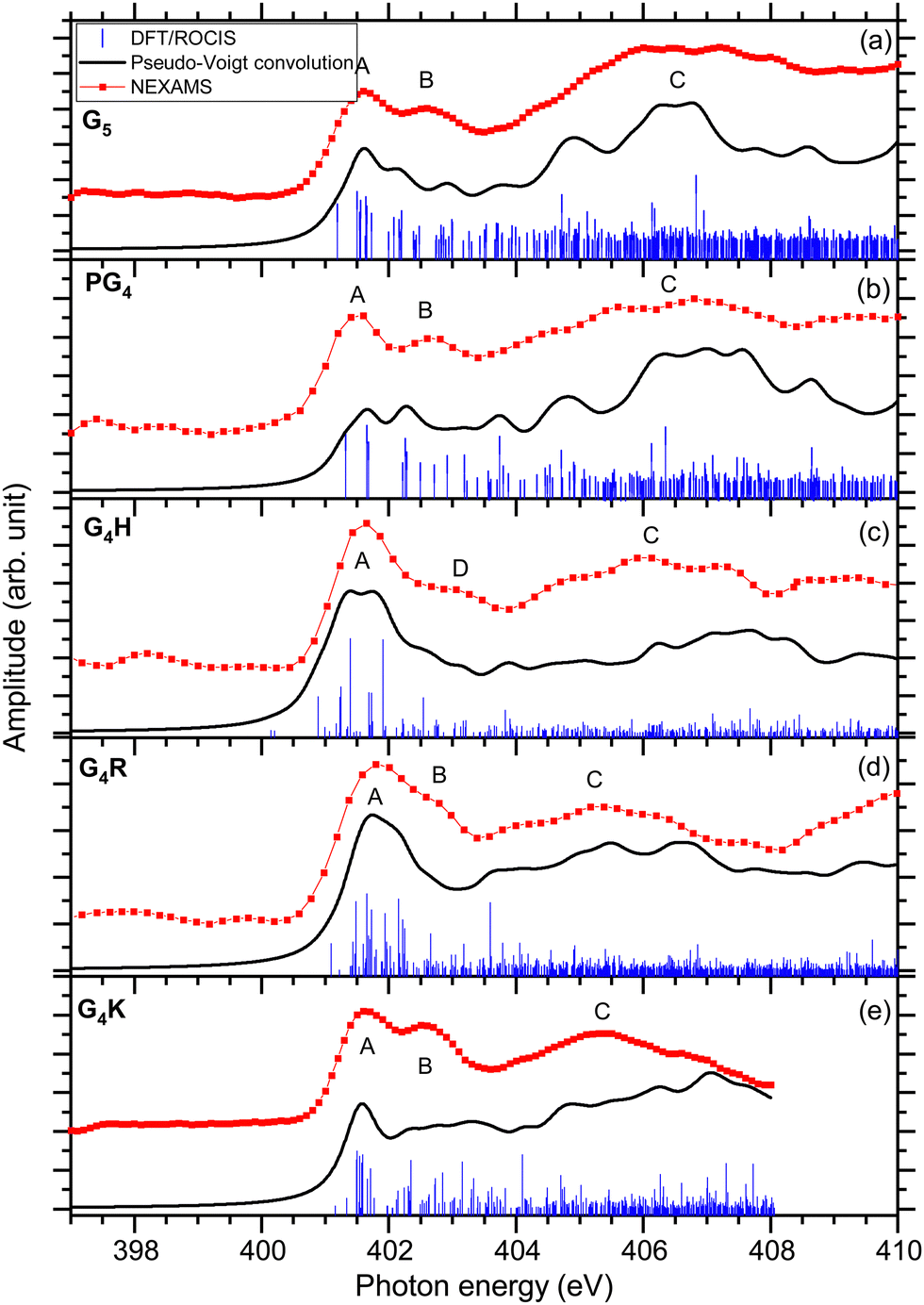 | ||
| Fig. 3 The experimental nitrogen K-edge total ion yield spectra for G5, PG4, G4H, G4R, and G4K, are shown with red dotted lines. In blue, the DFT/ROCIS X-ray absorption lines are displayed for each molecule. The absorption lines were shifted by ∼13 eV and were broadened using a Pseudo-Voigt profile with a 0.6 eV FWHM represented in black. | ||
The first resonance A at 401.6 eV is attributed to N 1s → π*(CONH) transitions within peptide bonds according to the DFT/ROCIS transition lines. This has already been reported for peptides in previous papers.15,21,23,63 The origin of resonance B at 402.6 eV is controversial. For gas-phase diglycine, Feyer and co-workers attributed this feature to an N 1s → π*(N-Cα) transition,26 as well as Dörner and co-workers for protonated methionine-enkephalin.15 Zubavichus and co-workers proposed that this feature is an N 1s → π*(CONH) transition, that is energetically shifted because of potentially different conformations existing in their studied homopolypeptides.63 Using time-dependent density functional theory (TDDFT) to calculate NEXAFS spectra of α-helix and β-turn models based on glycines, Buckley and Besley attributed the first two narrow resonances at the N K-edge to N 1s → π*(CONH), with the second band associated with π* orbitals in a neighbouring residue.50 Their computed spectra also suggest that this second feature is highly susceptible to the conformation of peptide, being more pronounced in an α-helix than in a β-turn. In the energy range of resonance B, the analysis of the lowest unoccupied molecular orbitals (LUMOs) of the conformers of G5 (LUMO+1 of conformer 1 and LUMO+3 of conformer 2) leads to transitions from the N 1s level in one peptide bond to unoccupied π*(CONH) orbitals in neighbouring peptide bonds. We finally attribute, according to our calculation and in agreement with the work of Buckley and Besley,50 resonance B to Ni 1s → π*(CONjH) where Ni and Nj are different nitrogen atoms in the peptide backbone.
The DFT/ROCIS calculations show two major transitions in the energy region of 404–408 eV, one resonance at 404.8 eV and one at 406.6 eV, that are contributing in the experimental spectrum to one broad spectral feature, noted C. The study of the molecular orbitals of the two main conformers of G5 (LUMO+1 and LUMO+2 of conformer 1, LUMO of conformer 2) assigned the first resonance to an N 1s → π*(CO) transition in the carboxylic group of the C-terminus, where the DFT/ROCIS calculations show that the transition is due to the excitation from the nitrogen atoms of the N-terminus to the carboxylic group. Such a transition occurs thanks to the folded conformation of G5 and the spatial proximity of the N and C-termini. The peak at 406.6 eV is assigned to transitions located at the protonated N-terminus, N 1s → σ*(NH3+) with the density of molecular orbitals at the N-terminus (LUMO+5 of conformer 1 and conformer 2). Similar transitions at 406.8 and 406.5 eV have been previously reported by Otero and Urquhart in solid-state NEXAFS spectra of zwitterionic glycine and glycine hydrochloride, respectively, and assigned to both N 1s → σ*(NH) and N 1s → σ*(CN).19
The first resonance A at 401.6 eV is attributed to N 1s → π*(CONH) transitions in a peptide bond according to the DFT/ROCIS transition lines (LUMO). In the energy range of resonance B, the DFT/ROCIS calculations show a resonance at 402.1 eV and one at 403.7 eV, involving the molecular orbitals LUMO+1 and LUMO+3, that can both be attributed to electronic transitions from one peptide bond to a neighboring peptide bond. However, experimentally, only a resonance at 402.6 eV has been observed. We therefore attribute resonance B to Ni 1s → π*(CONjH) transitions to neighboring peptide bonds, as discussed for G5 and consistent with our observations for the different protonated pentapeptides in this study.
Feature C is composed of different electronic transitions which explain the broadness of the peak. The DFT/ROCIS result gives a resonance at 404.7 eV mainly due to an N 1s → σ*(NH) transition from the nitrogen atoms in the backbone, whereas at 406.5 V, the calculated molecular orbital (LUMO+7) shows most of the density around the proline residue and describes an N 1s → σ*(CH) electronic transition due to the excitation of the protonated secondary amine nitrogen. Note that, for the immonium ion (m/z 70), shown in Fig. S8 (ESI†), the intensity of feature C is lower than for the other fragments contributing to the total ion yield spectrum.
The first resonance A is located at 401.6 eV and can be partly attributed to N 1s → π*(CONH) transitions in the peptide according to the analysis of the DFT/ROCIS transition lines at 401.2 eV. Additionally, the main resonance peak energetically overlaps with N 1s → π*(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) electronic transitions in the imidazole ring of the histidine side chain. The calculations give the transition lines at 400.8 eV, 401.4 eV, and 401.9 eV due to the double bond CN of the histidine imidazole ring (LUMO) and the transition lines at 401.2 eV, involving LUMO+1 and LUMO+2, stemming from the N 1s → π*(CONH) peptide bond transitions. These additional histidine-related transitions contribute to the increased intensity and broadening of resonance A in comparison with the other peptides. In contrast to the other studied pentapeptides, neither experimentally nor theoretically, does the absorption spectrum of protonated G4H show a resonance B at 402.6 eV, attributed to Ni 1s → π*(CONjH) transition in neighboring peptide bonds. Resonance B at 402.6 eV is only distinguishable in the partial ion yield spectrum of the immonium ion (m/z 110) (Fig. S9, ESI†) is. This could be attributed to the fact that the absorption in the histidine side chain in resonance A does not lead to the production of the immonium ion (similar to what was discussed for the proline immonium ion in PG4); thus, resonance A is relatively smaller in this fragment and resonance B can be observed. It is worth noting here that the PIY of G4H is dominated by the ion yield of fragment m/z 81 that involves H3CN loss from the histidine side chain. This fragment shows a very high yield at resonance A, which most probably correlates with the N 1s → π*(CN) electronic transitions within the histidine side chain.
N) electronic transitions in the imidazole ring of the histidine side chain. The calculations give the transition lines at 400.8 eV, 401.4 eV, and 401.9 eV due to the double bond CN of the histidine imidazole ring (LUMO) and the transition lines at 401.2 eV, involving LUMO+1 and LUMO+2, stemming from the N 1s → π*(CONH) peptide bond transitions. These additional histidine-related transitions contribute to the increased intensity and broadening of resonance A in comparison with the other peptides. In contrast to the other studied pentapeptides, neither experimentally nor theoretically, does the absorption spectrum of protonated G4H show a resonance B at 402.6 eV, attributed to Ni 1s → π*(CONjH) transition in neighboring peptide bonds. Resonance B at 402.6 eV is only distinguishable in the partial ion yield spectrum of the immonium ion (m/z 110) (Fig. S9, ESI†) is. This could be attributed to the fact that the absorption in the histidine side chain in resonance A does not lead to the production of the immonium ion (similar to what was discussed for the proline immonium ion in PG4); thus, resonance A is relatively smaller in this fragment and resonance B can be observed. It is worth noting here that the PIY of G4H is dominated by the ion yield of fragment m/z 81 that involves H3CN loss from the histidine side chain. This fragment shows a very high yield at resonance A, which most probably correlates with the N 1s → π*(CN) electronic transitions within the histidine side chain.
Resonance D at 403.1 eV is found, experimentally, at significantly higher energy than resonance B at 402.6 eV for the other protonated pentapeptides. The DFT/ROCIS calculation gives a resonance at 402.5 eV due to N 1s → σ*(CH) and N 1s → σ*(NH) electronic transitions (LUMO+6) in the histidine side chain where the protonation site is located. For G4H, the resonance at 403.1 eV is the signature of the protonation site which explains the low intensity of broad feature C compared to the intensity of feature C in G5. The broad spectral feature located in C between 404 and 408 eV remains unclear as it is due to different electronic transitions from the backbone of the protonated peptides. The DFT/ROCIS transitions are all of low intensity and cannot be attributed to one resonance because the molecular orbitals are highly delocalized. This feature has a contribution from LUMO+13 and LUMO+19 for the transition lines at 407.1 eV and from LUMO+34 for the transition lines around 407.9 eV. The analysis of the LUMO+13 and LUMO+19 orbitals shows that feature C includes contributions from σ* transitions from the backbone and the side chain.
In the experimental spectrum, the first two resonances are not well resolved compared to the other studied peptides. The first resonance A at 401.6 eV has been attributed to the peptide bond resonance N 1s → π*(CONH) arising from transitions to the first LUMOs of both conformers (LUMO to LUMO+6 for conformer 1 and from LUMO to LUMO+2 for conformer 2).
According to our DFT/ROCIS calculations, two resonances are responsible for the second feature B at 402.6 eV: Ni 1s → π*(CONjH) (LUMO of conformer 1) to a neighbouring peptide bond and N 1s → π*(CN) (LUMO+4 of conformer 2) in the arginine side chain. In the case of zwitterionic arginine, Zubavichus and co-workers observed a π* resonance located around 401.4 eV that they attributed to the guanidine group of arginine, whereas Leinweber and co-workers observed the π* resonance of arginine at 401.7 eV.13,14 Moreover, Stewart-Ornstein and co-workers attributed the resonance at 402.9 eV of a Sub6 solid sample, an arginine-containing antimicrobial peptide, to a π*(CN) transition in the arginine residue.64 In the case of protonated G4R, our calculations show that this resonance is shifted higher in energy to lie between the two peptide bond resonances at 402 eV. The overlap with this second transition explains why the resonance here is not clearly separated by one eV from the first peptide bond resonance, as some transition lines from the DFT/ROCIS calculations are related to the π*(CN) resonance. Interestingly, resonance B is more apparent in the PIY of the fragment m/z 73 (C2H7N3+) that is formed from the intact guanidine group of the arginine side chain. It can therefore be understood that N 1s → π*(CN) in the arginine would likely result in a CN bond cleavage explaining why these transitions are not observed in the PIY of m/z 73 (Fig. S10, ESI†) and thus better resolving the separation of resonances A and B. The DFT/ROCIS calculations show a transition at 403.6 eV that can possibly be responsible for the small feature before the broad one between 404 and 408 eV. This transition is attributed to N 1s → σ*(NH2+) transitions from the arginine side chain due to the additional proton for conformer 1 (LUMO+9). For conformer 2, it is more ambiguous as there are two main transition lines at 403.96 eV and 404.1 eV that are responsible for this small feature. The transition at 403.96 eV is due to the LUMO+2 and is attributed to N 1s → π*(CO) transitions, whereas at 404.1 eV, the molecular orbital (LUMO+13) shows most of the density around the arginine side chain, with some of the density in the backbone as well, and describes an N 1s → σ*(NH) transition due to an electronic excitation in the protonated side chain. Therefore, we concluded that the fingerprint of the protonation site is the first resonance of feature C, at 404 eV. We observed a broad feature, noted C between 404 eV and 408 eV which is less intense than feature C of the other protonated peptides in this study relative to feature A. As the spectra are normalized on the intensity of peak A, this effect could be explained by a higher intensity of peak A in the case of G4R because of the additional π*(CN) transition from the arginine side chain that can be found between the resonances A and B. The DFT/ROCIS calculations show that different electronic excitations are responsible for feature C with very low amplitude, which arises mainly from highly delocalized molecular orbitals. Comparing with the NEXAMS spectra of the other pentapeptides of this study, we conclude that the feature is mainly due to σ* transitions from the backbone.
The experimental spectrum of G4K presented in Fig. 3(e) shows the same three main features as the other peptides studied. The first peak A is the main peptide bond resonance N 1s → π*(CONH) at 401.6 eV (LUMO+1 of conformer 1 and LUMO+2 of conformer 2). This is consistent with our results obtained for the other peptides. As for protonated G4R, the second feature at 402.6 eV is due to different contributions. The DFT/ROCIS calculations of conformer 1 show that the peak at 402.4 eV only originates from Ni 1s → π*(CONjH) transitions to neighbouring peptide bonds (LUMO+5). On the other hand, the results of the DFT/ROCIS simulation for conformer 2 show that the resonance at 402.6 eV has major contributions from two different electronic transitions. The first one at 402.3 eV is attributed to N 1s → π*(COOH) (LUMO+5) in the C-terminus but with the excitation coming from the nitrogen of the fourth glycine rather than the nitrogen of the C-terminus connected to the lysine residue. The second electronic transition is N 1s → σ*(NH) in the protonated lysine side chain at 403.1 eV (LUMO+3). The third feature is due to N 1s → σ*(CH) electronic transitions from the backbone (LUMO+8 of conformer 1) around 404.5 eV, and N 1s → σ*(NH) from the backbone and the protonation site (LUMO+3 of conformer 1, LUMO+4 and LUMO+3, of conformer 2) at 407 eV. Relative to the yield of the N 1s → π*(CONH) resonance, the signature of the protonation site in G4K is less intense than for G5.
The study of these five protonated model pentapeptides at the N K-edge revealed some features that are common to any protonated peptide. The main results of the study are summarized in Table 1. As was reported previously, all peptides show a strong resonance related to electronic transitions in the peptide bonds, always located at 401.6 eV.13,23,26 In the NEXAMS spectra, all peptides show a resonance at 402.6 eV that we attribute to transitions from a nitrogen 1s level in one peptide bond to π*(CONH) antibonding orbitals in a neighbouring peptide bond, except for protonated G4H, which has a feature centred at 403.1 eV due to N 1s → σ*(CH) transitions in the histidine side chain where the protonation site is located.
| Peak | Exp (eV) | Calc (eV) | Assignment |
|---|---|---|---|
| G5 | |||
| A | 401.6 | 401.6 | N 1s → π*(CONH) |
| B | 402.6 | 402.1 | Ni 1s→ π*(CONjH) |
| C | 404–408 | 404.8 | N 1s → π*(CO) |
| 406.6 | N 1s → σ*(NH3+) | ||
| PG4 | |||
| A | 401.6 | 401.6 | N 1s → π*(CONH) |
| B | 402.6 | 402.1 | Ni 1s → π*(CONjH) |
| 403.7 | |||
| C | 404–408 | 404.7 | N 1s → σ*(NH) |
| 406.5 | N 1s → σ*(CH) | ||
| G4R | |||
| A | 401.6 | 401.7 | N 1s → π*(CONH) |
| B | 402.6 | 402.6 | Ni 1s → π*(CONjH) |
| N 1s → π*(CN) |
|||
| C | 404–408 | 403.6 | N 1s → σ*(NH2+) |
| 403.96 | N 1s → π*(CO) | ||
| 404.1 | N 1s → σ*(NH) | ||
| G4H | |||
| A | 401.6 | 400.8 | N 1s → π*(CONH) |
| 401.4 | N 1s → π*(CN) |
||
| D | 403.1 | 402.5 | N 1s → σ*(NH) |
| C | 404–408 | 407.1 | N 1s → σ* |
| 407.9 | |||
| G4K | |||
| A | 401.6 | 401.6 | N 1s → π*(CONH) |
| B | 402.6 | 402.4 | Ni 1s → π*(CONjH) |
| 402.3 | N 1s → π*(COOH) | ||
| 403.1 | N 1s → σ*(NH) | ||
| C | 404–408 | N 1s → σ*(NH) | |
| N 1s → σ*(CH) | |||
For protonated G4R, the π*(CN) transition from the arginine side chain contributes to the resonance at 401.6 eV and the one at 402.6 eV, which results in the resonance appearing broader than for the other peptides. Similarly, G4H shows π*(CN) transitions in the histidine's imidazole ring which contributes to the resonance at 401.6 eV. The NEXAMS spectrum of every peptide shows a broad feature between 404 and 408 eV with different intensities relative to the N 1s → π*(CONH) resonance. The comparison of the experimental spectra with DFT/ROCIS calculations shows that the feature comes from σ* resonances from the backbone of the peptide with a major contribution from σ*(NH) resonances located at the protonated nitrogen site of these peptides, both from the basic side chain and the N-terminus. In PG4 and G4H, we observed that the immonium ion exhibits a lower intensity of feature C, which can be explained by the resonant excitation in the peptide bond leading to more immonium ion production compared to the absorption of the photon in the side chain, around 406 eV. Interestingly, we did not observe any resonance from the unprotonated N-terminus in the experimental total ion yield spectra in the cases of G4H, G4R, and G4K. Furthermore, for peptides with a protonated side chain, we observed that resonance A is more intense than resonance C as compared to the peptides with a protonation site on the N-terminus. The additional transitions in the side chains energetically close to the resonances A and B lead to resonance A being more intense than for G5 or PG4. We could observe differences between G4H, G4R, and G4K NEXAMS spectra in the shape and the intensity of the resonances A and B. The total ion yield spectrum of G4H does not exhibit any resonance at 402.6 eV but one at 403.1 eV, resonance B appears as a shoulder on resonance A for G4R, while for G4K resonance B is almost as intense as resonance A. We discussed that these transitions are better resolved, or absent, in the PIY of certain fragments, especially when comparing e.g. immonium ion and fragments involving CN bond break. These differences allow distinguishing them from each other. As shown in the previous section, the high intensity of resonance C is due to the protonation site located on the N-terminus. For neutral diglycine in the gas phase, the feature observed at 406 eV is weaker and shows about the same intensity as the resonance at 401.6 eV,22 which agrees with our conclusion.
Protonation at the amide oxygen: the case of triglycine
Rodriquez and co-workers investigated the proton migration in protonated triglycine using density functional theory.65 In this study, they found that protonation of the N-terminal nitrogen atom is higher in free energy than the different isomers resulting from protonation at the carbonyl oxygen of the N-terminal residue. However, in a recent study, Li and co-workers10 calculated that the protonation site of triglycine is the N-terminal nitrogen rather than the amine oxygen, that they found to be at least 0.61 kcal mol−1 more stable than the other protonation sites possible for triglycine. They suggested that experimental studies using soft X-ray spectroscopy could provide a way to distinguish the various G3 isomers, since NEXAFS spectra should show unique peaks at nitrogen and oxygen K-edges for the different protomers.Using protonated pentaglycine, we were able to establish the N K-edge fingerprint of an N-terminal protonated peptide. Thus, comparing the NEXAMS spectra of G5 and G3 should show whether only conformers of G3 with the protonation site at the N-terminal nitrogen are present under our experimental conditions. As protonation of an amine oxygen is expected to influence the NEXAMS spectra at the oxygen K-edge, we present in the following results for both, the N and O K-edges. The carbon K-edge spectra of G5 and G3 have also been recorded, but as they exhibit only little differences, with a main resonance at 288.3 eV for both protonated peptides which come from C 1s → π*(CONH) transitions from the backbone, they are only shown in the ESI† (Fig. S33 to S35) and will not be discussed further in this manuscript. The partial ion yield spectra at the oxygen K-edge are shown in the ESI† (Fig. S36 and S37).
Comparison of the experimental spectra of G5 and G3
In this section, we first compare the experimental NEXAMS spectra of protonated pentaglycine and triglycine. We performed DFT/ROCIS calculations on different isomers and possible protonation sites of triglycine to interpret the results of the experimental spectra.As shown in the previous section and in previous theoretical and experimental studies,3,9 the protonation site of protonated pentaglycine is located at the N-terminus, i.e. on the amide nitrogen of the peptide. We have shown in the previous section that the electronic transition due to the protonation site of pentaglycine is located in the broad feature centred at 406 eV. The protonation site is responsible for the relatively high intensity of this feature as compared to the main N 1s → π*(CONH) resonance. Thus, we can use this resonance to map the difference between the two glycine-based peptides.
At the N K-edge, see Fig. 4, we observed clear differences and similarities. For both protonated peptides, a resonance is located at 401.6 eV and is due to an N 1s → π*(CONH) electronic transition in the peptide bonds blue-shifted by 0.1 eV for G3 compared to G5. The resonance at 402.6 eV is not observed for protonated triglycine but we observed a feature at 403.1 eV that has not been observed for protonated pentaglycine. For G4H, a resonance is observed at 403.1 eV for N 1s → σ*(NH). Note that in the NEXAMS spectrum of solid triglycine, no resonance between the one at 401.5 eV and the broad peak centred at 406 eV has been observed.22 In the work of Gordon and co-workers22 as well as of Zubavichus and co-workers21 on solid-state triglycine, they attributed the feature at 406 eV to σ*(NH) transitions from the amide group that is protonated in the zwitterionic form, which is consistent with our conclusion on the protonation site of peptides. This suggests that the peak we observe at 403.1 eV in the N K-edge spectrum of G3 could be due to an electronic transition from a different protonation site than the one of pentaglycine. This being said, both protonated peptides exhibit this broad feature between 404 and 408 eV that we attributed previously to excitations to the σ*(NH) and σ*(CH) orbitals, with an intense contribution from an –NH3+ protonated amino group. This suggests, in contrast, that triglycine would also be protonated at its N-terminus. Thus, from the experimental spectra only, we can highlight differences but cannot conclude whether triglycine has one or two protonation sites, as the experimental NEXAMS spectrum could suggest two different ones. A comparison with DFT/ROCIS calculations is shown in the next section for a better assignment of the different resonances.
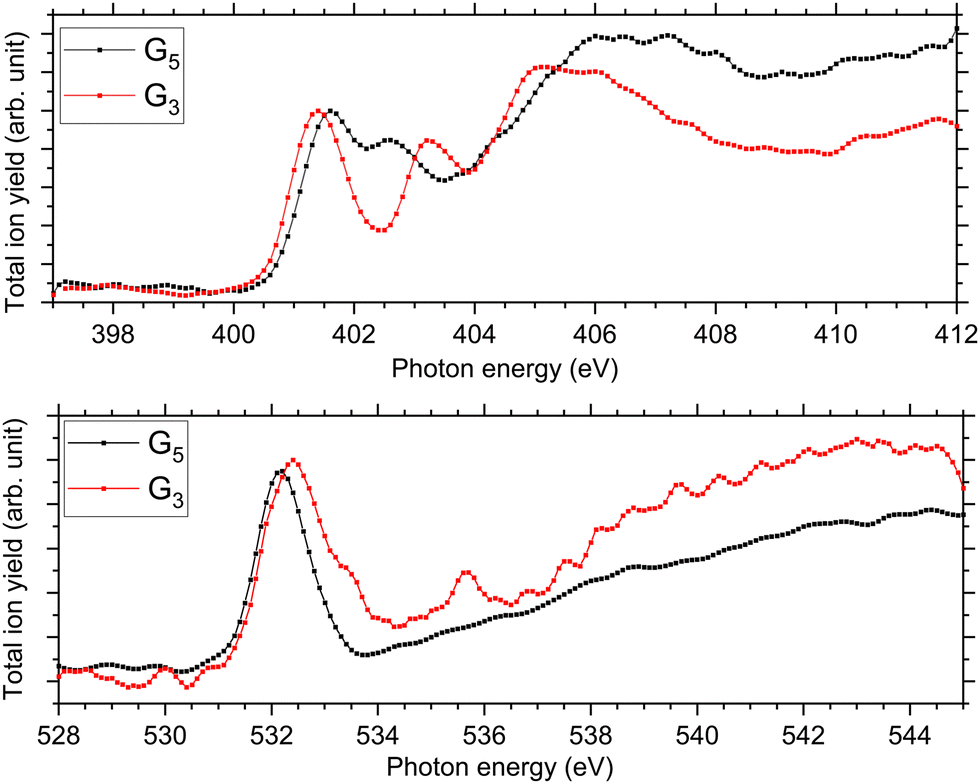 | ||
| Fig. 4 Experimental NEXAMS spectra at the N K-edge (top) and at the O K-edge (bottom) of G5 in black and G3 in red. | ||
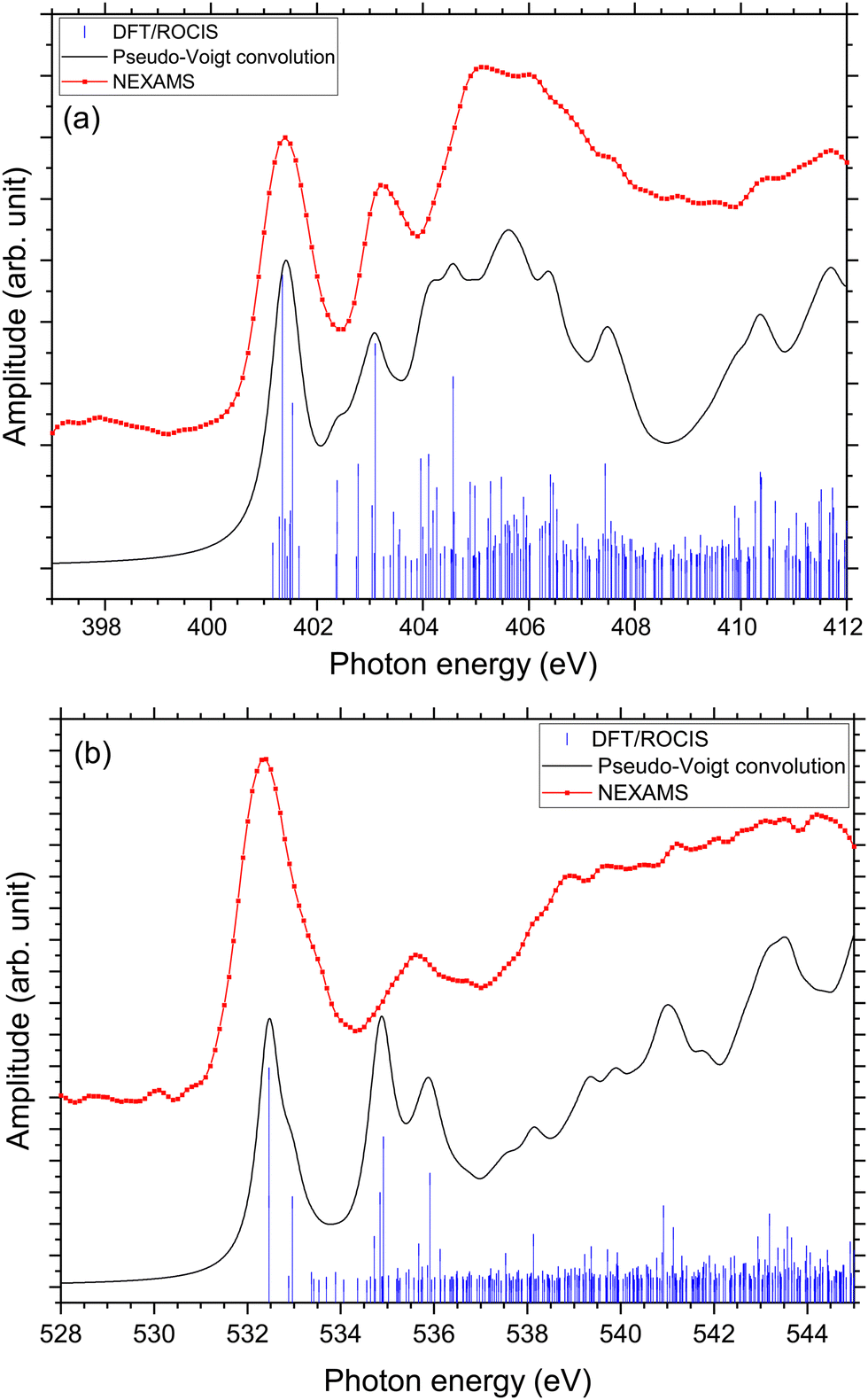
At the O K-edge, see Fig. 4, both spectra show an intense resonance centred at 532.2 eV, blue-shifted by 0.2 eV for G5 compared to G3. This feature has previously been observed in protonated peptides33,63,66 and proteins67,68 as well as for gas-phase diglycine by Feyer and co-workers.26 This resonance is attributed to an O 1s → π*(CONH) transition in the peptide bonds. In their study, Gordon and co-workers also attributed the resonance at 532.2 eV of solid diglycine and triglycine to an O 1s → π*(CONH) resonance with a small contribution from the O 1s → π*(COO−) electronic transition.22 G3 exhibits a shoulder at 533.4 eV that is not observed in the case of G5 and was not observed for gas-phase diglycine.26 Dörner and co-workers attributed this transition to an O 1s → π*(COOH) transition in the carboxylic group which was observed at 532.5 eV for leucine-enkephalin.15
In the case of protonated triglycine, a second resonance is observed at 535.6 eV that is not observed in the experimental spectrum of G5. This resonance has been attributed to an O 1s → π*(CO) transition in the carboxylic group by Feyer and co-workers for gas-phase glycine and diglycine.26 The spectrum of protonated triglycine at the oxygen K-edge does not exhibit any feature that at first glance would be associated with the protonation site.
To more deeply analyse the NEXAMS spectra for triglycine and understand better our experimental observation and the differences with pentaglycine, we compared the result of the NEXAMS experiment of protonated triglycine with DFT/ROCIS calculations.
N K-edge
In the case of protonated triglycine, a random structure search was performed in order to get different structures of this protonated peptide. Theoretically, the structure search has been performed at 0 K to probe the lowest-energy conformers of triglycine that should compare with our experimental conditions, where the ions in the trap were cryogenically cooled down to 8 K. The result of the conformer search and geometry optimization gives two main protomers, noted GGGH1 and GGGH2. The calculated structures of GGGH1 and GGGH2 are shown in Fig. 5. The first protomer GGGH1 has a more folded structure, with hydrogen bonds between the protonated N-terminus and the carboxyl oxygen atoms. In contrast, the GGGH2 structure is more linear, with the proton bound to the first amide oxygen forms a hydrogen bond with the terminal nitrogen.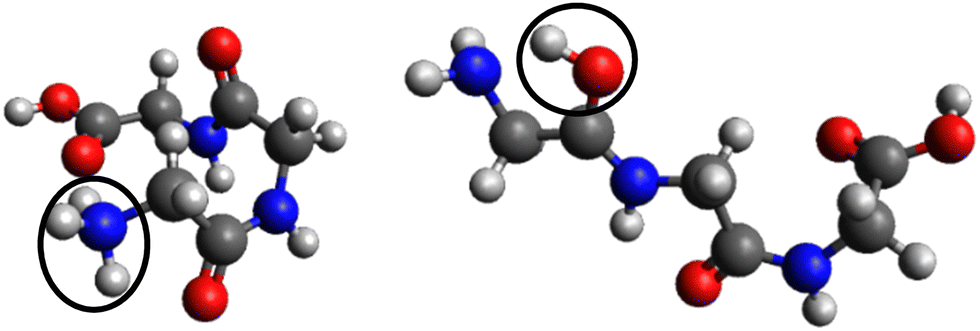 | ||
| Fig. 5 Calculated structures for GGGH1 (left) and GGGH2 (right) (B3LYP/ZORA-DEF2-TZVP). The dark circles highlight the position of the protonation site. | ||
In the theoretical study of Li and co-workers, they found that the most stable conformer of the traditional N-terminal protonation site of triglycine is 2.53 kcal mol−1 more stable than the most stable one of the O-protonated conformers at the highly accurate coupled cluster singles and doubles (CCSD) level of theory.10 Thus, it is expected that the experimental spectrum will be reproduced by mainly the calculated spectrum of GGGH1.
Fig. 6 presents the results of the DFT/ROCIS calculations for the absorption spectra of both isomers at the nitrogen K-edge.
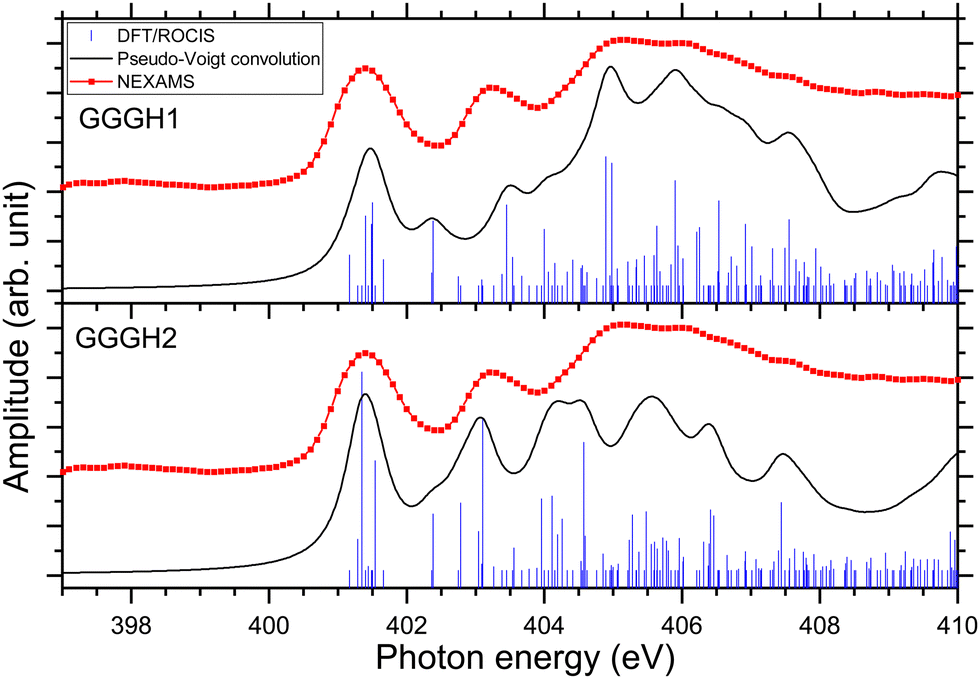 | ||
| Fig. 6 Experimental nitrogen K-edge ion yield spectrum in red of G3 as well as DFT/ROCIS spectrum for the calculated structure GGGH1 (shifted +12.07 eV) and for the calculated structure GGGH2 (shifted +12.56 eV). The absorption lines were broadened using a pseudo-Voigt profile with a 0.6 eV FWHM represented in black. | ||
Fig. S29 and S31 (ESI†) show the respective calculated DFT spectrum of GGGH1 and GGGH2 at the N K-edge with the different molecular orbitals responsible for the main transitions. At first glance, the two calculated spectra are showing strong differences, especially in the region from 402 eV to 408 eV and neither spectrum reproduces the experimental spectrum well overall. As for the other peptides, the DFT/ROCIS transition lines of the first resonance at 401.6 eV stem from N 1s → π*(CONH) transitions in the peptide bonds for both protomers. According to our calculations, in the case of GGGH1, the resonance at 403.4 eV is attributed to a transition from N 1s to σ*(CH) orbitals (LUMO+4) in the backbone of triglycine, whereas in the case of GGGH2, it is an N 1s → σ*(NH) transition in the unprotonated terminal-NH2 (LUMO+2). Interestingly, the resonance calculated for the GGGH2 conformer is more intense than it is in the case of GGGH1 and reproduces better the peak observed at this energy in the experimental spectrum. The broad feature centred at 406 eV is due to many transitions as described previously for the protonated pentapeptides. For GGGH1, the DFT/ROCIS calculations reproduce well the broad feature, and the analysis of the different electronic transitions shows that, for this isomer, the feature is mainly due to N 1s → σ*(CH) transitions from the backbone and N 1s → σ*(NH) transitions from the protonated terminal-NH3+. In the case of GGGH2, the calculated spectrum does not reproduce the high intensity of the feature between 404 and 408 eV. The main transition calculated at 404.8 eV (LUMO+10) is highly delocalized and can be due to N 1s → σ*(CH) or N 1s → σ*(NH) transitions from the backbone. The main resonances observed for G3 at the nitrogen and oxygen K-edges are summarized in Table 2. Ultimately, the experimental spectrum cannot be reproduced with sufficient confidence by either of the two calculated isomers, since only the spectrum of GGGH2 can explain the resonance at 403.1 eV while only the spectrum of GGGH1 reproduces the broader feature centred at 405 eV. As in the work of Wu and McMahon,9 the comparison of the experimental spectrum with the calculated ones suggests that both isomers might be present under our experimental conditions and contribute to the measured NEXAMS spectrum. In this experiment only the linear ion trap was cooled down to 8 K, which was conducted at the Ion Trap end-station. The other parts of the setup were at room temperature, such that we can expect to have different stable isomers of protonated triglycine transferred and cooled down in the trap, as explained by Li and co-workers10 and already shown for biomolecular ions by Warnke and co-workers using ion mobility spectrometry combined with a cryogenic ion trap.69
| Exp (eV) | Calc (eV) | Assignment |
|---|---|---|
| G3 | ||
| 401.6 | 401.5 | N 1s → π*(CONH) |
| 403.2 | 403.1 | N 1s → σ*(NH2) |
| 404–408 | 404.8 | N 1s → σ*(CH) |
| N 1s → σ*(NH3+) | ||
| 532.3 | 532.4 | O 1s → π*(CONH) |
| 535.5 | 534.7 | O 1s → π*(CO) |
| 535.6 | O 1s → σ*(OH) |
We have therefore attempted to estimate the relative populations of the two isomers present in our experiment by mixing the contribution of both isomers into a new calculated spectrum and by comparing the peak intensities with the experimental spectrum. The two resonances that we considered as markers for the comparison are the one at 403.1 eV to obtain the contribution of GGGH2 to the experimental spectrum and the broad feature centred at 405.2 eV to obtain the contribution of GGGH1, both of which are related to the protonation state of the N-terminus. To do this, we normalized the experimental spectrum and the calculated spectra to the first resonance, which is the peptide bond resonance and is similar in both calculated spectra. After normalization, we compared, for different isomer mixing ratios, the relative intensity between the calculated and experimental peak height for both resonances. The results are shown in Fig. 7. On the basis of the spectral comparison, we concluded that our experimental spectrum arises from a relative population of N-protonated triglycine of 30% with 70% of O-protonated triglycine. This ratio is the closest one to the experimental spectrum with 90% of the relative intensity for both resonances.
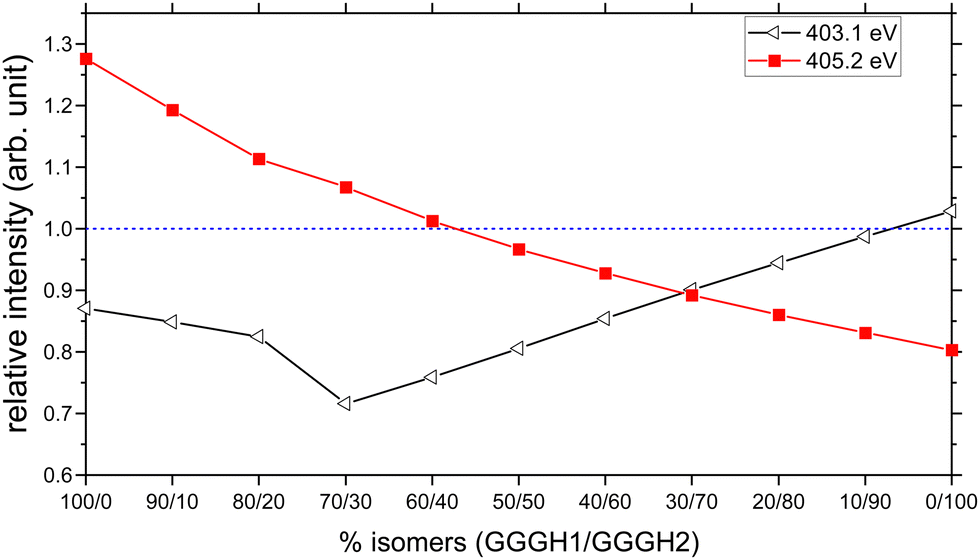 | ||
| Fig. 7 Relative intensities of the calculated resonances at 403.1 eV (black, σ*(NH) of the unprotonated – NH2 group) and at 405.2 eV (red, σ*(CH) from the backbone), compared to the experimental ones, as a function of the isomer mixing ratio GGGH1 and GGGH2. The dashed blue line represents a relative intensity of 1. | ||
The result of the isomer mixing on the calculated absorption spectra is shown in Fig. 9(a). This is in very good agreement with the results of Voss and co-workers who performed IR-IR double resonances of protonated triglycine in an ion trap at 10 K and concluded with a relative population of 35% N-protonated and 65% O-protonated.70 The authors also point out that this population is more representative of the isomers present at 300 K originating from the ESI source rather than the 0 K population distribution.
O K-edge
Fig. 8 shows the NEXAMS spectrum obtained at the oxygen K-edge and its comparison with DFT/ROCIS calculations for the two main isomers of protonated triglycine. Fig. S30 and S32 (ESI†) show the respective calculated DFT spectrum of GGGH1 and GGGH2 at the O K-edge with the different molecular orbitals responsible for the main transitions. The different resonances are summarized in Table 2. For GGGH1, the first resonance at 532.4 eV is attributed to O 1s → π*(CONH) transitions (LUMO and LUMO+2) in the peptide bond. The other main transition line at 533 eV, which is shown in the calculated spectrum, can be attributed to O 1s → π*(CO) (LUMO+1) in the carboxylic group starting from the oxygen atom of the –CO group. In contrast, in the case of GGGH2, we observed only the first resonance related to the O 1s → π*(CONH) transition. For GGGH1, the Pseudo-Voigt convolution of the DFT/ROCIS spectrum results in a much broader peak, closer to the experimental result.
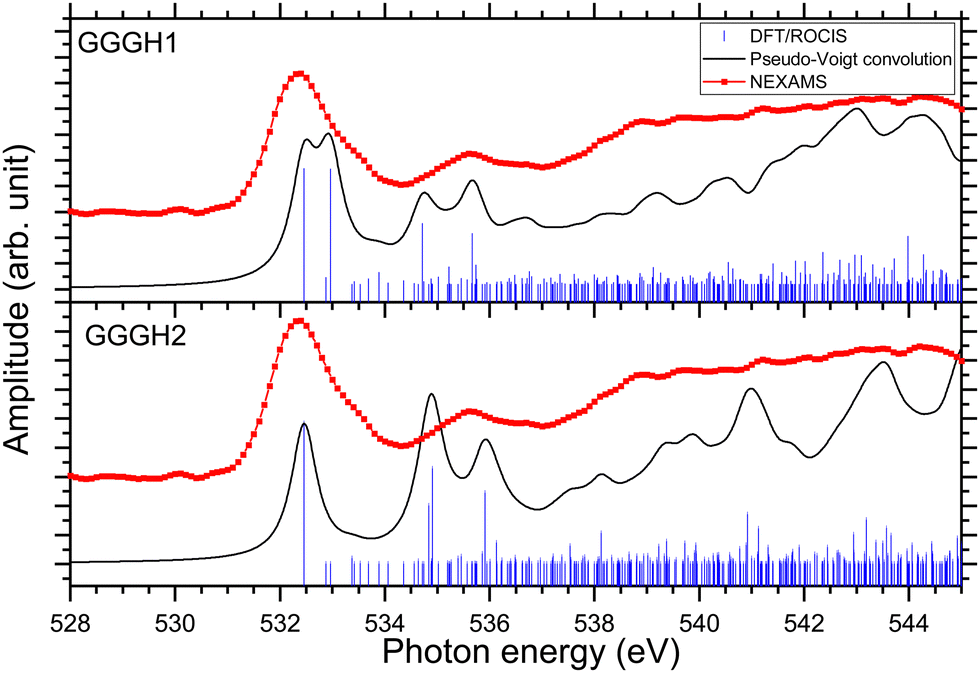 | ||
| Fig. 8 Experimental oxygen K-edge ion yield spectrum in red of G3 as well as DFT/ROCIS spectrum (shifted +14.15 eV) for the calculated structure GGGH1 and for the calculated structure GGGH2 (shifted +14.76 eV). The absorption lines were broadened using a pseudo-Voigt profile with a 0.6 eV FWHM represented in black. | ||
For both calculated spectra, the second feature includes two resonances, at 534.7 eV and 535.7 eV for GGGH1 (534.9 eV and 535.9 eV for GGGH2), but only one was observed experimentally, centred at 535.6 eV. In the case of GGGH1, the first resonance at 534.7 eV is due to an O 1s → π*(CO) transition to the LUMO+1 in the carboxylic group but starting from the oxygen atom of the –OH group, and the second is attributed to O 1s → σ*(OH) transitions (LUMO+5) in the carboxylic group of protonated triglycine. For GGGH2, the first resonance can be attributed to the peptide bond resonance O 1s → π*(CONH) in the molecular orbitals LUMO and LUMO+1, whereas the resonance at 535.9 eV is due to O 1s → σ*(OH) transitions (LUMO+8) at the C-terminus of the peptide, similar to GGGH1. At higher energy, the spectra are mainly constituted of excitations to σ* orbitals. These results are in good agreement with the previous study of Feyer and co-workers for neutral gas-phase diglycine.26
To confirm the results obtained at the nitrogen K-edge, we combined the two calculated spectra at the oxygen K-edge with the same factor, 30/70, for GGGH1 and GGGH2 as in the previous section. The resulting graph is shown in Fig. 9(b). Here, we achieve a good agreement with the experimental spectrum. We reproduced the different experimental resonances with the calculations. Despite the relative intensity of the resonance at 535 eV being higher for the calculated spectrum than the experimental spectrum, the attributed electronic transitions agree well with our results and the previous studies.22,23,26
 | ||
| Fig. 9 Comparison of, in red, the experimental NEXAMS spectra of protonated triglycine at the N-K edge (a) and O K-edge (b) and, in black, the result of the convolution of the two calculated spectra GGGH1 and GGGH2 with 30% of GGGH1 and 70% of GGGH2. | ||
In this section, we have shown that there are clear differences between a protonated amide nitrogen and a protonated amine oxygen and that the difference is clearly detectable in the N K-edge NEXAMS spectra. When the protonation site is located at the oxygen, we observed a resonance at 403.1 eV that is not observed for the case of a peptide protonated at a nitrogen, this resonance stems from an N 1s → σ*(NH) transition in the unprotonated –NH2 N-terminus. However, our experimental spectrum still contains the signature of a protonated N-terminus with a broad and intense feature centred around 405 eV, which can be explained by our experimental conditions. In fact, during this experimental campaign, only the ion trap was held at 8 K, whereas the other parts of the setup were at room temperature, which allows different conformers to coexist in the trap. Moreover, as shown in the study of Wu and Mc Mahon,9 smaller peptides of glycine are more stable in a linear structure whereas peptides containing more than three glycines are more stable in a folded structure. It can be assumed that as triglycine is of an intermediate size, the folded structure and the linear structure can coexist. According to the simulations, protonated triglycine is more stable in a folded structure when the proton is on the N-terminus and the linear structure is the most stable with the proton on the first amide oxygen. By comparing the DFT/ROCIS calculations with the N K-edge experimental spectrum, we could confirm that the latter is representative of a mixing of protomers with a relative population of N-protonated triglycine of 30% with 70% of O-protonated triglycine. At the oxygen K-edge, no striking difference appears between the two protomers on the calculated spectra, which indicates that the signature of the protonation site is best observed at the nitrogen K-edge. However, our measurement did not show most of the resonances expected by the theoretical work of Li and co-workers.10 Their simulated NEXAFS spectra, at the nitrogen K-edge, did not show any feature around the energy of the peptide bond excitation, and we did not observe any π* transition originating from the NH2 terminus expected by Li and co-workers but a σ* transition lying higher in energy than the expected transitions. They predicted many transitions in the region of 404–406 eV that we were not able to resolve experimentally. At the oxygen K-edge, we also observed striking differences between our experimental spectrum and their simulated one: we did not observe any peak at 531.3 eV or at 533.6 eV which they, respectively, attributed to an O 1s → π*(CO) transition only due to cyclic isomers or to linear isomers. One reason that could explain the differences between our results and their spectra can be the temperature used for the simulation, they calculated their spectra at a temperature of 498 K,10 while our ion trap was held at 8 K, which could result in different conformations of the protomers in the trap which would affect the absorption spectra. Overall, the NEXAMS technique has proven to be sensitive to the protonation site of peptides and we have shown that we can distinguish the different protomers present in the experimental setup and give an approximate ratio.
Conclusions
In conclusion, we have performed a NEXAMS study on five glycine-based protonated pentapeptides to investigate the effect of the protonation site on the electronic structure and to record its spectroscopic fingerprint in the soft X-ray range. We have shown that the combination of DFT/ROCIS calculations and soft X-ray spectroscopy allows mapping the electronic transitions of the protonation site. The influence of a protonation site on a nitrogen atom has only a moderate effect on the entire spectrum of protonated pentapeptides. Experimentally, the NEXAMS technique does not allow for straightforward identification of the resonances imparted to the protonation site of a peptide, essentially owing to the numerous overlapping transitions to σ* orbitals in these molecular systems and the intrinsic band broadening imparted to vibrational excitations in the core-hole excited state (Franck–Condon principle). However, we could observe that the high intensity of the feature centred at 406 eV was due to the protonation site and we discussed how certain changes in the spectral features, especially the shape and position of the resonances normally attributed to N 1s → π*(CONH) and Ni 1s → π*(CONjH), can be attributed to the different basic residues in the peptides, e.g. by additional transitions to π*(C = N) and σ*(NH) in the side chain of histidine and arginine. We could also identify that the production of immonium ions of a residue is quenched when the absorption takes place on its side. This coincides with the increased yield of side-chain-related fragments under the same condition, which suggests that fast bond cleavage in the side chain is induced after localized photon absorption, prior to energy redistribution in the system. In addition, we are able to answer an open question by Dörner and co-workers15 concerning the presence of a peak at 399.2 eV in protonated methionine enkephalin [YGGFM + H]+ but not in leucine enkephalin [YGGFL + H]+. The authors proposed, based on literature and calculation, that this peak could be attributed to either an N 1s → σ*(N–H) transition in an unprotonated terminal NH2 group or an N 1s → π*(CN) transition in a peptide bond in an enol form. In our set of experiments, peptides G4H, G4R and G4K have an unprotonated terminal NH2 group but do not show a prominent peak at 339.2 eV. The same is true for the calculations of O-protonated triglycine that do not show a transition at this energy. The assignment of this band to N 1s → σ*(N–H) can thus be excluded.
Moreover, we have demonstrated that the NEXAMS technique is sensitive to backbone protonation as demonstrated by studying protonated triglycine at the nitrogen K-edge and the oxygen K-edge. We have shown that a protonation site located at an amide oxygen leads to a resonance around 403 eV that is not present in the case of a peptide protonated at a nitrogen atom. However, the measurements did not reveal as striking differences between the protomers as expected from the work by Li and co-workers.10 We have shown that the combination of DFT/ROCIS calculations and NEXAMS can help estimate the mixing ratio between the protomers observed during the experiment. This work should also be extended to the search for deprotonation site signatures in peptides and in other organic molecules where the information on the (de)protonation site(s) could be revealed using NEXAMS. Additionally, it could be tested experimentally if signatures of conformational structures such as α-helix and β-turn can be resolved in the Ni 1s → π*(CONjH) transitions as proposed theoretically by Buckley and Besley.50 With this work, we can foresee that NEXAMS is perfectly suited to follow ultrafast proton transfer/migration in biomolecules, for example in pump–probe studies at X-ray free-electron laser facilities, where a pump pulse would trigger the proton transfer process and a probe X-ray pulse would interrogate the position of that proton on the molecule. In addition, NEXAMS will allow us to follow the protonation/hydrogenation change in nanohydrated peptides and locate the water molecules on the biomolecules. Finally, due to the ionizing properties of soft X-ray radiation, NEXAMS experiments do not suffer from the limitations of dissociation-based spectroscopy techniques with respect to “stable” molecules, Mn+, that would not break, but produce a minima intact photoionized precursor ions, M(n+1)+˙, from which an absorption spectrum can be retrieved.71 In these cases, NEXAMS is a valuable complementary technique to e.g. IRMPD.
Author contributions
L. S. and S. B. did the conceptualization and supervision. J. L. and L. S. did the formal analysis. A. K. and D. M. did the theoretical calculations under the supervision of R. M. and M. R. J. L., K. S., C. M., L. S., I. U., A. K., F. T., S. B. performed the experiment at the P04 beamline of Petra III. J. L., L. S., I. U., B. O., V. Z., T. L., K. H. and M. T. participated in the experiment at BESSY II. All authors participated to review and editing the original draft written by J. L.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Centre for Molecular Water Science (CMWS) in an Early Science Project. J. L. acknowledges the Région Normandie (France) for funding within the NANOHYDRAD project. L. S. and S. B. acknowledge funding by the Helmholtz Initiative and Networking Fund. This work is supported by the Cluster of Excellence ‘CUI: Advanced Imaging of Matter’ of the Deutsche Forschungsgemeinschaft (DFG) – EXC 2056 – project ID 390715994 and Grant-No. HIDSS-0002 DASHH (Data Science in Hamburg-HELMHOLTZ Graduate School for the Structure of Matter). F. T. acknowledges support by the MaxWater initiative of the Max-Planck-Gesellschaft. We thank Helmholtz-Zentrum Berlin for the allocation of synchrotron radiation beamtime at BESSY II at the beamline UE52-PGM. The Ion Trap end station at UE52-PGM is partially supported by the BMBF grant 05K16VF1. Parts of this research were carried out at PETRA III, at DESY, a member of the Helmholtz Association and we would like to thank Moritz Hoesch, Kai Bagschik and Frank Scholz for assistance in using the PETRA III beamline P04.Notes and references
- K. Zhang, C. J. Cassady and A. Chung-Phillips, Ab Initio Studies of Neutral and Protonated Triglycines: Comparison of Calculated and Experimental Gas-Phase Basicity, J. Am. Chem. Soc., 1994, 116, 11512–11521 CrossRef CAS.
- K. Zhang, D. M. Zimmerman, A. Chung-Phillips and C. J. Cassady, Experimental and ab Initio Studies of the Gas-Phase Basicities of Polyglycines, J. Am. Chem. Soc., 1993, 115, 10812–10822 CrossRef CAS.
- J. Wu and C. B. Lebrilla, Gas-Phase Basicities and Sites of Protonation of Glycine Oligomers (GLYn; n = 1–5), J. Am. Chem. Soc., 1993, 115, 3270–3275 CrossRef CAS.
- Z. Wu and C. Fenselau, Proton affinity of arginine measured by the kinetic approach, Rapid Commun. Mass Spectrom., 1992, 6, 403–405 CrossRef CAS.
- C. Bleiholder, S. Suhai and B. Paizs, Revising the Proton Affinity Scale of the Naturally Occurring α-Amino Acids, J. Am. Soc. Mass Spectrom., 2006, 17, 1275–1281 CrossRef CAS PubMed.
- B. Lucas, G. Grégoire, J. Lemaire, P. Maître, F. Glotin, J. P. Schermann and C. Desfrançois, Infrared multiphoton dissociation spectroscopy of protonated N-acetyl-alanine and alanyl-histidine, Int. J. Mass Spectrom., 2005, 243, 105–113 CrossRef CAS.
- G. Grégoire, M. P. Gaigeot, D. C. Marinica, J. Lemaire, J. P. Schermann and C. Desfrançois, Resonant infrared multiphoton dissociation spectroscopy of gas-phase protonated peptides. Experiments and Car-Parrinello dynamics at 300 K, Phys. Chem. Chem. Phys., 2007, 9, 3082–3097 RSC.
- A. Chung-Phillips, Polyglycine conformational analysis: Calculated vs. experimental gas-phase basicities and proton affinities, J. Phys. Chem. A, 2005, 109, 5917–5932 CrossRef CAS PubMed.
- R. Wu and T. B. McMahon, Protonation sites and conformations of peptides of glycine (Gly 1-5H+) by IRMPD spectroscopy, J. Phys. Chem. B, 2009, 113, 8767–8775 CrossRef CAS PubMed.
- H. Li, J. Jiang and Y. Luo, Identification of the protonation site of gaseous triglycine: the: cis-peptide bond conformation as the global minimum, Phys. Chem. Chem. Phys., 2017, 19, 15030–15038 RSC.
- P. T. Edwards, L. K. Saunders, D. C. Grinter, P. Ferrer, G. Held, E. J. Shotton and S. L. M. Schroeder, Determination of H-Atom Positions in Organic Crystal Structures by NEXAFS Combined with Density Functional Theory: A Study of Two-Component Systems Containing Isonicotinamide, J. Phys. Chem. A, 2022, 126(19), 2889–2898 CrossRef CAS PubMed.
- S. Eckert, P. S. Miedema, W. Quevedo, B. O’Cinneide, M. Fondell, M. Beye, A. Pietzsch, M. Ross, M. Khalil and A. Föhlisch, Molecular structures and protonation state of 2-Mercaptopyridine in aqueous solution, Chem. Phys. Lett., 2016, 647, 103–106 CrossRef CAS.
- Y. Zubavichus, A. Shaporenko, M. Grunze and M. Zharnikov, Innershell absorption spectroscopy of amino acids at all relevant absorption edges, J. Phys. Chem. A, 2005, 109, 6998–7000 CrossRef CAS PubMed.
- P. Leinweber, J. Kruse, F. L. Walley, A. Gillespie, K. U. Eckhardt, R. I. R. Blyth and T. Regier, Nitrogen K-edge XANES – An overview of reference compounds used to identify ‘unknown’ organic nitrogen in environmental samples, J. Synchrotron Radiat., 2007, 14, 500–511 CrossRef CAS PubMed.
- S. Dörner, L. Schwob, K. Atak, K. Schubert, R. Boll, T. Schlathölter, M. Timm, C. Bülow, V. Zamudio-Bayer, B. Von Issendorff, J. T. Lau, S. Techert and S. Bari, Probing Structural Information of Gas-Phase Peptides by Near-Edge X-ray Absorption Mass Spectrometry, J. Am. Soc. Mass Spectrom., 2021, 32, 670–684 CrossRef PubMed.
- A. R. Milosavljević, C. Nicolas, M. L. Ranković, F. Canon, C. Miron and A. Giuliani, K-Shell Excitation and Ionization of a Gas-Phase Protein: Interplay between Electronic Structure and Protein Folding, J. Phys. Chem. Lett., 2015, 6, 3132–3138 CrossRef.
- L. Schwob, S. Dörner, K. Atak, K. Schubert, M. Timm, C. Bülow, V. Zamudio-Bayer, B. Von Issendorff, J. T. Lau, S. Techert and S. Bari, Site-Selective Dissociation upon Sulfur L-Edge X-ray Absorption in a Gas-Phase Protonated Peptide, J. Phys. Chem. Lett., 2020, 11, 1215–1221 CrossRef CAS PubMed.
- X. Wang, S. Rathnachalam, K. Bijlsma, W. Li, R. Hoekstra, M. Kubin, M. Timm, B. von Issendorff, V. Zamudio-Bayer, J. T. Lau, S. Faraji and T. Schlathölter, Site-selective soft X-ray absorption as a tool to study protonation and electronic structure of gas-phase DNA, Phys. Chem. Chem. Phys., 2021, 23, 11900–11906 RSC.
- E. Otero and S. G. Urquhart, Nitrogen 1s near-edge X-ray absorption fine structure spectroscopy of amino acids: resolving zwitterionic effects, J. Phys. Chem. A, 2006, 110, 12121–12128 CrossRef CAS PubMed.
- B. M. Messer, C. D. Cappa, J. D. Smith, K. R. Wilson, M. K. Gilles, R. C. Cohen and R. J. Saykally, pH dependence of the electronic structure of glycine, J. Phys. Chem. B, 2005, 109, 5375–5382 CrossRef CAS PubMed.
- Y. Zubavichus, M. Zharnikov, A. Schaporenko and M. Grunze, NEXAFS study of glycine and glycine-based oligopeptides, J. Electron Spectrosc. Relat. Phenom., 2004, 134, 25–33 CrossRef CAS.
- M. L. Gordon, G. Cooper, C. Morin, T. Araki, C. C. Turci, K. Kaznatcheev and A. P. Hitchcock, Inner-shell excitation spectroscopy of the peptide bond: comparison of the C 1s, N 1s, and O 1s spectra of glycine, glycyl-glycine, and glycyl-glycyl-glycine, J. Phys. Chem. A, 2003, 107, 6144–6159 CrossRef CAS.
- G. Cooper, M. Gordon, D. Tulumello, C. Turci, K. Kaznatcheev and A. P. Hitchcock, J. Electron Spectrosc. Relat. Phenom., 2004, 137–140, 795–799 CrossRef CAS.
- O. Plekan, V. Feyer, R. Richter, M. Coreno, M. De Simone, K. C. Prince and V. Carravetta, Investigation of the amino acids glycine, proline, and methionine by photoemission spectroscopy, J. Phys. Chem. A, 2007, 111, 10998–11005 CrossRef CAS PubMed.
- R. Wu and T. B. McMahon, Infrared multiple-photon dissociation mechanisms of peptides of glycine, Chem. – Eur. J., 2008, 14, 7765–7770 CrossRef CAS PubMed.
- V. Feyer, O. Plekan, R. Richtery, M. Coreno, K. C. Prince and V. Carravetta, Photoemission and photoabsorption spectroscopy of glycyl-glycine in the gas phase, J. Phys. Chem. A, 2009, 113, 10726–10733 CrossRef CAS PubMed.
- S. Bari, D. Egorov, T. L. C. Jansen, R. Boll, R. Hoekstra, S. Techert, V. Zamudio-Bayer, C. Bülow, R. Lindblad, G. Leistner, A. Ławicki, K. Hirsch, P. S. Miedema, B. von Issendorff, J. T. Lau and T. Schlathölter, Soft X-ray Spectroscopy as a Probe for Gas-Phase Protein Structure: Electron Impact Ionization from Within, Chem. – Eur. J., 2018, 24, 7631–7636 CrossRef CAS PubMed.
- K. Hirsch, J. T. Lau, P. Klar, A. Langenberg, J. Probst, J. Rittmann, M. Vogel, V. Zamudio-Bayer, T. Möller and B. von Issendorff, X-ray spectroscopy on size-selected clusters in an ion trap: from the molecular limit to bulk properties, J. Phys. B: At., Mol. Opt. Phys., 2009, 42, 154029 CrossRef.
- A. R. Milosavljević, A. Giuliani and C. Nicolas, X-ray and Neutron Techniques for Nanomaterials Characterization, 2016, pp. 451–505 Search PubMed.
- K. Schubert, M. Guo, K. Atak, S. Dörner, C. Bülow, B. von Issendorff, S. Klumpp, J. T. Lau, P. S. Miedema, T. Schlathölter, S. Techert, M. Timm, X. Wang, V. Zamudio-Bayer, L. Schwob and S. Bari, The electronic structure and deexcitation pathways of an isolated metalloporphyrin ion resolved by metal L-edge spectroscopy, Chem. Sci., 2021, 12, 3966–3976 RSC.
- S. Dörner, L. Schwob, K. Schubert, M. Girod, L. MacAleese, C. L. Pieterse, T. Schlathölter, S. Techert and S. Bari, The influence of the methionine residue on the dissociation mechanisms of photoionized methionine-enkephalin probed by VUV action spectroscopy, Eur. Phys. J. D, 2021, 75, 142 CrossRef.
- G. Gopakumar, P. H. W. Svensson, O. Grånäs, B. Brena, L. Schwob, I. Unger, C. M. Saak, M. Timm, C. Bülow, M. Kubin, V. Zamudio-Bayer, J. T. Lau, B. von Issendorff, A. R. Abid, A. Lindblad, E. Danielsson, E. Koerfer, C. Caleman, O. Björneholm and R. Lindblad, X-ray Induced Fragmentation of Protonated Cystine, J. Phys. Chem. A, 2022, 126, 1496–1503 CrossRef CAS PubMed.
- Y. S. Lin, C. C. Tsai, H. R. Lin, T. L. Hsieh, J. L. Chen, W. P. Hu, C. K. Ni and C. L. Liu, Highly Selective Dissociation of a Peptide Bond Following Excitation of Core Electrons, J. Phys. Chem. A, 2015, 119, 6195–6202 CrossRef CAS PubMed.
- Y. J. Chiang, W. C. Huang, C. K. Ni, C. L. Liu, C. C. Tsai and W. P. Hu, NEXAFS spectra and specific dissociation of oligo-peptide model molecules, AIP Adv., 2019, 9, 085023 CrossRef.
- I. Ljubić, A. Kivimäki and M. Coreno, An experimental NEXAFS and computational TDDFT and ΔdFT study of the gas-phase core excitation spectra of nitroxide free radical TEMPO and its analogues, Phys. Chem. Chem. Phys., 2016, 18, 10207–10217 RSC.
- D. Maksimov and M. Rossi, GenSec: Generate and Search, 2021 DOI:10.5281/zenodo.5651513.
- D. Maksimov, PhD thesis, EPFL, 2022.
- C. Adamo and V. Barone, Toward reliable density functional methods without adjustable parameters: the PBE0 model, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- A. Tkatchenko and M. Scheffler, Accurate molecular van der Waals interactions from ground-state electron density and free-atom reference data, Phys. Rev. Lett., 2009, 102, 6–9 CrossRef PubMed.
- V. Blum, R. Gehrke, F. Hanke, P. Havu, V. Havu, X. Ren, K. Reuter and M. Scheffler, Ab initio molecular simulations with numeric atom-centered orbitals, Comput. Phys. Commun., 2009, 180, 2175–2196 CrossRef CAS.
- M. Roemelt, D. Maganas, S. Debeer and F. Neese, A combined DFT and restricted open-shell configuration interaction method including spin-orbit coupling: application to transition metal L-edge X-ray absorption spectroscopy, J. Chem. Phys., 2013, 138(20), 204101 CrossRef PubMed.
- R. R. Hudgins, Y. Mao, M. A. Ratner and M. F. Jarrold, Conformations of Gly(n)H+ and Ala(n)H+ peptides in the gas phase, Biophys. J., 1999, 76, 1591–1597 CrossRef CAS PubMed.
- F. Neese, F. Wennmohs, U. Becker and C. Riplinger, The ORCA quantum chemistry program package, J. Chem. Phys., 2020, 152, 224108 CrossRef CAS PubMed.
- J. A. Maier, C. Martinez, K. Kasavajhala, L. Wickstrom, K. E. Hauser and C. Simmerling, ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB, J. Chem. Theory Comput., 2015, 11, 3696–3713 CrossRef CAS PubMed.
- P. Gasparotto, R. H. Meißner and M. Ceriotti, Recognizing Local and Global Structural Motifs at the Atomic Scale, J. Chem. Theory Comput., 2018, 14, 486–498 CrossRef CAS PubMed.
- A. Glielmo, B. E. Husic, A. Rodriguez, C. Clementi, F. Noé and A. Laio, Unsupervised Learning Methods for Molecular Simulation Data, Chem. Rev., 2021, 121, 9722–9758 CrossRef CAS PubMed.
- A. Kotobi, L. Schwob, G. B. Vonbun-feldbauer, M. Rossi, P. Gasparotto, C. Feiler, G. Berden, J. Oomens, B. Oostenrijk, D. Scuderi, S. Bari and R. H. Meißner, Reconstructing the infrared spectrum of a peptide from representative conformers of the full canonical ensemble, Commun. Chem., 2023, 6, 1–10 CrossRef PubMed.
- M. F. Peintinger, D. V. Oliveira and T. Bredow, Consistent Gaussian basis sets of triple-zeta valence with polarization quality for solid-state calculations, J. Comput. Chem., 2013, 34, 451–459 CrossRef CAS PubMed.
- A. D. Becke, Density-functional exchange-energy approximation with correct asymptotic behavior, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 4, 276–282 Search PubMed.
- M. W. Buckley and N. A. Besley, A theoretical study of the near edge X-ray absorption fine structure of amino acids and proteins, Chem. Phys. Lett., 2011, 501, 540–546 CrossRef CAS.
- H. Imamura, Y. Otsuka and T. Nakai, Description of Core excitations by time-dependent density functional theory with local density approximation generalized gradient approximation meta-generalized gradient approximation and hybrid functionals, J. Comput. Chem., 2007, 28, 2067–2074 CrossRef PubMed.
- Y. Imamura and H. Nakai, Time-dependent density functional theory (TDDFT) calculations for core-excited states: assessment of an exchange functional combining the Becke88 and van Leeuwen-Baerends-type functionals, Chem. Phys. Lett., 2006, 419, 297–303 CrossRef CAS.
- P. J. Lestrange, P. D. Nguyen and X. Li, Calibration of Energy-Specific TDDFT for Modeling K-edge XAS Spectra of Light Elements, J. Chem. Theory Comput., 2015, 11, 2994–2999 CrossRef CAS PubMed.
- A. Chantzis, J. K. Kowalska, D. Maganas, S. Debeer and F. Neese, Ab Initio Wave Function-Based Determination of Element Specific Shifts for the Efficient Calculation of X-ray Absorption Spectra of Main Group Elements and First Row Transition Metals, J. Chem. Theory Comput., 2018, 14, 3686–3702 CrossRef CAS PubMed.
- R. A. Ingle, A. Banerjee, C. Bacellar, T. R. Barillot, L. Longetti, M. Coreno, M. De Simone, F. Zuccaro, L. Poletto, P. Miotti, A. Röder, A. Stolow, M. S. Schuurman, M. Odelius and M. Chergui, Carbon K-edge X-ray emission spectroscopy of gas phase ethylenic molecules, J. Phys. B: At., Mol. Opt. Phys., 2022, 55(4), 044001 CrossRef CAS.
- P. Roepstorff and J. Fohlman, Letter to the editors, Biol. Mass Spectrom., 1984, 11, 601 CrossRef CAS PubMed.
- K. Biemann, Contributions of mass spectrometry to peptide and protein structure, Biol. Mass Spectrom., 1988, 16, 99–111 Search PubMed.
- A. F. Lago, L. H. Coutinho, R. R. T. Marinho, A. Naves De Brito and G. G. B. De Souza, Ionic dissociation of glycine, alanine, valine and proline as induced by VUV (21.21 eV) photons, Chem. Phys., 2004, 307, 9–14 CrossRef CAS.
- J. R. Majer, B. I. Al-Ali and A. S. P. Azzouz, The mass spectrum of glycine, Org. Mass Spectrom., 1981, 16, 147–152 CrossRef CAS.
- P. Zhang, W. Chan, I. L. Ang, R. Wei, M. M. T. Lam, K. M. K. Lei and T. C. W. Poon, Revisiting Fragmentation Reactions of Protonated α-Amino Acids by High-Resolution Electrospray Ionization Tandem Mass Spectrometry with Collision-Induced Dissociation, Sci. Rep., 2019, 9, 6453 CrossRef PubMed.
- I. A. Papayannopoulos, Mass Spectrom. Rev., 1995, 14, 49–73 CrossRef CAS.
- R. Xiong, L. Xu, Y. Tang, M. Cao and H. Li, Identifying the protonation site and the scope of non-proline cis-peptide bond conformations: a first-principles study on protonated oligopeptides, Phys. Chem. Chem. Phys., 2023, 25, 13989–13998 RSC.
- Y. Zubavichus, A. Shaporenko, M. Grunze and M. Zharnikov, NEXAFS spectroscopy of homopolypeptides at all relevant absorption edges: polyisoleucine, polytyrosine, and polyhistidine, J. Phys. Chem. B, 2007, 111, 9803–9807 CrossRef CAS PubMed.
- J. Stewart-Ornstein, A. P. Hitchcock, D. H. Cruz, P. Henklein, J. Overhage, K. Hilpert, J. D. Hale and R. E. W. Hancock, Using intrinsic X-ray absorption spectral differences to identify and map peptides and proteins, J. Phys. Chem. B, 2007, 111, 7691–7699 CrossRef CAS PubMed.
- C. F. Rodriquez, A. Cunje, T. Shoeib, I. K. Chu, A. C. Hopkinson and K. W. M. Siu, Proton migration and tautomerism in protonated triglycine, J. Am. Chem. Soc., 2001, 123, 3006–3012 CrossRef CAS PubMed.
- A. R. Milosavljević, K. Jänkälä, M. L. Ranković, F. Canon, J. Bozek, C. Nicolas, A. Giuliani and A. Giuliani, Phys. Chem. Chem. Phys., 2020, 22, 12909–12917 RSC.
- D. Egorov, S. Bari, R. Boll, S. Dörner, S. Deinert, S. Techert, R. Hoekstra, V. Zamudio-Bayer, R. Lindblad, C. Bülow, M. Timm, B. von Issendorff, J. T. Lau and T. Schlathölter, Near-Edge Soft X-ray Absorption Mass Spectrometry of Protonated Melittin, J. Am. Soc. Mass Spectrom., 2018, 29, 2138–2151 CrossRef CAS PubMed.
- A. Giuliani, A. R. Milosavljević, K. Hinsen, F. Canon, C. Nicolas, M. Réfrégiers and L. Nahon, Structure and charge-state dependence of the gas-phase ionization energy of proteins, Angew. Chem., Int. Ed., 2012, 51, 9552–9556 CrossRef CAS PubMed.
- S. Warnke, A. Ben Faleh, R. P. Pellegrinelli, N. Yalovenko and T. R. Rizzo, Combining ultra-high resolution ion mobility spectrometry with cryogenic IR spectroscopy for the study of biomolecular ions, Faraday Discuss., 2019, 217, 114–125 RSC.
- J. M. Voss, K. C. Fischer and E. Garand, Revealing the structure of isolated peptides: IR-IR predissociation spectroscopy of protonated triglycine isomers, J. Mol. Spectrosc., 2018, 347, 28–34 CrossRef CAS.
- D. Egorov, L. Schwob, M. Lalande, R. Hoekstra and T. Schlathölter, Near edge X-ray absorption mass spectrometry of gas phase proteins: the influence of protein size, Phys. Chem. Chem. Phys., 2016, 18, 26213–26223 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp02524a |
| This journal is © the Owner Societies 2023 |