
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Decoding the infrared spectra changes upon formation of molecular complexes: the case of halogen bonding in pyridine⋯perfluorohaloarene complexes†
Alex
Iglesias-Reguant
 ab,
Heribert
Reis
c,
Miroslav
Medved’
de,
Borys
Ośmiałowski
a,
Robert
Zaleśny
*f and
Josep M.
Luis
*b
ab,
Heribert
Reis
c,
Miroslav
Medved’
de,
Borys
Ośmiałowski
a,
Robert
Zaleśny
*f and
Josep M.
Luis
*b
aFaculty of Chemistry, Nicolaus Copernicus University, Gagarina 7, PL-87100 Toruń, Poland
bInstitute of Computational Chemistry and Catalysis and Department of Chemistry, University of Girona, Campus de Montilivi, 17003, Girona, Catalonia, Spain. E-mail: josepm.luis@udg.edu
cInstitute of Chemical Biology, National Hellenic Research Foundation (NHRF), Vassileos Constantinou Ave 48th, 116 35 Athens, Greece
dDepartment of Chemistry, Faculty of Natural Sciences, Matej Bel University, Tajovského 40, SK-97400 Banská Bystrica, Slovak Republic
eRegional Centre of Advanced Technologies and Materials, Czech Advanced Technology and Research Institute (CATRIN), Palacký University in Olomouc, Šlechtitelu 27, 783 71 Olomouc, Czech Republic
fFaculty of Chemistry, Wroclaw University of Science and Technology, Wybrzeże Wyspiańskiego 27, 50-370 Wrocław, Poland. E-mail: robert.zalesny@pwr.edu.pl
First published on 13th July 2023
Abstract
A recently developed computational scheme is employed to interpret changes in the infrared spectra of halogen-bonded systems in terms of intermolecular interaction energy components (electrostatic, exchange, induction, dispersion) taking pyridine⋯perfluorohaloarene complexes as examples. For all complexes, we find a strong linear correlation between the different terms of the interaction-induced changes of the IR band associated with an intermolecular halogen bond stretching mode and the corresponding terms of the interaction energy, which implies that the interaction components play similar roles in both properties. This is not true for other vibrational modes localized in one of the monomers studied here, for which the corresponding interaction-induced changes in IR bands may present a completely different decomposition than the interaction energy.
Intermolecular interactions have a key role in the physicochemical properties of matter, such as solubility, reactivity, or all types of spectroscopic signatures, to name a few. Over the last decades, several theoretical frameworks have been developed to analyze the effect of the different intermolecular forces on the properties of the matter,1–10 allowing the understanding of their fundamental aspects in a plethora of molecular systems covering a wide palette of interaction types and strengths. Nowadays the methodology to analyze intermolecular interactions is an important part of modern interpretive chemistry and it is used to understand the physical origin of the experimental manifestations of intermolecular interactions. Myriads of atomic and molecular complexes have been studied using energy decomposition analyses, thus enriching the rationalization of the basic interaction types that govern the structure, stability, and properties of molecular complexes and their organization at larger scales.
Spectroscopic techniques, with infrared (IR) spectroscopy at the forefront, are frequently used to substantiate the formation of molecular complexes. The popularity of IR spectroscopy is due to its high interpretive power, e.g., the presence of hydrogen bonds in molecular complexes can be identified easily by the change of the frequency of the bands associated with the vibrational modes of functional groups involved in the intermolecular interaction,11,12 a feature listed by IUPAC13 as a criterion to call the interaction hydrogen bond (similar applies for halogen bonds).14 Spectroscopic IR studies are often paralleled by computational efforts in the framework of the theory of intermolecular interactions to link the strength of the interactions with specific interaction types (exchange, electrostatic, induction, dispersion).15,16 However, none of the performed studies attempted to analyze the changes in the intensity and frequency of the peaks of IR spectra in terms of interaction types since the available decomposition schemes were primarily developed for interaction energies. To overcome this limitation, we recently developed a new scheme to interpret the nature of the changes in infrared spectra occurring upon the formation of a molecular complex.17 This new scheme, called infrared spectra decomposition analysis (IRS-DA) allows the association of the simultaneous changes in the intensity and frequency of each peak of the IR spectra occurring upon complex formation with the different interaction types (i.e. exchange, electrostatic, induction, dispersion). Specifically, IRS-DA decomposes the changes in the ratio between intensity and frequency squared for each normal mode upon AB complex formation from monomers A and B. IRS-DA provides information regarding the interplay of interaction types, i (exchange, electrostatic, induction, dispersion), and their manifestation through changes in the band position and intensity of normal mode a:
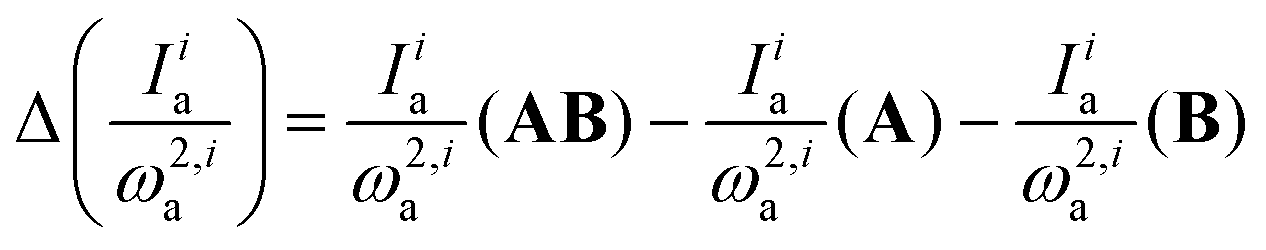 | (1) |
Note that for bands that undergo negligible frequency changes upon complex formation, or for which  , the above equation can be simplified to:
, the above equation can be simplified to:
| ΔIia = Iia(AB) − Iia(A) −Iia(B) | (2) |
In this communication, we report the first analysis of IR intensity changes upon the formation of halogen-bonded (XB) complexes, using the molecular complexes formed by pyridine (pyr) and perfluorohaloarenes (C6F5X, where X is Cl, Br, or I) as examples (Fig. 1). As demonstrated in our previous work, halogen bond formation in complexes containing pyridine can be identified using spectroscopic techniques, including IR spectroscopy.18 In particular, the measured IR spectrum of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of pyridine and C6F5I featured a new band at 195 cm−1 not present in the IR spectra of the pure liquids pyridine or C6F5I. This new band was assigned to the intermolecular C–I⋯N stretching, in agreement with the theoretical IR spectra obtained at the MN15/aug-cc-pVDZ(PP) level18 and previous measurements for the cocrystal.19 Thanks to the newly developed IRS-DA method it is now possible to link the formation of this band with individual intermolecular interaction types.17 XB complexes are nowadays in the limelight due to the unique properties of halogen bonding and its wide range of applications,20 such as in crystal engineering21 of emissive materials,22,23 sensing enhancement24 or in medicine.25–27 The term halogen bond is applied to a directional intermolecular interaction involving an electrophilic region (so-called σ-hole) associated with a halogen atom and a nucleophilic second atom. The σ-hole was described theoretically already a long time ago,28 and has been invoked as an explanation for many phenomena. However, it was only very recently possible to visualize the σ-hole directly by Kelvin probe force microscopy.29 Halogen bonding resembles hydrogen bonding (HB) in many aspects, including, in strongly interacting molecules, intermolecular migration of the heavy halogen atom, similar to proton transfer in certain hydrogen-bonded complexes.30 Given the widespread interest in XB complexes and their possible coexistence with hydrogen bonding, as exemplified in supramolecular synthons,31 we consider the analysis of changes in spectroscopic vibrational signatures in terms of interaction types very timely. Intermolecular interactions influence the location and topology of the potential energy minima and the electron density distribution of the molecules that form the complexes, which in turn affects both the position and the intensity of the bands in the IR spectra.
:1 mixture of pyridine and C6F5I featured a new band at 195 cm−1 not present in the IR spectra of the pure liquids pyridine or C6F5I. This new band was assigned to the intermolecular C–I⋯N stretching, in agreement with the theoretical IR spectra obtained at the MN15/aug-cc-pVDZ(PP) level18 and previous measurements for the cocrystal.19 Thanks to the newly developed IRS-DA method it is now possible to link the formation of this band with individual intermolecular interaction types.17 XB complexes are nowadays in the limelight due to the unique properties of halogen bonding and its wide range of applications,20 such as in crystal engineering21 of emissive materials,22,23 sensing enhancement24 or in medicine.25–27 The term halogen bond is applied to a directional intermolecular interaction involving an electrophilic region (so-called σ-hole) associated with a halogen atom and a nucleophilic second atom. The σ-hole was described theoretically already a long time ago,28 and has been invoked as an explanation for many phenomena. However, it was only very recently possible to visualize the σ-hole directly by Kelvin probe force microscopy.29 Halogen bonding resembles hydrogen bonding (HB) in many aspects, including, in strongly interacting molecules, intermolecular migration of the heavy halogen atom, similar to proton transfer in certain hydrogen-bonded complexes.30 Given the widespread interest in XB complexes and their possible coexistence with hydrogen bonding, as exemplified in supramolecular synthons,31 we consider the analysis of changes in spectroscopic vibrational signatures in terms of interaction types very timely. Intermolecular interactions influence the location and topology of the potential energy minima and the electron density distribution of the molecules that form the complexes, which in turn affects both the position and the intensity of the bands in the IR spectra.
 | ||
| Fig. 1 Optimized structures of the studied halogen-bonded complexes, composed of pyridine and (a) C6F5Cl, (b) C6F5Br or (c) C6F5I. | ||
In order to perform the analysis of ΔIa upon complex formation in terms of interaction types i we choose the general framework of decomposition of the interaction energy at the second-order Møller–Plesset perturbation theory (MP2) level based on the variational-perturbational decomposition scheme (VP-EDS).32–36 According to VP-EDS, the total interaction energy of a dimer is partitioned into several interaction energy terms as defined in the symmetry-adapted perturbation theory (SAPT).32–35 At the MP2 level, the total VP-EDS interaction energy of the complex calculated by a supermolecular approach in the dimer-centered-basis set (DCBS)37 is partitioned into the Hartree–Fock (HF) (ΔEHFint) and the electron correlation interaction energy correction (ΔEMP2corr):
 | (3) |
The HF term encompasses the electrostatic interactions of the charge densities of the unperturbed subsystems, ε(10)el, the exchange repulsion (ΔEHLex), and the charge delocalization (ΔEHFdel). The last two terms account for exchange effects due to the Pauli antisymmetry principle and induction, respectively. The second-order electron correlation term includes the electron correlation correction to the first-order electrostatic interaction, ε(12)el,r, the second-order dispersion interaction, ε(20)disp, and the remaining second-order electron correlation effects (ΔE(2)ex).34,36,38
We will start the analysis of the pyr:C6F5X complexes with the partitioning of the intermolecular interaction energy (ΔE). Fig. 2a and Table S1 (ESI†) show the MP2 interaction energies and their components for the three complexes ordered according to the increasing value of the total interaction energy. As expected, the absolute value of the VP-EDS/MP2 interaction energy is largest for the complex containing iodine, and it decreases with increasing electronegativity (χ) of atom X, (i.e., ΔE(I) > ΔE(Br) > ΔE(Cl)). The partitioning of the interaction energy for the three pyr:C6F5X complexes reveals a large destabilizing exchange-repulsion contribution, which is canceled by the sum of the electrostatic and delocalization terms, thus predicting only very weak binding at the uncorrelated level (Table S1, ESI†). Thus, the observed stability of the studied complexes is mostly due to the sum of the electron correlation terms, especially Δε(20)disp and ΔE(2)ex, to the total interaction energies.
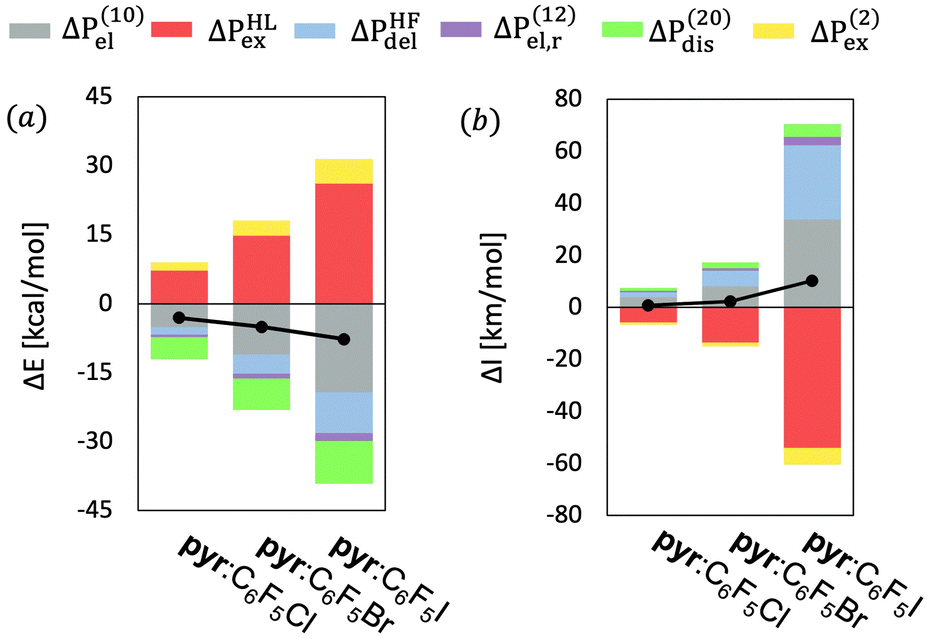 | ||
| Fig. 2 Partitioning of (a) the interaction energy (ΔP = ΔE) and (b) the excess IR intensity (ΔP = ΔI) of the intermolecular vibrational stretching mode (νinta) for the pyr:C6F5X complexes at the equilibrium geometries at the MP2/aug-cc-pVDZ-PP level of theory. The black dots combined with black lines indicate the total excess properties. | ||
Fig. 2a shows that the interplay of the interaction energy components for the three pyr:C6F5X complexes is very similar, i.e. the relative weight of the different interaction terms contributing to ΔE for the three different halogen bonds resembles each other to a large extent (see also Fig. S1, ESI†). We may thus conclude that the stability of these different XB complexes with the nitrogen atom in pyridine as the nucleophilic partner has a similar physical origin, in the sense that the different elements of the intermolecular interaction lead to comparable relative contributions to the total excess energy. We note that a similar conclusion can be drawn from the good linear fits with negligible y-intercept between the ΔE and ΔEi terms of D:C6F5X (where D represents a fluoroborate-based dye containing pyridine, see Fig. S2, ESI†)18 and the corresponding terms of pyr:C6F5I, as shown in Fig. S3 and Table S2 (ESI†).
Furthermore, the partition of ΔE for these six complexes follows the same trends observed in previous studies of small halogen-bonded dimers performed by our group, indicating that the nature of halogen bonding is similar for complexes of different chemical composition.39 Table S3 (ESI†) presents the R2 of the linear fits between partition terms of the intermolecular interaction energy of all the XB and HB complexes studied in this work and three complexes in π–π stacking arrangement studied in the ref. 17 The complexes presenting the same type of non-covalent interaction share a very similar physical origin (i.e., R2 > 0.89). On the contrary, this is not true for systems that present different types of non-covalent interactions (i.e., R2 as low as 0.39).
The difference IR spectrum of pyr:C6F5I displayed in Fig. 3 reveals significant changes in the IR spectra of the individual constituents upon complexation. Our IRS-DA analysis will be focussed on four representative bands attributed to (a) the intermolecular vibrational stretching mode (νinta); (b) the C–I stretching, which implies a change in the halogen bond distance (νC–Xb); and two vibrational modes that involve movements of the atoms of only one monomer: (c) a distortion of the ring of the C6F5 I, (νcC6F5I) and (d) a rocking in-plane bending of the hydrogens of the pyridine, (νpyrd). We have also analyzed the changes of the IR bands upon complex formation associated with the four equivalent vibrational modes of the complexes pyr:C6F5Br and pyr:C6F5Cl (Fig. S4 and S5, ESI†).
 | ||
| Fig. 3 Difference IR spectrum obtained by subtracting the simulated IR spectra of pyridine and C6F5I from the IR spectrum of pyr:C6F5I complex, as well as the displacements of: (a) an intermolecular vibrational stretching mode (νinta), (b) a C–X stretching mode of C6F5I, which involves a change in the halogen bond distance (νC–Xb), (c) one local vibrational mode of C6F5I (νcC6F5I) and (d) one local vibrational mode of pyridine (νpyrc). | ||
The mode νinta is only present in the complex and arises due to the halogen bonding. Therefore, we will first focus our analysis on the physical origin of ΔI for this peak. For νC–Xb, νcC6F5I and νpyrd, the frequency shifts upon complex formation are very small, which according to eqn (2) permits the direct decomposition of IR intensity changes. We note that the displacements of the two first modes include large variations of the halogen-bond distance, which is not the case for the latter two. Thus, νinta and νC–Xb have a direct effect on the intermolecular interaction, in contrast with νcC6F5I and νpyrd. Even though the observed vibrational frequency shifts upon complex formation observed for modes localized on C6F5X (νC–Xb, νcC6F5I) are much smaller than that for mode νinta, in all three cases we found a strong linear correlation between Δ![[small nu, Greek, macron]](https://www.rsc.org/images/entities/char_e0ce.gif) and intermolecular interaction energy terms (see Table S4, ESI†).
and intermolecular interaction energy terms (see Table S4, ESI†).
Fig. 2b shows the partition of ΔIa for the intermolecular vibrational stretching mode νinta of the three pyr:C6F5X complexes ordered according to increasing values of the total interaction energy (see also Tables S5–S7, ESI†). Similarly to the pattern found for the interaction energy, the largest IR intensity changes are found for the complex with X = I and they decrease with increasing electronegativity of the halogen atom X. Indeed, as shown in Table S8 (ESI†), for this normal mode a good linear correlation with a very small y-intercept exists between the computed values of ΔE and ΔIa for the three complexes (R2 = 0.93). Also, mimicking the decomposition of ΔE, the partitioning of the intensity of the band associated to νinta of pyr:C6F5I reveals that the increment of ΔIa is mainly due to the sum of electrostatic and delocalization terms partially canceled by a large negative Pauli exchange-repulsion contribution. But now the total ΔIa value is fairly well (up to ≈ 80%) described at the HF level. Nevertheless, as for ΔE, the electron correlation terms, especially the dispersion and second-order exchange contributions, also play an important role in ΔIa of this normal mode. However, we should point out that the relative weight of the dispersion contribution decreases when the electronegativity of the halogen increases, again in agreement with the interaction energies.
Fig. 4 and Fig. S6, S7 (ESI†) show that for νinta there is indeed a nice linear correlation with a small y-intercept between the values of the terms of the ΔE decomposition and those of the ΔIa decomposition (i.e. R2 > 0.89), indicating that both excess properties have the same physical nature in the sense defined above. The goodness-of-fit improves with the strength of the intermolecular interaction, giving the best linear correlation for the pyr:C6F5I complex. Similar good linear fits between the decomposition terms of the ΔE and ΔI are observed for the νC–Xb band of pyr:C6F5Br and pyr:C6F5I (see Table S8 and Fig. S8–S10, ESI†). On the contrary, we do not find a good linear correlation for pyr:C6F5Cl, possibly due to the strong deviation of the C–X–N angle from 180 (161), which reduces the coupling of νC–Xb vibration with the halogen bond distance. νcC6F5X and νpyrd bands also show a high linear correlation between the values of ΔE and ΔI. However, the decomposition of ΔI for those normal modes, which do not affect the halogen-bond distance, is very different from the decomposition of ΔI for νinta and νC–Xb. For νcC6F5X and νpyrd, the relative weights and even the signs of the terms of ΔI change depending on the complex and normal mode (Tables S5–S7 and Fig. S11, ESI†). For instance, the largest contribution for νcC6F5Cl, νcC6F5Br, and νcC6F5I is ΔIel(10), ΔIexHL and ΔIdelHF, respectively. Therefore, while for those vibrational modes that induce large variations of the halogen-bond distance the partitions of ΔI are strongly correlated with their ΔE counterparts, no such correlation is found for the other vibrational modes studied here.
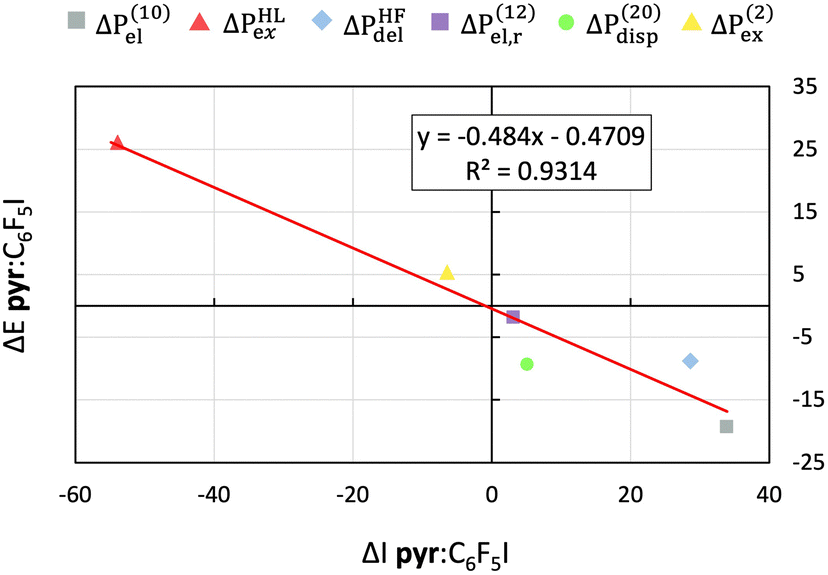 | ||
| Fig. 4 Linear fits between interaction-type contributions for the interaction energy (ΔP = ΔE) and the corresponding terms of the excess IR intensity (ΔP = ΔI) for νinta for the pyr:C6F5I complex. Energies are given in kcal mol−1 and intensities in km mol−1. | ||
To investigate if this observation can be generalized to other molecular complexes, the decompositions of ΔE and ΔI for three vibrational modes of two small halogen-bonded complexes (HCN:IF and HCN:BrF) and of three small hydrogen-bonded systems (HCN:HCl, HCN:HNC and HCN:HCN) were analyzed (Fig. S12–S16 and Tables S9–S14, ESI†). As summarized in Table S15 (ESI†), for the intermolecular stretching of the five complexes the decomposition terms of ΔI are linearly correlated with the decomposition terms of ΔE (R2 > 0.83). On the other hand, for the local vibrational modes the analogous linear correlation is usually not found, (i.e. the coefficients of determination (R2) are between 0.20 and 0.80).
In summary, thanks to our recent development of a new computational scheme to decompose the IR spectra of molecular complexes in terms of intermolecular interaction energy components, we have shown for different types of systems and intermolecular bonds that the partitions of ΔE and ΔI are strongly linearly correlated, with a very small y-intercept, for the vibrational modes which involve substantial variations of the intermolecular bond length. Therefore, for such modes, the physical origin of the changes in the corresponding IR bands in terms of interaction types is similar to the interaction energy, which implies that both excess properties have equivalent physical nature. This is not true for other vibrational modes localized in one of the monomers studied here, for which the corresponding interaction-induced changes in IR bands may present a completely different decomposition than the interaction energy.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. I. R., B. O., and R. Z. acknowledge support from the National Science Centre, Poland (Grant 2019/35/B/ST5/00656). J. M. L. thanks the funds from the Spanish government MICINN (PGC2018-098212-B-C22), Generalitat de Catalunya (2021SGR00623), and Donostia International Physics Center (DIPC-INV-003133). The COST Action CA21101 is also acknowledged. The authors thank Wroclaw Centre for Networking and Supercomputing for computational resources and Miss Elizaveta F. Petrusevich for the preparation of the GTOC.References
- K. Morokuma, J. Chem. Phys., 1971, 55, 1236–1244 CrossRef CAS.
- T. Ziegler and A. Rauk, Theor. Chim. Acta, 1977, 46, 1–10 CrossRef CAS.
- K. Kitaura and K. Morokuma, Int. J. Quantum Chem., 1976, 10, 325–340 CrossRef CAS.
- P. S. Bagus, K. Hermann and C. W. Bauschlicher, J. Chem. Phys., 1984, 80, 4378 CrossRef CAS.
- W. J. Stevens and W. H. Fink, Chem. Phys. Lett., 1987, 139, 15–22 CrossRef CAS.
- E. D. Glendening and A. Streitwieser, J. Chem. Phys., 1994, 100, 2900 CrossRef CAS.
- Y. Mo, J. Gao and S. D. Peyerimhoff, J. Chem. Phys., 2000, 112, 5530 CrossRef CAS.
- P. Su and H. Li, J. Chem. Phys., 2009, 131, 014102 CrossRef PubMed.
- P. R. Horn and M. Head-Gordon, Phys. Chem. Chem. Phys., 2016, 18, 23067 RSC.
- A. Aldossary, M. Gimferrer, Y. Mao, A. K. D. Honxia Hao, P. Salvador, T. Head-Gordon and M. Head-Gordon, J. Phys. Chem. A, 2023, 127, 1760–1774 CrossRef CAS PubMed.
- A. Buckingham, J. Del Bene and S. McDowell, Chem. Phys. Lett., 2008, 463, 1–10 CrossRef CAS.
- T. Fornaro, D. Burini, M. Biczysko and V. Barone, J. Phys. Chem. A, 2015, 119, 4224–4236 CrossRef CAS PubMed.
- E. Arunan, G. R. Desiraju, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R. H. Crabtree, J. J. Dannenberg, P. Hobza, H. G. Kjaergaard, A. C. Legon, B. Mennucci and D. J. Nesbitt, Pure Appl. Chem., 2011, 83, 1637–1641 CrossRef CAS.
- G. R. Desiraju, P. S. Ho, L. Kloo, A. C. Legon, R. Marquardt, P. Metrangolo, P. Politzer, G. Resnati and K. Rissanen, Pure Appl. Chem., 2013, 85, 1711–1713 CrossRef CAS.
- Q. Zhang, M. Chen, M. Zhou, D. M. Andrada and G. Frenking, J. Phys. Chem. A, 2015, 119, 2543–2552 CrossRef CAS PubMed.
- D. Ojha, K. Karhan and T. Kühne, Sci. Rep., 2018, 8, 2045–2322 CrossRef PubMed.
- A. Iglesias-Reguant, H. Reis, M. Medved, J. M. Luis and R. Zalesny, Phys. Chem. Chem. Phys., 2023, 25, 11658–11664 RSC.
- A. Iglesias-Reguant, J. Zielak-Milewska, T. Misiaszek, R. Zalesny, J. M. Luis and B. Osmialowski, J. Org. Chem., 2022, 87, 15159–15165 CrossRef CAS PubMed.
- V. Vasylyeva, L. Catalano, C. Nervi, R. Gobetto, P. Metrangolo and G. Resnati, CrystEngComm, 2016, 18, 2247–2250 RSC.
- F. Meyer and P. Dubois, CrystEngComm, 2013, 15, 3058–3071 RSC.
- G. R. Desiraju, J. Am. Chem. Soc., 2013, 135, 9952–9967 CrossRef CAS PubMed.
- W. Wang, Y. Zhang and W. J. Jin, Coord. Chem. Rev., 2020, 404, 213107 CrossRef CAS.
- J. K. Salunke, N. A. Durandin, T.-P. Ruoko, N. R. Candeias, P. Vivo, E. Vuorimaa-Laukkanen, T. Laaksonen and A. Priimagi, Sci. Rep., 2018, 8, 14431 CrossRef PubMed.
- K. M. Bak, S. C. Patrick, X. Li, P. D. Beer and J. J. Davis, Angew. Chem., Int. Ed., 2023, 62, e202300867 CrossRef CAS PubMed.
- P. Auffinger, F. A. Hays, E. Westhof and P. S. Ho, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 16789–16794 CrossRef CAS PubMed.
- R. Wilcken, M. O. Zimmermann, A. Lange, A. C. Joerger and F. M. Boeckler, J. Med. Chem., 2013, 56, 1363–1388 CrossRef CAS PubMed.
- W.-S. Zou, S. Lin, J.-Y. Li, H.-Q. Wei, X.-Q. Zhang, D.-X. Shen, J.-Q. Qiao, H.-Z. Lian, D.-Q. Xie and X. Ge, New J. Chem., 2015, 39, 262–272 RSC.
- T. Clark, M. Hennemann and J. Murray, et al. , J. Mol. Model., 2007, 13, 291–296 CrossRef CAS PubMed.
- B. Mallada, A. Gallardo, M. Lamanec, B. de la Torre, V. Spirko, P. Hobza and P. Jelinek, Science, 2021, 374, 863–867 CrossRef CAS PubMed.
- S. Yu, J. S. Ward, K.-N. Truong and K. Rissanen, Angew. Chem., Int. Ed., 2021, 60, 20739–20743 CrossRef CAS PubMed.
- N. Bedekovic, L. Fotovic, V. Stilinovic and D. Cincic, Cryst. Growth Des., 2022, 22, 987–992 CrossRef CAS PubMed.
- M. Gutowski, F. V. Duijneveldt, G. Chałasiński and L. Piela, Mol. Phys., 1987, 61, 233–247 CrossRef CAS.
- W. A. Sokalski, S. Roszak and K. Pecul, Chem. Phys. Lett., 1988, 153, 153–159 CrossRef CAS.
- S. M. Cybulski, G. Chałasiński and R. Moszyński, J. Chem. Phys., 1990, 92, 4357–4363 CrossRef CAS.
- G. Chałasiński and M. M. Szcześniak, Chem. Rev., 1994, 94, 1723–1765 CrossRef.
- G. Chałasiński and M. M. Szcześniak, Mol. Phys., 1988, 63, 205–224 CrossRef.
- F. B. van Duijneveldt, J. G. C. M. van Duijneveldt-van de Rijdt and J. H. Van Lenthe, Chem. Rev., 1994, 94, 1873–1885 CrossRef CAS.
- R. Moszyński, S. Rybak, S. Cybulski and G. Chałasiński, Chem. Phys. Lett., 1990, 166, 609–614 CrossRef.
- M. Medved, A. Iglesias, H. Reis, R. Góra, J. M. Luis and R. Zaleśny, Phys. Chem. Chem. Phys., 2020, 22, 4225–4234 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp02412a |
| This journal is © the Owner Societies 2023 |