
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photoinduced electron transfer in [10]CPP⊃C60 oligomers with stable and well-defined supramolecular structures†
A. J.
Stasyuk

Institut de Química Computacional i Catàlisi and Departament de Química, Universitat de Girona, C/Maria Aurèlia Capmany 69, 17003 Girona, Spain. E-mail: antony.stasuk@gmail.com
First published on 24th July 2023
Abstract
Recent synthesis of a new type of polymer containing conjugated cycloparaphenylene (CPP) macrocycles interconnected by a linear conjugated backbone opens up great potential of cyclic π-conjugated materials in organic photovoltaics. In this work, I report a theoretical study of the ground and excited state properties of such polymers and investigate an effect of inclusion of fullerene molecules into polymer chains. MD simulations reveal that oligomers ([10]CPP_Fused⊃C60)24 and ([10]CPP_Fused⊃C60)32 with π-extended CPPs tend to form stable, helix-like structures. I show that photoinduced electron transfer from the CPP-based polymer to C60 fullerene is favorable and occurs on a nanosecond time scale. The hole- and excess-electron transfer rates are found to be significantly higher than the corresponding charge recombination rates.
Introduction
Attention to molecules with radial π-conjugation has grown very rapidly in recent decades. Rare examples of cyclic structures with radial π-conjugation, such as paraphenylacetylenes,1,2 picotubes,3,4 or cyclocarbons,5–7 have been appearing in the literature since the early 1990s, but have been associated with extremely difficult and low-yield advanced organic synthesis. This fact significantly hindered the wide use of such cyclic structures. However, in 2008, Jasti and Bertozzi and co-workers invented a new strategy for the synthesis of cycloparaphenylenes (CPPs) in acceptable yields.8 The unusual hoop-shaped structure with radially conjugated π-electron systems of these cycloparaphenylenes leads to significant progress in the development of protocols for the synthesis of various CPPs.9,10 Modern approaches have made it possible to obtain CPPs with a diameter from 7 to 28 Å, precisely controlled by the numbers of units in the macrocycle.11,12 Nanosized concave cavities of CPPs have been prone to the formation of multiple non-covalent interactions and widely used in supramolecular chemistry.13 The first host–guest complex of CPP was reported by Iwamoto et al. in 2011.14 The authors found that [10]CPP with 10 phenylene units has a nearly ideal diameter (13.8 Å) to accommodate C60 fullerene. Considering that tuning the fullerene properties by functionalization is a non-trivial task, a supramolecular approach to changing the fullerene behavior seems to be a very attractive method for obtaining new fullerene-based systems. Thus, special attention has been paid to CPP's donor–acceptor complexes due to their potential in organic photovoltaic applications.13,15,16However, the practical usage of such systems in organic electronics is somewhat difficult due to issues typical for small molecules. They suffer from inhomogeneous film formation and create sharp grain boundaries as effective charge carrier traps due to their high crystallinity.17–19 In contrast, conjugated polymers form layered structures with a large domain size, as well as the orientation of crystallites with respect to neighboring domains and transport directions.19,20 This, in turn, makes the transport properties much less sensitive to the relative lattice orientation and promotes conduction in the device. Despite all the intriguing features of CPPs, they have not previously been utilized in polymeric materials. In 2019, Du and co-workers21 reported the first example of a π-conjugated polymer composed of functionalized [8]CPP. The synthetic strategy used is based on the generation of a cyclic molecule from an appropriate curved and bifunctionalized fragment using Pd-catalyzed Suzuki coupling. The subsequent polymerization was carried out using a Ni-mediated Yamamoto reaction. The resulting polymer had a low polydispersity index of 1.44 and consisted of approximately 22 repeating units. Measurements have shown good carrier mobility when using this polymer as a hole- and electron-transport layer.21
Peters et al.22 also described the synthesis and electronic properties of π-conjugated polymers that combine the linear intramolecular conjugation pathways (phenyl/thienylactylene) with the emerging properties of radial [6]/[8] CPP segments. Gel permeation chromatography data for most of the synthesized polymers indicated the presence of 7 to 27 repeating units. Photophysical measurements demonstrated that the combination of linear and radial π-conjugations provides more additive properties, but creates new electronic processes. The effect of the communication of CPP units in polymers was also considered. Authors demonstrate that the electronic properties of polymeric [8]CPPs with disjointed pi-conjugated substituents arise from multiple operative radial/linear conjugation pathways, as the disjoint pattern results in both ortho and meta connections to the CPP ring.23
More recently, Wang et al.24 have developed a novel all-carbon polymeric structure containing conjugated macrocycles linked together by a linear poly(para-phenylene) backbone acting as a host for the C60 fullerene. The procedure for synthesizing a new [10]CPP-based polymer was the same as that for the original [8]CPP-based polymer. Fullerene was incorporated into polymeric [10]CPP at the last stage. The polydispersity index of the [10]CPP polymer was found to be 2.66, indicating a broad molecular weight distribution. MALDI-TOF mass spectrometry detected fractions containing up to 17 repeating units. Transient absorption measurements showed that, upon photoirradiation, an electron is transferred from the photoexcited polymeric unit to the C60 fullerene. In addition, the measurement of electron- and hole-mobility clearly showed that the incorporation of C60 into a [10]CPP-based polymer leads to an improvement in the conductive properties.
Herein, I discuss the effect of a linker between CPPs and the inclusion of C60 fullerene into the polymer chain of CPPs on the spatial structure of ([10]CPP)n and quantitatively characterize photoinduced electron transfer (PET), hole transfer (HT), and excessive electron transfer (EET) processes occurring in the all-carbon conjugated ([10]CPP_Fused⊃C60)n polymer.
Results and discussion
Polymeric complex ([10]CPP⊃C60)n
In photovoltaic devices, the absorption of photons creates excitons (coupled electron–hole pairs). Their subsequent charge separation and carrier mobility determine the overall performance of the device. Thus, not only charge separation/recombination rates, but also hole and electron transfer rates are extremely important characteristics for solar cells and artificial photosynthetic systems. The possession of the unique structural and physical properties of long π-extended [10]CPP-based polymers encourages the investigation of their response to photoexcitation. Being a rather flexible polymeric chain where monomers are interconnected by a single bond, ([10]CPP)n and ([10]CPP⊃C60)n can have a significant conformational diversity. To gain insight into the dynamic shape of these polymers, I ran a series of molecular dynamics (MD) simulations for 8-unit oligomers. The simulations were performed using a classical Merck molecular force field (MMFF) with a step size of 0.5 fs up to a total of 5 ns at 300 K within the NVT ensemble as implemented in the Tinker-HP program25,26 (for details see the ESI†). The performed MD simulations revealed interesting features of the ([10]CPP)8 and ([10]CPP⊃C60)8 chains. In particular, the studied oligomers formed separate domains in which the units are in the syn or anti position to each other (Fig. 1a and b). As seen in the ([10]CPP)8 oligomer, syn and anti units alternate. There are two conformations were observed for this oligomer: conformation 1 was from 0.7 to 3.9 ns and conformation 2 was from 3.9 to 5 ns. They differ in the orientation of the terminal [10]CPP unit. In conformation 1, this unit is in syn orientation with the two adjacent units, while in conformation 2, this terminal unit adopts anti orientation. The representative structures of both conformers are shown in Fig. S1 (ESI†). The incorporation of the C60 fullerene leads to an increase in the size of syn-oriented units. In the ([10]CPP⊃C60)8 oligomer, this domain increases to 3 monomeric units. | ||
Fig. 1 Structure of monomeric units, 8-unit oligomers and RMSD diagrams for 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ns of MD simulation runs for the (a) fullerene free ([10]CPP)8 oligomer, (b) fullerene encapsulated ([10]CPP⊃C60)8 oligomer and (c) ([10]CPP_Fused⊃C60)8 oligomer containing an π-extended fragment. ns of MD simulation runs for the (a) fullerene free ([10]CPP)8 oligomer, (b) fullerene encapsulated ([10]CPP⊃C60)8 oligomer and (c) ([10]CPP_Fused⊃C60)8 oligomer containing an π-extended fragment. | ||
The mobility of electrons and holes are determined by the value of electronic coupling. It can be assumed that a large distance between the units in the anti configuration compared to those in the syn configuration greatly reduces the electronic coupling and thus affects the charge transport rate. Thus, the size of the syn domain and the number of units in the anti conformation are very important for efficient charge transport in such a polymer. To validate the aforementioned statement, electronic couplings were calculated for 20 randomly selected structures (10 syn and 10 anti conformations) extracted from the MD trajectory of the ([10]CPP)8 oligomer. The results revealed that the average coupling value for the syn conformers is twice higher than the average coupling value for the anti conformers.
Based on the above, it can be assumed that an increase in the electron and hole mobility of the ([10]CPP⊃C60)n polymer compared to the ([10]CPP)n polymer21,24 is due to an increase in the number of syn-oriented units. I hypothesized that the cyclodehydrogenation of the arylated CPP (monomeric unit), leading to the formation of the π-extended fragment (Fig. 1c), would increase the rigidity of the entire system and facilitate the syn conformation. To test this assumption, I performed MD simulations for the ([10]CPP_Fused⊃C60)8 oligomer. Already after 1.5 ns, the system formed a stable semicircular structure, in which all 8 monomeric units are in the syn conformation. When comparing the three considered 8-unit oligomers, a significant fluctuation of the RMSD values for ([10]CPP_Fused⊃C60)8 attracts attention. It was found that the frames with higher RMSD values correspond to a more compressed semicircular structure with high curvature. In turn, the frames with smaller RMSD values correspond to a more open structure with low curvature. The corresponding structures are shown in Fig. S2 (ESI†).
Inspired by the results, I ran simulations for polymers whose size is similar to that measured experimentally.21,24 Two polymers, ([10]CPP_Fused⊃C60)24 and ([10]CPP_Fused⊃C60)32, containing 24 and 32 monomeric units were analyzed. The simulations were performed under the same conditions as for oligomers containing 8 units, up to 10 ns in total. It is interesting to note that both systems formed stable helical structures (Fig. 2). The formation of the helical structures can be seen in Movies 1 and 2 (ESI†).
As seen in Fig. 2, the ([10]CPP_Fused⊃C60)24 polymer acquires a stable helical configuration after about 4 ns, while the ([10]CPP_Fused⊃C60)32 system takes about 7 ns to complete the formation process. As soon as the helical structure is formed, both considered systems retain it throughout the simulation time. RMSD graphs with the zoomed region of interest are presented in Fig. S3 (ESI†). It is important to note that the helical structures are very stable. To examine the thermal stability of the formed structure, MD simulations were performed for 5 ns at 500 K on each of two helical structures independently obtained by MD simulations at 300 K. High temperature did not lead to the disruption of the formed structures or their noticeable distortion (see Fig. S4, ESI†).
 | ||
| Fig. 2 Representative structure of the ([10]CPP_Fused)24 and ([10]CPP_Fused⊃C60)32 oligomers and RMSD diagrams for 10ns of MD simulation runs. | ||
Ground state properties of the [10]CPP_Fused⊃C60 monomer
The possibility of photoinduced electron transfer (PET) in non-covalently bound donor–acceptor dyads consisting of C60 as an electron acceptor and cycloparaphenylene (CPP) as an electron donor has been previously reported for several systems.13,27–29 To study the possible PET processes in the polymer systems, a monomeric inclusion complex [10]CPP_Fused⊃C60 (U1) and its dimer ([10]CPP_Fused⊃C60)2 (U2) were considered. Geometries of U1 and U2 complexes (Fig. 3) were optimized using the BLYP-D3(BJ)/def2-SVP level of theory.30–35 (The full computational details are provided in the ESI†). For the U1 system, the interaction energy is −61.0 kcal mol−1 (Table 1). This value is similar to the energy estimated for the complexes of fullerene with unmodified [10]CPP at the same level of theory.36,37 It is worth mentioning that the interaction energy in the U2 complex was found to be superadditive. The energy is −145.9 kcal mol−1, which is greater than the sum of energies of pair interactions (between the host dimer and each of fullerenes). The observed superadditivity can be explained by the favorable interaction of fullerenes with each other. For details see Table S2 and Fig. S5 (ESI†). A similar effect was observed earlier for carbon nano-onions and other carbon-rich complexes.38,39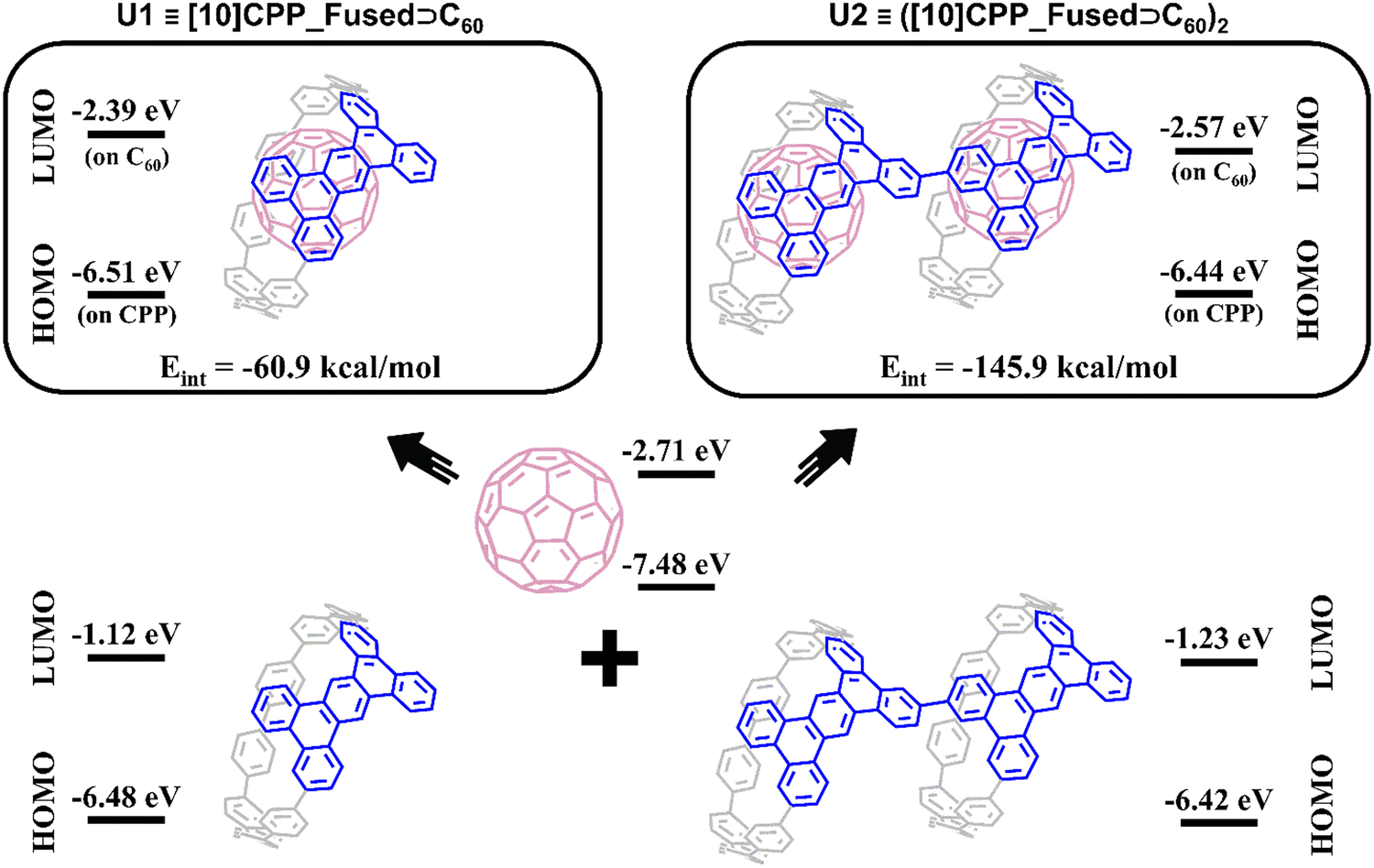 | ||
| Fig. 3 Structure and HOMO/LUMO energies of U1 and U2 complexes and their subunits. | ||
| Complex | Interaction scheme | Energy termsa, kcal mol−1 | ΔEint | |||
|---|---|---|---|---|---|---|
| ΔEPauli | ΔEelstat | ΔEoi | ΔEdisp | |||
| a The percentage contributions to the sum of attracting energies (ΔEelstat + ΔEoi + ΔEdisp) are given in parentheses. | ||||||
| U1 | E TOTALint | 83.00 | −34.28 (23.8%) | −17.44 (12.1%) | −92.23 (64.1%) | −60.95 |
| U2 |

|
102.42 | −41.60 (24.0%) | −22.60 (13.0%) | −109.39 (63.0%) | −71.16 |

|
99.35 | −39.32 (23.6%) | −22.16 (13.3%) | −105.39 (63.2%) | −67.52 | |
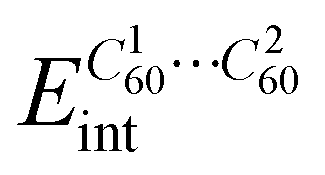
|
20.38 | −7.81(28.2%) | −4.89 (17.6%) | −15.04 (54.2%) | −7.36 | |
| E TOTALint | −145.85 | |||||
As seen in Fig. 3, the HOMO and LUMO in both U1 and U2 complexes are localized on [10]CPP_Fused and C60 units, respectively. The formation of complexes has a rather weak effect on the HOMO and LUMO energies of the fragments. The HOMO energy changes within 0.05 eV, whereas the increase in the LUMO energies was found to be 0.32 and 0.14 eV for U1 and U2, respectively. The small differences in the HOMO and LUMO energies of complexes and individual fragments indicate the absence of charge separation in the ground state of the complexes. The performed population analysis does not reveal any significant charge transfer between the host [10]CPP_Fused unit and C60 guest molecules (Table S3, ESI†).
To analyze the nature of the host–guest interactions, I performed the Morokuma-like energy decomposition analysis (EDA)40–42 implemented in the ADF program at the BLYP-D3(BJ)/TZP level of theory. The EDA decomposes the interaction energy into four components: electrostatic (ΔEelstat), Pauli repulsion (ΔEPauli), orbital interactions (ΔEoi), and dispersion correction (ΔEdisp). This allows one to estimate the role of specific interactions. The EDA results listed in Table 1 indicate that the host–guest interactions in the complexes are quite similar. Among the binding forces (ΔEelstat + ΔEoi + ΔEdisp), the dispersion term prevails with a contribution of 63–64%. The second largest term is electrostatic attraction with a contribution of 23–24%. Finally, the orbital interactions provide only 12–13% of the total stabilization interactions.
To gain access to the host–guest interaction topology, I performed a series of QTAIM (Bader's Atoms in Molecules theory) and NCI (non-covalent interaction index) calculations for the U1 and U2 systems. Various QTAIM characteristics at the bond critical points (BCPs) can provide information on the nature of particular interaction in supramolecular complexes.43 In the case of U1, the analysis revealed only C⋯C interactions (see Table S4, ESI†). The observed intermolecular interactions have ρ(r) values in the range of 0.005–0.008 a.u., while the Laplacian ∇2ρ(r) values are positive, indicating that these interactions are of closed-shell type. Moreover, −G(r)/V(r) values at all BCPs are greater than 1, demonstrating the non-covalent nature of the interaction. Taking into account that the host and guest units are the π-conjugated systems, the C⋯C interactions can be assigned to π⋯π interactions. In the case of the U2 complex, two types of interactions were observed: interactions between the CPP and C60 within one unit, intraunit interactions, and interactions between CPPs and fullerenes of different units – interunit interactions. Intraunit interactions in U2 are very similar to those in the U1 complex and can be assigned to pure π⋯π interactions. For interunit interactions, the C⋯C interactions between fullerenes and the CH⋯C interactions between CPPs and fullerenes of different units were observed, which can be assigned to π⋯π and C–H⋯π interactions. QTAIM molecular graphs for the U1 and U2 complexes are given in Fig. S6 (ESI†). NCI confirms the QTAIM data. The green NCI isosurfaces between CPP and C60 of the same unit are evenly distributed throughout the cavity and have the similar shape for both complexes, indicating weak van der Waals interactions. In addition, the green isosurface between fullerenes and CPPs of different units was found. Thus, the results of QTAIM and NCI analyses are fully coincident and confirm a set of weak van der Waals interactions between neighboring units. The reduced density gradient (RDG) plots and NCI isosurfaces are presented in Fig. S7 and S8 (ESI†).
Singlet excited states
Simulations of excited states were performed using the TD-DFT method with the CAM-B3LYP-D3(BJ)/def2-SVP scheme.32–35,44 The CAM-B3LYP functional is very well suited for modeling charge transfer processes in fullerene-based complexes.45 To characterize the properties of excited states, the U1 and U2 systems were divided into 2 and 4 fragments, respectively: host [10]CPP and guest C60. The electron density distribution was analyzed for the 80 lowest excited states. Three types of excited states were identified: (1) locally excited (LE) states, where excitation is mostly localized either on the guest or the host molecule, with charge separation smaller than 0.1e (CS < 0.1e); (2) charge transfer (CT) states showing significant charge separation (CS > 0.8e); and (3) mixed states, where both LE and CT states contribute substantially (0.1e < CS < 0.8e).46,47In the gas phase, the 80 lowest vertical singlet excitation energies of the U1 complex are found in the range from 2.51 to 4.41 eV. Table 2 contains the characteristics of the lowest excited states of each type. The LE1 state localized on C60 has the lowest energy, and is associated with a HOMO−4 to LUMO+1 transition. The LE2 state with the exciton localized on [10]CPP_Fused lies by 0.82 eV higher in energy at 3.33 eV and corresponds to the HOMO to LUMO+3 transition. The lowest CT1 state with 0.98e transferred from [10]CPP_Fused to C60 lies at 2.74 eV, and corresponds to the HOMO → LUMO transition. Such CT state can be described as [10]CPP_Fused+⊃C60−. Among the computed excited states, only this type of charge separated state was observed. The molecular orbitals representing the LE1, LE2, and CT1 states for the U1 complex are depicted in Fig. 4.
| Supramolecular host–guest system | ||||
|---|---|---|---|---|
| U1 | U2 | |||
| VAC | DCM | VAC | DCM | |
| a Mixed states with significant contributions of both LE and CT. b n/a – states of interest are not possible for the U1 complex. | ||||
| LE1 (fullerene C60) | ||||
| Ex | 2.511 | 2.503 | 2.473 | 2.474 |
| Transition (W) | H−4–L (0.53) | H−4–L+1 (0.35) | H−8–L (0.28) | H−8–L (0.28) |
| f | <0.001 | <0.001 | <0.001 | <0.001 |
| X | 0.947 | 0.946 | 0.625 | 0.616 |
| LE2 (host [10]CPP_Fused)a | ||||
| Ex | 3.330 | 3.335 | 3.222 | 3.232 |
| Transition (W) | H–L+3 (0.47) | H–L+3 (0.43) | H–L+6 (0.24) | H–L+6 (0.25) |
| f | 0.066 | 0.102 | 0.008 | 0.012 |
| X | 0.890 | 0.867 | 0.880 | 0.682 |
| Most absorptive transition (localized on [10]CPP_Fused) | ||||
| Ex | 3.802 | 3.805 | 3.832 | 3.834 |
| Transition (W) | H−2–L+3 (0.31) | H−2–L+3 (0.31) | H−1–L+6 (0.17) | H–L+6 (0.11) |
| f | 1.587 | 2.224 | 2.248 | 2.518 |
| X | 0.811 | 0.881 | 0.989 | 0.990 |
| CT1 intraunit ([10]CPP_Fused → fullerene C60) | ||||
| Ex | 2.741 | 2.449 | 2.667 | 2.429 |
| Transition (W) | H–L (0.93) | H–L (0.89) | H−1–L (0.66) | H−1–L (0.55) |
| f | 0.002 | 0.002 | <0.001 | <0.001 |
| CT | 0.976 | 0.978 | 0.979 | 0.978 |
| CT2 interunit ([10]CPP_Fused → fullerene C60) | ||||
| Ex | n/ab | 3.112 | 2.578 | |
| Transition (W) | H–L+2 (0.57) | H–L+2 (0.51) | ||
| f | <0.001 | <0.001 | ||
| CT | 0.992 | 0.987 | ||
 | ||
| Fig. 4 Molecular orbitals representing the LE1, LE2, and CT1 states for the U1 complex. | ||
The energies of the 80 lowest-lying singlet excited states of U2 due to the higher density of state found in a narrower range of energies compared to the U1 complex and vary from 2.47 to 3.59 eV. The energies of LE1 and LE2 states in both complexes are almost identical. In U2, two types of CT states were identified, unlike U1. The first CT type, similar to U1, corresponds to the electron transfer from [10]CPP_Fused to C60 within the same monomeric unit. The energy of the CT1 state in U2 is very similar to that in U1 and equals to 2.64 eV. The second type of the CT state, corresponding to the electron transfer from [10]CPP_Fused of one monomer to C60 of another monomer, was found at 3.11 eV (0.45 eV higher than CT1). This state can be described as ([10]CPP_Fused+⊃C60)-([10]CPP_Fused⊃C60−). For the U2 complex, within the computed 80 excited states, there are no states with a high oscillator strength. To get a clue about highly absorptive states, an additional 20 states were calculated. The state of interest has been found at 3.83 eV.
Effects of environment on excited states and electron transfer rates
To investigate the influence of the polar environment on electronic excitations, a well-proven COSMO-like model28,36,37,39,48,49 with dichloromethane (DCM) as a solvent was applied. DCM is a commonly used solvent in the measurement of the photophysical properties of carbon-based compounds. The dipole moments of the U1 and U2 complexes were calculated to be 0.09 and 0.76 D, respectively. The small dipole moment of the studied complexes can be explained by their high symmetry. The ground state (GS) solvation energies are −0.49 and −0.89 eV for U1 and U2, respectively (Fig. 5). Expectedly, the solvation energies for the LE states are very similar to the GS. In turn, the solvation energies for intraunit CT1 states were found to be −0.78 and −1.13 eV. The relatively small increase in the solvation energies can be explained by a high degree of charge delocalization in both fragments and their close proximity. The interunit CT2 state observed only in the U2 complex has a solvation energy of −1.43 eV. The computed CT2 solvation energy is almost twice as stronger as for the CT1 state, which is rationalized by the noticeable spatial separation of donor and acceptor units in the CT2 state. The dipole moment differences between the GS and CT1/CT2 states in the U2 complex were found to be 5.9 and 32.6 D, respectively. Detailed solvation data for both complexes are given in Table S5 (ESI†). | ||
| Fig. 5 Solvation diagram showing the energies of LE and CT states computed in vacuum (VAC) and dichloromethane (DCM) for U1 and U2 complexes and energy levels and ET/CR rates for the complexes in DCM. | ||
It is important to note that stabilization of the CT1 state by the solvent in both U1 and U2 complexes is sufficient to reorder LE1 and CT1 states, making the latter the lowest excited state (Fig. 5). In the case of the CT2 state in the U2 complex, the stabilization is not enough to reorder this state with LE1, but the energy difference between LE1 and CT2 states in DCM has become less than 0.1 eV. Because of the structural similarity, it seems interesting to compare the behavior of U1 and the well-studied [10]CPP⊃C60. As was shown earlier for original [10]CPP, the charge transfer reaction is characterized by a small positive Gibbs energy. Thus, a more extended π-system of the [10]CPP_Fused nanoring leads to the stabilization of the energy of the CT state and makes charge separation thermodynamically favorable.
For both complexes under consideration, the CT states are characterized by a very weak oscillator strength and therefore cannot be directly populated by light absorption. However, they can be generated by the decay of the lowest LE states. To estimate the efficiency of a such population, I computed the electron transfer rates using the semi-classical method proposed by Ulstrup and Jortner.50,51 Within this approach, the intramolecular relaxation associated with ET is described by an effective vibrational mode, and the rate is controlled by four parameters: electronic coupling (Vij) of the initial and final states, solvation reorganization energy λs, reaction Gibbs energy ΔG0, and effective Huang–Rhys factor Seff (for details see the ESI†). The computed parameters and rates in DCM are listed in Table 3. The rates were estimated using an effective frequency of 1600 cm−1, which corresponds to the stretching of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds. Note that for similar systems the calculated rates of charge separation do not change significantly by varying the effective frequency from 1400 to 1800 cm−1.52,53 For the U1 and U2 systems, the effect of frequency selection on the estimated ET rate was found to be negligible (Table S6, ESI†).
C bonds. Note that for similar systems the calculated rates of charge separation do not change significantly by varying the effective frequency from 1400 to 1800 cm−1.52,53 For the U1 and U2 systems, the effect of frequency selection on the estimated ET rate was found to be negligible (Table S6, ESI†).
| Complex | Transition | ΔG0a |
|Vij| | Reorg. energy | S eff | k X | |
|---|---|---|---|---|---|---|---|
| λ i | λ s | ||||||
| a Gibbs energy difference between the given states. b Effective value of the Huang–Rhys factor Seff = λi/ħωeff, where ħωeff is set to 1600 cm−1. | |||||||
| U1 | LE1 → CT1 | −0.054 | 1.92 × 10−4 | 0.200 | 0.161 | 1.008 | 7.75 × 108 |
| CT1 → GS | −2.449 | 9.73 × 10−3 | 0.181 | 0.161 | 0.837 | 3.63 × 103 | |
| U2 | LE1 → CT1 | −0.045 | 5.39 × 10−4 | 0.198 | 0.150 | 0.998 | 6.18 × 109 |
| LE1 → CT2 | 0.104 | 2.44 × 10−3 | 0.210 | 0.288 | 1.059 | 1.04 × 109 | |
| CT1 → GS | −2.429 | 1.14 × 10−2 | 0.170 | 0.150 | 0.857 | 6.86 × 103 | |
| CT2 → GS | −2.578 | 1.22 × 10−2 | 0.155 | 0.288 | 0.781 | 4.78 × 103 | |
In the studied complexes, the intraunit charge separation is characterized by a small negative Gibbs energy and reaction takes place in the normal Marcus region. The estimated charge separation rates were found to be 7.75 × 108 and 6.18 × 109 in U1 and U2, respectively. The rate of the interunit charge transfer reaction for U2 equals to 1.04 × 109, which is slightly lower compared to the intraunit CT. The charge recombination reactions take place in a deep inverted Marcus region, and their rates are significantly lower compared to the charge separation rates.
In photovoltaic applications, the efficient rate of hole- and electron-extraction is equally important for the generation of long-lived CT states with a high quantum yield. The hole transfer (HT) and excess electron transfer (EET) rates were computed for the U2 complex (Table 4). The wave function of a hole in a good approximation can be written as a linear superposition of HOMOs on the individual [10]CPP_Fused fragments. In turn, for the description of the excess electron, a liner superposition of LUMOs on the C60 fullerene was used. The HT/EET rates were estimated by using Marcus theory.54
| Complex | Transition | ΔG0 | |Vij| | Reorg. energy | S eff | k X | τ | |
|---|---|---|---|---|---|---|---|---|
| λ i | λ s | |||||||
| a Effective value of the Huang–Rhys factor Seff = λi/ℏωeff, where ℏωeff is set to 1600 cm−1. | ||||||||
| U2 | HT | 0.022 | 3.54 × 10−3 | 0.232 | 0.140 | 1.117 | 9.08 × 1010 | 11 |
| EET | 0.085 | 1.36 × 10−2 | 0.192 | 0.165 | 0.918 | 1.92 × 1011 | 5 | |
It is interesting to compare the charge transport properties of the originally reported (C60⊃[10]CPP)n polymeric conjugate and the studied ([10]CPP_Fused⊃C60) fused system in this work. Du and co-workers24 reported that the incorporation of C60 into a polymer dramatically increases hole mobility, making it even higher than electron mobility (μH = 3.64 × 10−4 cm2 V−1 s−1vs. μE = 8.06 × 10−5 cm2 V−1 s−1). Our calculations predict that in the fused (C60⊃[10]CPP_Fused)2 complex the rate of EET is higher than the corresponding HT rate. Their predicted characteristic times are equal to 11 and 5 ps, respectively. The EET process acceleration for the fused system is apparently associated with its supramolecular structure. As I showed at the beginning, the structure of ([10]CPP_Fused⊃C60)8 consists of units only in the syn conformation, whereas ([10]CPP⊃C60)8 contains the units in both syn- and anti-conformations. As seen in Fig. 1, the distance between fullerenes in the syn-oriented units is much smaller than that in the anti, and therefore the excess electron transfer via fullerene units in ([10]CPP_Fused⊃C60)n is expected to be faster than in ([10]CPP⊃C60)n. It is important to note that the HT/EET rates are much higher than the corresponding charge recombination rates, allowing for efficient electron and hole extraction in a polymer solar cell.
Conclusions
In this work, the molecular dynamics simulations and DFT/TD-DFT approaches were combined to study the ground and excited state properties of the supramolecular system consisting of π-conjugated CPP-based polymers and C60 guests. I demonstrated that π-extension in [10]CPP_Fused⊃C60 increases the rigidity of the entire system, but most importantly facilitates the syn conformation of the monomeric units in the polymer that accelerates the charge transport through oligomer. MD simulations suggest that ([10]CPP_Fused⊃C60)24 and ([10]CPP_Fused⊃C60)32 oligomers tend to form stable, helix-like structures. The TD-DFT results obtained for the ([10]CPP_Fused⊃C60)1 and ([10]CPP_Fused⊃C60)2 host–guest complexes indicate the efficient PET from the [10]CPP unit to the C60 fullerene. The ET occurs in the normal Marcus region on the nanosecond timescale. The hole transfer and excess electron transfer processes occur much faster than the corresponding charge recombination, which ensures their sufficient extraction. Overall, the results suggest that the proposed ([10]CPP_Fused⊃C60)n oligomer is a promising material for the ordered photoactive layer with the regular distribution of donor and acceptor phases for efficient charge separation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
I am grateful for financial support from the Spanish MINECO (Network RED2018-102815-T, project PID2020-13711GB-I00, and Juan de la Cierva contract IJC2019-039846-I). I gratefully acknowledge the Polish high-performance computing infrastructure PLGrid (HPC Centers: ACK Cyfronet AGH) for providing computer facilities and support within computational grant no. PLG/2022/015756 and PLG/2022/015981. I am also very grateful to Dr Olga Stasyuk and Prof. Alexander Voityuk for a fruitful discussion and their support.References
- T. Kawase, H. R. Darabi and M. Oda, Cyclic [6]- and [8]Paraphenylacetylenes, Angew. Chem., Int. Ed. Engl., 1996, 35, 2664–2666 CrossRef CAS.
- T. Kawase, N. Ueda, K. Tanaka, Y. Seirai and M. Oda, The newly modified McMurry reaction toward the improved synthesis of cyclic paraphenylacetylenes, Tetrahedron Lett., 2001, 42, 5509–5511 CrossRef CAS.
- S. Kammermeier, P. G. Jones and R. Herges, Ring-Expanding Metathesis of Tetradehydro-anthracene—Synthesis and Structure of a Tubelike, Fully Conjugated Hydrocarbon, Angew. Chem., Int. Ed. Engl., 1996, 35, 2669–2671 CrossRef CAS.
- M. Machón, S. Reich, J. Maultzsch, H. Okudera, A. Simon, R. Herges and C. Thomsen, Structural, electronic, and vibrational properties of (4,4) picotube crystals, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 155402 CrossRef.
- F. Diederich, Y. Rubin, C. B. Knobler, R. L. Whetten, K. E. Schriver, K. N. Houk and Y. Li, All-Carbon Molecules: Evidence for the Generation of Cyclo[18]carbon from a Stable Organic Precursor, Science, 1989, 245, 1088–1090 CrossRef CAS PubMed.
- K. Kaiser, L. M. Scriven, F. Schulz, P. Gawel, L. Gross and H. L. Anderson, An sp-hybridized molecular carbon allotrope, cyclo[18]carbon, Science, 2019, 365, 1299–1301 CrossRef CAS PubMed.
- H. L. Anderson, C. W. Patrick, L. M. Scriven and S. L. Woltering, A Short History of Cyclocarbons, Bull. Chem. Soc. Jpn., 2021, 94, 798–811 CrossRef CAS.
- R. Jasti, J. Bhattacharjee, J. B. Neaton and C. R. Bertozzi, Synthesis, Characterization, and Theory of [9]-, [12]-, and [18]Cycloparaphenylene: Carbon Nanohoop Structures, J. Am. Chem. Soc., 2008, 130, 17646–17647 CrossRef CAS PubMed.
- S. E. Lewis, Cycloparaphenylenes and related nanohoops, Chem. Soc. Rev., 2015, 44, 2221–2304 RSC.
- H. Omachi, Y. Segawa and K. Itami, Synthesis of Cycloparaphenylenes and Related Carbon Nanorings: A Step toward the Controlled Synthesis of Carbon Nanotubes, Acc. Chem. Res., 2012, 45, 1378–1389 CrossRef CAS PubMed.
- E. R. Darzi and R. Jasti, The dynamic, size-dependent properties of [5]–[12]cycloparaphenylenes, Chem. Soc. Rev., 2015, 44, 6401–6410 RSC.
- D. Wu, W. Cheng, X. Ban and J. Xia, Cycloparaphenylenes (CPPs): An Overview of Synthesis, Properties, and Potential Applications, Asian J. Org. Chem., 2018, 7, 2161–2181 CrossRef CAS.
- D. Lu, Q. Huang, S. Wang, J. Wang, P. Huang and P. Du, The Supramolecular Chemistry of Cycloparaphenylenes and Their Analogs, Front. Chem., 2019, 7, 668 CrossRef CAS PubMed.
- T. Iwamoto, Y. Watanabe, T. Sadahiro, T. Haino and S. Yamago, Size-Selective Encapsulation of C60 by [10]Cycloparaphenylene: Formation of the Shortest Fullerene-Peapod, Angew. Chem., Int. Ed., 2011, 50, 8342–8344 CrossRef CAS PubMed.
- E. J. Leonhardt and R. Jasti, Emerging applications of carbon nanohoops, Nat. Rev. Chem., 2019, 3, 672–686 CrossRef CAS.
- Y. Xu and M. von Delius, The Supramolecular Chemistry of Strained Carbon Nanohoops, Angew. Chem., Int. Ed., 2020, 59, 559–573 CrossRef CAS PubMed.
- A. Zen, M. Saphiannikova, D. Neher, J. Grenzer, S. Grigorian, U. Pietsch, U. Asawapirom, S. Janietz, U. Scherf, I. Lieberwirth and G. Wegner, Effect of Molecular Weight on the Structure and Crystallinity of Poly(3-hexylthiophene), Macromolecules, 2006, 39, 2162–2171 CrossRef CAS.
- B. J. Kim, H.-S. Lee, J. S. Lee, S. Cho, H. Kim, H. J. Son, H. Kim, M. J. Ko, S. Park, M. S. Kang, S. Y. Oh, B. Kim and J. H. Cho, Correlation between Crystallinity, Charge Transport, and Electrical Stability in an Ambipolar Polymer Field-Effect Transistor Based on Poly(naphthalene-alt-diketopyrrolopyrrole), J. Phys. Chem. C, 2013, 117, 11479–11486 CrossRef CAS.
- D. Spoltore, W. D. Oosterbaan, S. Khelifi, J. N. Clifford, A. Viterisi, E. Palomares, M. Burgelman, L. Lutsen, D. Vanderzande and J. Manca, Effect of Polymer Crystallinity in P3HT:PCBM Solar Cells on Band Gap Trap States and Apparent Recombination Order, Adv. Energy Mater., 2013, 3, 466–471 CrossRef CAS.
- O. Alqahtani, M. Babics, J. Gorenflot, V. Savikhin, T. Ferron, A. H. Balawi, A. Paulke, Z. Kan, M. Pope, A. J. Clulow, J. Wolf, P. L. Burn, I. R. Gentle, D. Neher, M. F. Toney, F. Laquai, P. M. Beaujuge and B. A. Collins, Mixed Domains Enhance Charge Generation and Extraction in Bulk-Heterojunction Solar Cells with Small-Molecule Donors, Adv. Energy Mater., 2018, 8, 1702941 CrossRef.
- Q. Huang, G. Zhuang, M. Zhang, J. Wang, S. Wang, Y. Wu, S. Yang and P. Du, A Long π-Conjugated Poly(para-Phenylene)-Based Polymeric Segment of Single-Walled Carbon Nanotubes, J. Am. Chem. Soc., 2019, 141, 18938–18943 CrossRef CAS PubMed.
- G. M. Peters, G. Grover, R. L. Maust, C. E. Colwell, H. Bates, W. A. Edgell, R. Jasti, M. Kertesz and J. D. Tovar, Linear and Radial Conjugation in Extended π-Electron Systems, J. Am. Chem. Soc., 2020, 142, 2293–2300 CrossRef CAS PubMed.
- E. Peterson, R. L. Maust, R. Jasti, M. Kertesz and J. D. Tovar, Splitting the Ring: Impact of Ortho and Meta Pi Conjugation Pathways through Disjointed [8]Cycloparaphenylene Electronic Materials, J. Am. Chem. Soc., 2022, 144, 4611–4622 CrossRef CAS PubMed.
- S. Wang, X. Li, X. Zhang, P. Huang, P. Fang, J. Wang, S. Yang, K. Wu and P. Du, A supramolecular polymeric heterojunction composed of an all-carbon conjugated polymer and fullerenes, Chem. Sci., 2021, 12, 10506–10513 RSC.
- L. Lagardère, L.-H. Jolly, F. Lipparini, F. Aviat, B. Stamm, Z. F. Jing, M. Harger, H. Torabifard, G. A. Cisneros, M. J. Schnieders, N. Gresh, Y. Maday, P. Y. Ren, J. W. Ponder and J.-P. Piquemal, Tinker-HP: a massively parallel molecular dynamics package for multiscale simulations of large complex systems with advanced point dipole polarizable force fields, Chem. Sci., 2018, 9, 956–972 RSC.
- TINKER-HP 1.1 Software Tools for Molecular Design.
- A. J. Stasyuk, O. A. Stasyuk, M. Solà and A. A. Voityuk, Hypsochromic solvent shift of the charge separation band in ionic donor–acceptor Li + @C60 ⊂ [10]CPP, Chem. Commun., 2019, 55, 11195–11198 RSC.
- A. J. Stasyuk, O. A. Stasyuk, M. Solà and A. A. Voityuk, Electron Transfer in a Li+-Doped Zn-Porphyrin–[10]CPP⊃Fullerene Junction and Charge-Separated Bands with Opposite Response to Polar Environments, J. Phys. Chem. B, 2020, 124, 9095–9102 CrossRef CAS PubMed.
- J. Wang, X. Zhang, H. Jia, S. Wang and P. Du, Large π-Extended and Curved Carbon Nanorings as Carbon Nanotube Segments, Acc. Chem. Res., 2021, 54, 4178–4190 CrossRef CAS PubMed.
- A. D. Becke, Density-functional exchange-energy approximation with correct asymptotic behavior, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Weigend, Accurate Coulomb-fitting basis sets for H to Rn, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the damping function in dispersion corrected density functional theory, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- O. A. Stasyuk, A. J. Stasyuk, M. Solà and A. A. Voityuk, [10]CPP-Based Inclusion Complexes of Charged Fulleropyrrolidines. Effect of the Charge Location on the Photoinduced Electron Transfer, Chem. – Eur. J., 2021, 27, 8737–8744 CrossRef CAS PubMed.
- O. A. Stasyuk, A. J. Stasyuk, M. Solà and A. A. Voityuk, The Hunter Falls Prey: Photoinduced Oxidation of C60 in Inclusion Complex with Perfluorocycloparaphenylene, Chem. Phys. Chem., 2022, 23, e202200226 CrossRef CAS PubMed.
- A. J. Stasyuk, O. A. Stasyuk, M. Solà and A. A. Voityuk, Photoinduced Charge Shift in Li+-Doped Giant Nested Fullerenes, J. Phys. Chem. C, 2019, 123, 16525–16532 CrossRef CAS.
- S. Zank, J. M. Fernández-García, A. J. Stasyuk, A. A. Voityuk, M. Krug, M. Solà, D. M. Guldi and N. Martín, Initiating Electron Transfer in Doubly Curved Nanographene Upon Supramolecular Complexation of C60, Angew. Chem., Int. Ed., 2022, 61, e202112834 CrossRef CAS PubMed.
- K. Morokuma, Molecular Orbital Studies of Hydrogen Bonds. III. CO⋯H–O Hydrogen Bond in H2CO⋯H2O and H2CO⋯2H2O, J. Chem. Phys., 1971, 55, 1236–1244 CrossRef CAS.
- T. Ziegler and A. Rauk, On the calculation of bonding energies by the Hartree Fock Slater method, Theor. Chim. Acta, 1977, 46, 1–10 CrossRef CAS.
- L. Zhao, M. von Hopffgarten, D. M. Andrada and G. Frenking, Energy decomposition analysis, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2018, 8, e1345 Search PubMed.
- S. J. Grabowski, What Is the Covalency of Hydrogen Bonding?, Chem. Rev., 2011, 111, 2597–2625 CrossRef CAS PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP), Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- P. Besalú-Sala, A. A. Voityuk, J. M. Luis and M. Solà, Evaluation of charge-transfer rates in fullerene-based donor–acceptor dyads with different density functional approximations, Phys. Chem. Chem. Phys., 2021, 23, 5376–5384 RSC.
- M. Izquierdo, B. Platzer, A. J. Stasyuk, O. A. Stasyuk, M. Sola, A. A. Voityuk, S. Cuesta, M. Solá, D. M. Guldi and N. Martin, All-Fullerene Electron Donor–Acceptor Conjugates, Angew. Chem., Int. Ed., 2019, 131, 7006–7011 CrossRef.
- S. Zank, J. M. Fernández-García, A. J. Stasyuk, A. A. Voityuk, M. Krug, M. Solà, D. Guldi and N. Martin, Initiating Electron Transfer in Doubly-curved Nanographene Upon Supramolecular Complexation of C60, Angew. Chem., Int. Ed., 2022, 61, e202112834 CrossRef CAS PubMed.
- A. Klamt and G. Schüürmann, COSMO: a new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient, J. Chem. Soc., Perkin Trans. 2, 1993, 799–805, 10.1039/P29930000799.
- A. A. Voityuk and S. F. Vyboishchikov, Fast and accurate calculation of hydration energies of molecules and ions, Phys. Chem. Chem. Phys., 2020, 22, 14591–14598 RSC.
- J. Ulstrup and J. Jortner, The effect of intramolecular quantum modes on free energy relationships for electron transfer reactions, J. Chem. Phys., 1975, 63, 4358–4368 CrossRef CAS.
- J. Jortner, Temperature dependent activation energy for electron transfer between biological molecules, J. Chem. Phys., 1976, 64, 4860–4867 CrossRef CAS.
- A. J. Stasyuk, O. A. Stasyuk, M. Solà and A. A. Voityuk, Triquinoline-versus Fullerene-Based Cycloparaphenylene Ionic Complexes: Comparison of Photoinduced Charge-Shift Reactions, Chem. – Eur. J., 2020, 26, 10896–10902 CrossRef CAS PubMed.
- O. A. Stasyuk, A. J. Stasyuk, M. Solà and A. A. Voityuk, Photoinduced electron transfer in nano-Saturn complexes of fullerene, Phys. Chem. Chem. Phys., 2021, 23, 2126–2133 RSC.
- R. A. Marcus and N. Sutin, Electron transfers in chemistry and biology, Biochim. Biophys. Acta, Rev. Bioenergy, 1985, 811, 265–322 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp02233a |
| This journal is © the Owner Societies 2023 |