
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRules governing metal coordination in Aβ–Zn(II) complex models from quantum mechanical calculations†
Julen
Aduriz-Arrizabalaga
,
Jose M.
Mercero
 ,
David
De Sancho
and
Xabier
Lopez
*
,
David
De Sancho
and
Xabier
Lopez
*
Polimero eta Material Aurreratuak: Fisika, Kimika eta Teknologia, Kimika Fakultatea, UPV/EHU & Donostia International Physics Center (DIPC), PK 1072, 20018 Donostia-San Sebastian, Euskadi, Spain. E-mail: xabier.lopez@ehu.eus
First published on 9th October 2023
Abstract
Transition metals directly contribute to the neurotoxicity of the aggregates of the amyloid-forming Aβ peptide. The understanding and rationalization of the coordination modes of metals to Aβ amyloid is, therefore, of paramount importance to understand the capacity of a given metal to promote peptide aggregation. Experimentally, multiple Aβ–metal structures have been resolved, which exhibit different modes of coordination in both the monomeric and oligomeric forms of Aβ. Although Zn(II) metalloproteins are very abundant and often involve cysteine residues in the first coordination shell, in the case of Aβ–Zn(II), though, Zn(II) is coordinated by glutamic/aspartic acid and/or histidine residues exclusively, making for an interesting case study. Here we present a systematic analysis of the underlying chemistry on Aβ–Zn(II) coordination, where relative stabilities of different coordination arrangements indicate that a mixture of Glu/Asp and His residues is favored. A detailed comparison between different coordination shell geometries shows that tetrahedral coordination is generally favored in the aqueous phase. Our calculations show an interplay between dative covalent interactions and electrostatics which explains the observed trends. Multiple structures deposited in the Protein Data Bank support our findings, suggesting that the trends found in our work may be transferable to other Zn(II) metalloproteins with this type of coordination.
1. Introduction
Alzheimer's disease is the leading cause of senile dementia, with over 55 million cases reported by the World Health Organization in 2021.1 Even though the cause of Alzheimer's is not completely known, a relation with aggregation and deposition of Amyloid β (Aβ) in neural tissue is widely accepted as a contributing factor on the onset. Transition metal ions and oxidative metabolism have been proposed to play fundamental roles in the process of aggregation and deposition of Aβ in Alzheimer's disease.2 The binding of divalent metals, such as copper (Cu(II)), iron (Fe(II)) and zinc (Zn(II)), with disordered fibrillogenic proteins, such as Aβ, influences the aggregation process of the protein, contributing directly to the neurotoxicity of the fibrils,3 and thus, to the severity of the neurodegenerative disease.4 It has been reported that both the monomeric and oligomeric forms of Aβ are neurotoxic.5 Interestingly, the concentration of Zn(II) in the brain, which varies from 150 to 200 µM in the neocortex, is one order of magnitude higher than the ion concentration in blood. Furthermore, although Zn(II) concentration stay relatively constant throughout adult life, a significantly elevated concentration has been found in the brains of patients affected by Alzheimer's disease.6 Therefore, the role Zn(II) plays in Alzheimer's disease has become of great interest.A detailed characterization of Aβ–Zn(II) coordination is essential to understand the effects these ions have on protein chains and, therefore, on the aggregation process. In proteins, Zn(II) can play both structural and/or catalytic roles. Zn(II) displays high flexibility with respect to the number of ligands it can coordinate in its first coordination shell. In aqueous solution, Zn(II) is found to bind six different ligands in octahedral coordination.7 However, inside proteins, it is usually found coordinating four ligands adopting a tetrahedral geometry. In some catalytic binding sites, Zn(II) appears pentacoordinated and, rarely, hexacoordinated.8 Within proteins, Zn(II) coordinates to different amino acid types, with cysteine (33%), histidine (31%) and aspartic/glutamic acid (11% and 7%, respectively) being the most prevalent.9 In the specific case of Aβ–Zn(II) systems, coordination exclusively happens via multiple combinations of histidine and glutamic/aspartic acid residues10 (see Fig. 1).
 | ||
| Fig. 1 Schematic representations of coordination shells for different experimentally characterized metal centers in Aβ–Zn(II) complexes. PDB IDs: (A) 1ZE9, (B) 5LFY, (C) 2LI9 and (D) 2MGT. | ||
Efforts have been made in the past to establish binding energies of Aβ–Zn(II) systems11 and, the overall coordination of Aβ–Zn(II) has also been studied.12 Dudev and Lim studied the preference for Zn(II) complexes to arrange in tetrahedral vs. octahedral arrangements,13 where they were able to establish that tetrahedral coordinations are favored in the solvent phase. Similarly, in a posterior work, they were able to elucidate some relevant points for metal complexes,14 such as establishing the maximum number of monodentately bound Asp/Glu ligands to the metal. Nevertheless, a work where the chemical propensities for such conclusions are explained has not been performed.
In this work, we perform a systematic study of five different Zn(II) coordination shells, that include all histidine and/or aspartic/glutamic acid combinations. Relative stabilities of the different coordination shell arrangements and relative stabilities of the different coordination shell geometries, where tetrahedral and octahedral coordinations are studied, have been calculated both in the gas phase and the aqueous phase. In order to gain more insights into the contributing factors of the preferred coordination shell arrangements, we have used various state-of-the-art methods. Our results indicate that tetrahedral geometries are favored over octahedral geometries in the solvent phase, and that a mixture of histidine and aspartic/glutamic acid is preferred in the coordination shell. Multiple structures deposited in the Protein Data Bank support our findings which suggest that our trends are not exclusive to Aβ–Zn(II) systems but to all Zn(II) metalloproteins where coordination is given in these unique residue arrangements.
2 Methods
2.1 Model systems
In order to study the different Aβ–Zn(II) coordinations, we take different coordination arrangements into consideration. To do so, we propose five different models, which were constructed using experimentally characterized Aβ–Zn(II) systems as reference15–18 (see Fig. 1). In order to construct our models, we used acetate groups (Ac−) to represent the sidechains of glutamic/aspartic acids and methylated imidazole (Im) groups to represent the sidechains of histidines, as shown in Fig. 2. The models that we consider here range from systems that are solely formed by acetate groups to systems that are solely formed by imidazole groups: [Zn(Ac)4]2−, [Zn(Im)1(Ac)3]−, [Zn(Im)2(Ac)2], [Zn(Im)3(Ac)1]+ and [Zn(Im)4]2+. Additionally, in order to compare the different possible geometries for the first coordination shell, we consider both tetrahedral and octahedral arrangements for each model (see Fig. 2). | ||
| Fig. 2 Schematic representations of the (A) FCS tetrahedral clusters, (B) FCS + 2W tetrahedral clusters and (C) octahedral clusters. | ||
2.2 Quantum mechanical calculations
To study the relative stabilities of the different Aβ–Zn(II) clusters in various coordinations, we performed quantum mechanical calculations using the Gaussian 16 program.19 All calculations were made at the density functional theory (DFT) level, using a variety of different functionals: B3LYP,20,21 CAM-B3LYP,22 M062X,23 PBE024 and ωB97XD.25 We used Pople's 6-31++G(d,p) basis set26 and dispersion interactions were considered with the empirical D3 version of Grimme's dispersion with Becke–Johnson damping when needed.27 All geometry optimizations were carried in the gas phase as well as the solvent phase, using the Polarizable Continuum Model approach to take solvation effects into account.28 All solvent phase calculations were performed considering a dielectric of ε = 78.2.3 Evaluation of formation energies of Zn(II) clusters
In order to establish the preferred residue combination to form Aβ–Zn(II) coordination shells we have calculated formation enthalpy (ΔH) and free energy (ΔG) terms. The formation reactions are defined according to the following reaction,| [Zn(H2O)6]2+ + mIm + nAc−→ [Zn(Im)m(Ac)n](2−n) + xH2O |
In order to establish the preferred geometry of each cluster, we directly compare the formation enthalpy and free energy of the different geometries,
| ΔΔHT/O = ΔHtetra − ΔHocta |
| ΔΔGT/O = ΔGtetra − ΔGocta |
There are, however, additional complications for a fair comparison between coordination geometries, due to the different number of ligands in the different coordination. For this reason, here we consider two different approaches.
Tetrahedral reaction:
[Zn(H2O)6]2+ + m![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) Im + nAc− → [Zn(Im)m(Ac)n](2−n) + 6H2O Im + nAc− → [Zn(Im)m(Ac)n](2−n) + 6H2O |
Octahedral reaction:
| [Zn(H2O)6]2+ + mIm + nAc− → [Zn(Im)m(Ac)n(H2O)2](2−n) + 4H2O |
Various studies have been performed to correct this over-stabilization.29–33 Several approaches have been proposed to introduce corrections to avoid unphysical results related to this phenomenon. One possibility, proposed by Mejias and Lago,30 is to neglect translational and rotational entropies, and hence redefine the free energy as
| G = H − T(Svib) |
| G = H − T(Svib + 1/2(Strans + Srot)) |
| [Zn(Im)m(Ac)n(H2O)2](2−n) → [Zn(Im)m(Ac)n](2−n)·2H2O |
| [Zn(Im)m(Ac)n](2−n)·2H2O → [Zn(Im)m(Ac)n](2−n) + 2H2O |
2.4 Per-ligand decomposition scheme
To gain insight into the stabilization each acetate/imidazole ligand provides in a given coordination shell (CS) around Zn(II), we have applied a per-ligand decomposition protocol of the total interaction between the Zn(II) and a given ligand in a coordination sphere. First, the overall interaction with Zn(II) is decomposed into two contributions: the interaction with Zn(II) in a preorganized coordination shell (CS), and the energy penalty paid for forming that coordination shell (CS) without the metal.Here, EZn@CS stands for the energy of a given Zn(II) cluster, ECS for the energy or a preorganized coordination shell without Zn(II), EZn is the energy of a Zn(II) atom and ELi is the energy of the isolated ligands. This scheme is similar to the one previously used to evaluate metal-binding energies for serum-transferrin,34 albeit more complete since we introduce the penalty paid for forming a given coordination shell. A particular feature of our calculations is that the basis sets of the Zn(II) cluster are always present in all calculations. That is, when removing atoms from the cluster to take into account the different terms specified before we use “ghost atoms”, namely, we keep their normal basis functions and numerical integration grid points but no nuclear charge or electrons19 for the ghost atoms.

To decompose these interactions in per-ligand contributions, we apply the following protocol. First, we remove a ligand Li from the coordination shell and reevaluate both terms described above, namely, the Zn(II) interaction energy and the coordination shell formation energy in the absence of that ligand, ΔEZnInt(Li) and ΔECSf(Li).

In general, the first term ΔEZnInt(Li) is a stabilizing contribution whereas the second term ΔECSf(Li) implies an energy penalty for the formation of a coordination shell.
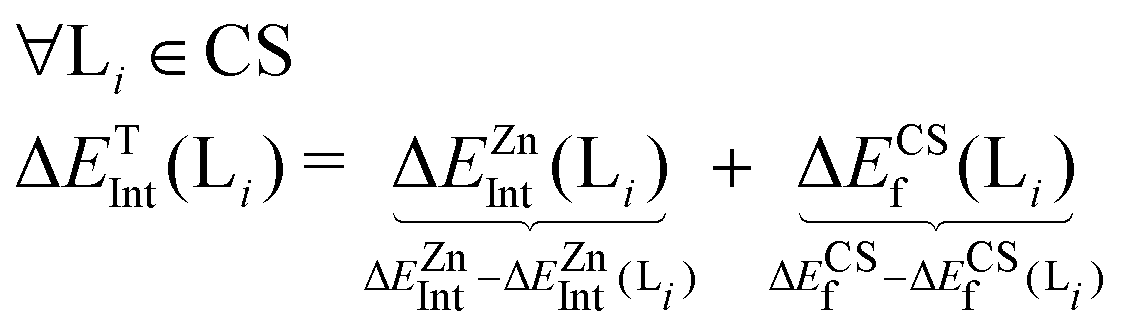
2.5 Analysis of the nature of Zn(II)–ligand interactions
As mentioned, in Aβ–Zn(II) complexes, coordination is almost exclusively given through glutamic/aspartic acid and histidine amino acids, which are charged and neutral amino acids. While charged amino acids primarily interact through electrostatic interactions, neutral amino acids mostly interact through covalent interactions. Although hard metals like copper and iron favour electrostatic interactions over covalent interactions and soft metals favor covalent interactions, Zn(II), an in-between metal, can present both interactions.To obtain an in-depth understanding of the nature of Zn(II)-ligand interactions we have therefore performed several analyses. Thus, we have performed Natural Bond Orbital (NBO) analysis using the Gaussian 16 software package.19 In addition, we have calculated delocalization indexes using the Quantum Theory of Atoms and Molecules (QTAIM) analysis with the AIMALL software package35 and the ESI-3D program.36,37 Lastly, in order to measure qualitatively electrostatic contributions, we have performed Natural Coulomb Electrostatics (NCE) analysis using the Gaussian 16 software package including the Natural Bond Orbital 6.0 program.38
3. Results
3.1 Relative stabilities of Aβ–Zn(II) metal centers
In this work, we have used multiple functionals to calculate energies, both in the gas phase and in the solvent phase (see Fig. S1 to S4, ESI†). Using multiple functionals in DFT calculations is recommended, as different functionals have varying levels of accuracy and are suitable for different chemical systems. By comparing the results obtained from multiple functionals, we can assess the level of agreement and identify possible discrepancies. The results obtained using all the functionals considered in this work follow the same general trends, with the exception of the M062X functional. Specifically, great agreement was obtained for the relative stabilities of the different clusters and the preferred geometrical arrangements. Following the consensus obtained by the majority of the functionals used and for clarity's sake, we have decided to exclusively report results obtained with a single functional. In the manuscript, we report results obtained with the state-of-the-art functional ωB97XD, which includes long-range corrections.3.2 Gas phase calculations
Results obtained from gas phase calculations are shown in Table 1. When measuring the relative stabilities of the different clusters proposed, we find a tendency towards favouring clusters with a higher number of acetate ligands. This trend is related to the expected propensity to neutralize the charge in the gas phase. Interestingly though, the most stable clusters are not those with the highest possible number of acetates (i.e., four ligands). Instead, the most stable clusters contain three acetate ligands and a single imidazole (i.e., [Zn(Im)1(Ac)3]−). This tendency is captured for tetrahedral and octahedral clusters, and agree with the results obtained by Dudev and Lim.14 In their work, they proposed the q + 1 rule, which states that the most stable coordination of a metal with a given charge q, is that formed by q + 1 monodentately bound Glu/Asp (in our case represented by the acetate moiety).| Cluster | Tetrahedral | Octahedral | Tetrahedral versus octahedral | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| FCS | FCS | FCS + 2W | ||||||||
| ΔH | ΔG | ΔH | ΔG | ΔΔHT/O | ΔΔGT/O | ΔΔGT/O1/2 | ΔΔHT/O | ΔΔGT/O | ΔΔG1/2T/O | |
| Gas phase | ||||||||||
| [Zn(Ac)4]−2 | −305.0 | −313.9 | −317.2 | −306.5 | 12.2 | −7.4 | 5.9 | −18.7 | −22.2 | −22.1 |
| [Zn(Im)(Ac)3]−1 | −336.7 | −347.1 | −346.2 | −335.2 | 9.5 | −11.9 | 1.4 | −22.1 | −24.8 | −24.7 |
| [Zn(Im)2(Ac)2] | −321.2 | −331.5 | −334.6 | −322.9 | 13.4 | −8.7 | 4.7 | −9.8 | −14.7 | −14.7 |
| [Zn(Im)3(Ac)1]+1 | −240.0 | −250.7 | −254.8 | −243.0 | 14.8 | −7.7 | 5.7 | −4.4 | −7.7 | −7.6 |
| [Zn(Im)4]+ 2 | −91.4 | −101.4 | −97.10 | −85.6 | 5.7 | −14.5 | −1.1 | −11.1 | −16.7 | −16.6 |
| Aqueous phase | ||||||||||
| [Zn(Ac)4]−2 | −54.1 | −64.7 | −50.5 | −37.7 | −3.6 | −27.0 | −13.6 | −14.7 | −22.5 | −22.4 |
| [Zn(Im)(Ac)3]−1 | −55.5 | −63.7 | −51.3 | −40.2 | −4.2 | −23.5 | −10.2 | −20.7 | −24.5 | −24.5 |
| [Zn(Im)2(Ac)2] | −56.4 | −67.1 | −59.6 | −47.2 | 3.2 | −19.9 | −6.5 | −8.7 | −17.7 | −17.6 |
| [Zn(Im)3(Ac)1]+1 | −56.4 | −66.1 | −59.5 | −47.3 | 3.1 | −18.8 | −5.5 | −6.2 | −9.5 | −9.4 |
| [Zn(Im)4]+ 2 | −56.2 | −59.8 | −47.5 | −34.3 | −8.7 | −25.5 | −12.2 | −6.7 | −15.1 | −15.0 |
Additionally, in Table 1 we show results obtained when comparing relative stabilities between tetrahedral and octahedral geometries for each cluster. Results from the first coordination shell approach (FCS) and the alternative where we consider of two additional water molecules (FCS + 2W, see Methods) point in different directions. On one hand, in the FCS approach, ΔΔHT/O energies show that octahedral coordination is favored, whereas ΔΔGT/O energies show that tetrahedral coordination is favored. However, after introducing the correction in ΔΔGT/O to obtain ΔΔGT/O1/2 energies, the latter also show that octahedral geometries are favored for all clusters. We should replace this in the gas phase, with the exception of the [Zn(Im)4]2+ cluster. Conversely, if the FCS + 2W approach is considered, both ΔΔHT/O and ΔΔGT/O energies point to tetrahedral coordination as the one favored in the gas phase. It is noteworthy that as mentioned, the FCS + 2W approach overestabilizes tetrahedral coordination. Nevertheless, if this overestabilization is calculated and energies are recalculated taking the overestabilization into consideration (see Table S1, ESI†), we see that both the ΔH and ΔG terms of the FCS and FCS + 2W approaches are now extremely similar. The FCS + 2W predicts a preference for tetrahedral arrangements in the gas phase, which is believed to be caused by the overestabilization introduced by the water molecules present in the second coordination shell.
3.3 Solvent phase calculations
Although results obtained in the gas phase are interesting, we care more about Aβ–Zn(II) systems in the physiological environment. Looking at our reference systems (see Fig. 1), we see that the metal centers of these proteins are fully exposed to the solvent, therefore, we have performed calculations in the solvent phase at ε = 78. Results obtained in the solvent phase are shown in Fig. 3 and Table 1. Once again, we start comparing the relative stabilities of the different clusters (see Fig. 3A). Considering solvent effects, there is an attenuation of the difference in stabilities among the different clusters, but with a shift in the relative stabilities of clusters. Thus, unlike in the gas phase, the q + 1 rule is no longer fulfilled, and instead, the most stable clusters have fewer acetate ligands, e.g., either [Zn(Im)2 (Ac)2] or [Zn(Im)3 (Ac)1]+. | ||
| Fig. 3 Results obtained in the solvent phase. (A) ΔH and ΔG energies obtained for tetrahedral (blue) and octahedral (red) clusters. (B) ΔΔHT/O and ΔΔGT/O1/2 energies are shown for the FCS (bold purple) and FCS + 2W (light purple) approaches. | ||
In Fig. 3B, we show the relative energies between tetrahedral and octahedral clusters. ΔΔHT/O energies using both the FCS and FCS + 2W approaches indicate that the tetrahedral geometry is favored in the solvent phase except for the [Zn(Im)2(Ac)2] and [Zn(Im)3(Ac)1]+ clusters in the FCS approach. On the other hand, both ΔΔGT/O and ΔΔGT/O1/2 energies clearly favour tetrahedral coordination for all clusters (see Table 1). Therefore, from these calculations we conclude that the preferred Aβ–Zn(II) systems in the solvent phase are the tetrahedrally coordinated [Zn(Im)2(Ac)2] and [Zn(Im)3(Ac)1]+ clusters. Interestingly these two clusters exhibit the lowest difference in energy between octahedral and tetrahedral coordination.
3.4 Contributions of each ligand to the stabilization energy
To understand the contribution of each ligand to the stability of a given cluster, we have applied the protocol specified in Section 2.4. We have estimated two contributions to the overall interaction energy (ΔETint(Li)) of a given ligand Li. We first evaluate the stabilizing contribution of the interaction of the ligand in a specific coordination shell (ΔEZnInt(Li)) with Zn(II). Second, we evaluate a destabilizing factor (ΔECSf(Li))) that represents the energy penalty required for the inclusion of a ligand into a coordination shell in the absence of the metal. In other words, ΔECSf(Li) represents the energy required to form a specific coordination shell. We show all the results from these calculations in Tables 2 and 3.| Cluster | Ligand | ΔEZnInt (kcal mol−1) | ΔECSf (kcal mol−1) | ΔETInt (kcal mol−1) | E(2) (kcal mol−1) | Delocalization index (a.u) |
|---|---|---|---|---|---|---|
| [Zn(Ac)4]2− | Ac− | −60.1 | 19.1 | −41.1 | −47.5 | 0.4283 |
| Im | — | — | — | — | — | |
| [Zn(Im)1 (Ac)3]− | Ac− | −59.5 | 15.5 | −44.0 | −47.1 | 0.4355 |
| Im | −49.9 | 10.6 | −39.3 | −57.7 | 0.4259 | |
| [Zn(Im)2 (Ac)2] | Ac− | −63.5 | 16.4 | −47.1 | −46.4 | 0.4275 |
| Im | −48.9 | 8.1 | −40.7 | −59.0 | 0.4476 | |
| [Zn(Im)3 (Ac)1]+ | Ac− | −71.1 | 16.7 | −54.4 | −53.2 | 0.4479 |
| Im | −50.5 | 7.6 | −42.9 | −56.9 | 0.4431 | |
| [Zn(Im)4]2+ | Ac− | — | — | — | — | — |
| Im | −55.6 | 6.0 | −49.7 | −60.7 | 0.4523 | |
| Cluster | Ligand | ΔEZnInt (kcal mol−1) | ΔECSf (kcal mol−1) | ΔETInt (kcal mol−1) | E(2) (kcal mol−1) | Delocalization index (a.u.) |
|---|---|---|---|---|---|---|
| [Zn(Ac)4]2− | Ac− | −51.3 | 14.4 | −36.9 | −42.3 | 0.3329 |
| Im | — | — | — | — | — | |
| Wat | 0.13 | −11.1 | −10.9 | −12.1 | 0.1286 | |
| [Zn(Im)1 (Ac)3]− | Ac− | −52.3 | 16.1 | −36.2 | −39.1 | 0.3063 |
| Im | −42.1 | 10.0 | −32.1 | −57.9 | 0.3561 | |
| Wat | −9.8 | −2.2 | −12.0 | −20.2 | 0.1887 | |
| [Zn(Im)2 (Ac)2] | Ac− | −63.1 | 14.2 | −49.0 | −41.2 | 0.3006 |
| Im | −39.8 | 11.4 | −28.3 | −51.7 | 0.3305 | |
| Wat | −15.8 | 0.0 | −15.8 | −29.9 | 0.2154 | |
| [Zn(Im)3 (Ac)1]+ | Ac− | −58.7 | 9.2 | −49.6 | −38.2 | 0.2596 |
| Im | −41.4 | 10.3 | −31.1 | −52.7 | 0.3395 | |
| Wat | −18.2 | 1.0 | −17.2 | −35.5 | 0.2234 | |
| [Zn(Im)4]2+ | Ac− | — | — | — | — | — |
| Im | −42.9 | 9.5 | −33.4 | −51.8 | 0.3324 | |
| Wat | −22.4 | 9.0 | −13.3 | −38.3 | 0.1859 | |
In Fig. 4B, we report the values of ΔEZnInt, which indicate that the stabilization introduced by acetates is higher than the stabilization introduced by imidazole ligands. This is expected because acetates are negatively charged while imidazoles are neutral. Nevertheless, the energy penalty of a ligand to be incorporated in the first coordination shell of Zn(II) (captured by the ΔECSf term), is also higher for acetates than for imidazoles. These two effects partially compensate each other, and the overall contribution of each acetate/imidazole ligand, ΔETint, is much more similar, as shown in Fig. 4A and C. For octahedral complexes, there is still a larger ΔETint interaction detected for acetates with an average of 42.9 kcal mol−1versus 31.2 kcal mol−1 for imidazoles. However, in the case of tetrahedral structures, imidazole and acetate ligands become interchangeable, with similar ΔETint of 46.6 and 43.1 kcal mol−1 on average for acetates and imidazoles, respectively.
 | ||
| Fig. 4 Decomposition of per-ligand interactions: (A) distribution of ΔETInt(Li) of each ligand for tetrahedral and octahedral clusters; (B) correlation between ΔECSf(Li) and ΔEZnInt(Li); (C) and mean ΔETInt(Li) values of each ligand type in each cluster. | ||
In Fig. 4C, we show the average total interaction energy for different ligand types and coordinations. There is a more effective interaction per ligand in tetrahedral environments than in octahedral ones, with an overall tendency to augment as the numbers of acetates decrease. In addition, when there is a mixture of acetate/imidazole ligands in the coordination shell, there is a slight preference for acetates, enhanced as the number of acetates decreases in the coordination shell. In this way, the most efficient interaction with acetates occurs in the [Zn(Im)3 (Ac)1]+ cluster for tetrahedral environments, and [Zn(Im)3 (Ac)1]+ and [Zn(Im)2 (Ac)2] for octahedral ones. The reason for this behavior is the energy penalties paid by acetates to be incorporated in the coordination shell around Zn(II), when other negatively charged ligands are present.
3.5 Covalent interactions
The similar interaction energies of imidazole and acetate, especially in tetrahedral compounds, are remarkable since imidazole is a neutral ligand and acetate is a negatively charged one. This behavior suggests the importance of dative covalent interactions from imidazole ligands to Zn(II), which can partially balance the more favourable electrostatics of acetates. The relatively soft nature of Zn(II) may favour these dative covalent donation compensations and explain the propensity for Zn(II)–imidazole interactions. To get more insight into the specific features of Zn(II)–ligand interactions and their difference between imidazole and acetates, we have performed additional analysis to characterize the covalent and electrostatic interactions between the metal and the ligands.We have performed two types of calculations to measure the covalent interactions in Aβ–Zn(II) coordination. NBO analysis, in which we have determined the delocalization energy, denoted as E(2), from each ligand to Zn(II). The most significant delocalizations are from the nO/nN lone pairs into the formally empty valence orbitals of Zn(II). In addition, we have calculated electron delocalization indexes between Zn(II) and O/N based on QTAIM,39δX–Zn with X = O, N, which is taken as a measure of the electron-pair sharing between two atoms. In Fig. 5A we show that there is a strong correlation between both quantities, suggesting a consistent trend in the degree of covalency along the different Zn(II)–ligand interactions. Imidazole ligands have the highest delocalization indexes and E(2) energies in tetrahedral and octahedral clusters. On the other hand, acetate ligands show lower delocalization indexes and E(2) energies, with water molecules in octahedral complexes having the lowest delocalization indexes and E(2) energies. In all cases, ligands in tetrahedral clusters show higher E(2) values and delocalization indexes (see Tables 2 and 3).
 | ||
| Fig. 5 (A) Correlation between E(2) energies (nx → Zn) and delocalization indexes (δZn–X) in a.u., where X = [oxygen, nitrogen] for Ac− and Im respectively, (B) total delocalization of each cluster calculated by summing all delocalization indexes, (C) distribution of E(2) energies of each ligand, and (D) distribution of delocalization indexes of each ligand. | ||
These results suggest that tetrahedral arrangements favour dative covalent interactions. In fact, when we calculate the total delocalization indexes by summing all δX–Zn (Fig. 5B), tetrahedral clusters show in all cases a higher value than octahedral ones, indicating a higher covalent nature of the interaction in tetrahedral arrangements than in octahedral ones. We also observe that as the number of imidazoles increases, the total delocalization of each cluster also increases. Nevertheless, a slight decrease of the total delocalization is observed in the case of the octahedral [Zn(Im)4]2+ cluster with respect to [Zn(Im)3 (Ac)1]+.
3.6 Electrostatic interactions
We have analyzed the qualitative trends anticipated from pure electrostatic interactions based on the NCE analysis.40 In this analysis, the classical electrostatics contributions are derived from natural atomic charges. First, we analyze the information of Fig. 6A, where the NCE interaction of each ligand with Zn(II) (Zn–Li interaction) and the NCE repulsion between ligands (Li–Lj NCE repulsion) are depicted. The Lewis and non-Lewis atomic charges lead to a similar qualitative picture. The attractive interaction with Zn(II) decreases in the following order: acetate > imidazole > water (Fig. 6C). However, the repulsion between ligands follows this order and partially balances the favourable interaction between Zn(II) and the acetates. Nevertheless, when both contributions sum to give the total NCE interaction, there is still a clear preference for acetates, irrespective of the type of coordination. | ||
| Fig. 6 (A) Li–Lj NCE repulsion and Zn–Li NCE interaction correlation are shown, where values calculated with non-Lewis atomic charges are shown in bold and values calculated with Lewis atomic charges are shown in light color, (B) total NCE interactions, (C) distributions of total NCE interactions, (D) Zn–Li NCE interactions, and (E) total NCE interactions of each cluster divided by ligand type are shown. | ||
Interestingly, and contrary to the behavior of Zn(II)-acetate interactions, the repulsive term between acetate and the rest of the ligands shows a wide range of values reaching up to 100 kcal mol−1. This indicates that the repulsive electrostatic interactions to accommodate an acetate in a given coordination shell can differ substantially among the clusters. In contrast, the attractive interactions with Zn(II) are always of a similar amount. Due to this, the total NCE interaction of acetate is strongly dependent on the number of acetates present in a coordination shell (Fig. 6E) for both tetrahedral and octahedral coordination. The origin of this behavior is the electrostatic repulsion between negatively charged acetates, which increases with the number of acetates in a given coordination shell, making the progressive addition of acetates less stabilizing. On the other hand, imidazoles and water ligands display similar values for all the clusters.
Finally, in Fig. 6B, we sum up all the electrostatic contributions per cluster. In general, electrostatics tends to slightly favour octahedral complexes with a larger number of acetates. Interestingly, a peak of total NCE interactions is seen for the [Zn(Im)1(Ac)3]− cluster as it reaches a saturation point. This saturation point is dictated by the lower electrostatic repulsion each acetate experiences in the coordination shell, thus favoring it over the [Zn(Ac)4]2− cluster. The slightly higher total NCE interaction seen for octahedral clusters is related to the geometry of the clusters, which more effectively distributes the same amount of charges surrounding the Zn(II) metal resulting in slightly lower repulsion amongst acetates. This event is better captured for the [Zn(Ac)4]2− cluster, where electrostatic repulsion is at its maximum, and the geometry difference plays a significant role. As expected, the total NCE interaction decreases as the number of acetates also decreases, obtaining a clear minimum for the [Zn(Im)4]2+ cluster for both tetrahedral and octahedral clusters.
4 Discussion
In this work, we have applied different quantum chemical methods and analysis tools to determine the overall tendencies in Zn(II) coordination with acetate/imidazole ligands in Aβ–Zn(II) complexes in order to elucidate the preferred Zn(II) coordination modes. Through our calculations, we have been able to indicate that a preference for tetrahedral arrangements and a mixture of acetates and histidines is present. Therefore, it is interesting to determine how much of the trend from quantum calculations recapitulates what is found experimentally in Aβ–Zn(II) systems. In Table 4 we list ten PDB entries for Aβ–Zn(II) complexes, which we divide in two groups, depending on whether the metal is coordinated to Aβ exclusively or including other proteins. We find that the most likely coordinations are those with to 1/2 acetate-type sidechains (Glu or Asp) and 2/3 imidazole sidechains from histidines, in excellent agreement with our predictions. Thus, the inherent stability of the first coordination shells which we have determined seems the primary leading factor determining the coordination of Aβ–Zn(II) structures.Additionally, to further contrast the results obtained from our study, we compare the most common coordination shells present in enzymes studied by Laitaoja et al. in their statistical study on Zn(II)-containing protein structures deposited in the PDB9 (see Table S5, ESI†). Cysteine is the most common residue to coordinate Zn(II) in proteins, making for 33% of the residues, followed by histidine (31%). Aspartic acid and glutamic acid make for 11% and 7%, respectively. Apart from those containing cysteine residues, the most common coordination spheres are those that correspond to mixed histidine and glutamic/aspartic acid residues, primarily the [Zn(Im)3(Ac)1]+ and [Zn(Im)2(Ac)2] coordinations, which come in perfect agreement with our findings. Moreover, Laitaoja et al. report that up to 65% of the structures characterized via X-ray crystallography and 98% of structures characterized using NMR are tetrahedral, which further supports the preference towards the tetrahedral arrangement of Zn(II) coordination shells found in this work. Since the study performed by Laitaoja et al. included all Zn(II)-containing protein systems, we conclude that the tendencies we have captured in our reduced quantum models reproduce the preferred coordination not only in Aβ–Zn(II) systems but also more generally to Zn(II)-containing metalloproteins.
5 Conclusions
We have studied the relative stabilities of five different clusters inspired by the Zn(II) coordination found in Aβ–Zn(II) complexes, with different numbers of histidines and glutamic/aspartic acid residues in the first coordination shells, considering tetrahedral and octahedral environments. We have found that in aqueous phase there is a preference for tetrahedral [Zn(Im)2(Ac)2] and [Zn(Im)3(Ac)1]+ clusters.Our results can be rationalized by a delicate equilibrium of diverse interactions. Thus, per-ligand decomposition of the interaction energies suggests that although the affinity towards Zn(II) is stronger for acetate ligands than for imidazole, the larger energy cost associated with incorporating negatively charged acetates within a specific coordination environment mitigates these disparities. Besides, the presence of significant dative covalent interactions with imidazole in tetrahedral clusters, as shown by both NBO and QTAIM analyses, partially offsets the more favorable electrostatic interaction of the negatively charged acetate with the metal ion.
Our trends explain the tendency found in the PDB for tetrahedral environments of Zn(II) and the interaction with mixed His and Glu/Asp coordination shells both in Aβ–Zn(II) and other protein environments in a relative number fully consistent with the trends determined in the present work. Therefore, the coordination of Zn(II) in these systems is highly dictated by the inherent properties and affinities of Zn(II) towards its first coordination residues.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the financing of the MINECO project (PID2021-127907NB-I00) founded by the Spanish Ministry of Science and Innovation, and the financing from the Basque Government (IT1584-22). The authors also thank the IZO-SGI SGIker (UPV/EHU/ERDF,EU) and DIPC for technical and human support and for the allocation of computational resources. D. D. S. receives support from a Ramón y Cajal contract (RYC-2016-19590) from the Spanish Ministry of Science and Innovation. J. A. thankfully acknowledges the University of the Basque Country for the scholarship for the completion of a masters degree in the academic years 2020/2021–2021/2022 and the Donostia International Physics Center for a summer internship.Notes and references
- Dementia, https://www.who.int/news-room/fact-sheets/detail/dementia, Accessed: 2023-02-14.
- K. J. Barnham and A. I. Bush, Curr. Opin. Chem. Biol., 2008, 12, 222–228 CrossRef CAS PubMed.
- T. Chen, X. Wang, Y. He, C. Zhang, Z. Wu, K. Liao, J. Wang and Z. Guo, Inorg., 2009, 48, 5801–5809 CrossRef CAS PubMed.
- O. Wise-Scira, L. Xu, G. Perry and O. Coskuner, J. Biol. Inorg. Chem., 2012, 17, 927–938 CrossRef CAS PubMed.
- J. Hardy and D. J. Selkoe, Science, 2002, 297, 353–356 CrossRef CAS PubMed.
- M. Deibel, W. Ehmann and W. Markesbery, J. Neurol. Sci., 1996, 143, 137–142 CrossRef CAS PubMed.
- Y. Marcus, Chem. Rev., 1988, 88, 1475–1498 CrossRef CAS.
- I. L. Alberts, K. Nadassy and S. J. Wodak, Protein Sci., 1998, 7, 1700–1716 CrossRef CAS PubMed.
- M. Laitaoja, J. Valjakka and J. Janis, Inorg., 2013, 52, 10983–10991 CrossRef CAS PubMed.
- Y. Miller, B. Ma and R. Nussinov, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 9490–9495 CrossRef CAS PubMed.
- T. Marino, N. Russo, M. Toscano and M. Pavelka, Interdiscip. Sci.: Comput. Life Sci., 2010, 2, 57–69 CrossRef CAS PubMed.
- A. Maiorana, T. Marino, V. Minicozzi, S. Morante and N. Russo, Biophys. Chem., 2013, 182, 86–93 CrossRef CAS PubMed.
- T. Dudev and C. Lim, J. Am. Chem. Soc., 2000, 122, 11146–11153 CrossRef CAS.
- T. Dudev and C. Lim, Annu. Rev. Biophys., 2008, 37, 97–116 CrossRef CAS PubMed.
- S. Zirah, S. A. Kozin, A. K. Mazur, A. Blond, M. Cheminant, I. Ségalas-Milazzo, P. Debey and S. Rebuffat, J. Biol. Chem., 2006, 281, 2151–2161 CrossRef CAS PubMed.
- V. I. Polshakov, A. B. Mantsyzov, S. A. Kozin, A. A. Adzhubei, S. S. Zhokhov, W. van Beek, A. A. Kulikova, M. I. Indeykina, V. A. Mitkevich and A. A. Makarov, Angew. Chem., 2017, 129, 11896–11901 CrossRef.
- A. N. Istrate, P. O. Tsvetkov, A. B. Mantsyzov, A. A. Kulikova, S. A. Kozin, A. A. Makarov and V. I. Polshakov, Biophys. J., 2012, 102, 136–143 CrossRef CAS PubMed.
- A. N. Istrate, S. A. Kozin, S. S. Zhokhov, A. B. Mantsyzov, O. I. Kechko, A. Pastore, A. A. Makarov and V. I. Polshakov, Sci. Rep., 2016, 6, 1–14 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Revision C.01, 2016, Gaussian Inc., Wallingford CT Search PubMed.
- A. D. Becke, Chem. Phys., 1993, 98, 1372–1377 CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- C. Adamo and V. Barone, Chem. Phys., 1999, 110, 6158–6170 CAS.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- W. J. Hehre, R. Ditchfield and J. A. Pople, Chem. Phys., 1972, 56, 2257–2261 CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS PubMed.
- A. V. Finkelstein and J. Janin, Protein Eng., Des. Sel., 1989, 3, 1–3 CrossRef CAS PubMed.
- J. Mejas and S. Lago, Chem. Phys., 2000, 113, 7306–7316 Search PubMed.
- Y. B. Yu, P. L. Privalov and R. S. Hodges, Biophys. J., 2001, 81, 1632–1642 CrossRef CAS PubMed.
- Y.-y Ohnishi, Y. Nakao, H. Sato, Y. Nakao, T. Hiyama and S. Sakaki, Organometallics, 2009, 28, 2583–2594 CrossRef CAS.
- H. Nakai and A. Ishikawa, Chem. Phys., 2014, 141, 174106 Search PubMed.
- T. Sakajiri, H. Yajima and T. Yamamura, Int. Sch. Res. Notices, 2012, 2012, 124803 Search PubMed.
- T. A. Keith, TK Gristmill Software, Overland Park, KS, USA, 2013 Search PubMed.
- E. Matito, ESI-3D: Electron Sharing Indices Program for 3D Molecular Space Partitioning, Institute of Computational Chemistry and Catalysis (IQCC), University of Girona, Catalonia, Spain, 2006.
- E. Matito, M. Solà, P. Salvador and M. Duran, Faraday Discuss., 2007, 135, 325–345 RSC.
- E. Glendening, J. Badenhoop, A. Reed, J. Carpenter, J. Bohmann, C. Morales, C. Landis and F. Weinhold, Theoretical Chemistry Institute, University of Wisconsin, Madison, 2013 Search PubMed.
- R. Bader et al. , Atoms in Molecules: A Quantum Theory, Clarendon, Oxford, UK, 1990 Search PubMed.
- F. Weinhold, Discovering chemistry with natural bond orbitals, John Wiley & Sons, 2012 Search PubMed.
- Q. Guo, M. Manolopoulou, Y. Bian, A. B. Schilling and W.-J. Tang, J. Mol. Biol., 2010, 395, 430–443 CrossRef CAS PubMed.
- H. Hashimoto, T. Takeuchi, K. Komatsu, K. Miyazaki, M. Sato and S. Higashi, J. Biol. Chem., 2011, 286, 33236–33243 CrossRef CAS PubMed.
- J. V. King, W. G. Liang, K. P. Scherpelz, A. B. Schilling, S. C. Meredith and W.-J. Tang, Structure, 2014, 22, 996–1007 CrossRef CAS PubMed.
- P. F. Teixeira, G. Masuyer, C. M. Pinho, R. M. Branca, B. Kmiec, C. Wallin, S. K. Wärmländer, R. P.-A. Berntsson, M. Ankarcrona and A. Gräslund, et al. , J. Mol. Biol., 2018, 430, 348–362 CrossRef CAS PubMed.
- J. P. Leite and L. Gales, FEBS Lett., 2019, 593, 128–137 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp02070c |
| This journal is © the Owner Societies 2023 |