
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photodissociation dynamics of tetrahydrofuran at 193 nm
Dennis
Milešević
 ,
Joseph
Stimson
,
Divya
Popat
,
Patrick
Robertson
and
Claire
Vallance
*
,
Joseph
Stimson
,
Divya
Popat
,
Patrick
Robertson
and
Claire
Vallance
*
Department of Chemistry, University of Oxford, Chemistry Research Laboratory, 12 Mansfield Rd, Oxford OX1 3TA, UK. E-mail: claire.vallance@chem.ox.ac.uk
First published on 25th August 2023
Abstract
Tetrahydrofuran (THF), a cyclic ether with the chemical formula C4H8O, can be considered the simplest analog of the deoxyribose backbone component of deoxyribonucleic acid (DNA). As such, it provides a useful model for probing the photochemistry of such biomolecular motifs. We present a velocity-map imaging study into the ultraviolet dissociation of THF at a wavelength of 193 nm. Excitation to the S1 state occurs via a 3s ← n transition involving a lone-pair electron on the oxygen atom, and has been shown by other authors to result in rapid ring opening via cleavage of one of the C–O bonds to form a ring-opened C4H8O diradical, followed by C–C bond cleavage over a longer timescale to form either OCH2 + C3H6 products (Channel 1a), HOCH2 + C2H5 products (Channel 1b), or OCH2CH2 + C2H4 products (Channel 2). The C2H4O products formed via Channel 2 are unstable on the timescale of our experiment and dissociate further to form CH3 and CHO. We also observe a number of minor products resulting from H or H2 loss from the primary photofragments. The speed distributions observed for all photofragments are broad, indicating excitation of a range of rotational and vibrational states of the products. The angular distributions of the photofragments show an interesting speed dependence: the slowest products have almost isotropic angular distributions, but the magnitude of the recoil anisotropy increases monotonically with photofragment speed. The fastest products exhibit highly anisotropic angular distributions, with the recoil anisotropy parameter β approaching its limiting value of −1 (−0.75 for Channel 1 and −0.5 for Channel 2). This behaviour is attributed to the range of timescales over which the diradical intermediate dissociates into the observed photofragments. Rapid dissociation leads to fast photofragments which retain the correlation between the transition dipole moment for the S1 ← S0 excitation (which lies perpendicular to the ring) and the photofragment velocities (which lie predominantly in the plane of the ring). Slow dissociation results in a high degree of energy redistribution into internal modes, slower photofragments, and loss of correlation between the photofragment velocities and the transition dipole. The higher barrier associated with dissociation via Channel 2 suggests somewhat longer lifetimes for the diradical intermediate and is consistent with a corresponding reduction in the maximum observed value for β.
1. Introduction
Extensive exposure of the human body to ultraviolet (UV) radiation can lead to damage of deoxyribonucleic acid (DNA), causing diseases such as skin cancer.1–3 Probing the effect of radiation on the molecular structure of DNA is therefore of fundamental interest. Tetrahydrofuran (c-C4H8O), henceforth referred to as THF, is a cyclic either, which has been considered previously as a model compound for the DNA backbone constituent ribose.4–10 THF also plays a vital role in polymerisation reactions11,12 and is used as a polar solvent for organic synthesis. As a prototypical oxygen-containing heterocycle, investigations into the photostability and photofragmentation behaviour of THF are also of more general interest, and the molecule has previously been the subject of studies involving excitation at a variety of different wavelengths.13,14Several authors have reported gas-phase absorption spectra of THF.15–18 The lowest energy (S1 ← S0) absorption band lies between 180 and 200 nm, and has been assigned to a 3s ← n transition involving one of the oxygen lone-pair electrons. The transition has been shown to form part of an s-type Rydberg series converging to the ground state of the ion at 9.41 eV.17 Giuliani and co-workers19 have probed the absorption spectrum in this region at high resolution, revealing separate but overlapping contributions to the band from the two near-degenerate ground-state conformers of THF, which have C2 and CS symmetry, respectively (see Fig. 5 for geometry-optimised structures of the two conformers). While the global energy minimum of ground-state THF is generally considered to correspond to the CS conformer,20–24 interconversion between the two near-degenerate conformers is almost barrierless via pseudorotation through a C1 conformation.23,25–27
When excited at the shorter wavelength of 160 nm, Röder et al.28 assigned a 3d ← n transition followed by a 3d → 3p → 3s Rydberg cascade. The authors reported cuts through excited-state potential energy surfaces (PESs) of THF along the C–O, Cα–H, and Cα–Cβ stretch coordinates. The S1 state of THF is bound along both the Cα–H, and Cα–Cβ coordinates. However, along the C–O stretching coordinate, the S1 state is repulsive and changes its electronic character from Rydberg (3s) in the Franck–Condon region to valence (nπ*) at larger C–O separations, leading eventually to a conical intersection with the S0 ground state along the ring-opening coordinate. In addition to direct dissociation on S1, the conical intersection provides a non-adiabatic pathway for populating excited vibrational levels of the electronic ground state, from which fragmentation can also occur. A similar picture of S1 ← S0 excitation, C–O bond elongation, and internal conversion to the ground-state followed by fragmentation has also been observed in oxetane29 and oxirane.30 Asymmetric ring expansion leading to coupling of excited-state and ground-state surfaces has also been explored in other systems such as furan,31,32 thiophene,33,34 and tetrahydrothiophene.35
The ring opening described above implies that THF dissociates in a stepwise manner following photoexcitation to the S1 state. Fig. 1 summarises the main observed fragmentation pathways in THF following excitation at 193 nm. Ring opening forms the linear diradical C4H8O, which then dissociates along either the Cα–Cβ (‘Channel 1a’) or the Cβ–Cγ (‘Channel 2’) bond coordinate, leading to the product pairs C3H6 + OCH2 and C2H4 + OCH2CH2, respectively.36 It is interesting to note that Kramer37 observed these same two channels by inducing decomposition of THF on the S0 surface via infrared multiphoton dissociation. The C3H6 product of the first channel has the stable isomers propylene and cyclopropane. However, the diradical C2H4O product of the first channel, formed initially as the O–CH2–CH2 isomer, rapidly rearranges via a hydrogen-atom transfer to internally excited (CH3–CHO)*, which decomposes into HCO and CH3 on the sub-nanosecond timescale38 in the absence of collisions to stabilise the excited intermediate. An intramolecular H atom migration can accompany the Cα–Cβ cleavage (Channel 1b), leading to the products HO–CH2 + CH2CHCH2.
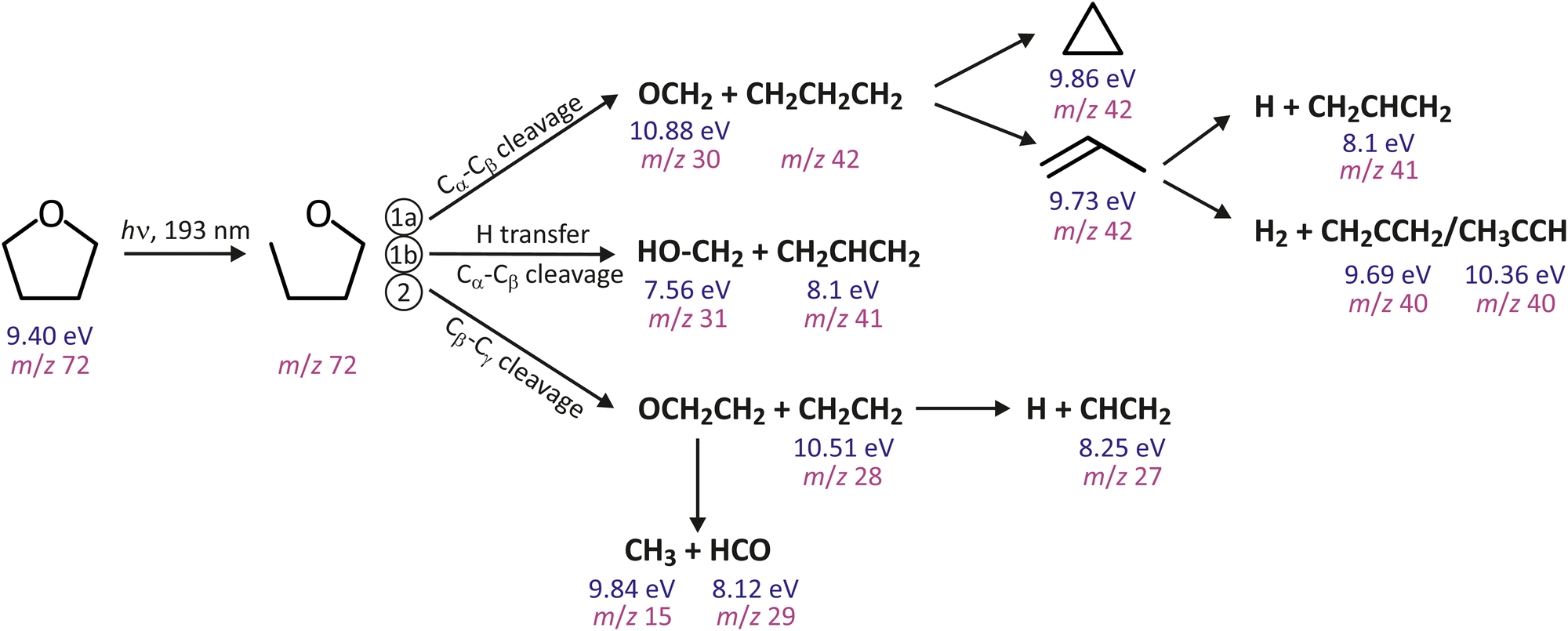 | ||
| Fig. 1 Reaction scheme for THF following 193 nm excitation. Absorption of a 193 nm photon leads to rapid cleavage of one of the C–O bonds to form the ring-opened diradical OCH2CH2CH2CH2. The diradical dissociates over a longer timescale via cleavage of either the Cα–Cβ or Cβ–Cγ bond to form C3H6 + OCH2 (Channel 1a) and C2H4 + OCH2CH2 (Channel 2) products, respectively. An internal H-atom migration can accompany the Cα–Cβ cleavage (Channel 1b). Several of the primary dissociation products undergo isomerisation and/or further dissociation, e.g. by loss of H or H2. Shown beneath each chemical species is the respective ionisation energy and mass-to-charge ratio. | ||
Lee39 investigated the photofragmentation of THF following photoexcitation via the S1 ← S0 transition at 193.3 nm, the same wavelength employed in the present study. Based on an analysis of the photofragment time-of-flight (ToF) distributions, Lee determined branching ratios and (total) translational kinetic energy releases (TKER) for five different fragmentation channels. The three dissociation channels with significant branching ratios were those described above, leading to C2H4 + HCO + CH3, C3H6 + OCH2, and C3H5 + HOCH2. The photofragment angular distributions were all reported to be nearly isotropic, which is somewhat surprising given that the fragmentations are believed to occur on the femtosecond timescale.13,28 Lee offered two possible explanations for the lack of angular anisotropy: (i) that the non-planar structure of THF causes the fragmentation products to recoil at angles near to the magic angle40 of 54.7° to the transition dipole; or (ii) that internal rotations within the diradical intermediate result in the dissociation products recoiling over a wide range of angles.
In the following, we present an experimental study into the photodissociation of THF at 193 nm in which we employ multi-mass velocity-map imaging to characterise the scattering distributions of the photofragments. We also report calculated transition dipole moments for the S1 ← S0 transition originating from the two possible ground-state conformers, and consider the relationship between these transition dipole moments, the angular dependence of the observed product scattering distributions, and the lifetime of the intermediate diradical C4H8O.
2. Methods
2.1 Experimental methods
The velocity-map imaging time-of-flight (ToF) mass spectrometer used in this work has been described in detail elsewhere.41 Briefly, helium (BOC, 99.9%) at a pressure of 1 bar was bubbled through liquid tetrahydrofuran (Sigma-Aldrich, ≥99.9%). The resulting gas mixture underwent supersonic expansion through a pulsed Solenoid Valve (Parker Hannifin, Series 9, 10 Hz) before being collimated by a 1.5 mm diameter skimmer and entering the interaction region, where the UV photolysis beam and VUV probe beam intersected the molecular beam at right angles.The 193.3 nm (from hereon referred to as 193 nm) UV photolysis beam was generated by a Neweks PSX-100 ArF excimer laser (7 ns, ∼1.5 mJ pulses). A Rochon polarizer was used to polarise the photolysis beam linearly along a polarisation axis parallel to the detector plane (perpendicular to the time-of-flight axis). The pulse energy of the photolysis light at the entrance to the vacuum chamber after passing through the polariser and beam steering optics was measured to be between 0.1 and 0.6 mJ, and the beam was focused down to a diameter of around 5–10 microns at the interaction region. The estimated fluence is therefore in the range ∼1 × 105 to 3 × 106 mJ cm−2.
After a delay of 20 ns, the neutral photofragments were ionized by the 118.2 nm (10.49 eV) vacuum-ultraviolet (VUV) probe beam, generated by frequency tripling the third harmonic of a Nd:YAG laser (Continuum Surelite I) in a phase-matched mixture of 298 mbar Argon (BOC, 99.9%) and 27 mbar Xenon (BOC, 99.9%).42–45 The probe laser beam was linearly polarized along the time-of-flight axis (perpendicular to the imaging plane) to ensure that all anisotropy observed in the measured photofragment angular distributions results from the photolysis step. However, preliminary experiments performed with the probe laser polarised either parallel or perpendicular to the imaging plane revealed no differences in the recorded images.
The ionised photoproducts were accelerated towards the detector by the electric field maintained within the velocity-map imaging ion lens, which projects the three-dimensional scattering distribution for each ion mass-to-charge (m/z) ratio into a two-dimensional image on the detector plane.46,47 The detector, consisting of a pair of microchannel plates (MCPs) coupled to a P47 phosphor screen, generates an optical signal on the phosphor for each incident ion. A Pixel Imaging Mass Spectrometry (PImMS2) camera48–50 records an (x,y,t)-data point for each incident ion. The full multi-mass imaging data set was acquired over 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 pump–probe cycles.
000 pump–probe cycles.
In addition to the two-colour pump–probe signals, one-laser signals (pump only and probe only) were recorded. In the raw data sets the signal from a single ion generally extends over several adjacent pixels and time bins, so to improve the spatial and temporal resolution as well as to reduce the size of the data set, the signal from each ion was centroided to a single (x,y,t) point before further analysis. The one-laser signals were then subtracted from the pump–probe signals in order to obtain the true pump–probe signal. The resulting data set can then be integrated over the (x,y) coordinates to yield the product time-of-flight spectrum, and velocity-map images can be generated for each photofragment by integrating over the relevant range of arrival times. The photofragment scattering distributions are cylindrically symmetric about the photolysis laser polarisation axis, allowing us to Abel invert the images for each fragment ion using a Python version of the Basex software package.51 From the inverted images, we extracted radial and angular distributions and converted the radial distributions into kinetic energy distributions using a pixel-to-velocity calibration determined in previous measurements on the 355 nm photolysis of Cl2.
2.2 Computational methods
The geometries of the C2 and CS ground-state conformers of THF were optimised in a B3LYP/aug-cc-pVQZ calculation using the Gaussian 09 software package.52 Transition dipole moments for the 3s ← n transition to the S1 state were calculated for each conformer using EOM-CCSD/aug-cc-pVTZ.3. Results
3.1 Time-of flight spectrum
Fig. 2 shows the mass-calibrated two-colour pump–probe ToF spectrum of THF and its photofragments following excitation at 193 nm and probe laser irradiation at 118 nm. The one-laser contributions have been subtracted from the two-colour signal, which leads to a negative peak in the ToF spectrum for the parent ion. This arises due to the depletion of neutral THF through dissociation by the pump laser, with the result that fewer intact molecules are available to be ionised by the probe laser in the pump–probe experiments than in the probe-only experiments. Note that because the parent ion velocites are essentially those of the neutral molecules in the molecular beam, the parent ions all arrive in a small spot at the centre of the detector (rather than being spread across the detector as in the case of fragment ions with non-zero kinetic energies) and often saturate the signal response of the microchannel plate to some extent. We therefore do not expect to observe a quantitative match between the intensity of this negative peak and the summed intensity of the fragment peaks.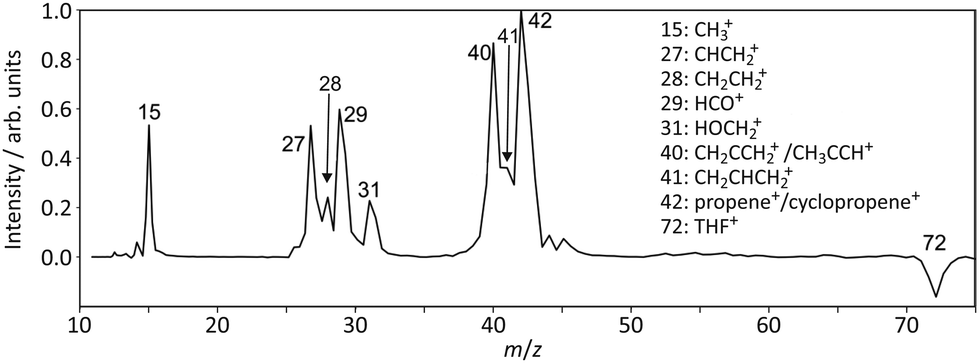 | ||
| Fig. 2 Background-subtracted time-of-flight mass spectrum for the 193 nm photolysis products of THF. | ||
Based on the 10.49 eV photon energy of our probe laser and the ionisation energies given for the various molecular fragments in Fig. 1, we might expect to observe the following species from each of the three main dissociation channels:
1. Channel 1a: propene (m/z 42), cyclopropane (m/z 42), CH2CHCH2 (m/z 41), and CH2CCH2/CH3CCH (m/z 40);
2. Channel 1b: HO–CH2 (m/z 31) and CH2CHCH2 (m/z 41);
3. Channel 2: CH3 (m/z 15), HCO(m/z 29), possibly CH2CH2 (m/z 28),and CHCH2 (m/z 27).
Inspecting the time-of-flight spectrum in Fig. 2, we indeed see peaks at all of the predicted m/z values, indicating that we observe the same three major dissociation channels as reported by previous authors37,39 and discussed in the Introduction. We do not see evidence in the ToF spectrum for cleavage of both C–O bonds as reported by SenGupta and co-workers,53 although we note that the atomic oxygen formed in such a process would not be ionised by our probe laser and would therefore not be detected in our experiment.
3.2 Product speed and angular distributions
Our multi-mass velocity-map imaging detection system enables us to record scattering distributions for all of the reaction products appearing in the ToF spectrum. The measured velocity-map images are shown in Fig. 3. As explained in Section 2, Abel inversion of the images allows the speed and angular distributions to be determined for each fragment. | ||
| Fig. 3 Symmetrised velocity-map images for the observed photodissociation products of THF. The direction of linear polarisation of the 193 nm photolysis laser is indicated by the double-headed arrow in the first panel. | ||
Inspection of the reaction scheme in Fig. 1 allows us to determine how the measured scattering distributions can best be used to probe the dynamics of the three dissociation channels. The most straightforward channel from this point of view is Channel 1b, which forms two momentum-matched products, HO–CH2 and CH2CHCH2, neither of which undergo any further fragmentation or rearrangement. The HO–CH2 product is uniquely assigned to m/z 31, and we can therefore use the corresponding image to determine the speed and angular distributions for the products of this channel. These are shown in Fig. 4(b).
 | ||
| Fig. 4 Speed distributions, P(v), and recoil anisotropy distributions, β(v), for the dissociation products of (a) Channel 1a, (b) Channel 1b, and (c) Channel 2. Speed and recoil anisotropy distributions are for the primary products indicated in red text, determined as explained in the text. The kinetic energy distributions determined in the present work are compared with the equivalent result from Lee,39 and the maximum accessible kinetic energy is indicated by a dashed line. | ||
The CH2CHCH2 product, appearing at m/z 41 in the ToF spectrum, is not unique to Channel 1b, since it is also formed as a secondary H-atom loss product in Channel 1a. The m/z 41 signal therefore contains contributions from both of these pathways.† However, as noted above, the component arising from Channel 1b is momentum matched with the HO–CH2 product, allowing the components arising from the two channels to be disentangled: after appropriate correction for the branching ratios and ionisation efficiencies using data from Lee,39 the momentum-matched distribution arising from Channel 1b can simply be subtracted from the total signal to leave the component arising from Channel 1a.
Moving now to Channel 1a, the O–CH2 product is unique to this channel and its scattering distribution would provide all of the required information on the dynamics of this channel. Unfortunately, this scattering distribution is not available to us, since the ionisation energy of O–CH2 lies above the 10.49 eV photon energy of our ionisation laser. Instead, we must consider the CH2CH2CH2 product. The initially formed linear CH2CH2CH2 primary product of Cα–Cβ bond cleavage is unstable, rearranging on the femtosecond timescale54,55 to either propene or cyclopropane.37 The propene product of this rearrangement can undergo H2 elimination to form CH2CCH2/CH3CCH or loss of a single H atom to produce CH2CHCH2. The scattering distribution of the primary CH2CH2CH2 product is unchanged by unimolecular rearrangement to propene or cyclopropane, so we do not lose any information in the isomerisation step. Additionally, the fact that linear momentum must be conserved during H or H2 loss from propene, coupled with the much lighter mass of H/H2 relative to the partner fragment, means that any kinetic energy released in the secondary dissociation step will almost all be carried away by the H or H2 product, leaving the velocity distributions of the partner fragments unchanged to a very good approximation. As a result, summing the velocity distributions recorded for the m/z 40, 41, and 42 products of Channel 1a (noting that for the m/z 41 fragment we use the Channel 1a component described earlier), yields a very good approximation to the velocity distribution of the nascent primary CH2CH2CH2 product. The speed and angular distributions determined using this approach are shown in Fig. 4(a).
Finally, we consider Channel 2. As noted earlier, the OCH2CH2 product (m/z 44) of this channel is known from previous work38 to dissociate rapidly into CH3 + HCO under the collision-free conditions of our experiment, consistent with the fact that we see virtually no signal at m/z 44. We therefore cannot use this product to learn about the primary dissociation step. Some of the partner fragment, CH2CH2, also dissociates further via H atom loss to give H + CHCH2. However, as noted above, the recoil momentum imparted to the CHCH2 product by the departing H atom is very small, and is expected to have an almost negligible effect on its velocity distribution. We can therefore sum the velocity distributions for the CH2CH2 and CHCH2 products to give a good approximation to the velocity distribution of the nascent primary CH2CH2 product‡ The resulting speed and angular distributions associated with Channel 2 are shown in Fig. 4(c).
Previous time-of-flight measurements carried out by Lee39 as a function of photolysis laser polarisation suggested that the average β parameter is close to zero, so our results do not agree with the conclusions drawn from these earlier measurements. The β(v) distributions measured in the present experiments lead to only a small asymmetry in the images. However, we have checked carefully that this anisotropy is real and is not caused by any artefacts of the experiment or the data processing. For example, we have made measurements on the same instrument of scattering distributions which by symmetry must be isotropic, and confirmed that they give rise to β values of zero across the full range of product velocities, as well as checking that we obtain β parameters in line with literature reports for processes such as the 355 nm photolysis of Cl2. Having carried out these checks, and given that we are using VMI detection and can therefore clearly resolve β as a function of product recoil velocity, we believe our values are accurate, and that the discrepancy arises from the different velocity resolution achievable in our velocity-map imaging measurements and Lee's time-of-flight measurements. If we project our images along the x and y axes, equivalent to Lee's measurements with the photolysis laser linearly polarised parallel and perpendicular to the time-of-flight axis, there is a small but clear difference in the widths of the resulting distributions. The velocity-dependent anisotropy we measure is therefore easily resolved within the spatial resolution of our velocity-map images, but would be very difficult to resolve with the typical temporal resolution achievable in a time-of-flight measurement. This is consistent with the fact that while our β(v) distribution does not agree with that reported by Lee, our translational kinetic energy distribution for the products of Channel 1a is in excellent agreement.
Fig. 5 shows geometry-optimised structures for the C2 and CS conformers of ground-state THF, which we expect to be present at similar number densities within our molecular beam, together with the transition dipole moments (red arrows) associated with the S1 ← S0 transition in each conformer. For both conformers the transition dipole moment lies perpendicular to the plane of the ring. In the axial recoil limit, both the ring-opening step and the subsequent C–C bond cleavage occur in the molecular plane. We therefore expect the products to recoil in this same plane, perpendicular to the transition dipole moment, giving rise to a β parameter close to its limiting value of −1.
 | ||
| Fig. 5 Optimised structures and S1 ← S0 transition dipoles (red arrows) for the C2 (top) and CS (bottom) conformers of THF. For visualisation purposes the transition dipole moments have been elongated by factors of five and four for the C2 and CS conformers, respectively. | ||
While the highest velocity products do have β parameters close to −1, we observe a strong dependence of β on the product recoil velocity. The slowest products have near-zero values, which decrease monotonically to a near-limiting value at the highest velocities. We ascribe this behaviour to the finite lifetime of the C4H8O diradical formed in the initial ring-opening step and to the distribution of internal energies with which the diradical is formed, noting from the discussion in Section 1 that dissociation can occur either directly on S1 or following internal conversion to S0. Following photoexcitation, the repulsive character of the S1 surface leads to rapid C–O bond cleavage to form the diradical. When the ring-opened diradical dissociates on a timescale that is short relative to intramolecular vibrational relaxation and rotation, most of the available energy will be released into product translation. In this case the product recoil velocity is high and β approaches its limiting value determined by the perpendicular orientation of the transition dipole moment relative to the molecular plane, as described above. As the lifetime of the diradical increases, perhaps following internal conversion to S1, the molecule has more time to undergo rotation and for energy to be redistributed into internal modes. This leads to a decrease both in the photofragment velocities and in the correlation between the transition dipole moment and the ejection direction of the photoproducts, explaining the reduction in β for slower fragments. The TKER distribution is consistent with this picture. The distribution peaks at low kinetic energies, indicating that a significant portion of the excess energy is partitioned into internal degrees of freedom in the photofragments. Taken together with the behaviour of β(v) described above, this is consistent wtih dissociation of the diradical over a relatively broad range of timescales.
3.3 Channel 2: THF → OCH2CH2 + CH2CH2
The photofragment speed distribution for the products of Channel 2 is very similar to that observed earlier for the products of Channels 1a and 1b, comprising a broadly-peaked distribution extending out to maximum speeds of around 3000 m s−1. As for the other channels, we can infer that the products are formed in a range of rotational and vibrational states. We also see similar behaviour for the velocity-dependence of the recoil anisotropy parameter β, which has values close to zero at low velocities (high internal excitation), but decreases monotonically to a value of around −0.5 at the highest velocities. We believe the explanation for this behaviour to be analogous to that for Channels 1a and 1b. Following the initial ring-opening step, dissociation of the diradical intermediates with the shortest lifetimes retains the highest degree of correlation between the transition dipole moment, which lies perpendicular to the plane of the ring, and the photofragment velocities, which lie in the plane of the ring. Dissociation on longer timescales degrades the correlation through rotational and vibrational motions prior to C–C bond cleavage, leading to a reduction in β for the slower products. The limiting value of β observed for Channel 2 of around −0.5 is significantly lower than the value of around −0.75 determined for Channels 1a and 1b. This perhaps implies a longer lifetime for C4H8O intermediates dissociating via Channel 2, which would be consistent with the higher barrier to dissociation for this channel reported by Scala et al.,13 and also with the lower branching ratio for Channel 2 relative to Channels 1a and 1b determined by Lee.39 The velocity-averaged value for β of −0.23 is again in disagreement with the near-zero value determined by Lee.39The TKER distribution for the primary dissociation products of Channel 2 is again in excellent agreement with that reported by Lee.39 The distribution peaks at around 0.15 eV and tails off by around 1 eV, intermediate between the behaviour for Channel 1a and Channel 1b, and well below the available energy of 2.30 eV. This is consistent with the lifetime arguments evoked above to explain the behaviour of β in the two channels. The longer lifetimes associated with dissociation via Channel 2 result in a greater degree of energy redistribution into internal modes, and therefore a smaller fraction of energy released into translation.
4. Conclusions
We have presented the results of a velocity-map imaging study into the 193 nm photodissociation of tetrahydrofuran. Excitation of THF at this wavelength leads to population of the S1 state via an 3s ← n transition involving a lone-pair electron on the oxygen atom. The associated transition dipole lies perpendicular to the plane of the ring for both the C2 and CS conformers of THF. The S1 state is repulsive along the C–O bond stretching coordinate, resulting in rapid bond scission to form a ring-opened diradical intermediate species. The diradical undergoes further dissociation over a broad range of timescales via cleavage of either the Cα–Cβ bond to form OCH2 + C3H6 or HOCH2 + C2H5 products (denoted Channel 1a and Channel 1b, respectively) or cleavage of the Cβ–Cγ bond to form OCH2CH2 + C2H4 (Channel 2). The C3H6 products of Channel 1a isomerise rapidly to cyclopropane and propene, the latter of which undergoes secondary decay via loss of H or H2. The OCH2CH2 products of Channel 2 are also unstable on the timescale of our experiment, and dissociate to form CH3 and CHO, while some of the CH2CH2 products decay via H loss to give CHCH2.The translational kinetic energy releases measured in the present work for all three channels are in excellent agreement with previous measurements by Lee.39 However, Lee reported near-zero angular anisotropy for both channels, whereas we observe a recoil anisotropy parameter β that has near-zero values for products formed with low velocities, but decreases to around −0.75 for Channels 1a and 1b and −0.5 for Channel 2, respectively, for products formed with the highest recoil velocities. We attribute this behaviour to the extent of internal excitation of the C4H8O diradical intermediate and the range of lifetimes over which it survives prior to cleavage of a C–C bond. Diradicals which dissociate on a rapid timescale have the highest translational energies and retain the strongest correlation between the transition diople moment for the S1 ← S0 excitation and the recoil velocities of the photofragments. Since the transition dipole lies perpendicular to the plane of the ring and the recoil velocities of the photofragments lie preferentially in the plane of the ring, this results in negative β parameters which approach the limiting value of −1. In contrast, diradicals with longer lifetimes undergo both rotation and redistribution of energy into internal degrees of freedom prior to dissociation, both of which reduce both the magnitude of the photofragment velocities and their correlation with the transition dipole moment. The highest values of β observed for Channel 2 are significantly lower than those observed for Channels 1a and 1b, which is consistent with the higher barrier to dissociation via Channel 2 leading to dissociation on a slower timescale than via Channels 1a and 1b.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the EPSRC for funding this work via Programme Grants EP/V026690/1 and EP/T021675/1.References
- B. Boudaïffa, P. Cloutier, D. Hunting, M. A. Huels and L. Sanche, Science, 2000, 287, 1658–1660 CrossRef PubMed.
- B. Boudaïffa, P. Cloutier, D. Hunting, M. A. Huels and L. Sanche, Radiat. Res., 2002, 157, 227–234 CrossRef.
- J. Cadet and T. Douki, Photochem. Photobiol. Sci., 2018, 17, 1816–1841 CrossRef CAS PubMed.
- S. Tonzani and C. H. Greene, J. Chem. Phys., 2006, 125, 094504 CrossRef PubMed.
- C. J. Colyer, S. M. Bellm, B. Lohmann, G. F. Hanne, O. Al-Hagan, D. H. Madison and C. G. Ning, J. Chem. Phys., 2010, 133, 124302 CrossRef CAS PubMed.
- X. Ren, E. Wang, A. D. Skitnevskaya, A. B. Trofimov, K. Gokhberg and A. Dorn, Nat. Phys., 2018, 14, 1062–1066 Search PubMed.
- N. Y. Aouina and Z. E. A. Chaoui, Surf. Interface Anal., 2018, 50, 939–946 CrossRef CAS.
- W. Wolff, B. Rudek, L. A. Da Silva, G. Hilgers, E. C. Montenegro and M. G. Homem, J. Chem. Phys., 2019, 151, 064304 CrossRef.
- H. J. Lüdde, M. Horbatsch and T. Kirchner, J. Phys. B: At., Mol. Opt. Phys., 2019, 52, 195203 CrossRef.
- A. García-Abenza, A. I. Lozano, L. Álvarez, J. C. Oller, F. Blanco, P. Stokes, R. D. White, J. de Urquijo, P. Limão-Vieira, D. B. Jones, M. J. Brunger and G. García, Eur. Phys. J. D, 2021, 75, 303 CrossRef.
- Y. Yagci and A. Ledwith, J. Polym. Sci. Part A Polym. Chem., 1988, 26, 1911–1918 CrossRef CAS.
- M. R. Rodrigues and M. G. Neumann, Macromol. Chem. Phys., 2001, 202, 2776–2782 CrossRef CAS.
- A. A. Scala, E. W. Diau, Z. H. Kim and A. H. Zewail, J. Chem. Phys., 1998, 108, 7933–7936 CrossRef CAS.
- T. J. Wasowicz, A. Kivimäki, M. Dampc, M. Coreno, M. De Simone and M. Zubek, Phys. Rev. A - At. Mol. Opt. Phys., 2011, 83, 033411 CrossRef.
- B. L. W. Pickett, N. J. Hoeflich and T.-C. Liu, J. Am. Chem. Soc., 1951, 73, 4865–4869 CrossRef.
- G. J. Hernandez, J. Chem. Phys., 1963, 38, 2233–2242 CrossRef CAS.
- J. Doucet, P. Sauvageau and C. Sandokfy, Chem. Phys. Lett., 1972, 17, 316–319 CrossRef CAS.
- L. J. Bremner, M. G. Curtis and I. C. Walker, J. Chem. Soc., Faraday Trans., 1991, 87, 1049–1055 RSC.
- A. Giuliani, P. Limão-Vieira, D. Duflot, A. R. Milosavljevic, B. P. Marinkovic, S. V. Hoffmann, N. Mason, J. Delwiche and M. J. Hubin-Franskin, Eur. Phys. J. D, 2009, 51, 97–108 CrossRef CAS.
- B. Cadioli, E. Gallinella, C. Coulombeau, H. Jobic and G. Berthier, J. Phys. Chem., 1993, 97, 7844–7856 CrossRef CAS.
- A. H. Mamleev, L. N. Gunderova and R. V. Galeev, J. Struct. Chem., 2001, 42, 365–370 CrossRef CAS.
- D. G. Melnik, S. Gopalakrishnan, T. A. Miller and F. C. De Lucia, J. Chem. Phys., 2003, 118, 3589–3599 CrossRef CAS.
- V. M. Rayón and J. A. Sordo, J. Chem. Phys., 2005, 122, 204303–204311 CrossRef PubMed.
- T. Yang, G. Su, C. Ning, J. Deng, F. Wang, S. Zhang, X. Ren and Y. Huang, J. Phys. Chem. A, 2007, 111, 4927–4933 CrossRef CAS PubMed.
- P. Duffy, J. A. Sordo and F. Wang, J. Chem. Phys., 2008, 128, 125102 CrossRef PubMed.
- S. M. Park, Y. R. Lee, D. W. Kang, H. L. Kim and C. H. Kwon, Phys. Chem. Chem. Phys., 2017, 19, 30362–30369 RSC.
- L. Paoloni, S. Rampino and V. Barone, J. Chem. Theory Comput., 2019, 15, 4280–4294 CrossRef CAS PubMed.
- A. Röder, A. B. Skov, A. E. Boguslavskiy, R. Lausten and A. Stolow, Phys. Chem. Chem. Phys., 2020, 22, 26241–26254 RSC.
- S. H. Lee, J. Chem. Phys., 2009, 131, 224309 CrossRef PubMed.
- E. Tapavicza, I. Tavernelli, U. Rothlisberger, C. Filippi and M. E. Casida, J. Chem. Phys., 2008, 129, 124108 CrossRef PubMed.
- N. Gavrilov, S. Salzmann and C. M. Marian, Chem. Phys., 2008, 349, 269–277 CrossRef CAS.
- S. Oesterling, O. Schalk, T. Geng, R. D. Thomas, T. Hansson and R. De Vivie-Riedle, Phys. Chem. Chem. Phys., 2017, 19, 2025–2035 RSC.
- M. Stenrup, Chem. Phys., 2012, 397, 18–25 CrossRef CAS.
- M. N. Ashfold, M. Bain, C. S. Hansen, R. A. Ingle, T. N. Karsili, B. Marchetti and D. Murdock, J. Phys. Chem. Lett., 2017, 8, 3440–3451 CrossRef CAS PubMed.
- N. Kotsina, S. L. Jackson, T. Malcomson, M. J. Paterson and D. Townsend, Phys. Chem. Chem. Phys., 2022, 24, 29423–29436 RSC.
- B. C. Roquitte, J. Am. Chem. Soc., 1969, 91, 7664–7667 CrossRef CAS.
- J. Kramer, J. Phys. Chem., 1982, 86, 26–35 CrossRef CAS.
- S. W. Benson, J. Chem. Phys., 1964, 40, 105–111 CrossRef CAS.
- S. H. Lee, Phys. Chem. Chem. Phys., 2010, 12, 2655–2663 RSC.
- C. H. Green and R. N. Zare, Ann. Rev. Phys. Chem., 1982, 33, 119–150 CrossRef.
- W. S. Hopkins, M. L. Lipciuc, S. H. Gardiner and C. Vallance, J. Chem. Phys., 2011, 135, 034308 CrossRef PubMed.
- A. H. Kung, J. F. Young and S. E. Harris, Appl. Phys. Lett., 1973, 22, 301–302 CrossRef CAS.
- A. H. Kung, J. F. Young and S. E. Harris, Appl. Phys. Lett., 1976, 28, 294 CrossRef.
- N. P. Lockyer and J. C. Vickerman, Laser Chem., 1997, 17, 139–159 CrossRef CAS.
- J. M. Gray, J. Bossert, Y. Shyur, B. Saarel, T. C. Briles and H. J. Lewandowski, J. Chem. Phys., 2021, 154, 024201 CrossRef CAS PubMed.
- D. W. Chandler and P. L. Houston, J. Chem. Phys., 1987, 87, 1445–1447 CrossRef CAS.
- A. T. Eppink and D. H. Parker, Rev. Sci. Instrum., 1997, 68, 3477–3484 CrossRef CAS.
- A. Nomerotski, M. Brouard, E. Campbell, A. Clark, J. Crooks, J. Fopma, J. J. John, A. J. Johnsen, C. Slater, R. Turchetta, C. Vallance, E. Wilman and W. H. Yuen, J. Instrum., 2010, 5, C07007 Search PubMed.
- E. S. Wilman, S. H. Gardiner, A. Nomerotski, R. Turchetta, M. Brouard and C. Vallance, Rev. Sci. Instrum., 2012, 83, 013304 CrossRef PubMed.
- A. T. Clark, J. P. Crooks, I. Sedgwick, R. Turchetta, J. W. Lee, J. J. John, E. S. Wilman, L. Hill, E. Halford, C. S. Slater, B. Winter, W. H. Yuen, S. H. Gardiner, M. L. Lipciuc, M. Brouard, A. Nomerotski and C. Vallance, J. Phys. Chem. A, 2012, 116, 10897–10903 CrossRef CAS PubMed.
- V. Dribinski, A. Ossadtchi, V. A. Mandelshtam and H. Reisler, Rev. Sci. Instrum., 2002, 73, 2634–2642 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, J. R. M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, M. C. X. Li, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, 2016 Search PubMed.
- S. SenGupta, H. P. Upadhyaya, A. Kumar, P. D. Naik and P. Bajaj, J. Chem. Phys., 2005, 122, 124309 CrossRef PubMed.
- S. Pedersen, J. K. Herek and A. H. Zewail, Science, 1994, 266, 1359–1364 CrossRef CAS PubMed.
- C. Doubleday, J. Phys. Chem., 1996, 100, 3520–3526 CrossRef CAS.
Footnotes |
| † We note that within the mass resolution of our experiment, there may also be some contributions to the ‘m/z 41’ signal from the higher velocity components of the m/z 40 and 42 signals. However, given the analysis we are about to describe, we do not believe that this is likely to introduce any significant error to the TKER and β(v) distributions reported for this channel, and does not change any of our conclusions. |
| ‡ Interestingly, but not surprisingly, comparison of the velocity-map image fors the CHCH2 and CH2CH2 products reveals more intensity in the centre of the image for the H-loss product. This implies that the slower-moving, more highly internally excited primary products are more likely to undergo H loss than the faster moving, less internally excited products. |
| This journal is © the Owner Societies 2023 |