
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpin–vibronic mechanism at work in a luminescent square-planar cyclometalated tridentate Pt(II) complex: absorption and ultrafast photophysics†
Souvik
Mandal
 and
Chantal
Daniel
*
and
Chantal
Daniel
*
Laboratoire de Chimie Quantique Université de Strasbourg, CNRS UMR7177, Institut Le Bel 4 Rue Blaise Pascal, Strasbourg 67000, France. E-mail: c.daniel@unistra.fr
First published on 21st June 2023
Abstract
The absorption spectrum of [Pt(dpybMe)Cl] (dpyb = 2,6-di-(2-pyridyl)benzene), representative of luminescent halide-substituted tridentate cyclometalated square planar Pt(II) neutral complexes, has been revisited by means of non-adiabatic wavepacket quantum dynamics. The early photophysics has been investigated on the basis of four singlet and five triplet excited states, namely nineteen “spin–orbit states”, coupled with both vibronic and spin–orbit couplings, and includes eighteen normal modes. It is shown that in-plane scissoring and rocking normal modes of the cyclometalated tridentate ligand are responsible for the vibronic structure observed at around 400 nm in the experimental spectrum of the complex. The ultrafast decay of [Pt(dpybMe)Cl], within 1 ps, follows a spin–vibronic mechanism governed by excited state electronic characters, spin–orbit, and active tuning mode interplay. Both spin–orbit coupling and Pt(II) coordination sphere stretching modes and in-plane scissoring/rocking of the cyclometalated ligand activate the ultrafast decay within 20 fs of absorption. At longer time-scales (>100 fs) an asynchronous stretching of the Pt–C and Pt–N bonds activates the depopulation of the upper “reservoir” electronic states to populate the two lowest luminescent T1 and T2 electronic states. The in-plane rocking motion of the ligand controls the T1/T2 population exchange which is equilibrated at about 1 ps. Stabilization of the upper non-radiative metal-centered (MC) states by out-of-plane ligand distortion of low frequency is not competitive with the ultrafast spin–vibronic mechanism discovered here for [Pt(dpybMe)Cl]. Modifying the Pt–C covalent bond position and rigidifying the cyclometalated ligand will have a dramatic influence on the spin–vibronic mechanism and consequently on the luminescence properties of this class of molecules.
1 Introduction
Since the discovery of emissive halide-substituted Pt(II) α-diimine complexes thirty years ago,1 a myriad of square-planar Pt(II) complexes with various ligands have been synthesized to optimize the luminescent properties and to control the branching ratio between radiative and non-radiative decays.2–6 The energy gap between lowest triplet metal-centered (MC) and charge-transfer (CT) excited states, and their coupling with the electronic ground state and with the environment (solution and solid) were particularly scrutinized. The introduction of multidentate ligands, with or without Pt–C bonds via pincer cyclometalated substitution, paved the way to a variety of applications such as functional materials, chromic devices, memory storage devices, supramolecular assemblies and probes or ion sensors.7–12 More recently, time-resolved (ps–fs) spectroscopy experiments were applied to mono- and bi-metallic Pt complexes in order to probe the interplay between nuclear and electronic motion, intersystem crossing and vibrational coherence in ultrafast processes.13–20 This field of research opens the way to fine-tuning of the photoluminescent and charge transfer properties under stimuli such as light, electric field or a specific environment for the design of building blocks in the development of new functional and switchable materials.21–24Standard theoretical investigations, based on routine static density functional theory (DFT) and time-dependent DFT (TD-DFT) calculations, are not up to the experimental challenges. Indeed, most of the time they focus on the structural and electronic properties of the electronic ground state and lowest singlet S1 and triplet T1 excited states with no account of spin–vibronic coupling effects and neglect the role of the upper electronic states.25–32
Consequently, it is very difficult to rationalize the luminescent behavior and to extract realistic radiative vs. non-radiative decay rate constants, on the basis of the qualitative Kasha rules, within a series of complexes because these observables are controlled by a subtle change in electronic densities associated with small structural distortions themselves correlated with the metal/ligand/substituent/medium/temperature interplay. This has been illustrated for a class of cyclometalated and non-cyclometalated bidentate and tridentate Pt(II) complexes,33–35 representative of a number of molecules synthesized for their luminescence properties over a wide range of energy. It has been shown that the presence of metal-centered (MC) states, easily stabilized by out-of-plane distortion in the non-cyclometalated compounds, is responsible for their poor luminescence, whereas the effectiveness of luminescence is optimized in the axially cyclometalated complex (Scheme 1) by efficient population of the low-lying T1 and T2 CT states and destabilization of the nearly degenerate T3 and T4 MC states.33
 | ||
| Scheme 1 DFT-optimized structure of [Pt(dpybMe)Cl] (dpyb = 2,6-di-(2-pyridyl)benzene) 1 (hydrogen atoms have been omitted for the sake of clarity). | ||
Moreover, even though the TD-DFT absorption spectra reproduce qualitatively the main features of the experimental spectra for this ensemble of Pt(II) square planar complexes, a quantitative agreement is out of reach without including spin–orbit couplings (SOCs) and vibronic effects.
Herein, the absorption spectroscopy of [[Pt(dpybMe)Cl] (dpyb = 2,6-di-(2-pyridyl)benzene), representative of halide-substituted cyclometalated tridentate Pt(II) square planar complexes, is revisited on the basis of non-adiabatic quantum dynamics including the four lowest singlet (S1–S4) and five lowest triplet (T1–T5) electronic excited states and 18 nuclear degrees of freedom (DOFs) within the linear vibronic coupling (LVC) model. The ultrafast excited state dynamics, simulated by nuclear wavepacket propagation, exhibits an efficient population of the active charge transfer T1 and T2 emissive states within 50 fs detrimental to the concurrent activation of the upper MC states in the decay process.
2 Theoretical background
2.1 Method
The S0 electronic ground state geometry of 1, depicted in Scheme 1, has been optimized by means of Kohn–Sham DFT with the B3LYP functional36,37 under Cs symmetry constraint, using the all-electrons scheme and a triple-ζ polarized basis set (TZP).38 The scalar relativistic effects have been introduced by the zeroth order regular approximation ZORA,39 the spin–orbit corrections being included as a perturbation. The nature of the stationary state was checked through a complete set of real frequencies.The transition energies to the low-lying singlet and triplet excited states have been computed by means of a time-dependent DFT (TD-DFT) method40,41 including solvent corrections for dichloromethane (ε = 8.9, rad = 2.94 Å) through the conductor like screening model (COSMO)42–44 as implemented in ADF.45,46 The calculations have been performed using ADF2019 quantum chemistry software,47 and the analysis by TheoDORE, a package for theoretical density, orbital relaxation and excitation analysis.48
The time-dependent (TD) Schrödinger equation within the diabatic Hamiltonian approach is solved by means of the multi-configuration time-dependent Hartree (MCTDH) method49 where the TD wavefunction is expressed as linear combination of Hartree products, the so-called single particle functions (SPFs). The SPFs are represented by multi-set formulation50 to adopt the present non-adiabatic correction problem. Here the primitive basis sets are chosen as harmonic oscillator wavefunctions in discrete variable representation (DVR). The initial wavefunction is taken as the product of harmonic wavefunctions at their vibrational ground state of the S0 electronic ground state. The simulation of the absorption is based on the oscillator strengths weighted S1, S2 and S3 allowed electronic transitions at time zero. The calculations have been performed using the Heidelberg MCTDH package version 8.4.13.51
As far as the computation of the absorption spectrum is concerned, the present approach is competitive as compared to other computationally expensive methods based on vibronic calculations over the whole vibrational space52 or on nuclear ensemble approach.53 Moreover, the LVC-based models give access to the interplay between active normal modes, involved electronic densities and spin–orbit effects.
2.2 Vibronic and spin–orbit couplings
Vibronic coupling theory has been developed in connection with a model Hamiltonian based on a diabatic representation of the electronic states.54 The diabatic Hamiltonian describing ηel coupled electronic states is written as| H(Q) = (TN + V0(Q))Π + W(Q) | (1) |
The intrastate κ(n)i and inter-state linear λ(n,m)i vibronic coupling constants generated by the vibrational molecular activity regulated by molecular symmetry rules are obtained using an analytical formula when only two electronic states are involved within the linear vibronic coupling model.50 The coupling constants can be deduced from electronic structure calculations using the first and second derivatives of the adiabatic potential energy surfaces Vn(Q) with respect to Qi at the ground state equilibrium geometry. Alternatively, and in order to go beyond the pair of states approximation and the linear formalism, λ(n,m)i can be computed on the basis of the overlap matrix between the electronic wavefunctions at close-lying geometries56 as an adiabatic-to-diabatic transformation matrix, such that the linear vibronic coupling (LVC) constants can be obtained by means of numerical differentiation.
 | (2) |
The method is applicable to wavefunction-based methods as well as to TD-DFT, as used in the present study. In the latter case the wavefunctions are replaced by auxiliary many-electron wavefunctions.57 The theory and construction of the W(Q) coupling matrix, including the 9 lowest S1–S4 and T1–T5 electronic excited state eigenvalues, SOC, intra- and inter-state vibronic couplings is detailed in the ESI.† The Sn and Tn notations are kept all along the manuscript. This is justified by the use of a model Hamiltonian based on a diabatic representation of the electronic states, both singlet and triplet. Of course, “spin–orbit” states could be retrieved by diagonalization of the W(Q) coupling matrix.
3 Results and discussion
3.1 Structural and electronic properties
The optimized structure of [Pt(dpybMe)Cl] is depicted in Scheme 1 and compared to the experimental one in Table S1 (ESI†) where important bond lengths and bond angles are reported. The normal modes of the ground state have been obtained on the optimized structure. The calculated frequencies and SOC are reported in Tables S2 and S3 (ESI†), respectively.The TD-DFT excited state transition energies calculated at the optimized ground state structure, the associated oscillator strengths and the “spin–orbit” states are reported in Tables S4 and S5 (ESI†). Fig. 1 displays the TD-DFT absorption spectra of 1 with and without SOC.
 | ||
| Fig. 1 Calculated TD-DFT absorption spectra of [Pt(dpybMe)Cl] with (in red) and without (in black) SOC. The experimental spectrum58 is given as an inset for comparison. | ||
As already analyzed in our previous work33 the calculated SOC spectrum agrees qualitatively well with the experimental one with a maximum absorption at 407 nm. However, it does not reproduce the vibronic structure observed experimentally. The double peak of the lowest experimental band centered at 407 nm is not reproduced by the TD-DFT calculations with or without SOC.
In order to obtain a quantitative absorption spectrum directly comparable to the experimental one, the spin–vibronic Hamiltonian model, defined above and in the ESI,† has been applied to a sub-set of 9 electronic states, namely 4 singlets and 5 triplets corresponding to 19 “spin–orbit” states (Table 1).
| Intra-state coupling terms | |||||
|---|---|---|---|---|---|
| κ S1 | κ S2 | κ T3 | κ T1 | κ T2 | |
| Q15 | 0.031 | 0.027 | −0.022 | 0.029 | 0.028 |
| Q83 | 0.064 | −0.057 | −0.057 | 0.009 | −0.070 |
| Q81 | 0.051 | −0.024 | −0.179 | 0.042 | −0.040 |
| Inter-state coupling terms | ||||
|---|---|---|---|---|
| λ S1,S2 | λ T1,T3 | λ T1,T2 | λ T2,T3 | |
| Q15 | 0.016 | |||
| Q77 | 0.071 | 0.085 | ||
| Q78 | 0.075 | 0.113 | ||
| Q83 | 0.039 | |||
| Q80 | 0.077 | 0.071 | ||
| Spin–orbit coupling terms | |
|---|---|
| η S2/T3 | 0.0 + 0.038i |
| η S1/T5 | 0.0 + 0.234i |
| η T3/T2 | 0.0 + 0.036i |
| η T1/T5 | 0.0 + 0.207i |
| η T3/T5 | 0.0 − 0.095i |
The choice of the electronic states has been based on the 350–500 nm energy window, the transition dipole moments and spin–orbit coupling values. At Franck–Condon the four lowest triplet T1–T4 are predominantly ligand-centered LC whereas T5 is mainly metal-to-ligand-charge-transfer MLCT as illustrated by TheoDORE analysis (Fig. S1, ESI†). S1, S2 and S4 have mixed LC/MLCT character while S3, which generates a shoulder at 340 nm on the absorption spectrum (Fig. 1), is purely MLCT (Fig. S1, ESI†). As previously reported,33 the MC excited states are very high in energy in this molecule and need costly out-of-plane distortion to be stabilized.
The Kohn–Sham orbitals contributing to the electronic excitations that generate S1–S4 and T1–T5 excited states are represented in Scheme 2.
 | ||
| Scheme 2 Kohn–Sham frontier orbitals of [Pt(dpybMe)Cl] 1. (Energies are given in eV.) | ||
The investigated complex 1 possesses 96 vibrational normal modes on which 12 A′ tuning modes and 6 A′′ coupling modes have been selected on the basis of significant intra-state κ(n)i/w coupling amplitudes and large inter-state λ(nm)i coupling constants (Table S6, ESI†). The number of primitive basis functions and SPFs for different mode combinations used in MCTDH for this 19 “spin–orbit” electronic states/18 normal modes model Hamiltonian are reported in Table S7 (ESI†).
The 18 selected vibrational normal modes are depicted in Fig. S2 (ESI†) and the corresponding frequencies given in Tables S2 and S6 (ESI†). We can distinguish between (i) the normal modes associated with the metal coordination sphere, namely metal–carbon and metal–nitrogen bond stretching accompanied by negligible in-plane bending of the ligand (Q15, Q49, Q50, Q81 and Q83) and (ii) the ligand localized normal modes associated with the in-plane bending of the pyridyl and benzene rings, via either scissoring or rocking.
Table 1 reports the most important coupling terms associated with the modes represented in Fig. S2 and extracted from Tables S3 and S6, ESI.†
The high frequency Q83 and Q81 normal modes (4.0–5.0 × 1013 Hz or 20–25 fs) correspond to the symmetric Pt–N and Pt–C bond stretching, respectively, accompanied by small ligand in-plane breathing. They generate significant intra-state vibronic coupling in the low-lying LC/MLCT S1, S2 and T1, T2 excited states corresponding to 68a′ → 69a′ and 68a′ → 45a′′ excitations (Scheme 2) and in the nearly pure LC T3 state (44a′′ → 45a′′ and Fig. S1, ESI†). We may anticipate important shifts in position and energy of the potentials associated with these excited states. Interestingly, the potentials of S1, T1 will be shifted in position and in energy under these nuclear perturbations in an opposite direction of S2, T2 and T3 as expected from the signs of the κ(n)i terms reported in Table 1. Consequently, we may expect the occurrence of crossings between S2 and S1 and between S1 and T3 diabatic potentials along the corresponding nuclear coordinates. The electronic transition involving the metal–benzene ring bonding and metal–pyridyl anti-bonding orbital (44a′′) (Scheme 2), namely T3, is particularly affected by the mode Q81 (κT3i = −0.179 eV) which combines Pt–C stretching and ligand scissoring. In addition Q83 contributes to a significant T3/T1 inter-state coupling λT1,T3.
Three other high frequency modes, namely ligand-localized Q80, Q77 and Q78 associated with antisymmetric in-plane bending of the three rings, generate large S1/S2, T1/T2 (Q77, Q78) and T1/T2, T2/T3 (Q80) inter-state vibronic couplings.
One low-frequency normal mode, namely Q15 (8.7 × 1012 Hz or 115 fs), corresponding to asynchronous Pt–C and Pt–N symmetric bond stretching, generates large intra-state vibronic couplings in S1, S2, T1, T2 (κ(n)i > 0) and T3 (κ(n)i < 0) (Table 1). The activity of this normal mode is correlated to the electronic structure of these excited states that involve excitations either from the metal–benzene anti-bonding 68a′ orbital or from the metal–benzene bonding and metal–pyridyl anti-bonding orbital (44a′′) (Scheme 2). The Q15 normal mode is also responsible for a significant T1/T3 inter-state vibronic coupling (Table 1).
One-D cuts of the diabatic potentials associated with S1–S4 and T1–T5 electronic states along selected normal modes inducing significant κ(n)i intra-state couplings are represented in Fig. S3, ESI.† An analysis of the curves related to Q81, Q83 and Q15 normal mode vibrations confirms a number of curve crossings between S1/S2 and T3 nearby Franck–Condon and between T3 and T1/T2 generated by the symmetric Pt–C stretching/ligand scissoring (Q81) mode. This supports the key role of the nearly pure LC electronic state T3 as a “reservoir” of the population. Indeed, the occurrence of curve crossings combined with large values of inter-state couplings controls the excited state decay at an early time. The One-D cut along the low-frequency mode Q15 associated with the asynchronous stretching of the Pt–C and Pt–N bonds exhibits, at distance of the FC region, T3/T1/T2 crossings that should play a role in the final population of the lowest radiative T1 and T2 states.
In addition, the results presented in Table 1 point out a particularly large SOC of the nearly pure MLCT T5 state (67a′ → 69a′ and Fig. S1, ESI†) with S1 and T1, and with T3 to a lesser extent. The manifestation of T5/S2/T3 crossings along the Q83 vibration corresponding to the symmetric N–Pt–N stretching/ligand scissoring (Fig. S3, ESI†) together with a large value of SOC suggests a key role of T5, as well.
From this qualitative analysis we may expect a spin-vibronic mechanism with efficient transfer of population from the absorbing S2 state to the nearly degenerate S1 and T3 states at an early time (20 fs). The upper T5 MLCT state should play a role in the transfer of population to S1 and T1 by SOC. At longer time scale (>100 fs) the low frequency mode, associated with asynchronous Pt–C and Pt–N symmetric bond stretching accompanied by small in-plane bending of the ligand, should participate actively in the population of the nearly degenerate luminescent LC/MLCT T1 and T2 excited states. This analysis highlights again the correlation between the activity of some specific normal modes and the character of the electronic transitions.59
3.2 Spin–vibronic absorption spectrum
The absorption spectrum of [Pt(dpybMe)Cl] generated by non-adiabatic quantum dynamics including 18 vibrational normal modes acting on the four lowest singlet and five lowest triplet electronic states, coupled vibronically and by spin–orbit, is represented in Fig. 2.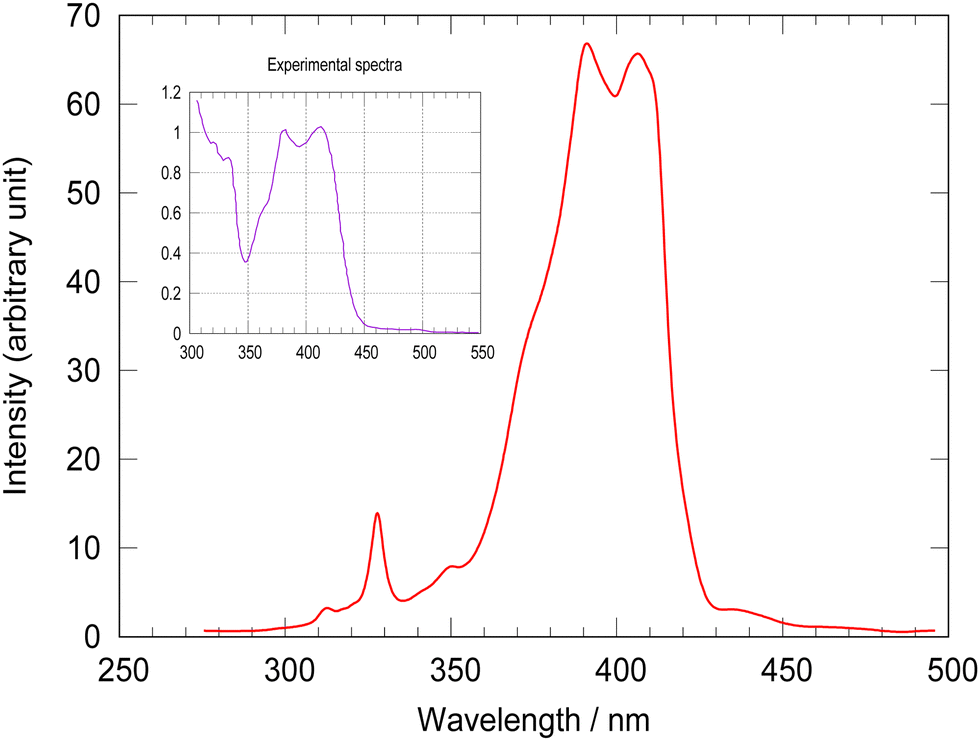 | ||
| Fig. 2 MCTDH calculated absorption spectrum of [Pt(dpybMe)Cl] as compared to the experimental one58 (inset). | ||
The simulated absorption spectrum reproduced perfectly the main features of the experimental one with a double peak maximum centered at 400 nm and a shoulder at 375 nm. The small peak at 330 nm is assigned to the S3 MLCT state. The theoretical spacing between the two peaks is slightly underestimated (by 343 cm−1 when SOC is not included) as compared to the experimental one. The TD-DFT absorption spectrum without SOC, including inter-state vibronic effects and the whole space of normal modes suffers from the same discrepancy (Fig. S4, ESI†). This could be attributed to the shortcoming of the environment treatment and/or to the accuracy of the electronic structure calculations. Neglecting of SOC between the active electronic states included in the LVC Hamiltonian and upper electronic states could also control this spacing.
In order to discover the role of the various couplings on the spin–vibronic structure of the absorption spectrum, four simulations have been performed by switching on/off spin–orbit and/or vibronic couplings and focusing on the S2 mainly absorbing state. The resulting absorption spectra are shown in Fig. S5, ESI.† Switching off SOC, keeping all active modes, leads to one intense peak at 410 nm and a second minor peak at 380 nm. This confirms that the structure of absorption spectrum is fully vibronic. The deactivation of the important normal modes discussed above (Table 1), gives slightly more intense peaks at 400 nm and 380 nm. Tuning off the modes Q62, Q68, Q75 and Q76 associated with the in-plane scissoring and rocking motions of the ligand (Fig. S2, ESI†), that generate large intra-state couplings (Table S6, ESI†), destroys the vibronic structure.
3.3 Ultrafast photophysics
Fig. 3 shows the evolution of the diabatic excited state population as function of time within 500 fs. The initial population of the S2 absorbing state decreases rapidly to populate the S1 state (40%) within less than 20 fs by intra- (Q81, Q83) and inter-state (Q77, Q78) vibronic couplings. Significant S2/T3, T5/T3 and S1/T5 SOC (Tables S3 and S6, ESI†) ensures a nearly instantaneous modest population of T3 (18%) and T5 (10%) after absorption within about 10 fs. In the first tens fs we observe and exchange of population between T5 (<10%) and T3 (about 23%) by SOC. At the same time, an increase of population of the lowest triplet LC/MLCT T1 state at the expense of T5 by SOC, and of T3 by inter-state vibronic coupling (Q83) is noticeable. At 50 fs the population of T1 and T2 reflects the decrease in population of S2, S1 and T3. This is explained by the role of “reservoir” of T3 which pumps population from the upper states (T5, S2, S1) and transfers it to T2 by both SOC and inter-state vibronic coupling (Q80) and to T1 by inter-state vibronic coupling (Q83).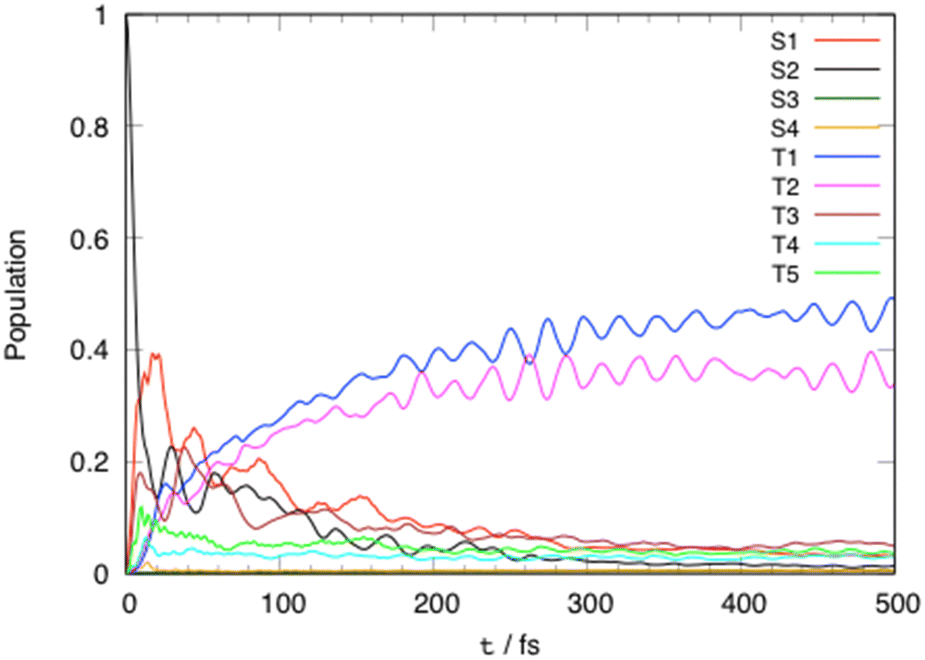 | ||
| Fig. 3 Time evolution of the diabatic excited state population of [Pt(dpybMe)Cl] within 500 fs after visible light irradiation. | ||
At a longer time scale (>50 fs) the population of S2, S1 and T3 decreases slowly from 20% to 10% at 100 fs. The depopulation of the “reservoir” state T3 is stimulated at about 100 fs by the low frequency mode Q15 associated with the asynchronous motion of the Pt–C and Pt–N bonds which generates T1/T3 inter-state coupling and crossings between the T3, T1 and T2 potentials (Fig. S3, ESI†). The population of the two low-lying potentially luminescent triplet states of [Pt(dpybMe)Cl] reaches 40% in 200 fs.
This ultrafast population of the luminescent states prevents the concurrent energetically demanding stabilization of the upper non-radiative MC states driven by out-of-plane distortion.33 This motion activated by low frequency modes should occur at longer time scales not simulated here.
The T1 and T2 populations are equilibrated within 1 ps (Fig. S6, ESI†).
4 Conclusions
The ultrafast non-adiabatic dynamics of [Pt(dpybMe)Cl], the reference molecule for square-planar Pt(II) luminescent complexes with tri-dentate cyclometalated ligands, has been investigated by nuclear wavepacket propagation within the LVC approach including SOC on the basis of electronic structure data calculated at the DFT level with solvent correction. Four singlet and five triplet electronic excited states, namely nineteen “spin–orbit” states, have been included in the simulation together with eighteen vibrational normal modes. The simulation has been validated by the construction of the absorption spectrum which reproduces quantitatively the vibronic structure of the experimental one. It is shown that the in-plane scissoring and rocking of the cyclometalated tridentate ligand are responsible for the vibronic structure characterized by a double peak at the absorption maximum around 400 nm, not reproduced at the TD-DFT level.The ultrafast decay of [Pt(dpybMe)Cl], within less than 1 ps, follows a spin–vibronic mechanism (Scheme 3) governed by the electronic/nuclear dynamics interplay, breaking the standard concept of the cascade model. In the first 20 fs following absorption to the singlet LC/MLCT (S2) state, both the lowest singlet LC/MLCT (S1) state and the upper triplet MLCT (T5) and LC (T3) excited states are significantly populated, vibronically and by SOC, respectively. The Pt–carbon and Pt–nitrogen bond stretching, accompanied by the in-plane scissoring and rocking of the cyclometalated tridentate ligand, activates the efficient exchange of population between the two nearly degenerate singlet LC/MLCT states, S1 and S2. The lowest triplet LC/MLCT state (T1) is populated within a few tens of fs by SOC from the upper MLCT state T5 and vibronically from the triplet LC (T3) state under the influence of the Pt–nitrogen bond stretching and of the in-plane scissoring of the cyclometalated ligand. The antisymmetric in-plane rocking motion of the ligand controls the transfer of population between the two lowest LC/MLCT T1 and T2 states that equilibrate within 1 ps.
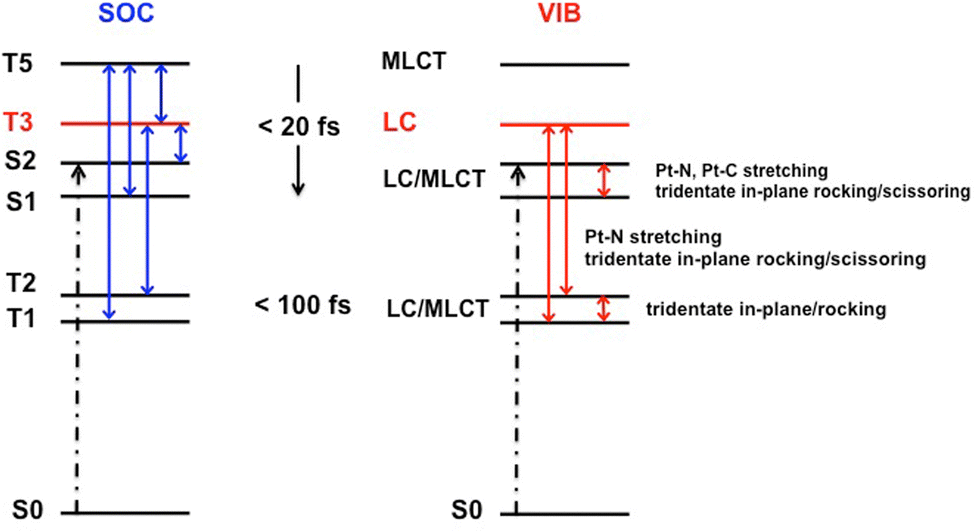 | ||
| Scheme 3 Schematic representation of the spin–vibronic mechanism of ultrafast decay within the low-lying electronic excited state manifold of [Pt(dpybMe)Cl] via the “reservoir” LC (T3) transient state. | ||
The potential stabilization of upper MC excited states, that could quench the radiative decay in this class of complexes,33 activated by low frequency out-of-plane bending modes of the ligand is not competitive with the ultrafast population of the luminescent T1 and T2 states described here.
The role of the triplet LC (T3) “reservoir” state is very important for transferring the population from the upper T5, S2 and S1 states to T1 and T2 within the first 50 fs. At longer time-scale, the depopulation of T3 is stimulated by a low frequency mode associated with the symmetric asynchronous motion of the Pt–carbon and Pt–nitrogen bond stretching.
As previously demonstrated for other coordination compounds59 the correlation between the electronic density characters in play with both the spin–orbit effects and the active normal modes that drive the spin–vibronic mechanism is manifest. This is illustrated here by the large SOC inherent to the upper triplet MLCT state or by the importance of the vibrations of the metal sphere coordination associated with the tuning mode dynamics of the 1,3LC/MLCT states. In contrast, ligand-localized anti-symmetric rocking modes are essential for coupling the LC/MLCT states together and with the pure LC states. This correlation allows for qualitative predictions, extremely useful for engaging a dialog with experimentalists and for limiting the size of the “quantum dynamics box” within the context of hybrid quantum/semi-classical dynamics.60,61 The correlation between the electronic character of the excited states engaged in the photophysical process, the spin–orbit coupling effects and the activity of the molecular normal modes will have dramatic consequences on the ultrafast photophysics of this class of compounds and afterward on their luminescence properties.
The key role of an intermediate 3LC state in ultrafast excited state processes of bi-metallic Pt(II) complexes has been largely discussed recently on the basis of ultrafast spectroscopy experiments by Chen et al.14 It has been shown that the ultrafast metal–metal–ligand–charge transfer 1MMLCT → 3MMLCT intersystem crossing is mediated by a 3LC state on a time scale shorter than a period of the Pt–Pt stretching motion. Similarly, the ultrafast 1LC/MLCT → 3LC/MLCT ISC in [Pt(dpybMe)Cl] is mediated by the nearly pure LC triplet state on a time scale shorter than the periods of the bending motion of the cyclometalated tridentate ligand and of the stretching of the metal coordination sphere.
The rationalization of the ultrafast excited state decay in Pt(dpybMe)Cl], reference molecule for square planar Pt(II) cyclometalated complex, paves the way to the control of the branching ratio between different pathways selectively activated by molecular vibrations that will perturb specifically some electronic densities to produce the desired photophysics and photochemistry, as proposed experimentally by Weinstein et al.21 in large donor–acceptor Pt(II) complexes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
SM acknowledges the Frontier Research in Chemistry Foundation and Labex CSC Chemistry of Complex Systems (Project VRO-CSC-0007) for funding support. The authors thank Etienne Gindensperger for helpful preliminary discussions. The calculations have been performed at the regional HPC, Strasbourg, through a grant of computer time.References
- V. M. Miskowski, V. H. Houlding, C.-M. Che and Y. Wang, Inorg. Chem., 1993, 32, 2518–2524 CrossRef CAS.
- J. A. G. Williams, Top. Curr. Chem., 2007, 281, 205–268 CrossRef CAS.
- J. A. G. Williams, S. Develay, D. L. Rochester and L. Murphy, Coord. Chem. Rev., 2008, 252, 2596–2611 CrossRef.
- J. A. G. Williams, Chem. Soc. Rev., 2009, 38, 1783–1801 RSC.
- L. Murphy and J. A. G. Williams, Top. Organomet. Chem., 2010, 28, 75–111 CrossRef CAS PubMed.
- J. E. Yarnell, I. Davydenko, P. V. Dorovatovskii, V. N. Khrustalev, T. V. Timofeeva, F. N. Castellano, S. R. Marder, C. Risko and S. Barlow, J. Phys. Chem. C, 2018, 122, 13848–13862 CrossRef CAS.
- A. Haque, L. Xu, R. A. Al-Balushi, M. K. Al-Suti, R. Ilmi, Z. Guo, M. S. Kahn, W.-Y. Wong and P. R. Raithby, Chem. Soc. Rev., 2019, 48, 5547–5563 RSC.
- Y. Han, Z. Gao, C. Wang, R. Zhong and F. Wang, Coord. Chem. Rev., 2020, 414, 213300 CrossRef CAS.
- V. W.-W. Yam and A. S.-Y. Law, Coord. Chem. Rev., 2020, 414, 213298 CrossRef CAS.
- M. A. Soto, R. Kandel and M. J. MacLachlan, Eur. J. Inorg. Chem., 2021, 894–906 CrossRef CAS.
- A. Haque, H. El Moll, K. M. Alenezi, M. S. Kahn and W.-Y. Wong, Materials, 2021, 14, 4236 CrossRef CAS PubMed.
- V. W.-W. Yam and Y.-H. Cheng, Bull. Chem. Soc. Jpn., 2022, 95, 846–854 CrossRef CAS.
- R. Monni, G. Auböck, D. Kinschel, K. M. Azi-Lange, H. B. Gray, A. Vlček and M. Chergui, Chem. Phys. Lett., 2017, 683, 112–120 CrossRef CAS.
- P. Kim, A. J. S. Valentine, S. Roy, A. W. Mills, F. N. Castellano, X. Li and L. X. Chen, Faraday Discuss., 2022, 237, 259 RSC.
- A. J. S. Valentine, J. J. Radler, P. Kim, F. N. Castellano and L. X. Chen, J. Chem. Phys., 2019, 151, 114303 CrossRef PubMed.
- E. O. Danilov, A. A. Rachford, S. Goeb and F. N. Castellano, J. Phys. Chem. A, 2009, 113, 5763–5768 CrossRef CAS PubMed.
- P. Kim, M. S. Kelley, A. Chakraborty, N. L. Wong, R. P. Van Duyne, G. C. Schatz, F. N. Castellano and L. X. Chen, J. Phys. Chem. C, 2018, 122, 14195–14204 CrossRef CAS.
- T. W. Kim, P. Kim, A. W. Mills, A. Chakraborty, S. Kromer, A. J. S. Valentine, F. N. Castellano, X. Li and L. X. Chen, J. Phys. Chem. C, 2022, 126, 11487–11497 CrossRef CAS.
- P. Kim, A. J. S. Valentine, S. Roy, A. W. Mills, A. Chakraborty, F. N. Castellano, X. Li and L. X. Chen, J. Phys. Chem. Lett., 2021, 12, 6794–6803 CrossRef CAS PubMed.
- G. A. Farrow, M. Quick, S. A. Kovalenko, G. Wu, A. Sadler, D. Chekulaev, A. A. P. Chauvet, J. Weinstein and N. P. Ernsting, Phys. Chem. Chem. Phys., 2021, 23, 21652 RSC.
- M. Delor, I. V. Sazanovich, M. Towrie and J. A. Weinstein, Acc. Chem. Res., 2015, 48, 1131–1139 CrossRef CAS PubMed.
- S. Archer and J. A. Weinstein, Coord. Chem. Rev., 2012, 256, 2530–2561 CrossRef CAS.
- M. Delor, T. Keane, P. A. Scattergood, I. V. Sazanovich, G. M. Greetham, M. Towrie, A. J. H. M. Meijer and J. A. Weinstein, Nat. Chem., 2015, 7, 689–695 CrossRef CAS PubMed.
- M. Delor, S. A. Archer, T. Keane, A. J. H. M. Meijer, I. V. Sazanovich, G. M. Greetham, M. Towrie and J. A. Weinstein, Nat. Chem., 2017, 9, 1099–1104 CrossRef CAS PubMed.
- Y. Kataoka, Y. Kitagawa, T. Kawakami and M. Okumura, J. Organomet. Chem., 2013, 743, 163–169 CrossRef CAS.
- X.-N. Li, Z.-J. Wu, L. Zhou, X.-J. Liu and H.-J. Zhang, Org. Electron., 2011, 12, 51–61 CrossRef CAS.
- S. Y.-L. Leung, E. S.-H. Lam, W. H. Lam, K. M.-C. Wong, W.-T. Wong and V. W.-W. Yam, Chem. – Eur. J., 2013, 19, 10360–10369 CrossRef CAS PubMed.
- H. Guo, S. Ji, W. Wu, W. Wu, J. Shao and J. Zhao, Analyst, 2010, 135, 2832–2840 RSC.
- H.-M. Wen, Y.-H. Wu, L.-J. Xu, L.-Y. Zhang, C.-N. Chen and Z.-N. Chen, Dalton Trans., 2011, 40, 6929–6938 RSC.
- Y. Zhang, J. A. Garg, C. Michelin, T. fox, O. Blacque and K. Venkatesan, Inorg. Chem., 2011, 50, 1220–1228 CrossRef CAS PubMed.
- W. H. Lam, E. S.-H. Lam and V. W.-W. Yam, J. Am. Chem. Soc., 2013, 135, 15135–15143 CrossRef CAS PubMed.
- D. Escudero and D. Jacquemin, Dalton Trans., 2015, 44, 8346–8355 RSC.
- C. Gourlaouen and C. Daniel, Dalton Trans., 2014, 43, 17806–17819 RSC.
- D. Escudero and W. Thiel, Inorg. Chem., 2014, 53, 11015–11019 CrossRef CAS PubMed.
- P. Kumar and D. Escudero, Inorg. Chem., 2021, 60, 17230–17240 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- E. van Lenthe and E. J. Baerends, J. Comp. Chem., 2003, 24, 1142–1156 CrossRef CAS PubMed.
- E. van Lenthe, R. van Leeuwen, E. J. Baerends and J. G. Snijders, Int. J. Quantum Chem., 1996, 57, 281–293 CrossRef CAS.
- E. Runge and E. K. U. Gross, Phys. Rev. Lett., 1984, 52, 997 CrossRef CAS.
- M. Petersilka, U. J. Gossmann and E. K. U. Gross, Phys. Rev. Lett., 1996, 76, 1212 CrossRef CAS PubMed.
- A. Klamt and G. Schüürmann, J. Chem. Soc.: Perkin Trans., 1993, 2, 799–805 Search PubMed.
- A. Klamt, J. Phys. Chem., 1995, 99, 2224–2235 CrossRef CAS.
- A. Klamt and V. Jones, J. Chem. Phys., 1996, 105, 9972–9981 CrossRef CAS.
- A. Rosa, E. J. Baerends, S. J. A. van Gisbergen, E. van Lenthe, J. A. Groeneveld and J. G. Snijders, J. Am. Chem. Soc., 1999, 121, 10356–10365 CrossRef CAS.
- C. C. Pye and T. Ziegler, Theor. Chem. Acc., 1999, 101, 396–408 Search PubMed.
- ADF, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands, 2019, https://www.scm.com.
- F. Plasser, J. Chem. Phys., 2020, 152, 084108 CrossRef CAS PubMed.
- Multidimensional Quantum Dynamics: MCTDH Theory and Applications, ed. H.-D. Meyer, F. Gatti, and G. A. Worth, Wiley-VCH, Weinheim, 2009 Search PubMed.
- U. Manthe, H.-D. Meyer and L. S. Cederbaum, J. Chem. Phys., 1992, 97, 9062–9071 CrossRef CAS.
- H.-D. Meyer, G. A. Worth, M. H. Beck, A. Jackle, U. Manthe, M. Ehara, A. Raab, M.-C. Heitz, S. Sukiasyan, C. Cattarius, S. Wefing, F. Gatti, M. Nest, F. Otto, M. R. Brill, O. Vendrell, M. Schröder and D. Pelaez, The MCTDH Package, Version 8.4, ( 2007), see http://mctdh.uni-hd.de/ Search PubMed.
- M. Dierksen and S. Grimme, J. Chem. Phys., 2004, 120, 3544–3554 CrossRef CAS PubMed; F. J. A. Ferrer, J. Cerezo, J. Soto, R. Improta and F. Santoro, Comput. Theor. Chem., 2014, 1040–1041, 328–337 CrossRef.
- R. Crespo-Otero and M. Barbatti, Theor. Chem. Acc., 2012, 131, 1237 Search PubMed; Š. Sršeň, J. Sita, P. Slavíček, V. Ladányi and D. Heger, J. Chem. Theory Comput., 2020, 16, 6428–6438 CrossRef PubMed; L. Cerdán and D. Roca-Sanjuán, J. Chem. Theory Comput., 2022, 18, 3052–3064 CrossRef PubMed.
- H. Köppel, W. Domcke and L. S. Cederbaum, Adv. Chem. Phys., 1984, 57, 59–246 Search PubMed.
- T. J. Penfold, E. Gindensperger, C. Daniel and C. M. Marian, Chem. Rev., 2018, 118, 6975–7625 CrossRef CAS PubMed.
- F. Plasser, M. Ruckenbauer, S. Mai, M. Oppel, P. Marquetand and L. Gonzalez, J. Chem. Theory Comput., 2016, 12, 1207–1219 CrossRef CAS PubMed.
- M. Fumanal, F. Plasser, S. Mai, C. Daniel and E. Gindensperger, J. Chem. Phys., 2018, 148, 124119 CrossRef PubMed.
- J. A. G. Williams, A. Beeby, E. S. Davies, J. A. Weinstein and C. Wilson, Inorg. Chem., 2003, 42, 8609–8611 CrossRef CAS PubMed.
- C. Daniel, Phys. Chem. Chem. Phys., 2021, 23, 43–58 RSC.
- F. Plasser, S. Mai, M. Fumanal, E. Gindensperger, C. Daniel and L. Gonzalez, J. Chem. Theory Comput., 2019, 15, 5031–5045 CrossRef CAS PubMed.
- S. Mai and L. Gonzalez, J. Chem. Phys., 2019, 151, 244115 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp01890c |
| This journal is © the Owner Societies 2023 |