Hydrogen-bonding and resonance stabilisation effects in cationic bis(iminium) phenoxide diacids†
Received
24th April 2023
, Accepted 18th May 2023
First published on 30th May 2023
Abstract
The synthesis and characterisation of a bis(iminium)phenoxide diacid cation [4-tBu-C6H2-2,6-(HC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N(H)Dipp)-1-O]+ ([H2tBu,DippL]+), is discussed. [H2tBu,DippL][BF4] (1) and [H2tBu,DippL][H2N{B(C6F5)3}2] (2) were synthesised in high yields via protonation of the bis(imino)phenol conjugate base with ethereal HBF4 or Bochmann's acid ([H(OEt2)2][H2N{B(C6F5)3}2]). Both species were fully characterised using NMR and IR spectroscopy as well as X-ray crystallography. The cationic fragment adopts an unusual tautomeric form in which both acidic protons are located on the nitrogen atoms: [HN〈O〉NH]+. This bis(iminium) phenoxide tautomer is stabilised by delocalisation of electron density from oxygen, into the extended π-system of the planar cation, and was found to be 22.6 and 263.1 kJ mol−1 lower in energy (ΔG) than the alternative [N〈OH〉NH]+ and [N〈OH2〉N]+ tautomers respectively. Topological analysis confirmed the presence of two electrostatic N+H⋯O− hydrogen bonds which contribute −111.2 kJ mol−1 towards the stabilisation of the diacid. The pKa values of the cations were estimated, from NMR experiments, to be 4.2 in THF (1) and 11.4 in acetonitrile (2).
N(H)Dipp)-1-O]+ ([H2tBu,DippL]+), is discussed. [H2tBu,DippL][BF4] (1) and [H2tBu,DippL][H2N{B(C6F5)3}2] (2) were synthesised in high yields via protonation of the bis(imino)phenol conjugate base with ethereal HBF4 or Bochmann's acid ([H(OEt2)2][H2N{B(C6F5)3}2]). Both species were fully characterised using NMR and IR spectroscopy as well as X-ray crystallography. The cationic fragment adopts an unusual tautomeric form in which both acidic protons are located on the nitrogen atoms: [HN〈O〉NH]+. This bis(iminium) phenoxide tautomer is stabilised by delocalisation of electron density from oxygen, into the extended π-system of the planar cation, and was found to be 22.6 and 263.1 kJ mol−1 lower in energy (ΔG) than the alternative [N〈OH〉NH]+ and [N〈OH2〉N]+ tautomers respectively. Topological analysis confirmed the presence of two electrostatic N+H⋯O− hydrogen bonds which contribute −111.2 kJ mol−1 towards the stabilisation of the diacid. The pKa values of the cations were estimated, from NMR experiments, to be 4.2 in THF (1) and 11.4 in acetonitrile (2).
Introduction
The first examples of bis(iminium) phenoxide diacids were reported by Brzezinski and co-workers in 1998.1,2 The salts were prepared via mixing of equimolar amounts of the bis(imino) phenol conjugate base and HClO4, and studied spectroscopically.1,2 The presence of weak absorbance continua (3000–400 cm−1) in the IR spectra led the authors to propose the occurrence of fast, collective, proton tunnelling between the electronegative atoms, in what must be an intramolecularly hydrogen-bonded system.1 More recent reports of bis(imininium) phenoxide salts involve the isolation of similar monomeric species and macrocyclic analogues.3–11 The monomeric NON diacids were typically produced upon treatment of the conjugate base with excess metal chloride reagent (MClx; M = Fe,3 Ga,4 Mn5) or as the product of sodium oxalate addition to a solution of dinickel catalyst.6 Contrastingly, the macrocyclic variants (i.e. two NON fragments joined together by aliphatic bridges) were synthesised using one-pot, acid-catalysed condensations between phenol- and diamino-derivatives in the presence of NaClO4.9–11 In all cases the authors use X-ray crystallographic analysis to identify, within the cationic fragment, deprotonated phenoxide group(s) alongside freely refinable protonated imines (–HCNH–).3–11 The localisation of these acidic protons in solution was barely discussed and the possibility of proton shuttling, as proposed by Brzezinski, has not been investigated.1 Chakraborty et al. reported that the protonation of the imine groups results in elongation of the CN bonds;4 this was attributed to the electrostatic/van der Waals attraction between the ion pairs.3,4 Sun and co-workers used density functional theory (DFT) studies to probe the strength of these anticipated intermolecular forces.3 By varying the position of the anion within the ion pair, the authors were able to calculate the interaction energy to lie within the range 36–54 kcal mol−1.3 No investigations or direct evidence of these stabilising forces acting intramolecularly has been performed, even though most reports assume that N+H⋯O− hydrogen-bonding is present in the cations of these systems.3,7–11 Hence, herein we report the synthesis of two new bis(iminium) phenoxide diacids, alongside a more in-depth study, that utilises advanced spectroscopy, isotope labelling and DFT, to investigate the inherent structure and possible N+H⋯O− bonding/proton shuttling in this class of compound.
Results and discussion
Diacid synthesis; [H2tBu,DippL][A]
Doubly protonated, bis(iminium) phenoxide salts were prepared via three-step syntheses in overall yields of 71% (1) and 77% (2) (Scheme 1). The conjugate base [HtBu,DippL] was prepared according to modified literature procedures via diformylation of 4-tert-butyl phenol with HMTA12–14 followed by condensation with two equivalents of diisopropyl aniline (DippNH2).15 Subsequent protonation with either ethereal HBF4 or Bochmann's acid ([H(OEt2)2][H2N{B(C6F5)3}2])16 yielded the corresponding NON diacids: [4-tBu-C6H2-2,6-(HCN(H)Dipp)-1-O)][A] ([HN 〈O〉 NH][A]; A = BF4− (1) or [H2N{B(C6F5)3}2]− (2)) as air-sensitive orange powders (See ESI† for details).
 |
| | Scheme 1 Three-step syntheses of bis(iminium)phenoxide salts [H2tBu,DippL][BF4] (1) and [H2tBu,DippL][H2N{B(C6F5)3}2] (2). Dipp = 2,6-iPr2-C6H3. | |
Spectroscopic analysis
The 1H NMR spectrum of 1 in THF-d8 is indicative of a C2v symmetric cation on the NMR timescale (see ESI†). A highly deshielded doublet at δ 15 ppm, with a coupling constant of 13.5 Hz, represents the two acidic protons. The four aromatic resonances (δ 7.3–9.1 ppm) correspond to the imine, central benzene ring and N-aryl CH protons. The isopropyl methine- and methyl-protons are represented by the multiplets at δ 3.4 and δ 1.3 ppm respectively, and the nine tert-butyl protons by a singlet at δ 1.4 ppm. Similar spectral features were published by Dutta et al. in their spectroscopic study of macrocyclic tetraiminodiphenol perchlorate salts.11 Variable temperature 1H NMR studies of 1 confirm retention of the observed C2v symmetry even at temperatures as low as 223 K (see ESI†). This suggests that the signal at δ 15 ppm is not the coalescent of two distinct NH, OH resonances therefore implying that the acidic protons must be chemically equivalent. The retention of solution symmetry also indicates that possible fluxional interactions between hydroxyl and imine groups, as seen in the unsymmetrical 1H NMR spectra of the conjugate base at low temperatures (see ESI†), are not occurring. This suggests that either both imine arms are fixed in space away from the oxygen centre, or that there is no hydroxyl group present at all. IR spectroscopic analysis of 1 supports the latter as no absorbances were recorded in the typical OH stretching region (3200–3700 cm−1; Fig. 1).17 NH stretching modes, however, were observed, implying that the cation instead contains two localised acidic iminium protons. The signal corresponding to the NH stretches (3060 cm−1) appears at lower-than-expected wavenumbers; this red-shift is typically caused by the presence of NH⋯O hydrogen bonds, thus indicating the presence of similar electrostatic attractions within these systems.1–3,7–11,18–20 Assignment of the NH absorption was further confirmed by partial-deuteration of 1. After stirring a sample in either methanol-d1 or ethanol-d1, the absorbance around 3100 cm−1 was supressed and a new signal, corresponding to ND stretches, appeared around 2360 cm−1 (Fig. 1). This value aligns with other reported ND signals and the predicted vibrational frequency (2400 cm−1) when using the harmonic oscillator approximation.21–24 Complete NH deuteration was also confirmed by 1H NMR spectroscopy (see ESI†). In the spectrum of the deuterated species, the highly deshielded doublet, at δ 15 ppm, is suppressed and its mutually coupling doublet at δ 9 ppm, corresponding to the imine protons, is converted into a broad singlet. The linewidth of this peak exceeds the expected 3JHD coupling and therefore the assumed 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 triplet remains unresolved. Compound 2 is spectroscopically similar to 1 aside from slight variations in chemical shift. An additional broad singlet at δ 5.8 ppm in the 1H NMR spectrum and new sharp IR stretching modes at 3300 cm−1 are also present due to the NH2 environment of the aminodiboronate anion (see ESI†).16
:1:1 triplet remains unresolved. Compound 2 is spectroscopically similar to 1 aside from slight variations in chemical shift. An additional broad singlet at δ 5.8 ppm in the 1H NMR spectrum and new sharp IR stretching modes at 3300 cm−1 are also present due to the NH2 environment of the aminodiboronate anion (see ESI†).16
 |
| | Fig. 1 IR spectra of 1 (blue) and its deuterated analogue: [D2tBu,DippL][BF4] (red). The corresponding NH and ND signals are highlighted. | |
X-ray crystallographic structural analysis
Single crystals of 1 were grown at room temperature from a saturated benzene solution and solved in the P21 space group. Compound 2 on the other hand, crystallised in the triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group, from a −25 °C dichloromethane/n-pentane mixture. In the solid-state, both compounds exist as charge-separated ion pairs with near-identical cationic fragments. The asymmetric unit of 1 contains two crystallographically independent, yet essentially-identical cations, alongside their corresponding anions and four solvent molecules. The following discussion therefore relates solely to the O1 containing cation (Fig. 2; see ESI† for experimental metrical parameters of the alternate cation). Like the previous reports of monomeric bis(iminum) phenoxide salts, the solid-state structures of the cations in 1/2 adopt a C2v symmetric “closed-conformation” in which both imine groups are co-planar with the central π-ring and orientated towards the oxygen centre.3–6 This symmetric form aligns with the solution 1H NMR data for this compound, but contrasts to the X-ray crystal structure of the bis(imino)phenol conjugate base, as well as that of other NON-containing ligands and metal complexes. In these systems, one imine is permanently rotated away from the oxygen to relieve steric strain and enable stabilising imine-CH⋯O interactions.15,25–28 Adoption of the unique conformation observed in the cationic conjugate diacid therefore implies that the typical steric constraint of the system is outweighed by alternative stabilising interactions. One such stabilising force is the enhanced delocalisation of electron density from the oxygen atom into the central π-ring and pendent iminium groups. This is confirmed by analysis of the experimental metrical parameters of the diacid cation (Table 1). Protonation of the conjugate base leads to a reduction in the C(1)–O(1) bond length.10 In the neutral species, this bond measures 1.37 Å whereas in the doubly protonated systems this bond (1.28 Å) lies much closer to the value, reported by Pyykkö, of a CO double bond (1.24 Å).29 This reflects an increase in bond order and thus an increase in transfer of electron density between the two atoms. An analogous shortening is also observed for the C–Cimine bonds (1.47 vs. 1.42 Å). Conversely, a slight elongation of the imine CN bonds is observed (1.27 vs. 1.29 Å); this agrees with previous reports of bis(iminium) cations, and highlights a partial decrease in bond order caused by the reduction of electron density on the nitrogen atoms.3,4,6,8 This increased delocalisation implies that this “closed-conformation” system is able to access a greater number of, phenoxide-like, resonance forms (Fig. 3). These are unachievable in the presence of a hydroxyl group thus reaffirming that, in both solution and solid-state, the cations do in fact only contain acidic NH protons. The bis(iminium) tautomer of the cation, [HN 〈O〉 NH]+, was further supported during structural analysis as the acidic protons were freely refined as being localised on the nitrogen atoms. Further stabilisation of this conformer may be due to the presence of two electrostatic hydrogen bonds.3,7–11 Previous reports detail that these may either be asymmetric (N+H⋯OH⋯N) or symmetric (N+H⋯O−⋯HN+);9–11 the lack of a phenoxy group indicated by spectroscopic and crystallographic studies, therefore means that if present, these hydrogen bonds must take the symmetric form. These attractions are potentially evidenced by the NH⋯O distance (av. 1.84 Å for 1 and 1.94 Å for 2) being longer than the NH bond lengths (av. 0.91 Å for 1 and 0.86 Å for 2) yet considerably shorter than the sum of the van der Waals radii (2.72 Å).26,30,31 In their publication on neutral NON-nickel catalyst protonations, Mecking and co-workers detail analogous N-bonded acidic protons alongside similar structural changes between the neutral and cationic species.26 This suggests that the stabilising effects of increased electron delocalisation, and possible hydrogen bonds, as seen in 1/2 are also in effect in their systems.
space group, from a −25 °C dichloromethane/n-pentane mixture. In the solid-state, both compounds exist as charge-separated ion pairs with near-identical cationic fragments. The asymmetric unit of 1 contains two crystallographically independent, yet essentially-identical cations, alongside their corresponding anions and four solvent molecules. The following discussion therefore relates solely to the O1 containing cation (Fig. 2; see ESI† for experimental metrical parameters of the alternate cation). Like the previous reports of monomeric bis(iminum) phenoxide salts, the solid-state structures of the cations in 1/2 adopt a C2v symmetric “closed-conformation” in which both imine groups are co-planar with the central π-ring and orientated towards the oxygen centre.3–6 This symmetric form aligns with the solution 1H NMR data for this compound, but contrasts to the X-ray crystal structure of the bis(imino)phenol conjugate base, as well as that of other NON-containing ligands and metal complexes. In these systems, one imine is permanently rotated away from the oxygen to relieve steric strain and enable stabilising imine-CH⋯O interactions.15,25–28 Adoption of the unique conformation observed in the cationic conjugate diacid therefore implies that the typical steric constraint of the system is outweighed by alternative stabilising interactions. One such stabilising force is the enhanced delocalisation of electron density from the oxygen atom into the central π-ring and pendent iminium groups. This is confirmed by analysis of the experimental metrical parameters of the diacid cation (Table 1). Protonation of the conjugate base leads to a reduction in the C(1)–O(1) bond length.10 In the neutral species, this bond measures 1.37 Å whereas in the doubly protonated systems this bond (1.28 Å) lies much closer to the value, reported by Pyykkö, of a CO double bond (1.24 Å).29 This reflects an increase in bond order and thus an increase in transfer of electron density between the two atoms. An analogous shortening is also observed for the C–Cimine bonds (1.47 vs. 1.42 Å). Conversely, a slight elongation of the imine CN bonds is observed (1.27 vs. 1.29 Å); this agrees with previous reports of bis(iminium) cations, and highlights a partial decrease in bond order caused by the reduction of electron density on the nitrogen atoms.3,4,6,8 This increased delocalisation implies that this “closed-conformation” system is able to access a greater number of, phenoxide-like, resonance forms (Fig. 3). These are unachievable in the presence of a hydroxyl group thus reaffirming that, in both solution and solid-state, the cations do in fact only contain acidic NH protons. The bis(iminium) tautomer of the cation, [HN 〈O〉 NH]+, was further supported during structural analysis as the acidic protons were freely refined as being localised on the nitrogen atoms. Further stabilisation of this conformer may be due to the presence of two electrostatic hydrogen bonds.3,7–11 Previous reports detail that these may either be asymmetric (N+H⋯OH⋯N) or symmetric (N+H⋯O−⋯HN+);9–11 the lack of a phenoxy group indicated by spectroscopic and crystallographic studies, therefore means that if present, these hydrogen bonds must take the symmetric form. These attractions are potentially evidenced by the NH⋯O distance (av. 1.84 Å for 1 and 1.94 Å for 2) being longer than the NH bond lengths (av. 0.91 Å for 1 and 0.86 Å for 2) yet considerably shorter than the sum of the van der Waals radii (2.72 Å).26,30,31 In their publication on neutral NON-nickel catalyst protonations, Mecking and co-workers detail analogous N-bonded acidic protons alongside similar structural changes between the neutral and cationic species.26 This suggests that the stabilising effects of increased electron delocalisation, and possible hydrogen bonds, as seen in 1/2 are also in effect in their systems.
 |
| | Fig. 2 Thermal displacement ellipsoid drawings (30% probability) of the cationic fragment of 1. All hydrogen atoms apart from those on N1, N2, C7 and C20 have been omitted for clarity. Only one of two independent molecules in the asymmetric unit is shown for clarity. | |
Table 1 Experimental metrical parameters for HtBu,DippL, 1 and 2 (bond lengths reported in Å, esds are in parentheses)
|
|
HtBu,DippLa |
1
|
2
|
|
X-ray crystal structure of conjugate base reported by Sun and co-workers.15
|
| C(1)–O(1) |
1.3477(18) |
1.284(3) |
1.278(3) |
| C(1)–C(2) |
1.408(2) |
1.434(3) |
1.443(3) |
| C(1)–C(6) |
1.398(2) |
1.435(3) |
1.438(3) |
| C(2)–C(7) |
1.457(2) |
1.424(3) |
1.419(3) |
| C(6)–C(20) |
1.470(2) |
1.429(3) |
1.424(3) |
| N(1)–C(7) |
1.269(2) |
1.295(4) |
1.291(3) |
| N(2)–C(20) |
1.266(2) |
1.298(3) |
1.297(3) |
| N(1)–H(1) |
— |
0.86(5) |
0.86(7) |
| N(2)–H(2) |
— |
0.95(4) |
0.85(6) |
 |
| | Fig. 3 The accessible resonance forms of the bis-iminium phenoxy-imine cation. | |
Density functional theory (DFT) calculations
To further understand the structure and inherent nature of the diacids, density functional theory (DFT) calculations were conducted. The optimised cationic fragment is of C2v symmetry and in good agreement with the experimental crystal structures. Calculated NH distances of 1.03 Å are consistent with the assigned bis(iminium)phenolate tautomer. The computed molecular orbital maps of 1 indicate that the proposed resonance-stabilised “closed” arrangement of the cation does indeed facilitate a high degree of π-electron delocalisation (Fig. 4). The calculated HOMO was found to be delocalised over the entire π-system of the cation, including a significant contribution from the oxygen centre. The LUMO is represented by π* orbitals delocalised over the central aromatic ring and both iminium groups. By contrast, the HOMO of the conjugate base is localised on a DippNC unit, with negligible involvement of the oxygen atom. This strong redistribution of electron density is also evidenced in the computed delocalisation indices (δ(A,B); Fig. 5).32 Upon formation of 1, the changes to these direct electron sharing indicators reflect the proposed resonance forms (Fig. 3) of the bis(iminium) cation. More quantitatively, the values of δ(C,O) and δ(C,N) are increased and decreased (on average) by 0.24 respectively. In this way, the delocalised electrons become more evenly distributed across the conjugated system which can only be the result of enhanced resonance.
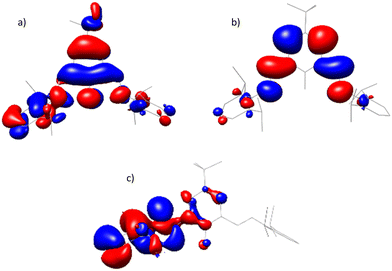 |
| | Fig. 4 DFT-computed (B3LYP) molecular orbitals: (a) and (b) HOMO and LUMO of [H2tBu,DippL]+ – the cation of diacids 1 and 2 and (c) HOMO of mono-acid – H2tBu,DippL. | |
 |
| | Fig. 5 Computed delocalisation indices – δ(A,B) – for HtBu,DippL (left) and 1 (right); reported in red are the values that increase upon formation of 1, whereas those reported in blue are seen to decrease. | |
Geometry optimisations of alternative cation tautomers, in which the two acidic protons are either localised one on oxygen and one on nitrogen ([N〈OH〉NH]+) or both on oxygen ([N〈OH2〉N]+), were then performed. These species converged at energy (ΔG) minima 22.6 kJ mol−1 and 263.1 kJ mol−1 higher relative to the observed structure (Fig. 6; see ESI† for more information regarding computational details). In fact, the [N〈OH2〉N]+ isomer only successfully converged to the desired species when the unprotonated imine arm(s) were rotated away from the phenolic oxygen. When the dihedral angles between the imines and central benzene ring were fixed at 180°, the molecule directly optimised to the [HN〈O〉NH]+ tautomer. These results imply that it is unfavourable for even a single acidic proton to be located on the oxygen atom therefore confirming the bis(iminium) system as the thermodynamically favoured conformation. This agrees with experimental observations that never detected the presence of a hydroxyl group (both in solution or solid-state). As a result, it seems unlikely that the proton shuttling, described by Brzezinski and co-workers, is taking place.1,2
 |
| | Fig. 6 DFT-calculated potential energy diagram displaying the energy difference between the possible cationic tautomers: [HN〈O〉NH]+, [N〈OH〉NH]+ and [N〈OH2〉N]+. | |
Direct evidence for the presence of supposed N+H⋯O− hydrogen bonds was sought through topological analysis. This indicated the presence of (3,−1) bonding critical points (BCPs) between both the N,H and H,O atoms (Fig. 7). In the case of the N,H BCPs, the high electron density (0.325/0.326 a.u.) coincides with negative Laplacian values (∇2ρ(r): −0.186/−0.189 a.u. - see contour plot in ESI†). This indicates concentration of charge at these point and thus represent regions of covalent bonding.33 In contrast, the O,H BCPs have low electron density (0.0367/0.0368 a.u.) and positive Laplacian values (0.120/0.121 a.u.) both of which imply a depletion of charge at these locations.33,34 Thus, these bonding critical points do not represent covalent interactions between atoms but instead indicate weaker intramolecular electrostatic attractions i.e. hydrogen bonds.33–35 These results corroborate the presence of the N+H⋯O− hydrogen bonding interactions postulated in the spectroscopic and structural analysis and again imply that the oxygen atom interacts with the acidic protons in a purely electrostatic fashion. Similar hydrogen bonding critical points were observed for the [N〈OH〉NH]+ tautomer however none were detected for the [N〈OH2〉N]+ system as the geometry only converged when the nitrogen and oxygen atoms were too far apart in space to facilitate such intramolecular electrostatic attractions (see ESI†). The lack of stabilising hydrogen bonding interactions helps to rationalise why the bis(hydroxyl) structure is so much higher in energy than the other two tautomers.
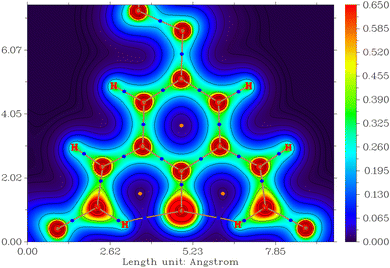 |
| | Fig. 7 Contour map of the electron density of [H2tBu,DippL]+ in its thermodynamically favoured conformation: [HN 〈O〉 NH]+ - plotted in the plane of the central benzene ring. The blue ( ) and orange ( ) and orange ( ) dots mark bonding (3,−1) and ring (3, +1) critical points respectively. ) dots mark bonding (3,−1) and ring (3, +1) critical points respectively. | |
The energy associated with these hydrogen bonds was then estimated using the methods reported by Lu et al. (see ESI† for more details). In 1, the average contribution of each charged N+H⋯O− interaction was −55.6 kJ mol−1 therefore resulting in a total hydrogen bonding stabilisation energy of −111.2 kJ mol−1. In comparison, the stabilisation imparted on the [N〈OH〉NH]+ tautomer, from the neutral N⋯HO and charged N+H⋯O hydrogen bonds, was −112.2 kJ mol−1 (1.0 kJ mol−1 more than in the thermodynamically favoured system). This suggests that the extra stabilisation of the bis(iminium) tautomer must result from the increased resonance; for this to be the case, the number of delocalised electrons in the [HN〈O〉NH]+ system must exceed that of the [N〈OH〉NH]+ conformer. To quantify this, the sum of the delocalisation indices of the possible isomers were calculated and compared to that of HtBu,DippL as a reference (see ESI†); the increase in number of delocalised electrons followed the trend: [HN〈O〉NH]+ (+0.16) > [N〈OH〉NH]+ (+ 0.11) > [N〈OH2〉N]+ (+0.08). These results map those of the geometry optimisations and indicate the greatest impact of resonance is indeed found in the [HN〈O〉NH] system. It can therefore be inferred that the increase in delocalisation is somewhat responsible for the 23.6 kJ mol−1 lowering in energy of the bis(iminium) system (when also accounting for the difference in hydrogen bond stabilisation).
pKa studies
The ΔpKa values of the diacids, relative to the reference base pyrazole, were determined using the NMR techniques reported by Koppel and co-workers.36 The pKa of 1, calculated in THF due to its partial solubility in acetonitrile, was computed to be 4.2 ± 0.008. This is similar to other borate containing salts such as [HPPh3]BPh4 (pKa = 3 in THF).37 Use of the alternative weakly coordinating anion meant that 2 was completely soluble in acetonitrile. Its pKa was therefore calculated in this solvent to be 11.4 ± 0.09. This value is comparable to other nitrogen-based acids such as (C6F5-SO2)2NH (pKa = 11.35 in acetonitrile).38,39 It also indicates that 2 is a much stronger acid than phenol (pKa = 29.14 in acetonitrile);40 this enhanced acidity can be attributed to the improved stabilisation of the conjugate base as a result of the expanded electron delocalisation through the NON-system.
Conclusions
A bis(iminium)phenolate cation [HN〈O〉NH]+ has been synthesised as salts of weakly coordinating anions [HN〈O〉NH][A] ([A] = [BF4]− (1), [H2N{B(C6F5)3}2]− (2)) by protonation of the bis(imino)phenol conjugate base [N〈OH〉N]. Characterisation by NMR and IR spectroscopy, single-crystal X-ray diffraction, and DFT calculations, show that the cation exists as a C2v symmetric tautomer with two iminium groups and a terminal phenoxy-oxygen atom [HN〈O〉NH]+ (acidic protons have pKa = 4.2 (THF), 11.4 (acetonitrile)). Topological analysis confirmed the presence of two N+H⋯O− hydrogen bonds which contribute −111.2 kJ mol−1 towards the stabilisation of the diacid. Similar hydrogen bond stabilisation was observed in the alternate [N〈OH〉NH]+ tautomer and thus the thermodynamic favourability of the bis(iminium) structural motif may be rationalised as an extreme example of textbook resonance stabilisation of a phenoxy group through π-delocalisation. Future studies will investigate the inherent reactivity of these diacids, as well as their use as precursors to cationic metal complexes supported by the NON ligand framework.
Author contributions
R. L. J. carried out the experimental work, contributed to conception and design of experiments, data collection, analysis and interpretation, and drafting/revision of the manuscript. L. J. M. contributed to conception and design of experiments, analysis and interpretation and drafting/critical revision of the manuscript. C. G. C. R. and Z. R. T. contributed to crystallographic data collection, analysis and interpretation. D. O’H contributed to conception and design of experiments, analysis and interpretation, and drafting/critical revision of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
R. L. J., L. J. M., C. G. C. R., and Z. R. T. would like to thank SCG Chemicals Co., Ltd (Thailand) for financial support. Chemical Crystallography (University of Oxford) is thanked for the use of the diffractometers. John E. McGrady is thanked for thoughtful discussions and suggestions.
Notes and references
- B. Brzezinski, Z. Rozwadowski, T. Dziembowska and G. Zundel, J. Mol. Struct., 1998, 440, 73–79 CrossRef CAS.
- Z. Rozwadowski, T. Dziembowska, G. Schroeder and B. Brzezinski, J. Mol. Struct., 1998, 444, 221–225 CrossRef CAS.
- L. Han, J. Du, H. Yang, H. Wang, X. Leng, A. Galstyan, S. Zarić and W.-H. Sun, Inorg. Chem. Commun., 2003, 6, 5–9 CrossRef CAS.
- S. Ghosh, R. R. Gowda, R. Jagan and D. Chakraborty, Dalton Trans., 2015, 44, 10410–10422 RSC.
- W. Yang, K.-Q. Zhao, B.-Q. Wang, C. Redshaw, M. R. J. Elsegood, J.-L. Zhao and T. Yamato, Dalton Trans., 2016, 45, 226–236 RSC.
- A. Patra, S. Chakraborty, S. Lohar, E. Zangrando and P. Chattopadhyay, Inorg. Chim. Acta, 2021, 525, 120493 CrossRef CAS.
- D. Esteban-Gómez, C. Platas-Iglesias, F. Avecilla, A. de Blas and T. Rodríguez-Blas, Eur. J. Inorg. Chem., 2007, 1635–1643 CrossRef.
- X. Wang, K.-Q. Zhao, M. R. J. Elsegood and C. Redshaw, Supramol. Chem., 2018, 30, 404–410 CrossRef CAS.
- S. Majumder, M. Fleck, C. R. Lucas and S. Mohanta, J. Mol. Struct., 2012, 1020, 127–133 CrossRef CAS.
- S. Hazra, S. Majumder, M. Fleck, R. Koner and S. Mohanta, Polyhedron, 2009, 28, 2871–2878 CrossRef CAS.
- B. Dutta, P. Bag, B. Adhikary, U. Flörke and K. Nag, J. Org. Chem., 2004, 69, 5419–5427 CrossRef CAS PubMed.
- L. F. Lindoy, G. V. Meehan and N. Svenstrup, Synthesis, 1998, 1029–1032 CrossRef CAS.
- P. D. Knight, A. J. P. White and C. K. Williams, Inorg. Chem., 2008, 47, 11711–11719 CrossRef CAS PubMed.
-
J. March, Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, Wiley, New York, 3rd edn, 1985 Search PubMed.
- L. Wang, W.-H. Sun, L. Han, Z. Li, Y. Hu, C. He and C. Yan, J. Organomet. Chem., 2002, 650, 59–64 CrossRef CAS.
- S. J. Lancaster, A. Rodriguez, A. Lara-Sanchez, M. D. Hannant, D. A. Walker, D. H. Hughes and M. Bochmann, Organometallics, 2002, 21, 451–453 CrossRef CAS.
-
G. Socrates, Infrared and Raman characteristic group frequencies: tables and charts, John Wiley & Sons, 2004 Search PubMed.
- E. Arunan, G. R. Desiraju, R. A. Klein, J. Sadlej, S. Scheiner, I. Alkorta, D. C. Clary, R. H. Crabtree, J. J. Dannenberg, P. Hobza, H. G. Kjaergaard, A. C. Legon, B. Mennucci and D. J. Nesbitt, Pure Appl. Chem., 2011, 83, 1637–1641 CrossRef CAS.
- P. E. Hansen, M. Vakili, F. S. Kamounah and J. Spanget-Larsen, Molecules, 2021, 26, 7651 CrossRef CAS PubMed.
- V. Vasylyeva, L. Catalano, C. Nervi, R. Gobetto, P. Metrangolo and G. Resnati, CrystEngComm, 2016, 18, 2247–2250 RSC.
- J. T. Burke, J. Chem. Educ., 1997, 74, 1213 CrossRef CAS.
- L. Sobczyk, M. Obrzud and A. Filarowski, Molecules, 2013, 18, 4467–4476 CrossRef CAS PubMed.
- H. H. Limbach, J. Hennig and J. Stulz, J. Chem. Phys., 1983, 78, 5432–5436 CrossRef CAS.
- M. Rozenberg, G. Shoham, I. Reva and R. Fausto, Spectrochim. Acta, Part A, 2004, 60, 2323–2336 CrossRef CAS PubMed.
- L. Qu, T. Roisnel, M. Cordier, D. Yuan, Y. Yao, B. Zhao and E. Kirillov, Inorg. Chem., 2020, 59, 16976–16987 CrossRef CAS PubMed.
- E. Schiebel, M. Voccia, L. Falivene, L. Caporaso and S. Mecking, ACS Catal., 2021, 11, 5358–5368 CrossRef CAS.
- E. Schiebel, M. Voccia, L. Falivene, I. Göttker-Schnetmann, L. Caporaso and S. Mecking, Angew. Chem., Int. Ed., 2021, 60, 18472–18477 CrossRef CAS PubMed.
- H.-C. Chiu, A. Koley, P. L. Dunn, R. J. Hue and I. A. Tonks, Dalton Trans., 2017, 46, 5513–5517 RSC.
- P. Pyykkö, J. Phys. Chem. A, 2015, 119, 2326–2337 CrossRef PubMed.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- R. S. Rowland and R. Taylor, J. Phys. Chem., 1996, 100, 7384–7391 CrossRef CAS.
- J. M. Guevara-Vela, E. Romero-Montalvo, A. Costales, Á. M. Pendás and T. Rocha-Rinza, Phys. Chem. Chem. Phys., 2016, 18, 26383–26390 RSC.
- R. F. W. Bader and H. Essén, J. Chem. Phys., 1984, 80, 1943–1960 CrossRef CAS.
- P. S. V. Kumar, V. Raghavendra and V. Subramanian, J. Chem. Sci., 2016, 128, 1527–1536 CrossRef CAS.
- R. Parthasarathi, V. Subramanian and N. Sathyamurthy, J. Phys. Chem. A, 2006, 110, 3349–3351 CrossRef CAS PubMed.
- T. Rodima, V. Mäemets and I. Koppel, J. Chem. Soc., Perkin Trans. 1, 2000, 2637–2644 RSC.
- K. Abdur-Rashid, T. P. Fong, B. Greaves, D. G. Gusev, J. G. Hinman, S. E. Landau, A. J. Lough and R. H. Morris, J. Am. Chem. Soc., 2000, 122, 9155–9171 CrossRef CAS.
- A. Kütt, S. Tshepelevitsh, J. Saame, M. Lõkov, I. Kaljurand, S. Selberg and I. Leito, Eur. J. Org. Chem., 2021, 1407–1419 CrossRef.
- A. Kütt, I. Leito, I. Kaljurand, L. Sooväli, V. M. Vlasov, L. M. Yagupolskii and I. A. Koppel, J. Org. Chem., 2006, 71, 2829–2838 CrossRef PubMed.
- E. Raamat, K. Kaupmees, G. Ovsjannikov, A. Trummal, A. Kütt, J. Saame, I. Koppel, I. Kaljurand, L. Lipping, T. Rodima, V. Pihl, I. A. Koppel and I. Leito, J. Phys. Org. Chem., 2013, 26, 162–170 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, NMR data, X-ray data and computational details. CCDC 2235648 and 2235649. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3cp01881d |
|
| This journal is © the Owner Societies 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  ,
Louis J.
Morris
,
Louis J.
Morris








 ) and orange (
) and orange ( ) dots mark bonding (3,−1) and ring (3, +1) critical points respectively.
) dots mark bonding (3,−1) and ring (3, +1) critical points respectively.