
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tunable J-type aggregation of silicon phthalocyanines in a surface-anchored metal–organic framework thin film†
Hongye
Chen
a,
Luis
Martín-Gomis
b,
Zhiyun
Xu
a,
Jan C
Fischer
c,
Ian A
Howard
 c,
David
Herrero
b,
Víctor
Sobrino-Bastán
b,
Ángela
Sastre-Santos
*b,
Ritesh
Haldar
*d and
Christof
Wöll
*a
c,
David
Herrero
b,
Víctor
Sobrino-Bastán
b,
Ángela
Sastre-Santos
*b,
Ritesh
Haldar
*d and
Christof
Wöll
*a
aInstitute of Functional Interfaces (IFG), Karlsruhe Institute of Technology (KIT), 76344 Eggenstein-Leopoldshafen, Germany. E-mail: christof.woell@kit.edu
bÁrea de Química Orgánica, Instituto de Bioingeniería, Universidad Miguel Hernández, Avda Universidad S/N, 03202, Elche, Spain. E-mail: asastre@umh.es
cInstitute of Microstructure Technology, Karlsruhe Institute of Technology (KIT), 76344 Eggenstein-Leopoldshafen, Germany
dTata Institute of Fundamental Research Hyderabad, Gopanpally, Hyderabad 500046, India. E-mail: riteshhaldar@tifrh.res.in
First published on 3rd July 2023
Abstract
Organic chromophores and semiconductors, like anthracene, pentacene, perylene, and porphyrin, are prone to aggregation, and their packing in the solid state is often hard to predict and difficult to control. As the condensed phase structures of these chromophores and semiconductors are of crucial importance for their optoelectronic functionality, strategies to control their assembly and provide new structural motifs are important. One such approach uses metal–organic frameworks (MOFs); the organic chromophore is converted into a linker and connected by metal ions or nodes. The spatial arrangement of the organic linkers can be well-defined in a MOF, and hence optoelectronic functions can be adjusted accordingly. We have used such a strategy to assemble a phthalocyanine chromophore and illustrated that the electronic inter-phthalocyanine coupling can be rationally tuned by introducing bulky side grounds to increase steric hindrance. We have designed new phthalocyanine linkers and using a layer-by-layer liquid-phase epitaxy strategy thin films of phthalocyanine-based MOFs have been fabricated and their photophysical properties explored. It was found that increasing the steric hindrance around the phthalocyanine reduced the effect of J-aggregation in the thin film structures.
Introduction
Metal–organic frameworks (MOFs) are crystalline, permanently porous solids.1 MOFs, also known as porous coordination polymers (PCPs), are obtained by linking metal ions or metal-oxo nodes with functionalized organic linkers. Since huge libraries exist for each of these building blocks, the number of metal and organic linker combinations is virtually infinite and many different topologies of MOFs have been reported till date.2 Based on the connectivities and geometries of the constituents, the porosity in MOFs can vary and surface areas as high as ∼7000 m2 g−1 have been reported.3 This provides the basis for many applications; e.g. gas storage, separation,4,5 catalysis6,7 and sensing.8,9 In addition to their high intrinsic porosity, MOFs also offer the possibility to assemble functional organic molecules into structures with predefined packing and spatial geometry. In this context, isoreticular strategies are of pronounced interest.10,11 In the context of photoactive materials, recently in particular MOF-based optoelectronic applications have received substantial attention.12–15The crystalline structure of MOFs allows to precisely determining the positions of the organic linkers and the metal ion nodes. Hence, standard quantum chemistry codes can be applied in a straightforward fashion to interpret experimental findings, a task which is substantially more complicated for amorphous materials. In addition, possible structures can be investigated in silico prior to the experimental realization.16,17 The inter-linker and metal ion distances as well as the overall geometry are crucial parameters to tune the electronic coupling between the MOF building blocks. Properties like electron transfer, energy transfer, charge mobility, and recombination rate can show pronounced variations for even slight variation of structural parameters. Hence MOFs can provide excellent model systems to study electronic and optical properties of chromophoric assemblies. In addition, these materials can be integrated into devices in a straightforward fashion, as has been demonstrated in a number of cases, including photovoltaics, light emitting diodes, and transistor-related applications.18,19 In recent years, the following properties and applications have been explored for MOFs; metallic conductivity,20,21 high charge carrier mobilities,22 charge transfer,23 light emitting diodes,24 photovoltaics,25 efficient and anisotropic energy transfer,26 photodiodes,27 integration of electric fields,28 and thermoelectric effects.29 For many of these applications, the most common form of MOFs, polycrystalline powder, is not well suited since the integration of particulate material into devices represents a major challenge. For numerous types of model systems and devices, the fabrication of MOF thin films deposited on a conducting or transparent surface is required. For this reason, a number of different methods have been developed for MOF thin film deposition. In the present paper, we have employed the surface-anchored MOF (SURMOF) approach to fabricate optically active silicon-phthalocyanine-based MOF thin films exhibiting J-type electronic coupling.
Different organic semiconductors, e.g. anthracene, perylene, pentacene, perylenediimides, naphthalenediimide, porphyrin and phthalocyanine have been assembled into MOF structures, and also as MOF thin films.16,30–37 Phthalocyanines, with their unique macrocyclic structure, exhibit strong π–π interactions and can self-assemble into supramolecular aggregates. However, this type of aggregation is not as common as in other chromophores due to the planar and rigid structure of phthalocyanine molecules, which restricts the formation of the required face-to-face stacking necessary for J-aggregate formation. Different strategies have been designed in order to achieve this kind of aggregation,38 but the number of known cases is still rather limited. Since due to their rather special characteristics phthalocyanine J aggregates are very promising candidates for applications in various optoelectronic devices, including organic solar cells, light-emitting diodes, and photodetectors, more rational strategies are required to assemble these highly interesting macrocycles into ordered arrays.
Here, we focus on silicon phthalocyanines (SiPcs) (see Fig. 1), chromophores which are known to exhibit strong absorption in the visible range (far-red) of the absorption spectrum. Along with an intense visible color, for these SiPc compounds the tendency towards aggregation (and thus to unwanted quenching of excitations by intermolecular coupling) is strongly reduced due to the presence of axial substitution of the Si. Hence, these SiPc molecules have received pronounced interest in the context of biological as well as optical applications.
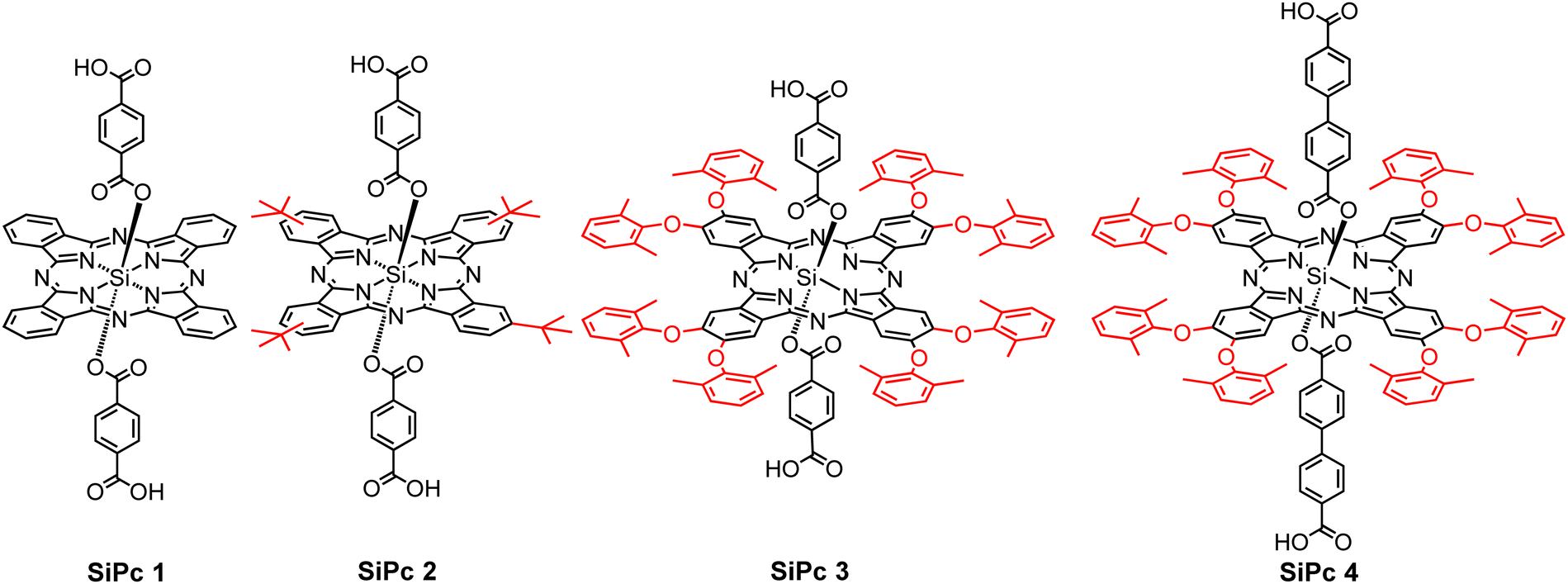 | ||
| Fig. 1 Chemical structure of the four ditopic silicon phthalocyanine linkers. | ||
For the integration of these chromophores into MOFs, the corresponding organic compounds need to be equipped with coupling groups, thus yielding a MOF linker. Hence, we have functionalized the SiPc compounds by attaching –COOH groups at the axial positions. Although at first sight the resulting ditopic linkers appear to be very bulky, we have demonstrated in previous work that MOF thin films can be successfully assembled from the SiPc 1 as linkers, with the resulting SURMOFs showing the formation of an interesting type of polaritons.39 In this contribution, we demonstrate that the optical properties of SiPc-based SURMOFs can be varied in a rational fashion by tuning the intermolecular electronic coupling. We have designed three new sterically modulated silicon phthalocyanine dicarboxylate linkers (SiPc 2–4) and synthesized isostructural MOF thin films using the layer-by-layer liquid-phase epitaxy method. Introduction of the bulky tert-butyl and dimethylphenoxy groups in the SiPc linkers changes the linker–linker distance in the MOF structure, and thus the electronic coupling of the chromophores is modulated (Scheme 1). On the other hand, we have incorporated phenyl (SiPc 3) and biphenyl (SiPc 4) linkers in the axial SiPc position to study the effect of the axial linker in the SURMOF organization. A photophysical characterization of these new SiPc SURMOFs revealed that the SiPc linkers in the MOF structure exhibit J-type coupling. In the followings we have demonstrated how MOF-based design approach can be useful to systemically tune the J-type electronic coupling, which is important for many optoelectronics applications, including photovoltaics, and light emitting diodes.
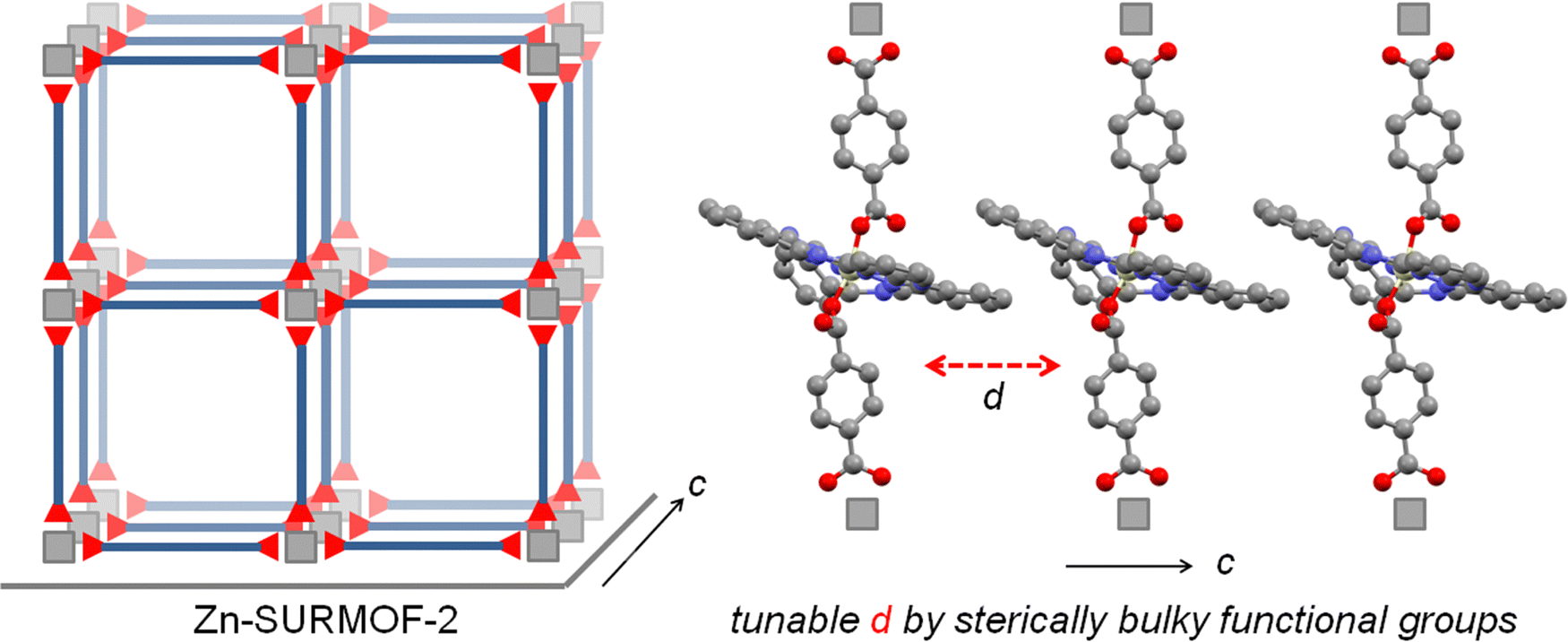 | ||
| Scheme 1 Schematic illustration of the 3D assembly of SiPc linkers as a SURMOF-2 (left) and control of the inter-linker distance by sterically demanding functional group introduction to tune electronic coupling. | ||
Results and discussion
SiPc 2 was directly obtained from (tBu)4SiPc(ArCHO)2540 through a mild oxidation step, employing a mixture of sulfamic acid and sodium chlorite in THF/H2O at RT during 12 h (Scheme 2). On the other hand, SiPc 3 and SiPc 4 were obtained employing the already described mild oxidation protocol (Scheme 3), over both formyl groups of (ArO)8SiPc(ArCHO)2 6 and (ArO)8SiPc(Ar2CHO)2 7, respectively.41 All the new compounds were unambiguously characterized by 1H NMR and UV-vis absorption spectroscopy, and HR MALDI-TOF mass spectrometry (see Experimental section and Fig. S1–S9, ESI†). | ||
| Scheme 2 Synthesis scheme of SiPc 2. | ||
 | ||
| Scheme 3 Synthesis scheme of SiPc 3 and SiPc 4. | ||
The designed SiPc linkers are linear and ditopic. These are thus suited, in principle, to coordinate to Zn-based paddle-wheel type nodes to produce a SURMOF-2 type structure, as demonstrated in our previous works.26,32,42 The SiPc linker-based SURMOF-2 structure was fabricated using a layer-by-layer synthesis (Scheme 1).43 The out-of-plane and in-plane X-ray diffraction (XRD) patterns suggested the presence of a 2D square grid of sheets stacked along the substrate plane ([010] direction) with a regular spacing of 1.1 nm,39 as reported for similar Zn-based porphyrin-based SURMOFs.25 In such a structure, the phthalocyanine aromatic plane is oriented orthogonal to the metal-coordination axis, and two phthalocyanines of the neighboring 2D sheets interact in an edge-to-edge fashion, as illustrated in Scheme 1. The newly synthesized SiPc 2, SiPc 3 and SiPc 4 linkers have bulkier substituents in comparison to the SiPc 1, and these are marked in red in Fig. 1. Due to the presence of the sterically demanding functional groups, we expected that the inter-layer spacing will increase16 for these three new SiPc linkers, leading to a different electronic coupling in the corresponding MOF structures.
The out-of-plane and in-plane XRD patterns of Zn-SiPc 2 are very similar to that obtained from the Zn-SiPc 1, indicating the presence of a similar structure (Fig. 2b and Fig. S10, ESI†). This is expected as the length of the SiPc 2 linker (i.e. distance between the two carboxylic acid groups) is identical to SiPc 1. In contrast, for the Zn-SiPc 3, most likely due to the hindrance of the dimethylphenoxy groups, no crystalline thin film was obtained. For Zn-SiPc 4, the XRD pattern revealed that the square grid unit cell dimension is larger, ∼2.9 nm. This is due to the increased length of the SiPc 4 linker along the metal-coordination axis. A detailed characterization of the SiPc-based SURMOFs using infra-red reflection absorption spectroscopy was fully consistent with the presence of paddle-wheel nodes (Fig. S11, ESI†). The scanning electron microscopy (SEM) images of the Zn-SiPc 2 and Zn-SiPc 4 confirmed the continuous thin film formation (Fig. S12, ESI†).
 | ||
| Fig. 2 (a) A model structure of Zn-SiPc 1, (b) out-of-plane XRD patterns of Zn-SiPc 2 and 4. | ||
After the structural and spectroscopic characterization of the Zn-SiPc SURMOFs, we have carried out a photophysical characterization of the thin films. In Fig. 3, we present the absorption spectra of the three SiPc linkers (SiPc 1, SiPc 2 and SiPc 4) recorded for their solvated state and for the SURMOFs. In the solvated state, the SiPc 1 linker shows a sharp intense peak at 683 nm, corresponding to the Q-band. The Soret band transition appears at ∼350 nm. As a result of the tertiary butyl and phenoxy substitution of the SiPc, the Q-band maxima is shifted to higher wavelengths, 691 and 694 nm, respectively. After assembly into the SURMOF, all the Q-band peaks shift to longer wavelength compared to the solvated linkers. The red shift is largest for Zn-SiPc 1, ∼57 nm. For SiPc 2 and 4, the shifts are smaller and amount to ∼29 and ∼10 nm, respectively. Evidently, the different packing of the chromophores resulting from the coordination to the metal and to yield the SURMOF-2 structure enforces an edge-to-edge SiPc stacking and yields a change in the Q-band transitions. The transition dipoles governing the corresponding excitation are aligned in a head-to-tail fashion and the lowering of the electronic transition energy indicates a J-type electronic coupling between the SiPc linkers of neighboring 2D layers. The solvatochromic shift,44i.e. the difference of the Q-band maximum observed for the solvated state and the SURMOF state, can be considered as a measure of the strength of the J-type electronic coupling. As in Zn-SiPc 1 the linkers of the neighboring 2D layers are closer in space than in the cases of Zn-SiPc 2 and 4, the electronic coupling is strongest, and hence the solvatochromic shift is largest. For Zn-SiPc 3, because of the very bulky substitution on SiPc, the 2D layers are not close enough to induce strong electronic coupling. Hence, using this sterically bulky substitution approach, it is possible to fine tune electronic coupling. Note, that while inducing such changes in the chromophore–chromophore spacing via adding steric control units, the overall topology of the MOF structure does not change.
 | ||
| Fig. 3 Absorption spectra of the three Si-Pc linkers in solvated state and as Zn-SURMOF-2, (a) SiPc 1, (b) SiPc 2 and (c) SiPc 4. | ||
To gain further information on the photophysical properties of these aggregated SiPc linkers, we have carried out temperature dependent absorption spectra measurements for non-substituted Zn-SiPc 1 (Fig. 4a and b) and for the bulkiest substitution, Zn-SiPc 4 (Fig. 4c and d). For both SURMOFs, a ∼4 nm blue shift was observed upon lowering the temperature. Similar spectral change indicates that the electronic coupling nature may be similar. As we have observed very small bathochromic shift for Zn-SiPc 4 compared to the monomer state of SiPc 4, the electronic coupling can be considered weak. However similar temperature dependency confirms that for both of the structures J-coupling is present.
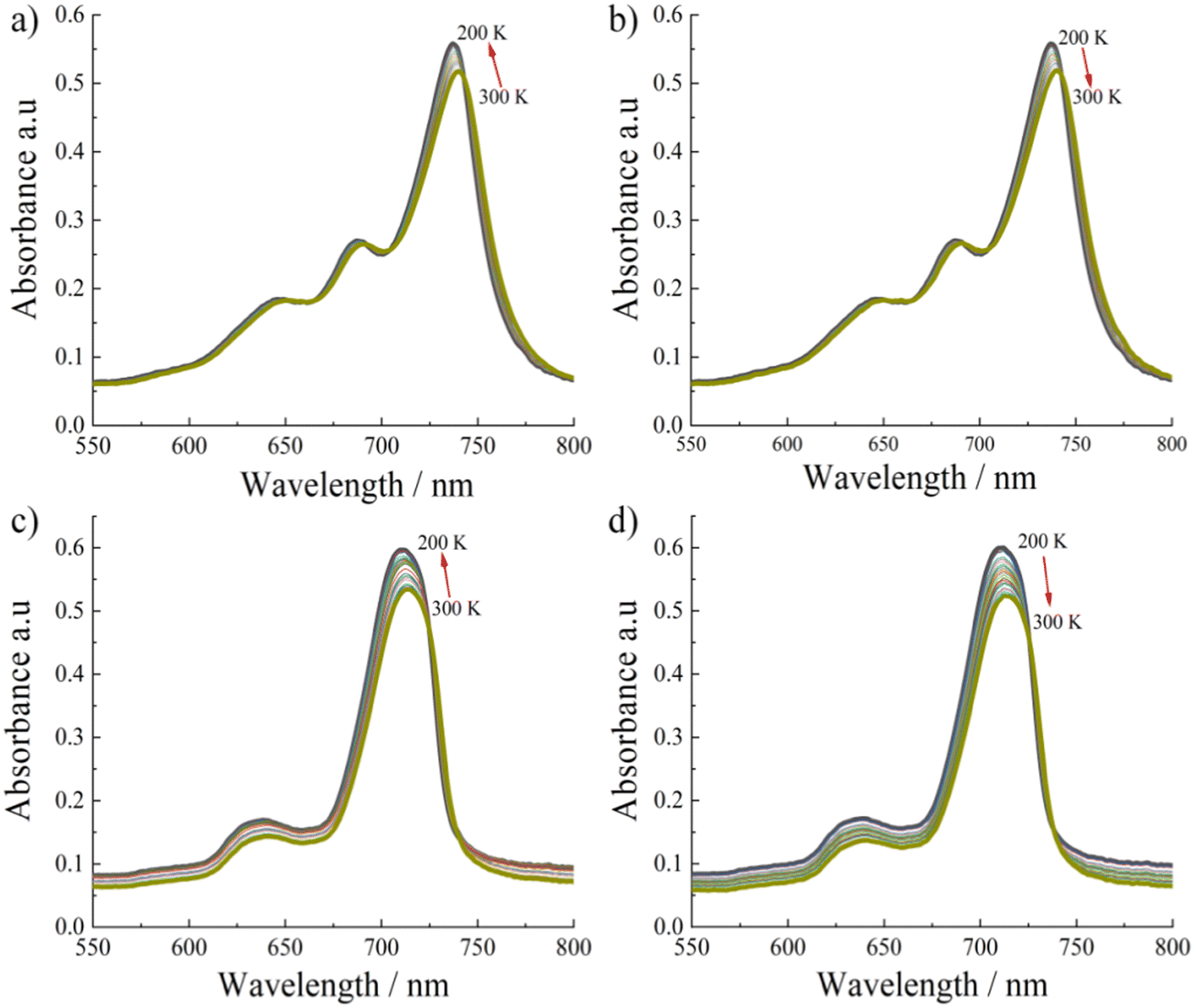 | ||
| Fig. 4 Temperature dependent absorption spectra for (a), (b) Zn-SiPc 1 and (c), (d) Zn-SiPc 4. | ||
Conclusion
In conclusion, we have synthesized three new SiPc dicarboxylic acids and demonstrated that two of them can be used as organic linkers to fabricate MOF thin films, or SURMOFs, using a layer-by-layer approach. We propose a SURMOF-2 type structure for the corresponding MOF thin films, where the SiPc linkers are connected by Zn-paddle-wheel nodes to form a 2D layer. These 2D layers are then arranged to yield stacks with the individual planes oriented perpendicular to the substrate. Within this 3D structure, the SiPc linkers of the neighboring 2D layers interact in an edge-to-edge fashion. From a photophysical characterization we conclude that this interaction leads to J-type electronic coupling. By using the isoreticular MOF design principle, we have designed MOF films with different extent of J-coupling interactions, as evidenced by the absorption spectra measurements. Temperature dependent measurements indicated that even with the bulky substitution, J-type coupling remains intact. The strategy presented here illustrates that in MOF electronic interactions can be modulated with fine control, and the consequent electronic and optical properties can be studied in a straightforward fashion using the thin film structure.Experimental section
Zinc acetate dihydrate was purchased from Merck Millipore. 16-Mercaptohexadecanoic acid (MHDA, 97%), was purchased from Sigma-Aldrich (Germany). Absolute ethanol was purchased from VWR (Germany). All other chemicals were reagent grade, purchased from commercial sources, and were used as received unless otherwise specified.NMR spectra were acquired on a Bruker 400 MHz spectrometer. UV-vis spectra were recorded on a PerkinElmer LAMBDA 365 UV-WinLab spectrophotometer (for the solution). The thin film (grown on quartz glass) spectra were recorded in transmission mode, using a Cary 5000 spectrometer with a UMA unit from Agilent.
The SEM measurements were carried out using a Field Emission Gun (FEI) Philips XL SERIES 30 ESEM-FEG (FEI Co., Eindhoven, NL). Thin films were coated with a ∼5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) nm thick gold/palladium film before recording the SEM micrographs. High-Vacuum (1.5torr) was applied with all specimen, using 20 keV acceleration voltage.
nm thick gold/palladium film before recording the SEM micrographs. High-Vacuum (1.5torr) was applied with all specimen, using 20 keV acceleration voltage.
High-resolution mass spectra were obtained from a Bruker Reflex II matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) spectrometer using dithranol as matrix.
The XRD measurements for out-of-plane (co-planar orientation) were carried out using a Bruker D8-Advance diffractometer equipped with a position sensitive detector Lynxeye in geometry, variable divergence slit and 2.3° Soller-slit was used on the secondary side. The Cu-anodes which utilize the Cu Kα1,2-radiation (λ = 0.154018 nm) were used as source.
The temperature-dependent absorption measurements were carried out with the sample situated in a closed-cycle Helium cryostat (Oxford Instruments OptistatDry TLEX). The cryostat sample chamber was evacuated to a pressure of <10−4 hPa. In a temperature range from 300 K to 200 K, the sample was illuminated with collimated white-light that was then captured by a spectrometer. A fiber-coupled Ocean Optics DH-2000-BAL Deuterium–Halogen lamp served as a white-light source using one output port of a bifurcated optical fiber (QBIF600-UV-BX). The transmitted spectrum was recorded with an Avantes AvaSpec-2048L-USB2 spectrometer with a Thorlabs FP1000URT optical fiber attached.
Preparation of SiPc 2
(tBu)4SiPc(ArCHO)24 (50 mg, 0.048 mmol), sulfamic acid (H3NO3S, 35 mg, 0.361 mmol) and sodium chlorite (25 mg, 0.276 mmol) in a 5:1 THF/H2O mixture (18 mL) were stirred at room temperature, and avoiding direct light exposure, for 12 h. The crude product was evaporated to dryness, and repeatedly washed with 1:1 MeOH/H2O mixture, in order to obtain 42 mg of pure product (82%) as blue-green solid. HR-MS (MALDI-TOF, dithranol) m/z [M]− calcd for C64H58N8O8Si 1094.4251; found 1094.4257. 1H NMR (400 MHz, DMSO-d6) δ 9.79–9.58 (m, 8H, Pc-Ar-H), 8.70–8.60 (m, 4H, Pc-Ar-H), 6.87 (d, J = 8.4 Hz, 4H, Ar-H), 5.13 (d, J = 8.5 Hz, 4H, Ar-H), 1.79–1.73 (m, 36H, 4×tBu). UV-vis (THF): λmax, nm (logε) 360 (5.21), 620 (5.18), 690 (5.89).
Preparation of SiPc 3
(ArO)8SiPc(ArCHO)26 (70 mg, 0.039 mmol), sulfamic acid (H3NO3S, 59 mg, 0.608 mmol) and sodium chlorite (42 mg, 0.464 mmol) in a 4:1 THF/H2O mixture (25 mL) were stirred at room temperature, and avoiding direct light exposure, for 24 h. The crude product was evaporated to dryness, and repeatedly washed with 1:1 MeOH/H2O mixture, to obtain 66 mg of pure product (93%) as blue-green solid. HR-MS (MALDI-TOF, dithranol): m/z [M]− calcd for C112H90N8O16Si 1831.6278; found 1831.6441. 1H NMR (400 MHz, DMSO-d6) δ 8.02 (8H, m, H-Pc), 7.62–7.45 (24H, m, H-Ar), 6.78 (4H, d, J = 8.3Hz, H-Ar), 4.86 (4H, d, J = 8.3Hz, H-Ar), 2.36 (48H, s, 16 × CH3). UV-vis (THF): λmax, nm (logε) 366 (4.98), 426 (4.54), 624 (4.56), 665 (4.55), 694 (5.42).
Preparation of SiPc 4
(ArO)8SiPc(Ar2CHO)27 (35 mg, 0.018 mmol), sulfamic acid (H3NO3S, 28 mg, 0.288 mmol) and sodium chlorite (20 mg, 0.288 mmol) in a 6:1 THF/H2O mixture (12 mL) were stirred at room temperature, and avoiding direct light exposure, for 12 h. The crude product was evaporated to dryness, and repeatedly washed with 1:1 MeOH/H2O mixture, to obtain 30 mg of pure product (84%) as blue-green solid. HR-MS (MALDI-TOF, dithranol): m/z [M]+ calcd for C124H98N8O16Si 1983.6904; found 1983.6744. 1H NMR (400 MHz, CDCl3) δ 8.20 (8H, m, H-Pc), 7.79 (4H, d, J: 8.3 Hz, H-Ar), 7.36 (24H, brs, H-Ar), 7.02 (4H, d, J: 8.3 Hz, H-Ar), 6.46 (4H, d, J: 8.3 Hz, H-Ar), 5.14 (4H, d, J: 8.3 Hz, H-Ar), 2.42 (48H, s, 16 × CH3). UV-vis (THF): λmax, nm (logε) 366 (4.89), 424 (4.46), 623 (4.49), 666 (4.52), 692 (5.33).
Preparation of Zn-SiPc 2 and Zn-SiPc 4
Syntheses of the SURMOFs are carried out following the previous procedure. Ethanolic solution of 1 mM zinc acetate and 20 μM SiPc 2/4 solutions (in ethanol) were sequentially deposited onto the precleaned substrates (cleaned by UV-ozone, Si or quartz glass) using spin coating method in a layer-by-layer fashion. After the metal or linker coating, the samples were rinsed with ethanol to remove unreacted metal/linker or by-products from the surface. For metal and linker both, the spin coating time is fixed as 10 s with rpm of 2000.Conflicts of interest
There are no conflicts to declare.Acknowledgements
H. C. acknowledges financial support from China Scholarship Council (CSC, no. 201806830055). A. S. S. thanks the European Regional Development Fund “A way to make Europe” and the Spanish Ministerio de Ciencia e Innovación/Agencia Estatal de Investigación (PID2020-117855 RB-I00) and the Advanced Materials program by MCIN with funding from European Union NextGenerationEU (PRTR-C17.I1) and Generalitat Valenciana (MFA/2022/028) for funding. R. H. acknowledges intramural funds at TIFR Hyderabad from the Department of Atomic Energy (DAE), India, under project identification number RTI 4007. C. W. acknowledges support from Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy – 2082/1 – 390761711.References
- H. Furukawa, K. E. Cordova, M. O’Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- N. Stock and S. Biswas, Chem. Rev., 2012, 112, 933–969 CrossRef CAS PubMed.
- O. K. Farha, I. Eryazici, N. C. Jeong, B. G. Hauser, C. E. Wilmer, A. A. Sarjeant, R. Q. Snurr, S. T. Nguyen, A. Ö. Yazaydın and J. T. Hupp, J. Am. Chem. Soc., 2012, 134, 15016–15021 CrossRef CAS PubMed.
- J.-R. Li, J. Sculley and H.-C. Zhou, Chem. Rev., 2012, 112, 869–932 CrossRef CAS PubMed.
- Q. Qian, P. A. Asinger, M. J. Lee, G. Han, K. Mizrahi Rodriguez, S. Lin, F. M. Benedetti, A. X. Wu, W. S. Chi and Z. P. Smith, Chem. Rev., 2020, 120, 8161–8266 CrossRef CAS PubMed.
- V. Pascanu, G. González Miera, A. K. Inge and B. Martín-Matute, J. Am. Chem. Soc., 2019, 141, 7223–7234 CrossRef CAS PubMed.
- A. Bavykina, N. Kolobov, I. S. Khan, J. A. Bau, A. Ramirez and J. Gascon, Chem. Rev., 2020, 120, 8468–8535 CrossRef CAS PubMed.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105–1125 CrossRef CAS PubMed.
- H.-Y. Li, S.-N. Zhao, S.-Q. Zang and J. Li, Chem. Soc. Rev., 2020, 49, 6364–6401 RSC.
- Z. Mai and D. Liu, Cryst. Growth Des., 2019, 19, 7439–7462 CrossRef CAS.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469–472 CrossRef CAS PubMed.
- R. Haldar, L. Heinke and C. Wöll, Adv. Mater., 2020, 32, 1905227 CrossRef CAS PubMed.
- R. Haldar, A. Ghosh and T. K. Maji, Chem. Commun., 2023, 59, 1569–1588 RSC.
- V. Stavila, A. A. Talin and M. D. Allendorf, Chem. Soc. Rev., 2014, 43, 5994–6010 RSC.
- G. A. Leith, C. R. Martin, J. M. Mayers, P. Kittikhunnatham, R. W. Larsen and N. B. Shustova, Chem. Soc. Rev., 2021, 50, 4382–4410 RSC.
- R. Haldar, A. Mazel, M. Krstić, Q. Zhang, M. Jakoby, I. A. Howard, B. S. Richards, N. Jung, D. Jacquemin, S. Diring, W. Wenzel, F. Odobel and C. Wöll, Nat. Commun., 2019, 10, 2048 CrossRef PubMed.
- M. Mostaghimi, C. R. C. Rêgo, R. Haldar, C. Wöll, W. Wenzel and M. Kozlowska, Front. Mater., 2022, 9, 840644 CrossRef.
- G. Wu, J. Huang, Y. Zang, J. He and G. Xu, J. Am. Chem. Soc., 2017, 139, 1360–1363 CrossRef CAS PubMed.
- D.-H. Chen, H. Gliemann and C. Wöll, Chem. Phys. Rev., 2023, 4, 011305 CrossRef CAS.
- A. J. Clough, N. M. Orchanian, J. M. Skelton, A. J. Neer, S. A. Howard, C. A. Downes, L. F. J. Piper, A. Walsh, B. C. Melot and S. C. Marinescu, J. Am. Chem. Soc., 2019, 141, 16323–16330 CrossRef CAS PubMed.
- L. S. Xie, G. Skorupskii and M. Dincă, Chem. Rev., 2020, 120, 8536–8580 CrossRef CAS PubMed.
- R. Dong, P. Han, H. Arora, M. Ballabio, M. Karakus, Z. Zhang, C. Shekhar, P. Adler, P. S. Petkov, A. Erbe, S. C. B. Mannsfeld, C. Felser, T. Heine, M. Bonn, X. Feng and E. Cánovas, Nat. Mater., 2018, 17, 1027–1032 CrossRef CAS PubMed.
- B. Liu and V. S. Thoi, Chem. Mater., 2020, 32, 8450–8459 CrossRef CAS.
- M. Gutiérrez, C. Martín, M. Van der Auweraer, J. Hofkens and J.-C. Tan, Adv. Opt. Mater., 2020, 8, 2000670 CrossRef.
- J. Liu, W. Zhou, J. Liu, I. Howard, G. Kilibarda, S. Schlabach, D. Coupry, M. Addicoat, S. Yoneda, Y. Tsutsui, T. Sakurai, S. Seki, Z. Wang, P. Lindemann, E. Redel, T. Heine and C. Wöll, Angew. Chem., Int. Ed., 2015, 54, 7441–7445 CrossRef CAS PubMed.
- R. Haldar, M. Jakoby, A. Mazel, Q. Zhang, A. Welle, T. Mohamed, P. Krolla, W. Wenzel, S. Diring, F. Odobel, B. S. Richards, I. A. Howard and C. Wöll, Nat. Commun., 2018, 9, 4332 CrossRef PubMed.
- H. Arora, R. Dong, T. Venanzi, J. Zscharschuch, H. Schneider, M. Helm, X. Feng, E. Cánovas and A. Erbe, Adv. Mater., 2020, 32, 1907063 CrossRef CAS PubMed.
- A. Nefedov, R. Haldar, Z. Xu, H. Kühner, D. Hofmann, D. Goll, B. Sapotta, S. Hecht, M. Krstić, C. Rockstuhl, W. Wenzel, S. Bräse, P. Tegeder, E. Zojer and C. Wöll, Adv. Mater., 2021, 33, 2103287 CrossRef CAS PubMed.
- Y. He, C. D. Spataru, F. Léonard, R. E. Jones, M. E. Foster, M. D. Allendorf and A. Alec Talin, Phys. Chem. Chem. Phys., 2017, 19, 19461–19467 RSC.
- R. Haldar, M. Kozlowska, M. Ganschow, S. Ghosh, M. Jakoby, H. Chen, F. Ghalami, W. Xie, S. Heidrich, Y. Tsutsui, J. Freudenberg, S. Seki, I. A. Howard, B. S. Richards, U. H. F. Bunz, M. Elstner, W. Wenzel and C. Wöll, Chem. Sci., 2021, 12, 4477–4483 RSC.
- M. Kozlowska, Y. Pramudya, M. Jakoby, S. Heidrich, L. Pan, B. S. Richards, I. A. Howard, C. Wöll, R. Haldar and W. Wenzel, J. Phys.: Condens. Matter, 2021, 33, 034001 CrossRef CAS PubMed.
- R. Haldar, K. Batra, S. M. Marschner, A. B. Kuc, S. Zahn, R. A. Fischer, S. Bräse, T. Heine and C. Wöll, Chem. - Eur. J., 2019, 25, 7847–7851 CrossRef CAS PubMed.
- S. S. Rajasree, X. Li and P. Deria, Commun. Chem., 2021, 4, 47 CrossRef CAS PubMed.
- S. De, T. Devic and A. Fateeva, Dalton Trans., 2021, 50, 1166–1188 RSC.
- M. Wang, Z. Zhang, H. Zhong, X. Huang, W. Li, M. Hambsch, P. Zhang, Z. Wang, P. Petkov, T. Heine, S. C. B. Mannsfeld, X. Feng and R. Dong, Angew. Chem., Int. Ed., 2021, 60, 18666–18672 CrossRef CAS PubMed.
- J.-R. Zhang, H.-Y. Zhang, J.-H. Guo, Z.-H. Liu, C.-Y. Ma, X.-G. Yang, X.-Y. Lu, J.-H. Qin and L.-F. Ma, Dalton Trans., 2022, 51, 1769–1774 RSC.
- P. I. Scheurle, A. Biewald, A. Mähringer, A. Hartschuh, D. D. Medina and T. Bein, Small Struct., 2022, 3, 2100195 CrossRef CAS.
- Z. Chen, C. Zhong, Z. Zhang, Z. Li, L. Niu, Y. Bin and F. Zhang, J. Phys. Chem. B, 2008, 112, 7387–7394 CrossRef CAS PubMed.
- R. Haldar, Z. Fu, R. Joseph, D. Herrero, L. Martín-Gomis, B. S. Richards, I. A. Howard, A. Sastre-Santos and C. Wöll, Chem. Sci., 2020, 11, 7972–7978 RSC.
- L. Martín-Gomis, K. Ohkubo, F. Fernández-Lázaro, S. Fukuzumi and Á. Sastre-Santos, Org. Lett., 2007, 9, 3441–3444 CrossRef PubMed.
- L. Martín-Gomis, S. Seetharaman, D. Herrero, P. A. Karr, F. Fernández-Lázaro, F. D'Souza and Á. Sastre-Santos, Chem. Phys. Chem., 2020, 21, 2254–2262 CrossRef PubMed.
- J. Liu, B. Lukose, O. Shekhah, H. K. Arslan, P. Weidler, H. Gliemann, S. Bräse, S. Grosjean, A. Godt, X. Feng, K. Müllen, I.-B. Magdau, T. Heine and C. Wöll, Sci. Rep., 2012, 2, 921 CrossRef PubMed.
- O. Shekhah, H. Wang, S. Kowarik, F. Schreiber, M. Paulus, M. Tolan, C. Sternemann, F. Evers, D. Zacher, R. A. Fischer and C. Wöll, J. Am. Chem. Soc., 2007, 129, 15118–15119 CrossRef CAS PubMed.
- A. Ferencz, D. Neher, M. Schulze, G. Wegner, L. Viaene and F. C. De Schryver, Chem. Phys. Lett., 1995, 245, 23–29 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp01865b |
| This journal is © the Owner Societies 2023 |