
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA combined crossed molecular beam and theorerical study of the O(3P,1D) + acrylonitrile (CH2CHCN) reactions and implications for combustion and extraterrestrial environments†
Giacomo
Pannacci
 a,
Luca
Mancini
a,
Gianmarco
Vanuzzo
a,
Pengxiao
Liang
a,
Demian
Marchione
a,
Marzio
Rosi
b,
Piergiorgio
Casavecchia
a and
Nadia
Balucani
*a
a,
Luca
Mancini
a,
Gianmarco
Vanuzzo
a,
Pengxiao
Liang
a,
Demian
Marchione
a,
Marzio
Rosi
b,
Piergiorgio
Casavecchia
a and
Nadia
Balucani
*a
aDipartimento di Chimica, Biologia e Biotecnologie, Università degli Studi di Perugia, Perugia, Italy. E-mail: nadia.balucani@unipg.it
bDipartimento di Ingegneria Civile e Ambientale, Università degli Studi di Perugia, Perugia, Italy
First published on 5th June 2023
Abstract
Acrylonitrile (CH2CHCN) is ubiquitous in space (molecular clouds, solar-type star forming regions, and circumstellar envelopes) and is also abundant in the upper atmosphere of Titan. The reaction O(3P) + CH2CHCN can be of relevance in the chemistry of the interstellar medium because of the abundance of atomic oxygen. The oxidation of acrylonitrile is also important in combustion as the thermal decomposition of pyrrolic and pyridinic structures present in fuel-bound nitrogen generates many nitrogen-bearing compounds, including acrylonitrile. Despite its relevance, limited information exists on this reaction. We report a combined experimental and theoretical investigation of the reactions of acrylonitrile with both ground 3P and excited 1D atomic oxygen. From product angular and time-of-flight distributions in crossed molecular beam experiments with mass spectrometric detection at a collision energy, Ec, of 31.4 kJ mol−1, we have identified the primary reaction products and determined their branching fractions (BFs). Theoretical calculations of the relevant triplet and singlet potential energy surfaces (PESs) were performed to interpret the experimental results and elucidate the reaction mechanism. Adiabatic statistical calculations of product BFs for the decomposition of the main triplet and singlet intermediates have been carried out. Combining the experimental and theoretical results, we conclude that the O(3P) reaction leads to two main product channels: (i) CH2CNH (ketenimine) + CO (dominant with a BF of 0.87 ± 0.05), formed via efficient intersystem crossing from the entrance triplet PES to the underlying singlet PES, and (ii) HCOCHCN + H (minor, with a BF of 0.13 ± 0.05), occurring adiabatically on the triplet PES. Our study suggests the inclusion of this reaction as a possible destruction pathway of CH2CHCN and a possible formation route of CH2CNH in the interstellar medium. The O(1D) + CH2CHCN reaction mainly leads to the formation of CH2CNH + CO adiabatically on the singlet PES. This result can improve models related to the chemistry of interstellar ice and cometary comas, where O(1D) reactions can play a role. Overall, our results are expected to be useful for improving the models of combustion and extraterrestrial environments.
1. Introduction
The oxidative processes of acrylonitrile, CH2CHCN, are of considerable importance in a variety of fields, ranging from astrochemistry to combustion.1–3 Acrylonitrile (also called cyanoethene or vinyl cyanide) is widespread in textile and plastics industries, with countless applications. The polymerization of acrylonitrile results in the formation of polyacrylonitrile (PAN), the precursor of acrylic and carbon fibers; furthermore, its combination with other repeat units, such as styrene and 1,3-butadiene, is the basis for the synthesis of many copolymers, such as SAN (styrene acrylonitrile), ABS (acrylonitrile butadiene styrene), and ASA (acrylonitrile styrene acrylate).4,5 Acrylonitrile is also classified as a “volatile organic compound” (VOC) and it has hazardous properties, including toxicity and carcinogenicity.6 Therefore, the regulation on the emission of this compound into the atmosphere is very strict and many efforts have been made to optimize sustainable methods to convert acrylonitrile into clean species. Thermal combustion at temperatures higher than 850 °C results in the emission of secondary pollutants (nitrogen oxides, NOx),7 while selective catalytic combustion (SCC), which is characterized by an operating temperature of 300–500 K, has a lower environmental impact.8–12 Moreover, the inclusion of the oxidation process of acrylonitrile in models that simulate the combustion of coal and other low-rank fossil fuels is important to account for dangerous emissions. This is because the nitrogen content of many fuels is essentially ascribed to the presence of pyrrolic and pyridinic structures,13–15 but their thermal decomposition generates many nitrogen-bearing compounds, including acrylonitrile,16–19 that can undergo subsequent oxidation to NOx.20 For the above reasons, it is worth investigating the reactions of acrylonitrile with common oxidant species in combustion, including atomic oxygen.In addition, the O(3P) + acrylonitrile reaction can also be relevant in the chemistry of the interstellar medium (ISM), with acrylonitrile being the first molecule with a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond detected in the ISM via its rotational transition at 1372 MHz (211–212),21,22 and atomic oxygen being quite abundant in various regions of the ISM (where it represents the third most abundant element after H and He). After its first detection in Sagittarius (Sgr) B2,21 acrylonitrile has been identified in a variety of interstellar environments, such as the hot molecular core Sgr B2(N),23,24 Orion-KL,25 the TMC-1 dark cloud,26 the circumstellar envelope of the C-rich star IRC + 10216,27 and the L1544 prototypical prestellar core,28 as well as in Titan's atmosphere.29–35 Additionally, atomic oxygen in its first electronically excited state, O(1D), can play a role in the chemistry of extraterrestrial environments, and several reactions involving O(1D) have been considered in the chemistry of cometary comae36 and of interstellar ice.37 Therefore, the study of the reaction of acrylonitrile with oxygen atoms can contribute to enrich current astrochemical models, as was done previously in our group with the CN(X2Σ+) + CH2CHCN38 and N(2D) + CH2CHCN reactions.39
C double bond detected in the ISM via its rotational transition at 1372 MHz (211–212),21,22 and atomic oxygen being quite abundant in various regions of the ISM (where it represents the third most abundant element after H and He). After its first detection in Sagittarius (Sgr) B2,21 acrylonitrile has been identified in a variety of interstellar environments, such as the hot molecular core Sgr B2(N),23,24 Orion-KL,25 the TMC-1 dark cloud,26 the circumstellar envelope of the C-rich star IRC + 10216,27 and the L1544 prototypical prestellar core,28 as well as in Titan's atmosphere.29–35 Additionally, atomic oxygen in its first electronically excited state, O(1D), can play a role in the chemistry of extraterrestrial environments, and several reactions involving O(1D) have been considered in the chemistry of cometary comae36 and of interstellar ice.37 Therefore, the study of the reaction of acrylonitrile with oxygen atoms can contribute to enrich current astrochemical models, as was done previously in our group with the CN(X2Σ+) + CH2CHCN38 and N(2D) + CH2CHCN reactions.39
Despite its possible relevance, the O(3P) + CH2CHCN reaction has been the subject of only one kinetic study and one theoretical investigation so far. The rate constant at 298 K and 1.2–1.7 Torr was determined to be (4.9 ± 1.0) × 10−13 cm3 molecule−1 s−1 by Upadhyaya et al. in a fast-flow experiment with a derived activation energy of 7.1 kJ mol−1.40 The authors suggested that the reaction occurs via electrophilic addition of the oxygen atom to the CC double bond, resulting in the formation of a resonantly stabilized intermediate OCH2CHCN, which can be cyclized to cyanoethyleneoxide.40 The same conclusion was reached by Kimber et al.41 by experimentally studying the heterogeneous reaction between oxygen atoms and acrylonitrile on highly oriented pyrolytic graphite (HOPG) kept at 14–100 K. The experimental rate constant40 was corroborated by theoretical computations carried out by Sun et al.42 using a statistical approach on the calculated triplet potential energy surface (PES) (ktot = 1.79 × 10−13 cm3 mol−1 s−1 at 298 K and at a pressure of 1.2 Torr of Ar). Branching fractions (BFs) for addition and abstraction channels on the adiabatic triplet PES were estimated as a function of temperature and pressure. The dominant product channel was predicted to be H + HCOCHCN (BF ∼ 0.75), followed by CH2O + HCCN (BF ∼ 0.25), while other channels became significant only with temperature rising above 500 K.42 In this theoretical study, intersystem-crossing (ISC) to the underlying singlet PES was not considered, even though it is well known that ISC is quite common in most reactions involving unsaturated hydrocarbons and O(3P).43–49 To improve current combustion and astrochemical models, it is important to explore the role of ISC and its effect on the product branching fractions.
Here, we report the first investigation of the dynamics of the O(3P,1D) + acrylonitrile reactions using the crossed-molecular beam (CMB) scattering technique with mass spectrometric (MS) detection and time-of-flight (TOF) analysis, which is arguably the most powerful method to reveal the primary products and determine their BFs for elementary multichannel reactions (see ref. 50, 51, and references therein). The experimental work has been complemented by theoretical calculations of the triplet/singlet PESs and statistical RRKM/ME (Rice–Ramsperger–Kassel–Marcus/Master equation) computations of the product BFs as a function of temperature, considering separately the triplet and the singlet PESs.
The paper is organized as follows. In Section 2 the experimental technique is presented, while in Section 3 the computational method is described. Section 4 presents the experimental and theoretical results, while the discussion is presented in Section 5. Finally, Section 6 is about the implications of the findings on extraterrestrial environments and Section 7 provides a summary and conclusions.
2. Experimental method
The dynamics of the O(3P) + CH2CHCN reaction was investigated by exploiting the CMB technique based on an apparatus equipped with a rotatable electron-impact quadrupole MS detector and TOF analysis system. The basis of the method and the technical specifications of our apparatus have been extensively described elsewhere.50–55 Briefly, two supersonic beams of the reactants were crossed at the angle γ = 90° inside a scattering chamber kept at a base pressure of 2 × 10−7 hPa, which increased up to 1 × 10−6 hPa under the operating conditions. The aim of identifying the primary reaction products is supported by the “single-collision conditions” of the experiment. The scattered products at the collision center enter a triply differentially pumped, ultra-high vacuum (UHV) chamber; in the inner region of this chamber, ionization takes place through an electron impact ionizer, featuring tunable electron energy; the ions are then selected by a quadrupole mass filter and collected in a Daly-type detector.56 The detector angular resolution for a point collision zone is about 1°.The intensity of the products as a function of the laboratory (LAB) scattering angle Θ, namely, the product angular distribution N(Θ), is recorded using a mass spectrometer that can be rotated in the collision plane, around an axis orthogonal to the plane containing the colliding beams and passing through their intersection axis. During the angular distribution measurements, the molecular beam is modulated at 160 Hz using a tuning fork chopper for background subtraction. The distribution of the products as a function of time (or velocity) is obtained by setting the detector at selected angles and employing the pseudo-random TOF technique based on a pseudo-random wheel containing four identical sequences of 127 open/closed elements, spinning at 328.1 Hz (corresponding to a dwell time of 6 μs/channel) in front of the entrance of the detector.
The supersonic oxygen beam was generated using a radio-frequency (RF) discharge beam source57–59 in which 85 mbar of a diluted gas mixture of (5%) He/O2 was discharged at 300 W of RF power, through a 0.48 mm diameter water-cooled quartz nozzle followed by a 0.8 mm diameter boron nitride skimmer and a further collimating aperture. The resulting beam was characterized by a predominance (≥90%) of atomic oxygen in its ground electronic state (3P), with a small fraction (≤10%) of atomic oxygen in its first electronically excited state (1D).57 The supersonic beam of acrylonitrile was produced using the neat species. Commercial acrylonitrile (CH2CHCN, Merck 99% purity, containing 35–45 ppm of mequinol, C7H8O2, as inhibitor) was stored inside a reservoir kept at 20 °C; 87 torr, corresponding to the vapor pressure of liquid acrylonitrile at the temperature of the container, undergo an expansion through a 0.1 mm diameter stainless steel (S.S.) nozzle, followed by a 0.8 mm S. S. skimmer, and a further collimating aperture. The peak velocity of the resulting atomic and molecular beams, as obtained from single-shot TOF analysis, was 2162 m s−1 with a speed ratio of 4.4 and 663 m s−1 with a speed ratio of 3.8, respectively. The resulting collision energy, Ec, was 31.4 kJ mol−1 and the center-of-mass angle, ΘCM, was 45°.
In order to gain a detailed understanding of the reaction dynamics with a quantitative and physical interpretation of the scattering event, it is necessary to move from the LAB reference frame to the center-of-mass (CM) one.51–54 The LAB number density, NLAB(Θ), and the CM flux, ICM(θ,u), are related through the equation: 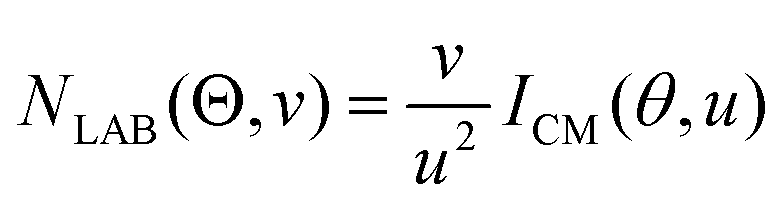 (where v and u are the velocity in the LAB and in the CM frame, respectively, and the term v/u2 is the transformation Jacobian).60 Because of the finite resolution of the experimental conditions, the CM flux, which can be factorized into the product of the angular (T(θ)) and translational energy
(where v and u are the velocity in the LAB and in the CM frame, respectively, and the term v/u2 is the transformation Jacobian).60 Because of the finite resolution of the experimental conditions, the CM flux, which can be factorized into the product of the angular (T(θ)) and translational energy 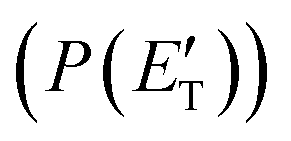 distributions, is derived through a forward convolution fit of the total product LAB angular and TOF distributions at a certain mass to charge (m/z) ratio value50,51 according to the relation:
distributions, is derived through a forward convolution fit of the total product LAB angular and TOF distributions at a certain mass to charge (m/z) ratio value50,51 according to the relation:
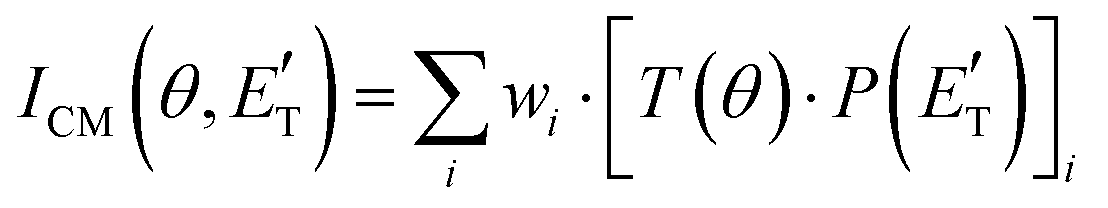
3. Computational methods
3.1 Quantum chemical calculations
The theoretical investigation of the reactions between O(3P,1D) and acrylonitrile was performed adopting a well-established computational strategy previously used for the investigation of many other bimolecular reactions.61–65 In particular, both the triplet and the singlet PESs were characterized by locating the lowest stationary points using density functional theory (DFT), with the hybrid B3LYP functional66,67 in conjunction with the correlation consistent valence polarized set aug-cc-pVTZ.68–70 Harmonic vibrational frequencies were computed at the same level of theory, in order to check the nature of the stationary points, i.e., minimum if all the frequencies are real, while a saddle point if there is one, and only one, imaginary frequency. Intrinsic reaction coordinate (IRC) calculations71,72 were performed in order to ensure the correct assignment of the identified transition states. The more accurate coupled cluster theory, including single and double excitations as well as a perturbative estimate of connected triples [CCSD(T)],73–75 was used in conjunction with the same basis set aug-cc-pVTZ to calculate the energy of each stationary point. Both B3LYP and CCSD(T) energies were corrected to 0 K by adding the zero-point energy correction computed using the scaled harmonic vibrational frequencies evaluated at the B3LYP/aug-cc-pVTZ level.Following a computational strategy already used by some of the present authors for different reactions involving atomic oxygen,43,46,47,76–78 more accurate calculations for the stationary points involved in the main reaction channels were performed at the CCSD(T) level corrected with a density fitted (DF) MP2 extrapolation to the complete basis set (CBS) and with corrections for core electrons excitations. In particular, we computed the energies as:
| E = E(CCSD(T)/aug-cc-pVTZ) + [E(CCSD(T,core)/cc-pVTZ) − E(CCSD(T)/cc-pVTZ)] + [E(DF-MP2/CBS)-E(DF-MP2/aug-cc-pVTZ)] |
| E(DF-MP2/CBS) = E[(DF-MP2)/aug-cc-pVQZ)] + 0.5772 × [E(DF-MP2/aug-cc-pVQZ) − E(DF-MP2/aug-cc-pVTZ)] |
3.2 RRKM calculations of the product branching fractions
RRKM calculations were carried out on the two separated PESs to derive the BFs of the products for the O(3P,1D) + CH2CHCN reactions separately on the triplet and singlet PESs, that is, the extent of ISC was not evaluated theoretically. In detail, RRKM calculations were performed using a code developed by our group for this purpose.85–88 At a specific total energy, the microcanonical rate constant k(E) for a specific reaction is given by the following expression:where N(E) denotes the sum of states in the transition state at energy E, ρ(E) is the reactant density of states at the energy E, and h is Planck's constant. N(E) is obtained by integrating the relevant density of states up to energy E, using the rigid rotor/harmonic oscillator model. Tunneling effects are included in the present RRKM calculations using the imaginary vibrational frequency of the transition states and calculating the tunneling probability for the corresponding Eckart barrier. For the cases where no clear transition state barrier was found, the corresponding microcanonical rate constants were obtained through the variational RRKM approach, evaluating k(E) at various points along the reaction coordinate and choosing the point which minimizes the rate constant, in accordance with the variational approach (variational transition state theory-VTST).89 For the dissociation steps in which no intermediates point were available, due to difficulties in the electronic structure calculations, the products at infinite separation were taken into account as possible “transition states”. In this way it was possible to avoid problems arising from the different number of degrees of freedom between the reactants and the transition state by not including the 2D part of the overall rotation in the RRKM treatment of the reactants (leaving only the “prolate” 1D contribution). After the calculation of all microcanonical rate constants, a Markov (stochastic) matrix was set up for all intermediates and final channels of the reaction containing all product BFs derived from the rate constants, separately on the triplet PES and the singlet PES. k(E) was subsequently Boltzmann averaged for each temperature of interest to yield k(T).
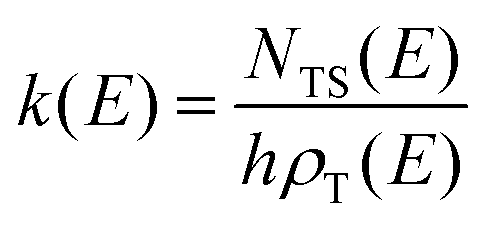
4. Results
According to our electronic structure calculations, there are many possible open reactive channels (for the complete list, see the ESI†) which can originate from the triplet and singlet PESs, of which the most relevant ones are:| O(3P) + CH2CHCN | → 1CH3CN + CO |
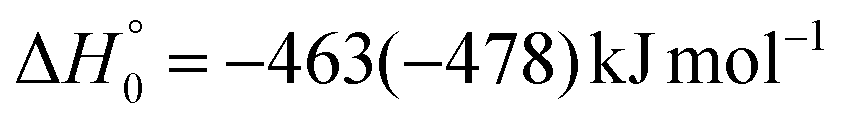
|
(1a) |
| → 1CH2CNH + CO |
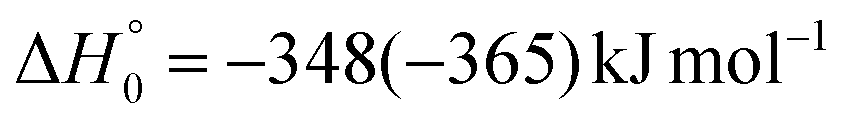
|
(1b) | |
| → 1OCCHCN + H2 |
|
(1c) | |
| → 2CH2CN + HCO |
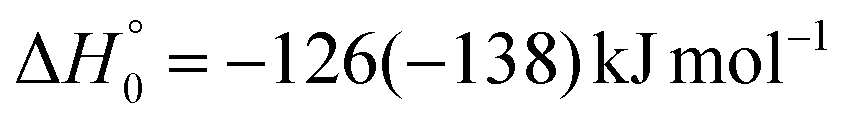
|
(1d) | |
| → 2COCN + 2CH3 |
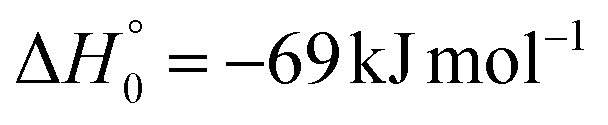
|
(1e) | |
| → 2HCOCHCN + H |
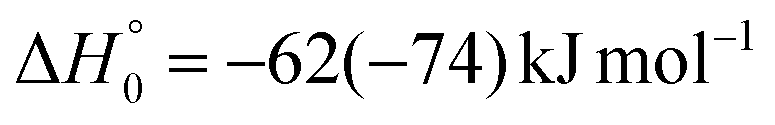
|
(1f) | |
| → 1CH2O + 3HCCN |
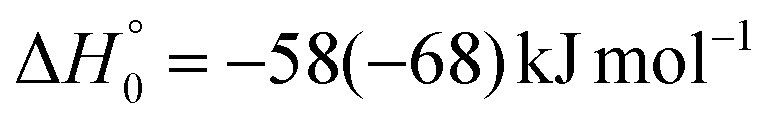
|
(1g) | |
| → 2CH2COCN + H |
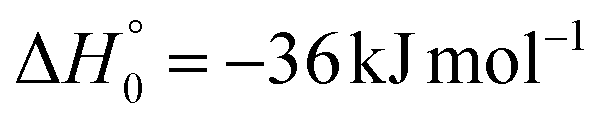
|
(1h) | |
| → 2CH2CH + 2NCO |
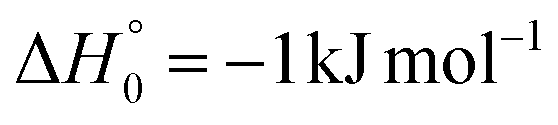
|
(1i) | |
| → 1OCHCN + 3CH2 |
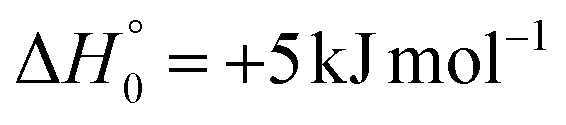
|
(1j) | |
| O(1D) + CH2CHCN | → 1CH3CN + CO |
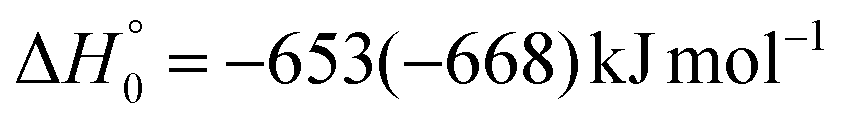
|
(2a) |
| → 1CH2CNH + CO |
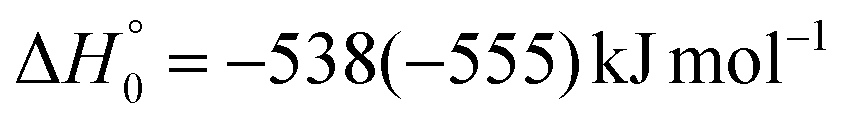
|
(2b) | |
| → 1OCCHCN + H2 |
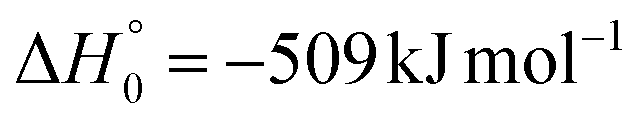
|
(2c) | |
| → 2CH2CN + HCO |
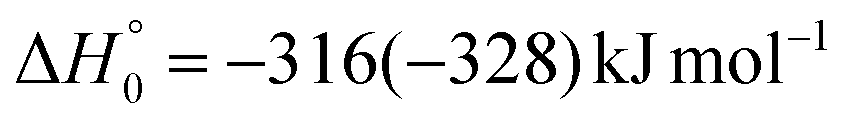
|
(2d) | |
| → 2COCN + 2CH3 |
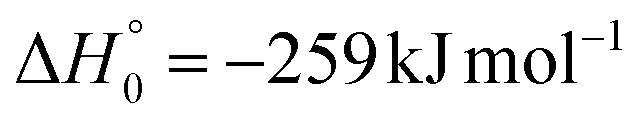
|
(2e) | |
| → 2HCOCHCN + H |
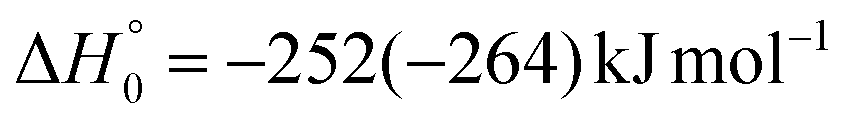
|
(2f) | |
The enthalpies of reaction reported are those calculated in the present work at the CCSD(T)/aug-cc-pVTZ level and at the CCSD(T)/CBS level (values in parentheses) (only for the most relevant channels). Regarding the O(3P) + acrylonitrile reaction, channels (1g)–(1j) correlate adiabatically with the triplet PES, while channels (1a)–(1e) are expected to occur on the singlet PES after ISC. Channel (1f) can occur both adiabatically on the triplet PES and via ISC to the singlet PES. All the channels (2a)–(2f) of the O(1D) + acrylonitrile reaction correlate adiabatically with the singlet PES.
4.1. Experimental results: LAB product angular and TOF distributions
Experimentally, reactive scattering signals were detected at m/z = 68 (C3H2NO+), 67 (C3HNO+), 66 (C3NO+), 41 (C2H3N+), 40 (C2H2N+), 39 (C2HN+), and 29 (HCO+) with relative intensities of 0.08, 0.56, 0.04, 0.16, 1.00, 0.64, and 0.34, respectively, at Θ = 44° (which is very close to the CM angle of 45°), when using an electron energy of 28 eV. Due to considerations regarding the signal-to-noise (S/N) ratio, product LAB angular distributions, N(Θ), and TOF distributions, N(Θ, t), were measured in detail only for m/z = 67 and m/z = 40, and they are shown in Fig. 1 and 2, respectively. The angular distribution at m/z = 67 was obtained by counting the reactive signal for 50 s per angle during each angular scan (total five scans), while the accumulation time for the TOF spectra at Θ = 44° and 32° was 150 and 240 minutes, respectively. It should be noted that while measurements at m/z = 67 could be performed using both hard (70 eV) and soft (28 eV) electron ionization, measurements at m/z = 40 needed to be performed using soft ionization to suppress/mitigate the problem of dissociative ionization of interfering species.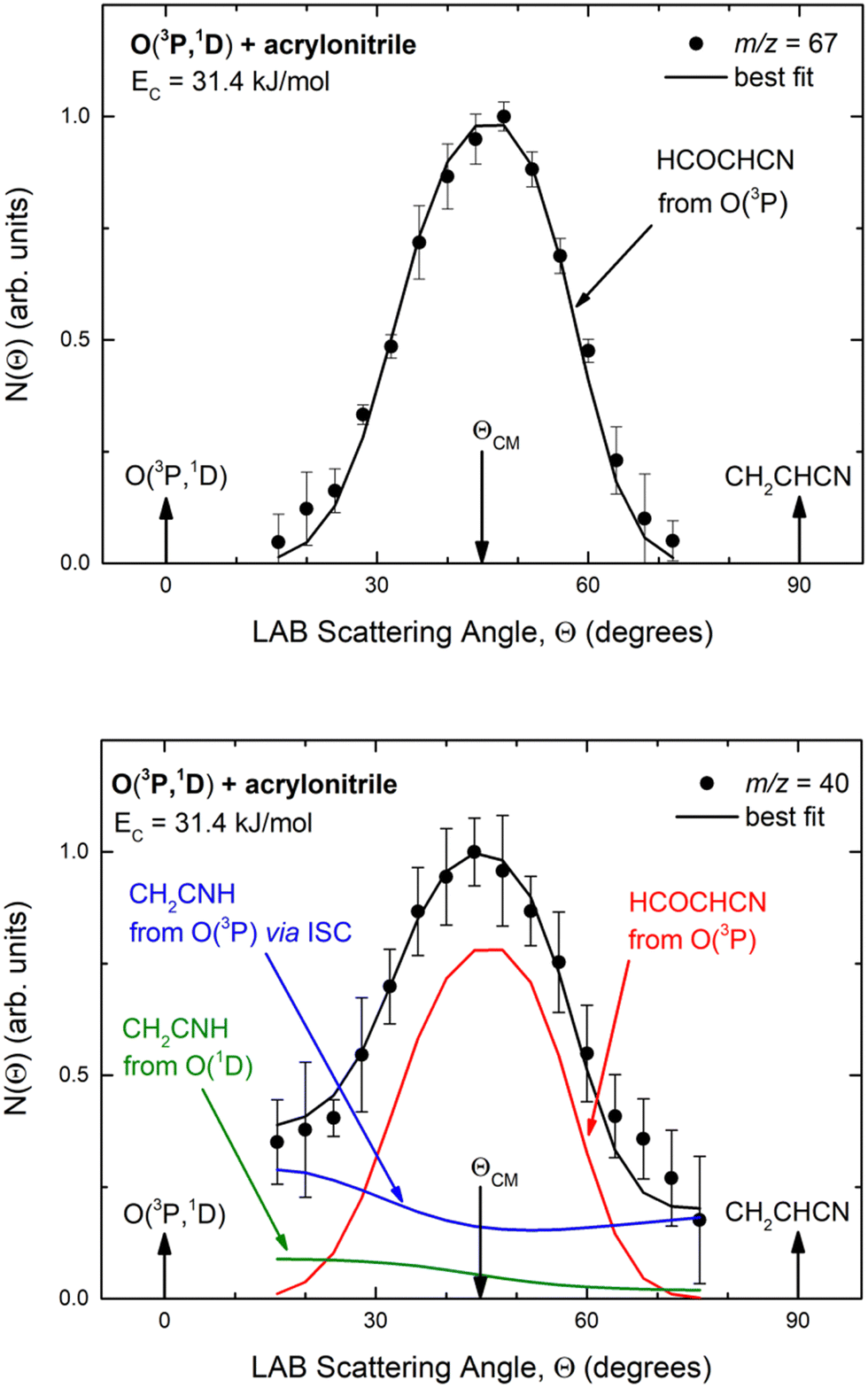 | ||
| Fig. 1 LAB angular distribution of (top) the HCOCHCN product (from channel (1f)) detected at m/z = 67 (C3HNO+), and (bottom) of the HCOCHCN and CH2CNH products detected at m/z = 40 (C2H2N+) for the O(3P,1D) + acrylonitrile reactions at Ec = 31.4 kJ mol−1. The filled circles are the experimental data (with ±1σ error bars indicated), while the heavy solid black curve represents the calculated total distribution when using the best-fit functions shown in Fig. 4. (bottom): The red, blue, and green solid curves (bottom panel) represent the separate contributions of the HCOCHCN and CH2CNH products from channel (1f), (1b), and (2b), respectively, to the calculated total LAB angular distribution. The black curve of HCOCHCN from O(3P) in the top panel corresponds to the red curve in the bottom panel. | ||
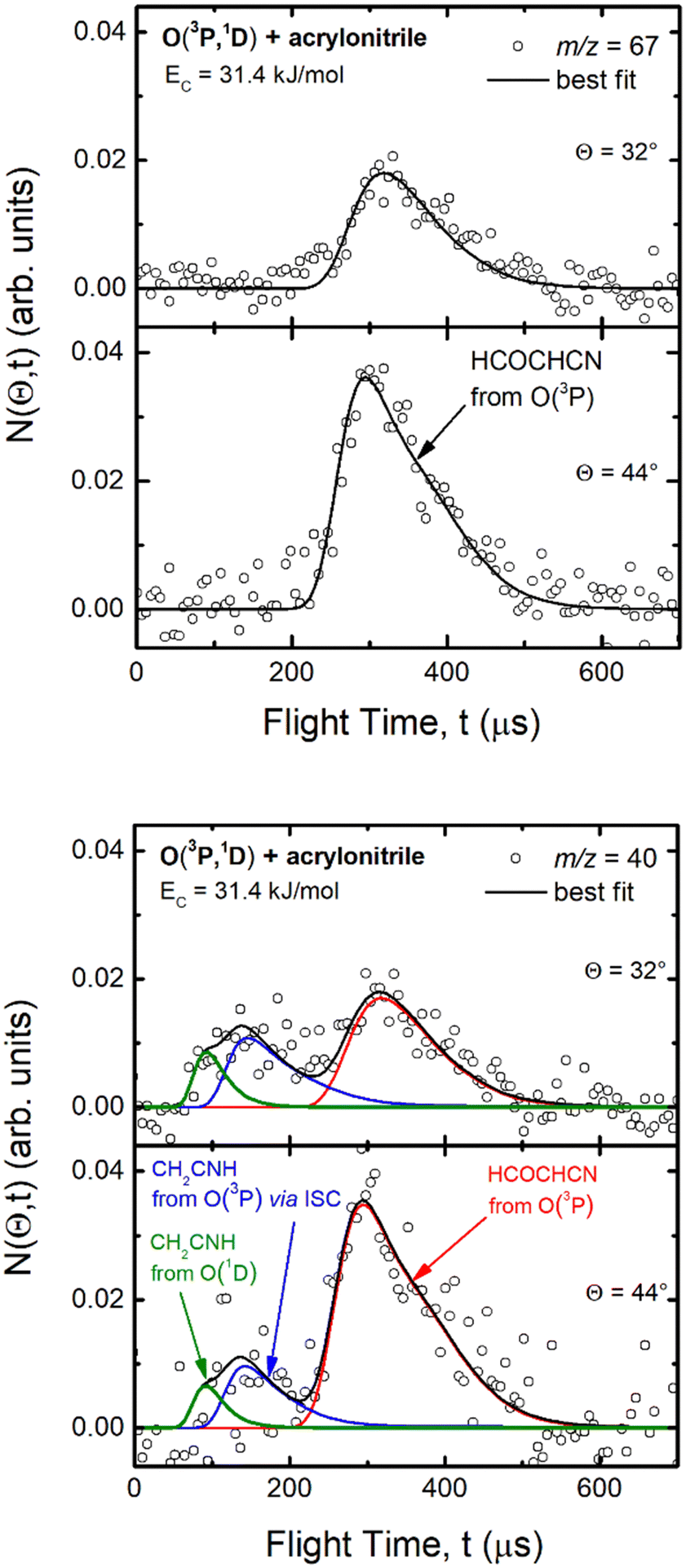 | ||
| Fig. 2 Time-of-flight distributions at the indicated LAB angles of (top) the HCOCHCN product detected at m/z = 67 (C3HNO+), and (bottom) the HCOCHCN and CH2CNH products detected at m/z = 40 (C2H2N+) for the O(3P,1D) + acrylonitrile reactions at Ec = 31.4 kJ mol−1. The empty circles are experimental data, while the solid black curve represents the calculated (total) distribution when using the best-fit functions shown in Fig. 4. (bottom): The red, blue, and green solid curves represent the separate contributions of the indicated products to the calculated global TOF distribution. The black curve of HCOCHCN from O(3P) in the top panel corresponds to the red curve in the bottom panel. | ||
As discussed below, from the experimental data we have characterized channels (1f), (1b), and (2b) by measuring angular and TOF distributions at m/z = 67 and 40, respectively and we have determined their relative yields (BFs).
The velocity vector (so-called “Newton”) diagram of the experiment is depicted in Fig. 3, where only the limiting circles of the observed products are indicated. The circle associated to the CH2O (+ HCCN) product from the O(3P) reaction (channel (1g)) was also added to the Newton diagram because it will be useful for the discussion of the results.
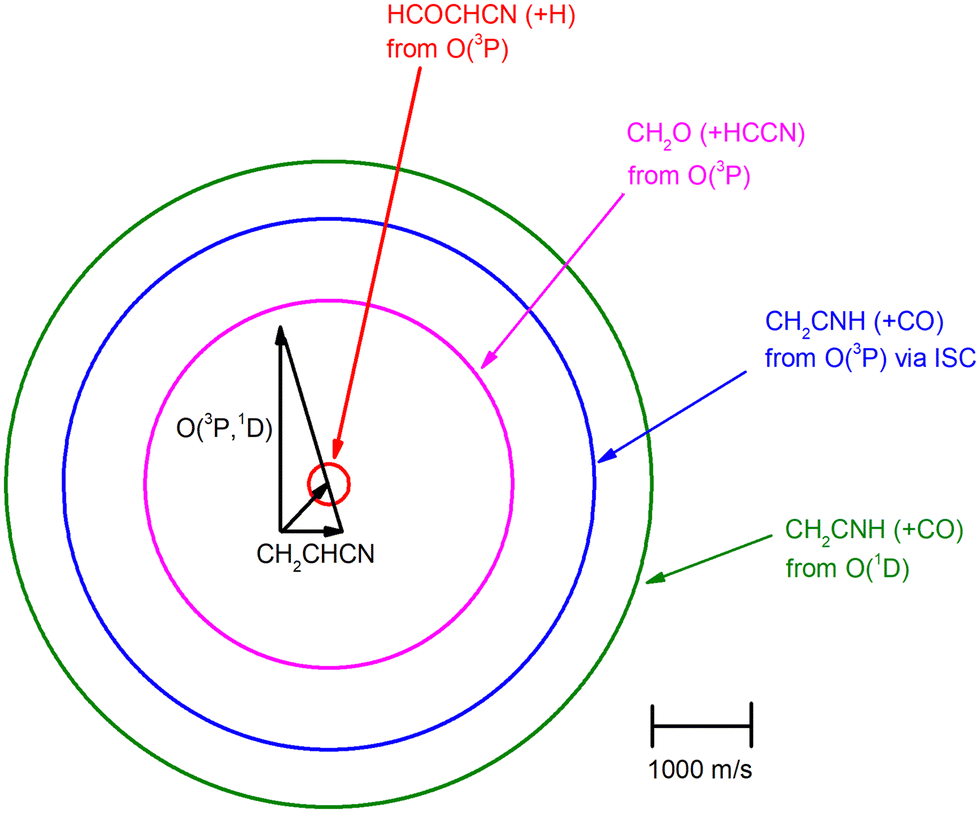 | ||
| Fig. 3 Velocity vector (Newton) diagram of the experiment. The radius of each circle represents the maximum velocity that the indicated products HCOCHCN, CH2CNH, and CH2O can attain in the CM system if all available energy is channeled into product recoil energy (see text). | ||
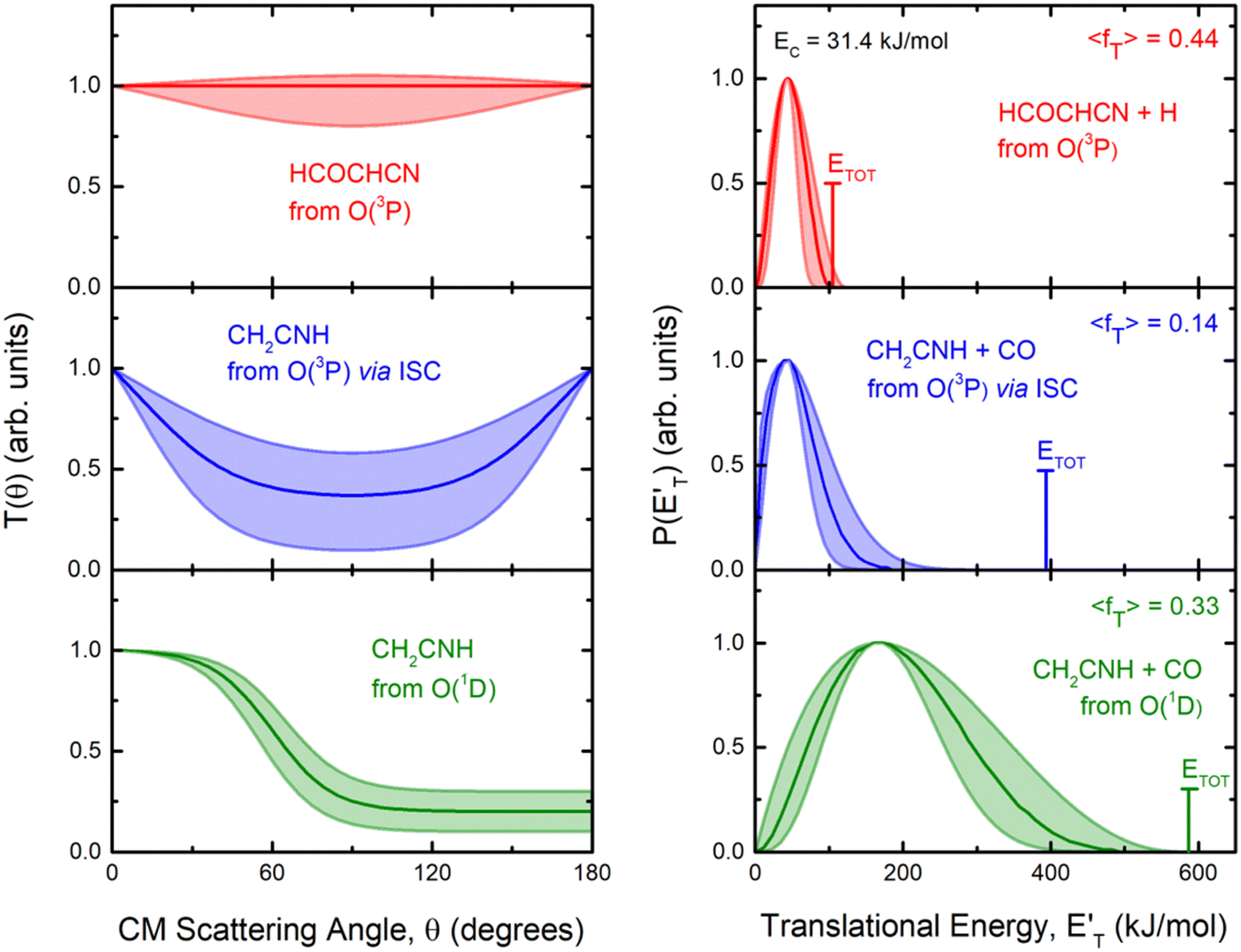 | ||
Fig. 4 Best-fit product angular, T(θ), (left) and translational energy, 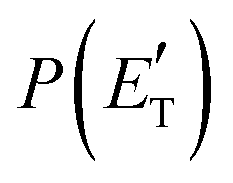 , (right) distributions in the CM reference system for the O(3P,1D) + acrylonitrile reactions. The shaded areas represent the error bars determined for the best-fit CM functions. The vertical arrow in the graphs of , (right) distributions in the CM reference system for the O(3P,1D) + acrylonitrile reactions. The shaded areas represent the error bars determined for the best-fit CM functions. The vertical arrow in the graphs of 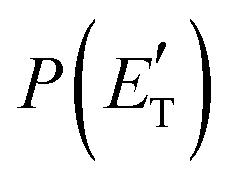 indicates the total energy indicates the total energy 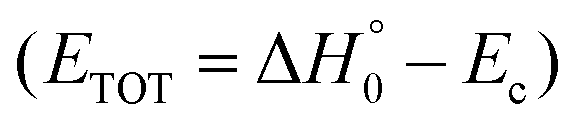 for the corresponding reactive channel. for the corresponding reactive channel. | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) K = 7.3 × 10−13 cm3 mol−1 s−1 for the O(3P) + C2H4 reaction92 and k298K = 4.9 × 10−13 cm3 mol−1 s−1 for the O(3P) + CH2CHCN reaction40). For the system involving ethylene, pulsed CMB studies at Ec = 6 kcal mol−1 and 3 kcal mol−1 with VUV photoionization detection93,94 revealed that the contribution of O(1D) cannot be neglected and its role was also confirmed in the CMB experiment performed at Ec = 35.1 kJ mol−1 by our research group.90 The second reason is that the system involving atomic oxygen and cyanoacetylene (another nitrile molecule, with gross formula HC3N), recently studied by our research group at Ec = 31.1 kJ mol−1,91 characterized by an entrance energy barrier on the triplet PES similar to that of O(3P) + acrylonitrile (9 kJ mol−1vs. 7 kJ mol−1, at CBS level), had shown unambiguous evidence of the O(1D) reaction in the TOF spectra of the HCCN coproduct of CO registered at the daughter ion m/z = 38. So, it is reasonable to state that, in the present experimental investigation also, the O(1D) + acrylonitrile reaction contributes to the total reactive signal registered at m/z = 40. Therefore, in order to correctly account for the contribution of CH2CNH formed adiabatically on the singlet PES to the signal recorded at m/z = 40, we have taken the O(3P,1D) + cyanoacetylene reactions91 as reference because the two systems, having a comparable entrance potential energy barrier on the triplet PES and having been studied at nearly equal collision energy, have been investigated by using an oxygen beam generated under the same conditions (in the experimental study of the O(3P,1D) + ethylene reactions the diameter of the nozzle was different) and because the reaction pathway under analysis is the same, i.e., a CO-forming channel (in the experiment involving ethylene, however, the CO-forming channel was not observed to occur90,93,94).
K = 7.3 × 10−13 cm3 mol−1 s−1 for the O(3P) + C2H4 reaction92 and k298K = 4.9 × 10−13 cm3 mol−1 s−1 for the O(3P) + CH2CHCN reaction40). For the system involving ethylene, pulsed CMB studies at Ec = 6 kcal mol−1 and 3 kcal mol−1 with VUV photoionization detection93,94 revealed that the contribution of O(1D) cannot be neglected and its role was also confirmed in the CMB experiment performed at Ec = 35.1 kJ mol−1 by our research group.90 The second reason is that the system involving atomic oxygen and cyanoacetylene (another nitrile molecule, with gross formula HC3N), recently studied by our research group at Ec = 31.1 kJ mol−1,91 characterized by an entrance energy barrier on the triplet PES similar to that of O(3P) + acrylonitrile (9 kJ mol−1vs. 7 kJ mol−1, at CBS level), had shown unambiguous evidence of the O(1D) reaction in the TOF spectra of the HCCN coproduct of CO registered at the daughter ion m/z = 38. So, it is reasonable to state that, in the present experimental investigation also, the O(1D) + acrylonitrile reaction contributes to the total reactive signal registered at m/z = 40. Therefore, in order to correctly account for the contribution of CH2CNH formed adiabatically on the singlet PES to the signal recorded at m/z = 40, we have taken the O(3P,1D) + cyanoacetylene reactions91 as reference because the two systems, having a comparable entrance potential energy barrier on the triplet PES and having been studied at nearly equal collision energy, have been investigated by using an oxygen beam generated under the same conditions (in the experimental study of the O(3P,1D) + ethylene reactions the diameter of the nozzle was different) and because the reaction pathway under analysis is the same, i.e., a CO-forming channel (in the experiment involving ethylene, however, the CO-forming channel was not observed to occur90,93,94).
Analyzing the TOF spectra, it should be noted that the fingerprint of the CH2CNH + CO channel from the reaction of O(3P) and O(1D) is clearer at Θ = 32° than at the angle Θ = 44° (that is, very near to the center of mass angle of 45°), because at ΘCM the relative contribution of the heavy coproduct of the H-displacement channel (1f) is strongly amplified for kinematic reasons (by the CM → LAB transformation Jacobian60), being scattered over a much narrower LAB angular range (see the Newton diagram in Fig. 3). The fact that the angular distribution of CH2CNH (formed in channels (1b) and (2b)) is much wider than that of HCOCHCN (formed in channel (1f)), as well as that the TOF peaks associated with the two contributions of CH2CNH are much faster than those associated with HCOCHCN, is due to two reasons: (i) the main one is that the CH2CHCN product is scattered over a much larger angular range (see the Newton circles in Fig. 3) compared to the HCOCHCN product because CO (the coproduct of CH2CNH) is much heavier than H (the coproduct of HCOCHCN); (ii) in addition, channels (1b) and (2b) are much more exothermic than channel (1f) (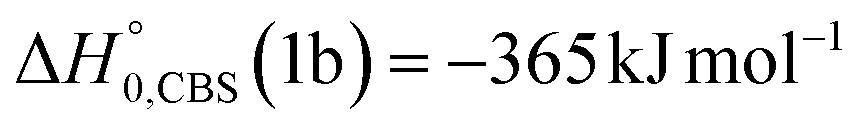 ,
, 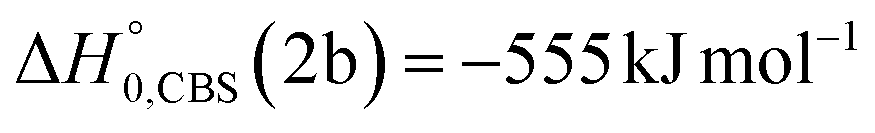 vs.
vs.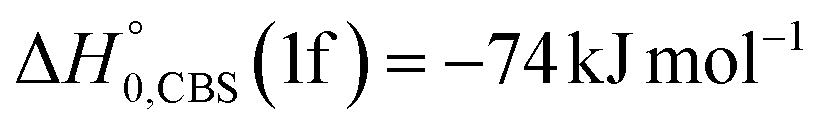 ).
).
In order to characterize channel (1g) (CH2O + HCCN), we attempted to measure the angular distribution at m/z = 30, corresponding to the parent ion of formaldehyde (CH2O), but there was no detectable signal; at the same time, the angular distribution at m/z = 39, corresponding to the parent ion of HCCN (the coproduct of formaldehyde), resulted to be perfectly superimposable (within the error bars) to the angular distribution measured at m/z = 67: therefore, the signal at m/z = 39 can only be associated to a fragment of the heavy coproduct of the H-displacement channel, whose dynamics was already characterized from measurements at m/z = 67.
Formaldehyde could also undergo dissociative ionization to HCO+ (m/z = 29), which could also be the parent ion of the HCO product of channels (1d)/(2d). However, our attempt to measure reliable angular and TOF distributions at m/z = 29 failed because the S/N ratio was too low due to the unfavorable kinematics and the high inherent detector background at m/z = 28 connected with residual CO in the HUV chamber of the MS detector, that significantly affects m/z = 29 also because of the natural isotopic abundance of 13C. Furthermore, we could not subtract the contribution associated with the elastically/inelastically scattered acrylonitrile beam, that still dissociates in the ionizer to a fragment with m/z = 28 (that influences also the signal at m/z = 29 due to 13C). So, we must rely on the RRKM statistical estimates of the BFs in order to learn if channels (1g) and (1d)/(2d) are both present or not and what eventually is their relative importance to the collision energy of the experiment (see below).
Regarding channel (1e)/(2e) (COCN + CH3), there was no signal at m/z = 15 (CH3+). Hence, we can rule out, within our sensitivity (i.e., BF ≤ 2–3%), the occurrence of reactive channels (1e)/(2e).
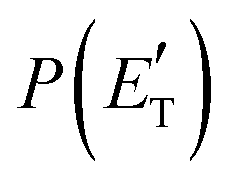 functions and reaction mechanism.
Quantitative information on the reaction dynamics is obtained by moving from the LAB frame to the CM frame and analyzing the product angular, T(θ), and translational energy,
functions and reaction mechanism.
Quantitative information on the reaction dynamics is obtained by moving from the LAB frame to the CM frame and analyzing the product angular, T(θ), and translational energy, 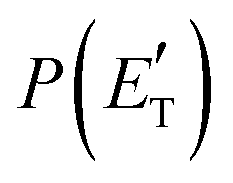 , distributions, into which the total CM product flux can be factorized (see Section 2). The black solid curves superimposed on the experimental results in Fig. 1 and 2 are the total calculated LAB angular and TOF distributions (at the indicated m/z) when using the best-fit CM functions T(θ) and
, distributions, into which the total CM product flux can be factorized (see Section 2). The black solid curves superimposed on the experimental results in Fig. 1 and 2 are the total calculated LAB angular and TOF distributions (at the indicated m/z) when using the best-fit CM functions T(θ) and 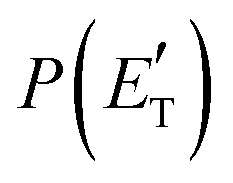 depicted in Fig. 4, for the contributing reaction channels. The partial contributions at a given m/z value are indicated with color coded lines.
depicted in Fig. 4, for the contributing reaction channels. The partial contributions at a given m/z value are indicated with color coded lines.
By examining the best-fit T(θ) and 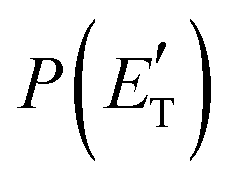 functions for the main competing channels (1f), (1b), and (2b) shown in Fig. 4, we can note that the two best-fit T(θ) functions related to the channels of the O(3P) + acrylonitrile reaction are backward–forward symmetric; however, while the CM angular distribution of the HCOCHCN radical is found to be isotropic, i.e., constant within the error bars in the whole CM angular range (θ = 0°–180°), the T(θ) of ketenimine is fairly polarized at the poles (θ = 0° and θ = 180°). These characteristics are consistent with the occurrence of a long-lived complex mechanism (i.e., the intermediate complex lifetime is longer than 5–6 rotational periods).95–97 The different degree of polarization of the two T(θ) functions can be rationalized in terms of the different partitioning of the total angular momentum for the two different product channels.95–98 In fact, the total momentum conservation law can be written in the form Ji= Li+ ji= Jf= Lf+ jf, with Ji, Li, ji, JfLf, and jf, being the initial and final total, orbital, and rotational angular momenta, respectively. We remind that ji can usually be neglected in a crossed molecular beam experiment because the supersonic expansion causes a strong rotational cooling of the molecular reactant. For the H-displacement channel, it is expected to have jf ≫ Lf because L is directly proportional to the reduced mass μ of the products, and is therefore small as μ is small (L = μvb, where b is the exit impact parameter); so, from the total momentum conservation law, one has Lf ≪ Li, for the H-displacement channel and the products can be scattered isotropically, which implies a high rotational excitation of the molecular product HCOCHCN.99 In contrast, when Lf ≅ Li (as for the CO-forming channel) (and therefore jf is small), and the plane of the relative velocity vector
functions for the main competing channels (1f), (1b), and (2b) shown in Fig. 4, we can note that the two best-fit T(θ) functions related to the channels of the O(3P) + acrylonitrile reaction are backward–forward symmetric; however, while the CM angular distribution of the HCOCHCN radical is found to be isotropic, i.e., constant within the error bars in the whole CM angular range (θ = 0°–180°), the T(θ) of ketenimine is fairly polarized at the poles (θ = 0° and θ = 180°). These characteristics are consistent with the occurrence of a long-lived complex mechanism (i.e., the intermediate complex lifetime is longer than 5–6 rotational periods).95–97 The different degree of polarization of the two T(θ) functions can be rationalized in terms of the different partitioning of the total angular momentum for the two different product channels.95–98 In fact, the total momentum conservation law can be written in the form Ji= Li+ ji= Jf= Lf+ jf, with Ji, Li, ji, JfLf, and jf, being the initial and final total, orbital, and rotational angular momenta, respectively. We remind that ji can usually be neglected in a crossed molecular beam experiment because the supersonic expansion causes a strong rotational cooling of the molecular reactant. For the H-displacement channel, it is expected to have jf ≫ Lf because L is directly proportional to the reduced mass μ of the products, and is therefore small as μ is small (L = μvb, where b is the exit impact parameter); so, from the total momentum conservation law, one has Lf ≪ Li, for the H-displacement channel and the products can be scattered isotropically, which implies a high rotational excitation of the molecular product HCOCHCN.99 In contrast, when Lf ≅ Li (as for the CO-forming channel) (and therefore jf is small), and the plane of the relative velocity vector 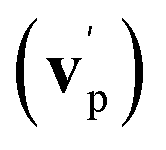 of the products is the same of that of the reagents (vr), the T(θ) function is backward–forward symmetric with a maximum at θ = 0° and θ = 180°.95–99 Regarding the O(1D) + CH2CHCN reaction, the T(θ) function for the CO-forming channel (2b) was found to be strongly forward biased, as in the case of the O(1D) + HCCCN (cyanoacetylene) reaction,91 indicating an osculating complex mechanism.97
of the products is the same of that of the reagents (vr), the T(θ) function is backward–forward symmetric with a maximum at θ = 0° and θ = 180°.95–99 Regarding the O(1D) + CH2CHCN reaction, the T(θ) function for the CO-forming channel (2b) was found to be strongly forward biased, as in the case of the O(1D) + HCCCN (cyanoacetylene) reaction,91 indicating an osculating complex mechanism.97
Regarding the product translational energy distributions, the 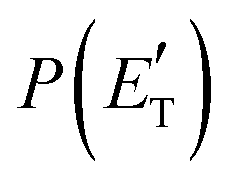 functions of the two reactive channels of the O(3P) + acrylonitrile reaction exhibit a peak well away from zero (at ∼44 kJ mol−1 for both channels), an indication that the two reactive channels have a sizeable exit potential energy barrier. The
functions of the two reactive channels of the O(3P) + acrylonitrile reaction exhibit a peak well away from zero (at ∼44 kJ mol−1 for both channels), an indication that the two reactive channels have a sizeable exit potential energy barrier. The 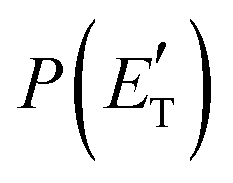 of channel (1f) extends up to the total available energy
of channel (1f) extends up to the total available energy 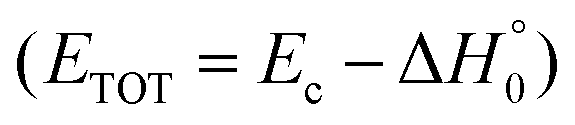 of the channel and the average product translational energy, defined as
of the channel and the average product translational energy, defined as 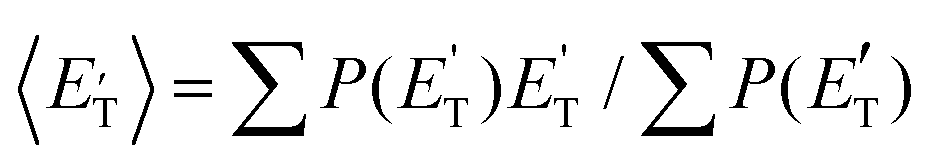 , is 47 kJ mol−1 which corresponds to a fraction,
, is 47 kJ mol−1 which corresponds to a fraction, 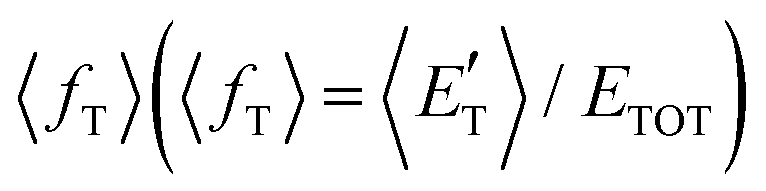 , of the total available energy released in translation of 0.44. This means that the energy of the products is partitioned nearly equally between the translational and internal (ro-vibrational) degrees of freedom. In contrast, the
, of the total available energy released in translation of 0.44. This means that the energy of the products is partitioned nearly equally between the translational and internal (ro-vibrational) degrees of freedom. In contrast, the 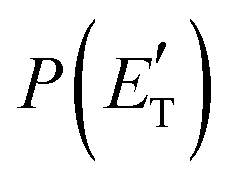 of channel (1b) has a cut-off at about 190 kJ mol−1, which is much less than the total energy available for the reaction channel (1b) (∼396 kJ mol−1, which is the energy calculated at the CCSD(T)/CBS level of theory), and the molecular product (CH2CNH) is highly internal excited, with
of channel (1b) has a cut-off at about 190 kJ mol−1, which is much less than the total energy available for the reaction channel (1b) (∼396 kJ mol−1, which is the energy calculated at the CCSD(T)/CBS level of theory), and the molecular product (CH2CNH) is highly internal excited, with 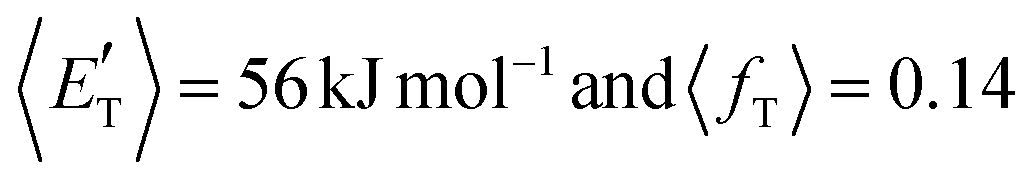 . This indicates that about 85% of the total available energy is channeled into internal ro-vibrational excitation of the CH2CNH and CO products. Notably, as will be shown in Section 4.2, the peaking of the
. This indicates that about 85% of the total available energy is channeled into internal ro-vibrational excitation of the CH2CNH and CO products. Notably, as will be shown in Section 4.2, the peaking of the 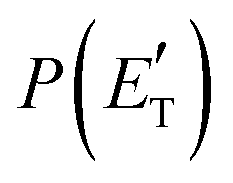 for the CH2CNH + CO channel is in line with the high (about 130 kJ mol−1 with respect to products) exit barrier for this channel (see Fig. 6), and much less with the very high (340 kJ mol−1 with respect to products) exit barrier to the competing, more exothermic, isomeric channel (1a) (CH3CN + CO), which, because of the very high transition state 1TS4 (from 1MIN3), is not expected to have a large BF, despite being more exothermic than channel (1b), as discussed further below. Finally, the
for the CH2CNH + CO channel is in line with the high (about 130 kJ mol−1 with respect to products) exit barrier for this channel (see Fig. 6), and much less with the very high (340 kJ mol−1 with respect to products) exit barrier to the competing, more exothermic, isomeric channel (1a) (CH3CN + CO), which, because of the very high transition state 1TS4 (from 1MIN3), is not expected to have a large BF, despite being more exothermic than channel (1b), as discussed further below. Finally, the 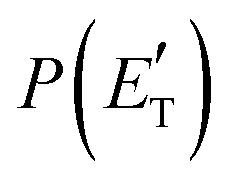 distribution of the CH2CNH-forming channel (2b) peaks very far away from zero, at 167 kJ mol−1, and dies off at ∼490 kJ mol−1, which is less than the total energy available for the reaction channel (2b) (∼586 kJ mol−1). The corresponding average product translational energy and the fraction of the total energy channeled into translation are 195 kJ mol−1 and 0.33, respectively.
distribution of the CH2CNH-forming channel (2b) peaks very far away from zero, at 167 kJ mol−1, and dies off at ∼490 kJ mol−1, which is less than the total energy available for the reaction channel (2b) (∼586 kJ mol−1). The corresponding average product translational energy and the fraction of the total energy channeled into translation are 195 kJ mol−1 and 0.33, respectively.
| Reactants | Primary products | PES involved | Global BFs | BFs for the O(3P) + CH2CHCN reaction |
|---|---|---|---|---|
| O(3P) + CH2CHCN | HCOCHCN + H | Triplet | 8 ± 3% | 13 ± 5% |
| CH2CNH + CO | Singlet via ISC | 55 ± 17% | 87 ± 5% | |
| O(1D) + CH2CHCN | CH2CNH + CO | Singlet | 37 ± 13% | — |
The BFs for the O(3P) + acrylonitrile reaction reported in the last column of Table 1 are obtained from the global BFs (second last column of Table 1) by normalizing the sum of BFs of the two O(3P) channels to unity. The derived BFs for the O(3P) + acrylonitrile reaction indicate a very strong preference for formation of CH2CNH + CO (BF = 87%), while the competing HCOCHCN + H-displacement channel has BF = 13%. Uncertainties in the derived BFs vary from about ±30% to ±40% and are indicated in Table 1.
Because the calculated yield of the H channel from the singlet PES is almost negligible (BF = 1.2%, see Section 4.3), we can conclude that the extent of ISC in the O(3P) + CH2CHCN reactions at Ec = 31.4 kJ mol−1 is, therefore, about 87%.
4.2 Triplet and singlet OCH2CHCN potential energy surfaces
The overall triplet/singlet PES obtained theoretically for the reactions O(3P,1D) + CH2CHCN shows 12 different minima and 34 possible transition states. A total number of 13 possible product channels, energetically located below the experimental collision energy, can be formed. For the sake of clarity, in Fig. 5 we have reported a simplified version of the triplet PES, considering only the main channels, leading to appreciable values of product BFs, with the energies reported at the CCSD(T)/aug-cc-pVTZ level of theory. The energies computed at the CCSD(T)/CBS level of theory for the most relevant channels are given in parentheses. A global overview of the triplet PES including all possible isomerization processes of the intermediates, together with all the energetically accessible product channels, is reported in Fig. S1 of the ESI.†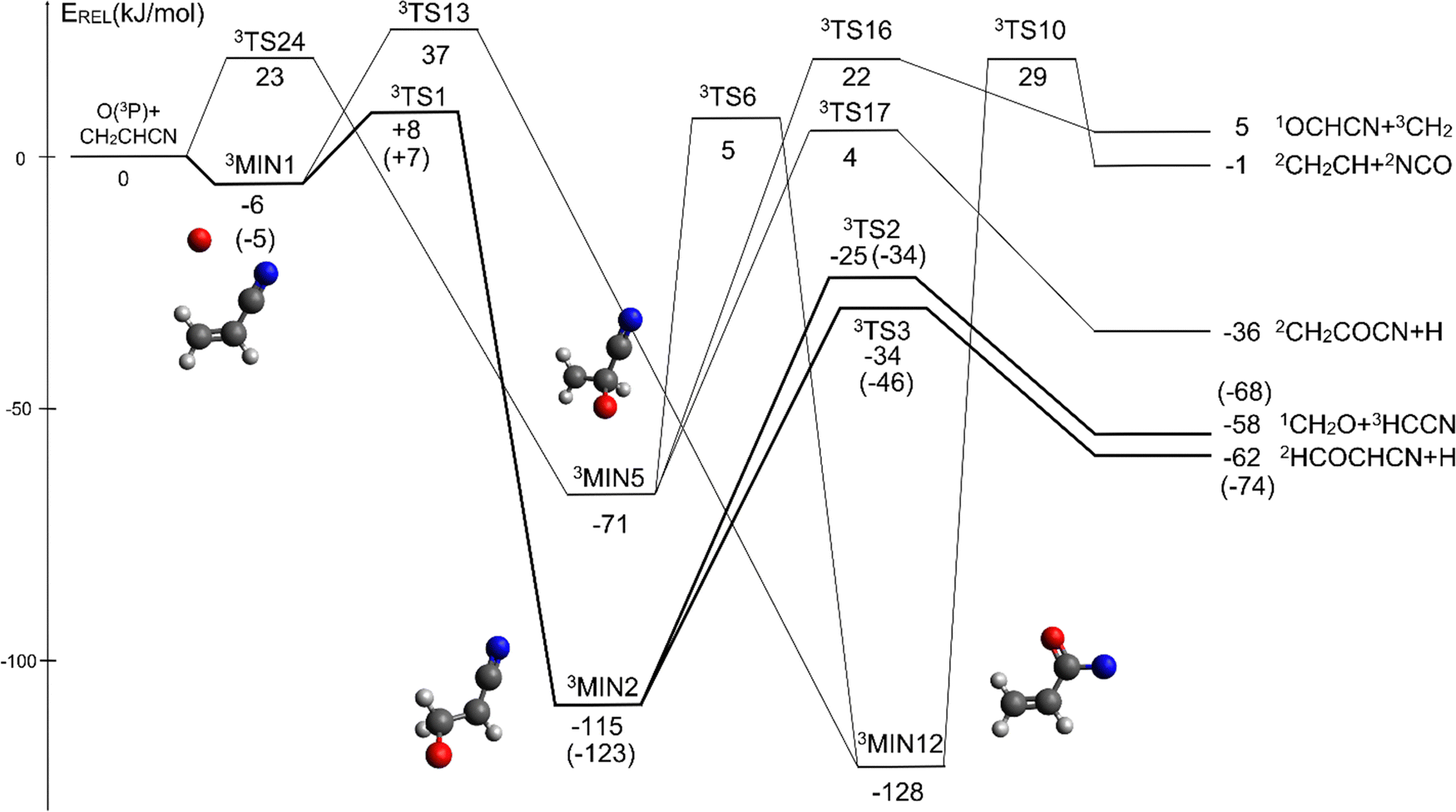 | ||
| Fig. 5 Schematic representation of the simplified triplet potential energy surface for the O(3P) + acrylonitrile reaction with energies evaluated at the CCSD(T)/aug-cc-pVTZ level of theory; the energies computed at the CCSD(T)/CBS level of theory are reported in parentheses for the most relevant channels (see text). | ||
The reaction of O(3P) with CH2CHCN starts with the initial formation of the weakly bound van der Waals (vdW) adduct 3MIN1, where the oxygen atom interacts with the acrylonitrile molecule at long distances, larger than 3 Å. The so formed weakly bound complex is located 6 (5) kJ mol−1 below the reactant energy asymptote. A potential energy barrier (3TS1), whose relative energy is +8 (+7) kJ mol−1, must be overcome in order to form the intermediate 3MIN2, located 115 (123) kJ mol−1 below the reactant energy asymptote, in which a new C1–O bond is formed. Addition on C1 is the most favorable site of attack of O(3P).
Once formed, MIN2 can dissociate to form HCOCHCN + H (channel (1f)), the most exothermic channel on the triplet PES, through an exit barrier (3TS3) of 81 kJ mol−1 (77 kJ mol−1 at the CBS level) from 3MIN2, that is of 28 kJ mol−1 (at the CBS level) with respect to products (see Fig. 5). The related transition state 3TS3 clearly shows the breaking of the C1-H bond, with a bond distance of 1.831 Å (see Fig. S6 in the ESI†). In addition, the fission of the C1–C2 σ bond allows the formation of formaldehyde (CH2O), together with the 3HCCN (cyanomethylene) coproduct (channel (1g)). Also in this case, an exit barrier (3TS2) of 90 (89) kJ mol−1 from 3MIN2 (that is, + 33 (34) kJ mol−1 with respect to the product asymptote), is present and the related transition state, 3TS2, shows a C1–C2 distance of 2.108 Å (see Fig. S6 in the ESI†). Alternatively, the oxygen atom can add to the C3 atom of the CN group of the acrylonitrile molecule, leading to the formation of 3MIN12 via3TS13 (located 37 kJ mol−1 above the reactants) (see Fig. 5). The breaking of the C2–C3 bond in 3MIN12 would lead to formation of CH2CH (vinyl) + NCO (channel (1i)), via the transition state 3TS10 located 29 kJ mol−1 above the energy of the reactants. However, because of the high energy of 3TS13 and 3TS10, this pathway is expected to contribute negligibly to the product yield under our experimental conditions.
A different reaction mechanism is represented by the attack of O(3P) on the C2 of the acrylonitrile molecule. In this case the related transition state (3TS24) is located 23 kJ mol−1 above the energy of the reactants (see Fig. 5). Once formed, the intermediate 3MIN5, which shows a relative energy of −71 kJ mol−1, can isomerize to the above 3MIN12, through a barrier (3TS6) of 76 kJ mol−1. Alternatively, a hydrogen elimination process can take place from 3MIN5, leading to the formation of CH2COCN + H (channel (1h)), through a barrier of 75 kJ mol−1, located 4 kJ mol−1 above the reactants and represented by the transition state 3TS17. Also in this case, the fission of the C1–C2 σ bond is possible, leading to the formation of a slightly endothermic (by 5 kJ mol−1) 3CH2 + 1OCHCN product channel (1j). However, channels (1j), (1h), and (1i) are expected, on energetic grounds, to be minor at Ec = 31.4 kJ mol−1 because of the high barriers involved, and only the channels (1f) and (1g) are likely significant on the triplet PES.
The singlet PES shows 13 different minima and 21 possible transition states, leading to 18 energetically possible products at the experimental collision energy. Following the same approach described for the triplet PES, in Fig. 6 we have reported a simplified version of the singlet PES, while a global picture is portrayed in Fig. S2 of the ESI.† In Fig. 6 the singlet PES is reported in blue lines, with energies evaluated at the CCSD(T)/aug-cc-pVTZ level of theory for all stationary points and at the CCSD(T)/CBS level of theory (values in parentheses) for the main reactive channels. In the same figure, the most relevant stationary points of the triplet PES are also reported in red, together with the qualitative indication (dashed ellipse) of the region of ISC between the triplet/singlet PESs. The energy of all identified stationary points, including the possible products, in the singlet PES is referred to the reactants O(3P) + CH2CHCN.
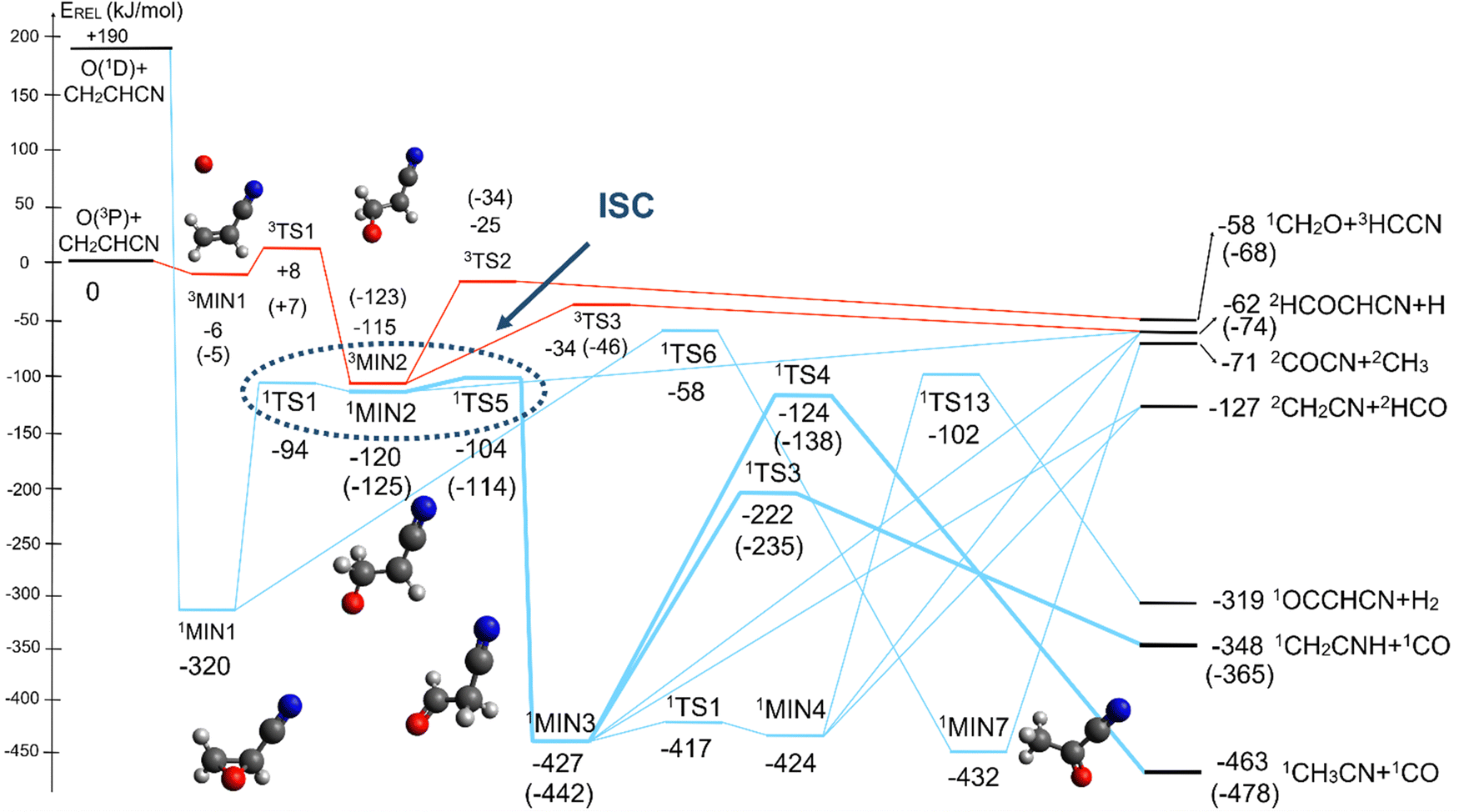 | ||
| Fig. 6 Schematic representation of the simplified singlet potential energy surface (in blue) for the O(3P,1D) + acrylonitrile reactions with energy evaluated at the CCSD(T)/aug-cc-pVTZ level of theory and at the CCSD(T)/CBS level of theory (data in parentheses) for the most relevant channels (see text). In both cases the zero of the energy is represented by the O(3P) + CH2CHCN reactants. A simplified representation of the two most important channels of the triplet PES (in red) is also reported, together with a possible identification of the region of ISC (blue dashed ellipse). | ||
The barrierless addition of O(1D) to the acrylonitrile molecule leads to the formation of the cyclic intermediate 1MIN1, located 320 kJ mol−1 below the O(3P) reactant energy asymptote. 1MIN1, by migration of a hydrogen atom from the C2 to the C1 carbon, together with the breaking of the O–C1 bond, can lead to the formation of the intermediate 1MIN7, located 432 kJ mol−1 below the reactant energy asymptote. The related transition state (1TS6) shows a relative energy of −58 kJ mol−1 with respect to reactants.
The breaking of the C1–C2 σ bond in 1MIN7 can lead to the formation of COCN + CH3 (channel (2e)) through a barrierless process. 1MIN1 can also isomerize to the 1MIN2 intermediate, by overcoming a barrier (1TS1) of 214 (211) kJ mol−1 from 1MIN1. It should be noted that 1MIN2 can be reached also from a nonadiabatic transition (ISC) from the triplet to the singlet PES, because of the closeness in energy of 3MIN2 and 1MIN2 (see Fig. 6). Once formed, 1MIN2 can rapidly undergo two different competitive processes. The migration of a hydrogen atom via the transition state 1TS5 from the C1 to the C2 carbon atom of 1MIN2 leads to formation of a very stable intermediate 1MIN3 (located at −427 (−442) kJ mol−1 with respect to the reactants). Competitively, a barrierless H-displacement process is possible, leading to the formation of the HCOCHCN + H products (channels (1f)/2(f)). Two main products can be formed starting from 1MIN3. The breaking of the C1–C2 bond, together with the migration of the H atom from C1 to the N atom of the CH2CN moiety, leads to the formation of CH2CNH (ketenimine) + CO (channels (1b)/(2b)), through an exit barrier, represented by 1TS3, located −222 kJ mol−1 below the reactant energy asymptote (−235 kJ mol−1 at the CBS level), of 126 (130) kJ mol−1 (with respect to products), showing a global channel exothermicity of 348 (365) kJ mol−1. This is the lowest energy pathway to products on the singlet PES. Alternatively, a barrier (1TS4, located at −124 (−138) kJ mol−1) of 303 (304) kJ mol−1 (from 1MIN3) must be overcome in order to have the C1–C2 hydrogen migration, together with the fission of the C1–C2 σ bond, allowing the formation of the CH3CN + CO isomeric product channel ((1a)/(2a)), which is the most exothermic of all channels 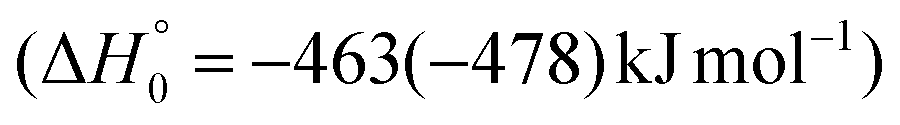 . However, the very high energy of the transition state 1TS4 (98 (97) kJ mol−1 higher than 1TS3) should make channels (1a)/(2a) much less likely than channels (1b)/(2b). Competitively, 1MIN3 can also undergo isomerization. The rotation around the C1–C2 bond leads to the formation of the intermediate 1MIN4, located 424 kJ mol−1 below the reactant energy asymptote. The barrierless C1–C2 σ bond breaking in 1MIN4 can allow the formation of CH2CN + HCO (channels (1d)/(2d))
. However, the very high energy of the transition state 1TS4 (98 (97) kJ mol−1 higher than 1TS3) should make channels (1a)/(2a) much less likely than channels (1b)/(2b). Competitively, 1MIN3 can also undergo isomerization. The rotation around the C1–C2 bond leads to the formation of the intermediate 1MIN4, located 424 kJ mol−1 below the reactant energy asymptote. The barrierless C1–C2 σ bond breaking in 1MIN4 can allow the formation of CH2CN + HCO (channels (1d)/(2d)) 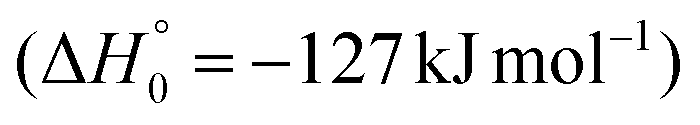 . In addition, together with the barrierless formation of H + HCOCHCN (channels (1f)/(2f)), 1MIN4 can also lead, via1TS13, to the formation of 1OCCHCN + H2 (channels (1c)/(2c))
. In addition, together with the barrierless formation of H + HCOCHCN (channels (1f)/(2f)), 1MIN4 can also lead, via1TS13, to the formation of 1OCCHCN + H2 (channels (1c)/(2c)) 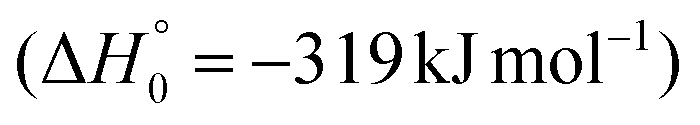 . Again, channels (1d)/(2d) and (1c)/(2c) are expected, on energetic grounds, to be less favorable than channel (1b)/(2b) (see Fig. 6).
. Again, channels (1d)/(2d) and (1c)/(2c) are expected, on energetic grounds, to be less favorable than channel (1b)/(2b) (see Fig. 6).
It is interesting to compare the theoretical data of the energies for the different stationary points for the main reactive channels obtained at the CCSD(T) level with those obtained when using the aug-cc-pVTZ basis set and the complete basis set (CBS) algorithm presented in Section 3.1. Some differences can be appreciated and, in particular, the global exothermicity values evaluated with the CBS method are lower than the respective aug-cc-pVTZ values. In general, all stationary points at the CBS level appear to be lower in energy, with respect to those at the aug-cc-pVTZ level, by about 8–16 kJ mol−1. It is noteworthy that the most significant variations in energy are related to some of the stationary points identified in the singlet PES. The main reason of the discrepancies is the multi-reference character of the intermediates and stationary points, evaluated through the T1 factor, which assumes values of 0.023, 0.030, 0.014 and 0.016 for 3MIN2, 3TS2, 1MIN3 and 1TS3, respectively. As already suggested,46 one of the main processes showing a strong multireference character is the isomerization of the oxiranyl singlet intermediate to aldehydes, similar to the case of the isomerization of 1MIN1 to 1MIN2 and 1MIN3. In this case, multireference calculations would be the best solution to provide accurate energy values. In the present work, as a consequence of the topology of the PES, no particular deviations in the values of the final BFs (see below) were experienced when using CCSD(T) and CCSD(T)-CBS values. Therefore, it is possible to conclude that the calculations performed at the CCSD(T) level of theory afford, for our purpose, a good estimation of the energy of the stationary points.
The geometry of the most relevant stationary points for both PESs (including intermediates, transition states and products) is reported in Fig. S6–S8 in the ESI.†
4.3 Statistical branching fractions
The kinetic calculations were performed using the energies obtained from the CCSD(T)/CBS extrapolations. These calculations (see Section 3.2) were performed at four different temperatures (10 K, 100 K, 200 K, and 298 K (room temperature)), as well as at the collision energy of the present CMB experiment.According to the present RRKM calculations, the main reaction channel on the computed triplet PES is the H-displacement channel (1f) starting from intermediate 3MIN2, leading to the formation of HCOCHCN + H, with a value of BF, at the Ec of the experiment, of 62.4%, while the fission of the C–C bond followed by the formation of CH2O (formaldehyde) + 3HCCN represents the second most favorable channel ((1g)), with BF = 37.6% (see Table 2). Due to the presence of high barriers, whose energy lies above the reactant energy asymptote, the other reaction pathways identified on the triplet PES (see Fig. 5), leading to the formation of 1OCHCN + 3CH2 (channel (1j)), CH2CH + NCO (channel (1i)), and CH2COCN + H (channel (1h)), appear to have no contribution to the value of the global branching fractions (see Table 2). On the other hand, as can be seen from Table 3, according to the present RRKM analysis, the main reaction channel on the singlet PES is, by far, the molecular channel leading to formation of CH2CNH (ketenimine) + CO (channel (2b)), with BF = 94.8% at the experimental collision energy. The second and the third most abundant channels are associated with the formation of CH3CN + CO (channel (2a)) and HCOCHCN + H (channel (2f)) with BFs of only 2.5% and 1.2%, respectively. At the experimental Ec, the formation of CO accounts for 97.3% of the total reactive flux on the singlet PES. Finally, on this PES, the fourth most abundant channel is formation of CH3 + COCN (channel (2e)), with BF = 1.1%, while the formation of HCO + CH2CN (channel (2d)) and OCCHCN + H2 (channel (2c)) accounts for only 0.3% and 0.03%, respectively, of the total reactive flux.
| Reaction channel | Products | 10 K | 100 K | 200 K | 298 K | E c = 31.4 kJ mol−1 |
|---|---|---|---|---|---|---|
| 1j | 1OCHCN + 3CH2 | ∼0.0% | ∼0.0% | ∼0.0% | ∼0.0% | ∼0.0% |
| 1i | CH2CH + NCO | ∼0.0% | ∼0.0% | ∼0.0% | ∼0.0% | ∼0.0% |
| 1h | CH2COCN + H | ∼0.0% | ∼0.0% | ∼0.0% | ∼0.0% | ∼0.0% |
| 1g | 1CH2O + 3HCCN | 23.6% | 25.3% | 27.6% | 30.2% | 37.6% |
| 1f | HCOCHCN + H | 76.4% | 74.7% | 72.4% | 69.8% | 62.4% |
| Reaction channel | Products | 10 K | 100 K | 200 K | 298 K | E c = 31.4 kJ mol−1 |
|---|---|---|---|---|---|---|
| 2f | HCOCHCN + H | 0.6% | 0.6% | 0.7% | 0.8% | 1.2% |
| 2e | COCN + CH3 | 0.4% | 0.5% | 0.6% | 0.7% | 1.1% |
| 2d | CH2CN + HCO | 0.2% | 0.2% | 0.2% | 0.2% | 0.3% |
| 2c | OCCHCN + H2 | ∼0.0% | ∼0.0% | ∼0.0% | ∼0.0% | 0.03% |
| 2b | 1CH2CNH + CO | 97.6% | 97.4% | 97.0% | 96.6% | 94.8% |
| 2a | 1CH3CN + CO | 1.2% | 1.2% | 1.4% | 1.6% | 2.5% |
If we analyze the variation of the BFs with temperature (see Table 3), we note that on the singlet PES, the decrease in temperature leads to a mild increase in the BF of the dominant, spin-forbidden, channel (2b) (CH2CN + CO) from 94.8% at 31.4 kJ mol−1 (experimental Ec) to 97.6% at 10 K, while the branching fraction of the second and the third most important channels ((2a) and (2f)) decreases from 1.2% and 2.5% to 0.6% and 1.2%, respectively. Analogously, on the triplet PES (see Table 2), the main channel (1f) HCOCHCN + H increases with decreasing energy (in the same energy range) from 62.4% to 76.4%, while in contrast the less exothermic CH2O + HCCN channel (1g) decreases from 37.6% to 23.6%.
Overall, the results in Tables 2 and 3, reporting the values of the distinct BFs on the triplet and singlet PES, respectively, at different temperatures, show that there is little dependence of the BFs on the energy (temperature) available to the system. Since in this work the extent of ISC has not been evaluated theoretically, we will use the experimental BFs to estimate the extent of ISC in the reaction by comparing the experimental BFs with the statistically predicted BFs, separately on the triplet PES and on the singlet PES.
5. Discussion
5.1 Dynamics of the O(3P) + acrylonitrile reaction
The treatment of the experimental and theoretical results in an integrated way allows us to provide a detailed description of the dynamics of the title reactions.Regarding the O(3P) + acrylonitrile reaction, the possibility to fit the angular and TOF distributions at m/z = 67 with a single set of CM functions characterized, in particular, by a 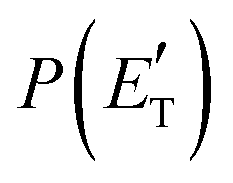 peaking at 44 kJ mol−1, allows us to ascertain the presence of channel (1f) occurring on the triplet PES with an exit energy barrier that is theoretically predicted to be 28 kJ mol−1 (with respect to products) and rule out (within our sensitivity) the barrierless pathways to the same products from the singlet PES via ISC. These findings are corroborated by the statistical estimates of the BFs at the collision energy of the experiment, which have revealed that, among the various H-displacement channels, the most important is the one leading to H + HCOCHCN (channel (1f)) on the triplet PES with a BF = 62.4%, having its analogue on the singlet PES a BF of 1.2% and the others negligible BFs (see Tables 2 and 3). It should be noted that the isotropic CM angular distribution of the HCOCHCN product is perfectly in line with the considerable stability (about −120 kJ mol−1) of 3MIN2 on the triplet PES that can be defined as a long-lived complex at Ec = 31.4 kJ mol−1. Furthermore, the absence of clear traces of the H2-elimination channel in the experimental data registered at m/z = 67 is consistent with the low yield of this channel statistically calculated on the singlet PES at the collision energy of the experiment (BF = 0.03%) (see Table 3).
peaking at 44 kJ mol−1, allows us to ascertain the presence of channel (1f) occurring on the triplet PES with an exit energy barrier that is theoretically predicted to be 28 kJ mol−1 (with respect to products) and rule out (within our sensitivity) the barrierless pathways to the same products from the singlet PES via ISC. These findings are corroborated by the statistical estimates of the BFs at the collision energy of the experiment, which have revealed that, among the various H-displacement channels, the most important is the one leading to H + HCOCHCN (channel (1f)) on the triplet PES with a BF = 62.4%, having its analogue on the singlet PES a BF of 1.2% and the others negligible BFs (see Tables 2 and 3). It should be noted that the isotropic CM angular distribution of the HCOCHCN product is perfectly in line with the considerable stability (about −120 kJ mol−1) of 3MIN2 on the triplet PES that can be defined as a long-lived complex at Ec = 31.4 kJ mol−1. Furthermore, the absence of clear traces of the H2-elimination channel in the experimental data registered at m/z = 67 is consistent with the low yield of this channel statistically calculated on the singlet PES at the collision energy of the experiment (BF = 0.03%) (see Table 3).
It is noteworthy that the RRKM calculations on the triplet PES have predicted a yield of 37.6% for the CH2O-forming channel (Table 2); however, despite this fact, we have ruled out this channel from an experimental point of view due to the absence of reactive signals at m/z = 30 and the total overlap (within the error bars) of the angular distribution recorded at m/z = 39, corresponding to the coproduct of formaldehyde (HCCN), with that obtained at m/z = 67. This apparent contradiction, instead, supports our hypothesis of considering, in the fitting of the two wings of the angular distribution and the fast peak of the TOF spectra at m/z = 40, not only the contribution resulting from CH2CNH formed adiabatically on the singlet PES, but also a contribution associated to the O(3P) + acrylonitrile reaction, that can lead to the formation of CH2CNH via ISC from the triplet to the singlet PES. As a matter of fact, having succeeded in revealing the presence of the HCOCHN + H reactive pathway of the O(3P) + acrylonitrile reaction, if the ketenimine product was formed only from the O(1D) + acrylonitrile reaction, our experimental sensitivity would have been sufficient to detect (despite the less favorable kinematics) CH2O (or HCCN) also, which was predicted to have a BF of ∼38% through statistical computations, as was carried out in other systems in the past.43,45,76 So, considering that 87% of the reactive flux (see Table 1) related to the O(3P) + acrylonitrile reaction is channeled into the singlet PES (via ISC) leading to CH2CNH + CO, the relative yield of the CH2O + HCCN reactive channel, that correlates only with the triplet PES, will be a fraction of the minor H-displacement channel (BF = 13%). Furthermore, the intensity of formaldehyde (and, to a lesser extent, of HCCN) in the LAB frame is strongly reduced because of the unfavorable CM → LAB Jacobian transformation.60 These two combined factors make the sensitivity of the experimental method insufficient to speculate on the CH2O + HCCN channel.
Although the theoretical calculations did not consider the evaluation of the nonadiabatic effect of ISC, its importance in the dynamics of the title reaction is appreciable, as suggested by the experimental results. As a matter of fact, experimentally, the main reaction channel of the O(3P) + acrylonitrile reaction was found to be CH2CNH + CO (channel (1b)), which was accessible only after ISC from the triplet to the singlet PES, with a BF = 87% (see Table 1). The attribution of the experimental signal registered at m/z = 40 to ketenimine (formed by also the O(1D) + acrylonitrile reaction) was supported by two pieces of theoretical evidence: (i) the relatively small BFs for the CH2CN + HCO (BF = 0.3%) and CH3CN + CO (BF = 2.5%) channels, with respect to the channel that involves ketenimine (BF = 94.8%) on the singlet PES and (ii) the presence, for the CH2CNH + CO channel (channel (1b)), of a potential energy barrier that must be overcome to reach the transition state 1TS3 from 1MIN3 which is much lower (205 vs. 303 kJ mol−1) than that involving 1TS4 and leading to the formation of CH3CN + CO (channel (1a)) products. Notably, the possibility of disentangling the two isomeric products with a gross formula C2H3N from the CM functions used to obtain the best-fit of the experimental data is inhibited due to the fact that both functions are in principle consistent with the micro-mechanism and the energetics of the two channels. In fact, the backward–forward symmetric T(θ) distribution is consistent with the occurrence of a long lived complex, that is, 1MIN3 which is common between the two reactive pathways. Then, the fitting is not sensitive to the different exothermicity of CH2CNH + CO and CH3CN + CO channels because the CM translational energy distribution 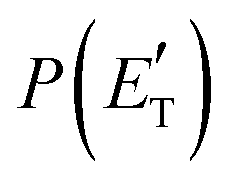 dies off at about 190 kJ mol−1, a value which is much lower than the total energy of both reactive channels (ETOTCBS = 509 kJ mol−1 for CH3CN + CO and ETOTCBS = 396 kJ mol−1 for CO + CH2CNH) (see Fig. 4). However, the fact that the peak of
dies off at about 190 kJ mol−1, a value which is much lower than the total energy of both reactive channels (ETOTCBS = 509 kJ mol−1 for CH3CN + CO and ETOTCBS = 396 kJ mol−1 for CO + CH2CNH) (see Fig. 4). However, the fact that the peak of 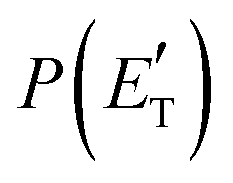 is much closer to the height of the exit barrier of the CH2CNH channel than to that of the CH3CN channel (130 kJ mol−1vs. 340 kJ mol−1) is an indication that the observed reaction dynamics align more closely with the ketenimine channel. Moreover, we would like to point out that a CO-forming channel (3CH2CHN + CO) can occur adiabatically on the triplet PES from the O(3P) reaction, as can be seen from the complete PES scheme reported in Fig. S1 of the ESI.† However, in order to form these products, the 3MIN3 intermediate has to be reached while its formation is precluded by the presence of high barriers, located above the energy of the reactants (3TS24 and 3TS34 are +23 kJ mol−1 and +37 kJ mol−1 above the reactant energy asymptote, respectively). This leads to a negligible contribution of the 3CH2CHN + CO product channel to the global branching fractions.
is much closer to the height of the exit barrier of the CH2CNH channel than to that of the CH3CN channel (130 kJ mol−1vs. 340 kJ mol−1) is an indication that the observed reaction dynamics align more closely with the ketenimine channel. Moreover, we would like to point out that a CO-forming channel (3CH2CHN + CO) can occur adiabatically on the triplet PES from the O(3P) reaction, as can be seen from the complete PES scheme reported in Fig. S1 of the ESI.† However, in order to form these products, the 3MIN3 intermediate has to be reached while its formation is precluded by the presence of high barriers, located above the energy of the reactants (3TS24 and 3TS34 are +23 kJ mol−1 and +37 kJ mol−1 above the reactant energy asymptote, respectively). This leads to a negligible contribution of the 3CH2CHN + CO product channel to the global branching fractions.
The CH2CH + NCO and OCHCN + CH2 channels ((1i) and (1j), respectively) have been included in the simplified triplet PES (Fig. 5) for completeness, but they are negligible due to the presence of high energy barriers, as demonstrated by their calculated BFs that are close to zero (see Tables 2 and 3). Finally, the relatively low BF of the COCN + CH3 channel on the singlet PES (BF = 1.1%) is in line with the experimental outcome (absence of a signal at m/z = 15 (CH3+)) that has excluded the occurrence of this reactive pathway (that, in principle, could have derived from also the O(1D) + acrylonitrile reaction).
It is interesting to note that the yields of the various reaction channels obtained from our RRKM/ME calculations on the triplet PES vary with temperature (see Table 2). Indeed, although the HCOCHCN + H channel is always the preferential route (its BF increases from 62.4% at Ec = 31.4 kJ mol−1 up to 76.4% at 10 K), its relative importance with respect to the CH2O + HCCN channel increases as the collision energy (or temperature) decreases, with the ratio [yield HCOCHCN + H]/[yield CH2O + HCCN] being 1.7 at Ec = 31.4 kJ mol−1 and 3.2 at 10 K.
If we compare the above results with the theoretical results of Sun et al.,42 some differences are present in the values of the product BFs, even though the trends are similar. In fact, Sun et al.42 estimated that at 298 K and 10−7 Torr (i.e., under single-collision conditions), the ratio [yield HCOCHCN + H]/[yield CH2O + HCCN] is more or less the same as that at 1.2 Torr, that is, about 3. This could be compared to our predicted ratio of 69.8/30.2 = 2.3 (see Table 2). However, it should be noted that Sun et al., having neglected the singlet PES and the intersystem crossing, did not predict the occurrence of the spin-forbidden 1CH2CNH + CO channel, which, according to the present results, has a BF that is about seven times larger than that of the adiabatic H-forming channel at Ec= 31.3 kJ mol−1 (see Table 1).
5.2 Dynamics of the O(1D) + acrylonitrile reaction
Regarding the O(1D) + acrylonitrile reaction, the attribution of the signal registered at m/z = 40 to the ketenimine product (channel (2b)) was made on the basis of the topology of the singlet PES and the statistical RRKM/ME estimates of product BFs, as described in the previous section on the dynamics of the O(3P) + acrylonitrile reaction.The best-fit CM angular distribution for the CH2CNH + CO channel (2b) is characterized by a noticeable forward bias, that reflects the occurrence of an osculating complex mechanism. In fact, the electronic energy of O(1D) (the O(3P)–O(1D) energy separation is ∼190 kJ mol−1) is channeled into the internal energy of the intermediate 1MIN3, which is formed via the isomerization of the cyclic intermediate 1MIN1 obtained through the barrierless addition of O(1D) to acrylonitrile. The high internal energy content of 1MIN3 reduces its lifetime, τ, making it shorter than its rotational period (τr). By using the approximate osculating model of chemical reactions,95,97,98 it is possible to estimate that τ/τr is smaller than unity, with the backward–forward asymmetry of the T(θ) being equal to 0.2.
Analyzing the CM translational energy distribution for the CH2CNH + CO channel (2b), it should be noted that the peaking of the 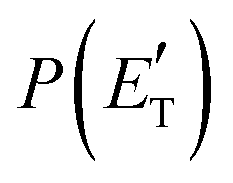 function (at ∼167 kJ mol−1) is consistent with the high exit energy barrier that separates the products and 1TS3, which is theoretically predicted to be 128 kJ mol−1. However, the peak of the best-fit CM
function (at ∼167 kJ mol−1) is consistent with the high exit energy barrier that separates the products and 1TS3, which is theoretically predicted to be 128 kJ mol−1. However, the peak of the best-fit CM 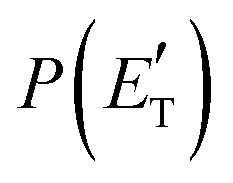 for channel (2b) is substantially shifted toward higher energy compared to that of the CH2CNH + CO channel (1b) resulting from the O(3P) + acrylonitrile reaction, despite the intermediate (1MIN3), the transition state (1TS3), and the exit potential energy barrier (128 kJ mol−1) being the same. This could be rationalized with two different explanations: (i) the electronic energy of O(1D) is largely channeled into product translational motion, much more so than in the O(3P) + acrylonitrile reaction, and/or (ii) in the O(1D) + acrylonitrile reaction, the energy randomization of the intermediate is not complete because of its short lifetime.
for channel (2b) is substantially shifted toward higher energy compared to that of the CH2CNH + CO channel (1b) resulting from the O(3P) + acrylonitrile reaction, despite the intermediate (1MIN3), the transition state (1TS3), and the exit potential energy barrier (128 kJ mol−1) being the same. This could be rationalized with two different explanations: (i) the electronic energy of O(1D) is largely channeled into product translational motion, much more so than in the O(3P) + acrylonitrile reaction, and/or (ii) in the O(1D) + acrylonitrile reaction, the energy randomization of the intermediate is not complete because of its short lifetime.
The description of the dynamics of the O(1D) + acrylonitrile reaction is totally in line with that of the O(1D) + cyanoacetylene reaction,91 based on the analogy between the two systems which has been justified and described in Section 4.1.2. In the present system, however, the ratio between the sum of the relative yields of the O(3P) reaction channels ((1b) and (1f)) and the yield of the O(1D) reaction channel (2b) was found to be [yield O(3P) reaction]/[yield O(1D) reaction] = 0.63/0.37 (= 1.7), which is higher than that found in the system involving cyanoacetylene (the ratio was [yield O(3P) reaction]/[yield O(1D) reaction] = 0.38/0.62 (= 0.6)).91 The higher reactivity of O(3P) when reacting with acrylonitrile compared to the system involving cyanoacetylene is a direct consequence of the lower entrance potential energy barrier on the triplet PES, which was calculated to be 7 kJ mol−1 (at the CCSD(T)/CBS level of theory) for the O(3P) + acrylonitrile reaction and 9 kJ mol−1 (at the CCSD(T)/CBS level of theory) for the O(3P) + cyanoacetylene reaction.91
5.3 Comparison with the O(3P) + ethylene (CH2CH2) and O(3P) + propene (CH2CHCH3) reactions
The interpretation of the results of this work can be enriched by comparison with the related O(3P) + C2H4 and O(3P) + C3H6 reactions that have been studied in our laboratory at a comparable collision energy.43,90,102,103 From a chemical point of view, ethylene and propene can be considered analogous to an acrylonitrile molecule where the CN group is substituted with H and CH3, respectively. The role of substituents in determining a change in the reactivity of the species is of utmost importance in the development of realistic models and, in this case, we can rationalize the effect of a nitrile group in place of one of the four hydrogen atoms of ethylene and of the methyl group of propene. To make a consistent experimental comparison between the three systems, we focus only on the reaction involving O(3P). The topology of the triplet/singlet PESs of the three systems presents many similarities but a substantial difference arises in the topology of their singlet PESs. In particular, in the case of the reactions involving acrylonitrile, the hydrogen atom bound to the CO unit of 1MIN3 (see Fig. 6) can migrate to the other two carbon atoms and to the nitrogen atom, leading to the formation of CH3CN + CO and CH2CNH + CO, respectively. The first pathway has a barrier (1TS4) at −138 kJ mol−1 (that is, 304 kJ mol−1 above 1MIN3), while the second one is characterized by a transition state (1TS3) at −235 kJ mol−1 (that is, located 207 kJ mol−1 above 1MIN3) (see Fig. 6 and also Fig. S6 in the ESI† for the structure of 1TS4 and 1TS3). In contrast, in the case of ethylene and propene, if we consider the evolution of the CH3CHO (for the O(3P) + ethylene reaction) and CH3CH2CHO (for the O(3P) + propene reaction) intermediates, which are the equivalent to 1MIN3 of the OCH2CHCN system, the only possibility is the migration of the H atom from the terminal carbon atom to the directly bound carbon atom with very large potential energy barriers toward CO elimination and formation of CH4 + CO (in O(3P) + ethylene) and of C2H6 + CO (in O(3P) + propene). This results in the predominance of the HCO-forming channels in these two O(3P) + alkene reactions and the absence of the CO-forming channels on the singlet PES. So, the presence of an additional, energetically favored reactive pathway, following H migration to the N atom of acrylonitrile on the singlet PES after ISC, is the main difference between the three systems that justifies the significant prevalence of the CH2CNH + CO channel over the CH2CN + HCO channel in the O(3P) + acrylonitrile reaction (see Table 3). The extent of ISC is inferred to be largest (87%) in O(3P) + acrylonitrile, while it is lower (50%) in O(3P) + ethylene, and even lower (20%) in O(3P) + propene. Finally, the easy channeling of the reactive flux, via ISC, toward the CH2CNH + CO products explains why, in the O(3P) + CH2CHCN reaction, the CH2O + HCCN, OCCH2CN + H, CH2CN + HCO, and OCCHCN + H2 channels are negligible (see Table 3), in contrast, for instance, to the equivalent channels in the O(3P) + C2H4 reaction.90,102,1036. Implications for extraterrestrial environments
The main result of our combined experimental and theoretical study is that the channel leading to the formation of ketenimine (CH2CNH) + CO from the O(3P) + acrylonitrile reaction is by far the dominant reaction pathway. Since the reaction is characterized by an entrance potential energy barrier of 7 kJ mol−1 (at CCSD(T)/CBS level), in line with a rate constant of (4.9 ± 1.0) × 10−13 cm3 molecule−1 s−1 at 298 K,40 it can play a significant role only in warm extraterrestrial environments, such as the Sagittarius B2(N) hot molecular core,23,24 the circumstellar envelope of C-rich star IRC + 10216,27 or, possibly, the upper atmosphere of Titan.29–35,104Ketenimine and acrylonitrile have both been detected toward the Sagittarius B2(N) hot core (see Introduction) in comparable abundance. Ketenimine abundance has been estimated to be a hundredth of that of its constitutional isomer acetonitrile (CH3CN).105 Quan and Herbst106 were able to reproduce the abundance of ketenimine by assuming that it is formed from the electron ion recombination of CH3CNH+ in competition with the formation of CH3CN and CH2CN. There is experimental evidence that electron–ion recombination of CH3CNH+ predominantly preserves the heavy atom skeleton107 while recent theoretical estimates have actually found that the formation of ketenimine is the dominant channel by far.108 The present reaction adds to the list of ketenimine formation routes especially in those regions where atomic oxygen and acrylonitrile are both abundant, as the O-rich Sagittarius B2(N) hot core. Ketenimine is neglected in the most common astrochemical databases, such as KIDA109 and UMIST,110 that also overlook the reaction between acrylonitrile and oxygen atoms. To improve the accuracy of current astrochemical models, we recommend including the O(3P) + acrylonitrile reaction both as a possible destruction pathway of CH2CHCN and a possible formation route of CH2CNH.
Furthermore, the same conclusions can be extended to the O(1D) + acrylonitrile reaction because our theoretically predicted BF of the CH2CNH + CO channel on the singlet PES is very high (BF > 90%, see Table 3) at all the investigated temperatures. O(1D) is extremely reactive and its bimolecular reactions have been invoked to explain the formation of complex species on the icy mantle of interstellar grains37 or in the comas of comets.36
In conclusion, the present work can enrich our knowledge of the gas-phase chemistry of nitrile compounds and imines that are key intermediates in the formation of many species with biological potential, such as nucleobases and amino acids.111
7. Conclusions
We have reported a combined CMB and theoretical study of the O(3P,1D) + acrylonitrile reactions that are processes of considerable relevance in combustion systems as well as in a variety of extraterrestrial environments (from Sagittarius B2(N) hot molecular cores23,24 to Titan's atmosphere29–35,104). We have determined that the O(3P) + acrylonitrile reaction exhibits two main product channels, among a large variety of possible open channels. Specifically, at a collision energy of 31.4 kJ mol−1, it was found that the main reaction channel is the one leading to the formation of spin-forbidden CH2CNH (ketenimine) + CO products (BF = 0.87 ± 0.05), which are formed via efficient intersystem crossing from the entrance triplet PES to the underlying singlet PES, while the spin-allowed H + HCOCHCN product channel, occurring adiabatically on the triplet PES, is minor (BF = 0.13 ± 0.05). The theoretical results have indicated that the dominant reaction mechanism is addition of atomic oxygen to the C1 carbon of the double bond, occurring after overcoming an entrance potential energy barrier of 7 kJ mol−1 (at CCSD(T)/CBS level), and this makes this reaction not only relevant in combustion environments, but also in warm extraterrestrial environments, where it could represent an efficient mechanism of formation of ketenimine. Notably, the BF of the CH2CNH + CO channel actually increases with decreasing temperature (Table 3), as expected, because the probability of ISC increases with decreasing temperature (i.e., with increasing lifetime of the initial triplet diradical intermediate, which facilitates the possibility of ISC).112 We note that the main product of the title reaction, ketenimine, has been detected toward the same star-forming region of acrylonitrile as in Sagittarius B2(N) hot core. 23,24,105 Ketenimine is often neglected by modelers, with respect to its more abundant isomer CH3CN, and also the reaction between O(3P) and acrylonitrile is overlooked in models. This study leads us to propose the inclusion of the O(3P) + acrylonitrile reaction both as a possible destruction pathway of CH2CHCN and a possible formation route of CH2CNH.We have also characterized the dynamics of the O(1D) + acrylonitrile reaction that mainly leads to the formation of CH2CNH + CO adiabatically on the singlet PES. This can improve models related to the chemistry of interstellar ice37 and cometary comas,36 where O(1D) reactions are believed to play an important role. We remind that nitriles and imines are key intermediates in the formation of species with biological potential, such as nucleobases and amino acids.111 Finally, the results of this study are expected to be also useful for improving combustion models involving the oxidation of acrylonitrile.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the Italian MUR (Ministero dell’Università e della Ricerca) for “PRIN 2017” funds, project “Modeling and Analysis of carbon nanoparticles for innovative applications Generated dIrectly and Collected DUring combuSTion (MAGIC DUST)”, grant number 2017PJ5XXX. This work was also supported by the Italian Space Agency (ASI, DC-VUM-2017-034, grant no. 2019-3 U.0 Life in Space). We also acknowledge the support from Italian MUR, University of Perugia within the program “Department of Excellence-2018-2022-Project AMIS”, and “Dipartimento di Ingegneria Civile ed Ambientale” (DICA) of the University of Perugia for allocated computing time within the project “Dipartimenti di Eccellenza 2018–2022”. P. L. thanks the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement no. 811312 for the project “Astro-Chemical Origins” (ACO).References
- A. Belloche, H. S. P. Müller, K. M. Menten, P. Schilke and C. Comito, Astron. Astrophys., 2013, 559, A47 CrossRef (187 pp).
- R. Zhang, D. Shi, N. Liu, B. Chen, L. Wu, L. Wu and W. Yang, Catal. Today, 2015, 258, 17–27 CrossRef CAS.
- P. Wyman, V. Crook, J. Ebdon, B. Hunt and P. Joseph, Polym. Int., 2006, 55, 764–771 CrossRef CAS.
- R. Zhang, N. Liu, Z. Lei and B. Chen, Chem. Rev., 2016, 116, 3658–3721 CrossRef CAS PubMed.
- L. T. Groet, Encyclopedia of Chemical Technology, Wiley, New York, 3rd edn, 1978 Search PubMed.
- S. P. Felter and J. S. Dollarhide, Regul. Toxicol. Pharm., 1997, 26, 281–287 CrossRef CAS PubMed.
- D. R. van der Vaart, W. M. Vatvuk and A. H. Wehe, J. Air Waste Manage. Assoc., 1991, 41, 92–98 CrossRef CAS PubMed.
- T. Namba, S. Masukawa, J. Uchisawa and A. Obuchi, Catal. Lett., 2004, 93, 195–201 CrossRef.
- N. Liu, D. Shi, R. Zhang, Y. Li and B. Chen, Catal. Today, 2019, 332, 201–213 CrossRef CAS.
- A. Gervasini, G. C. Vezzoli and V. Ragani, Catal. Today, 1996, 29, 449–455 CrossRef CAS.
- S. Y. Li, S. L. Li and B. L. Li, React. Kinet. Catal. Lett., 1997, 62, 89–95 CrossRef CAS.
- T. Namba, S. Masukawa, A. Ogata, J. Uchisawa and A. Obuci, Appl. Catal., B, 2005, 61, 288–296 CrossRef.
- L. R. Snyder, Anal. Chem., 1969, 41, 314–323 CrossRef CAS.
- C. F. Brandenburg and D. R. Latham, J. Chem. Eng. Data, 1968, 13, 391–394 CrossRef CAS.
- S. Wallace, K. D. Bartle and D. L. Perry, Fuel, 1989, 68, 1450–1455 CrossRef CAS.
- J. C. Mackie, M. B. Colket III and P. F. Nelson, J. Phys. Chem., 1990, 94, 4099–4106 CrossRef CAS.
- A. Lifshitz, C. Tamburu and A. Suslensky, J. Phys. Chem., 1989, 93, 5802–5808 CrossRef CAS.
- N. R. Hore and D. K. Russell, J. Chem. Soc., Perkin Trans., 1998, 2, 269–276 RSC.
- A. Terentis, A. Doughty and J. C. Mackie, J. Phys. Chem., 1992, 96, 10334–10339 CrossRef CAS.
- B. J. Finlayson-Pitts and J. N. Pitts Jr, Atmospheric Chemistry – Fundamentals and Experimental Techniques, Wiley, New York, 1986 Search PubMed.
- F. F. Gardner and G. Winnewisser, Astrophys. J., 1975, 195, L127–L130 CrossRef CAS.
- M. C. L. Gerry, K. Yamada and G. Winnewisser, J. Phys. Chem. Ref. Data, 1979, 8, 107 CrossRef CAS.
- A. Nummelin and P. Bergman, Astron. Astrophys., 1999, 341, L59–L62 CAS.
- H. S. Müller, A. Belloche, K. M. Menten, C. Comito and P. Schilke, J. Mol. Spectrosc., 2008, 251, 319–325 CrossRef.
- A. López, B. Tercero, Z. Kisiel, A. M. Daly, C. Bermúdez, H. Calcutt, N. Marcelino, S. Viti, B. J. Drouin, I. R. Medvedev, C. F. Neese, L. Pszczólkowski, J. L. Alonso and J. Cernicharo, Astron. Astrophys., 2014, 572, A44 CrossRef (39 pp).
- H. E. Matthews and T. J. Sears, Astrophys. J., 1983, 272, 149–153 CrossRef CAS.
- M. Agúndez, J. P. Fonfría, J. Cernicharo, J. R. Pardo and M. Guélin, Astron. Astrophys., 2008, 479, 493–501 CrossRef.
- C. Vastel, J. C. Loison, V. Wakelam and B. Lefloch, Astron. Astrophys., 2019, 625, A91 CrossRef CAS (19 pp).
- J. Cui, R. Yelle, V. Vuitton, J. Waite, W. Kasprzak, D. Gell, H. Niemann, I. Müller-Wodarg, N. Borggren and G. Fletcher, et al. , Icarus, 2009, 200, 581–615 CrossRef CAS.
- V. Vuitton, R. V. Yelle and M. J. McEwan, Icarus, 2007, 191, 722–742 CrossRef.
- B. A. Magee, J. H. Waite, K. E. Mandt, J. Westlake, J. Bell and D. A. Gell, Planet. Space Sci., 2009, 57, 1895–1916 CrossRef CAS.
- B. Bézard, R. V. Yelle and C. A. Nixon, in Titan: Interior, Surface, Atmosphere, and Space Environment, ed. I. Müller-Wodarg, C. A. Griffith, E. Lellouch, T. E. Cravens, Cambridge Planetary Science, Cambridge University Press, 2014, vol. 5, pp. 158–189 Search PubMed.
- M. Y. Palmer, M. A. Cordiner, C. A. Nixon, S. B. Charnley, N. A. Teanby, Z. Kisiel, P. G. J. Irwin and M. J. Mumma, Sci. Adv., 2017, 3, e1700022 CrossRef PubMed.
- J. C.-Y. Lai, M. A. Cordiner, C. A. Nixon, R. K. Achterberg, E. M. Molter, N. A. Teanby, M. Y. Palmer, S. B. Charnley, J. E. Lindberg, Z. Kisiel, M. J. Mumma and P. G. J. Irwin, Astron. J., 2017, 154, 206 CrossRef (10 pp).
- M. A. Cordiner, N. A. Teanby, C. A. Nixon, V. Vuitton, A. E. Thelen and S. B. Charnley, Astron. J., 2019, 158, 76 CrossRef CAS (14 pp).
- M. A. Cordiner and S. B. Charnley, MNRAS, 2021, 504, 5401–5408 CrossRef CAS.
- J. B. Bergner, K. I. Öberg and M. Rajappan, Astophys. J., 2019, 874, 115 CrossRef CAS (16 pp).
- D. Marchione, L. Mancini, P. Liang, G. Vanuzzo, F. Pirani, D. Skouteris, M. Rosi, P. Casavecchia and N. Balucani, J. Phys. Chem. A, 2022, 126, 3569–3582 CrossRef CAS PubMed.
- G. Vanuzzo, D. Marchione, L. Mancini, P. Liang, G. Pannacci, P. Recio, Y. Tan, M. Rosi, D. Skouteris, P. Casavecchia and N. Balucani, J. Phys. Chem. A, 2022, 126, 6110–6123 CrossRef CAS PubMed.
- H. P. Upadhyaya, P. D. Naik, U. B. Pavanaja, A. Kumar, R. K. Vatsa, A. V. Sapre and J. P. Mittal, Chem. Phys. Lett., 1997, 274, 383–389 CrossRef CAS.
- H. J. Kimber, J. Toscano and S. D. Price, Mon. Notices Royal Astron. Soc., 2018, 476, 5332–5340 CrossRef CAS.
- J. Sun, W. Wu, Y. Zhang, Y. Tang, H. Yi, J. Lu and R. Wang, Comput. Theor. Chem., 2015, 1052, 17–25 CrossRef CAS.
- F. Leonori, N. Balucani, V. Nevrly, A. Bergeat, S. Falcinelli, G. Vanuzzo, P. Casavecchia and C. Cavallotti, J. Phys. Chem. C, 2015, 119, 14632–14652 CrossRef CAS.
- G. Vanuzzo, N. Balucani, F. Leonori, D. Stranges, V. Nevrly, S. Falcinelli, A. Bergeat, P. Casavecchia and C. Cavallotti, J. Phys. Chem. A, 2016, 120, 4603–4618 CrossRef CAS PubMed.
- A. Caracciolo, G. Vanuzzo, N. Balucani, D. Stranges, P. Casavecchia, L. Pratali Maffei and C. Cavallotti, J. Phys. Chem. A, 2019, 123, 9934–9956 CrossRef CAS PubMed.
- A. Caracciolo, G. Vanuzzo, N. Balucani, D. Stranges, S. Tanteri, C. Cavallotti and P. Casavecchia, Chin. J. Chem. Phys., 2019, 32, 113–122 CrossRef CAS.
- C. Cavallotti, C. De Falco, L. Pratali Maffei, A. Caracciolo, G. Vanuzzo, N. Balucani and P. Casavecchia, J. Phys. Chem. Lett., 2020, 11, 9621–9628 CrossRef CAS PubMed.
- G. Vanuzzo, A. Caracciolo, T. K. Minton, N. Balucani, P. Casavecchia, C. de Falco, A. Baggioli and C. Cavallotti, J. Phys. Chem. A, 2021, 125, 8434–8453 CrossRef CAS PubMed.
- P. Recio, S. Alessandrini, G. Vanuzzo, G. Pannacci, A. Baggioli, D. Marchione, A. Caracciolo, V. J. Murray, P. Casavecchia, N. Balucani, C. Cavallotti, C. Puzzarini and V. Barone, Nat. Chem., 2022, 14, 1405–1412 CrossRef CAS PubMed.
- P. Casavecchia, F. Leonori and N. Balucani, Int. Rev. Phys. Chem., 2015, 34, 161–204 Search PubMed.
- P. Casavecchia, F. Leonori, N. Balucani, R. Petrucci, G. Capozza and E. Segoloni, Phys. Chem. Chem. Phys., 2009, 11, 46–65 RSC.
- Y. T. Lee, Science, 1987, 236, 793–798 CrossRef CAS PubMed.
- P. Casavecchia, Rep. Prog. Phys., 2000, 63, 355–414 CrossRef CAS.
- P. Casavecchia, K. Liu and X. Yang, in Reactive Scattering: Reactions in Three Dimensions. In Tutorials in Molecular Reaction Dynamics, ed. M. Brouard, C. Vallance, Royal Society of Chemistry, Cambridge, UK, 2010, ch. VI Search PubMed.
- M. Alagia, N. Balucani, P. Casavecchia, D. Stranges and G. G. Volpi, J. Chem. Soc., Faraday Trans., 1995, 91, 575–596 RSC.
- N. R. Daly, Rev. Sci. Instrum., 1960, 31, 264–267 CrossRef CAS.
- M. Alagia, V. Aquilanti, D. Ascenzi, N. Balucani, D. Cappelletti, L. Cartechini, P. Casavecchia, F. Pirani, G. Sanchini and G. G. Volpi, Isr. J. Chem., 1997, 37, 329–342 CrossRef CAS.
- F. Leonori, K. M. Hickson, S. D. Le Picard, X. Wang, R. Petrucci, P. Foggi, N. Balucani and P. Casavecchia, Mol. Phys., 2010, 108, 1097–1113 CrossRef CAS.
- S. J. Sibener, R. J. Buss, C. Y. Ng and Y. T. Lee, Rev. Sci. Instrum., 1980, 51, 167–182 CrossRef CAS.
- Y. T. Lee, Reactive Scattering I: Nonoptical Methods, in Atomic and Molecular Beam Methods, ed. G. Scoles, Oxford University Press, New York, USA, 1987, vol. 1, pp. 553–568 Search PubMed.
- F. Leonori, R. Petrucci, N. Balucani, P. Casavecchia, M. Rosi, C. Berteloite, S. D. Le Picard, A. Canosa and I. R. Sims, Phys. Chem. Chem. Phys., 2009, 11, 4701–4706 RSC.
- C. Sleiman, G. El Dib, M. Rosi, D. Skouteris, N. Balucani and A. Canosa, Phys. Chem. Chem. Phys., 2018, 20, 5478–5489 RSC.
- C. Berteloite, S. D. Le Picard, I. R. Sims, M. Rosi, F. Leonori, R. Petrucci, N. Balucani, X. Wang and P. Casavecchia, Phys. Chem. Chem. Phys., 2011, 13, 8485–8501 RSC.
- N. Balucani, A. Bergeat, L. Cartechini, G. G. Volpi, P. Casavecchia, D. Skouteris and M. Rosi, J. Phys. Chem. A, 2009, 113, 11138–11152 CrossRef CAS PubMed.
- N. Balucani, M. Alagia, L. Cartechini, P. Casavecchia, G. G. Volpi, K. Sato, T. Takayanagi and Y. Kurosaki, J. Am. Chem. Soc., 2000, 122, 4443–4450 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- T. H. Dunning Jr., J. Chem. Phys., 1989, 90, 1007–1023 CrossRef.
- D. E. Woon and T. H. Dunning Jr., J. Chem. Phys., 1993, 98, 1358–1371 CrossRef CAS.
- R. A. Kendall, T. H. Dunning Jr. and J. R. Harrison, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS.
- C. Gonzalez and H. B. Schlegel, J. Chem. Phys., 1989, 90, 2154–2161 CrossRef CAS.
- C. Gonzalez and H. B. Schlegel, J. Phys. Chem., 1990, 94, 5523–5527 CrossRef CAS.
- R. J. Bartlett, Annu. Rev. Phys. Chem., 1981, 32, 359–401 CrossRef CAS.
- K. Raghavachari, G. W. Trucks, J. A. Pople and M. Head-Gordon, Chem. Phys. Lett., 1989, 157, 479–483 CrossRef CAS.
- J. Olsen, P. Jorgensen, H. Koch, A. Balkova and R. J. Bartlett, J. Chem. Phys., 1996, 104, 8007–8015 CrossRef CAS.
- C. Cavallotti, F. Leonori, N. Balucani, V. Nevrly, A. Bergeat, S. Falcinelli, G. Vanuzzo and P. Casavecchia, J. Phys. Chem. Lett., 2014, 5, 4213–4218 CrossRef CAS PubMed.
- I. Gimondi, C. Cavallotti, G. Vanuzzo, N. Balucani and P. Casavecchia, J. Phys. Chem. A, 2016, 120, 4619–4633 CrossRef CAS PubMed.
- C. Cavallotti, A. Della Libera, C.-W. Zhou, P. Recio, A. Caracciolo, N. Balucani and P. Casavecchia, Faraday Discuss., 2022, 238, 161–182 RSC.
- J. M. L. Martin, Chem. Phys. Lett., 1996, 259, 669–678 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. A. Petersson, et al., Gaussian 09, rev. A.02, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby and M. Schütz, WIREs Comput. Mol. Sci., 2012, 2, 242–253 CrossRef CAS.
- H.-J. Werner, P. J. Knowles, F. R. Manby, J. A. Black, K. Doll, A. Heßelmann, D. Kats, A. Köhn, T. Korona and D. A. Kreplin, et al. , J. Chem. Phys., 2020, 152, 144107 CrossRef CAS PubMed (24 pp).
- Avogadro: an open-source molecular builder and visualization tool. Ver. 1.2.0. https://avogadro.cc/.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeersch, E. Zurek and G. R. Hutchison, J. Cheminform., 2012, 4, 17 CrossRef CAS PubMed.
- D. Skouteris, N. Balucani, C. Ceccarelli, F. Vazart, C. Puzzarini, V. Barone, C. Codella and B. Lefloch, Astrophys. J., 2018, 854, 135 CrossRef (10 pp).
- N. Balucani, L. Cartechini, M. Alagia, P. Casavecchia and G. G. Volpi, J. Phys. Chem. A, 2000, 104, 5655–5659 CrossRef CAS.
- N. Balucani, F. Leonori, R. Petrucci, M. Stazi, D. Skouteris, M. Rosi and P. Casavecchia, Faraday Discuss., 2010, 147, 189–216 RSC.
- N. Balucani, D. Skouteris, F. Leonori, R. Petrucci, M. Hamberg, W. D. Geppert, P. Casavecchia and M. Rosi, J. Phys. Chem. A, 2012, 116, 10467–10479 CrossRef CAS PubMed.
- S. J. Klippenstein, J. Chem. Phys., 1992, 96, 367–371 CrossRef.
- B. Fu, Y.-C. Han, J. M. Bowman, F. Leonori, N. Balucani, L. Angelucci, A. Occhiogrosso, R. Petrucci and P. Casavecchia, J. Chem. Phys., 2012, 137, 22A532 CrossRef PubMed (22 pp).
- P. Liang, E. V. F. de Aragão, G. Pannacci, G. Vanuzzo, A. Giustini, D. Marchione, P. Recio, F. Ferlin, D. Stranges and N. Faginas Lago, et al. , J. Phys. Chem. A, 2023, 127, 685–703 CrossRef CAS PubMed.
- D. L. Baulch, C. T. Bowman, C. J. Cobos, R. A. Cox, T. Just, J. A. Kerr, M. J. Pilling, D. Stocker, J. Troe, W. Tsang, R. W. Walker and J. Warnatz, J. Phys. Chem. Ref. Data, 2005, 34, 757–1397 CrossRef CAS ; and references therein.
- S.-H. Lee, W. J. Huang and W. K. Chen, Chem. Phys. Lett., 2007, 446, 276–280 CrossRef CAS.
- S.-H. Lee, W. K. Chen and W. J. Huang, J. Chem. Phys., 2009, 130, 054301 CrossRef PubMed.
- W. B. Miller, S. A. Safron and D. R. Herschbach, Discuss. Faraday Soc., 1967, 44, 108–122 RSC.
- J. I. Steinfeld, G. A. Fisk, J. D. McDonald and D. R. Herschbach, et al. , Discuss. Faraday Soc., 1967, 44, 226–240 RSC.
- R. D. Levine and R. B. Bernstein, Molecular reaction dynamics and chemical reactivity, Oxford University Press, New York, 1987 Search PubMed.
- G. A. Fisk, J. D. McDonald and D. R. Herschbach, Discuss. Faraday Soc., 1967, 44, 228–229 Search PubMed.
- D. R. Herschbach, Adv. Chem. Phys., 1966, 10, 319–393 Search PubMed.
- A. M. Schmoltner, P. M. Chu and Y. T. Lee, J. Chem. Phys., 1989, 91, 5365–5373 CrossRef CAS.
- W. L. Fitch and A. D. Sauter, Anal. Chem., 1983, 55, 832–835 CrossRef CAS.
- B. Fu, Y.-C. Han, J. M. Bowman, L. Angelucci, N. Balucani, F. Leonori and P. Casavecchia, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9733–9738 CrossRef CAS PubMed.
- N. Balucani, F. Leonori, P. Casavecchia, B. Fu and J. M. Bowman, J. Phys. Chem. A, 2015, 119, 12498–12511 CrossRef CAS PubMed.
- H. R. Martens, D. B. Reisenfeld, J. D. Williams, R. E. Johnson and H. D. Smith, Geophys. Res. Lett., 2008, 35, L20103 CrossRef (4pp).
- F. J. Lovas, J. M. Hollis, A. J. Remijan and P. R. Jewell, Astrophys. J., 2006, 645, L137–L140 CrossRef CAS.
- D. Quan and E. Herbst, Astron. Astrophys., 2007, 474, 521–527 CrossRef CAS.
- E. Vigren, M. Kamińska, M. Hamberg, V. Zhaunerchyk, R. D. Thomas, M. Danielsson, J. Semaniak, P. U. Andersson, M. Larsson and W. D. Geppert, Phys. Chem. Chem. Phys., 2008, 10, 4014–4019 RSC.
- L. Giani, C. Ceccarelli, L. Mancini, E. Bianchi, F. Pirani, M. Rosi and N. Balucani, Interstellar methyl cyanide and methanol connection: the revised CH3CN gas-phase chemical network, MNRAS Search PubMed.
- V. Wakelam, E. Herbst, J. C. Loison, I. W. M. Smith, V. Chandrasekaran, B. Pavone, N. G. Adams, M. C. Bacchus-Montabonel and A. Bergeat, et al. , Astrophys. J., Suppl. Ser., 2012, 199, 21 CrossRef (10 pp).
- J. Woodall, M. Agúndez, A. J. Markwick-Kemper and T. J. Millar, Astron. Astrophys., 2007, 466, 1197–1204 CrossRef CAS.
- D. E. Woon, Astrophys. J., 2002, 571, L177–L180 CrossRef CAS.
- H. Pan, K. Liu, A. Caracciolo and P. Casavecchia, Chem. Soc. Rev., 2017, 46, 7517–7547 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: List of all possible reaction channels of the O(3P,1D) + acrylonitrile reactions, with reaction enthalpies. Detailed global triplet and singlet PESs, with reaction enthalpies and barrier heights (kJ mol−1, 0 K) computed at the CCSD(T)/aug-cc-pVTZ level of theory, considering the geometries obtained at the B3LYP/aug-cc-pVTZ level, for dissociation and isomerization processes for the O(3P,1D) + CH2CHCN reactions. Energies at the CCSD(T)/CBS level of theory for the most relevant reaction channels are reported (in parentheses) only in Fig. 5 of the main text. See DOI: https://doi.org/10.1039/d3cp01558k |
| This journal is © the Owner Societies 2023 |