
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencepH Dependence of the speciation and optical properties of 4-benzoylbenzoic acid†
Natalia
Karimova
 a,
Onita
Alija
b,
Stephanie L.
Mora García
c,
Vicki H.
Grassian
c,
R. Benny
Gerber
ad and
Juan G.
Navea
*b
a,
Onita
Alija
b,
Stephanie L.
Mora García
c,
Vicki H.
Grassian
c,
R. Benny
Gerber
ad and
Juan G.
Navea
*b
aDepartment of Chemistry, University of California Irvine, Irvine, CA 92697, USA
bChemistry Department, Skidmore College, Saratoga Springs, NY 12866-1632, USA. E-mail: jnavea@skidmore.edu
cDepartment of Chemistry, University of California San Diego, La Jolla, CA 92093, USA
dThe Institute of Chemistry and Fritz Haber Research Center, The Hebrew University of Jerusalem, 91904 Jerusalem, Israel
First published on 5th June 2023
Abstract
Organic chromophores initiate much of daytime aqueous phase chemistry in the environment. Thus, studying the absorption spectra of commonly used organic photosensitizers is paramount to fully understand their relevance in environmental processes. In this work, we combined UV-Vis spectroscopy, 1H-NMR spectroscopy, quantum chemical calculations, and molecular dynamics simulations to investigate the absorption spectra of 4-benzoyl benzoic acid (4BBA), a widely used photosensitizer and a common proxy of environmentally relevant chromophores. Solutions of 4BBA at different pH values show that protonated and deprotonated species have an effect on its absorbance spectra. Theoretical calculations of these species in water clusters provide physical and chemical insights into the spectra. Quantum chemical calculations were conducted to analyze the UV-Vis absorbance spectra of 4BBA species using various cluster sizes, such as C6H5COC6H4COOH·(H2O)n, where n = 8 for relatively small clusters and n = 30 for larger clusters. While relatively small clusters have been successfully used for smaller chromophores, our results indicate that simulations of protonated species of 4BBA require relatively larger clusters of n = 30. A comparison between the experimental and theoretical results shows good agreement in the pH-dependent spectral shift between the hydrated cluster model and the experimental data. Overall, the theoretical and empirical results indicate that the experimental optical spectra of aqueous phase 4BBA can be represented by the acid–base equilibrium of the keto-forms, with a spectroscopically measured pKa of 3.41 ± 0.04. The results summarized here contribute to a molecular-level understanding of solvated organic molecules through calculations restricted to cluster models, and thereby, broader insight into environmentally relevant chromophores.
Introduction
Light-absorbing organic acids are ubiquitous chromophores in natural systems and play a significant role in the daytime chemistry of the environment.1–7 These organic acids are important fractions of dissolved organic matter (DOM), one of the most abundant and strongest light-absorbing compounds in environmental aqueous systems.8–14 The fraction of DOM that absorbs ultraviolet (UV) and visible light is referred as chromophoric dissolved organic matter (CDOM), commonly found in the sea surface microlayer and, more recently, within sea spray aerosols.14–18 These chromophores are effective photosensitizers,19,20 exhibiting strong absorption bands that extend into the visible spectral region, overlapping with the solar spectral flux.3,21–30 Given that naturally occurring photosensitizers are known to induce photochemical reactions in aqueous environments, both in bulk and interfaces, the understanding of these photochemical processes is significant from an atmospheric point of view.31–33 In particular, studies that combine experimental techniques with theoretical simulations have provided deeper insight into the processes of photoactivation of molecules at interfaces.1,34–36Present knowledge on this topic is incomplete due to the complex composition of CDOM. A recent analysis by Alves et al. shows that CDOM is rich in aromatic ketones, aldehydes, carboxyl alicyclic molecules, among other classes of compounds.37 Theoretical and experimental investigations of small molecules matching these molecular moieties can provide insights into the optical properties of these more complex light absorbing substances and the role of environmental factors (pH, hydration state etc.) in these properties.1,36,38,39 For instance, studies of the optical properties of simple isolated molecules, such as benzoic1 and pyruvic acids,36 have been shown to mimic some of the photochemistry and photophysics properties of CDOM using a simple-to-complex approach, leading to a possible molecular structure of CDOM presented through a large realistic simulation, where aromaticity is shown to play a significant role.34 4-Benzoylbenzoic acid (4BBA), a well-known photosensitizer, has been used as a model to mimic the optical properties of more complex marine CDOM.5,40–43 Aromatic carboxylic acids have strong absorption bands in the UV spectral region above 290 nm, overlapping with the solar spectral region.44,45 With relatively low solubility, 4BBA can be considered as one possible model of marine CDOM and terrestrial humic substances that mimic the aromatic fraction of the chromophores. Yet, little is known about the optical properties and excited states of this widely used photosensitizer, especially in the context of environmental factors such as pH.
For example, the photoactivity of 4BBA, like those of other carboxylic acids and chromophores in CDOM, can change with pH.1,36 Protonation and deprotonation of keto organic acids such as 4BBA, a common moiety within CDOM, can establish hydrate under acidic conditions, leading to the possible keto-diol equilibrium, as shown in Scheme 1. This possible increase in keto-acid speciation has the potential to impact the optical properties and photoactivation of photosensitizers.36 This is particularly important considering the wide range of pH variations in the marine atmosphere, with a basic sea surface microlayer (SSML) of pH ≈ 8, and a nascent sea spray aerosol (nSSA) reaching acidic conditions of pH ≈ 2,46 a more typical pH for the aerosol deliquescent layer.3,47–51
 | ||
| Scheme 1 Possible equilibria of species found in aqueous solutions of keto acids. R represents an organic group, with [R] including the possible absence of it. | ||
Thus, any keto acid molecular model system needs to consider the more complex speciation arising from hydrate equilibria. Acidification of aqueous phase molecular models, such as 4BBA, impacts photophysics and photochemistry by shifting the speciated forms of an organic molecule from neutral to protonated forms.36,52 It is, thus, paramount to investigate the pH-induced changes in the photoactivity of molecular models of environmental photosensitizers to better understand their role in daytime chemistry. Although the 4BBA molecule is a relatively simple system, the possibility of hydration in the keto group, leading to a geminal diol-form of protonated and deprotonated species, has not been investigated.
In this work, we carried out experimental spectroscopic measurements combined with theoretical calculations to study the optical properties of 4BBA in aqueous solutions at different pHs, providing insight into both the acid form and its conjugate base. We elucidate the speciation of 4BBA as a function of pH and show the effects of protonation in its optical properties. Titrations of 100 μM 4BBA solutions were carried out from pH = 1.0 to pH = 12.0, with the UV-Vis spectral data collected at each pH value in order to determine the pKa of the system. A major challenge of the current study is the complexity of the photophysical and photochemical properties of 4BBA in solution. These challenges arise from (i) medium effects – solvent molecules have been shown to have an effect on the structure of chromophoric compounds in environmental interfaces;1,36,53 (ii) the contribution of different speciated forms to the optical absorption spectrum has not been investigated; and (iii) 4BBA, the molecule of interest here, is a relatively large molecule compared to what has been done in previous studies. Along with aromaticity, larger molecular species make the calculations computationally challenging, especially when solvent molecules are included (the largest system contains around 120 atoms). Here, our calculations provide insight into (i) the nature of 4BBA excited states, (ii) solvent effects, (iii) orbitals participated in the electron transition, and (iv) the contribution of speciated forms to the 4BBA optical spectrum at different pHs.
Experimental methods and materials
Optical properties and speciation analysis
Chromophore 4-benzoyl benzoic acid (4BBA) was purchased from Sigma Aldrich and used without further purification. Aqueous solutions of 4BBA were prepared using 18 MΩ Milli-Q water at a concentration of 100 μM. All the pH adjustments were carried out by adding either hydrochloric acid (1 N stock solution) or sodium hydroxide (1 N stock solution), both solutions from Fisher Chemical. Potentiometric titration for the measurement of pKa and pH measurements were carried out using an ACUMET AB15 pH meter. All UV-Vis spectral data were acquired using a PerkinElmer Lambda 35 spectrophotometer. Speciation and further pKa studies were carried out by 1H-NMR spectroscopy performed using a 500 Jeol ECA NMR spectrometer with solvent suppression in the acquisition parameters. The solution containing 10% D2O was used for instrument locking and 5 μM sodium trimethylsilylpropanesulfonate (DSS) purchased from Sigma Aldrich was used as the internal standard for peak locking.In order to determine if the observed optical changes correspond to acid–base equilibria of the keto acid of 4BBA, or more complex speciation is responsible for the changes in the optical spectra of 4BBA, a combination of potentiometric titration and solvent-suppression 1H-NMR spectra was carried out. The UV-Vis spectra for the 100 μM solution of 4BBA at pHs were acquired at pHs ranging from 1.0 to 12.0. Similarly, 1H-NMR spectra were collected for solutions at pHs from 1.0 to 7.0.
Theoretical models and methods
To better understand the water–4BBA interaction as a function of pH, the theoretical spectra of all possible forms of 4BBA, as described in Scheme 1, were simulated and compared with the UV-Vis spectral data of 4BBA. To confirm the NMR spectral data, the protonated and deprotonated keto- and diol-forms of water–4BBA were also calculated and its absorption spectra simulated. In this work, the theoretical results of neutral keto- and diol-forms of 4BBA were associated with the experimental spectrum measured at pH = 1, whereas the deprotonated keto- and diol-forms of 4BBA were considered as possible models for the experimental spectrum of 4BBA at pH = 6. This pH range around pKa,1 was used to model the protonated and deprotonated species, allowing the study of which the speciated form of 4BBA is favored at low and high pHs. | ||
| Fig. 1 Geometrical structures of hydrated 4BBA molecules: (A) neutral keto-form of 4BBA, (B) neutral diol-form of 4BBA, (C) deprotonated keto-form of 4BBA, and (D) deprotonated diol-form of 4BBA. | ||
All theoretical calculations for small clusters 4BBA·(H2O)8 were performed using density functional theory (DFT) and time-dependent DFT (TD-DFT) methods within the Q-Chem program.54 Geometry optimizations employed the B3LYP functional55 and the basis set 6-31+G*, with dispersion corrections from Grimme's DFT-D2.56 For calculations of excited states, the explicit solvent molecules (water clusters in our suggested models) were combined with the polarizable continuum model (C-PCM).57 Solute cavities are constructed from a union of atom-centered spheres whose radii are 1.2 times the atomic van der Waals radii suggested by Bondi.58 The choice of the functional and the basis set is crucial in accurately studying molecular systems. Here, the B3LYP/6-311++G** method is selected. This functional and basis set combination has been successfully applied before to study benzoic acid (BA).1 In this study, the B3LYP functional was assessed with various standard basis sets, including Pople (6-31+G*, 6-311+G*, 6-311++G**) and Dunning (aug-cc-pVTZ and aug-cc-PVQZ) basis sets. The results revealed that the aug-cc-pVTZ and 6-311++G** basis sets provided the closest agreement to the aug-cc-pVQZ basis set, which is known for its accuracy. Therefore, using the Pople's basis set 6-311++G** can be a reasonable alternative that saves computational time compared to the more expensive augmented Dunning's basis sets. This finding suggests that by employing the appropriate basis set, reliable results can be obtained while reducing the computational cost.
To assess the accuracy of the B3LYP functional in predicting the optical absorption spectra of 4BBA systems, we performed a comprehensive comparative analysis involving various functionals such as B3LYP, CAM-B3LYP, LRC-wPBEh, and wB97X. The calculations were focused on the neutral keto-form of the 4BBA·(H2O)8 complex, and the obtained spectra were compared to the experimental data obtained at pH = 1. The findings of this investigation will be discussed in detail in the forthcoming “Results and discussion” section.
This relatively small size of this water cluster limits the solvent interaction with 4BBA to the carboxyl group, leaving the keto/diol groups with no solvating water interactions. To gain deeper insights into the nature of the excited states responsible for the I- and II-bands, we conducted calculations of hole/particle natural transition orbital (NTO) pairs using the B3LYP/6-311++G** method. Additionally, we explored the NTOs for the neutral keto-form of 4BBA·(H2O)8 using the CAM-B3LYP/6-311++G** method in the gas phase to evaluate if there is an extent of orbital delocalization and investigate the potential contribution of water molecules in the excited states if any. This allowed us to examine if there is the possibility of charge transfer phenomena as the system's cluster size increased, which can be missed due to the B3LYP functional. CAM-B3LYP is known to be superior to B3LYP for studying charge transfer phenomena due to its ability to capture long-range electron transfer interactions accurately. Unlike B3LYP, which treats all electron–electron interactions equally, CAM-B3LYP incorporates a long-range correction that improves the description of charge transfer states. This correction accounts for the fraction of Hartree–Fock exchange that contributes to long-range electron–electron interactions, which are crucial for charge transfer phenomena. The outcomes of these calculations will be extensively discussed, shedding light on the underlying electronic characteristics of the specific excited states under investigation.
For large systems, all calculations were performed using the DFTB+(18.2 version) package.62 Calculations were performed using the DFTB3-D3H5 formalism60,63 and the 3ob-3-1 parameter set.64–66 DFTB-D3H5 was a variant of DFTB3 with additional corrections for non-covalent interactions, including dispersion and hydrogen bonds. For the DFTB3-D3H5 calculation, the specific parametrization of the dispersion correction (DftD3) was used: sr6 = 1.25; alpha6 = 29.62; s6 = 1.0; and s8 = 0.49.65 In addition, hydrogen–hydrogen repulsion was also activated.67 The maximum angular momentum was set to: H = s; C = p; O = p; and N = p. The Hubbard derivatives (in atomic units) were specified for the selected parameter set: H = −0.1857; C = −0.1492; O = −0.1575; and N = −0.1535. The excited states of the molecule 4BBA in a water cluster were calculated using the TD-DFTB method62 as implemented in the DFTB+ package. To generate initial structures for four distinct forms of 4BBA, the most energetically preferable geometries of 4BBA (obtained in a gas phase using the B3LYP/6-31+G* method) were surrounded by 30 water molecules. The Packmol68 program was used to randomly distribute these 30 water molecules around the 4BBA molecule, specifically placed at the center of the cluster sphere (radius 8 Å). The obtained geometries were optimized by DFTB and then used as an initial structure for MD-DFTB calculations.
All MD calculations were performed at constant energy. Initial velocities were sampled for the equilibrium structure of interest from the Boltzmann distribution at 298 K. The time-step was 0.4 fs; with a total of 40 trajectories, 10 of these trajectories were simulated for 4BBA·(H2O)30 per isomer. The total calculation time was around 10 ps per trajectory. Each of these trajectories was used for the calculation of the total optical absorption spectrum: we extracted structures every 250 fs of the simulation, and their vertical excitation energies and oscillator strengths were calculated using the TD-DFTB method. All the resulting Gaussian distributions were added to yield the excitation spectrum. This approach has been successfully implemented for the simulation of β-hydroxyalkyl nitrates, pyruvic and benzoic acids.1,38,61
To assess the performance of the TD-DFTB method, we conducted a comparative analysis of the optical absorption spectra obtained using the tight-binding approach and the TD-DFT method. Specifically, we examined the optical spectra of the neutral and ionized keto-forms of 4BBA·(H2O)30, which were selected from the DFTB-MD trajectories. Our findings indicate that there is no significant difference in the curve shape of the optical spectra obtained using both methods. In the case of the neutral structure, the maximum difference in peak positions between TD-DFTB and TD-DFT is only 3 nm, as depicted in Fig. S2 of the ESI.† However, for the anionic structure, there is a notable redshift of approximately 20 nm in TD-DFTB compared to TD-DFT. Overall, the results demonstrate that TD-DFTB yields reasonable agreement with TD-DFT in reproducing the optical absorption spectra for both the neutral and anionic structures of 4BBA. This suggests that the TD-DFTB method can be a reliable alternative method for studying the optical properties of these systems.
The optical spectra were simulated by convoluting the vertical transitions at each excitation energy with a Gaussian line shape. The selection of the full width at half-maximum (FWHM) for the Gaussian distribution was based on the experimental FWHM, which ranged between 0.39 and 0.42 eV. To simplify the calculations, the FWHM value was approximated as 0.4 eV. Subsequently, all spectra were convolved in energy units (eV). Then, the result was converted to wavelength units (nm) using the formula λ (nm) = 1239.9/E (eV), where λ represents the wavelength and E denotes the energy.
Results and discussion
Experimental results
The UV-Vis spectra of the 100 μM solutions of 4BBA at pHs 1.0 to 12.0 are shown in Fig. 2A. At low pHs, two bands are clearly observed, highlighted as minima in the first-order derivative of the UV-Vis spectra (Fig. 2B): an intense I-band centered at 260 nm and a II-band centered at 333 nm. The I-band has a long absorbance tail across all pHs examined, reaching the visible spectral region. Between pHs 1.0 and 3.0, there are no significant changes in the spectral features of the solution. As the pH increases from 3.0 to 4.0, a small redshift of 4 nm in the I-band is observed, after which the intensity of the I-band decreases and remains relatively constant up to pH 12.0. Above pH 4.0, the II-band is no longer observable.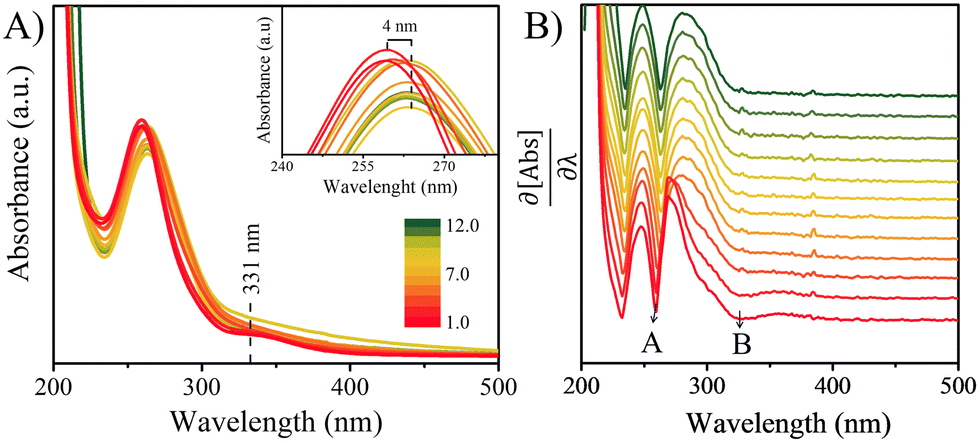 | ||
| Fig. 2 (A) Representative UV-vis absorption spectra of 100 μM 4BBA solutions over a range of pH conditions from 1.0 to 12.0. The inset shows the redshift of band I. (B) First order derivative of spectral data, showing minima for bands I and II. | ||
As shown in Scheme 1, keto acids can be found in protonated and deprotonated forms.69 In order to determine whether the diol- or keto-forms of 4BBA are present in the aqueous phase, the pH-dependent 1H-NMR analysis was carried out at 22 °C (ESI,† Fig. S1). This keto–diol equilibrium has been observed in other environmentally relevant keto acids. While the diol-form is not stably isolated, its presence in solution is possible and can increase the speciation of the system. This 1H-NMR analysis shows no detectable formation of the diol in the solution phase at any of the pHs examined. Thus, the speciation of 4BBA is primarily the acid and the conjugate base of the keto-form, without any diol species detected.
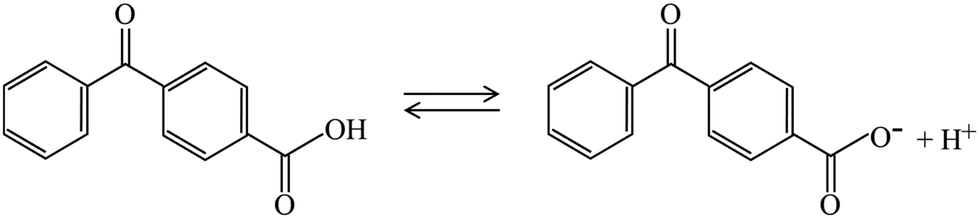
Aromatic rings will tend to make the carbon in the keto group more electrophilic by induction, which would favor hydration and the formation of gem-diol species in the aqueous phase.69 However, resonance effects make the carbon atom in the keto group less electrophile, driving the charge density and thus preventing the formation of diols for 4BBA in aqueous solutions.691H-NMR only shows the keto-form of 4BBA, with the expected shifts in the spectra as the protonation/deprotonation of carboxylic acid takes place.
Fitting of the NMR spectral data shifts in response to the acid and conjugate base equilibrium yielded a pKa of 3.41 ± 0.02 (ESI,† Fig. S1B and C). Previous 4BBA pKa measurements were performed at a pKa near 4.85;70 yet, these measurements were performed in 50% ethanol solutions, in order to increase the solubility of 4BBA. In addition to 1H-NMR, the acid–base equilibrium of 4BBA was investigated in 18 MΩ water via potentiometric titration, resulting in a pKa of 3.41 ± 0.04 (ESI,† Fig. S1A), a statistically similar value than that reported via1H-NMR analysis. Thus, the UV-Vis absorption spectra below pH 3.0 represent that of neutral 4BBA, with over 98% of species in the neutral form. Absorption spectra taken above pH 6 represent that of anionic species as the system is more than 99% deprotonated. At an acidic pH, neutral 4BBA shows higher absorbance in the I-band and II-band. As the pH increases, forming deprotonated species, the I-band at 260 nm redshifts to 264 nm and the II-band is not observable above pH 4.
The NMR analysis and pH-dependent changes in the optical properties of 4BBA in solutions indicate slight variations in the electronic structure of the protonated and deprotonated forms of 4BBA. The redshift, observed as the pH increases, occurs at the reported pKa of 4BBA, suggesting that deprotonation plays a role in the absorbance spectral shift. These variations may alter the solvating effects and stability of 4BBA, as well as its optical properties in the aqueous phase.
Comparison of experimental results with theoretical calculations
As noted above, the experimental spectrum of 4BBA at pH = 1 shows two bands: a strong I-band at 260 nm and a weak broad II-band between 320 and 360 nm (Fig. 2). However, the spectrum at high pH has only one strong I-band centered at 264 nm. Above pH 4, the I-band on the UV-Vis spectra of aqueous 4BBA is redshifted by 4 nm and becomes significantly intense with increasing pH. The ratio of experimental I-band intensities between pH 1 and 6, pH values that represent the theoretical models, is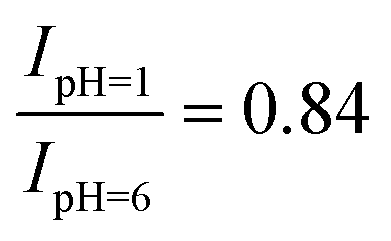 .
.
Spectral changes in aqueous 4BBA with respect to pH, summarized in Fig. 3, suggest that the increase in pH, with the concomitant deprotonation of 4BBA, results in two competing effects that ultimately lead to a slight redshift of the I-band: First, the electron delocalization of the deprotonated species (higher pH) decreases the electron transition energy, leading to the redshift in the absorption spectrum as the pH increases. Second, the interaction between water and the more polar anionic 4BBA is relatively more favorable, which stabilizes the ground state of the deprotonated species (higher pH) compared to that of neutral species (lower pH). This last solvent effect increases the electron transition energy, resulting in a slight blueshift. This last effect is not significant, leading to an overall redshift with the pH increase. Understanding the relative magnitude of these two competing effects is examined using theoretical clusters (vide infra). Yet, the experimental results suggest that the relatively low solubility of 4BBA, interpreted here as a less favored interaction between water and 4BBA compared to smaller organic acids, minimizes the second effect, leading primarily to the redshift in the absorption spectrum as the pH increases primarily due to electron delocalization. The water interaction with 4BBA, even though smaller than other organic acids, is enough to counterbalance the redshift due to delocalization, resulting in only a slight redshift as the pH increases, as shown in Fig. 2 and 3. The relative combination of these two competing effects is more complex, as it involves 4BBA speciation, energy changes in the excited state, and the intermolecular interaction of each speciated form with water molecules. Overall, the small redshift observed in 4BBA solutions indicates that the electronic structures of the anions and neutral species are relatively similar.
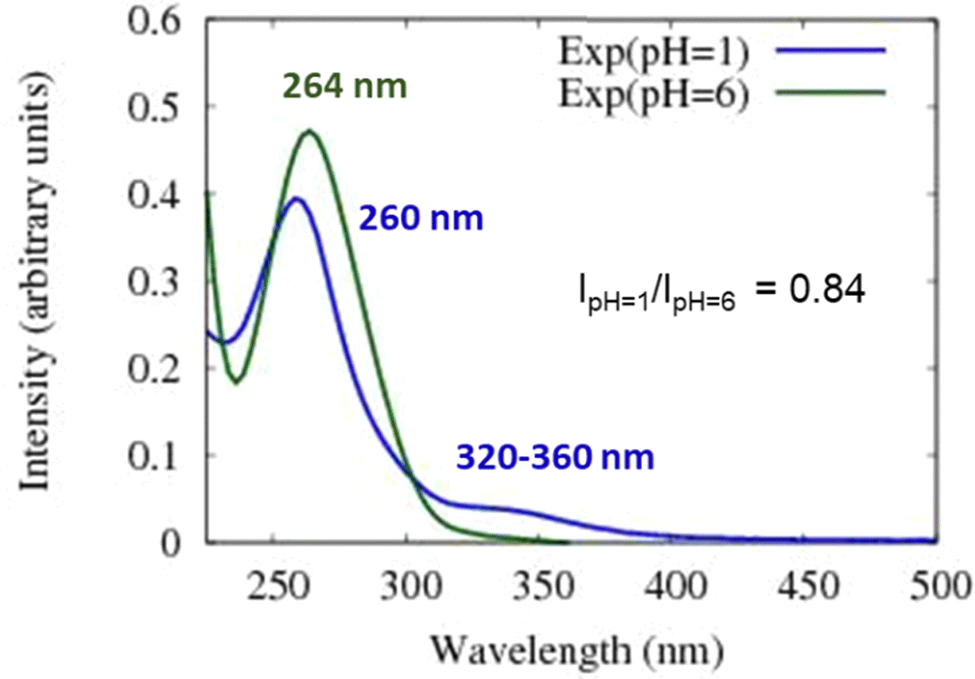 | ||
| Fig. 3 Experimental UV-Vis absorption spectra of 4BBA at both pH = 1 (blue line) and pH = 6 (green line). These two spectra were used to compare the theoretical results of protonated and deprotonated 4BBA. | ||
Using the DFT method, we calculated the populations of the keto- and diol-forms of 4BBA in a water solution at various pH values by evaluating the free Gibbs energy change between these forms (see Fig. S3a and b, ESI†). Our results revealed that the keto-form is energetically favored in both cases, with ΔG values of −8.31 kcal mol−1 for the neutral state and −13.69 kcal mol−1 for the ionized state. Consequently, at room temperature in low pH water, it is expected that the neutral keto-form will dominate (91.76%), while the neutral diol-form will constitute a smaller fraction (8.24%). Similarly, at high pH, the ionized keto-form is expected to be overwhelmingly predominant (99.98%), while the ionized diol-form will be present in negligible quantities (0.02%). These findings provide valuable insights into the relative populations of the different forms of 4BBA under specific pH conditions.
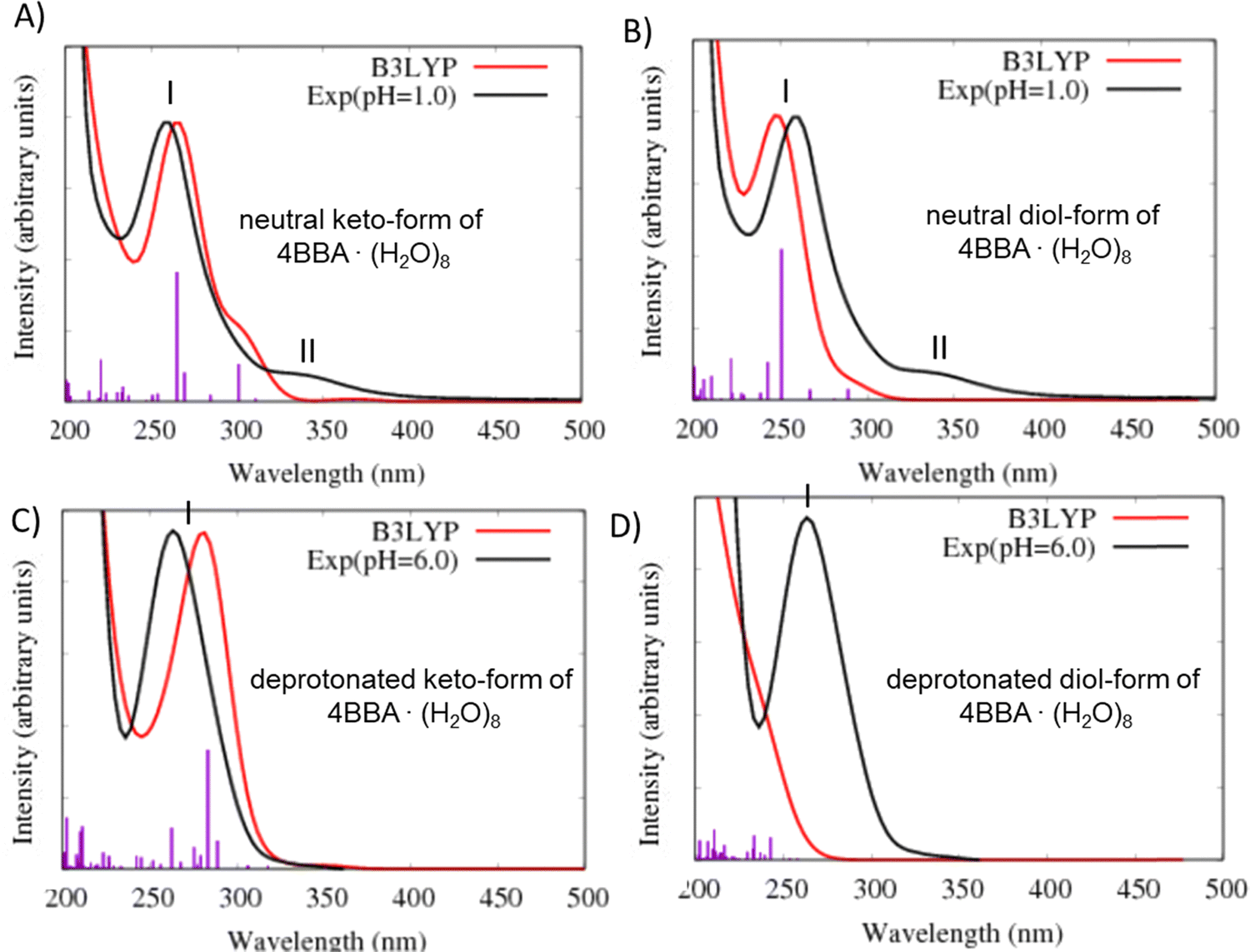 | ||
| Fig. 4 Comparison of the theoretical (red line) and experimental (black line) optical absorption spectra at different pHs: (A) theoretical complex of neutral keto-form of 4BBA with 8 water molecules vs. experiment at pH = 1; (B) theoretical complex of neutral diol-form of 4BBA with 8 water molecules vs. experiment at pH = 1; (C) theoretical complex of deprotonated keto-form of 4BBA with 8 water molecules vs. experiment at pH = 6; and (D) theoretical complex of deprotonated diol-form of 4BBA with 8 water molecules vs. experiment at pH = 6. Level of theory is B3LYP/6-311++G** (gas phase). | ||
In the theoretical spectrum of neutral keto-4BBA, the strong band at 265 nm can be considered as the experimental I-band and the second broad low-energy band at 300 nm can be assigned with the experimental II-band. This small cluster model provides a reasonable agreement between the experiment and theory in the case of the strong I-band, with a difference of 5 nm in the peak position between theory and the experiment. However, according to the theoretical results, the experimental II-band appeared at significantly lower wavelengths (Table 1). Additional solvent effects, such as the polarizable continuum model (C-PCM), do not make any significant improvements in the calculation (Fig. S4, ESI†).
| Bands | Experiment (pH = 1) | TD-DFT neutral 4BBA·(H2O)8 | TD-DFTB neutral 4BBA·(H2O)30 | ||
|---|---|---|---|---|---|
| Keto-form | Diol-form | Keto-form | Diol-form | ||
| I-band | 260 nm | 265 nm | 251 nm | 267 nm | 274 nm |
| 4.77 eV | 4.68 eV | 4.94 eV | 4.64 eV | 4.53 eV | |
| II-bands | 320–360 nm | 300 nm | 289 nm | 315–375 nm | 325–375 nm |
| 3.44–3.89 eV | 4.13 eV | 4.29 eV | 3.31–3.94 eV | 3.31–3.82 | |
| Bands | Experiment (pH = 6) | TD-DFT deprotonated 4BBA·(H2O)8 | TD-DFTB deprotonated 4BBA·(H2O)30 | ||
|---|---|---|---|---|---|
| Keto-form | Diol-form | Keto-form | Diol-form | ||
| I-band | 264 nm | 283 nm | <250 nm | 309 nm | 261 nm |
| 4.67 eV | 4.38 eV | >4.96 eV | 4.01 eV | >4.74 eV | |
The calculation of the optical spectrum for the neutral keto-form of 4BBA·(H2O)8 using different functionals, including CAM-B3LYP, LRC-wPBEh, and wB97X, revealed their inability to reproduce the low-energy band accurately (Fig. S5a, ESI†). This is consistent with our previous studies on benzoic acid (BA) at low pH, where the low-energy band was also challenging to reproduce using the TD-DFT method.1 In contrast, the ADC(2) method successfully reproduced the intensities and position of the peaks of the BA spectrum, with good agreement between theory and the experiment (Fig. S5b, ESI†). Despite the fact that the ADC(n) level of theory was able to address these issues, its computational demand makes it impractical for the current system. Also, it is important to note that in the study of benzoic acid (BA), TD-DFT could accurately reproduce the nature of the involved orbitals for both bands, achieving a level of accuracy comparable to the more advanced ADC(2) method (Fig. S5b, ESI†). Therefore, in the current work, we can confidently conclude that the theoretical broad low-energy band observed in the spectrum of neutral keto-4BBA is indeed related to the II-band observed in the experimental spectrum. This suggests that TD-DFT can capture the electronic characteristics of the excited states involved in this specific system.
The optical spectrum of the neutral diol-form also exhibits two spectral bands at 251 and 289 nm (Fig. 4B and Table 1). However, the difference with the experimental data is more significant in comparison to the keto-form: the I-band of this theoretical spectrum is blue-shifted at 9 nm and the II-band is shifted at 51 nm. The experimental energy gap between these two bands is around 1.1 eV, whereas the theoretical calculation yields a significantly smaller value of 0.65 eV (Table 1).
Two additional models were considered to simulate the optical spectrum of 4BBA at high pH: deprotonated keto- and diol-forms of 4BBA with 8 water molecules. The shape of the theoretical spectrum of the deprotonated keto-form of 4BBA agrees with the experimental results (Fig. 4C and Table 1). However, the calculated spectrum of the deprotonated diol-form of 4BBA exhibits a shoulder band blue-shifted with respect to experimental peak II. There is only a weak shoulder band above 250 nm (Fig. 4D). The calculated spectra for four speciated forms of 4BBA allowed us to conclude that the molecule of 4BBA at pH = 1 exists in the neutral keto-form, whereas at pH = 6, it should have a deprotonated keto-structure. These theoretical results agree with the experimental findings and further suggest that the hydrate form of 4BBA is not present in the aqueous phase.
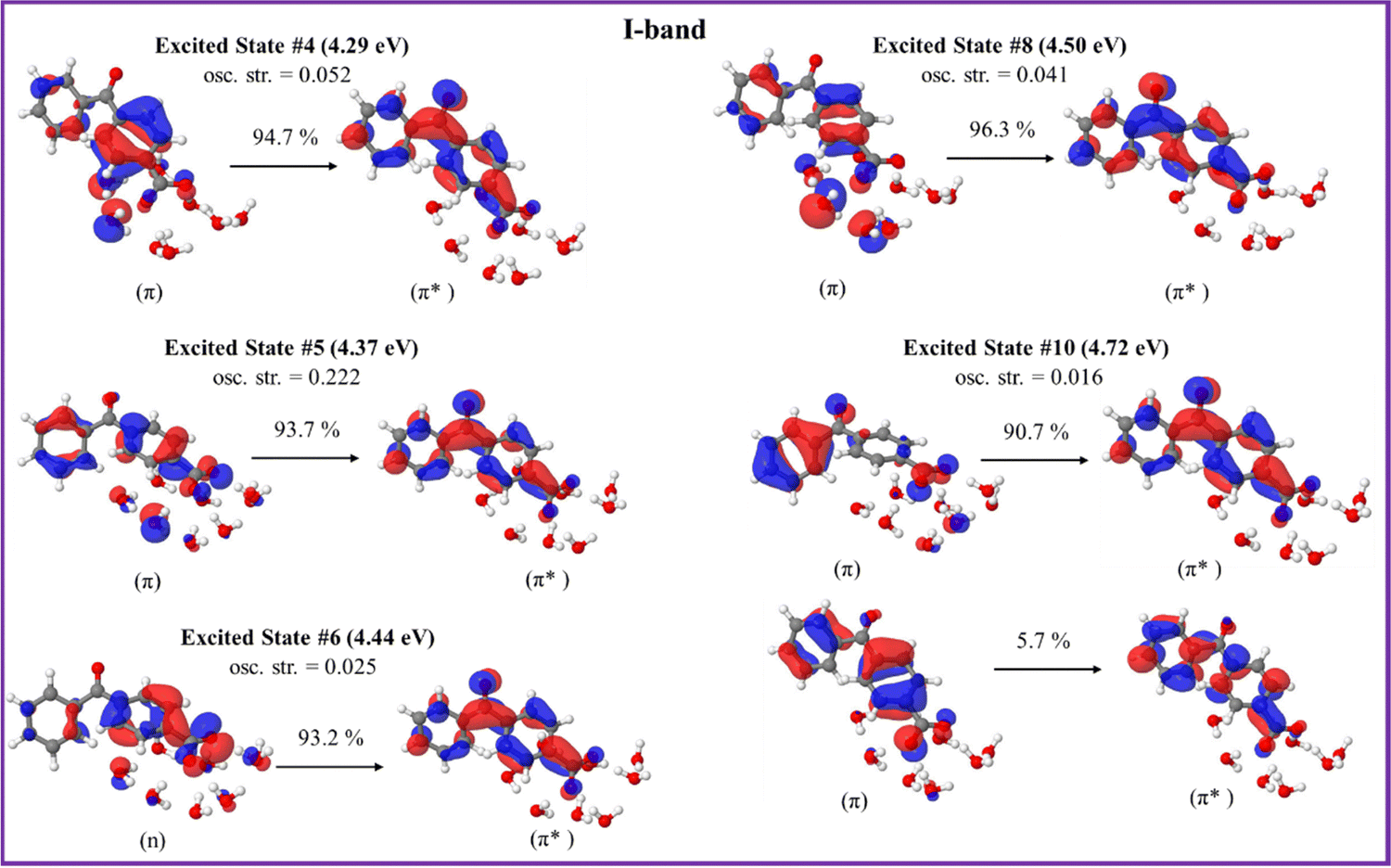
The orbitals involved in the electron transitions in both protonated and deprotonated clusters are shown in Fig. 5 and 6. The low-energy II-band of the neutral keto-form arises due to the π → π* transitions (Fig. 5). However, the strong I-band appears due to the mixture of n → π* and π → π* transitions for both speciated forms: neutral- and deprotonated keto-4BBA (Fig. 5 and 6).
 | ||
| Fig. 5 Natural transition orbitals of the selected excited states of the neutral keto-form of 4BBA·(H2O)8 calculated at the B3LYP/6-311++G** (gas phase). | ||
 | ||
| Fig. 6 Natural transition orbitals of the selected excited states of the deprotonated keto-form of 4BBA·(H2O)8 calculated at the B3LYP/6-311++G** (gas phase). | ||
The comparison of orbitals corresponding to the neutral and deprotonated keto-clusters indicates clear similarities. For example, the I-band of both structures arises due to the mixture of n → π* and π → π* transitions (Fig. 5 and 6). However, there are certain differences that impact the spectral features of aqueous 4BBA. In particular, the natural transition orbitals (NTOs) of the deprotonated structure are located on the entire 4BBA fragment, whereas in the case of the neutral keto-form, these orbitals are only located on the C6H5–C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)– or –C6H4–COO fragments only (Fig. 5 and 6). Additionally, we can see those orbitals on the water molecules participate in the electron transitions of the I-band of both the neutral- and the deprotonated keto-forms of 4BBA. This suggests that both organic acid and water molecules within the cluster contribute to excitation and further emphasizes the relevance of water cluster modeling around the chromophore. Thus, it is paramount to examine the effect of water solvation on the optical properties of 4BBA.
O)– or –C6H4–COO fragments only (Fig. 5 and 6). Additionally, we can see those orbitals on the water molecules participate in the electron transitions of the I-band of both the neutral- and the deprotonated keto-forms of 4BBA. This suggests that both organic acid and water molecules within the cluster contribute to excitation and further emphasizes the relevance of water cluster modeling around the chromophore. Thus, it is paramount to examine the effect of water solvation on the optical properties of 4BBA.
Calculated NTOs for the neutral keto-form of 4BBA·(H2O)8 using the CAM-B3LYP/6-311++G** level of theory are found in Fig. S6, ESI.† The results obtained from both B3LYP and CAM-B3LYP functionals exhibit striking similarity, with all highest occupied molecular orbitals (HOMOs) and lowest unoccupied molecular orbitals (LUMOs) being localized solely on the 4BBA molecules. Notably, there are no orbitals involving the water molecules, indicating the absence of any detected charge transfer process.
The optical spectra of the neutral forms of 4BBA (low pH models) were found to be significantly influenced by the contribution of different speciated forms and solvent effects, as evidenced by the results obtained for larger clusters 4BBA·(H2O)30 (Fig. 7A and B). In Fig. 7A, we can see some improvements in the theoretical spectrum of the neutral keto-form of 4BBA·(H2O)30 related to the position of the low-energy II-band. This band still has a higher amplitude than it has in the experiment, but the position of this band is closer to the experiment. The gap between the I and II peaks is closer to the experimental value for the larger complexes (Table 1).
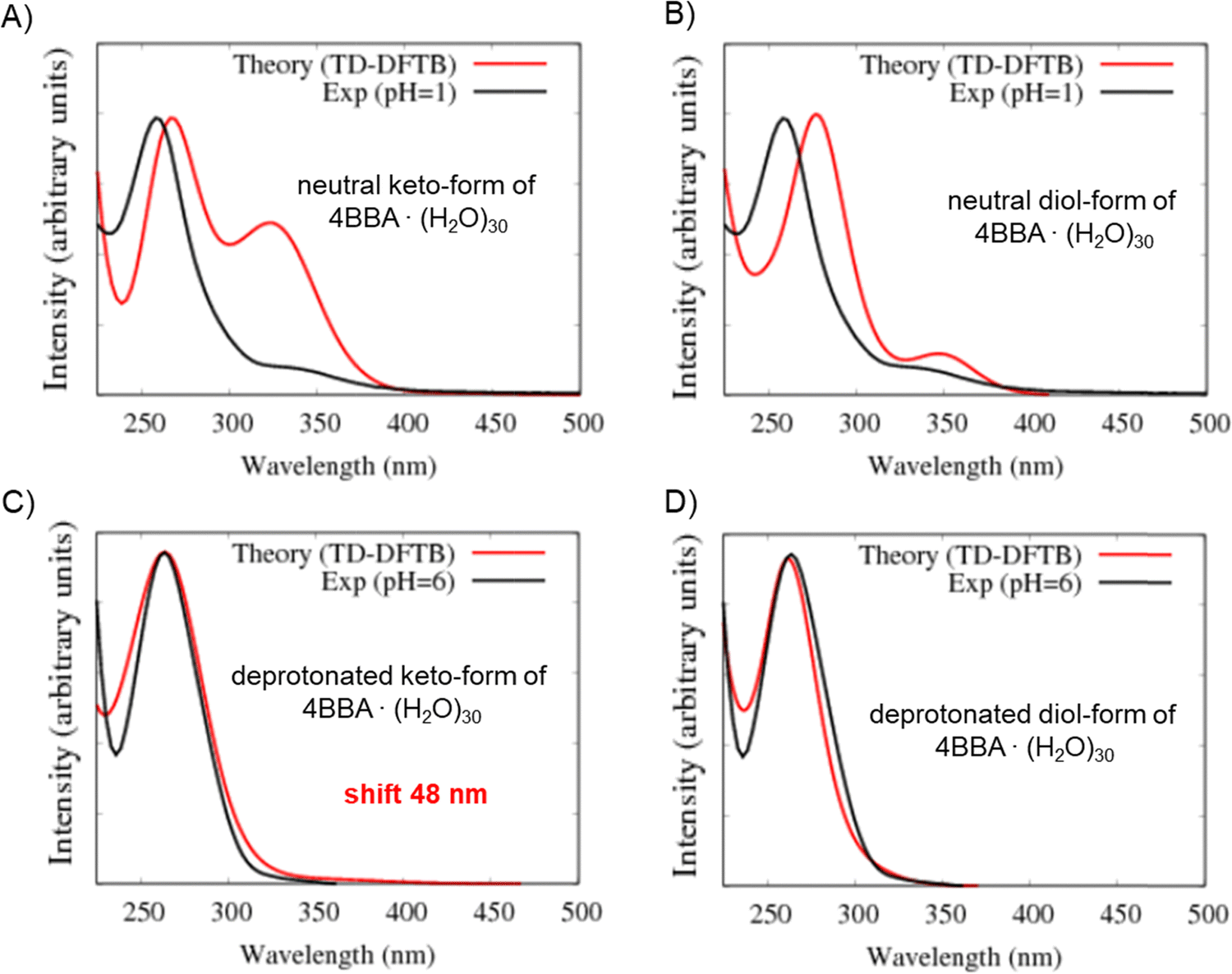 | ||
| Fig. 7 Comparison of the theoretical (red line) and experimental (black line) optical absorption spectra at low pH. The theoretical spectrum is an average spectrum of all structures along MD-DFTB trajectories: (A) theoretical complex of neutral keto-form of 4BBA with 30 water molecules vs. experiment at pH = 1; (B) theoretical complex of neutral diol-form of 4BBA with 30 water molecules vs. experiment at pH = 1; (C) theoretical complex of deprotonated keto-form of 4BBA with 30 water molecules vs. experiment at pH = 6; and (D) theoretical complex of deprotonated diol-form of 4BBA with 30 water molecules vs. experiment at pH = 6. The level of theory is TD-DFTB. | ||
The optical spectrum of the neutral diol form of 4BBA, calculated with 30 water molecules, exhibits a distinct feature not observed in the spectrum obtained with eight water molecules: the intensity of the low-energy broad peak (II-band between 325 and 375 nm) increases in the presence of the larger water cluster (Fig. 7B). Additionally, the energy gap between the I- and II-bands became larger, around 1.0 eV. This result is likely due to the solvating stabilization of the diol-forms, which can form hydrogen bonding with water molecules in geminal diol carbon. The separation between the carbonyl and the hypothetical diol functional groups in the neutral diol-form of 4BBA prevents the formation of intramolecular hydrogen bonding, leaving the diol-form and carbonyl groups available for intermolecular hydrogen-bonding with the water solvent. This effect is in stark contrast with smaller environmentally relevant chromophores, such as pyruvic acid, where intermolecular interactions tend to stabilize the protonated forms and weaken intermolecular hydrogen bonding with the surrounding water.35 This intramolecular H-bonding likely contributes to the stability of the diol-form of keto-acids in solution, the effect absent in 4BBA. This low-energy band on the spectrum of the neutral diol-form of 4BBA·(H2O)30 is very close (position and size of magnitude) to the experimental II-band of 4BBA at low pH.
The optical spectra of the deprotonated keto-form complexes (high pH models) exhibited a noticeable redshift compared to both the experimental data and the small water cluster system (Table 1). However, the overall shape of the theoretical curve closely matches the experimental results. Despite the I-band centered at a higher wavelength than the experimental band, it is consistent with the redshift experimentally observed upon pH increase across the pKa of 4BBA (Fig. 7C). This matching behavior from protonated to deprotonated forms suggests that 4BBA at pHs above the pKa exists in the deprotonated keto-form, in good agreement with the experimental findings. Overall, there is a 48 nm difference between the experimental and theoretical spectra of the deprotonated keto-forms due to the increasing effect of solvating water molecules in the deprotonated 4BBA, demanding more challenging computation. This significant spectral shift of the theoretical spectra of 4BBA complexes in the keto-form with respect to the experimental data can be related to the relatively short MD trajectories where the system did not have enough time for relaxation. Thus, extending the molecular dynamics trajectories should increase the agreement between theoretical simulations and experimental observations where the system will reach a more stable and equilibrated state.
In contrast to the small cluster, the calculated spectrum of the deprotonated diol-form of 4BBA·(H2O)30 exhibits a strong peak at 261 nm (Fig. 7D). The peak of the large cluster has stronger intensity and it is red-shifted compared to the small cluster. This phenomenon can be attributed to solvent effects, specifically the interaction between water molecules and the diol group, which leads to the stabilization of the diol-form of 4BBA.
The structures of all speciated forms of 4BBA were initially placed at the center of a 30-water cluster sphere. Diol- and keto-forms exhibit different water distributions during the MD simulations. In the case of the keto-forms (Fig. 8A and B), the keto-group coordinates with just 1 or 2 water molecules. For the neutral keto-structure, there are 60% of trajectories leading to the formation of structures with two water molecules coordinated nearby the keto-group (Fig. 8A.2 and 3), whereas, in the case of the deprotonated form, there are 90% of all trajectories where just one water molecule is located near the oxygen atom of the keto-group (Fig. 8B.2 and 3). The opposite is found for the diol-forms, and the diol-functional group increases the capability of hydrogen bonding with the surrounding water molecules compared to the keto-group. A strong diol-group and water molecule interaction is observed (Fig. 8C and D. Given the additional OH functional group in geminal carbon, the diol-group coordinates with 3 or 4 water molecules with an average hydrogen bond distance of 2 Å (Fig. 8C and D). These hydrogen-bond lengths do not significantly change during the MD simulation time (Fig. 8C.3 and D.3).
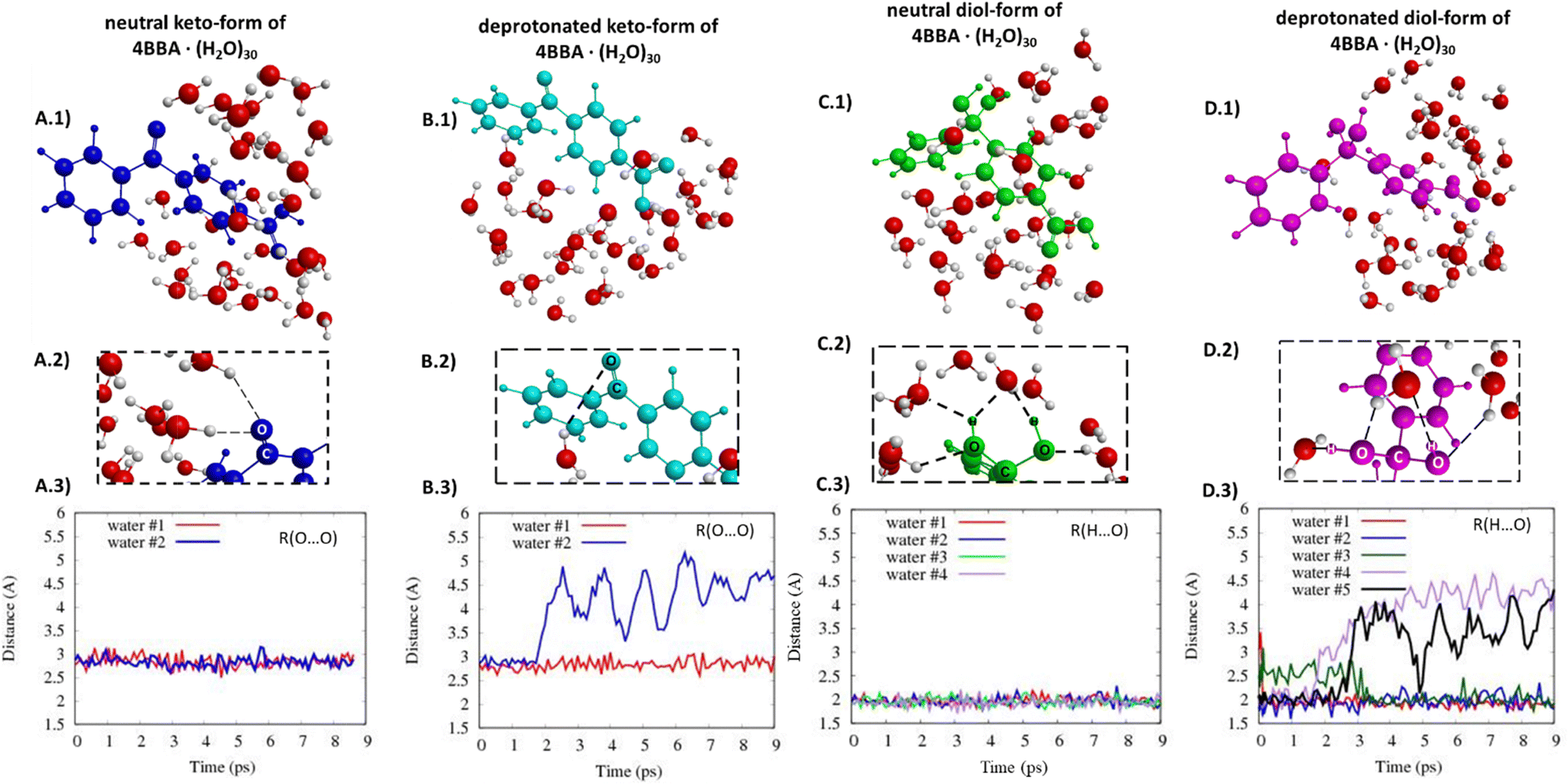 | ||
| Fig. 8 Selected structures of the (A.1) neutral keto-form of 4BBA, (B.1) deprotonated keto-form of 4BBA, (C.1) neutral diol-form of 4BBA, (D.1) deprotonated diol-form of 4BBA, and (A.2, B.2, C.2 and D.2) water molecules coordination around the keto- and diol-groups. Distances between the oxygen atom of the keto-group and oxygen atoms of the nearest water molecules (along the MD trajectory) for the neutral (A.3) and deprotonated (B.3) clusters of keto-form 4BBA. Hydrogen bonds along the MD trajectory between the diol-group and the nearest water molecules in the cluster of the neutral diol-form of 4BBA (C.3) and the deprotonated diol-form of 4BBA (D.3). Note: for the keto-form, we considered O⋯O distances, because some of the water molecules using different hydrogen atoms to coordinate by an oxygen atom of the keto-group along the trajectory. | ||
Overall, theoretical calculations and experimental findings indicate that the experimental optical spectra of the aqueous phase 4BBA at low pH can be represented by the neutral keto-form. Similarly, the theoretical spectrum of the deprotonated keto-form successfully represents the experimental spectra at high pH, as it models the experimentally observed redshift that occurs as the pH increases. Calculations confirm that the more electron-delocalized deprotonated keto-form form of 4BBA, observed at higher pHs, leads to a small decrease in the excitation energy observed as a redshift in Fig. 1. In contrast, the deprotonated keto-form leads to a shift to the blue as pH increases, likely due to water interactions. While the theoretical spectrum of the deprotonated keto-form form of 4BBA models the correct direction of the spectral shift with respect of pH, a more accurate simulation of water solvent effects at different pHs is needed to improve the agreement between theory and the experiment further.
Conclusions
Chromophoric organic acids containing aromatic groups are widely present on the sea surface and can partition into the atmosphere through bubble bursting and wave breaking.19,37 Thus, these chromophores can undergo significant pH changes from the slightly basic sea surface microlayer (SSML pH 7.8) to the more acidic sea spray aerosol (SSA pH 2 to 4).46 Molecules like 4BBA, often used as a proxy for more complex organic substances, such as CDOM and humic-like substances, can provide the molecular level understanding of light-induced environmental processes and the role of chromophores in the Earth's radiative energy balance.5,40–43 Therefore, the systematic investigation of the absorption of 4BBA carried out here provides the critical understanding of the optical properties as it is applied to represent more complex light-absorbing substances under specific environmental conditions.In the present work, spectroscopic measurements are combined with theoretical calculations to study the optical absorption spectra of 4BBA in an aqueous solution. 1H-NMR spectra were used to experimentally determine that no hydration diol-forms of 4BBA are detectable at the pHs examined, indicating that the speciation of aqueous phase 4BBA consists of keto-forms. These experimental data, combined with potentiometric titrations, were used to determine a pKa of 4BBA of 3.41 ± 0.04. The theoretical hydrated model agrees well with the experimental spectra, especially for neutral speciated forms. Calculated absorption spectra also indicate that the keto-forms of 4BBA are the predominant form of 4BBA in the aqueous phase. Overall, the work shown here suggests that 4BBA, when used as a moiety in environmental photosensitizers, has characteristic speciation and optical properties depending on the pH of the environment, primarily as a protonated keto-form when present in SSA to the deprotonated keto-form in SSML and bulk natural waters. For these calculations, solvent effects are an important parameter for reproducing the correct optical spectra of 4BBA in the water environment, especially at lower pHs. Our theoretical results agree with the experimental observations, with a lower pH I-band showing both experimentally (at 260 nm) and theoretically (at 265 nm, TD-DFT). At higher pHs, the effect of water becomes weaker, with the redshift upon pH increases at 4 nm experimentally and 18 nm (TD-DFT) in our simulations.
The experimental optical spectra of 4BBA at different pHs can be presented by the neutral keto-form in the case of low pH and the deprotonated keto-form in the case of high pH. Both the experimental and theoretical results indicate the presence of a high-energy I-band that undergoes a slight redshift as pH increases, moving it closer to the solar spectral region. In addition, a low energy II-band at 330 nm only shows the characteristic of sea spray aerosols at low pHs. Theoretical model clusters of 4BBA with different numbers of water molecules demonstrated that solvent effects are crucial in reproducing the correct optical spectra of 4BBA. Theoretical clusters can be successful in describing chromophores in solution. The theoretical results showed that the I-band of both speciated forms of 4BBA arises from the combination of n → π* and π →π* transitions. The orbital analysis of the theoretical clusters allowed us to see that the orbitals involved in the electron transitions of the I-band involve a combination of 4BBA and water molecules within the cluster. Combining the quantum chemical calculations of excited states with molecular dynamics simulations thoroughly interprets experimental results and demonstrates the possibility of microscopic insight into the absorption spectra of organic molecules in water by calculations restricted to cluster models. The results show that a good spectroscopic description of organics dissolved in bulk can be obtained by simulations for clusters of water of moderate sizes. It seems sufficient when water clusters roughly extend over the size of the organic species. Applications to other systems of this type should thus be feasible.
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors would like to thank Mark Young and Imon Mandal for helpful comments, Anthony Mrse for assistance with NMR spectra analysis, and Alma Montserrat Palacios Puga and Gilberto Daniel Partida Coria for solution preparation in the 1H-NMR experiments. The authors would like to gratefully acknowledge support from the National Science Foundation through the Center for Aerosol Impacts on Chemistry of the Environment funded under the Centers for Chemical Innovation Program Grant CHE1801971. Additionally, this work used the Extreme Science and Engineering Discovery Environment (XSEDE),71 and SDSC EXPANSE CPU through TG-CHE170064 allocations from the Advanced Cyberinfrastructure Coordination Ecosystem: Services & Support (ACCESS) program, which is supported by National Science Foundation grants #2138259, #2138286, #2138307, #2137603, #2018494, and #2138296, and the MERCURY consortium (https://mercuryconsortium.org/) under NSF grants CHE-1229354 and CHE-1662030.References
- N. Karimova, M. Luo, V. H. Grassian and R. Gerber, Absorption Spectra of Benzoic Acid in Water at Different pH and in the Presence of Salts: Insights from the Integration of Experimental Data and Theoretical Cluster Models, Phys. Chem. Chem. Phys., 2020, 22, 5046–5056 RSC.
- E. M. Thurman, Organic Geochemistry of Natural Waters, Springer, Netherlands, Dordrecht, 1985 Search PubMed.
- J. Borgatta and J. G. NaveaFate, of Aqueous Iron Leached from Tropospheric Aerosols during Atmospheric Acidic Processing: Study of the Effect of Humic-Like Substances, WIT Trans. Ecol. Environ., 2015, 198, 155–166 Search PubMed.
- D. Drapanauskaite, K. Buneviciene, R. Repsiene, R. Mazeika, J. Navea and J. Baltrusaitis, Physicochemical Characterization of Pelletized Lime Kiln Dust as Potential Liming Material for Acidic Soils, Waste Biomass Valorization, 2021, 12, 1267–1280 CrossRef CAS.
- S. L. Mora Garcia, S. Pandit, J. G. Navea and V. H. Grassian, Nitrous Acid (HONO) Formation from the Irradiation of Aqueous Nitrate Solutions in the Presence of Marine Chromophoric Dissolved Organic Matter: Comparison to Other Organic Photosensitizers, ACS Earth Space Chem., 2021, 5(11), 3056–3064 CrossRef CAS.
- H. M. Ricker, A. L. Leonardi and J. G. Navea, Reduction and Photoreduction of NO2 in Humic Acid Films as a Source of HONO, ClNO, N2O, NOX, and Organic Nitrogen, ACS Earth Space Chem., 2022, 6(12), 3066–3077 CrossRef CAS PubMed.
- K. Mopper and R. G. Zika, Natural Photosensitizers in Sea Water: Riboflavin and Its Breakdown Products. In Photochemistry of Environmental Aquatic Systems, Am. Chem. Soc., 1987, 327, 174–190 CAS.
- D. J. Hassett, M. S. Bisesi and R. Hartenstein, Bactericidal action of humic acids, Soil Biol. Biochem., 1987, 19, 111–113 CrossRef CAS.
- M. A. Young, in Environmental Photochemistry in Surface Waters, Water Encyclopedia, ed. J. H. Lehr and J. Keeley, Wiley, 2005, pp. 529–535 Search PubMed.
- M. Klavins and O. Purmalis, Humic substances as surfactants, Environ. Chem. Lett., 2010, 8, 349–354 CrossRef CAS.
- J. A. Rice and P. MacCarthy, Statistical evaluation of the elemental composition of humic substances, Org. Geochem., 1991, 17, 635–648 CrossRef CAS.
- C. E. J. Van Rensburg, The Antiinflammatory Properties of Humic Substances: A Mini Review, Phytother. Res., 2015, 29, 791–795 CrossRef CAS PubMed.
- M. Zark and T. Dittmar, Universal molecular structures in natural dissolved organic matter, Nat. Commun., 2018, 9, 3178 CrossRef PubMed.
- W. S. Wan Ngah, M. A. K. M. Hanafiah and S. S. Yong, Adsorption of humic acid from aqueous solutions on crosslinked chitosan–epichlorohydrin beads: Kinetics and isotherm studies, Coll. Surf. B: Biointerfaces, 2008, 65, 18–24 CrossRef CAS PubMed.
- D. Kwon, M. J. Sovers, V. H. Grassian, P. D. Kleiber and M. A. Young, Optical Properties of Humic Material Standards: Solution Phase and Aerosol Measurements, ACS Earth Space Chem, 2018, 2, 1102–1111 CrossRef CAS.
- S. Jayalath, H. Wu, S. C. Larsen and V. H. Grassian, Surface Adsorption of Suwannee River Humic Acid on TiO2 Nanoparticles: A Study of pH and Particle Size, Langmuir, 2018, 34, 3136–3145 CrossRef CAS PubMed.
- Y. Miyazaki, Y. Yamashita, K. Kawana, E. Tachibana, S. Kagami, M. Mochida, K. Suzuki and J. Nishioka, Chemical transfer of dissolved organic matter from surface seawater to sea spray water-soluble organic aerosol in the marine atmosphere, Sci. Rep., 2018, 8, 14861 CrossRef PubMed.
- M. V. Santander, J. M. Schiffer, C. Lee, J. L. Axson, M. J. Tauber and K. A. Prather, Factors controlling the transfer of biogenic organic species from seawater to sea spray aerosol, Sci. Rep., 2022, 12, 3580 CrossRef CAS PubMed.
- J. V. Trueblood, M. R. Alves, D. Power, M. V. Santander, R. E. Cochran, K. A. Prather and V. H. Grassian, Shedding Light on Photosensitized Reactions within Marine-Relevant Organic Thin Films, ACS Earth Space Chem, 2019, 3, 1614–1623 CrossRef CAS.
- K. McNeill and S. Canonica, Triplet state dissolved organic matter in aquatic photochemistry: reaction mechanisms, substrate scope, and photophysical properties, Environ. Sci.: Processes Impacts, 2016, 18, 1381–1399 RSC.
- C. Baduel, D. Voisin and J.-L. Jaffrezo, Seasonal variations of concentrations and optical properties of water soluble HULIS collected in urban environments, Atm. Chem. Phys., 2010, 10, 4085–4095 CrossRef CAS.
- A. Hoffer, A. Gelencsér, P. Guyon, G. Kiss, O. Schmid, G. P. Frank, P. Artaxo and M. O. Andreae, Optical properties of humic-like substances (HULIS) in biomass-burning aerosols, Atmos. Chem. Phys., 2006, 6, 3563–3570 CrossRef CAS.
- Y. Chin, G. Aiken and E. O’Loughlin, Molecular Weight, Polydispersity, and Spectroscopic Properties of Aquatic Humic Substances, Environ. Sci. Technol., 1994, 28, 1853–1858 CrossRef CAS PubMed.
- D. P. Veghte, S. China, J. Weis, L. Kovarik, M. K. Gilles and A. Laskin, Optical Properties of Airborne Soil Organic Particles, ACS Earth Space Chem, 2017, 1, 511–521 CrossRef CAS.
- K. Kumada, Absorption spectra of humic acids, Soil Sci. Plant Nutr, 1955, 1, 29–30 CrossRef.
- X. Lei, J. Pan and A. Devlin, Characteristics of Absorption Spectra of Chromophoric Dissolved Organic Matter in the Pearl River Estuary in Spring, Remote Sens., 2019, 11(3), 1533 CrossRef.
- J. Ma, R. Del Vecchio, K. S. Golanoski, E. S. Boyle and N. V. Blough, Optical Properties of Humic Substances and CDOM: Effects of Borohydride Reduction, Environ. Sci. Technol., 2010, 44, 5395–5402 CrossRef CAS PubMed.
- E. S. Boyle, N. Guerriero, A. Thiallet, R. D. Vecchio and N. V. Blough, Optical Properties of Humic Substances and CDOM: Relation to Structure, Environ. Sci. Technol., 2009, 43, 2262–2268 CrossRef CAS PubMed.
- M. Peacock, C. Evans, N. Fenner, C. Freeman, R. Gough, T. Jones and I. Lebron, UV-visible absorbance spectroscopy as a proxy for peatland dissolved organic carbon (DOC) quantity and quality: Considerations on wavelength and absorbance degradation, Environ. Sci.: Processes Impacts, 2014, 16, 1445–1461 RSC.
- H. E. Reader, C. A. Stedmon, N. J. Nielsen and E. S. Kritzberg, Mass and UV-visible spectral fingerprints of dissolved organic matter: sources and reactivity, Front. Mar. Sci., 2015, 2, 88 Search PubMed.
- A. Momzikoff, R. Santus and M. A. Giraud, A study of the photosensitizing properties of seawater, Mar. Chem., 1983, 12, 1–14 CrossRef CAS.
- A. Leonardi, H. M. Ricker, A. G. Gale, B. T. Ball, T. T. Odbadrakh, G. C. Shields and J. G. Navea, Particle formation and surface processes on atmospheric aerosols: A review of applied quantum chemical calculations, Int. J. Quantum Chem., 2020, e26350 CAS.
- J. G. Navea and V. H. Grassian, Photochemistry of Atmospheric Particles, Reference Module in Chemistry, Molecular Sciences and Chemical Engineering, Elsevier, 2017 Search PubMed.
- N. V. Karimova, M. Luo, V. H. Grassian and B. R. Gerber, Towards a microscopic model of light absorbing complex organic components an aqueous environments: Theoretical and experimental study, Phys. Chem. Chem. Phys., 2021, 23, 10487–10497 RSC.
- C. J. Ostaszewski, N. M. Stuart, D. M. B. Lesko, D. Kim, M. J. Lueckheide and J. G. Navea, Effects of Coadsorbed Water on the Heterogeneous Photochemistry of Nitrates Adsorbed on TiO2, J. Phys. Chem. A, 2018, 122, 6360–6371 CrossRef CAS PubMed.
- D. Shemesh, M. Luo, V. H. Grassian and R. B. Gerber, Absorption Spectra of Pyruvic Acid in Water: Insights from Calculations for Small Hydrates and Comparison to Experiment, Phys. Chem. Chem. Phys., 2020, 22, 12658–12670 RSC.
- M. R. Alves, E. K. Coward, D. Gonzales, J. S. Sauer, K. J. Mayer, K. A. Prather and V. H. Grassian, Changes in light absorption and composition of chromophoric marine-dissolved organic matter across a microbial bloom, Environ. Sci.: Processes Impacts, 2022, 24, 1923–1933 RSC.
- M. Luo, D. Shemesh, M. N. Sullivan, M. R. Alves, M. Song, R. B. Gerber and V. H. Grassian, Impact of pH and NaCl and CaCl2 Salts on the Speciation and Photochemistry of Pyruvic Acid in the Aqueous Phase, J Phys Chem A, 2020, 124, 5071–5080 CrossRef CAS PubMed.
- N. V. Karimova, M. Luo, V. H. Grassian and R. B. Gerber, Absorption spectra of benzoic acid in water at different pH and in the presence of salts: insights from the integration of experimental data and theoretical cluster models, Phys. Chem. Chem. Phys., 2020, 22, 5046–5056 RSC.
- L. Tinel, S. Rossignol, A. Bianco, M. Passananti, S. Perrier, X. Wang, M. Brigante, D. J. Donaldson and C. George, Mechanistic Insights on the Photosensitized Chemistry of a Fatty Acid at the Air/Water Interface, Environ. Sci. Technol., 2016, 50, 11041–11048 CrossRef CAS PubMed.
- P. Corral Arroyo, T. Bartels-Rausch, P. A. Alpert, S. Dumas, S. Perrier, C. George and M. Ammann, Particle-Phase Photosensitized Radical Production and Aerosol Aging, Environ. Sci. Technol., 2018, 52, 7680–7688 CrossRef CAS PubMed.
- K. Couch, F. Leresche, C. Farmer, G. McKay and F. Rosario-Ortiz, Assessing the source of the photochemical formation of hydroxylating species from dissolved organic matter using model sensitizers, Environ. Sci.: Processes Impacts, 2022, 24, 102–115 RSC.
- M. E. Monge, T. Rosenørn, O. Favez, M. Müller, G. Adler, A. Abo Riziq, Y. Rudich, H. Herrmann, C. George and B. D’Anna, Alternative pathway for atmospheric particles growth, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 6840–6844 CrossRef CAS PubMed.
- H. E. Ungnade and R. W. Lamb, The Absorption Spectra of Benzoic Acid and Esters, J. Am. Chem. Soc., 1952, 74, 3789–3794 CrossRef CAS.
- B. V. Kamath, J. D. Mehta and S. L. Bafna, Ultraviolet absorption spectra: Some substituted benzoic acids, J. Appl. Chem, 1975, 25, 743–751 CrossRef CAS.
- K. J. Angle, D. R. Crocker, R. M. C. Simpson, K. J. Mayer, L. A. Garofalo, A. N. Moore, S. L. Mora Garcia, V. W. Or, S. Srinivasan, M. Farhan, J. S. Sauer, C. Lee, M. A. Pothier, D. K. Farmer, T. R. Martz, T. H. Bertram, C. D. Cappa, K. A. Prather and V. H. Grassian, Acidity across the interface from the ocean surface to sea spray aerosol, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2018397118 CrossRef CAS PubMed.
- C. R. Usher, A. E. Michel and V. H. Grassian, Reactions on mineral dust, Chem. Rev., 2003, 103, 4883–4940 CrossRef CAS PubMed.
- M. A. Freedman, E. E. Ott and K. E. Marak, Role of pH in Aerosol Processes and Measurement Challenges, J Phys Chem A, 2019, 123, 1275–1284 CrossRef CAS PubMed.
- J. Borgatta, A. Paskavitz, D. Kim and J. G. Navea, Comparative evaluation of iron leach from fly ash from different source regions under atmospherically relevant conditions, Environ. Chem., 2016, 13, 902–912 CrossRef CAS.
- D. M. B. Lesko, E. M. Coddens, H. D. Swomley, R. M. Welch, J. Borgatta and J. G. Navea, Photochemistry of nitrate chemisorbed on various metal oxide surfaces, Phys. Chem. Chem. Phys., 2015, 17, 20775–20785 RSC.
- D. Kim, Y. Xiao, R. Karchere-Sun, E. Richmond, H. M. Ricker, A. Leonardi and J. G. Navea, Atmospheric processing of anthropogenic combustion particles: Iron mobility and nitrite formation from flyash, ACS Earth Space Chem., 2020, 4, 750–761 CrossRef CAS.
- M. Luo, N. A. Wauer, K. J. Angle, A. C. Dommer, M. Song, C. M. Nowak, R. E. Amaro and V. H. Grassian, Insights into the behavior of nonanoic acid and its conjugate base at the air/water interface through a combined experimental and theoretical approach, Chem. Sci., 2020, 11, 10647–10656 RSC.
- M. C. Almandoz, M. I. Sancho and S. E. Blanco, Spectroscopic and DFT study of solvent effects on the electronic absorption spectra of sulfamethoxazole in neat and binary solvent mixtures, Spectrochim. Acta, Part A, 2014, 118, 112–119 CrossRef CAS PubMed.
- Y. Shao, Z. Gan, E. Epifanovsky, A. T. B. Gilbert, M. Wormit, J. Kussmann, A. W. Lange, A. Behn, J. Deng, X. Feng, D. Ghosh, M. Goldey, P. R. Horn, L. D. Jacobson, I. Kaliman, R. Z. Khaliullin, T. Kuś, A. Landau, J. Liu, E. I. Proynov, Y. M. Rhee, R. Richard, M. A. Rohrdanz, R. P. Steele, E. J. Sundstrom, H. L. Woodcock, P. M. Zimmerman, D. Zuev, B. Albrecht, E. Alguire, B. Austin, G. J. O. Beran, Y. A. Bernard, E. Berquist, K. Brandhorst, K. B. Bravaya, S. T. Brown, D. Casanova, C. Chang, Y. Chen, S. H. Chien, K. D. Closser, D. L. Crittenden, M. Diedenhofen, R. A. DiStasio, H. Do, A. D. Dutoi, R. G. Edgar, S. Fatehi, L. Fusti-Molnar, A. Ghysels, A. Golubeva-Zadorozhnaya, J. Gomes, M. Hanson-Heine, P. H. P. Harbach, A. W. Hauser, E. G. Hohenstein, Z. C. Holden, T. Jagau, H. Ji, B. Kaduk, K. Khistyaev, J. Kim, J. Kim, R. A. King, P. Klunzinger, D. Kosenkov, T. Kowalczyk, C. M. Krauter, K. U. Lao, A. D. Laurent, K. V. Lawler, S. V. Levchenko, C. Y. Lin, F. Liu, E. Livshits, R. C. Lochan, A. Luenser, P. Manohar, S. F. Manzer, S. Mao, N. Mardirossian, A. V. Marenich, S. A. Maurer, N. J. Mayhall, E. Neuscamman, C. M. Oana, R. Olivares-Amaya, D. P. O’Neill, J. A. Parkhill, T. M. Perrine, R. Peverati, A. Prociuk, D. R. Rehn, E. Rosta, M. J. Russ, S. M. Sharada, S. Sharma, D. W. Small, A. Sodt, T. Stein, D. Stück, Y. Su, A. J. W. Thom, T. Tsuchimochi, V. Vanovschi, L. Vogt, O. Vydrov, T. Wang, M. A. Watson, J. Wenzel, A. White, C. F. Williams, J. Yang, S. Yeganeh, S. R. Yost, Z. You, I. Y. Zhang, X. Zhang, Y. Zhao, B. R. Brooks, G. K. L. Chan, D. M. Chipman, C. J. Cramer, W. A. Goddard, M. S. Gordon, W. J. Hehre, A. Klamt, H. F. Schaefer, M. W. Schmidt, C. D. Sherrill, D. G. Truhlar, A. Warshel, X. Xu, A. Aspuru-Guzik, R. Baer, A. T. Bell, N. A. Besley, J. Chai, A. Dreuw, B. D. Dunietz, T. R. Furlani, S. R. Gwaltney, C. Hsu, Y. Jung, J. Kong, D. S. Lambrecht, W. Liang, C. Ochsenfeld, V. A. Rassolov, L. V. Slipchenko, J. E. Subotnik, T. Van Voorhis, J. M. Herbert, A. I. Krylov, P. M. W. Gill and M. Head-Gordon, Advances in molecular quantum chemistry contained in the Q-Chem 4 program package, Mol. Phys., 2015, 113, 184–215 CrossRef CAS.
- A. D. Becke, Density-functional thermochemistry. III. The role of exact exchange, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- S. Grimme, Semiempirical GGA-type density functional constructed with a long-range dispersion correction, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- T. N. Truong and E. V. A. Stefanovich, New method for incorporating solvent effect into the classical, ab initio molecular orbital and density functional theory frameworks for arbitrary shape cavity, Chem. Phys. Lett., 1995, 240, 253–260 CrossRef CAS.
- A. Bondi, van der Waals Volumes and Radii, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- D. Porezag, T. Frauenheim, T. Köhler, G. Seifert and R. Kaschner, Construction of tight-binding-like potentials on the basis of density-functional theory: Application to carbon, Phys. Rev. B, 1995, 51, 12947–12957 CrossRef CAS PubMed.
- M. Gaus, Q. Cui and M. Elstner, DFTB3: Extension of the Self-Consistent-Charge Density-Functional Tight-Binding Method (SCC-DFTB), J. Chem. Theory Comput., 2011, 7, 931–948 CrossRef CAS PubMed.
- D. E. Romonosky, L. Q. Nguyen, D. Shemesh, T. B. Nguyen, S. A. Epstein, D. B. C. Martin, C. D. Vanderwal, R. B. Gerber and S. A. Nizkorodov, Absorption spectra and aqueous photochemistry of β-hydroxyalkyl nitrates of atmospheric interest, Mol. Phys., 2015, 113, 2179–2190 CrossRef CAS.
- B. Hourahine, B. Aradi, V. Blum, F. Bonafe, A. Buccheri, C. Camacho, C. Cevallos, M. Y. Deshaye, T. Dumitrică, A. Dominguez, S. Ehlert, M. Elstner, T. van der Heide, J. Hermann, S. Irle, J. J. Kranz, C. Köhler, T. Kowalczyk, T. Kubař, I. S. Lee, V. Lutsker, R. J. Maurer, S. K. Min, I. Mitchell, C. Negre, T. A. Niehaus, A. M. N. Niklasson, J. Page, A. Pecchia, G. Penazzi, M. P. Persson, J. Řezáč, C. G. Sánchez, M. Sternberg, M. Stöhr, F. Stuckenberg, A. Tkatchenko, V. W. Yu and T. Frauenheim. DFTB+, a software package for efficient approximate density functional theory based atomistic simulations; Sponsor Org.: USDOE Office of Science (SC), Basic Energy Sciences (BES) (SC-22). Chemical Sciences, Geosciences & Biosciences Division; National Science Foundation (NSF); CAREER Award; UKRI Future Leaders Fellowship; Engineering and Physical Sciences Research Council (EPSRC). 2020.
- J. Řezáč, Empirical Self-Consistent Correction for the Description of Hydrogen Bonds in DFTB3, J. Chem. Theory Comput., 2017, 13, 4804–4817 CrossRef PubMed.
- M. Kubillus, T. Kubař, M. Gaus, J. Řezáč and M. Elstner, Parameterization of the DFTB3 Method for Br, Ca, Cl, F, I, K, and Na in Organic and Biological Systems, J. Chem. Theory Comput., 2015, 11, 332–342 CrossRef CAS PubMed.
- M. Gaus, A. Goez and M. Elstner, Parametrization and Benchmark of DFTB3 for Organic Molecules, J. Chem. Theory Comp., 2013, 9, 338–354 CrossRef CAS PubMed.
- M. Gaus, X. Lu, M. Elstner and Q. Cui, Parameterization of DFTB3/3OB for Sulfur and Phosphorus for Chemical and Biological Applications, J. Chem. Theory Comp, 2014, 10, 1518–1537 CrossRef CAS PubMed.
- J. Řezáč and P. Hobza, Advanced Corrections of Hydrogen Bonding and Dispersion for Semiempirical Quantum Mechanical Methods, J. Chem. Theory Comp., 2012, 8, 141–151 CrossRef PubMed.
- L. Martínez, R. Andrade, E. Birgin and J. M. Martínez, PACKMOL: A package for building initial configurations for molecular dynamics simulations, J. Comp. Chem., 2009, 30, 2157–2164 CrossRef PubMed.
- F. Carey, R. Giuliano, N. Allison and B. Bane, Organic Chemistry, McGraw Hill, 2020 Search PubMed.
- S. J. Gumbley and R. Stewart, Effect of substituents at the 5-position on the first and second dissociation constants of isophthalic acid, J. Chem. Soc., Perkin Trans. 2, 1984, 529–531 RSC.
- J. Towns, T. Cockerill, M. Dahan, I. Foster, K. Gaither, A. Grimshaw, V. Hazlewood, S. Lathrop, D. Lifka, G. D. Peterson, R. Roskies, J. R. Scott and N. Wilkins-Diehr., XSEDE: Accelerating Scientific Discovery, Comput. Sci. Eng., 2014, 16, 62–74 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp01520c |
| This journal is © the Owner Societies 2023 |