
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unveiling the atomistic and electronic structure of NiII–NO adduct in a MOF-based catalyst by EPR spectroscopy and quantum chemical modelling†
Kavipriya
Thangavel‡
 ab,
Paolo Cleto
Bruzzese‡§
ac,
Matthias
Mendt¶
a,
Andrea
Folli
b,
Katharina
Knippen
d,
Dirk
Volkmer
d,
Damien M.
Murphy
b and
Andreas
Pöppl
*a
ab,
Paolo Cleto
Bruzzese‡§
ac,
Matthias
Mendt¶
a,
Andrea
Folli
b,
Katharina
Knippen
d,
Dirk
Volkmer
d,
Damien M.
Murphy
b and
Andreas
Pöppl
*a
aFelix Bloch Institute for Solid State Physics, Leipzig University, Linnéstraße 5, 04103 Leipzig, Germany. E-mail: poeppl@physik.uni-leipzig.de
bSchool of Chemistry, Main building, Cardiff University, CF10 3AT, Cardiff, UK
cDepartment of Chemistry and NIS Centre of Excellence, University of Turin, via Giuria 9, 10125 Torino, Italy
dInstitute of Physics, Chair of Solid State and Materials Chemistry, University of Augsburg, Universitätstraße 1, D-86159 Augsburg, Germany
First published on 22nd May 2023
Abstract
The nature of the chemical bonding between NO and open-shell NiII ions docked in a metal–organic framework is fully characterized by EPR spectroscopy and computational methods. High-frequency EPR experiments reveal the presence of unsaturated NiII ions displaying five-fold coordination. Upon NO adsorption, in conjunction with advanced EPR methodologies and DFT/CASSCF modelling, the covalency of the metal–NO and metal–framework bonds is directly quantified. This enables unravelling the complex electronic structure of NiII–NO species and retrieving their microscopic structure.
Introduction
The interaction of nitric oxide with transition metal ions (TMIs) supported on microporous systems has been abundantly studied with a view of finding the relationship among bonding, stability, and reactivity of the metal–nitrosyl group.1–6 Moreover, NO adsorption studies may reveal valuable information about the accessibility, number, chemical reactivity and electron pair acceptor strength4,7–12 of the TMI sites as well as provide fundamental insights into the mechanism of essential processes, e.g. abatement of NOx emissions.3,4,13,14Infrared (IR) spectroscopy has been successfully employed to probe metal–NO adducts encapsulated in microporous materials.2,3,15 This technique has been proved to be exceedingly powerful for revealing NO-adducts even in operando conditions.16,17 However, IR spectroscopy cannot provide direct insight into the intimate features of metal–nitrosyl chemical bonding, which is particularly nontrivial to unravel. This ambiguity arises from the close relative energy of the NO–π* orbitals compared to the d orbitals of first-row TMIs, which makes the accurate description of oxidation and/or spin state of such species difficult.18–20
Due to the paramagnetic nature of metal–NO adducts, electron paramagnetic resonance (EPR) spectroscopy is ideally suitable for obtaining exquisite details on the cryptic bonding of NO to transition metal centers.1,10,11,14,21 The application of sophisticated pulse EPR techniques allows assessing the degree of covalency and spin delocalization between the metal–NO bond as well as the one with all the other ligands magnetically active, offering additional complementary insight into the electronic structure of the NO–metal ion bonding with respect of IR spectroscopies.22,23 The subsequent reproduction of the EPR spectroscopic findings with electronic structure methods translates the experimental findings into microscopic structures enabling structure-function correlation of metal–NO species.24–27
While EPR investigations of NO adsorption over metal oxide surfaces and zeolites are abundant,4–6,10,14,22,23,25,28 only a few magnetic resonance studies of such species have been reported for the metal–organic framework (MOF) compounds,29–32 a class of microporous materials which has attracted substantial research interest within the last decades. In these systems, the coordination of NO with coordinatively unsaturated (CUS) metal ions has been probed by EPR methodologies. On one hand, weak physisorption of nitric oxide at closed-shell AlIII sites was detected in MIL-100 by observing the interaction of the unpaired electron of NO with the nuclear spin of 27Al nucleus.30 Analysis employing density functional theory (DFT) indicated that about 95–97% of the spin density is located at the NO molecule and only 2–4% on the aluminium ion, underlying the weak interaction of the probe molecule with the framework metal ion.
On the other hand, thermally stable paramagnetic EPR active NiII–NO adducts occurred upon adsorption of NO at defective open-shell NiII paddle-wheel species in DUT-8(Ni). Based on their g-tensor, two distinct NiII–NO moieties have been identified and interpreted in terms of an axially and equatorially binding nitroxide molecule31 and comparison with previously published investigations of NiII–NO complexes formed at the surface of Ni-doped MgO powders.33,34 However, direct proof of this coordination motive and a deeper understanding of the corresponding electronic structure have not been presented yet.
In this work, a NiII-substituted variant of the rigid MFU-4l(large) framework family35 comprising Ni–NO2 coordination units36 is adopted as a model case for the formation of NiII–NO species in a metal–organic framework. Through post-synthetic metal and side ligand exchange, the NiII ions substitute the peripheral ZnII sites in the pentanuclear “Kuratowski-type” SBU, displaying five-fold coordination with three nitrogen atoms (Nf) from the SBU and two oxygen atoms from coordinating nitrite ion (see Fig. 1).37 This leaves a potential sixth CUS site for the binding of an adsorbed nitric oxide molecule to form a stable six-fold octahedral-type coordination of the nickel ion.
 | ||
| Fig. 1 View along the (−1 −1 −1) face of Ni-MFU-4l-NO2 space-filling periodic model. An inset of the main subunit of the material is shown on the right. C, N, O, Ni, Zn and H are green, blue, red, yellow, violet, and white colour, respectively. | ||
First, high-field W-band continuous wave (CW) EPR spectroscopy is employed to verify the S = 1 electron spin state of the NiII ions in MFU-4l-NO2 prior to NO adsorption. Subsequently, conventional X-band CW-EPR experiments are employed to reveal the formation of NiII–NO complexes upon the exposure of NiII-containing MFU-4l-NO2 to gaseous nitric oxide. Pulse EPR experiments reveal the 14N hyperfine (hf) interactions with the nitrogen nuclei Nf belonging to the first and second coordination sphere of nickel ion and with the NO allowing to assess the nature of the chemical bonding between the nickel and the different nitrogen ligands. Cutting-edge quantum chemical computations of the magnetic parameters of the five-coordinated NiII ion in the parent Ni-MFU-4l-NO2 compound and of the NiII–NO species formed after NO adsorption translate the spectroscopic findings into atomistic structure unravelling the unique electronic structure of nickel–nitrosyl moieties supported on a MOF platform.
Material and methods
Ni-MFU-4l-NO2 sample preparation and structural characterization
The parent Ni-MFU-4l preparation and the side-ligand post-synthetic exchange modification were done similarly to a previously published procedure:37 First, Ni-MFU-4l was synthesized by a post-synthetic exchange of 150 mg MFU-4l with a solution of 12 mmol NiCl2.6H2O in 30 mL DMF at 60 °C for 20 h. The light greenish MOF was filtrated and washed with 2.5 mL DMF and MeOH. The success of the nickel exchange was proved by energy dispersive X-ray analysis (EDAX) measurement (chemical formula: [Zn4NiCl4(BTDD)3], where H2-BTDD is bis(1H-1,2,3-triazolo[4,5-b],[4′,5′-i])dibenzo[1,4]dioxin38). Then, a 1 M solution of LiNO2 in methanol (0.4 mL, 0.4 mmol) was added to the Ni-MFU-4l suspension (150 mg, approx. 0.12 mmol) in acetonitrile (30 mL). The mixture was stirred for 30 min at room temperature (RT), and the precipitate was filtered off and washed with methanol and CH2Cl2. Finally, the washed sample dried at 80 °C under vacuum, yielding 140 mg of Ni-MFU-4l-NO2 as a greenish-yellow product with an analytically determined chemical composition [Zn4Ni(NO2)3Cl1(BTDD)3].Powder X-ray diffraction (PXRD) pattern was recorded using Seifert XRD 3003 TT diffractometer equipped with a Meteor 1D detector at room temperature. The microstructure and stoichiometry were analysed using the scanning electron microscope (SEM – model Philips XL 30 FEG) and EDAX – model EDAX SiLi detector fitted with SEM), respectively. Fourier transform Infrared (FTIR) spectroscopy has been performed in the range 1600–400 cm−1 on a Bruker Equinox 55 FT-IR spectrometer.
EPR sample preparation
The CW Q- and W- band experiments on Ni-MFU-4l-NO2 were acquired in the hydrated state. Further, The CW X-band and pulse experiments were performed on the NO-adsorbed sample in the below-mentioned condition. 4.7 mg of parent MOF was transferred into a conventional quartz glass EPR tube, and the sample was activated at 120 °C for overnight to remove the extra framework solvent/water molecules before the NO gas adsorption. After the thermal activation, the colour of the sample changed from pale green to dark yellowish green colour. Then the sample was loaded with nitric oxide (0.2 mbar) using a vacuum line at 294 K, and the NO gas was condensed into the EPR tubes by applying a liquid nitrogen cold trap to ensure that the entire amount of loaded NO was trapped within the EPR tube. After NO gas loading, the sample was immediately sealed, keeping the NO adsorbed at the parent Ni-MFU-4l-NO2 sample in the EPR tube. Ultimately, the NO adsorbed sample was in a lite whitish-green colour.EPR spectroscopy
CW X-band (∼9.5 GHz) EPR spectra were measured at a temperature ranging from 10 K to 288 K employing a Bruker EMXmicro spectrometer fitted with a Bruker ER4119HS cylindrical cavity using a He cryostat ESR900, Oxford instruments. The CW Q-band (∼34 GHz) EPR spectrum was recorded using Bruker EMX 10–40 spectrometer fitted with a cylindrical cavity and an Oxford Instruments CF935 cryostat at T = 300 K. The high magnetic field of W-band (∼95 GHz) EPR requires a superconducting magnet, Bruker 6T SC and the W-band spectra were measured at T = 20 K using an Elexsys E600 spectrometer equipped with a Bruker E600-1021H TeraFlex resonator. The EPR intensities of the X-band signals ranging from T = 10 K to T = 288 K (Fig. S5b, ESI†) were extracted by taking double integration of the full-range EPR spectrum.The following spin Hamiltonian was used for the NiII species with spin S = 1 to interpret the Q- and W-band EPR data
 | (1) |
 , the electron g-tensor, and the applied external magnetic field
, the electron g-tensor, and the applied external magnetic field ![[B with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0042_20d1.gif) . The second term indicates the zero-field splitting (ZFS), and D and E are the axial and rhombic ZFS parameters, respectively.
. The second term indicates the zero-field splitting (ZFS), and D and E are the axial and rhombic ZFS parameters, respectively.
For the NO adsorption on the parent MOF, the spin Hamiltonian for the resulting NiII–NO species with spin S = 1/2 can be written as
 | (2) |
 the 14N nuclear spin operator, and ANi and QNi are the 14N hf and nuclear quadrupole (nq) interactions tensors of the nitrogen of the adsorbed NO molecule and of the nitrogen atoms in the first (Nf1-f3) and second (NS) coordination spheres of the triazole linkers coordinating to the NiII ion in the Kuratowski-type SBU (Fig. 7).
the 14N nuclear spin operator, and ANi and QNi are the 14N hf and nuclear quadrupole (nq) interactions tensors of the nitrogen of the adsorbed NO molecule and of the nitrogen atoms in the first (Nf1-f3) and second (NS) coordination spheres of the triazole linkers coordinating to the NiII ion in the Kuratowski-type SBU (Fig. 7).
The EPR data were simulated by MATLAB R2019b using the EasySpin toolbox (version 6.0.0-dev36), which is based on numerical diagonalization of the spin Hamiltonian.39 In the simulations of the CW-EPR spectra, the 14N hf and nq coupling has been neglected as no nitrogen hf spitting was resolved here.
The X-band electron-spin-echo (ESE) detected EPR spectra were recorded with the pulse sequence  . The lengths of microwave (mw) pulsed tπ/2 = 16 ns and tπ = 32 ns, a τ value of 120 ns and a shot repetition rate of 3.55 kHz were adopted.
. The lengths of microwave (mw) pulsed tπ/2 = 16 ns and tπ = 32 ns, a τ value of 120 ns and a shot repetition rate of 3.55 kHz were adopted.
X-band hyperfine sublevel correlation (HYSCORE)40 experiments were performed with the pulse sequence  , applying an eight-step phase cycle for deleting unwanted echoes. Pulse lengths of tπ/2 = 16 ns and tπ = 32 ns and a shot repetition rate of 1.77 kHz were used. The increment of the time intervals t1 and t2 was 16 ns giving a data matrix of 200 × 200 points; the pulse delay τ value was set to 146 ns. The time traces of HYSCORE spectra were baseline corrected with a third-order polynomial, apodized with a hamming window and zero-filled to 2048 points. After the 2D Fourier transformation, the absolute-value frequency spectra were obtained.
, applying an eight-step phase cycle for deleting unwanted echoes. Pulse lengths of tπ/2 = 16 ns and tπ = 32 ns and a shot repetition rate of 1.77 kHz were used. The increment of the time intervals t1 and t2 was 16 ns giving a data matrix of 200 × 200 points; the pulse delay τ value was set to 146 ns. The time traces of HYSCORE spectra were baseline corrected with a third-order polynomial, apodized with a hamming window and zero-filled to 2048 points. After the 2D Fourier transformation, the absolute-value frequency spectra were obtained.
X-band electron nuclear double resonance (ENDOR) spectra were recorded using the Davies ENDOR pulse sequence  .41 Mw pulse lengths tπ/2 = 100 ns and tπ = 200 ns, and a radiofrequency pulse length tπRF = 10 μs, together with the mw pulse delay τ = 820 ns were employed.
.41 Mw pulse lengths tπ/2 = 100 ns and tπ = 200 ns, and a radiofrequency pulse length tπRF = 10 μs, together with the mw pulse delay τ = 820 ns were employed.
Models and computational details
![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) invented by Volkmer et al.,42 one ZnII–Cl coordination unit among the four peripheral coordination sites of each Kuratwski-type SBU was substituted by one NiII–Cl coordination unit. In this way, one NiII site was introduced per SBU, removing the cubic space group symmetry in the model (space group P1). Subsequently, the Cl− anions were replaced by NO2− ligands in order to reproduce the experimental composition of the material. Adsorption of the nitric oxide molecule was simulated by positioning a NO molecule close to the peripheral NiII or ZnII sites of the previously optimized structures and reoptimizing the whole adduct.
m) invented by Volkmer et al.,42 one ZnII–Cl coordination unit among the four peripheral coordination sites of each Kuratwski-type SBU was substituted by one NiII–Cl coordination unit. In this way, one NiII site was introduced per SBU, removing the cubic space group symmetry in the model (space group P1). Subsequently, the Cl− anions were replaced by NO2− ligands in order to reproduce the experimental composition of the material. Adsorption of the nitric oxide molecule was simulated by positioning a NO molecule close to the peripheral NiII or ZnII sites of the previously optimized structures and reoptimizing the whole adduct.
Periodic calculations have been complemented with molecular cluster calculations to compute the g-tensor, the ZFS parameters D and E, 14N hf and nq coupling tensors ANi, QNi including the orientation of their principal axes frame with respect to the g-tensor principal axes frame. Cluster models were cut out from the corresponding optimized periodic structures. The dangling bonds were saturated with hydrogen atoms oriented along the broken bonds to keep the local environment as in the optimized periodic models. Thus, no further geometry optimization of the cluster models was performed: the EPR parameters were computed, maintaining the same atomic coordinates as the ones in the relaxed periodic structures. The resulting net charge on the cluster models was always set to 0.
A pruned grid consisting of 75 radial points and a maximum number of 974 angular points in regions relevant to chemical bonding has been adopted. The accuracy of the calculation of the two-electron integrals in the Coulomb and exchange series was controlled by setting truncation criteria at the values of 10−7 except for the pseudo-overlap of the Hartree–Fock (HF) exchange series, which was fixed to 10−25. Due to the large unit cell in the direct space, a shrink factor equal to 1 was used to diagonalize the Hamiltonian matrix in 1 k-point of the first Brillouin zone. The default value of mixing (30%) of the Kohn–Sham (KS) matrix at a cycle with the previous one was adopted. The threshold in energy variation of SCF cycles was set equal to 10−7 Hartree for geometry optimization and equal to 10−10 Hartree for frequency calculations. The number of unpaired electrons in the unit cell was locked to two for the case of NiII and to one for NiII–NO in order to guide the SCF procedure to converge to a triplet and doublet spin state of the system wavefunction, respectively.
Harmonic vibrational frequencies were computed at the center of the first Brillouin zone in the reciprocal space (Γ point) from the diagonalization of the mass-weighted Hessian matrix of the second energy derivatives with respect to atomic displacement.51–53 One displacement for each atom along each Cartesian direction was considered to numerically compute the second energy derivatives.
Molecular cluster calculations were carried out with ORCA (v5.0.3) code.54 The spin–orbit coupling (SOC) contribution (not negligible for Ni species)55 was explicitly treated by using a complete mean-field spin–orbit operator (SOMF).56 The potential was constructed to include one-electron terms, compute the Coulomb term in a semi-numeric way, incorporate exchange via one-centre exact integrals, including the spin-other orbit interaction and include local DFT correlation (SOCFlags 1,2,3,1 in ORCA). Concerning the Ni-MFU-4l-NO2 cluster model with NiII, ZFS and g-tensor were computed at the double-hybrid DFT level of theory by employing the B2PLYP functional.57 The def2-QZVP basis set was employed for the Ni nucleus, while the def2-TZVPP basis sets were employed for all the other atoms.58 Increased integration grids were employed (DefGrid3 keyword), and tight energy convergence settings were applied throughout (TightSCF keyword). The resolution of identity (RI)59 (in conjunction with the corresponding auxiliary basis sets was adopted.60 In case no auxiliary basis set was available, the AutoAux keyword was employed to automatically build the auxiliary basis set.61 The “relaxed” Møller-Plesset (MP2) density was used to compute the EPR parameters, and all the electrons were kept active (NoFrozenCore keyword). Both the spin–orbit and spin–spin contributions were taken into account for the computation of the ZFS interaction.
The ab initio prediction of the electronic structure for the NiII–NO adduct were based on single-point complete active space self-consistent field (CASSCF) calculations on the cluster model extracted from the optimized periodic structure. The def2-QZVP basis set was employed for Ni, EPR-III62 for the coordinating N nuclei, def2-TZVP58 for the coordinating O nuclei and def2-SVP58 for all the other atoms. The adopted active space (CAS) contains 11 electrons and 11 orbitals composed of five Ni 3d orbitals, two orbitals with predominant NO π* character (namely πz* and πy*, where the z-axis coincides with the Ni–NO bond), one σ-type orbital that describes covalent bonding between Ni and the nitrogen atoms from the linkers and three double-shell orbitals of Ni (to describe radial correlation effects). State-averaged (SA) CASSCF calculations, including 15 doublet and 10 quartet states were adopted to optimize the active orbitals and compute the g-tensor.
14N hyperfine and quadrupole couplings from the coordinating nitrogen nuclei of the first and second coordination spheres were obtained by performing a ground-state complete active space configuration interaction (CASCI) calculation of the previously optimized SA-CASSCF wavefunction. The Euler angles relative to the orientations of the 14N hyperfine and quadrupole tensors were instead obtain at PBE058/EPR-III level of theory.
Results and discussion
Structural analysis
PXRD obtained at 300 K (Fig. S1a, ESI†) confirms that post-synthetic ion exchanged Ni-MFU-4l-NO2 is in a single phase and crystalizes within space group Fmm (no. 225, cubic crystal system). SEM result ascertains the agglomerated particles ranging from 1 to 3 μm range (inset in Fig. S1a, ESI†). The EDAX result reveals the fraction of the post-synthetically incorporated NiII, and the ratio of Ni (20%) and Zn (80%) is found to be 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4. The IR spectra result recorded between 1600–400 cm−1 confirms the successful formation of Ni-MFU-4l-NO2 (Fig. S1b, ESI†).
:4. The IR spectra result recorded between 1600–400 cm−1 confirms the successful formation of Ni-MFU-4l-NO2 (Fig. S1b, ESI†).
CW-EPR spectroscopy and coordination geometry of NiII in Ni-MFU-4l-NO2
In general, non-Kramer (integer spin) systems like the NiII ion having a d8 electronic configuration with spin S = 1 are challenging to detect in EPR experiments at conventional X- and Q-band mw frequencies because of their large ZFS.63–65 As a consequence, the allowed EPR transitions (ΔMs = ±1) cannot be excited by mw quanta being too small. Only a very few NiII-containing materials were characterized employing X- and Q-band EPR spectroscopy for NiII species having smaller or comparable ZFS to the MW frequency.64–66 In order to overcome these complications, CW high-frequency EPR (HFEPR) spectroscopy techniques,67–69 (∼90 GHz to ∼611 GHz and magnetic fields up to ∼22 T) and even time-domain terahertz EPR measurements70 were utilized to acquire the complete triplet spectrum of the S = 1 NiII species. Furthermore, temperature- and field-dependent magnetic susceptibility measurements also provided spin Hamiltonian parameters for such high-spin NiII systems.63,67In our case, Ni-MFU-4l-NO2 was first measured at Q-band frequency (see Fig. S2b, ESI†), and a part of the triplet spectrum was observed at ∼300 mT. The indication of ZFS is ambiguous to conclude the value of the ZFS of NiII species as the energy of ZFS is expected to be larger than the MW quanta energy at Q-band. To obtain the ZFS along with other spin Hamiltonian parameters, a W-band CW-EPR spectrum was recorded at 20 K (see Fig. 2). Interestingly, ZFS energy of NiII ion is not so large, and an intense forbidden transition (ΔMs = ±2) arose at ∼1450 mT in the W-band spectrum, whereas some poorly resolved allowed transitions (ΔMs = ±1) were observed at high fields 2000–4000 mT. The spin Hamiltonian parameters of the NiII species gxx = 2.000, gyy = 2.025, gzz = 2.060, D = 35.5 GHz (1.18 cm−1), and E = 0.5 GHz (0.17 cm−1) are obtained by spectral simulation and suggests that the symmetry around NiII ion is slightly rhombic. Fig. S3 (ESI†) shows the angular dependences of the NiII EPR signals computed with the derived spin Hamiltonian parameters at W- and Q-band frequencies, confirming the assignment of the signals observed at about 1450 mT at W-band (Fig. S3a, ESI†) and 300 mT at Q-band (Fig. S3b, ESI†) to the ΔMs = ±2 transition and the consistency of both experiments.
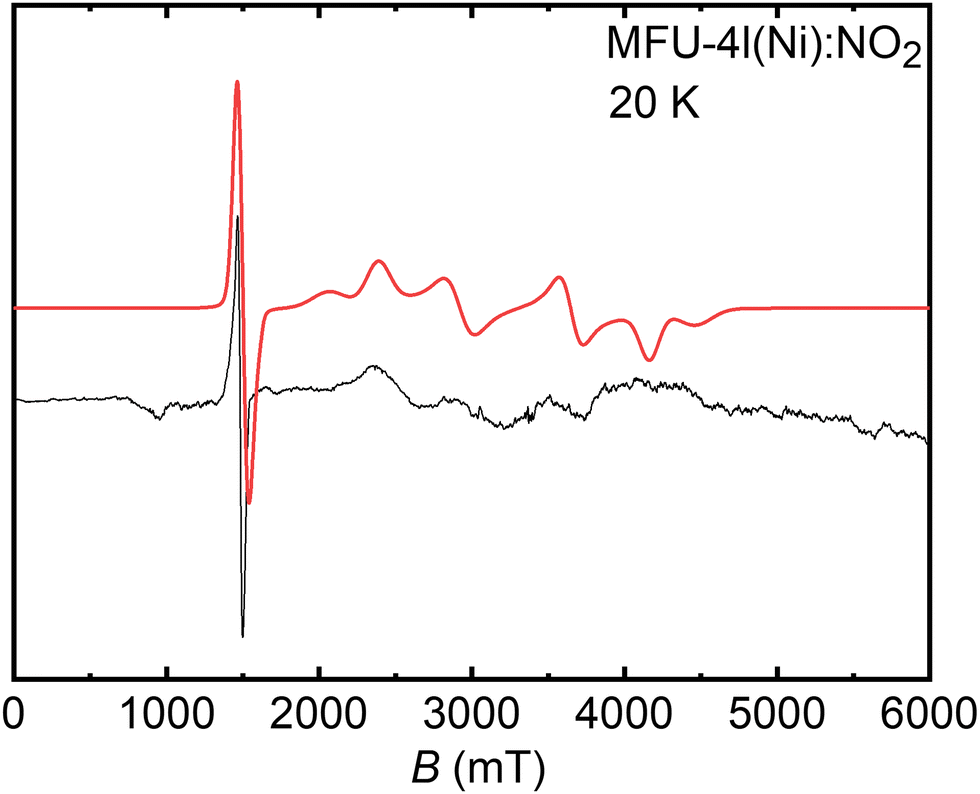 | ||
| Fig. 2 Experimental (black line) and simulated (red line) CW-EPR W-band spectrum of NiII ions having S = 1 in Ni-MFU-4l-NO2 obtained at 20 K. | ||
It is well-known that zero-field splitting is intrinsically connected with the geometric structure of NiII complexes and originated from the spin–spin interactions mediated by the ligand field and from the spin–orbit coupling.68,70 While relatively small ZFS values were reported for octahedral NiII complexes,71–74 larger ZFS parameters occur for tetrahedral coordination.69,75 Hence, the estimated values of D and E for NiII ions in Ni-MFU-4l-NO2 may be used to retrieve peculiar details on the five coordination-based atomistic structures of the NiII paramagnetic center, as discussed below.
To transpose the spectroscopic results extracted from the analysis of W-band experiment into a microscopic structure, ab initio calculations of the g-tensor and ZFS were performed on the optimized structure of NiII-MFU-4l reported in Fig. 3. The NiII ion occupies a single peripheral site of the Kuratowski-type SBU displaying a five-fold coordination with three lattice nitrogen atoms and two oxygen atoms from the NO2− ligand. The Ni–O bond lengths are slightly longer (≈0.27 nm) as compared to the Ni–N bond lengths (≈0.20 nm). A quantitative analysis from EDAX results indicates that the amount of Ni in the material is 22.5% in atomic weight (and 21% in molar weight) in comparison with ZnII centers, justifying the assumption of considering only one NiII site per one SBU in the model.
 | ||
| Fig. 3 Geometry optimized periodic structures at B3LYP-D3/pob-TZVP-rev2 of Ni-MFU-4l-NO2. The computed g- and D-tensor frames are also reported. | ||
Although DFT calculations of the ZFS often fail to arrive at the correct sign and magnitude of D and/or E parameters,76,77 the computed D and E parameters obtained at B2PLYP/def2-QZVP level of theory are in good agreement with the experimental ones (see Table 1). The superiority of double-hybrid functionals with respect to more common hybrid functionals lies in a better description of the excited states of different multiplicities, which contribute significantly to the ZFS parameters.78 The prevalent source of computed ZFS arises from the spin–orbit coupling effect, in agreement with other open-shell transition metal ions.79 The calculated spin–spin contribution accounts only for 0.3% for D and 9% for E parameters. For comparison, a tetrahedral NiII ion in the Ni-MFU-4l model with a Cl− ligand (see Fig. S4b, ESI† and Table 1) instead of NO2− provides an axially symmetric g- and ZFS tensor with D parameter that is further overestimated with respect to the experimental value, validating the five-coordinated structure presented in Fig. 3. Otherwise, a slightly rhombic g-tensor is predicted from the calculations for Ni-MFU-4l-NO2 model (Table 1) consistent with the experimental values.
| Geometry (ligand X) | g xx | g yy | g zz | D (cm−1) | E/D | |
|---|---|---|---|---|---|---|
| Computed | Four-coordinated (Cl) | 2.186 | 2.186 | 2.194 | 1.62 | 0.01 |
| Five-coordinated (NO2) | 2.133 | 2.175 | 2.191 | 1.53 | 0.09 | |
| Experimental | 2.000 | 2.025 | 2.060 | 1.18 | 0.14 |
To summarize, the analysis of the W-band spectrum evidences the presence of NiII species incorporated within the Ni-MFU-4l-NO2 framework via post-synthetic ion exchange modification. The microscopic structure of such NiII centers is retrieved by comparing the experimental spin Hamiltonian parameters, in particular the ZFS, with the computed ones, and it may be ascribed as five-coordinated NiII ion located on one of the peripheral sites of the SBUs of the Ni-MFU-4l-NO2. Additionally, in complement with the EPR analyses, IR spectra for the Ni-MFU-4l-NO2 and Ni-MFU-4l-Cl complexes are consistent with the spectra extracted from the DFT calculations (Fig. S10, ESI†).
CW and pulse EPR investigations of the NiII–NO adduct in Ni-MFU-4l-NO2
Interaction of the thermally activated Ni-MFU-4l-NO2 with adsorbed NO was initially monitored by CW-EPR. The recorded X-band EPR spectra at 10 K and 288 K are shown in Fig. 4 and display the appearance of an intense EPR signal upon adsorption of nitric oxide over Ni-MFU-4l-NO2. The complete set of temperature-dependent (ΔT = ∼25 K) EPR data ranging from 10 K up to 288 K is given in Fig. S5a and Table S1 (ESI†). The signal intensity and linewidth increase with higher NO loading (Fig. S12, ESI†). Spectral simulations reveal that the spectra in Fig. 4 are composed of a superposition of a major species A (93% signal contribution) with principal values of its g-tensor given in Table 2 and a minor species B (7% signal contributions) with principal values gxx,yy = 2.296, gzz < 2.224. The EPR signal intensity follows the expected 1/T behavior of a paramagnetic system according to Curie's law (Fig. S5b, ESI†). Both, gii-values and linewidths, exhibit a weak temperature dependence, which is presented and discussed in Fig. S6 and S7 (ESI†). The obtained g-values for species A and B, gii > ge, where ge = 2.0023 is the g-value of the free electron, indicate that the unpaired electron resides in a 3d9 orbital of the NiII ion33,34 of the Kuratowski-type SBU and is not localized in the antibonding πz* orbital of the adsorbed NO molecule. The latter case has been typically observed for nitric oxide physisorbed on metal oxide surfaces, in zeolites, and MOFs with CUS sites at closed-shell metal ions, where ge ≥ gii holds.1 Moreover, the EPR spectra of these NO adsorption complexes are usually not detectable at room temperature because of the small adsorption energies of the nitric oxide molecules.8,9,11–14,19–21 | ||
| Fig. 4 Experimental (black lines) and simulated (red lines) X-band CW-EPR spectra of the NiII–NO adduct in Ni-MFU-4l-NO2 formed upon NO adsorption and recorded at (a) 10 K and (b) 288 K. The simulation is composed of the sum of two different species A (dotted green line) and B (dotted blue line). The small signal around ∼340 mT corresponds to the radical. | ||
| g -Tensor | 14N A-tensor | 14N Q-tensor | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Experimental | g xx | g yy | g zz | a iso | |T| | |e2qQ/h| | η | ||
| 2.136 | 2.167 | 2.270 | Strongly coupled | 14N(1) | −3.0 | 13.0 | 2.7 | 0.8 | |
| 14N(2) | 11.0 | 1.5 | 3.2 | 0.6 | |||||
| Weakly coupled | Ns | 1.0 | 0.4 | 3.2 | 0.8 | ||||
| Computed | 2.226 | 2.255 | 2.276 | NNO | −3.9 | 11.0 | 5.0 | 0.3 | |
| Nf1 | 9.9 | 1.3 | 4.7 | 0.5 | |||||
| Nf2 | 9.6 | 1.3 | 4.6 | 0.4 | |||||
| Nf3 | 8.0 | 1.1 | 4.3 | 0.4 | |||||
| Ns | 0.6 | 0.3 | 5.8 | 0.5 | |||||
The observed paramagnetic 3d9 ground state of the NiII–NO adsorption complex has been interpreted in terms of a NiI–NO+ species33,34 or by an AFM coupled NiII–NO adduct,33,34 where the later assignment has been strongly supported by recent quantum chemical computations.33,34 In the following, we will restrict our discussion to the major species A and assign the minor species B to a NiII–NO adsorption complex, which is formed at nickel ion associated with a structural defect of the MOF framework or at a residual four-coordinated tetrahedral NiII ion with a Cl− ligand being left from the initially synthesized Ni-MFU-4l material. However, as this minor species B accounts for only 7% of the total NiII it cannot be identified in the W band spectra of Ni-MFU-4l-NO2 (Fig. 2), and unambiguous assignment is not possible.
According to second-order perturbation theory, a (dx2−y2) ground state leads to principal values of the g-tensor80–82
 | (3) |
The lack of 14N hyperfine structure in the CW-EPR spectra indicates that the spin density is predominantly based on the nickel ion. As a result of this, the hyperfine interactions from the N nuclei of the organic linker and NO are small, and the information is hidden in the inhomogeneously broadened line of the CW-EPR spectrum. To recover the missing couplings arising from 14N (I = 1), and obtain details on the local coordination environment of the NiII–NO species, pulse EPR measurements (HYSCORE and ENDOR) were carried out at X-band.
Orientation-selective 14N Davies ENDOR spectra of NiII–NO in MFU-4l-NO2 are reported in Fig. 5. An ENDOR signal represents an NMR absorption which is observed as a change in the echo signal intensity at a fixed resonant magnetic field, B0. The ENDOR pattern for the ΔmI = ±1 transitions for 14N (I =1), are expected to obey the following equation for the nuclear transition frequencies of the electron spin manifolds with α and β corresponding to Ms = ±1/2
| να,β(mI ↔ mI + 1) = |A/2 ± νI + 3Q(mI − 1/2)| | (4) |
 | ||
| Fig. 5 Experimental (black) and simulated (red) X-band 14N ENDOR spectra of NiII–NO adduct in Ni-MFU-4l-NO2 recorded at different magnetic field settings. The simulation of the 14N(2) signal was obtained by using one of the DFT-computed set of Euler angles for the triazole 14N. The ESE spectrum with the corresponding field positions at which the ENDOR spectra were taken is plotted on the left-hand side. All spectra were recorded at 10 K. | ||
The low field 14N ENDOR spectrum of NiII–NO in MFU-4l-NO2 (Fig. 5) corresponds to a single crystal-like orientation and is characterized by an unresolved set of 2I = 2 quadrupole lines separated by 2νI and centered at a frequency corresponding to A/2. The quadrupole splitting is partially resolved at higher fields generating complex ENDOR spectra. Spectral features at 320 mT and 325 mT suggest the existence of two sets of 14N nuclei, one with a larger hyperfine coupling (contributing especially to the high-frequency part of the spectra, hereafter named 14N(1)), the other with a smaller coupling responsible for the splitting structure in the low-frequency region (hereafter referred to as 14N(2)). This assignment was confirmed by a simulation analysis, which proved impossible to convincingly fit simultaneously the spectra recorded at three field positions with a single nitrogen species. The involvement of two interacting 14N species dramatically complicates the simulation procedure by increasing the number of unknown parameters. For this reason, the relative orientations of the quadrupole coupling and hyperfine coupling tensors with respect to the g-tensor principal axes frame were fixed from DFT calculations (vide infra). Careful scrutiny of the ENDOR spectra evinced that the 14N(2) signal is given by multiple nitrogen nuclei possessing comparable magnitude of hyperfine and quadrupole couplings but slightly different orientations of the corresponding AN1,2 and QN1,2 tensors with respect to the g-tensor. Nevertheless, the spectral resolution does not allow to completely disentangle nitrogen nuclei magnetically equivalent but with different orientations of the hyperfine and quadrupole tensors. Simulations of the field-dependent ENDOR spectra allowed to extract of the principal values of the 14N tensors AN1,2 and QN1,2. The 14N hf interaction tensors are found to be axially symmetric within the accuracy of the simulation procedure, and the corresponding isotropic Fermi contact (aiso) and dipolar (T) hf coupling parameters83 are given in Table 2 (for further details, see Table S2, ESI†). The sign of the principal values of tensors AN1,2 was assigned according to the ab initio calculations. The estimated nq interaction tensors are rhombic and presented in terms of the nq coupling parameter e2qQ/h and the rhombic distortion parameter η.83 The contribution from the different species was properly weighted in the simulation in order to fit better the experimental plot (14N(1) and 14N(2) species were considered in 1:1 ratio).
The decomposition of the 14N hfi tensors aiso and T components allows to the extraction of exquisite information on the nature of Ni–N chemical bonding. The dominant aiso contribution in the 14N(2) hf coupling tensor implies a large s-character of the Ni–14N(2) bonds diagnostic for a prevalent σ-type bonding. On the other hand, the 14N(1) hf interaction is dominated by the dipolar T contribution pinpointing to a main p-character of the Ni–14N(1) bond. The degree of spin delocalization in the 2s (ρs) and 2p (ρp) orbitals of the two different nitrogen species may be derived from the extracted hf couplings. By using the atomic parameters for nitrogen (a0 = 1540.33 MHz and b0 = 127.22 MHz)84 and considering a unitary spin density in the 2s and 2p orbitals, ρs = 0.003 and ρp = 0.10 for N(1) while ρs = 0.007 and ρp = 0.012 for N(2). These values clearly reflect the substantial p-character of the Ni–N(1) bond with respect to the Ni–N(2) bond.
The X-band HYSCORE spectra of NO adsorbed NiII-MFU-4l-NO2 recorded at three field positions are reported in Fig. 6. In 14N HYSCORE spectra, the correlation peaks (να, νβ) and (νβ, να) are further split into multiplets due to the nq interaction. In this case, the 14N hyperfine interaction detected by HYSCORE experiments is approximately twice the nitrogen Larmor frequency at X-band frequency, leading to the so-called cancellation regime.85 Therefore, the transitions detected are assigned to 14N nuclei weakly coupled to the NiII–NO adduct, likely located on the second coordination sphere. Cross peaks at (±1.6, +4.2) and (±4.2, +1.6) MHz are assigned to (ν−, νDQ) frequencies, the signals at (+3.2, +4.2) and (+4.2, +3.2) MHz correspond to (ν+, νDQ) and (νDQ, ν+) frequencies while the low-frequency ridges at (±0.6, +1.6) and (±1.6, +0.6) may be assigned to (ν0, ν−) frequencies. An additional feature appearing at about 4 MHz in the spectra is due to the nuclear double-quantum transition frequency (νDQ) of the other electron spin manifold. The full set of spin Hamiltonian parameters for such weakly coupled nitrogen nuclei (Ns) were recovered by fitting the HYSCORE spectra simultaneously at three magnetic fields and are likewise summarized in Table 2.
 | ||
| Fig. 6 Simulation (in red) of the X-band 14N HYSCORE spectrum (in black) of NiII–NO adduct in Ni-MFU-4l-NO2 recorded at (a) 325.0 mT, (b) 320.8 mT, and (c) 309.0 mT. The ESE detected EPR signal of NiII–NO is reported on the left side. Spectra were recorded at 10 K. | ||
Summarizing, the combination of CW-EPR and hyperfine techniques provide evidence that, upon NO adsorption on NiII-MFU-4l-NO2 material, a NiII–NO adduct is formed in which the spin density is prevalently located at the nickel center. The absence of resolved 14N hyperfine splitting in the CW-EPR spectra points out that only minute spin density is retained on the NO moiety and N ligands from the Ni-MFU-4l-NO2 framework. Therefore the NO binding mode to the NiII ion occurs via the following spin pairing mechanism NO↑ + NiII↑↑ → [↑NiII(↑ ↓)NO], as it was previously proposed on other systems containing metal–nitrosyl bonding.6,14,86–88 Most importantly, hyperfine techniques allowed us to detect the hidden 14N hf interaction from coordinating nitrogen ligands. In a complementary fashion, HYSCORE experiments indicate the presence of remote nitrogen atoms belonging to the second coordination sphere of the NiII–NO species. In contrast, ENDOR measurements indicate the presence of two magnetically inequivalent nitrogen species directly linked to the Ni ion, each of them displaying a different degree of covalency of the Ni–N chemical bond.
Geometric and electronic structure of NiII–NO in MFU-4l-NO2
It is widely established that NO binds transition metal centers through the nitrogen atom.18 In metal–nitrosyl complexes, the NO character may range from that of a nitrosyl cation (NO+), which binds to the metal with a metal–NO angle of about 180°, to that of a nitrosyl anion (NO−), for which a bond angle of about 120° might be predicted. The occurrence of the former case instead of the latter depends on the amount of electron density donated from the antibonding orbital of NO to the metal 3d orbital and vice versa (σ-donation/π-back donation). A generalized description of the metal–NO bonding mechanism is provided by the {MNO}n formulation proposed by Feltham and Enemark,89,90 where M is the metal center and n is the sum of the metal d-electrons and the nitrosyl π* electrons. For instance, for a six-coordinated complex with n = 9, like our case, the metal–N–O angle is predicted to be bent.89–91The adsorption of NO on the peripheral NiII site of Ni-MFU-4l-NO2 was modelled by exploiting periodic boundary conditions, and the optimized geometry is shown in Fig. 7. The computed absolute adsorption energy of NO to NiII site (ΔEads = 31.0 kJ mol−1) is higher in absolute value than that of NO on the peripheral ZnII ions (ΔEads = 12.0 kJ mol−1, see also Fig. S8, ESI†), validating the appearance of nickel species in the X-band EPR spectra upon NO adsorption. The computed adsorption energy ΔEads = 31.0 kJ mol−1 is in the range of the activation energy EA2 = 23(1) kJ mol−1 determined from the temperature dependence of the homogeneous line width of the CW-EPR signal of the NiII–NO adduct at higher temperatures (Fig. S7, ESI†). Thus, we may speculatively relate the homogeneous line broadening of the EPR signal at elevated temperatures to the onset of the desorption progress of the nitric oxide molecules from the NiII ions, as already observed for other NO adsorption complexes.33,34 The formation of NiII–NO adduct leads to a pseudo-octahedral geometry in which the Ni–N–O bond angle is slightly bent (122.5°), as predicted by Walsh-type diagrams.89,90 The parent Ni–Nf and Ni–O bonds are utterly preserved, and their length underwent a small increase with respect to the values for the five-coordinated nickel ion, especially the Ni–Nf3 distance (Fig. 7). The major elongation of the Ni–Nf3 bond is consistent with the weakening of the metal–ligand bond trans to the nitrosyl predicted by the {MNO}n model for a six-coordinated complex.92 Similar structural changes were reported for porphyrin systems.93,94 The N–O bond length (0.11 nm) of the nitric oxide ligand is slightly shorter than the one relative to the gas-phase value. The reduction of the N–O bond length is a clear reflection of the depopulation of the antibonding π* orbital, which contains the unpaired electron in the NO molecule.
 | ||
| Fig. 7 Atomistic structure of NiII–NO species in Ni-MFU-4l-NO2 as obtained after the geometry optimization of the periodic model. The labels of the significant nuclei are reported. The relevant bond lengths are given in nm. The computed g-tensor orientations are shown in red. | ||
Nevertheless, a detailed depiction of the electronic structure of nickel–nitrosyl complexes may not be accurately described by means of widely used approximate DFT methods. Indeed, the open-shell 3d8 configuration of NiII, along with the “non-innocent” NO ligand, generates a multiconfigurational character in the NiII–NO electronic structure, extensively observed in the case of the nitrosyl ligands.14,88,95–99 Therefore, CASSCF calculations have been employed adopting an active space composed of 11 electrons and 11 orbitals (11e,11o), which involves all the 3d Ni orbitals, the NO π* orbitals, the σ-bonding orbitals describing the covalent bonding with the framework of the Ni-MFU-4l-NO2 and the nitrite ligand and three 4d Ni orbitals.
A graphical representation of some of the CAS-optimized natural orbitals is given in Fig. 8 (see Fig. S11, ESI† for the visualization of the complete set of orbitals). The bonding between Ni and NO is based on a σ-type bond composed of the bonding (dz2 + πz*) and the antibonding (dz2 − πz*) molecular orbitals (the cluster model was oriented in order to have the z-axis passing through the NiII–NO bond). The σ-bonding orbital is mainly represented by the Ni 3dz2 (≈ 86%) orbital, while the σ-antibonding orbital is composed of the NO πz* (≈ 58%), πy* (≈ 23%) and Ni 3dz2 (≈ 5%) orbitals. The different contributions of the Ni- and NO-based fragment orbitals into the bonding and antibonding natural orbitals indicate the presence of non-negligible ionic components in the Ni–NO σ-bond.
 | ||
| Fig. 8 Contour plots of the most important natural orbitals (with predominant Ni 3d and NO π* character) optimized with the CASSCF(11e,11o) calculation and spin density map. Indicated qualitative nature and fractional occupation number (n) are reported. Contour values: ±0.03 a.u. for the orbitals and ± 0.003 electrons/a0 for the spin density (the positive sign is shown in cyan, the negative sign in dark blue). N, O, Ni, C and H atoms are reported in blue, red, yellow, green and white, respectively. | ||
CASSCF calculations correctly predicted a doublet (S = 1/2) ground state, in line with the experimental evidence. The most representative contribution to the NiII–NO electronic structure is provided by the NiII(S = 1)–NO0(S = 1/2) resonance structure (85.1%) with the following electronic configuration: (dyz)2(dxz)2(dxy)2(dx2−y2)↑(dz2)↑(πz*)↓. Such configuration describes the antiferromagnetic coupling between the unpaired electrons on the NiII 3dz2 orbital and the NO πz* orbital. Thereby, its dominant role entirely agrees with the proposed spin pairing mechanism of the NO binding. The remaining contributions to the NiII–NO ground state are given by NiI(S = 1/2)–NO+(S = 0) with an electronic configuration of (dyz)2(dxz)2(dxy)2(dz2)2(dx2−y2)↑ and NiIII(S = 1/2)–NO−(S = 0) with an electronic configuration of (dyz)2(dxz)2(dz2)2(dx2−y2)↑(dxy)↑(πz*)↓, which account for 8.9% and 1.3%, respectively. The larger contribution of the cationic resonance structure with respect to the anionic one agrees well with the NOδ+ formulation of the nitrosyl moiety, already reported in other precedented studies.14
The SOMO of the doublet spin state is mainly a Ni 3dx2−y2 orbital with a slight overlap with the hybrid sp orbitals of the Nf and O atoms of the NO2 ligand. The calculated spin density exhibits a positive region predominantly localized on the nickel center, with minute portions on the Nf and O atoms of the NO2 ligand. On the other hand, a negative spin density is predicted on the nitrosyl ligand (Fig. 8, at the bottom) due to the effective polarization induced by the unpaired electron spin density in the 3dx2−y2 orbital perpendicular to the Ni–NO bond. Given the positive gyromagnetic ratio γ of the 14N nuclear spin, a negative contribution of the spin distribution in the nitrogen 2s orbital corresponds to a negative hf interaction. This is indeed the case of the N atom of NO. On the contrary, a positive hf interaction is calculated for Nf atoms because of a direct spin density transfer via the overlap of the hybrid sp orbitals of Nf atoms with the Ni 3dx2−y2 orbital.
The rhombic g-tensor is correctly reproduced, and the trend gzz > gyy > gxx, is detected experimentally for the main NiII–NO species validating the microscopic structure proposed in Fig. 7. The computed orientation of the z principal axis of the g-tensor is approximately perpendicular with respect to the plane defined by the dx2−y2 orbital, as it typically happens when the unpaired electron is in the dx2−y2 orbital. Overall, the computed quadrupole interaction for the different nitrogen nuclei is in reasonable agreement with the experimental findings.
The computed spin Hamiltonian parameters for the nitrogen ligands directly bound to Ni may be grouped into two families of nitrogen nuclei, in agreement with ENDOR experiments. Nf1, Nf2 and Nf3 possess almost identical hyperfine and quadrupole couplings which nicely fit with the experimental values found for 14N(2). Moreover, they are characterized by Euler angles different from each other (Table S2, ESI†). A spectral simulation of the ENDOR spectra obtained by using the calculated Euler angles for Nf1, Nf2 and Nf3 is reported in Fig. S9 (ESI†) confirming that, by considering nitrogen nuclei with similar spin Hamiltonian parameters but different orientation of the hyperfine and quadrupole tensors (as predicted by quantum chemical calculations), a satisfying explanation of the features of the low-frequency spectra may be obtained. This permits to assign 14N(2) species to nitrogen atoms from the SBU, forming a σ-bond with the nickel center. On the other hand, 14N(1) signal is consistent with the computed 14N hyperfine couplings from the NO ligand, which correctly reproduces the large dipolar contribution. The weak 14N hf interaction detected by HYSCORE experiments is instead consistent with the ones calculated for nitrogen atoms of the benzobistriazolate immediately close to the nitrogen linked to the nickel (Ns in Fig. 7).
Although the binding mechanism of NO to NiII ion occurs through the same way (e.g. spin pairing mechanism) regardless the nature of the embedding considered, there are substantial structural and magnetic differences between the NiII–NO adduct described here and the ones reported for other microporous systems. Table 3 summarizes the main structural, electronic and magnetic differences between the nickel–nitrosyl complex in this work and the one recently characterized by Pietrzyk et al.14 in zeolite-type material. Apart from the different coordination geometry (pseudo-octahedral instead of square pyramidal), the Ni–NO bond distance is longer while the N–O bond length is slightly shorter compared to what is reported for Ni-ZSM-5 material.14 This is consistent with a weaker NO associative mechanism agreeing with the lower NO adsorption energy reported here compared to zeolite case. Such tiny structural details affect the electronic structure and, thus, the EPR parameters. Because of the shorter N–O bond, the NO ligand acquires a partial positive charge. The cationic resonance structure (NiI(S = 1/2) − NO+) has a higher contribution (8.9%) compared to the case in ZSM-5 (6.3%) in the description of the ground state. Consequently, the positive spin population on the Ni ion as well as the negative spin population on the NO ligand – induced by spin polarization – are larger than the ones reported by Pietrzyk (see Table 3). The experimental 14N hyperfine couplings of the nitrosyl ligand clearly reflect the changes in spin distribution whereby larger hf interactions are detected in MFU-4l-NO2 compared to ZSM-5 case.
| System | Geometry | d Ni–NO | d N–O | ρ Ni | ρ NO | N A max | Ref. | |
|---|---|---|---|---|---|---|---|---|
| NiII–NO in MFU-4l | MOF | Pseudo-octahedral | 0.230 | 0.114 | +1.55 | −0.35 | 29.0 | This work |
| NiII–NO in ZSM-5 | Zeolite | Square pyramidal | 0.190 | 0.117 | +1.22 | −0.27 | 14.0 | 14 |
Conclusion
EPR spectroscopy and quantum chemical calculations were carried out to assess the geometric and electronic structure of NiII–NO moieties in NiII-MFU-4l-NO2 material. W-band CW-EPR detected five-coordinated NiII species assigned to the peripheral sites of the parent Ni-MFU-4l-NO2, MOF in agreement with DFT calculations. Such divalent nickel centers are capable of chemoselective capture of gaseous NO-forming mononitrosyl complexes with electron spin S = 1/2, which can be easily identified by CW X-band EPR measurements. The nature of the NiII–NO bond and of the Ni–Nf bonds was ascertained by ENDOR studies and thoroughly accounted for by DFT/CASSCF calculations. While the bonding of Ni with Nf ligands from the SBU is characterized by a direct spin density transfer via overlap of the nitrogen sp orbitals with the nickel 3dx2−y2 orbital, the NO bonding is due to spin pairing mechanism NO↑ + NiII↑↑ → [↑NiII(↑ ↓)NO] whereby the transfer of spin density arises via spin polarization of the NO π orbital perpendicular to the Ni 3dx2−y2 orbital. The results presented here highlight the capabilities of sophisticated EPR techniques in combination with quantum chemical calculations in providing fundamental insights into the non-obvious electronic structure of open-shell species docked in metal–organic frameworks.Abbreviations
| EPR | Electron paramagnetic resonance |
| HYSCORE | Hyperfine sublevel correlation spectroscopy |
| DFT | Density functional theory |
| MOF | Metal–organic framework |
| ESEEM | Electron spin echo envelope modulation |
| ENDOR | Electron nuclear double resonance |
Author contributions
The manuscript was written through the contributions of all authors. All authors have given approval for the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has received funding from the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska-Curie Grant agreement no. 813209. The authors gratefully acknowledge the DFG (FOR2433. MOF Switches) for financial support and the Gauss Centre for Supercomputing e.V. (http://www.gauss-centre.eu) for funding this project by providing computing time on the GCS Supercomputer SUPERMUC-NG at Leibniz Supercomputing Centre (http://www.lrz.de). We kindly acknowledge also the Computing Center of Leipzig University.References
- M. Chiesa, E. Giamello and M. Che, Chem. Rev., 2010, 110(3), 1320 CrossRef CAS PubMed
.
- L. Andrews and A. Citra, Chem. Rev., 2002, 102(4), 885 CrossRef CAS PubMed
- K. Hadjiivanov, Catal. Rev.: Sci. Eng., 2000, 42(1–2), 71 CrossRef CAS
- Z. Sojka, M. Che and E. Giamello, J. Phys. Chem. B, 1997, 101(24), 4831 CrossRef CAS
- G. Spoto, A. Zecchina, S. Bordiga, G. Ricchiardi, G. Martra, G. Leofanti and G. Petrini, Appl. Catal., 1994, 3(2–3), 151 CrossRef CAS
- E. Giamello, D. Murphy, G. Magnacca, C. Morterra, Y. Shioya, T. Nomura and M. Anpo, J. Catal., 1992, 136(2), 510 CrossRef CAS
- F. Witzel, H. G. Karge, A. Gutsze and U. Hartel, Chem.-Ing.-Tech., 1991, 63, 744 CrossRef CAS
- T. Rudolf, W. Böhlmann and A. Pöppl, J. Magn. Reson., 2002, 155(1), 45 CrossRef CAS PubMed
- M.-T. Nechita, G. Berlier, G. Ricchiardi, S. Bordiga and A. Zecchina, Catal. Lett., 2005, 103(1–2), 33 CrossRef CAS
- A. Pöppl, T. Rudolf, P. Manikandan and D. Goldfarb, J. Am. Chem. Soc., 2000, 122(41), 10194 CrossRef
- A. Pöppl, T. Rudolf and D. Michel, J. Am. Chem. Soc., 1998, 120(19), 4879 CrossRef
- V. Umamaheswari, M. Hartmann and A. Pöppl, Magn. Reson. Chem., 2005, 43, S205–14 CrossRef CAS PubMed
- P. Fisicaro, E. Giamello, G. Berlier and C. Lamberti, Res. Chem. Intermed., 2003, 29(7–9), 805 CrossRef CAS
- P. Pietrzyk, K. Góra-Marek, T. Mazur, B. Mozgawa, M. Radoń, M. Chiesa, Z. Zhao and Z. Sojka, J. Catal., 2021, 394, 206 CrossRef CAS
- S. Jensen, K. Tan, L. Feng, J. Li, H.-C. Zhou and T. Thonhauser, J. Am. Chem. Soc., 2020, 142(39), 16562 CrossRef CAS PubMed
- M. Ahrens, O. Marie, P. Bazin and M. Daturi, J. Catal., 2010, 271(1), 1 CrossRef CAS
- W. Schießer, H. Vinek and A. Jentys, Appl. Catal., 2001, 33(3), 263 CrossRef
- P. C. Ford and I. M. Lorkovic, Chem. Rev., 2002, 102(4), 993 CrossRef CAS PubMed
- J. A. McCleverty, Chem. Rev., 2004, 104(2), 403 CrossRef CAS PubMed
- G. R. A. Wyllie and W. R. Scheidt, Chem. Rev., 2002, 102(4), 1067 CrossRef CAS PubMed
- V. Umamaheswari, M. Hartmann and A. Pöppl, J. Phys. Chem. B, 2005, 109(42), 19723 CrossRef CAS PubMed
- H. Yahiro, A. Lund and M. Shiotani, Spectrochim. Acta, Part A, 2004, 60(6), 1267 CrossRef PubMed
- M. Chiesa and E. Giamello, Catal. Lett., 2021, 151(12), 3417 CrossRef CAS
-
P. Pietrzyk and Z. Sojka, EPR spectroscopy and DFT calculations of the g tensors of {VO}1/ZSM-5, {CuNO}11/ZSM-5 and {NaNO}1/ZSM-5 intrazeolitic complexes, in Studies in Surface Science and Catalysis: Molecular Sieves: From Basic Research to Industrial Applications, ed. J. Čejka, N. Žilková, P. Nachtigall, Elsevier, 2005. pp. 617–624 Search PubMed
- Z. Sojka and P. Pietrzyk, Spectrochim. Acta, Part A, 2004, 60(6), 1257 CrossRef PubMed
- P. Pietrzyk and Z. Sojka, J. Phys. Chem. A, 2005, 109(46), 10571 CrossRef CAS PubMed
- K. Podolska-Serafin and P. Pietrzyk, J. Mol. Struct., 2019, 1180, 754 CrossRef CAS
- M. Gutjahr, R. Böttcher and A. Pöppl, J. Phys. Chem. B, 2002, 106(6), 1345 CrossRef CAS
- B. Barth, M. Mendt, A. Pöppl and M. Hartmann, Microporous Mesoporous Mater., 2015, 216, 97 CrossRef CAS
- M. Mendt, B. Barth, M. Hartmann and A. Pöppl, J. Chem. Phys., 2017, 147(22), 224701 CrossRef PubMed
- M. Mendt, F. Gutt, N. Kavoosi, V. Bon, I. Senkovska, S. Kaskel and A. Pöppl, J. Phys. Chem. C, 2016, 120(26), 14246 CrossRef CAS
- B. Jee, K. Koch, L. Moschkowitz, D. Himsl, M. Hartman and A. Pöppl, J. Phys. Chem. Lett., 2011, 2(5), 357 CrossRef CAS
- E. Giamello, E. Garrone, E. Guglielminotti and A. Zecchina, J. Mol. Catal., 1984, 24(1), 59 CrossRef CAS
- M. Chiesa, J. Mol. Catal. A: Chem., 2003, 204–205, 779 CrossRef CAS
- D. Denysenko, M. Grzywa, M. Tonigold, B. Streppel, I. Krkljus, M. Hirscher, E. Mugnaioli, U. Kolb, J. Hanss and D. Volkmer, Chem. – Eur. J., 2011, 17(6), 1837 CrossRef CAS PubMed
- S. Biswas, M. Grzywa, H. P. Nayek, S. Dehnen, I. Senkovska, S. Kaskel and D. Volkmer, Dalton Trans., 2009, 6487 RSC
- D. Denysenko, J. Jelic, K. Reuter and D. Volkmer, Chem. – Eur. J., 2015, 21(22), 8188 CrossRef CAS PubMed
- D. Denysenko and D. Volkmer, Faraday Tras., 2017, 201, 101 CAS
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178(1), 42 CrossRef CAS PubMed
- P. Höfer, A. Grupp, H. Nebenführ and M. Mehring, Chem. Phys. Lett., 1986, 132(3), 279 CrossRef
- E. R. Davies, Phys. Lett. A, 1974, 47(1), 1 CrossRef CAS
- D. Denysenko, M. Grzywa, J. Jelic, K. Reuter and D. Volkmer, Angew. Chem., 2014, 53(23), 5832 CrossRef CAS PubMed
- R. Dovesi, A. Erba, R. Orlando, C. M. Zicovich-Wilson, B. Civalleri, L. Maschio, M. Rérat, S. Casassa, J. Baima, S. Salustro and B. Kirtman, Comput. Mol. Sci., 2018, 8(4), e1360 Search PubMed
- A. Erba, J. Baima, I. Bush, R. Orlando and R. Dovesi, J. Chem. Theory Comput., 2017, 13(10), 5019 CrossRef CAS PubMed
- C. Lee, W. Yang and R. B. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37(2), 785 CrossRef CAS PubMed
- A. D. Becke, J. Chem. Phys., 1993, 98(7), 5648 CrossRef CAS
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132(15), 154104 CrossRef PubMed
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32(7), 1456 CrossRef CAS PubMed
- D. Vilela Oliveira, J. Laun, M. F. Peintinger and T. Bredow, J. Comput. Chem., 2019, 40(27), 2364 CrossRef CAS PubMed
- K. Eichkorn, F. Weigend, O. Treutler and R. Ahlrichs, Theor. Chem. Acta., 1997, 97(1–4), 119 CrossRef CAS
- C. M. Zicovich-Wilson, F. Pascale, C. Roetti, V. R. Saunders, R. Orlando and R. Dovesi, J. Comput. Chem., 2004, 25(15), 1873 CrossRef CAS PubMed
- F. Pascale, C. M. Zicovich-Wilson, F. López Gejo, B. Civalleri, R. Orlando and R. Dovesi, J. Comput. Chem., 2004, 25(6), 888 CrossRef CAS PubMed
- C. Carteret, M. de La Pierre, M. Dossot, F. Pascale, A. Erba and R. Dovesi, J. Chem. Phys., 2013, 138(1), 14201 CrossRef PubMed
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12(5) Search PubMed
- C. Remenyi, R. Reviakine, A. V. Arbuznikov, J. Vaara and M. Kaupp, J. Phys. Chem. A, 2004, 108(23), 5026 CrossRef CAS
- B. A. Heß, C. M. Marian, U. Wahlgren and O. Gropen, Chem. Phys. Lett., 1996, 251(5–6), 365 CrossRef
- S. Grimme, J. Chem. Phys., 2006, 124(3), 34108 CrossRef PubMed
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7(18), 3297 RSC
- J. L. Whitten, J. Chem. Phys., 1973, 58(10), 4496 CrossRef CAS
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8(9), 1057 RSC
- G. L. Stoychev, A. A. Auer and F. Neese, J. Chem. Theory Comput., 2017, 13(2), 554 CrossRef CAS PubMed
- V. Barone, J. Phys. Chem., 1995, 99(30), 11659 CrossRef CAS
- J. P. S. Walsh, S. Sproules, N. F. Chilton, A.-L. Barra, G. A. Timco, D. Collison, E. J. L. McInnes and R. E. P. Winpenny, Inorg. Chem., 2014, 53(16), 8464 CrossRef CAS PubMed
- K. Amrutha and V. Kathirvelu, J. Phys. Chem. Solids, 2021, 157, 110224 CrossRef
- C. R. Wilson, M. J. Riley, D. Wang and G. R. Hanson, Chem. Phys., 1997, 217(1), 63 CrossRef CAS
- K. Amrutha and V. Kathirvelu, Magn. Reson. Chem., 2022, 60(3), 414 CrossRef CAS PubMed
- J. Krzystek, J.-H. Park, M. W. Meisel, M. A. Hitchman, H. Stratemeier, L.-C. Brunel and J. Telser, Inorg. Chem., 2002, 41(17), 4478 CrossRef CAS PubMed
- J. Krzystek, A. Ozarowski and J. Telser, Coord. Chem. Rev., 2006, 250(17–18), 2308 CrossRef CAS
- J. Krzystek, S. A. Zvyagin, A. Ozarowski, S. Trofimenko and J. Telser, J. Magn. Reson., 2006, 178(2), 174 CrossRef CAS PubMed
- J. Lu, I. O. Ozel, C. A. Belvin, X. Li, G. Skorupskii, L. Sun, B. K. Ofori-Okai, M. Dincă, N. Gedik and K. A. Nelson, Chem. Sci., 2017, 8(11), 7312 RSC
- D. Collison, M. Helliwell, V. M. Jones, F. E. Mabbs, E. J. L. McInnes, P. C. Riedi, G. M. Smith, R. G. Pritchard and W. I. Cross, Faraday Tras., 1998, 94(19), 3019 RSC
- P. J. van Dam, A. A. K. Klaassen, E. J. Reijerse and W. R. Hagen, J. Magn. Reson., 1998, 130(1), 140 CrossRef CAS PubMed
- J. Mroziński, A. Skorupa, A. Pochaba, Y. Dromzée, M. Verdaguer, E. Goovaerts, H. Varcammen and B. Korybut-Daszkiewicz, J. Mol. Struct., 2001, 559(1–3), 107 CrossRef
- L. A. Pardi, A. K. Hassan, F. B. Hulsbergen, J. Reedijk, A. L. Spek and L. C. Brunel, Inorg. Chem., 2000, 39(2), 159 CrossRef CAS PubMed
- S.-D. Jiang, D. Maganas, N. Levesanos, E. Ferentinos, S. Haas, K. Thirunavukkuarasu, J. Krzystek, M. Dressel, L. Bogani, F. Neese and P. Kyritsis, J. Am. Chem. Soc., 2015, 137(40), 12923 CrossRef CAS PubMed
- A. Kubica, J. Kowalewski, D. Kruk and M. Odelius, J. Chem. Phys., 2013, 138(6), 64304 CrossRef CAS PubMed
- F. Neese, Coord. Chem. Rev., 2009, 253(5–6), 526 CrossRef CAS
- S. Ye and F. Neese, Inorg. Chem., 2010, 49(3), 772 CrossRef CAS PubMed
- F. Neese, J. Am. Chem. Soc., 2006, 128(31), 10213 CrossRef CAS PubMed
- W. Hayes and J. Wilkens, Proc. R. Soc. Lond. A, 1964, 281(1386), 340 CAS
-
A. Abragam and B. Bleaney, Electron paramagnetic resonance of transition ions, Dover Publications, New York, 1986 Search PubMed
-
N. M. Atherton, Ellis Horwood and PTR Prentice Hall, eMagRes, 1993 Search PubMed
-
J. R. Harmer, Hyperfine Spectroscopy – ENDOR, eMagRes, John Wiley & Sons, Ltd, 2016. pp. 1493–514 Search PubMed
- J. A. J. Fitzpatrick, F. R. Manby and C. M. Western, J. Chem. Phys., 2005, 122, 084312 CrossRef PubMed
- W. B. Mims and J. Peisach, J. Chem. Phys., 1978, 69(11), 4921 CrossRef CAS
- V. K. K. Praneeth, F. Neese and N. Lehnert, Inorg. Chem., 2005, 44(8), 2570 CrossRef CAS PubMed
- C. Wäckerlin, D. Chylarecka, A. Kleibert, K. Müller, C. Iacovita, F. Nolting, T. A. Jung and N. Ballav, Nat. Commun., 2010, 1(1), 61 CrossRef PubMed
- C. Daniel and C. Gourlaouen, Mol., 2019, 24(20), 3638 CrossRef CAS PubMed
- J. H. Enemark and R. D. Feltham, J. Am. Chem. Soc., 1974, 96(15), 5002 CrossRef CAS
-
R. D. Feltham and J. H. Enemark, Structures of Metal Nitrosyls, in Geoffroy, editor. Topics in inorganic and organometallic stereochemistry. Topics in Stereochemistry, John Wiley, 1981, vol. 12, pp. 155–215 Search PubMed
- J. H. Enemark and R. D. Feltham, Coord. Chem. Rev., 1974, 13(4), 339 CrossRef CAS
- R. Hoffmann, M. M. L. Chen, M. Elian, A. R. Rossi and D. M. P. Mingos, Inorg. Chem., 1974, 13(11), 2666 CrossRef CAS
- W. R. Scheidt, K. Hatano, G. A. Rupprecht and P. L. Piciulo, Inorg. Chem., 1979, 18(2), 292 CrossRef CAS
- E. A. Dierks, S. Hu, K. M. Vogel, A. E. Yu, T. G. Spiro and J. N. Burstyn, J. Am. Chem. Soc., 1997, 119(31), 7316 CrossRef CAS
- A. Stępniewski, M. Radoń, K. Góra-Marek and E. Broclawik, Phys. Chem. Chem. Phys., 2016, 18(5), 3716 RSC
- M. Radoń, E. Broclawik and K. Pierloot, J. Phys. Chem. B, 2010, 114(3), 1518 CrossRef PubMed
- L. Freitag, S. Knecht, C. Angeli and M. Reiher, J. Chem. Theory Comput., 2017, 13(2), 451 CrossRef CAS PubMed
- T. Ampßler, G. Monsch, J. Popp, T. Riggenmann, P. Salvador, D. Schröder and P. Klüfers, Angew. Chem., 2020, 59(30), 12381 CrossRef PubMed
- J. K. Bower, A. Y. Sokolov and S. Zhang, Angew. Chem., 2019, 58(30), 10225 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp01449e |
| ‡ These authors contributed equally. |
| § Present address: Paolo Cleto Bruzzese – Max Planck Institute for Chemical Energy Conversion, Stiftstr. 34-36, 45470 Mülheim an der Ruhr, Germany. |
| ¶ Present address: Matthias Mendt – SaxonQ GmbH, Emilienstr. 15, 04107 Leipzig, Germany. |
| This journal is © the Owner Societies 2023 |