
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotocyclization reaction and related photodynamics in the photoproducts of a tetraphenylethylene derivative with bulky substituents: unexpected solvent viscosity effect†
Mario
de la Hoz Tomás
 a,
Mao
Yamaguchi
b,
Boiko
Cohen
a,
Ichiro
Hisaki
b and
Abderrazzak
Douhal
*a
a,
Mao
Yamaguchi
b,
Boiko
Cohen
a,
Ichiro
Hisaki
b and
Abderrazzak
Douhal
*a
aDepartamento de Química Física, Facultad de Ciencias Ambientales y Bioquímica, and INAMOL, Universidad de Castilla-La Mancha, Avenida Carlos III, S/N, 45071 Toledo, Spain. E-mail: Abderrazzak.douhal@uclm.es
bGraduate School of Engineering Science, Osaka University, 1-3 Machikaneyama, Toyonaka, Osaka 560-8531, Japan
First published on 27th June 2023
Abstract
Tetraphenylethylene (TPE) derivatives are ones of the most versatile building blocks showing aggregation-induced emission (AIE). However, their applications are limited by the photophysical and photochemical processes that occur in their excited state. Herein, we report a detailed study of the photochemical behaviour of a new TPE derivative with bulky terphenyl groups (TTECOOBu) in solvents of different viscosities and in a PMMA film. UV light irradiation shows an efficient photocyclization reaction, which produces a 9,10-diphenylphenanthrene (DPP) derivative photoproduct. The emission spectra of the irradiated samples show intermediate (∼420 nm) and final (∼380 nm) species. The photocyclization events are more efficient in environments of higher viscosities or rigidity. We show that in a photoirradiated PMMA film containing TTECOOBu, it is possible to etch a message for more than 1 year. The kinetics is dictated by the motions of the phenyl rings and is faster when their motions are precluded or inhibited. We also elucidated the femto- to millisecond photodynamics of the intermediate and final photoproducts and provide a full picture of their relaxation, with the latter in ∼1 ns at S1 and ∼1 μs at T1. We also demonstrate that the kinetics of the bulky TTECOOBu is much slower than that of the TPE core. Our results also show that both photoevents are not reversible contrary to the case of TPE kinetics. We believe that these results will shed more light on the photochemical behaviour of TPE derivatives and should help in the development of novel TPE-based materials with improved photostability and photo-properties.
1. Introduction
Molecular rotors are molecules with photophysical properties that differ from those of the conventional fluorophores. In contrast to the latter, these compounds are generally non-emissive in dilute solutions. However, their emission becomes significantly enhanced in viscous solvents, aggregated form, or solid state.1–7 In the electronically excited state, these molecular systems undergo intramolecular twisting motions responsible for the observed strong non-radiative behaviour. When the rotations or twisting motions are prevented due to aggregation, the emission intensity of the molecular rotors increases. This phenomenon, known as aggregation-induced emission (AIE), has been extensively studied since 2001 and has proven to be in contradiction to the aggregation-caused quenching (ACQ).8–14 Many experimental and theorical studies have tried to understand the underlaying mechanisms of the AIE phenomenon with the aim to develop new AIE-luminogens (AIEgens) with improved properties.9,11–13,15Tetraphenylethylene (TPE) is the archetypal molecule with AIE properties.16–18 Like other AIEgens, it displays a very weak emission in solutions, while its fluorescence is enhanced in aggregated or solid state where the internal motions are impeded. In addition to aggregation, the fluorescence intensity could be increased suppressing these internal motions by modifying the TPE framework with bulky substituents,19–22 connecting their phenyl rings with hydrocarbon tethers23,24 or rigidifying their structure by constraining them in metal organic frameworks (MOFs)25,26 or hydrogen-bonded organic frameworks (HOFs).27,28 These AIE properties, together with its easy functionalization, have made TPE a versatile building block for many applications.29–34 However, its application is partially limited by the competing processes that occur in its excited state.
The non-radiative relaxation pathways of excited TPE in solutions have been interpreted in terms of internal conversion associated with an ethylene bond twisting35–41 and torsion of the phenyl rings,23,41,42 which may affect the photoisomerization and photocyclization process. Unlike the photoisomerization, the photocyclization reaction has received little attention. Under UV-light, TPE undergoes an efficient photochemical cyclization reaction when irradiated for long periods of time.21,43–50 The final photoproduct displays a characteristic blue emission band (reminiscent to phenanthrene) at ∼375 nm. Similar to that observed in stilbene,51–53 the photocyclization of TPE is a two-step reaction that goes through an intermediate prior to reaching the final product. The intermediate photoproduct is unstable and if not trapped it reverts back to the initial TPE molecule. However, if trapped by oxidation, it gives a new irreversible cyclized-TPE photoproduct – 9,10-diphenylphenanthrene (DPP).43,44,53 To increase the efficiency of the photocyclization reaction, different strategies have been developed such as functionalization of the TPE core or the use of different solvents and concentrations.21,45,48 It was reported that the photocyclization is faster in TPE derivatives functionalized with bulky groups,21 electron-donating ether group45 or with the phenyl rings connected by hydrocarbon tethers,48 while it is slower at higher concentrations or in aggregated state.45 These studies suggest that the torsion of the phenyl rings might play a key role in the photocyclization, which can be further affected in TPE derivatives, functionalized in the ortho position of the phenyl rings.47,48
Thus, studying the photo-induced behavior of TPE derivatives with bulky groups should shed more light on the working mechanism of their photochemical processes. Using bulky groups in TPE, one can also explore the effect of the viscosity on the photocyclization mechanism. Furthermore, it is of great interest to also understand the dynamical photobehaviour of the formed intermediate and final species. Femto- to millisecond spectroscopy will provide detailed information on their dynamics and give a full picture on the photocyclization process.
Herein, we report on a detailed photochemical and photophysical study of TTECOOBu, a symmetric TPE derivative functionalized with terphenyl groups.41 Steady-state absorption and emission experiments in different solvents following different times of irradiation under UV light (365 nm) show a photocyclization reaction to give two photocyclated derivatives in a stepwise fashion. In dichloromethane (DCM) and N,N-dimethylformamide (DMF) solutions the photoreaction occurs slowly, while in viscous media and rigid polymer matrix (polymethyl methacrylate, PMMA) it is more efficient and significantly faster. The emission spectra of irradiated solutions show the presence of three different emissive species: initial (∼540 nm), intermediate (∼410 nm) and final (∼370 nm). The intermediate species does not revert to the initial one, and in presence of oxygen it is rapidly photo-oxidized to the final DPP derivative. We also studied the photophysical behaviour of the intermediate and final species produced in this process using fs-, ps- and ms-time-resolved spectroscopic techniques. The lifetime of the intermediate (∼1.8 ns) and final (∼0.8 ns) species in DCM solutions are longer than those of the initial ones (∼67 ps),41 which can be explained in terms of restriction of the intramolecular motions due to the efficient photocyclization process. The fs-transient absorption spectra of the final photoproduct are narrow and rapidly decay to a constant contribution. The presence of a triplet state with a lifetime of 0.98 μs and absorption maximum at 530 nm is confirmed by the millisecond-time-resolved experiments in DCM solutions. Our results shed more light into the photochemical behaviour of TPE derivatives and could be used for a better design of new TPE-based materials with improved photostability or photochromism for applications in photonics or sensing.
2. Materials and methods
For the spectroscopic studies, dichloromethane (DCM, 99.9%), N,N-dimethylformamide (DMF, 99.8%), triacetin (TAC, 99%), tetrahydrofuran (THF, 99.9%) and poly(methyl methacrylate) (PMMA, Mw ∼ 996![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1) were obtained from Sigma-Aldrich (Merck) and used as received. To prepare TTECOOBu@PMMA film, 250 μL of a solution of 1 mg mL−1 of TTECOOBu in THF were added to a solution of PMMA in THF (15% w/w). The content of the dye in PMMA film was 0.05% (w/w). The films are stable for at least one year, and no significant changes in their photobehaviour was observed. For the steady-state UV-vis absorption and emissions spectra, time-correlated single-photon counting (TCSPC) and flash photolysis experiments, TTECOOBu was dissolved in the selected solvents adjusting the optical density (1 cm) to a value of 0.3 at the absorption at 325 nm. For the femtosecond (fs) transient UV-visible absorption experiments, we prepared concentrated solutions with an optical density of 0.3 at the maxima of absorption in a 1 mm cell. All the experiments were performed at 293 K.
000 g mol−1) were obtained from Sigma-Aldrich (Merck) and used as received. To prepare TTECOOBu@PMMA film, 250 μL of a solution of 1 mg mL−1 of TTECOOBu in THF were added to a solution of PMMA in THF (15% w/w). The content of the dye in PMMA film was 0.05% (w/w). The films are stable for at least one year, and no significant changes in their photobehaviour was observed. For the steady-state UV-vis absorption and emissions spectra, time-correlated single-photon counting (TCSPC) and flash photolysis experiments, TTECOOBu was dissolved in the selected solvents adjusting the optical density (1 cm) to a value of 0.3 at the absorption at 325 nm. For the femtosecond (fs) transient UV-visible absorption experiments, we prepared concentrated solutions with an optical density of 0.3 at the maxima of absorption in a 1 mm cell. All the experiments were performed at 293 K.
The steady-state UV-visible absorption and emission spectra have been recorded using JASCO V-670 and FluoroMax-4 (Jobin-Yvone) spectrophotometers, respectively. For the steady-state and TCSPC photoconversion experiments, the samples were irradiated with an UV flashlight (Alonefire SV38, 5 W) at 365 nm and ∼0.3 W cm−2. For the transient UV-visible absorption and flash photolysis experiments the samples were irradiated with a Nd:YAG laser (Brilliant, Quantel) at 355 nm and ∼3 mJ cm−2.
2.1 Time-correlated single-photon counting (TCSPC)
The picosecond (ps) time-resolved emission experiments were carried out by employing a TCSPC system.54 The samples were excited at 325 nm using the second harmonic of the output of a femtosecond (fs) optical parametric oscillator (OPO, Inspire Auto 100, Radiantis). The instrumental response function (IRF) of the setup is ∼35 ps. The fluorescence signal was gated at the magic angle (54.7°) and monitored at a 90° angle to the excitation beam at discrete emission wavelengths. The decays were deconvoluted and fitted to a multiexponential function using the FLUOFIT package (PicoQuant) allowing single and global fits. The quality of the fit was estimated by χ2, which was always below 1.2, and by the distribution of the residuals.2.2 Transient absorption
The fs-transient absorption experiments were performed using a chirped pulse amplification setup.55 The OPA (TOPAS, Positive Light) output at 325 nm was used as the pump and kept below 500 μW. The transient absorption measurements were performed in the spectral ranges of 490–710 (vis region). To avoid photo degradation and re-excitation by consecutive pulses, the samples were placed in a 0.8 mm thick rotating quartz cell. The IRF was measured in terms of ΔOD for DCM following excitation at 325 nm to give ∼120 fs. Data were analyzed using a multiexponential fit program (IGOR Pro v. 6.37, Wavemetrics). The quality of the fit was evaluated by examining the residual distributions.2.3 Flash photolysis
The nanosecond (ns) flash photolysis setup has been described previously.56 Briefly, it consists of a LKS.60 laser flash photolysis spectrometer (Applied Photophysics) and Vibrant (HE) 355 II laser (Opotek) as a pump pulse source (5 ns duration). We used the third harmonic of the Q-switched Nd:YAG laser (Brilliant, Quantel) at 355 nm. The pump fluence of the output was 70 mJ cm−2, which was attenuated to 2.5 mJ by a pair of a half-waveplate and a polarizer. As a probe source, we used the output of a 150 W Xenon arc lamp. The light transmitted through the 1 cm quartz cuvette containing the sample was then dispersed by a monochromator and detected by a visible photomultiplier (Applied Photophysics R928), coupled to a digital oscilloscope (Agilent Infiniium DS08064A, 600 MHz, 4 GSa s1). The measured IRF of the system was ∼8 ns.3. Results and discussion
3.1 Synthesis and characterization of TTECOOBu
Details of the synthesis procedure and structural characterization of TTECOOBu have been previously reported.41 Briefly, the compound was obtained using Suzuki coupling reaction between 1,1,2,2-tetrakis(4-bromophenyl) ethene and boron pinacolate derivative of biphenyl ester using Pd(PPh3)4 in THF. Scheme 1 shows the molecular structure of TTECOOBu.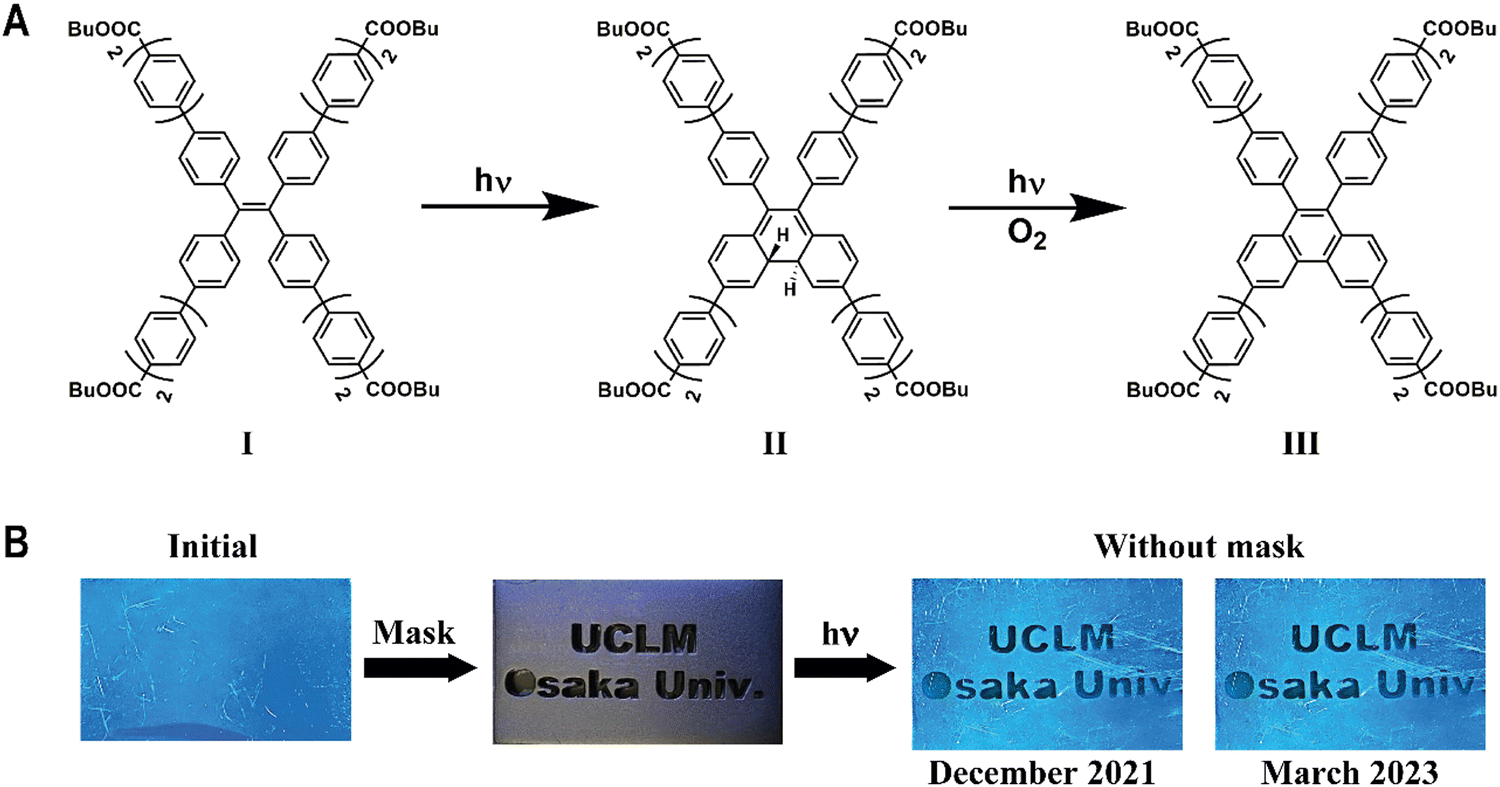 | ||
| Scheme 1 (A) Illustration of the photocyclization of TTECOOBu in solutions. (B) Photoetching of a message (UCLM and Osaka Univ.) on a PMMA film containing TTECOOBu upon UV irradiation. Note that the etched message is stable at least for more than one year. | ||
3.2 Photocyclization reaction in solutions and in a PMMA film
 | ||
| Fig. 1 Changes in the absorption and emission (λexc = 325 nm) spectra of TTECOOBu in (A) DCM, (B) DMF, (C) TAC and (D) PMMA film for different periods of irradiation using an UV-light at 365 nm (UVMAX = ∼0.3 W cm−2). (E) Photographs of TTECOOBu in a DCM solution following different periods of irradiation taken under UV light. The irradiation times of the solutions are indicated in the photographs. For PMMA film, the irradiation times were 0, 0.5, 2, 3, 4, 5, 7.5, 10, 20, 30, 40, 50, 60, 75, 90, 120 and 210 min. | ||
Before showing and discussing the results upon UV-irradiation, we shortly describe the previously reported steady-state absorption and emission spectra.41 Before UV-light irradiation, the absorption spectra in the used solvents show similar shape with a single band with an intensity maximum at 325 nm and shoulders at 290 and 370 nm. This band corresponds to the π–π* and S0 → S1 transition of the chromophore.41 Contrary to the absorption spectra, the emission ones show a solvent-dependent behaviour in which the emission intensity maximum appears at 543 (DCM), 541 (DMF), 530 (TAC) and 465 nm (PMMA film). The spectral change is explained in terms of the different degree of restriction of the twisting and torsional motion of the ethylenic bond and phenyl rings.41
Upon irradiation with UV light and depending on the used medium, the shape of the absorption spectrum becomes different. We observe an UV-shift at the intensity maximum and a decrease in the shoulder intensity at 370 nm (Fig. 1). While in DMF solutions the absorption spectrum exhibits a small change, in DCM and TAC solutions we recorded an UV-shift along with a clear decrease in the intensity of the shoulder at 370 nm. In the PMMA film these changes are more pronounced and the intensity maximum is further UV-shifted to 295 nm. Note that in PMMA film the irradiation time for the full photoconversion process was only 210 min while in other solvents it was 300 min and the photoconversion was not fully complete. Based on these observations, we suggest that the photoreaction is more efficient in TAC (highly viscous solvent, η = 17.4 cP at 298 K) and in PMMA film (rigid matrix) probably due to the difference in the conformations adopted by TTECOOBu in these media. To get more information on this photoreaction, we also recorded the changes in the emission spectra of the above samples.
The emission spectra vary significantly upon UV-light irradiation: the intensity around 550 (solvents) and 470 nm (PMMA film) becomes gradually weaker and a new emission band at around 370–400 nm simultaneously emerges (Fig. 1). The change clearly depends on the used solvent and irradiation time. To begin with, in DCM solutions, upon irradiation, a new emission band at around 425 nm emerges in the first minutes and it quickly becomes the predominant band at longer times of irradiation. From around 120 min of irradiation, this emission band gives rise to a new one at ∼375 nm. The observed spectral changes from yellow to blue can be observed by naked eye (Fig. 1E). In DMF solutions, the initial emission (∼550 nm) slightly decreases along with the formation of species with a weak emission at ∼410 nm, which is gradually converted to strong emission band at ∼380 nm (Fig. 1B). In TAC, we observed the appearance of a weak emission at ∼410 nm, which at longer irradiation times decreases in intensity and gives a strong band at ∼380 nm (Fig. 1C). The initial emission intensity (∼530 nm) decreases considerably in TAC indicating a faster photoreaction in comparison with the one occurring in DCM and DMF. While in DCM and TAC solutions we do not observe a clear isoemissive point, the emission spectra in DMF solutions display a clear isoemissive point around 500 nm mainly coming from two different emitters. Finally, for the PMMA film, the conversion from the initial (∼460 nm) to the final (∼375 nm) species occurs immediately (no intermediate band is recorded) and the photoreaction is faster than in solution (Fig. 1D).
Based on these observations, the photoconversion of TTECOOBu in solutions undergoes a two-step photoreaction that goes through an intermediate species emitting at ∼425 nm to give the final photoproduct with the maximum of emission intensity at ∼375 nm. In agreement with the changes in the absorption spectra upon irradiation, the excitation spectra recorded at the maxima of intensity of these three emitters are different (Fig. S1, ESI†). Thus, the excitation spectra show similar behaviour to the absorption ones characterized by a blue shift of the intensity maximum, and a shoulder located around 370 nm with decreasing emission intensity. The three different excitation spectra collected at representative times of irradiation and at the selected wavelengths corresponding to the three emission bands demonstrate the existence of three different absorbing and emissive species as a result of photoirradiation.
To further confirm the existence of three different emissive species involved in the photocyclization reaction, we carried out the photoirradiation experiments of TTECOOBu in a DCM solution under different atmosphere conditions. Fig. 2 shows the obtained results in ambient conditions (A), in presence of N2 (B) and in presence of O2 (C) at different irradiation times. Following previous studies of the photoreaction of TPE and its derivatives,43,44,48,53 the mechanism of the photocyclization reaction of TTECOOBu in solutions can be understood if one considers two possible scenarios: (1) a single step where O2 molecules are very close to the initial form to “directly” give the photoproduct. In this scenario, the mechanism will not depend on the oxygen molecule diffusion; (2) a two-step mechanism involving a long-living intermediate species that will depend on the concentration and diffusion of O2 molecules (Scheme 1). Based on the obtained results under different O2/N2 conditions (see the following results), we anticipate the nature and structure of species I, II and III as indicated in Scheme 1.
 | ||
| Fig. 2 Comparison of the time-dependent emission spectra of TTECOOBu in DCM solutions with different times of UV-light irradiation at 365 nm (UVMAX = ∼0.3 W cm−2): (A) air conditions; (B) in presence of N2 and (C) in presence of O2. The excitation wavelength for the emission spectra was 325 nm. | ||
Under ambient conditions, we observed the three emission bands, which we assign to the initial (I), intermediate (II) and final (III) species. However, when the sample is saturated with N2, the emission spectrum mainly displays the two emission bands corresponding to species I and II, while the one corresponding to the final photoproduct (III, ∼375 nm) is very weak even after irradiating for a longer time (Fig. S2, ESI†). On the other hand, for oxygen-saturated solutions, the formation of the final species occurs almost instantaneously without clear signature of the species II (Fig. 2C). These results clearly indicate that the second step of the photocyclization reaction requires the presence of an oxidative reagent (O2 in this case) and that the emission band at 425 nm corresponds to the intermediate species. Therefore, we suggest that, in similarity with the TPE photocyclization, TTECOOBu is converted into its corresponding DPP derivative upon UV-light irradiation and that the presence of oxygen in the solvent is enough to oxidize the intermediate species (II) toward the final photoproduct (III) (Scheme 1).44 This oxidation is also a photoinduced reaction from intermediate to final species, as no significant change in the emission spectra were recorded when the intermediate species was kept in a dark environment for 12 h in an O2-saturated atmosphere (Fig. S3, ESI†). Furthermore, our experiments using N2-saturated solutions strongly indicate that the initial form (I) still reacts in absence of oxygen to give species II, which does not evolve to species III (Fig. 2B). Therefore, a mechanism based on a single step without involving an intermediate does not explain the observations. However, under aerobic conditions, we can clearly see both intermediate (II) and final (III) species emission bands.
The explanation of the behaviour of TTECOOBu following irradiation with UV light is in agreement with previous reports on the photocyclization reaction in the TPE core, which converts to DPP.43,44,48,53 For the TTECOOBu, the absorption and emission spectra of the final form of the photocyclization reaction are comparable to those reported for DPP, thus suggesting that the final product is a derivative of DPP (note that due to the functionalization with the terphenyl rings we do not observe the vibrational resolution reminiscent of DPP). It was shown that the photochemical generation of DPP from TPE is a two-step reaction where the first one is a reversible process and the second one, the conversion from the intermediate to DPP, requires the presence of oxygen or another oxidant molecule.43,48,53 For the TPE core, the emission shifts rapidly from 550 to 375 nm during the irradiation indicating that the intermediate is unstable and either goes back to the initial structure or is rapidly oxidized to produce DPP.43,44,53 In contrast to this mechanism and interestingly, the emission spectrum of TTECOOBu displays an intermediate emission band, which is very stable in absence of irradiation and does not revert to the initial structure. Although there is a number of studies on the two-step photocyclization of TPE and its derivatives, there is little information for the back reaction from the TPE intermediate to the initial form, and the effect of bulky substituents on its efficiency. It is largely assumed that, in similarity with stilbene, the first step in the photocyclization is a reversible process followed by a fast photooxidation.21,43–45,48,50,57 Several works have reported on TPE derivatives with photochromic properties where the back reaction was produced following irradiation by visible light.58–60 These works suggest that the reverse process needs to overcome an energy barrier to revert to the initial form. On the other hand, the presence of bulky substituents in TPE derivatives that require larger changes in nuclear coordinates of the intermediate to revert to the initial form can further increase this energy barrier, making irreversible the first step. Furthermore, TPE derivatives with increased electronic conjugation in the molecule (more planar conformation) have been reported to show longer lifetime of the intermediate species.48 Weakly emissive intermediate species of other TPE derivatives were also observed in solid state, stable in darkness for several days.59,61 Thus, based on the above, we suggest that the bulky groups of TTECOOBu (two phenyl and ester groups) on each of the four arms should enhance the π-conjugation of the involved initial, intermediate and final species. The stabilization of the intermediate species will prevent the back reaction to the initial structure. It should be noted that leaving the UV-irradiated sample (under aerobic conditions) in darkness for several days, as well as upon its irradiation with visible light, we observed no changes in the UV-vis absorption and emission, which indicates stable intermediate species (Fig. S4, ESI†).
Kinetic model in solutions and in PMMA film. Based on the steady-state absorption and emission spectra, the photocyclization reaction is solvent-dependent. While in DCM and DMF solutions the reaction proceeds slowly, in TAC and PMMA film the photocyclization is fast. Taking into account that the photocyclization process of TTECOOBu in solutions is a two-step reaction and the intermediate species is stable and does not revert back to the initial ones, we applied a kinetic model for irreversible consecutive reactions (eqn (1)).43–45,48,53 While the kinetics model (eqn (1)) does not depend on the electronic nature of the involved species (singlet or triplet configuration), we believe that the photoreaction involving O2 (step 2) is most likely occurring in the triplet state of species II. Triplet states have much longer lifetimes (μs–ms regime) than singlets (ns regime) at S1/S2 and the molecular oxygen will have enough time to collide with the TTECOOBu molecules in the triplet state. However, we cannot exclude that the photocyclization reaction takes place in the singlet states, as it was reported for other molecular systems.62–64 The photocyclization reaction of TTECOOBu can be summarized according to the following equation:65
 | (1) |
 | (2) |
 | (3) |
 | (4) |
| Medium | k 1/10−3 (min−1) | k 2/10−3 (min−1) | η (cP) | [O2]/10−4 (XG) |
|---|---|---|---|---|
| a For PMMA film and THF/PMMA mixed solutions, we do not observe the intermediate species and hence, the photoconversion process follows the I* → III* reaction, thereby the rate constant k2 does not apply in this case. The two values obtained for k1 in PMMA film are due to two different conformations of the molecules trapped in the film. Estimated errors: ∼15%. | ||||
| DCM | 4.70 | 0.90 | 0.41 | 7.09 |
| DMF | 1.13 | 0.24 | 0.79 | 3.89 |
| TAC | 8.55 | 0.80 | 17.40 | — |
| PMMA filma | 193.71/19.03 | — | — | — |
| THF/PMMAa | 113.77 | — | 23.97 | — |
The first photocyclization process depends on the solvent properties (i.e., viscosity) and it is an unimolecular event, while the photo-oxidation reaction is oxygen dependent and it is a bimolecular reaction. Based on the experiments in solutions, we identify two behaviours related to the viscosity of the medium: one in low-viscosity solvents (DCM and DMF), and another one in high-viscosity solvent (TAC).
To begin with, we focus on the obtained results in low-viscosity solvents (DCM and DMF) (Table 1). Based on the kinetic behaviour and taking into account the values of k1 and k2 in non-viscous solvents (DCM and DMF), it is clear that both the unimolecular and bimolecular events are faster in DCM. The rate constant value obtained for the photocyclization process (first step) is k1 (10−3 min−1) = 4.7 and 1.13 for DCM and DMF, respectively (Table 1). The photocyclization reaction of TPE and its derivatives takes place via a co-rotative electrocyclization of the excited 6π-electronic system according to the Woodward–Hoffmann rules.46,49 Since it involves twisting motions of the phenyl rings, the reaction should be slower in viscous solvents where the rotation of these substituents would be restricted. Indeed, this effect is observed when the viscosity of the solvent changes only moderately, i.e. from DCM to DMF. The rate constant of photocyclization in DMF decreases in agreement with the expected moderation of the intramolecular motions to reach the most favorable configuration for the reaction to proceed towards the final photoproduct. On the other hand, the obtained values for the rate constant of the photo-oxidation process (step 2) is k2 (10−3 min−1) = 0.90 and 0.24 in DCM and DMF, respectively (Table 1). The value is smaller using DMF and can be explained in terms of oxygen concentration in these solvents, which is higher for DCM (XG = 7.09 × 10−4 for DCM and XG = 3.89 × 10−4 for DMF, at 298.2 K, XG is the molar fraction of O2 in the solvent).66
Interestingly, the photocyclization reaction becomes faster in a highly viscous solvent (TAC, η = 17.4 cP at 298 K), and now the rate constants are k1 (10−3 min−1) = 8.55 and k2 (10−3 min−1) = 0.80 (Table 1). Here, the effect is opposite to that previously observed, and the photoreaction becomes faster when the viscosity significantly increases. In similarity with the presence of bulkier substituents to the TPE core, the increased rate of the photoproduct formation for TTECOOBu in solutions having a higher viscosity can be explained in terms of a more favorable initial conformation adopted by the molecule.57 The higher viscosity of TAC traps a population of TTECOOBu molecules with a conformation that favors the photocyclization reaction (step 1) which is significantly faster in comparison to the two solvents with a lower viscosity (DCM and DMF) (Table 1).
However, as described above, the photochemical behaviour in a PMMA film differs from that observed in solutions (Fig. 1D). In this rigid media, we do not observe the emission of intermediate species and it seems like the reaction takes places in a single step. The photooxidation process depends on O2 concentration and diffusion. While in solution the reaction is expected to be diffusion limited, hence, dependent on both O2 concentration and diffusion, in PMMA, where O2 molecules mobility/diffusion is strongly restricted, for the reaction with O2 we have to consider that they are very close to the reaction center of TTECOOBu significantly increasing the photooxidation rate (step 2) and observing a “quasi” one-step photocyclization reaction. Thus, the changes observed in the emission spectra correspond to the transformation from the initial to the final photoproduct. However, we found that the photoreaction does not follow a simple one-step reaction, but the photoconversion of the trapped TTECOOBu molecules is rather described by two parallel single steps (eqn (5)):
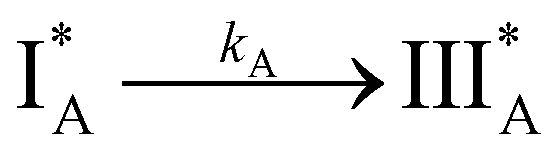 | (5a) |
 | (5b) |
 | (6a) |
 | (6b) |
 and
and  ) with different conformations of the phenyl groups and exhibiting different rate constants of the photoconversion reaction. The obtained values for the rate constants in PMMA film are higher than those observed in solutions indicating that the restricted or prohibited twisting motions of the phenyl groups promote the photocyclization reaction. The kinetic behavior of these stabilized structures in the PMMA film is thermodynamically favorable in this medium.
) with different conformations of the phenyl groups and exhibiting different rate constants of the photoconversion reaction. The obtained values for the rate constants in PMMA film are higher than those observed in solutions indicating that the restricted or prohibited twisting motions of the phenyl groups promote the photocyclization reaction. The kinetic behavior of these stabilized structures in the PMMA film is thermodynamically favorable in this medium.
To get further information on the effect of the intramolecular rotation/twisting of phenyl rings on the kinetics, we performed irradiation experiments increasing the viscosity of the medium. For this purpose, mixed solutions of TTECOOBu in THF and PMMA were prepared by increasing the viscosity of the mixture (from 0.46 cP to 23.97 cP at 295 K) (Fig. S7 and Table S2, ESI†). The observed changes can be divided into two families. Family I where the viscosity of the mixture is less than 0.5 cP, and family II where the viscosisty is higher than 0.5 cP. Fig. 3 shows the changes in absorption and emission spectra of the two families after UV irradiating at ambient conditions.
 | ||
| Fig. 3 Changes in the absorption and emission spectra of TTECOOBu in mixed THF/PMMA solutions with different viscosities of (A) 0.46 cP and (B) 23.97 cP at different irradiation times with UV-light at 365 nm (UVMAX = ∼0.3 W cm−2). The irradiation times with UV-light were 0, 0.5, 1, 1.5, 2, 3.5, 5, 7.5, 10, 15, 20, 30, 40 and 50 min. For the emission spectra, the excitation wavelength was 325 nm. | ||
The spectral change in the absorption spectra of family I is almost inexistent contrary to the one of family II, in which we recorded a clean intensity decreases of the 370 nm shoulder, an UV-shift and increase in the main UV-absorption band, with a clear isosbestic point at 320 nm. For the emission spectra, the behaviour is also different. For family I, the initial emission at 550 nm almost does not change in intensity while the ∼400 nm emission band quickly increases in intensity. For family II, the 500 nm emission intensity strongly decreases and vanishes at 50 min of irradiation. At the same time, the 375 nm strongly increases. For this family, we also observed an isoemissive point at 500 nm. We tried to apply a kinetic model to obtain the rate constants. For the changes in family I, we could not succeed to fit the change in the emission intensity at ∼400 nm, as it reflects three steps: (1) formation of II*, (2) disappearance of II* and (3) formation of III*. The emission intensity is not high, and it is a combination of the signals of I*, II* and II*. However, for family II the photoreaction is almost complete, and the intensity is large, and a single step (I* → III*) describe the observed change with a rate constant of k1 (10−3 min−1) = 113.77 (Fig. S8 and Table 1, Table S1, ESI†). This value is much higher than the one obtained for the other used solvents, which indicates that the viscosity of the medium plays a fundamental role in the photocyclization process by increasing the efficiency of the reaction. Similar to what we observed in the PMMA film, the emission spectra show only two bands (initial and final species). Thus, we suggest that the photooxidation in the second step is very fast, not leaving time to the intermediate to be stabilized.
To study the effect of the UV-light irradiation (continuous-wave flashlight source) on the efficiency of the photocyclization process, we carried out irradiation studies varying the viscosity of the medium and working with a lower power source (UVMAX = ∼0.18 W cm−2). Fig. 4A and B show the changes in the absorption and emission spectra. The photobehavior for both families are quite similar to that depicted in Fig. 3. For family II (η > 0.5 cP), the changes in the absorption and emission spectra are noticeable with a slight UV-shift in the absorption spectra, while the emission one shows a decrease of the initial species, and the 400 nm emission band quickly appears. However, for family I (η < 0.5 cP), no significant changes were observed. Because of the lower power of the irradiation source, the changes observed in both spectra for each family are smaller than those observed using a higher power light source (UVMAX = ∼0.30 W cm−2). These observations are explained in terms of incomplete photocyclization and photooxidation reactions as also reported for TPE and its derivatives.45 Interestingly, working at lower power we observed the 400 nm emission band corresponding to the intermediate species (Fig. 4B). Fig. 4C and D show the change in the emission intensity of the final photoproduct (species III) at different viscosities and irradiation times, respectively. The results clearly indicate that the viscosity of the medium increases the velocity of the photoreaction to photoproduce species III.
 | ||
| Fig. 4 (A and B) Changes in the absorption and emission spectra of TTECOOBu in mixed THF/PMMA solutions with different viscosities at different irradiation times with UV light at 365 nm (UVMAX = ∼0.18 W cm−2). (C) Photoproduct emission intensity at 375 nm vs. viscosity of the mixture as indicated in the inset. (D) Changes of emission intensity at 375 nm with irradiation time as indicated in the inset. For the emission spectra, the excitation wavelength was 325 nm. | ||
To compare the photoconversion of TTECOOBu with that of the TPE core, we carried out experiments under the same experimental conditions for TPE. We observed significant changes in both the absorption and emission spectra at low and high viscosities (Fig. S9, ESI†). The emission spectra show a characteristic band of the DPP photoproduct at ∼375 nm with a well-defined vibrational structure.43,44,53 It should be noted that the overall emission intensity of the final species is a function of its emission quantum yield and on its concentration, both dependent on the medium viscosity. Clearly, the photoreaction in the TPE core occurs faster than in TTECOOBu. Furthermore, while the emission intensity of the final photoproduct of TTECOOBu increases with the viscosity (experiments done up to 23.97 cP), the TPE core reaches its maximum conversion at a viscosity of only 3–5 cP. This clearly reflects the effect of the bulky phenyl groups and their twisting motions on the photocyclization kinetics. Previously, it has been described that the torsional motions of the phenyl rings in TPE take place in ∼19 ps,48 while for TTECOOBu (extended conjugation with three phenyl rings in each arm) this process is slower and it occurs in ∼50 ps.41 Therefore, we expect to observe a clear difference in the photocyclization process of both molecules. For TPE (one phenyl ring in each arm) a small increase in the viscosity (3–5 cP) is enough to reach a plateau of the photocyclization reaction, while for TTECOOBu, a significantly higher viscosity is required to reach a similar behaviour. We explain this difference in terms of several twisting/rotation of the three phenyl rings on each arm in TTECOOBu, which might be coupled resulting in a larger response to changes in the solvent viscosity. These observations, along with the results in TAC and PMMA film, demonstrate that the viscosity of the reaction medium plays a key role in the photoreaction efficiency by increasing the rate constant at higher viscosity, contrary to what one may expect at first guess.
To summarize this part (Scheme 1), the photocyclization reaction of TTECOOBu in solutions is a two-step process that depends on the solvent viscosity and oxygen content. The photoreaction in DCM and DMF solutions is slower than in highly viscous media and very fast in a rigid PMMA film. The difference resides in the molecular conformation adopted by the phenyl groups of TTECOOBu in these media and restriction of their motions. We also showed that by controlling the amount of O2 in the medium, one can photoproduce more or less of the final product. In normal conditions the two-step mechanism is not reversible and leads to the final photoproduct at shorter or longer times of irradiation depending on the viscosity of the medium.
Molecules with photochromism behaviour that can reversibly change their optical properties in response to UV light are being used in numerous applications, such as glasses, information storage, non-linear optics and organic optoelectronic devices.67–71 For example, a large number of organic photochromic materials based on diarylethene, azobenzene and TPE derivatives have been developed for this purpose.58,67–72 Based on the results presented in this work and taking into account the changes in the emission spectra observed in a PMMA film upon irradiation of the sample, we explored the possibility to use TTECOOBu@PMMA film as a materials composite to irreversibly print information upon its irradiation by UV light. For this purpose, we fabricated a polymer film of PMMA containing TTECOOBu and a mask with a hidden message (Scheme 1B). The film was irradiated with UV-light (365 nm) for 3 hours and the light passed through the used mask, recording the message on the film. When the mask was removed and, under UV light, the message etched on the film can be easily observed. The etched message is stable for a longtime (at least 15 months).
 | ||
| Fig. 5 Magic-angle emission decays of (A) initial (tirr. = 0 min), (B) intermediate (tirr. = 90 min) and (C) final species (tirr. = 240 min) of TTECOOBu in DCM solution upon excitation at 325 nm and observation as indicated in the inset. The solid lines are from the best multiexponential fits, and the IRF is the instrumental response function. Fig. 5A is adapted from ref. 41. | ||
Since the photodynamics of the initial species has been reported previously,41 here we only focus on the study of the intermediate and final species. Table 2 gives the parameters of the multiexponential fit of the decays of the three samples. We tried to fit the data of the irradiated samples using stretched exponential function model which is based on the response (in this case emission) of different and independent populations reflecting the heterogeneity of the samples.73–76 For the emission decays of the photoproduct, gated from 350–450 nm, we obtained a characteristic time of 0.72 ns and a dispersion parameter, β = 0.9. As β value is very close to unity, this analysis suggests a homogeneous sample containing different emitters. Therefore, we used multiexponential model to fit the data. The initial species shows a multiexponential and emission wavelength-dependent behaviour of the decays, which was explained in terms of formation of different emitters followed by intramolecular twisting and phenyl rotation motions.41
| Sample | λ obs/nm | τ 1/ns | a 1 | c 1 | τ 2/ns | a 2 | c 2 | τ 3/ns | a 3 | c 3 |
|---|---|---|---|---|---|---|---|---|---|---|
| a Data of initial species adapted from ref. 41. | ||||||||||
| Initial speciesa | 450 | 0.02 | 84 | 19 | 0.07 | 9 | 7 | 1.14 | 7 | 74 |
| 470 | 76 | 36 | 23 | 38 | 1 | 26 | ||||
| 610 | 0.04 | −100 | −100 | 100 | 100 | — | ||||
| 630 | −100 | −100 | 100 | 100 | ||||||
| Intermediate species | 380 | — | 0.74 | 44 | 28 | 1.48 | 56 | 72 | ||
| 400 | 11 | 6 | 89 | 94 | ||||||
| 420 | 6 | 3 | 94 | 97 | ||||||
| Final species | 350 | 0.14 | 20 | 4 | 0.83 | 81 | 96 | 1.83 | ||
| 370 | 19 | 4 | 80 | 95 | 1 | 1 | ||||
| 390 | 19 | 4 | 79 | 91 | 2 | 5 | ||||
| 410 | 17 | 3 | 78 | 85 | 5 | 12 | ||||
| 430 | 16 | 3 | 75 | 78 | 9 | 19 | ||||
| 450 | 16 | 3 | 68 | 68 | 16 | 29 | ||||
The decays of the intermediate and final species also exhibit a multiexponential behaviour with time constants of τ1 = 0.74 and τ2 =1.48 ns for the intermediate and τ1 = 0.14, τ2 = 0.83 and τ3 = 1.83 ns for the final species (Table 2). However, contrary to the complex behavior of the emission decays of the initial TTECOOBu form that are characterized by both decaying and rising components, for these species all components are decaying throughout the entire observation spectral range. The shortest component for the intermediate species (0.74 ns) contributes only at the bluest part of the emission spectrum and disappears at wavelengths longer than 400 nm. Considering that the emission of the final product is located at 370 nm and a component with a similar time constant (0.83 ns) was found in its emission decays, we suggest that the observed behavior corresponds to the contribution from both species and the lifetime of 0.74 ns is due to the presence of final species. Similarly, the longer component in the emission decays of the final species (1.83 ns) is only recorded in the blue part (390 to 450 nm, with the higher contribution at ∼450 nm) of the emission spectrum, where the emission of the intermediate is also observed and characterized by a similar lifetime (1.48 ns). The data suggests that the emission decays of the intermediate and the final products, have mixed contributions from both forms. However, based on the spectral behaviour of the time components, we suggest that the intermediate species decays monoexponentially with a time constant of 1.48 ns, while the final species exhibits a biexponential behaviour with time constants of 0.14 and 0.83 ns. We assign the two components observed in the decays of the final species to the contribution of two different populations – one that has a conformation of the phenyl rings that favors and undergoes fast non-radiative process (i.e. intersystem crossing, ISC), and a second one that relaxes by internal conversion to the ground state. Low yields of ISC for TPE and other phenylethylenes have been reported and suggested that the process depends on the conformation of the phenyl rings.77 Furthermore, as we show in the flash photolysis section (3.2.4), the experiments under O2, N2 and ambient conditions show the existence of triplet state in the relaxation of the final photoproduct.
To summarize this part, the longer components of both decays correspond to the lifetime of the intermediate and final species (1.48 ns and 0.83 ns, respectively), and the short component observed in the final species (∼140 ps) is an indication for the presence of an ISC to a non-emissive triplet state.
Next, we also studied the photodynamics of the final photoproduct in PMMA film. The emission decays are shown in Fig. S11 (ESI†). They exhibit a multiexponential behaviour with time constants of 0.12, 0.66 and 1.22 ns (Table S3, ESI†). The components are decaying throughout the entire observation spectral range. The complexity of the decays and the nature of the PMMA environments do not allow an easy assignment of these components. Based on the kinetics model proposing two different conformers in PMMA film susceptible to undergo photocyclization reaction, we assign the time constants of 0.66 and 1.22 ns to the emission lifetime of the two photocyclated conformers, while the 0.12 ns-component should reflect the presence of fast non-radiative process in the photoproducts of these structures.
 | ||
| Fig. 6 Time-resolved transient visible absorption spectra of the (A) initial, (B) intermediate and (C) final species of TTECOOBu in DCM at different delay times upon excitation at 325 nm. Representative transient absorption decays of the (D) initial, (E) intermediate and (F) final species probing as indicated in the inset. ΔOD is the change in the optical density upon electronic excitation at 325 nm. Fig. 6A and D are adapted from ref. 41. | ||
To begin with, the TAS of the final photoproduct consists of a single and narrow absorption band with a maximum of intensity at 580 nm along with a broad shoulder at 520 nm that decays to a constant signal in the first 25 ps (Fig. 6C). The TAS of the photoproduct does not present the same rich photodynamics observed for the initial structure where several interconnected bands were observed and explained in terms of intramolecular twisting and rotation of the phenyl rings (Fig. 6A).41 The TAS behaviour of the final photoproduct reflects that the recorded narrow band is mainly coming from the rigid core of DPP of the final species. The transient signal at 520 nm decays multiexponentially with time constants of 0.62 ps (35%), 5.7 ps (30%) and 153 ps (16%) (Table 3). We obtained similar values for the transient at 580 nm that was also fit adequately by a multiexponential function giving time components with values of 0.57 ps (38%), 6.1 ps (27%) and 141 ps (19%) (Table 3). Both signals show an offset of a time constant longer than 1 ns (Fig. S13, ESI†). Thus, the observed dynamics in the probed spectral range has the same origin and reflects an ultrafast relaxation of the final photoproduct that involves intramolecular vibrational-energy redistribution (IVR, ∼0.6 ps) and vibrational cooling (VC, ∼6 ps). Similar values for the time constants of these two processes have been reported for other comparable molecular systems.78,79 These values are also not different from those recorded for the initial species (Table 3). As we assigned in the TCSPC experiments, the ∼150 ps component corresponds to a population that most probably undergoes ISC (S1 → T1) to a non-emissive triplet state (vide infra). The longer, ns-component could reflect the lifetime of the final product (∼0.83 ns) combined with that of the intermediate one (∼1.83 ns), as we suggest in the emission lifetime part (Table 2).
| Sample | λ obs/nm | τ 1/ps | a 1 (%) | τ 2/ps | a 2 (%) | τ 3/ps | a 3 (%) | τ 4/ps | a 4 (%) |
|---|---|---|---|---|---|---|---|---|---|
| a Data of initial species adapted from ref. 41. | |||||||||
| Initial speciesa | 620 | 0.49 | −4 | — | 32 | −96 | 104 | 100 | |
| 730 | 0.56 | 16 | 8.70 | 25 | 27 | 23 | 98 | 36 | |
| Intermediate species | 520 | 0.41 | 21 | 0.61 | −100 | 141 | 25 | >1 ns | 54 |
| 580 | 0.47 | 18 | 0.54 | −100 | 166 | 30 | >1 ns | 52 | |
| Final species | 520 | 0.62 | 35 | 5.7 | 30 | 153 | 16 | >1 ns | 19 |
| 580 | 0.57 | 38 | 6.1 | 27 | 141 | 19 | >1 ns | 16 | |
Finally, the TAS of the solution irradiated to produce the intermediate form presents a behaviour that corresponds to mixed contributions from the initial form and of the final photoproduct (Fig. 6B). On one hand, we observed a broad spectrum with a narrow band at ∼575 nm. This later is reminiscent to the TAS of the final photoproduct, while the broad band might be due to the absorption of a population from the initial structure. This is further supported by the analysis of representative transient decays (Fig. 6E). The transients probed at 520 nm and 580 nm show the signature of the ultrafast dynamics of the final photoproduct, governed by IVR (∼0.5 ps) and ISC (140–160 ps) processes. Furthermore, the fit of these transients requires a rising component of ∼0.6 ps, which most probably reflects a combination of the long rising component observed in the transient absorption of the initial species (Fig. 6D) and the decaying one of the final structures (Fig. 6F). These transient also exhibit a ns-component as we observed for the final species.
 | ||
| Fig. 7 (A) Time-resolved transient absorption spectra (TAS) of the TTECOOBu photoproduct in an aerobic DCM solution at different delay times. (B) Transient absorption decays probed at 520 nm under different atmospheric conditions: air (blue), oxygen (red) and nitrogen (green). The excitation wavelength was 355 nm. | ||
| λ obs/nm | Air | O2 | N2 | |||
|---|---|---|---|---|---|---|
| τ 1/μs | a 1 (%) | τ 1/μs | a 1 (%) | τ 1/μs | a 1 (%) | |
| 450 | 0.98 | 100 | 0.18 | 100 | 14.73 | 100 |
| 500 | 100 | 100 | 100 | |||
| 550 | 100 | 100 | 100 | |||
| 600 | 100 | 100 | 100 | |||
| 650 | 100 | 100 | 100 | |||
At delay times longer than 10 ns, the TAS show a single positive band that covers the whole observation spectral range with maximum of intensity at 530 nm. At shorter times (<20 ns), we observe a negative band. Based on its spectral position which coincides with the emission spectrum (Fig. S12, ESI†), we assign it to the stimulated emission. The positive band arises from the transient absorption of TTECOOBu in T1. We also recorded the TAS of the TTECOOBu photoproduct under N2 conditions. The observed TAS is similar to that described above with a single positive band in the probed spectral range (Fig. S14, ESI†). The transient decays of the samples exhibit a monoexponential behaviour with time constants of 0.98, 0.18 and 14.73 μs equilibrated with air, O2, and N2, respectively (Table 4). The dependence of the lifetimes on the presence of oxygen in the system is evidence that the observed absorption corresponds to that of a triplet state (T1) of the final photoproduct. These results agree with previous report on the photobehaviour of phenanthrene and its derivatives, which show a triplet state with comparable time constants (μs timescale).80
4. Conclusions
In this work, we report on the photochemical behaviour of TTECOOBu in solvents of different viscosities and in a rigid PMMA film. Steady-state absorption and emission studies following UV-light irradiation of the samples, show the occurrence of a two-step photocyclization reaction to yield as a final photoproduct a 9,10-diphenylphenanthrene (DPP) derivative with an enhanced and blue shifted fluorescence band. While in DCM and DMF solutions the photoreaction occurs relatively slowly, in viscous solvents (TAC or mixed THF-PMMA solutions) and in a PMMA film, the reaction is fast due to a restriction of the phenyl rings motion. Taking advantage of the fast photocyclization in PMMA matrix and the irreversibility of the photoreaction, we show that the TTECOOBu@PMMA composite might be used for recording a message which is stable for a long time. The emission spectra of the irradiated solutions show the presence of three different emissive species which can be attributed to: initial (∼540 nm), intermediate (∼420 nm) and final (∼380 nm) structures (Scheme 1). Photoirradiation experiments of solutions in equilibrium with atmosphere of air, oxygen and nitrogen show that the formation of the final form needs the presence of oxygen. Thus, while the first step of the photocyclization is a unimolecular event, the subsequent event is a bimolecular one. The intermediate species is stable and do not revert to the initial structure, while in presence of oxygen and UV light it is rapidly photooxidized to a DPP derivative. The fluorescence lifetime in DCM solutions of intermediate (∼1.8 ns) and final (∼0.8 ns) species are relatively longer compared to the initial one (∼70 ps).41 Femtosecond (fs) transient absorption experiments of the final species in DCM solutions show a narrow band which exhibits ultrafast events: ∼0.6 ps (IVR), ∼6 ps (VC) and ∼150 ps (ISC, S1 → T1). Flash photolysis experiments demonstrate the existence of a triplet state with a lifetime of 0.98 μs (air equilibrated solutions) and an absorption maximum at 550 nm of the photoproduct. The present results provide clear insights into the photochemical and photophysical behaviour of a TPE derivative having bulky phenyl groups in solutions, viscous and rigid environments. The findings clearly show the relevance of motion of these groups in its photocyclization, when compared to the TPE core. These findings may help in the design of TPE-based materials with controllable photocyclization and improved photostability for photonic applications.Author contributions
MY and IH synthesized and characterized TTECOOBu and co-wrote the paper. MH, BC and AD performed and analysed data of the steady-state and time-resolved experiments and co-wrote the paper. AD planned the research, supervised the experiments, analyzed, and interpreted the data. All authors have discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the following grants: PID2020-116519RB-I00 funded by MCIN/AEI/10.13039/501100011033 and by the “European Union, EU”; SBPLY/19/180501/000212 funded by JCCM and by the EU through “Fondo Europeo de Desarollo Regional” (FEDER) and 2020-GRIN-28929 funded by UCLM (FEDER). MH thanks MCIN for the FPI fellowship PRE2021-099064 financed by MCIN/AEI/10.13059/501100011033 and by FSE+. IH thanks Multidisciplinary Research Laboratory System for Future Developments (MRL), Graduate School of Engineering Science, Osaka University.References
- R. O. Loutfy and B. A. Arnold, J. Phys. Chem., 1982, 86, 4205–4211 CrossRef CAS.
- M. S. A. Abdel-Mottaleb, R. O. Loutfy and R. Lapouyade, J. Photochem. Photobiol., A, 1989, 48, 87–93 CrossRef CAS.
- M. A. Haidekker, T. P. Brady, D. Lichlyter and E. A. Theodorakis, Bioorg. Chem., 2005, 33, 415–425 CrossRef CAS PubMed.
- M. A. Haidekker and E. A. Theodorakis, J. Biol. Eng., 2010, 4, 11 CrossRef PubMed.
- A. Vyšniauskas, M. Qurashi, N. Gallop, M. Balaz, H. L. Anderson and M. K. Kuimova, Chem. Sci., 2015, 6, 5773–5778 RSC.
- H. Qian, M. E. Cousins, E. H. Horak, A. Wakefield, M. D. Liptak and I. Aprahamian, Nat. Chem., 2017, 9, 83–87 CrossRef CAS PubMed.
- P. Minei, G. Iasilli, G. Ruggeri, V. Mattoli and A. Pucci, Chemosensors, 2021, 9, 3 CrossRef CAS.
- J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740–1741 RSC.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332–4353 RSC.
- Y. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Soc. Rev., 2011, 40, 5361–5388 RSC.
- A. Qin and B. Z. Tang, Aggregation-Induced Emission: Fundamentals and Applications, Wiley, New York, 2013 Search PubMed.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- Y. Chen, J. W. Y. Lam, R. T. K. Kwok, B. Liu and B. Z. Tang, Mater. Horiz., 2019, 6, 428–433 RSC.
- Z. Zhao, H. Zhang, J. W. Y. Lam and B. Z. Tang, Angew. Chem., Int. Ed., 2020, 59, 9888–9907 CrossRef CAS PubMed.
- Y. Tang and B. Z. Tang, Handbook of Aggregation-Induced Emission, Wiley, USA, 2022 Search PubMed.
- Y. Dong, J. W. Y. Lam, A. Qin, J. Liu, Z. Li, B. Z. Tang, J. Sun and H. S. Kwok, Appl. Phys. Lett., 2007, 91, 011111 CrossRef.
- Z. Zhao, J. W. Y. Lam and B. Zhong Tang, Curr. Org. Chem., 2010, 14, 2109–2132 CrossRef CAS.
- Z. Zhao, J. W. Y. Lam and B. Z. Tang, J. Mater. Chem., 2012, 22, 23726–23740 RSC.
- V. S. Vyas and R. Rathore, Chem. Commun., 2010, 46, 1065–1067 RSC.
- W. Wang, T. Lin, M. Wang, T.-X. Liu, L. Ren, D. Chen and S. Huang, J. Phys. Chem. B, 2010, 114, 5983–5988 CrossRef CAS PubMed.
- G. Huang, B. Ma, J. Chen, Q. Peng, G. Zhang, Q. Fan and D. Zhang, Chem. – Eur. J., 2012, 18, 3886–3892 CrossRef CAS PubMed.
- J. Zhou, Z. Chang, Y. Jiang, B. He, M. Du, P. Lu, Y. Hong, H. S. Kwok, A. Qin, H. Qiu, Z. Zhao and B. Z. Tang, Chem. Commun., 2013, 49, 2491–2493 RSC.
- D. A. Shultz and M. A. Fox, J. Am. Chem. Soc., 1989, 111, 6311–6320 CrossRef CAS.
- N. L. C. Leung, N. Xie, W. Yuan, Y. Liu, Q. Wu, Q. Peng, Q. Miao, J. W. Y. Lam and B. Z. Tang, Chem. – Eur. J., 2014, 20, 15349–15353 CrossRef CAS PubMed.
- N. B. Shustova, B. D. McCarthy and M. Dincă, J. Am. Chem. Soc., 2011, 133, 20126–20129 CrossRef CAS PubMed.
- Z. Wei, Z.-Y. Gu, R. K. Arvapally, Y.-P. Chen, R. N. McDougald, Jr., J. F. Ivy, A. A. Yakovenko, D. Feng, M. A. Omary and H.-C. Zhou, J. Am. Chem. Soc., 2014, 136, 8269–8276 CrossRef CAS PubMed.
- G. Guo, D. Wang, X. Zheng, X. Bi, S. Liu, L. Sun and Y. Zhao, Dyes Pigm., 2022, 197, 109881 CrossRef CAS.
- S. Cai, Z. An and W. Huang, Adv. Funct. Mater., 2022, 32, 2207145 CrossRef CAS.
- Y. Liu, C. Deng, L. Tang, A. Qin, R. Hu, J. Z. Sun and B. Z. Tang, J. Am. Chem. Soc., 2011, 133, 660–663 CrossRef CAS PubMed.
- H. Shi, R. T. K. Kwok, J. Liu, B. Xing, B. Z. Tang and B. Liu, J. Am. Chem. Soc., 2012, 134, 17972–17981 CrossRef CAS PubMed.
- J.-H. Wang, H.-T. Feng and Y.-S. Zheng, Chem. Commun., 2014, 50, 11407–11410 RSC.
- J. Li, Y. Zhang, J. Mei, J. W. Y. Lam, J. Hao and B. Z. Tang, Chem. – Eur. J., 2015, 21, 907–914 CrossRef CAS PubMed.
- J. Huang, N. Sun, Y. Dong, R. Tang, P. Lu, P. Cai, Q. Li, D. Ma, J. Qin and Z. Li, Adv. Funct. Mater., 2013, 23, 2329–2337 CrossRef CAS.
- J. Huang, R. Tang, T. Zhang, Q. Li, G. Yu, S. Xie, Y. Liu, S. Ye, J. Qin and Z. Li, Chem. – Eur. J., 2014, 20, 5317–5326 CrossRef CAS PubMed.
- P. F. Barbara, S. D. Rand and P. M. Rentzepis, J. Am. Chem. Soc., 1981, 103, 2156–2162 CrossRef CAS.
- B. I. Greene, Chem. Phys. Lett., 1981, 79, 51–53 CrossRef CAS.
- E. Lenderink, K. Duppen and D. A. Wiersma, J. Phys. Chem., 1995, 99, 8972–8977 CrossRef CAS.
- R. W. J. Zijlstra, P. T. van Duijnen, B. L. Feringa, T. Steffen, K. Duppen and D. A. Wiersma, J. Phys. Chem. A, 1997, 101, 9828–9836 CrossRef CAS.
- G.-J. Zhao, K.-L. Han, Y.-B. Lei and Y.-S. Dou, J. Chem. Phys., 2007, 127, 094307 CrossRef PubMed.
- K. Kokado, T. Machida, T. Iwasa, T. Taketsugu and K. Sada, J. Phys. Chem. C, 2018, 122, 245–251 CrossRef CAS.
- M. de la Hoz Tomás, M. Yamaguchi, B. Cohen, I. Hisaki and A. Douhal, Phys. Chem. Chem. Phys., 2023, 25, 1755–1767 RSC.
- N. B. Shustova, T.-C. Ong, A. F. Cozzolino, V. K. Michaelis, R. G. Griffin and M. Dincă, J. Am. Chem. Soc., 2012, 134, 15061–15070 CrossRef CAS PubMed.
- R. J. Olsen and R. E. Buckles, J. Photochem., 1979, 10, 215–220 CrossRef CAS.
- C. E. Bunker, N. B. Hamilton and Y. P. Sun, Anal. Chem., 1993, 65, 3460–3465 CrossRef CAS.
- M. P. Aldred, C. Li and M.-Q. Zhu, Chem. – Eur. J., 2012, 18, 16037–16045 CrossRef CAS PubMed.
- A. Prlj, N. Došlić and C. Corminboeuf, Phys. Chem. Chem. Phys., 2016, 18, 11606–11609 RSC.
- Y.-J. Gao, X.-P. Chang, X.-Y. Liu, Q.-S. Li, G. Cui and W. Thiel, J. Phys. Chem. A, 2017, 121, 2572–2579 CrossRef CAS PubMed.
- Y. Cai, L. Du, K. Samedov, X. Gu, F. Qi, H. H. Y. Sung, B. O. Patrick, Z. Yan, X. Jiang, H. Zhang, J. W. Y. Lam, I. D. Williams, D. Lee Phillips, A. Qin and B. Z. Tang, Chem. Sci., 2018, 9, 4662–4670 RSC.
- T. Tran, A. Prlj, K.-H. Lin, D. Hollas and C. Corminboeuf, Phys. Chem. Chem. Phys., 2019, 21, 9026–9035 RSC.
- J. Rouillon, C. Monnereau and C. Andraud, Chem. – Eur. J., 2021, 27, 8003–8007 CrossRef CAS PubMed.
- F. B. Mallory, C. S. Wood, J. T. Gordon, L. C. Lindquist and M. L. Savitz, J. Am. Chem. Soc., 1962, 84, 4361–4362 CrossRef CAS.
- F. B. Mallory, J. T. Gordon and C. S. Wood, J. Am. Chem. Soc., 1963, 85, 828–829 CrossRef CAS.
- F. B. Mallory, C. S. Wood and J. T. Gordon, J. Am. Chem. Soc., 1964, 86, 3094–3102 CrossRef CAS.
- J. A. Organero, L. Tormo and A. Douhal, Chem. Phys. Lett., 2002, 363, 409–414 CrossRef CAS.
- M. Gil and A. Douhal, Chem. Phys., 2008, 350, 179–185 CrossRef CAS.
- C. Randino, M. Ziółek, R. Gelabert, J. A. Organero, M. Gil, M. Moreno, J. M. Lluch and A. Douhal, Phys. Chem. Chem. Phys., 2011, 13, 14960–14972 RSC.
- J. Guan, R. Wei, A. Prlj, J. Peng, K. H. Lin, J. Liu, H. Han, C. Corminboeuf, D. Zhao and Z. Yu, Angew. Chem., Int. Ed., 2020, 59, 14903–14909 CrossRef CAS PubMed.
- S. Guo, S. Zhou, J. Chen, P. Guo, R. Ding, H. Sun, H. Feng and Z. Qian, ACS Appl. Mater. Interfaces, 2020, 12, 42410–42419 CrossRef CAS PubMed.
- Z. He, L. Shan, J. Mei, H. Wang, J. W. Y. Lam, H. H. Y. Sung, I. D. Williams, X. Gu, Q. Miao and B. Z. Tang, Chem. Sci., 2015, 6, 3538–3543 RSC.
- S. Zhou, S. Guo, W. Liu, Q. Yang, H. Sun, R. Ding, Z. Qian and H. Feng, J. Mater. Chem. C, 2020, 8, 13197–13204 RSC.
- L. Zhu, R. Wang, L. Tan, X. Liang, C. Zhong and F. Wu, Chem. – Asian J., 2016, 11, 2932–2937 CrossRef CAS PubMed.
- N. J. Turro, Molecular Photochemistry, W. A. Benjamin, Inc., New York, 1965 Search PubMed.
- M. Klessinger and J. Michl, Excited States and Photo-Chemistry of Organic Molecules, Wiley-VCH, Weinheim, 1996 Search PubMed.
- N. J. Turro, V. Ramamurthy and J. C. Scaiano, Principles of Molecular Photochemistry: An Introduction, University Science Books, USA, 2009 Search PubMed.
- W. J. Moore, Physical Chemistry, Prentice-Hall, USA, 1955 Search PubMed.
- T. Sato, Y. Hamada, M. Sumikawa, S. Araki and H. Yamamoto, Ind. Eng. Chem. Res., 2014, 53, 19331–19337 CrossRef CAS.
- S. Kawata and Y. Kawata, Chem. Rev., 2000, 100, 1777–1788 CrossRef CAS PubMed.
- M. Irie, T. Fukaminato, K. Matsuda and S. Kobatake, Chem. Rev., 2014, 114, 12174–12277 CrossRef CAS PubMed.
- N. Tamai and H. Miyasaka, Chem. Rev., 2000, 100, 1875–1890 CrossRef CAS PubMed.
- M. Irie, Chem. Rev., 2000, 100, 1685–1716 CrossRef CAS PubMed.
- J. A. Delaire and K. Nakatani, Chem. Rev., 2000, 100, 1817–1846 CrossRef CAS PubMed.
- Z. Xiong, X. Zhang, L. Liu, Q. Zhu, Z. Wang, H. Feng and Z. Qian, Chem. Sci., 2021, 12, 10710–10723 RSC.
- M. N. Berberan-Santos, E. N. Bodunov and B. Valeur, Chem. Phys., 2005, 315, 171–182 CrossRef CAS.
- M. N. Berberan-Santos and B. Valeur, J. Lumin., 2007, 126, 263–272 CrossRef CAS.
- S. E. Koops, B. C. O’Regan, P. R. F. Barnes and J. R. Durrant, J. Am. Chem. Soc., 2009, 131, 4808–4818 CrossRef CAS PubMed.
- N. Boens and M. Van der Auweraer, Photochem. Photobiol. Sci., 2014, 13, 422–430 CrossRef CAS PubMed.
- H. Goerner, J. Phys. Chem., 1982, 86, 2028–2035 CrossRef CAS.
- N. Alarcos, M. Gutierrez, M. Liras, F. Sánchez and A. Douhal, Phys. Chem. Chem. Phys., 2015, 17, 16257–16269 RSC.
- T. Kumpulainen, B. Lang, A. Rosspeintner and E. Vauthey, Chem. Rev., 2017, 117, 10826–10939 CrossRef CAS PubMed.
- A. Onkelinx, G. Schweitzer, F. C. De Schryver, H. Miyasaka, M. Van der Auweraer, T. Asahi, H. Masuhara, H. Fukumura, A. Yashima and K. Iwai, J. Phys. Chem. A, 1997, 101, 5054–5062 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp01295f |
| This journal is © the Owner Societies 2023 |