
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Maturation of amyloid β fibrils alters their molecular stability†
Stefan
Becker
c,
Karin
Giller
c,
Daniel
Sieme
c and
Nasrollah
Rezaei-Ghaleh
 *ab
*ab
aInstitute of Physical Biology, Heinrich Heine University Düsseldorf, Universitätsstraße 1, D-40225 Düsseldorf, Germany. E-mail: Nasrollah.Rezaie.Ghaleh@hhu.de
bInstitute of Biological Information Processing, IBI-7: Structural Biochemistry, Forschungszentrum Jülich, D-52428 Jülich, Germany
cDepartment of NMR-based Structural Biology, Max Planck Institute for Multidisciplinary Sciences, Am Fassberg 11, D-37077 Göttingen, Germany
First published on 23rd May 2023
Abstract
Little is known about how maturation of Alzheimer's disease-related amyloid β (Aβ) fibrils alters their stability and potentially influences their spreading in the brain. Using high-pressure NMR, we show that progression from early to late Aβ40 aggregates enhances the kinetic stability, while ageing during weeks to months enhances their thermodynamic stability.
A remarkable feature of neurodegenerative diseases such as Alzheimer's disease (AD) is their progressive clinical and neuropathological course.1,2 Two neuropathological hallmarks of AD are the extracellular deposition of amyloid β (Aβ) peptide as senile plaques and the intracellular deposition of hyperphosphorylated tau protein as neurofibrillary tangles.2 The Aβ and tau aggregation pathology starts several years earlier than the onset of clinical symptoms and spreads in the brains of AD patients following particular spatiotemporal patterns, potentially through prion-like propagation of protein aggregates.2–5 Several studies have shown that the amyloid fibrils undergo various biochemical modifications and structural rearrangements during the long prodromal phase of AD.6–9 Little, however, is known about how the maturation of AD-related amyloid fibrils alters their stability and thereby potentially affects their spreading in the brain.
Previous 2D IR spectroscopy-based studies of Aβ fibrils suggest slow structural changes likely due to the presence of mobile trapped water molecules in fresh fibrils, which are re-located and eventually lost over the course of years.8 The structural maturation of Aβ fibrils can also occur in vivo, as it was shown in transgenic mouse models of AD using fluorescence probes of the internal structure of fibrils.9 High-pressure NMR is a powerful technique to detect structural alterations in protein systems if they are associated with changes in the degree of compaction.10 Recently, this technique has been utilized to quantify the stability of wild-type and modified Aβ and other fibrils.11–14 Here, we exploit high-pressure NMR and investigate the effect of potential structural maturation spanning months on the thermodynamic and kinetic stability of Aβ40 fibrils.
First, the Aβ40 aggregation condition was set up. The 0.4 mg mL−1, i.e. ca. 95 μM, solution of uniformly 15N,13C-labeled recombinant Aβ40 was incubated at 37 °C under mild agitation. The pH was set at 7.4 and the solution contained 25 mM HEPES as buffer and 50 mM NaCl to partially screen out the net negative charges of Aβ40 molecules and accelerate aggregation. The aliquots were taken from the same Aβ40 sample at 6 h, 24 h, 48 h, one week and two months after starting incubation. The EM images demonstrated the presence of typical amyloid fibrils in all the studied samples (Fig. 1a). No significant difference in the morphology or size of the fibrils was observed at these incubation time points (the average diameter of individual fibrils were respectively 12.4 ± 1.6, 13.5 ± 1.6, 13.2 ± 1.9, 13.5 ± 3.6 and 13.9 ± 2.8 nm). In addition, the 1D 1H NMR spectra showed almost complete loss of Aβ40's NMR signals after incubation, confirming the conversion of nearly all Aβ40 monomers to NMR-invisible large aggregates (Fig. 1b). Overall, the EM and NMR data confirmed the predominant presence of Aβ40 fibrils in samples taken at all five time points.
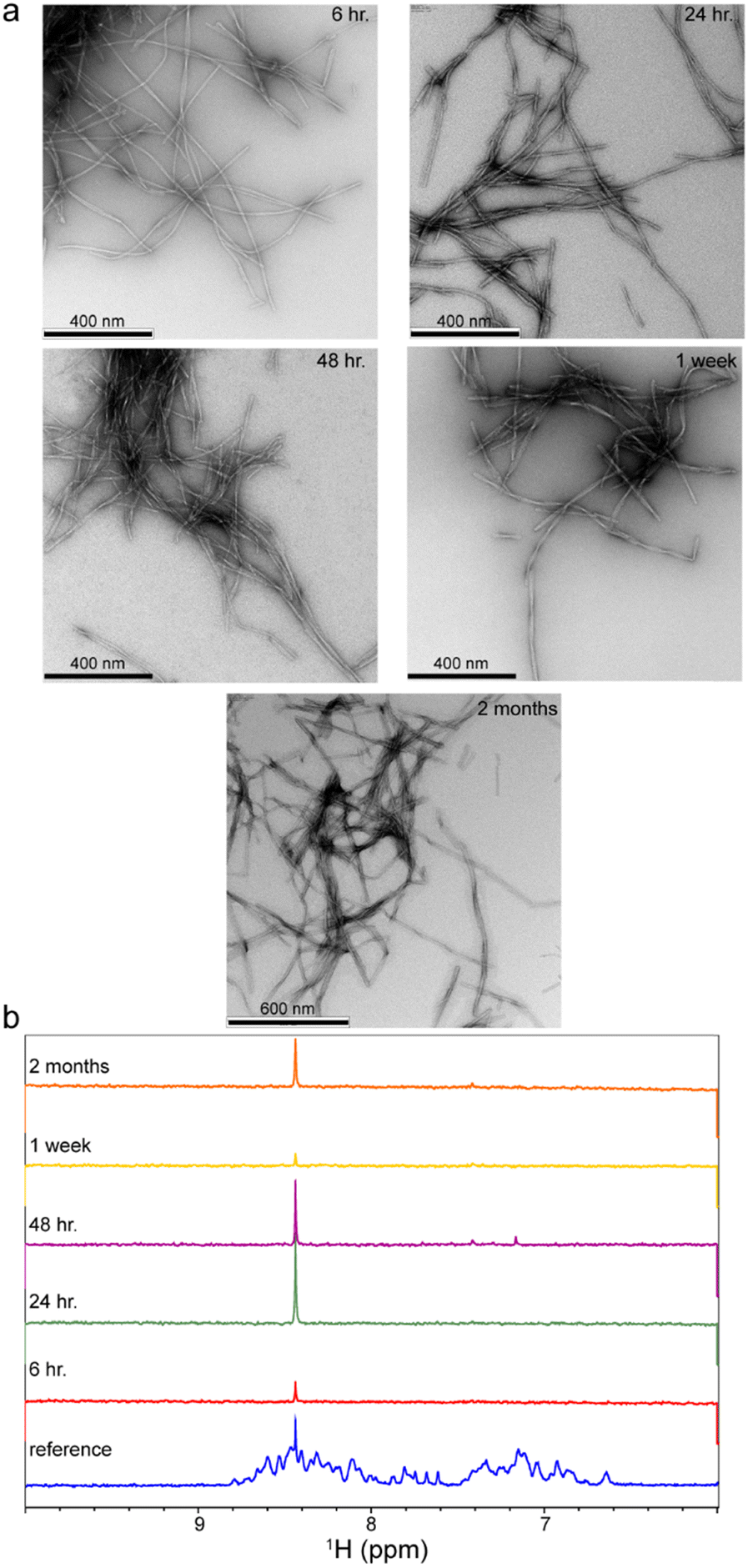 | ||
| Fig. 1 Aggregation of Aβ40 peptide into NMR-invisible fibrils after 6 h, 24 h, 48 h, 1 week and 2 months of incubation in aggregation condition, as revealed by EM images (a) and 1D 1H NMR spectra (b). In (b), the loss of amide and aromatic proton signals indicates nearly complete conversion of monomeric Aβ40 peptide into large aggregates. The remaining sharp signal at ∼8.45 ppm originates from an unknown small molecule existing in the studied Aβ samples. | ||
Amyloid fibrils are often less compact than the core of globular folded proteins and frequently contain internal water-excluded void volumes.15–17 Monomer dissociation from amyloid fibrils can therefore create access to the void volume and lead to filling of this space by the surrounding water molecules and consequently reduce the system volume.10,17,18 In addition, whenever monomer dissociation from amyloid fibrils is accompanied by salt bridge disruption, the electrostriction effect of separated charges on surrounding water can further reduce the system volume.19 The application of high pressure to amyloid fibrils is therefore expected to push the system towards the state with lower volume, i.e. promote monomer dissociation from fibrils.
To address how the stability of Aβ aggregates changes over the course of incubation in the aggregation condition, the fibrillated Aβ40 aliquots taken at the above-mentioned time points were transferred to pressure-resistant ceramic NMR tubes for NMR measurements at 2000 bar (200 MPa). As stated above, the measurement at ambient pressure of 1 bar did not show any significant NMR signal from Aβ40 monomers (Fig. 1b). However, upon a pressure rise to 2000 bar, the real-time 2D 15N,1H HSQC experiments revealed gradual increase in the intensity of the Aβ40 signals (Fig. 2a). The average time-dependent monomer release profiles were indistinguishable for samples aggregated for 6 or 24 hours (hence averaged and termed as “early-aggregate”) and for 48 hours and one week (hence averaged and termed as “late-aggregate”). As shown in Fig. 2b, the monomer release from early-aggregates was much faster than that of late-aggregates, whereas the final level of monomer release was nearly identical between them (around 65% and 62% of the freshly prepared Aβ sample). The characteristic time of monomer release from early-aggregates obtained through global fitting of residue-specific curves was 5.46 ± 0.17 h, while the corresponding value for late-aggregates was 25.29 ± 0.47 h, around 4-5 times longer than that of early-aggregates (see ESI,† Supplementary methods, for details on fitting). The Aβ40 sample aggregated for two months (hence termed “aged-aggregate”) showed slow monomer release with a characteristic time of 23.60 ± 0.56 h, very similar to that of late-aggregates. Strikingly, however, the final level of released monomer was considerably lower (around 40% of the freshly prepared Aβ sample). Overall, the time-dependent monomer release data clearly show that progression from early to late Aβ40 aggregates in the course of hours to days and from late to aged aggregates in the course of weeks to months differentially alters the stability of Aβ40 fibrils.
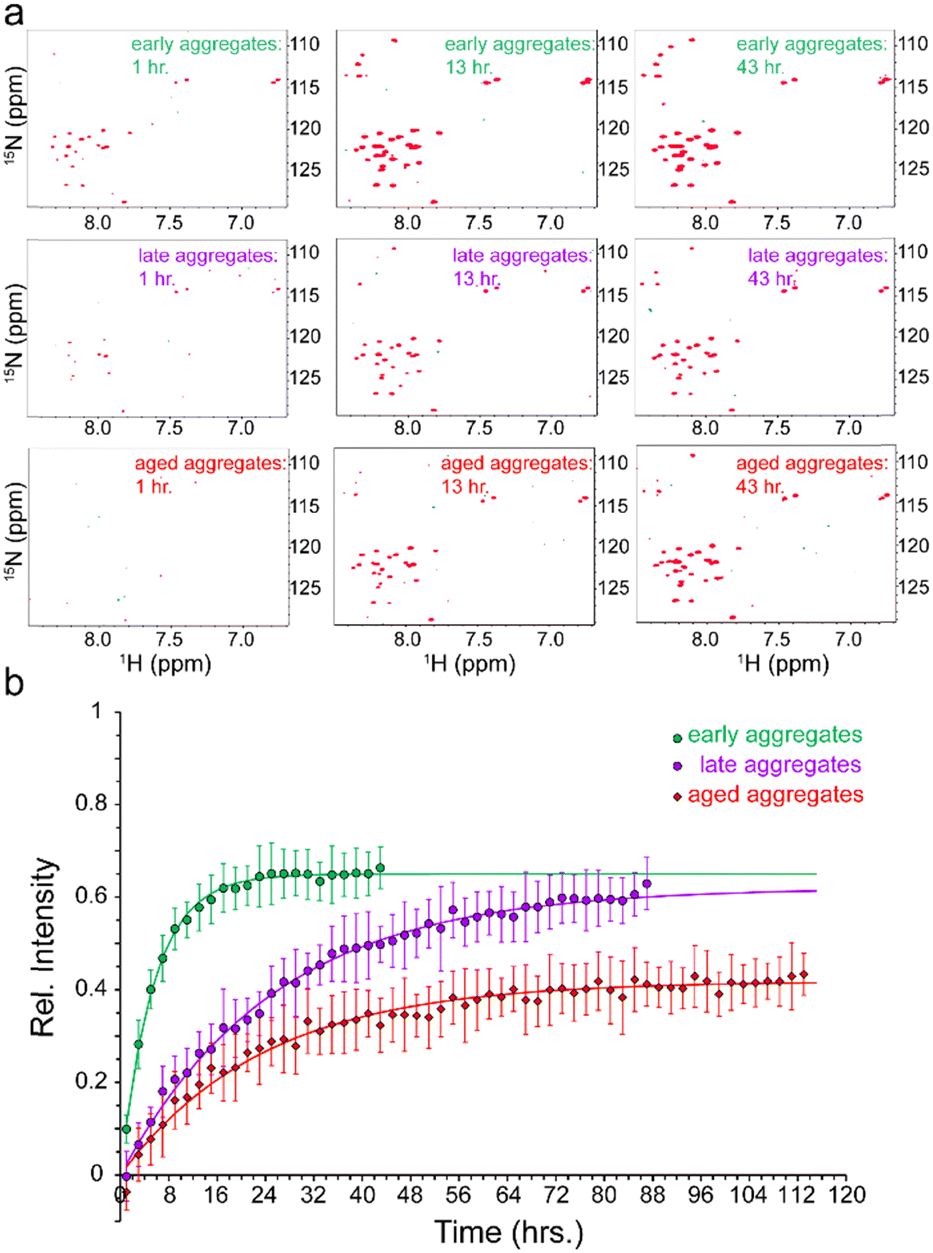 | ||
| Fig. 2 Pressure-induced monomer release from Aβ40 aggregates. (a) 2D 15N,1H HSQC of early- (top row), late- (middle row) and aged-aggregates (bottom row) of Aβ40, measured after ca. 1 h (left column), 12 h (middle column) and 43 h (right column) of incubation at high pressure (2000 bar). A time-dependent increase in peak intensities is observed, indicating pressure-induced monomer dissociation from large NMR-invisible Aβ40 aggregates. (b) Kinetics of pressure-induced monomer release from early- (green), late- (purple) and aged-aggregates (red) of Aβ40, shown as the average (±SD) relative intensity of HSQC peaks in dependence of incubation time at 2000 bar. The solid lines of the same colours represent the respective fitted curves. See text for the definition of early-, late- and aged-aggregates. | ||
The time-dependent monomer release data shown in Fig. 2b represent pressure-induced monomer dissociation from Aβ40 fibrils and their back-association to Aβ40 fibrils. After sufficiently long time, a new thermodynamic equilibrium between monomer and fibrillar states of Aβ40 is established at higher pressure. If we assume a simple two-state model consisting of monomers and fibrils (Fig. 3a), where Aβ40 monomer dissociation and back-association are respectively governed by first-order rate constants koff and kon,app, then Keq = kon,app/koff will represent the thermodynamic stability of Aβ40 fibrils. On the other hand, the kinetic stability of Aβ40 fibrils will be reflected in koff, which is inversely related to the lifetime of Aβ40 fibrils. Through fitting the monomer release data to the two-state model, the Keq values of 0.54, 0.61 and 1.5 were obtained respectively for early-aggregates, late-aggregates and aged-aggregates of Aβ40 (see ESI,† Supplementary methods, for details on fitting). The corresponding koff values were 33.1 ± 1.0 × 10−6, 6.8 ± 0.1 × 10−6 and 4.7 ± 0.1 × 10-6 s−1, respectively (Fig. 3b). The analysed data indicate prominent enhancement of the kinetic, but not thermodynamic, stability of Aβ40 fibrils during progression from early- to late-aggregates. Further progression from late- to aged-aggregates however involves more prominent enhancement in the thermodynamic stability when compared with the kinetic stability.
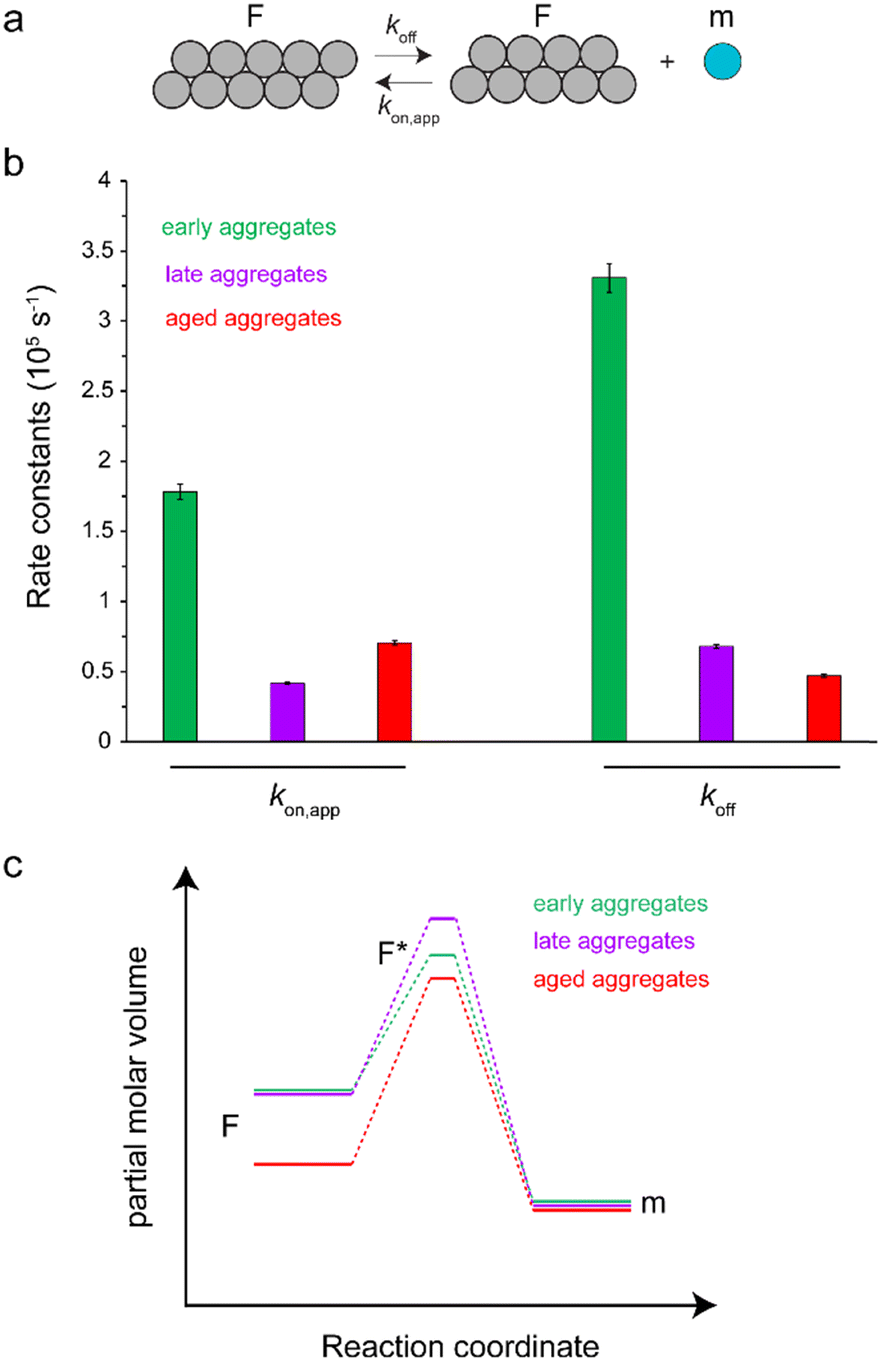 | ||
| Fig. 3 Kinetic analysis of pressure-induced monomer release from Aβ40 aggregates, according to a two-state model composed of a fibrillar (F) and monomeric (m) state as shown in (a). The obtained rate constants kon,app and koff governing the interconversion between F and m states are shown in (b), separately for early-, late- and aged-aggregates of Aβ40. The obtained rate constants suggest the scheme shown in (c) on how the partial molar volumes of F, m and the transition F* states are altered during maturation of Aβ40 aggregates. The maturation from early- to late-aggregates does not change the volume of the F state but increases that of the F* state. Further maturation to aged-aggregates decreases the volume of F* and particularly F states. These volume changes underlie the enhancement of the kinetic and thermodynamic stability of Aβ40 aggregates against high-pressure monomer dissociation, respectively during early and late maturation phases. | ||
Our results show that maturation of Aβ40 fibrils over the course of aggregation involves (at least) two phases: “early maturation” in the course of hours to days during which the kinetic stability of Aβ40 fibrils is enhanced, and “late maturation” in the course of weeks to months during which the thermodynamic stability of Aβ40 fibrils are affected. Thermodynamic stability of Aβ40 fibrils against pressure-induced monomer release is determined by the change in partial molar volume ΔVF→m from the fibrillar (F) to monomeric (m) state, while their kinetic stability is determined by the change in partial molar volume 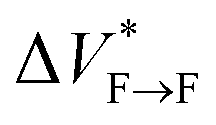 from the fibrillar state to an intermediate transition state (F*) existing on the disaggregation path. Accordingly, during early maturation of Aβ40 fibrils, the positive
from the fibrillar state to an intermediate transition state (F*) existing on the disaggregation path. Accordingly, during early maturation of Aβ40 fibrils, the positive 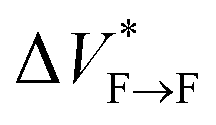 increases, indicating that the partial molar volume of the transition F* state is increased, i.e. it becomes less compact. Conversely, during late maturation, the negative ΔVF→m decreases, indicating that the partial molar volume of fibrils is decreased, i.e. they become more compact (Fig. 3c). It should however be emphasized that the picture of Aβ fibril maturation presented above is qualitative and the few number of data points used in this study does not allow rigorous determination of the exact number and timing of fibril maturation phases.
increases, indicating that the partial molar volume of the transition F* state is increased, i.e. it becomes less compact. Conversely, during late maturation, the negative ΔVF→m decreases, indicating that the partial molar volume of fibrils is decreased, i.e. they become more compact (Fig. 3c). It should however be emphasized that the picture of Aβ fibril maturation presented above is qualitative and the few number of data points used in this study does not allow rigorous determination of the exact number and timing of fibril maturation phases.
Aβ fibrils exhibit a significant degree of morphological and structural heterogeneity, sometimes even within the same Aβ sample.20–22 If different polymorphs of Aβ fibrils have different thermodynamic stability, then the reversible Aβ monomer dissociation from fibrils and its back-association will gradually shift the fibril population distribution towards fibrillar species with higher thermodynamic stability. In addition, slow structural rearrangements within single Aβ fibrils may underlie the time-dependent enhancement of their stability. Our data provide experimental support for maturation-dependent enhancement in thermodynamic stability of Aβ fibrils over the course of weeks to months, accompanied by higher compaction of Aβ fibrils. In addition to a gradual shift of fibril population distribution towards more stable compact species, the loss of water molecules trapped in the core of Aβ fibrils, as demonstrated by previous 2D IR studies,8,23 and its consequent structural adjustments may underlie the maturation-dependent changes in compaction level of Aβ fibrils and their thermodynamic stability. Further support for maturation-dependent changes in the hydration of fibrils can be obtained by direct observation of bound water molecules through solid-state NMR and deuterium-based magnetic relaxation dispersion (MRD) methods.24,25 The maturation-dependent changes in the stability of Aβ fibrils resembles the glass-like aging processes reported for proteins, including globular and intrinsically disordered proteins and protein condensates,26–28 and could in principle reflect the slow release of Aβ fibrils from the local kinetic traps (topological or energetic) existing in their energy landscapes.27
Neurodegeneration-related deposition of Aβ and other proteins as amyloid fibrils often starts years before the onset of the first clinical symptoms.1,2 During this long prodromal phase, biochemical modifications of amyloid deposits may alter their properties and potentially act as a switch in the pathological course of neurodegenerative diseases.6,7 In addition to biochemical maturation, amyloid fibrils may undergo structural maturation similar to what is demonstrated here in in vitro conditions.9 The resultant alteration in the kinetic and thermodynamic stability of amyloid fibrils has neuropathological importance, as it can influence the spreading of amyloid pathology in the brain. It should, however, be noted that the temporal dynamics of AD is a complex process and the structural (and biochemical) maturation of Aβ fibrils constitutes only one aspect of this multifaceted in vivo process. Furthermore, our data suggest the potential importance of considering the structural difference between fresh and mature Aβ fibrils in structure-based development of molecular imaging probes in AD and other neurodegenerative diseases.29
To conclude, we investigated how the maturation of Aβ40 fibrils affects their stability against pressure-induced dissociation. Through real-time high-pressure NMR experiments, it was shown that there are roughly two phases of Aβ fibril maturation, an early phase within the course of hours to days during which the kinetic stability of Aβ fibrils is enhanced, and a late phase within the course of weeks to months during which the thermodynamic stability of Aβ fibrils is enhanced. The higher stability of mature Aβ fibrils indicates their higher degree of compaction. Overall, our data demonstrate the structural and stability changes of Aβ fibrils during maturation and highlights the potential importance of Aβ fibril maturation in the pathological course of Alzheimer's disease.
Author contributions
Conceptualization, methodology, investigation, funding acquisition, project administration, supervision, writing – original draft: NRG; visualization, formal analysis: NRG, DS; resources, writing – review & editing: SB, KG, DS, NRG.Conflicts of interest
There are no conflicts of any sort to declare.Acknowledgements
N. R.-G. acknowledges the Deutsche Forschungsgemeinschaft (German Research Foundation, DFG) for research grants RE 3655/2-1 and 3655/2-3. We thank Dr Dietmar Riedel and Gudrun Heim for electron microscopy imaging.Notes and references
- A. A. Tahami Monfared, M. J. Byrnes, L. A. White and Q. Zhang, Neurol. Ther., 2022, 11, 553–569 CrossRef PubMed.
- M. A. DeTure and D. W. Dickson, Mol. Neurodegener., 2019, 14, 32 CrossRef PubMed.
- R. Morales, K. Callegari and C. Soto, Virus Res., 2015, 207, 106–112 CrossRef CAS PubMed.
- M. Jucker and L. C. Walker, Nature, 2013, 501, 45–51 CrossRef CAS PubMed.
- D. R. Thal, U. Rub, M. Orantes and H. Braak, Neurology, 2002, 58, 1791–1800 CrossRef PubMed.
- A. Rijal Upadhaya, I. Kosterin, S. Kumar, C. A. von Arnim, H. Yamaguchi, M. Fandrich, J. Walter and D. R. Thal, Brain, 2014, 137, 887–903 CrossRef PubMed.
- X. H. Li, S. Ospitalieri, T. Robberechts, L. Hofmann, C. Schmid, A. R. Upadhaya, M. J. Koper, C. A. F. von Arnim, S. Kumar, M. Willem, K. Gnoth, M. Ramakers, J. Schymkowitz, F. Rousseau, J. Walter, A. Ronisz, K. Balakrishnan and D. R. Thal, Brain, 2022, 145, 3558–3570 CrossRef PubMed.
- J. Q. Ma, H. Komatsu, Y. S. Kim, L. Liu, R. M. Hochstrasser and P. H. Axelsen, ACS Chem. Neurosci., 2013, 4, 1236–1243 CrossRef CAS PubMed.
- S. Nystrom, K. M. Psonka-Antonczyk, P. G. Ellingsen, L. B. G. Johansson, N. Reitan, S. Handrick, S. Prokop, F. L. Heppner, B. M. Wegenast-Braun, M. Jucker, M. Lindgren, B. T. Stokke, P. Hammarstrom and K. P. R. Nilsson, ACS Chem. Biol., 2013, 8, 1128–1133 CrossRef PubMed.
- J. A. Caro and A. J. Wand, Methods, 2018, 148, 67–80 CrossRef CAS PubMed.
- N. Rezaei-Ghaleh, M. Amininasab, S. Kumar, J. Walter and M. Zweckstetter, Nat. Commun., 2016, 7, 11359 CrossRef CAS.
- G. A. de Oliveira, M. A. Marques, C. Cruzeiro-Silva, Y. Cordeiro, C. Schuabb, A. H. Moraes, R. Winter, H. Oschkinat, D. Foguel, M. S. Freitas and J. L. Silva, Sci. Rep., 2016, 6, 37990 CrossRef CAS PubMed.
- F. Piccirilli, N. Plotegher, M. G. Ortore, I. Tessari, M. Brucale, F. Spinozzi, M. Beltramini, P. Mariani, V. Militello, S. Lupi, A. Perucchi and L. Bubacco, Biophys. J., 2017, 113, 1685–1696 CrossRef CAS.
- N. Rezaei-Ghaleh, M. Amininasab, K. Giller and S. Becker, J. Phys. Chem. Lett., 2023, 14, 1427–1435 CrossRef CAS PubMed.
- F. Meersman and C. M. Dobson, Biochim. Biophys. Acta, 2006, 1764, 452–460 CrossRef CAS.
- K. Akasaka, A. R. Latif, A. Nakamura, K. Matsuo, H. Tachibana and K. Gekko, Biochemistry, 2007, 46, 10444–10450 CrossRef CAS.
- R. Mishra and R. Winter, Angew. Chem., Int. Ed., 2008, 47, 6518–6521 CrossRef CAS PubMed.
- N. V. Nucci, B. Fuglestad, E. A. Athanasoula and A. J. Wand, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 13846–13851 CrossRef CAS PubMed.
- I. Danielewicz-Ferchmin, E. Banachowicz and A. R. Ferchmin, Biophys. Chem., 2003, 106, 147–153 CrossRef CAS.
- W. Close, M. Neumann, A. Schmidt, M. Hora, K. Annamalai, M. Schmidt, B. Reif, V. Schmidt, N. Grigorieff and M. Fandrich, Nat. Commun., 2018, 9, 699 CrossRef PubMed.
- J. Meinhardt, C. Sachse, P. Hortschansky, N. Grigorieff and M. Fandrich, J. Mol. Biol., 2009, 386, 869–877 CrossRef CAS.
- A. T. Petkova, R. D. Leapman, Z. Guo, W. M. Yau, M. P. Mattson and R. Tycko, Science, 2005, 307, 262–265 CrossRef CAS PubMed.
- Y. S. Kim, L. Liu, P. H. Axelsen and R. M. Hochstrasser, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 17751–17756 CrossRef CAS PubMed.
- K. T. Movellan, R. Dervisoglu, S. Becker and L. B. Andreas, Angew. Chem., Int. Ed., 2021, 60, 24075–24079 CrossRef CAS PubMed.
- E. Persson and B. Halle, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 6266–6271 CrossRef CAS PubMed.
- L. Herenyi, K. Szigeti, J. Fidy, T. Temesvari, J. Schlichter and J. Friedrich, Eur. Biophys. J., 2004, 33, 68–75 CrossRef CAS.
- I. L. Morgan, R. Avinery, G. Rahamim, R. Beck and O. A. Saleh, Phys. Rev. Lett., 2020, 125, 058001 CrossRef CAS.
- L. Jawerth, E. Fischer-Friedrich, S. Saha, J. Wang, T. Franzmann, X. Zhang, J. Sachweh, M. Ruer, M. Ijavi, S. Saha, J. Mahamid, A. A. Hyman and F. Julicher, Science, 2020, 370, 1317–1323 CrossRef CAS.
- M. Fandrich, S. Nystrom, K. P. R. Nilsson, A. Bockmann, H. LeVine, 3rd and P. Hammarstrom, J. Intern. Med., 2018, 283, 218–237 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary methods. See DOI: https://doi.org/10.1039/d3cp01276j |
| This journal is © the Owner Societies 2023 |