
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fluorine-free organic electrolytes for the stable electrodeposition of neodymium metal†
Pieter
Geysens
 a,
Da
Tie
b,
Alexandru
Vlad
b,
Jan
Fransaer
c and
Koen
Binnemans
*a
a,
Da
Tie
b,
Alexandru
Vlad
b,
Jan
Fransaer
c and
Koen
Binnemans
*a
aKU Leuven, Department of Chemistry, Celestijnenlaan 200F, P.O. box 2404, B-3001 Leuven, Belgium. E-mail: Koen.Binnemans@kuleuven.be
bUC Louvain, Institute of Condensed Matter and Nanosciences, Place L. Pasteur 1, Lavoisier Building, B-1348 Louvain-la-Neuve, Belgium
cKU Leuven, Department of Materials Engineering, Kasteelpark 44, P.O. box 2450, B-3001 Leuven, Belgium
First published on 26th July 2023
Abstract
Electrowinning is regarded as a clean process to recover neodymium metal from secondary sources such as spent Nd–Fe–B permanent magnets, but the current methods are severely limited by a high energy consumption (molten salts), or by the high costs and environmental impact of the electrolyte components (ionic liquids). Therefore, there is a demand for more sustainable electrowinning methods for the recovery of neodymium metal. Inspired by our own previous work and the work of others, we developed new fluorine-free organic electrolytes that enable the electrodeposition of neodymium metal at room temperature. The electrolytes consist of solvated neodymium borohydride, Nd(BH4)3, dissolved in the ether solvents tetrahydrofuran (THF), 2-methyltetrahydrofuran (2-MeTHF), 1,2-dimethoxyethane (DME) and diethylene glycol dimethyl ether (diglyme, G2), and these complexes can be prepared entirely from non-fluorinated precursors such as neodymium(III) chloride (NdCl3) and sodium borohydride (NaBH4). In contrast to our previous bis(trifluoromethylsulfonyl)imide-containing electrolytes, electrodeposition of neodymium proceeds over time without significant loss of current density, indicating a higher stability against unwanted side-reactions that lead to passivation of the deposit on the electrode. Characterization of the deposits by scanning electron microscopy (SEM), energy-dispersive X-ray fluorescence (EDX), and X-ray photoelectron spectroscopy (XPS) unambiguously indicated the presence of neodymium metal.
Introduction
Neodymium is economically the most important rare-earth element (REE) because it is one of the main components in Nd–Fe–B permanent magnets, which are used extensively in household electric appliances, hard disk drives, electric motors and generators.1 In order to meet the goals of the green energy transition, the demand of Nd–Fe–B magnets for (hybrid) electric vehicles and wind turbines is expected to increase sharply in the near future.2 Therefore, the development of sustainable methods for the recovery of neodymium metal from secondary sources such as end-of-life products is urgently needed.3,4 Electrowinning is widely regarded as a reliable, low-cost and clean method for the recovery of metals because an electric current is used to reduce the desired metal cations from solution, as opposed to a chemical reducing agent which generates additional waste.5 This process is also used for REEs, including neodymium, but additional challenges regarding the choice of electrolyte must be addressed. Due to the high chemical reactivity and very negative standard reduction potentials of these elements (ranging between −2.3 to −2.4 V vs. SHE), aqueous electrolytes are not suitable due to the hydrogen evolution reaction, and research has been mainly focussed on nonaqueous electrolytes such as molten salts and ionic liquids. In the industry, molten salt electrolysis is currently the most attractive recovery method because the electrolyte salts (e.g. LiCl–KCl eutectic) are inexpensive and high deposition current densities can be achieved.6–8 However, high temperatures (>500 °C) are required to operate these electrowinning baths, and the molten salts are corrosive to the infrastructure. This imposes a high energy requirement and important considerations on the maintenance and safety of the process. Therefore, there is a high demand for alternative electrolytes that are operable at moderate temperatures (≤150 °C) while still allowing high deposition current densities. Currently, ionic liquids (ILs), which are low-melting organic molten salts (mp ≤ 100 °C) and are non-volatile, are being investigated to fulfill this role.9–11 For electrowinning of neodymium metal, research has been focused mainly on ILs with phosphonium and ammonium cations and the bis(trifluoromethylsulfonyl)imide (bistriflimide, TFSI−, Tf2N−) anion because they have many desirable properties for electrodeposition, i.e. a very negative cathodic decomposition limit, good thermal stability, hydrophobicity, a relatively low viscosity, and a high conductivity.12–15 While these reports demonstrate the proof-of-concept for the achievability of low-temperature electrowinning of neodymium and other REEs, upscaling of these processes is not sustainable due to the fluorinated nature of the IL anions. Numerous studies have shown that strongly fluorinated organic compounds are very poorly (bio)degradable, and have a high degree of (eco)toxicity.16–18 Furthermore, the fluorinated bistriflimide anion is expensive because of its multi-step synthesis procedure and its niche application fields.19 Hence, its use should be avoided or at least minimized, especially for large-scale applications such as electrowinning.Recently, research on electrodeposition of REEs from ILs has shifted to more cost-effective and non-fluorinated ILs, in particular N-butyl-N-methylpyrrolidinium dicyanamide ([BMPyrr][DCA]).20–22 In an earlier report, it was found that electrolytes consisting of Ln(Tf2N)3 (Ln = La, Nd, Sm) in [BMPyrr][DCA] were not suitable for electrodeposition because the cathodic decomposition of the IL occurred simultaneously with the electrodeposition of metal.20 However, more recently it was discovered that using 0.5 mol kg−1 of the hydrated salt Nd(NO3)3·6H2O as the source of Nd3+ cations in this IL, resulted in a significant positive shift of the deposition onset potential (−1.75 V vs. Fc+/Fc) and an increase in the deposition current density (−40 mA cm−2).21 The obtained deposits consisted of a mixture of metallic neodymium and Nd2O3. The presence of water in the coordination sphere around Nd3+ was proven to be essential to achieve neodymium electrodeposition.21,22 However, while water might improve the thermodynamics and kinetics of the deposition process and represent a “more realistic” (i.e. not rigorously dried) environment, these wet electrolytes also provide an inherently unstable environment for the deposits, leading to contamination of the metallic REEs with oxides and hydroxides, and a decreased current efficiency.23
Organic electrolytes are composed of salts which are dissolved in an organic solvent and they could be a less expensive and sustainable alternative to ILs, provided that they are sufficiently electrochemically stable to support REE electrodeposition. This category of electrolytes has been much less explored than ILs. “Neutral-ligand” ionic liquids (NLILs), composed of RE(Tf2N)3 dissolved in neutral ligands (L) such as trimethyl phosphate or dihexyl octanamide, resulting in a cationic complex [RE(L)3][Tf2N]3, were briefly investigated by Bagri et al. and Krishna et al.24,25 However, the authors did not characterize the obtained deposits in detail. Similarly, Zhang and co-workers have recently proposed two different systems, NdCl3-AlCl3 and Nd(SO3CF3)3-LiNO3 dissolved in the green aprotic organic solvent 1,3-dimethyl-2-imidazolidinone, for the electrodeposition of neodymium at ambient temperature.26,27 Evidence for the presence of metallic neodymium in the deposits was provided by X-ray photoelectron spectroscopy (XPS) and X-ray diffraction (XRD) analysis.
In our previous work, we have reported the successful electrodeposition of neodymium- and dysprosium-containing layers from organic electrolytes consisting of Ln(Tf2N)3 (Ln = Nd, Dy) and a commercially available borohydride salt, dissolved in 1,2-dimethoxyethane (DME, G1).28 Due to the presence of strongly electron-withdrawing CF3SO2-functionalities, the bistriflimide anion is prone to reductive side-reactions, leading to the formation of passivating surface films.29–31 This was evidenced by a fast decrease in deposition current density during chronoamperometry (CA) experiments, and by the presence of fluorine- and sulfur- containing species in the EDX spectra of the deposits. Therefore, in order to achieve electrodeposition of pure metallic REE at a constant and high current density, it is necessary to avoid the use of this anion completely.
In this Paper, we describe a new strategy to prepare organic electrolytes for neodymium electrodeposition at ambient temperatures, without relying on fluorinated salts. Commercially available neodymium(III) chloride and sodium, lithium and tetrabutylammonium borohydride are used as the precursors. Alternatively, neodymium(III) methanesulfonate, which is conveniently synthesized from Nd2O3 and methanesulfonic acid can also be used as a starting salt. Moreover, because the electrolytes do not contain any reactive impurities, electrodeposition proceeds at a constant current density, as no passivation occurs.
Experimental
Products
Sodium borohydride (NaBH4, 99%, powder), lithium borohydride (LiBH4, 95%), tetrabutylammonium borohydride (TBABH4, 98%), 1,2-dimethoxyethane (ethylene glycol dimethyl ether, DME, 99+%, extra dry over molecular sieves), bis(2-methoxyethyl) ether (diethylene glycol dimethyl ether, diglyme, 99+%, extra dry over molecular sieves), tetrahydrofuran (THF, 99.5%, extra dry over molecular sieves), 2-methyltetrahydrofuran (2-MeTHF, 99+%, extra dry over molecular sieves), ferrocene (Fc, 98%), and neodymium(III) chloride (NdCl3, 99.9% trace metal base, anhydrous) were purchased from Acros Organics (Geel, Belgium). Ferrocenium hexafluorophosphate (FcPF6, 97%) was purchased from Sigma Aldrich (Diegem, Belgium). Hydrogen bis(trifluoromethylsulfonyl)imide (HTf2N, 98%, 80 wt% aqueous solution), lithium bis(trifluoromethylsulfonyl)imide (LiTf2N, 99%), and 1-butyl-1-methylpyrrolidinium bis(trifluoromethylsulfonyl)imide ([BMP][Tf2N], 99%) were purchased from IoLiTec (Heilbronn, Germany). Methanesulfonic acid (HOMs, ≥99.5%, for synthesis) was purchased from Carl Roth (Karlsruhe, Germany). Neodymium(III) oxide (Nd2O3, 99.99%) was purchased from Rare Earth Products ltd. (Beverly, USA). Neodymium(III) bis(trifluoromethylsulfonyl)imide (Nd(Tf2N)3),32 neodymium(III) methanesulfonate (Nd(OMs)3),33 sodium bis(trifluoromethylsulfonyl)imide (NaTf2N)34 were synthesized according to previously reported procedures.All chemicals were used as received without any further purification, with the exception of the bis(trifluoromethylsulfonyl)imide and methanesulfonate salts, BMPTf2N, and neodymium(III) borohydride complexes which were dried prior to use on a Schlenk line for 12 hours at 220 °C, 120 °C, and ambient temperature, respectively. All the products used in the electrolytes were stored and manipulated inside an argon-filled glovebox with water and oxygen concentrations below 1 ppm.
Characterization
Powder XRD diffractograms were recorded on a Bruker D2 phaser in a 2θ range of 5°–85°, using a Cu Kα X-ray source. UV-Vis absorption spectra were recorded on a Varian Cary 6000i UV-VIS-NIR spectrophotometer at a scan rate of 10 nm s−1 with 1 nm intervals. The instrument was used in double-beam mode. Samples were measured in quartz glass cuvettes with a path length of 5 mm. Blanks consisted of the same solvent used in the samples, and were measured in a matched pair of cuvettes. Scanning electron microscopy (SEM) was performed on an XL30 FEG scanning electron microscope. Electrodepositions were made on platinum-coated silicon wafers and were thoroughly rinsed with anhydrous THF, and were allowed to dry at ambient glovebox conditions before they were attached to the SEM sample holder with conductive carbon tape. Energy dispersive X-ray fluorescence (EDX) analysis was performed in the same setup, using an octane elite super silicon drift detector (Ametek EDAX), and using TEAM software. Scans were recorded over a total time of 200 s to achieve a sufficient signal-to-noise ratio. The acceleration voltage was 20 kV or 30 kV for both imaging and EDX. During the sample preparation, exposure of the deposits to air was unavoidable, causing oxidation. Electrodepositions for XPS analysis were made and treated identically to the SEM samples, but were transported in air-tight vials to another glovebox, where they were transferred to a vessel which was directly attached to the ultra-high-vacuum system to avoid exposure to air. The XPS measurement and etching was conducted by a PHI5000 Versaprobe-II, UlVAC-PHI with an Al target working at 1486.8 kV. The surface layer of the deposits was etched with Ar+ beams. The etching area was 2 mm × 2 mm and the etching time was 30 s for a total of 10 times. The XPS measurement was conducted during the interval between two etchings. The XPS spectra were calibrated by shifting the low binding energy component of the C 1s peak to 284.8 eV. The peak fitting and quantitative analysis of the spectra was performed with CasaXPS software. The spectra were fitted with Doniach–Sunjic functions convoluted with a Gaussian/Lorentzian distribution and a Shirley background.Electrochemical experiments
All the electrochemical measurements were performed inside an argon-filled glovebox with water and oxygen concentrations below 1 ppm, at ambient temperature (28 °C). The electrolyte solutions were prepared by mixing the appropriate amounts of products and solvents in 20 mL glass vials and stirring for 5 minutes at ambient temperature. In many cases, precipitation was observed in the electrolyte solutions, which was allowed to settle to the bottom of the vessel before proceeding with the measurement. All the electrochemical measurements were performed in three-electrode setup, using an Autolab PGSTAT302N potentiostat and Nova 2.1 software. The working electrodes for cyclic voltammetry (CV) were pieces of silicon wafer, coated with 500 nm of silica, 10 nm of titanium and 100 nm of platinum (Imec, Belgium) (surface area of 0.3 cm2), the counter electrode a larger piece of platinum-coated silicon wafer (surface area of approx. 1.0 cm2) and the scan rate was 10 mV s−1, unless stated otherwise. The reference electrode consisted of a platinum wire, submerged in a mixture of ferrocene (0.005 mol L−1) and ferrocenium hexafluorophosphate (0.005 mol L−1) dissolved in the ionic liquid 1-butyl-1-methylpyrrolidinium bis(trifluoromethylsulfonyl)imide, contained inside a fritted glass tube (referred to as Fc+/Fc). The CVs were started at open circuit potential (OCP) and are uncorrected for Ohmic drop. Electrodepositions were performed at constant potential, using pieces of platinum-coated silicon wafers (surface area of approx. 0.3 cm2) as substrates.Synthesis of salts/complexes
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1301, 1199 (νSO), 1150, 1123, 1055, 974, 797, 782, 553, 534, 517, 506, 409.
O), 1301, 1199 (νSO), 1150, 1123, 1055, 974, 797, 782, 553, 534, 517, 506, 409.
Results and discussion
Synthesis and characterization of complexes
In our previous work, an ether-soluble neodymium(III) salt, Nd(Tf2N)3, was combined with a borohydride salt, NaBH4, LiBH4, or TBABH4 to give a soluble anionic neodymium borohydride complex, according to eqn (1):28| Nd(Tf2N)3 + 4Q(BH4) → Q[Nd(BH4)4] + 3Q(Tf2N) | (1) |
| NdCl3 + 3NaBH4 + 3THF → Nd(BH4)3 (THF)3 + 3NaCl | (2) |
| NdCl3 + 4NaBH4 + 4DME → Na[Nd(BH4)4](DME)4 + 3NaCl | (3) |
After removal of NaCl by filtration, the solvated neodymium(III) complex can be isolated by crystallization (cooling) or solvent removal. These reactions afford a convenient strategy to isolate the redox-active complexes in a pure form without relying on a bistriflimide salt. We also found that anhydrous neodymium(III) methanesulfonate (Nd(OMs)3) can be used as a substitute for the chloride in reaction (3). This salt is prepared conveniently via the reaction of neodymium(III) oxide with environmentally benign methanesulfonic acid (MSA, HOMs) (eqn 4), followed by a thermal dehydration reaction (eqn 5).33
 | (4) |
 | (5) |
The neutral complex Nd(BH4)3(THF)3 (eqn (2)) can be used as a precursor for the synthesis of anionic complexes with different counter cations by the addition of one equivalent of a borohydride salt (eqn (6)).
| Nd(BH4)3 (THF)3 + Q(BH4) + (x − 3)THF → Q[Nd(BH4)4](THF)x | (6) |
 | ||
| Fig. 1 UV-Vis absorption spectra of 0.1 mol L−1 Nd(Tf2N)3 + 4 molar equivalents of TBABH4 in THF, 0.1 mol L−1 isolated Na[Nd(BH4)4](DME)4 in THF, 0.1 mol L−1 isolated Nd(BH4)3(THF)3 in THF, and 0.1 mol L−1 isolated Nd(BH4)3(THF)3 + 1 molar equivalent of LiBH4, NaBH4 and TBABH4 in THF. | ||
The most intense transition in the absorption spectra is the 4G5/2 ← 4I9/2 transition with an absorption maximum at about 586 nm. This is a so-called “hypersensitive” transition, which means that its intensity and crystal-field fine structure are very dependent on the ligand environment around the Nd3+ ion, not only on the nature of the atoms in the first coordination sphere, but also on the symmetry of the coordination polyhedron.40 Since this transition reflects small changes in the coordination sphere, it can be used as a “spectroscopic probe” to monitor complex formation upon addition of a ligand. Alternatively, if two neodymium(III) complexes have a 4G5/2 ← 4I9/2 transition with the same intensity and crystal-field fine structure, one can conclude that the local environment of Nd3+ in the 2 complexes is very similar. When Nd(Tf2N)3 is combined with 4 molar equivalents of TBABH4 in THF, the violet-colored anionic complex [Nd(BH4)4]− is formed, which gives a very characteristic hypersensitive transition with a maximum at 586 nm.28 The spectrum is very similar to those of the isolated Na[Nd(BH4)4](DME)4 in THF, and the spectra of isolated Nd(BH4)3(THF)3 + 1 molar equivalent of LiBH4, NaBH4 and TBABH4 in THF. This indicates that the same anionic complex is present in these solutions. Only in the spectrum of neat Nd(BH4)3(THF)3 in THF, the 4G5/2 ← 4I9/2 transition has a different intensity and fine structure, indicating that the speciation of the neutral complex is indeed different from that of the anionic complex. As expected, the other absorption bands are very similar for all the complexes, and this can be expected for the non-hypersensitive transitions of Nd3+.
Cyclic voltammetry
An often overlooked property of electrolytes for the electrodeposition of reactive metals is how the current density of electrodeposition evolves over multiple scans in a cyclic voltammogram (CV). Due to the reactivity of the deposits towards electrolyte components, non-conductive surface films can be formed which passivate the electrode, resulting in a significantly decreased current density over subsequent scans. Fig. 2 shows the CVs (three subsequent scans) of a Nd(Tf2N)3 + NaBH4-containing electrolyte and an electrolyte based on the pure isolated Na[Nd(BH4)4](DME)4 complex. | ||
| Fig. 2 Cyclic voltammograms (three scans) of (a) 0.1 mol L−1 Nd(Tf2N)3 + 4 molar equivalents of NaBH4 in THF, and (b) 0.1 mol L−1 Na[Nd(BH4)4](DME)4 in THF, recorded at ambient temperature at a scan rate of 10 mV s−1. The scan direction is indicated with arrows. The working and counter electrodes were pieces of platinum-coated silicon wafers with a surface area of 0.3 cm2 and 1.0 cm2, respectively. The reference electrode was Fc+/Fc (0.005 mol L−1 each) dissolved in [BMP][Tf2N]. | ||
In the first scan of the CV recorded in the bistriflimide-containing electrolyte, a high current density cathodic wave was observed with an onset potential around −2.8 V vs. Fc+/Fc. In the backwards scan, nucleation hysteresis was observed indicating an electrodeposition process, but a stripping peak was absent. After the experiment, a black deposit was observed on the working electrode. This cathodic wave corresponds to the electrodeposition of neodymium, because the deposition of sodium metal is only observed at a lower potential of −3.2 V vs. Fc+/Fc (Fig. S2, ESI†).28 In the second and third scan, the electrodeposition wave is shifted to significantly more negative potentials and the current density is significantly lower. On the other hand, for the bistriflimide-free electrolyte, the electrodeposition onset potential was also observed around −2.8 V vs. Fc+/Fc, but shifted to less negative potentials in subsequent cycles and the current density is not decreased. In the backwards scan, a distinct stripping peak was observed but the electrodeposition process is clearly not completely reversible, as residual deposits were also observed post-experiment on the working electrode in this case. This stripping peak definitely corresponds to the electrodeposition wave, as its size scales with the cathodic vertex potential in a broad range (Fig. S3, ESI†). The shift of the electrodeposition onset to less negative potentials is probably also caused by the incomplete stripping, as in the second and third cycle the electrodeposition occurred on the residual deposit from the first cycle, as opposed to a clean platinum substrate. These results indicate that passivation of the deposit occurs in the bistriflimide-containing electrolyte, whereas it occurs to at least a much lesser degree in the bistriflimide-free electrolyte. This is also supported by the scan rate-dependent behavior of the current density in both electrolytes (Fig. 3).
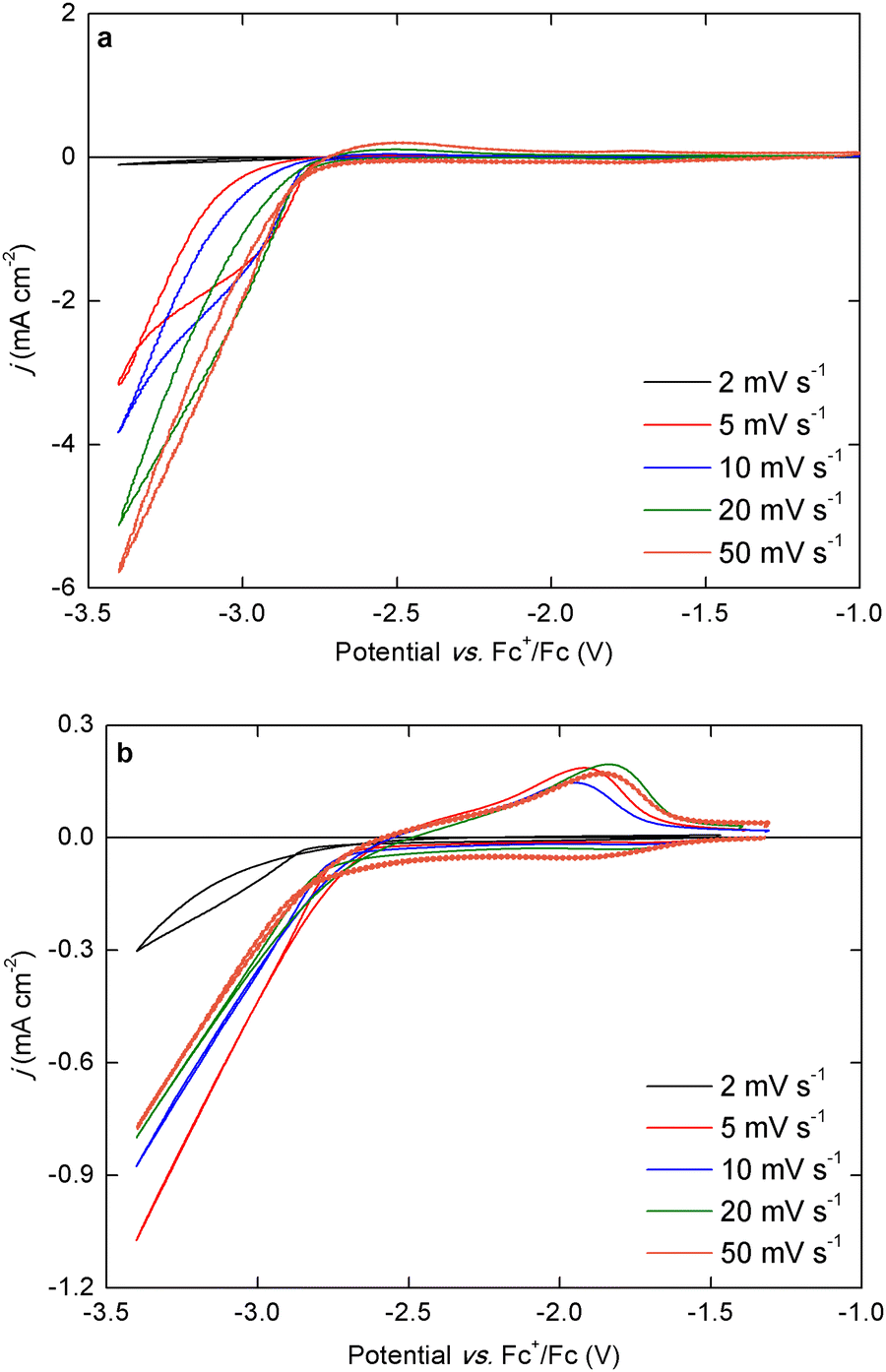 | ||
| Fig. 3 Cyclic voltammograms (first scan) of (a) 0.1 mol L−1 Nd(Tf2N)3 + 4 molar equivalents of NaBH4 in THF, and (b) 0.1 mol L−1 Na[Nd(BH4)4](DME)4 in THF, recorded at ambient temperature at varying scan rate. The working and counter electrodes were pieces of platinum-coated silicon wafers with a surface area of 0.3 cm2 and 1.0 cm2, respectively. The reference electrode was Fc+/Fc (0.005 mol L−1 each) dissolved in [BMP][Tf2N]. | ||
For the bistriflimide-containing electrolyte, the electrodeposition current density was relatively high (approx. −6.0 mA cm−2) at 50 mV s−1, but a negative trend was observed as the scan rate was decreased, and the current density almost reached a limiting value at 5 mV s−1. At the lowest scan rate (2 mV s−1), only a very small current could flow in the electrolyte as the deposited metal was fully passivated before the negative vertex potential was reached. For the bistriflimide-free electrolyte, the current density was initially lower because the total concentration of charge carriers was lower than for the bistriflimide-containing electrolyte. However, from 50 mV s−1 to 5 mV s−1, there was no observed negative trend for the current density. Only at 2 mV s−1 the current density was lower, but still significantly higher than for the analogue CV in the bistriflimide-containing electrolyte, indicating a smaller degree of passivation. For the 50 mV s−1 CV, a small pre-wave is observed starting around −1.9 V vs. Fc+/Fc. It is not entirely clear what the origin of this feature is and possibly, it's related to adsorption processes of the borohydride anion on the working electrode surface. The electrodeposition of neodymium from the isolated Na[Nd(BH4)4](DME)4 complex was also studied in other ether solvents, such as 2-methyltetrahydrofuran (2-MeTHF), 1,2-dimethoxyethane (DME), and diglyme (G2). In each solvent, the overall electrodeposition behavior was very similar to that observed in THF, with an onset at approx. −2.8 V vs. Fc+/Fc in the first cycle, which shifted to less negative potentials in the later cycles (Fig. S4, ESI†). However, the stripping behavior was notably different. Compared to the CV recorded in THF, the stripping peak in the 2-MeTHF electrolyte was larger and broader, indicating a more complete stripping of the deposit. On the other hand, in the DME and G2 electrolytes, the stripping peak was smaller and seemed to consist of two successive separate broad peaks. Because Na+ cations are also present in these electrolytes, and the cathodic vertex potential is sufficiently negative (−3.4 V vs. Fc+/Fc), it is possible that co-deposition of sodium metal occurred. The peak at more negative potentials probably corresponds to the stripping of sodium while the second peak corresponds to the stripping of neodymium.
Neodymium electrodeposition was also investigated in electrolytes consisting of the neutral complex Nd(BH4)3(THF)3 and one molar equivalent of LiBH4 or TBABH4 (Fig. S5, ESI†). This allowed for the introduction of cations other than Na+ in the electrolytes, thereby avoiding possible co-deposition of sodium. Similarly to the Na[Nd(BH4)4](DME)4-containing electrolytes, the onset for neodymium electrodeposition in the first scan is observed at approx. −2.8 V vs. Fc+/Fc for the LiBH4 electrolyte, whereas it is observed at a significantly more negative −3.1 V vs. Fc+/Fc for the TBABH4 electrolyte. In the second and third scan, the onset is shifted to less negative potentials for both electrolytes. Interestingly, the electrodeposition wave is also observed at −2.8 V vs. Fc+/Fc for an electrolyte consisting of neat Nd(BH4)3(THF)3 dissolved in THF but the current density was lower due to the neutral character of this complex, resulting in a lower conductivity (Fig. S6, ESI†). However, for both the neat Nd(BH4)3(THF)3 electrolyte and the one with added TBABH4, a distinct stripping peak is observed. Because these electrolytes do not contain any Na+ or Li+ cations, this is evidence that this anodic process corresponds to the stripping of deposited neodymium, and not just stripping of co-deposited sodium or lithium metal. The occurrence of neodymium stripping peaks in the CVs is in contrast with the findings in our previous work, where the electrodeposition of neodymium was claimed to be completely irreversible.28 However, in those bistriflimide-containing electrolytes, it is possible that the formation of passivating surface layers inhibits the stripping of the deposits, while in these bistriflimide-free electrolytes, passivation is much less problematic.
An additional advantage of the LiBH4- and TBABH4-electrolytes is that higher neodymium concentrations can be achieved compared to the Na[Nd(BH4)4](DME)4-containing electrolytes which seem to saturate at approx. 0.1 mol L−1 in DME, THF or 2-MeTHF. Because Li+ is more strongly solvated than Na+, it is possible to prepare electrolytes with a Nd3+ concentration of 0.5 mol L−1, and this is also the case for the organic TBA+ cation (Fig. 4). The electrolytes seem to be saturated at this concentration, as no more of the complex would dissolve.
 | ||
| Fig. 4 Cyclic voltammograms (first scan) of (a) Nd(BH4)3(THF)3 + one molar equivalent of LiBH4 in THF at different concentrations, and (b) Nd(BH4)3(THF)3 + one molar equivalent of TBABH4 in THF at different concentrations, recorded at ambient temperature at a scan rate of 10 mV s−1. The working and counter electrodes were pieces of platinum-coated silicon wafers with a surface area of 0.3 cm2 and 1.0 cm2, respectively. The reference electrode was Fc+/Fc (0.005 mol L−1 each) dissolved in [BMP][Tf2N]. | ||
In both electrolytes, a positive trend was observed for the current density as the concentration of the neodymium complex is increased, with a value of almost −10.0 mA cm−2 reached for the most concentrated LiBH4-electrolyte at a cathodic vertex potential of −3.4 V vs. Fc+/Fc. This is a very promising result for reaching sufficiently high current densities during potentiostatic electrodeposition without relying on supporting electrolyte salts as in the case of the bistriflimide-containing electrolytes. As the amount of electroactive species is increased, the current should increase linearly with the concentration, whether the polarization is steady or unsteady. It is evident from Fig. 4 that this is not the case. This implies that the neodymium metal that is being electrodeposited, reacts with the electrolyte, reducing the electroactive part of the electrode. As the concentration of electro-active species in solution is increased, neodymium metal is depositing faster than it can react with the electrolyte and the current is increasing faster than the increase in concentration. This also explains why the anodic currents are larger after crossing the zero-current line. It also explains why in the CVs in Fig. 3, the cathodic current decreases with increasing scan rates for the same concentration of electroactive species. As the scan rate decreases, more time is available for the deposited neodymium metal to react with the electrolyte, which results in the partial passivation of the working electrode.
Potentiostatic electrodeposition
Electrodeposition of neodymium was performed potentiostatically at −3.0 V or −3.1 V vs. Fc+/Fc for the Na+- and Li+-containing electrolytes, respectively in order to avoid excessive co-deposition of sodium or lithium metal (Fig. 5). For the TBABH4 electrolytes, the potential was −3.3 V vs. Fc+/Fc to compensate for the higher deposition overpotential. | ||
| Fig. 5 Chronoamperograms recorded at ambient temperature for a potentiostatic deposition at −3.0 V vs. Fc+/Fc for 3600 s in a static solution of 0.1 mol L−1 Na[Nd(BH4)4](DME)4 in THF (black curve), and 0.1 mol L−1 Nd(Tf2N)3 + 4 molar equivalents of NaBH4 in THF (blue curve). The working and counter electrodes were pieces of platinum-coated silicon wafers with a surface area of 0.3 cm2 and 1.0 cm2, respectively. The reference electrode was Fc+/Fc (0.005 mol L−1 each) dissolved in [BMP][Tf2N]. | ||
Similarly to the cyclic voltammetry experiments, the electrodeposition current density was initially higher for the bistriflimide-containing electrolytes at approx. −2.6 mA cm−2, but quickly decreased during the first 500 s of the experiment to less than 4% of this initial value. For the rest of the duration of the experiment, a plateau was observed at a negligibly small current density (<−0.1 mA cm−2). This behavior is again indicative of the formation of passivating surface films due to side reactions of the neodymium deposit with the bistriflimide anion. On the other hand, for the bistriflimide-free electrolyte, the current density was initially lower at approx. −1.0 mA cm−2 but remained nearly constant over the duration of the experiment, indicating that the deposit was not passivated during electrodeposition. As a result, it was possible to deposit much more charge in the bistriflimide-free electrolyte (−1.03 C, as opposed to −0.14 C for the bistrifimide-containing electrolyte), and to achieve thicker deposits. More concentrated electrolytes consisting of 0.5 M of the neutral complex Nd(BH4)3(THF)3 + one molar equivalent of LiBH4 or TBABH4 in THF were studied as well (Fig. S7, ESI†). Due to the higher concentration, the electrodeposition current density was initially higher for the LiBH4 electrolyte (approx. −6.4 mA cm−2), but a gradual decrease was observed over the duration of the experiment, to 20% of the initial value after one hour. For the TBABH4 electrolyte, the current density was initially lower (approx. −2.5 mA cm−2), but remained much more constant over time. A scanning electron micrograph and corresponding EDX analysis of a neodymium deposit is shown in Fig. 6.
 | ||
| Fig. 6 (a) SEM micrograph of a neodymium deposit (theoretical thickness = 2.8 μm) at 2000× magnification, obtained after potentiostatic electrodeposition from 0.1 mol L−1 Na[Nd(BH4)4] in THF at −3.0 V vs. Fc+/Fc for 2.5 hours at ambient temperature. The substrate was a piece of platinum-coated silicon wafer with an approximate surface area of 0.3 cm2. (b) EDX spectrum in the range 0–12 keV corresponding to the spot indicated on the SEM micrograph, recorded at an acceleration voltage of 30 kV. | ||
A spongy, nodular morphology was observed, with deep cracks dividing the deposit. This is in contrast to the smooth shard-like morphology observed in previous work.28 Some crystalline particles are observed on top of the cracked layer, which are most likely crystals of electrolyte salts (NaBH4) that were not rinsed off completely, judging by EDX analysis (Fig. S8, ESI†). According to the EDX analysis of the deposit itself, neodymium (Lα = 5.23 keV, M = 0.98 keV) is present as the major constituent at an estimated 75 atomic %, while oxygen (Kα = 0.53 keV) is only present at 3.5 at%. The main impurity of the deposit is co-deposited sodium (Kα = 1.04 keV) at an estimated 21 at%. It should also be noted that typical impurities such as fluorine (Kα = 0.68 keV), sulfur (Kα = 2.31 keV), chlorine (Kα = 2.62 keV) and carbon (Kα = 0.28 keV) are not detected in deposits originating from the bistriflimide-free electrolytes, providing further evidence that no passivating films are formed. However, this elemental composition is not representative for the entire deposit, as EDX analysis of other spots on the surface showed a significantly higher oxygen content, up to 67 at% (Fig. S8, ESI†). During transfer to the SEM vacuum chamber it was not possible to avoid short exposure of the deposit to air, therefore resulting in partial oxidation, although the increased layer thickness seems to have somewhat compensated for this. Nevertheless, this is a very promising result, giving evidence that the electrodeposition of pure metallic neodymium layers is possible.
Motivated by this positive result, the black deposits resulting from 0.1 mol L−1 Na[Nd(BH4)4](DME)4 in THF were further analyzed by X-ray photoelectron spectroscopy (XPS) surface and depth profiling to investigate the composition changes in the inner layers of the deposits. The deposits were transported under argon and placed inside an air-tight vessel which was directly attached to the ultra-high vacuum line of the XPS instrument in order to avoid exposure to air. The Nd 3d5/2 spectra for the sample at various depths are shown in Fig. 7.
 | ||
| Fig. 7 (a)–(d) Curve-fitted Nd 3d5/2 region XPS spectra after 0 s, 90 s, 180 s and 270 s of etching time from a neodymium deposit obtained from potentiostatic deposition in 0.1 mol L−1 Na[Nd(BH4)4] in THF electrolyte at −3.0 V vs. Fc+/Fc for 2.5 hours at ambient temperature. (e) Comparison of neodymium metal and Nd2O3 content after different etching times. | ||
The Nd 3d5/2 region of the XPS spectra was deconvoluted into three peaks: Nd2O3 (>982 eV), neodymium metal (979–980 eV), and the O KL23L23 auger electron line.13,21,41 At the surface, the lines of Nd2O3 and neodymium metal are observed, giving evidence for the presence of both species. As the etching progressed from the surface (i.e. at 0 s etching) to the inner layers, an increase in the Nd 3d5/2 peak intensity was observed, indicating an increased amount of both species present. The relative atomic concentrations of Nd2O3 and neodymium metal determined from the spectra at each etching time are summarized in Fig. 7(e). At the surface, neodymium metal is present at 51% of the total neodymium content and Nd2O3 at 49%. Surprisingly, as the etching progresses, the content of neodymium metal is slightly decreased to approx. 47%, whereas the content of Nd2O3 is increased to approx. 53%, which is opposite to the expected trend in case of surface oxidation. Furthermore, the relative content of Nd2O3 and neodymium metal remains constant over the entire depth of the deposit. However, this could be a result of the rough, spongy morphology of the deposit. This was also observed by Periyapperuma et al., who hypothesized that the presence of water in their ionic liquid electrolytes was responsible for constant side-reactions during electrodeposition, resulting in oxide formation throughout the entire deposit.21 However, the organic electrolytes in this work are completely water-free, due to the reactivity of the borohydride anions. Therefore, the origin of Nd2O3 throughout the entire thickness of the deposit is unclear, other than maybe a side-reaction with the THF solvent. It is also surprising that the presence of oxides does not result in passivation during electrodeposition, as was observed during the CV and CA experiments. Nevertheless, both Nd2O3 and Nd metal are considered valuable materials to recover due to their many applications. The survey spectrum of the surface of the deposit is shown in Fig. S9 (ESI†). Other than the peaks corresponding to neodymium, the most prominent other peaks correspond to oxygen (O 1s) and carbon (C 1s), whereas peaks of typical bistriflimide-derived elements such as fluorine or sulfur are completely absent.
Conclusions
A new class of fluorine-free organic electrolytes was developed for the electrodeposition of neodymium metal. The electrolytes consist of an isolated solvated neodymium borohydride active complex, Na[Nd(BH4)4](DME)4 or Nd(BH4)3(THF)3, dissolved in an ether solvent. These complexes can be prepared from commercially available NdCl3 or the conveniently prepared Nd(OMs)3, and the borohydride salt NaBH4, without relying on any salts with fluorinated anions such as bis(trifluoromethylsulfonyl)imide (bistriflimide). Because the electrolytes do not contain this reactive anion, they are much more stable against the metallic neodymium deposits, resulting in the possibility to perform potentiostatic electrodeposition at constant current density, without the interference of passivating surface films. The deposits were characterized by different analytical techniques, giving evidence for the presence of both Nd2O3 and Nd metal throughout the entire deposit. Despite these promising results, there are some obvious limitations associated with this system that must be addressed in the context of electrowinning of neodymium. Firstly, the borohydride-based organic electrolytes are highly moisture-sensitive, which requires handling in a rigorously controlled dry and inert atmosphere. Secondly, the electrical conductivity of organic electrolytes is always lower than that of any molten salt electrolyte, resulting in lower deposition current densities. Thirdly, the reaction at the counter electrode must be considered as well. The anode reaction is probably oxidation of the borohydride anion in this case, which results in gradual contamination of the electrolyte by decomposition products. Nevertheless, we are convinced that this system is a leap towards low-temperature electrowinning of neodymium and other rare earths in fluorine-free electrolytes and that it can be significantly improved by further research.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project (EOS 40007515) has received funding from the FWO and F.R.S.-FNRS under the Excellence of Science (EOS) programme.References
- K. Binnemans, P. T. Jones, B. Blanpain, T. Van Gerven, Y. Yang, A. Walton and M. Buchert, Recycling of rare earths: a critical review, J. Cleaner Prod., 2013, 51, 1–22, DOI:10.1016/j.jclepro.2012.12.037.
- K. Binnemans, P. T. Jones, T. Müller and L. Yurramendi, Rare earths and the balance problem: How to deal with changing markets?, J. Sustainable Metall., 2018, 4, 126–146, DOI:10.1007/s40831-018-0162-8.
- M. A. R. Önal, S. Dewilde, M. Degri, L. Pickering, B. Saje, S. Riaño, A. Walton and K. Binnemans, Recycling of bonded NdFeB permanent magnets using ionic liquids, Green Chem., 2020, 22, 2821–2830, 10.1039/d0gc00647e.
- M. Orefice and K. Binnemans, Solvometallurgical process for the recovery of rare-earth elements from Nd-Fe-B magnets, Sep. Purif. Technol., 2021, 258, 117800, DOI:10.1016/j.seppur.2020.117800.
- R. AliAkbari, Y. Marfavi, E. Kowsari and S. Ramakrishna, Recent Studies on Ionic Liquids in Metal Recovery from E-Waste and Secondary Sources by Liquid-Liquid Extraction and Electrodeposition: a Review, Mater. Circ. Econ., 2020, 2, 10, DOI:10.1007/s42824-020-00010-2.
- E. Stefanidaki, C. Hasiotis and C. Kontoyannis, Electrodeposition of neodymium from LiF–NdF3–Nd2O3 Melts, Electrochim. Acta, 2001, 46, 2665–2670, DOI:10.1016/S0013-4686(01)00489-3.
- D. Shen and R. Akolkar, Electrodeposition of Neodymium from NdCl3-Containing Eutectic LiCl–KCl Melts Investigated Using Voltammetry and Diffusion-Reaction Modeling, J. Electrochem. Soc., 2017, 164, H5292–H5298, DOI:10.1149/2.0451708jes.
- Y. Jeon, J. Hur, G. Y. Jeong, S. Sohn and J. park, Selective and efficient extraction of Nd from NdFeB magnets via ionization in LiCl-KCl-CdCl2 melt, J. Alloys Compd., 2021, 860, 158424, DOI:10.1016/j.jallcom.2020.158424.
- K. Binnemans, Lanthanides and Actinides in Ionic Liquids, Chem. Rev., 2007, 107, 2592–2614, DOI:10.1021/cr050979c.
- A. V. Rudnev, Electrodeposition of lanthanides from ionic liquids and deep eutectic solvents, Russ. Chem. Rev., 2020, 89, 1463–1482, DOI:10.1070/RCR4970.
- E. Bourbos, I. Giannopoulou, A. Karantonis, I. Paspaliaris and D. Panias, Reduction of Light Rare Earths and a Proposed Process for Nd Electrorecovery Based on Ionic Liquids, J. Sustainable Metall., 2018, 4, 395–406, DOI:10.1007/s40831-018-0186-0.
- D. R. MacFarlane, N. Tachikawa, M. Forsyth, J. M. Pringle, P. C. Howlett, G. D. Elliott, J. H. Davis, M. Watanabe, S. Patrice and A. C. Austen, Energy applications of ionic liquids, Energy Environ. Sci., 2014, 7, 232–250, 10.1039/c3ee42099j.
- H. Kondo, M. Matsumiya, K. Tsunashima and S. Kodama, Attempts to the Electrodeposition of Nd from Ionic Liquids at Elevated Temperatures, Electrochim. Acta, 2012, 66, 313–319, DOI:10.1016/j.electacta.2012.01.101.
- L. Sanchez-Cupido, J. M. Pringle, A. L. Siriwardana, A. Unzurrunzaga, M. Hilder, M. Forsyth and C. Pozo-Gonzalo, Water-Facilitated Electrodeposition of Neodymium in a Phosphonium-Based Ionic Liquid, J. Phys. Chem. Lett., 2019, 10, 289–294, DOI:10.1021/acs.jpclett.8b03203.
- L. Sanchez-Cupido, J. M. Pringle, A. I. Siriwardana, M. Hilder, M. Forsyth and C. Pozo-Gonzalo, Correlating Electrochemical Behavior and Speciation in Neodymium Ionic Liquid Electrolyte Mixtures in the Presence of Water, ACS Sustainable Chem. Eng., 2020, 8, 14047–14057, DOI:10.1021/acssuschemeng.0c04288.
- D. Coleman and N. Gathergood, Biodegradation studies of ionic liquids, Chem. Soc. Rev., 2010, 39, 600–637, 10.1039/b817717c.
- S. P. F. Costa, A. M. O. Azevedo, P. C. A. G. Pinto and M. L. M. F. S. Saraiva, Environmental Impact of Ionic Liquids: Recent Advances in (Eco)toxicology and (Bio)degradability, ChemSusChem, 2017, 10, 2321–2347, DOI:10.1002/cssc.201700261.
- M. C. Bubalo, K. Radošević, I. R. Redovniković, J. Halambek and V. G. Srček, A brief overview of the potential environmental hazards of ionic liquids, Ecotoxicol. Environ. Saf., 2014, 99, 1–12, DOI:10.1016/j.ecoenv.2013.10.019.
- W. Zhao and J. Sun, Triflimide (HNTf2) in Organic Synthesis, Chem. Rev., 2018, 118, 10349–10392, DOI:10.1021/acs.chemrev.8b00279.
- M. Razo-Negrete, R. Ortega-Borges, V. Zinovyeva, C. Cannes, C. Le Naour, G. Trejo-Córdova and Y. Meas, Comparison of the electrochemical behavior of some Rare Earth Elements in butyl methylpyrrolidinium dicyanamide ionic liquid, Int. J. Electrochem. Sci., 2019, 14, 10431–10447, DOI:10.20964/2019.11.16.
- K. Periyapperuma, J. M. Pringle, L. Sanchez-Cupido, M. Forsyth and C. Pozo-Gonzalo, Fluorine-free ionic liquid electrolytes for sustainable neodymium recovery using an electrochemical approach, Green Chem., 2021, 23, 3410–3419, 10.1039/d1gc00361e.
- E. B. Molodkina, M. R. Ehrenburg, I. A. Arkhipushkin and A. V. Rudnev, Interfacial effects in the electro(co)deposition of Nd, Fe, and Nd-Fe from an ionic liquid with controlled amount of water, Electrochim. Acta, 2021, 398, 139342, DOI:10.1016/j.electacta.2021.139342.
- K. Orme, D. L. Baek, R. V. Fox and A. Atifi, Water Interplays during Dysprosium Electrodeposition in Pyrrolidinium Ionic Liquid: Deconvoluting the Pros and Cons for Rare Earth Metallization, ACS Sustainable Chem. Eng., 2021, 9, 14631–14643, DOI:10.1021/acssuschemeng.1c06189.
- P. Bagri, H. Luo, I. Popovsa, B. P. Thapaliyac, J. Dehaudta and S. Dai, Trimethyl phosphate based neutral ligand room temperature ionic liquids for electrodeposition of rare earth elements, Electrochem. Commun., 2018, 96, 88–92, DOI:10.1016/j.elecom.2018.10.001.
- G. M. Krishna, A. Routa and K. A. Venkatesan, Voltammetric investigation of some lanthanides in neutral ligand-ionic liquid, J. Electroanal. Chem., 2020, 856, 113671, DOI:10.1016/j.jelechem.2019.113671.
- B. Zhang, L. Wang, Y. Liu, Y. Zhang, L. Zhang and Z. Shi, AlCl3-assisted dissolution of NdCl3 in organic solvents for Nd refining, Sep. Purif. Technol., 2021, 276, 119416, DOI:10.1016/j.seppur.2021.119416.
- B. Zhang, L. Wang, K. Pan, W. Zhang, Y. Liu, Y. Zhang, L. Zhang and Z. Shi, LiNO3-Supported Electrodeposition of Metallic Nd from Nd-Containing Solvate Ionic Liquid, J. Phys. Chem. C, 2021, 125, 20798–20805, DOI:10.1021/acs.jpcc.1c06335.
- P. Geysens, P.-C. Lin, J. Fransaer and K. Binnemans, Electrodeposition of neodymium and dysprosium from organic electrolytes, Phys. Chem. Chem. Phys., 2021, 23, 9070–9079, 10.1039/d0cp06606k.
- N. de Vos, C. Maton and C. V. Stevens, Electrochemical Stability of Ionic Liquids: General Influences and Degradation Mechanisms, ChemElectroChem, 2014, 1, 1258–1270, DOI:10.1002/celc.201402086.
- M. C. Kroon, W. Buijs, C. J. Peters and G.-J. Witkamp, Decomposition of ionic liquids in electrochemical processing, Green Chem., 2006, 8, 241–245, 10.1039/b512724f.
- P. C. Howlett, E. I. Izgorodina, M. Forsyth and D. R. MacFarlane, Electrochemistry at Negative Potentials in Bis(trifluoromethanesulfonyl)amide Ionic Liquids, Z. Phys. Chem., 2006, 220, 1483–1498, DOI:10.1524/zpch.2006.220.10.1483.
- T. Vander Hoogerstraete, N. R. Brooks, B. Onghena, L. Van Meervelt and K. Binnemans, Crystal structures of hydrated rare-earth bis(trifluoromethylsulfonyl)imide salts, CrystEngComm, 2015, 17, 7142–7149, 10.1039/c5ce01270h.
- E. M. Aricó, L. B. Zinner, B. Kanellakopulos, E. Dornberger, J. Rebizante and C. Apostolidis, Structure and properties of hydrated La(III), Nd(III) and Er(III) methanesulfonates, J. Alloys Compd., 2001, 323–324, 39–44, DOI:10.1016/S0925-8388(01)00975-6.
- L. Xue, C. W. Padgett, D. D. DesMarteau and W. T. Pennington, Synthesis and structures of alkali metal salts of bis[(trifluoromethyl)sulfonyl]imide, Solid State Sci., 2002, 4, 1535–1545, DOI:10.1016/S1293-2558(02)00050-X.
- V. D. Makhaev, A. H. Borisov, B. P. Tarasov and B. I. Semenenko, Synthesis and physico-chemical properties of anionic borohydride complexes of cerium group rare-earth metals, Russ. J. Inorg. Chem., 1981, 26, 2645–2651 CAS.
- V. D. Makhaev, A. H. Borisov, B. P. Tarasov and B. I. Semenenko, Synthesis and physico-chemical properties of anionic borohydride complexes of yttrium group rare-earth metals, Russ. J. Inorg. Chem., 1983, 28, 340–344 CAS.
- V. D. Makhaev, A. H. Borisov and E. B. Lobkovsky, Scandium- and yttrium borohydride complexes solvated with dimethoxyethane, Russ. J. Inorg. Chem., 1984, 29, 1160–1163 CAS.
- V. D. Makhaev, A. H. Borisov and B. I. Semenenko, Borohydride complexes of the yttrium group rare-earth elements solvated with dimethoxyethane, Ln(BH4)3.DME, Russ. J. Inorg. Chem., 1986, 31, 1586–1588 CAS.
- V. D. Makhaev, A. P. Borisov and B. P. Tarasov, Complexes of Tetrahydroborates of Rare-Earth Elements with Tetrabutylammonium Tetrahydroborate (Bu4N)[Ln(BH4)4·nDME] (n = 0,1), Russ. J. Inorg. Chem., 2000, 45, 40–45 Search PubMed.
- C. Görller-Walrand and K. Binnemans, in Handbook on the Physics and Chemistry of Rare Earths, ed. K. A. Gschneidner Jr. and L. Eyring, Elsevier, Amsterdam, 1998, ch. 167, vol. 25, pp. 101–264 DOI:10.1016/S0168-1273(98)25006-9.
- M. Ishii, M. Matsumiya and S. Kawakami, Development of Recycling Process for Rare Earth Magnets by Electrodeposition Using Ionic Liquids Media, ECS Trans., 2012, 50, 549–560, DOI:10.1149/05011.0549ecst.
Footnote |
| † Electronic supplementary information (ESI) available: Powder XRD diffractograms of Na[Nd(BH4)4][(DME)4 and Nd(BH4)3(THF)3. CVs of Li(Tf2N) + 4 molar equivalents of LiBH4, and NaTf2N + saturated NaBH4 in THF. CVs of Na[Nd(BH4)4](DME)4 in THF with varying cathodic vertex potential. CVs of Na[Nd(BH4)4](DME)4 in MeTHF, DME, and G2. CVs of Nd(BH4)3(THF)3 + LiBH4 in THF, and Nd(BH4)3(THF)3 + TBABH4 in THF. CV of Nd(BH4)3(THF)3 in THF. Chronoamperograms for a potentiostatic deposition in Nd(BH4)3(THF)3 + one molar equivalent of LiBH4 or TBABH4 in THF. SEM micrograph of a neodymium deposit at 500× magnification + corresponding EDX analysis in several spots. XPS survey scan in the range 0–1100 eV of a neodymium deposit obtained from potentiostatic deposition in 0.1 mol L−1 Na[Nd(BH4)4] in THF electrolyte. See DOI: https://doi.org/10.1039/d3cp01262j |
| This journal is © the Owner Societies 2023 |