
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Topological effects in ultrafast photoinduced processes between flurbiprofen and tryptophan in linked dyads and within human serum albumin†
Lorena
Tamarit
 ,
Laura
García-Gabarda
,
M. Consuelo
Jiménez
,
Miguel A.
Miranda
* and
Ignacio
Vayá
*
,
Laura
García-Gabarda
,
M. Consuelo
Jiménez
,
Miguel A.
Miranda
* and
Ignacio
Vayá
*
Departamento de Química/Instituto de Tecnología Química UPV-CSIC, Universitat Politècnica de València, Camino de Vera s/n, 46022 València, Spain. E-mail: mmiranda@qim.upv.es; igvapre@qim.upv.es
First published on 3rd June 2023
Abstract
The interaction dynamics between flurbiprofen (FBP) and tryptophan (Trp) has been studied in covalently linked dyads and within human serum albumin (HSA) by means of fluorescence and ultrafast transient absorption spectroscopy. The dyads have proven to be excellent models to investigate photoinduced processes such as energy and/or electron transfer that may occur in proteins and other biological media. Since the relative spatial arrangement of the interacting units may affect the yield and kinetics of the photoinduced processes, two spacers consisting of amino and carboxylic groups separated by a cyclic or a long linear hydrocarbon chain (1 and 2, respectively) have been used to link the (S)- or (R)-FBP with the (S)-Trp moieties. The main feature observed in the dyads was a strong intramolecular quenching of the fluorescence, which was more important for the (S,S)- than for the (R,S)- diastereomer in dyads 1, whereas the reverse was true for dyads 2. This was consistent with the results obtained by simple molecular modelling (PM3). The observed stereodifferentiation in (S,S)-1 and (R,S)-1 arises from the deactivation of 1Trp*, while in (S,S)-2 and (R,S)-2 it is associated with 1FBP*. The mechanistic nature of 1FBP* quenching is ascribed to energy transfer, while for 1Trp* it is attributed to electron transfer and/or exciplex formation. These results are consistent with those obtained by ultrafast transient absorption spectroscopy, where 1FBP* was detected as a band with a maximum at ca. 425 nm and a shoulder at ∼375 nm, whereas Trp did not give rise to any noticeable transient. Interestingly, similar photoprocesses were observed in the dyads and in the supramolecular FBP@HSA complexes. Overall, these results may aid to gain a deeper understanding of the photoinduced processes occurring in protein-bound drugs, which may shed light on the mechanistic pathways involved in photobiological damage.
Introduction
Binding of photoactive drugs to biomolecules has received considerable attention since disorders including photogenotoxicity or photoallergy may arise from interaction of the resulting complexes with UV light.1,2 In this context, photoinduced energy or electron transfer, as well as exciplex formation, can be responsible for the biological damage. These processes are known to be highly influenced by the surroundings of the drug,3 especially when bound to a protein, where the conformational arrangement of the excited chromophore is determinant for its photoreactivity.4,5Molecular dyads consisting of a drug moiety covalently linked to an amino acid derivative have proven to be useful models to investigate in detail the photoprocesses that occur in the real protein-bound drug complexes.6–9 As an example, marked similarities have previously been observed between the fundamental photoprocesses occurring in fenofibric acid, the active metabolite of the hypolipidemic drug fenofibrate,10 upon binding to human serum albumin (HSA),11 the most abundant transport protein in plasma,12 and the photobehavior of model dyads composed of fenofibric acid linked to tyrosine (Tyr) or tryptophan (Trp). Likewise, the photophysical properties of HSA-bound flurbiprofen (FBP), a non-steroidal anti-inflammatory drug used for the treatment of different diseases,13–17 are comparable to those observed for diastereomeric dyads where the drug and Trp are directly linked (FBP-Trp) through an amide bond.18
In the particular case of FBP-Trp, fluorescence spectroscopy reveals a strong stereodifferentiation in the quenching of the singlet excited state for both the drug (1FBP*) and the amino acid (1Trp*); the latter has been attributed to an electron transfer process and/or exciplex formation, whereas the drug quenching has been proposed to occur through an energy transfer process from 1FBP* to Trp, although this hypothesis has not been confirmed.8,18 In this regard, even though fluorescence spectroscopy, both in the steady-state and time-resolved modes, is a powerful tool to investigate the photoinduced processes operating in these photoactive systems,18–22 there are some limitations to analyze them in detail due to the strong overlap of FBP and Trp emission.
An alternative technique that can in principle be used to investigate the behavior of drugs in solution and in biological media is femtosecond transient absorption spectroscopy.23–31 This technique has proven to be a very sensitive, selective and precise tool that allows investigating processes occurring at the very early stages after excitation, including ultrafast energy and electron transfer or charge separation.32,33 In this context, it has recently been observed an ultrafast energy transfer from HSA to bound lapatinib (LAP),25 a drug currently used to treat breast and lung cancer.34–37 It has also been proven that ultrafast electron transfer is markedly influenced by the relative orientation and the distance between the individual chromophores in bichromophoric linked systems.38–40 In general, the location and spatial arrangement of a linked or bound drug are highly relevant to understand the kinetics and yields of the photoinduced processes occurring both in covalently attached drug-amino acid systems and in non-covalent drug–protein complexes.
With this background, the present work aims to study in depth the photobehavior of diastereomeric dyads composed of FBP and Trp separated by linking bridges of different length and nature (Fig. 1), where the excited state processes are expected to be strongly influenced by the environment and the distance between the involved chromophores, using a combination of fluorescence and ultrafast transient absorption spectroscopies. In this regard, (S)- and/or (R)-FBP are connected to Trp through a short and rigid cyclic spacer (CSp) or using a long and flexible linear one (LSp). The former displays a cis configuration which in principle would allow closer interaction between the two chromophores, while the latter permits greater freedom of movement of both subunits and hence lower probability of interaction. In connection with this, the conformational arrangement adopted by the drug and the amino acid units in the different dyads have been approached by means of simple molecular modelling. In parallel, the photobehavior of FBP@HSA complexes has also been studied for comparison, in order to draw an improved picture of the photoinduced processes taking place in the supramolecular media.
 | ||
| Fig. 1 Chemical structure of the investigated systems. | ||
Results and discussion
The synthesis of the diastereomeric dyads (S,S)- and (R,S)-1, or (S,S)- and (R,S)-2 was performed by conventional methods. In the former, FBP and Trp are separated by a short and rigid cyclic spacer; here, the synthesis of the intermediates (S)- and (R)-FBP-CSp can be found elsewhere.7 In the case of dyads 2, both chromophores are linked through a flexible linear spacer longer than the cyclic one. The syntheses of (S)- and (R)-FBP-LSp as well as those of dyads 1 and 2, in addition to the chemical characterization of the compounds, can be found in the experimental section of ESI† (Fig. S1 to S6, ESI†). Briefly, the drug was first linked to the cyclic or linear spacer through an amide bond leading to the intermediates FBP-CSp and FBP-LSp, respectively. Then, these intermediates were coupled to the methyl ester of Trp (TrpMe) in the presence of EDC and BtOH to afford the desired dyads.The photobehavior of the intermediates and the dyads was investigated in acetonitrile, since their solubility in aqueous media was very low. In general, the presence of the spacer (cyclic or linear) did not affect significantly the photophysical properties of the parent drug, so FBP was used as reference. The absorption spectra of (S,S)-1 and (R,S)-1 matched with those of the addition spectra of the isolated FBP and Trp at the same concentration; this points to little if any interactions between both moieties in the ground state (see Fig. 2). By contrast, a different picture was obtained from the fluorescence studies. Here, isoabsorptive solutions (A266 ∼ 0.1) at the excitation wavelength (λexc = 266 nm) were employed in order to compare the fluorescence quantum yields (ϕF).
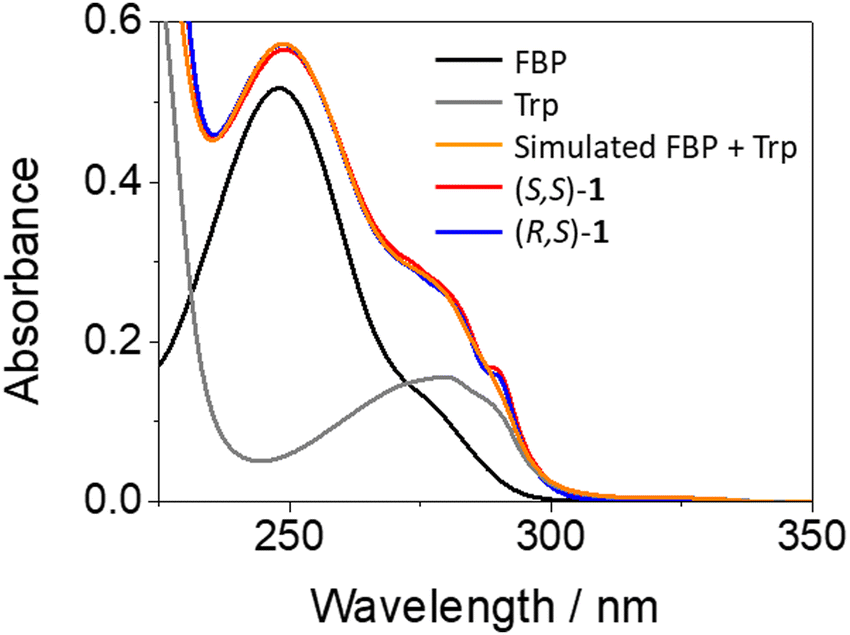 | ||
| Fig. 2 UV absorption spectra for (S)-FBP (black), (S)-TrpMe (gray), (S,S)-1 (red) and (R,S)-1 (blue). The simulated spectrum resulting from the addition of isolated FBP and Trp is shown in orange. The concentration of all samples was 20 μM in acetonitrile. | ||
As it can be observed from Fig. 3A, emission from isolated FBP (ϕF = 0.21)41 was much stronger than that observed for (S,S)-1 (ϕF = 0.04) or (R,S)-1 (ϕF = 0.05). From the shape and position of the spectra, both the drug and the amino acid contribute to the fluorescence of the dyads, since two weak shoulders were observed at ca. 300 and 315 nm, arising from 1FBP*, and the relative maximum was closer to the emission of 1Trp*. This is not surprising since both chromophores absorb at 266 nm (see Fig. 2). Interestingly, the simulated emission of the dyad (violet spectra in Fig. 3A) was much stronger than the experimental observations; it can be calculated using the relation (1), considering the percentage of photons absorbed by FBP (60%) and Trp (40%) at 266 nm and assuming independent emission of the two chromophores due to the absence of significant interactions in their excited states.
| AF(simulated) = 0.6 × AF(FBP) + 0.4 × AF(Trp) | (1) |
| AF((S,S)-1) = 0.06 × AF(FBP) + 0.16 × AF(Trp) | (2) |
| AF((R,S)-1) = 0.06 × AF(FBP) + 0.20 × AF(Trp) | (3) |
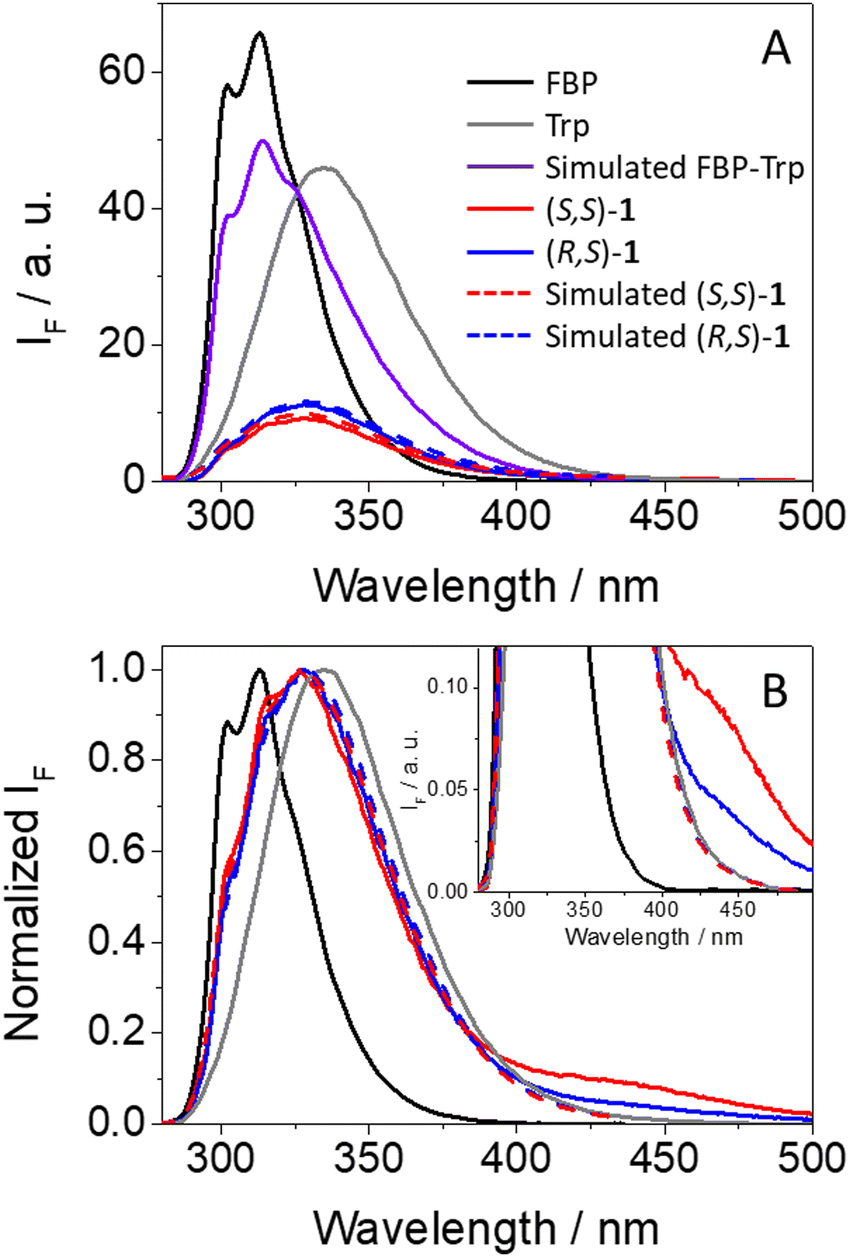 | ||
| Fig. 3 (A) Fluorescence spectra (λexc = 266 nm) for (S)-FBP (black), (S)-TrpMe (gray), (S,S)-1 (red) and (R,S)-1 (blue) in acetonitrile. The simulated spectrum that is obtained considering the percentage of photons absorbed by isolated FBP and TrpMe at 266 nm and assuming no interactions between them in their excited states is shown in violet. The simulated emissions for (S,S)-1 and (R,S)-1 considering a quenching process as explained in the text are shown in dashed red and dashed blue, respectively (both spectra were slightly moved upwards to be distinguished). (B) Normalized spectra at the maximum emission. The inset shows a zoom of the lower energy emitting states. | ||
Here, strong fluorescence quenching was determined for both chromophores, ca. 90% for 1FBP* in both (S,S)-1 and (R,S)-1, whereas for 1Trp* ca. 60% reduction in the emission intensity was noticed in (S,S)-1 and ∼50% in (R,S)-1. Hence, some stereodifferentiation was observed along the deactivation of 1Trp* in the dyads.
An additional outcome to highlight is the weak but clearly detectable emission above 425 nm displayed by both dyads (see Fig. 3B), which according to previous observations for related systems can be ascribed to emission from exciplexes.7,8,18 Formation of these transients was also stereoselective, being more favored in the case of (S,S)-1; thus, the stronger the fluorescence quenching, the higher exciplex formation. Such species are commonly associated with charge transfer character,42 in agreement with the electron donor nature of Trp.43 The exciplex nature of this long-wavelength emission is supported by the excitation spectra at 400 nm, where FBP does not emit and Trp displays low emission; in this regard, the spectra of dyads 1 are basically superimposable to the absorption spectra, in line with the formation of exciplex-like species. The same was true for the excitation spectra at λem = 450 nm, where there is still contribution of both FBP and Trp (see Fig. S7 in ESI†).
In this framework, an interesting point for discussion is the mechanism of fluorescence quenching in the dyads (S,S)-1 and (R,S)-1. In the flurbiprofen unit, it can arise from an energy transfer process from 1FBP* to Trp, in accordance with the relative energies of the excited singlet states (99 and 95 kcal mol−1, respectively).41,44 This process was previously proposed for directly linked FBP-Trp dyads,8,18 but due to the massive fluorescence quenching, in addition to the absorption of both the drug and the amino acid at 266 nm and the strong overlap of their emission spectra, it was difficult to prove this hypothesis by means of fluorescence spectroscopy. However, the proposed energy transfer can be observed even to a higher extent upon irradiation at 250 nm, where the drug absorbs practically all the incident light (ca. 90%, see Fig. 2). As expected, a more marked decrease of 1FBP* emission compared with the simulated one in the absence of any interaction between the drug and Trp was noticed for both (S,S)-1 and (R,S)-1 (see Fig. S8 in ESI†).
As regards the quenching of 1Trp*, it can only be attributed to a charge transfer process (exciplex formation, EXC, and/or electron transfer, eT). In fact, application of the Weller equation45 showed that the two processes are exergonic (ΔGEXC = −10 kcal mol−1 and ΔGeT = −15 kcal mol−1; eqn 4 and 5, respectively),8,18 and experimental formation of exciplex-like species has been mentioned above for both (S,S)-1 and (R,S)-1.
 | (4) |
 | (5) |
Since the discussed photophysical processes are dynamic in nature, a kinetic analysis was undertaken. As anticipated, the kinetic traces of (S,S)-1 and (R,S)-1 at the emission maximum decayed faster than for FBP (see Fig. 4A), displaying fluorescence lifetimes (τF) of ca. 0.8, 0.9 and 1.8 ns, respectively; these values were obtained upon fitting the decay traces by a non-linear fitting/deconvolution procedure using a one-exponential function F(t) = Σai·exp(−t/τi). However, due to the limitations in the time-resolution of our setup, it was difficult to make a reliable analysis of the kinetics in the nanosecond time scale, since a simple monoexponential law would be insufficient to explain in detail all the involved photoinduced processes, i. e. quenching of 1FBP* and 1Trp* as well as exciplex formation and decay (kinetic traces shown in Fig. 4B). In the case of the exciplexes, the (S,S)- diastereomer displayed longer τF values than its (R,S)- counterpart (3.7 vs. 2.6 ns, respectively), in line with the results from steady-state experiments. Here, a two-exponential function was necessary to get a good fitting for both decays; the obtained τF values are average lifetimes determined as <τF> = a1τ1+ a2τ2.
 | ||
| Fig. 4 Decay traces at the maximum emission (A) and at wavelengths longer than 420 nm (B) for (S)-FBP (black), (S,S)-1 (red) and (R,S)-1 (blue) after excitation at 265 nm in acetonitrile. The best fitting curves are shown in dashed. The instrumental response function is shown in light gray. | ||
To investigate the photobehavior of dyads in more detail, femtosecond transient absorption spectroscopy was applied to the study of (S,S)-1 and (R,S)-1. This is a powerful tool to elucidate the nature of processes occurring at the very early steps after excitation, such as energy and/or electron transfer.32,33 Isolated FBP and Trp were first investigated as references. All measurements were performed at λexc = 250 nm, since absorption of the drug is about ten times higher than that of the amino acid (see Fig. 2). Thus, excitation of FBP led to a rapid formation of a band with maximum at ca. 425 nm and a shoulder at ∼385 nm (see Fig. S9 in ESI†), which was previously assigned to 1FBP*.29 This species decayed following a second order law with lifetimes of ca. 52 and 1700 ps. The longer component is assigned to deactivation of 1FBP* to the ground state, while the shorter one is associated to intersystem crossing (ISC) to form the triplet excited state of the drug (3FBP*). These results totally agree with those previously reported by Su et al.29 Accordingly, the band peaking at 425 nm evolved towards the formation of a new one with maximum at ∼375 nm, which survived in the microsecond time scale and was assigned to 3FBP*.29,41 By contrast, excitation of Trp under the same experimental conditions did not result in the formation of any noticeable transient species (see Fig. S10 in ESI†). Hence, femtosecond transient absorption spectroscopy seemed a more appropriate technique than fluorescence to investigate ultrafast photoinduced processes of the dyads, since Trp does not produce any interference under the used experimental conditions.
In agreement with expectations, excitation of (S,S)-1 gave rise to transient species (see Fig. 5A) whose spectra were similar to those of isolated FBP. Thus, an instantaneous formation of 1FBP* (λmax = 425 nm) that evolved towards 3FBP* (λmax = 375 nm) was also observed. Analogous results were obtained for the (R,S)- diastereomer (see Fig. S11 in ESI†). However, the kinetics of the dyads at 425 nm (see Table S1, ESI†) were different from those of the isolated drug (see Fig. 5B): both (S,S)-1 and (R,S)-1 decayed faster than FBP in the first 20 ps, to continue their deactivation in parallel to that of the drug up to the nanosecond time scale. The ultrafast dynamics observed for both diastereomers (τ ∼ 10 ps) are assigned to an energy transfer from 1FBP* to Trp, since this process would be energetically favorable in view of the excited singlet energy values of both chromophores (99 vs. 95 kcal mol−1).41,44 The components with lifetimes of about 50 and 1400 ps (see Table S1, ESI†) are assigned to ISC and to deactivation of 1FBP* to the ground state. In relation to the energy transfer process, it is worth to recall that the cyclic spacer in its cis configuration may provide a closer orientation for FBP and Trp to interact with each other in both (S,S)-1 and (R,S)-1. Hence, immediately after excitation, the fraction of molecules displaying the appropriate conformation in their excited states may undergo energy transfer through a Dexter mechanism,46,47 which involves collision between the donor and acceptor partners. Concerning the longer component reaching the ns time domain, it may correspond to the fraction of excited molecules with a less favorable arrangement to facilitate the energy transfer process, thus promoting radiative deactivation through fluorescence. It is worth to mention that no stereodifferentiation was detected at this stage, which totally agrees with the results discussed above from the fluorescence measurements, where quenching of 1FBP* was identical for both (S,S)-1 and (R,S)-1. Accordingly, the observed stereodifferentiation was attributed to deactivation of 1Trp* through an electron transfer process and/or exciplex formation; these processes might take place at time-scales much longer than energy transfer. Indeed, it was previously observed for related systems that exciplex formation occur at times longer than 100 ps.18 Likewise, exciplex detection could be expected by femtosecond transient absorption; however, their absorption bands should have the signature of both FBP (with some FBP·− character,48 peaking at ca. 408 nm) and Trp (with some Trp·+ character,11,49 peaking at ca. 500 nm). These signals are probably much weaker than the strong absorption of 1FBP* and 3FBP*, which hinders their detection in these time and spectral windows.
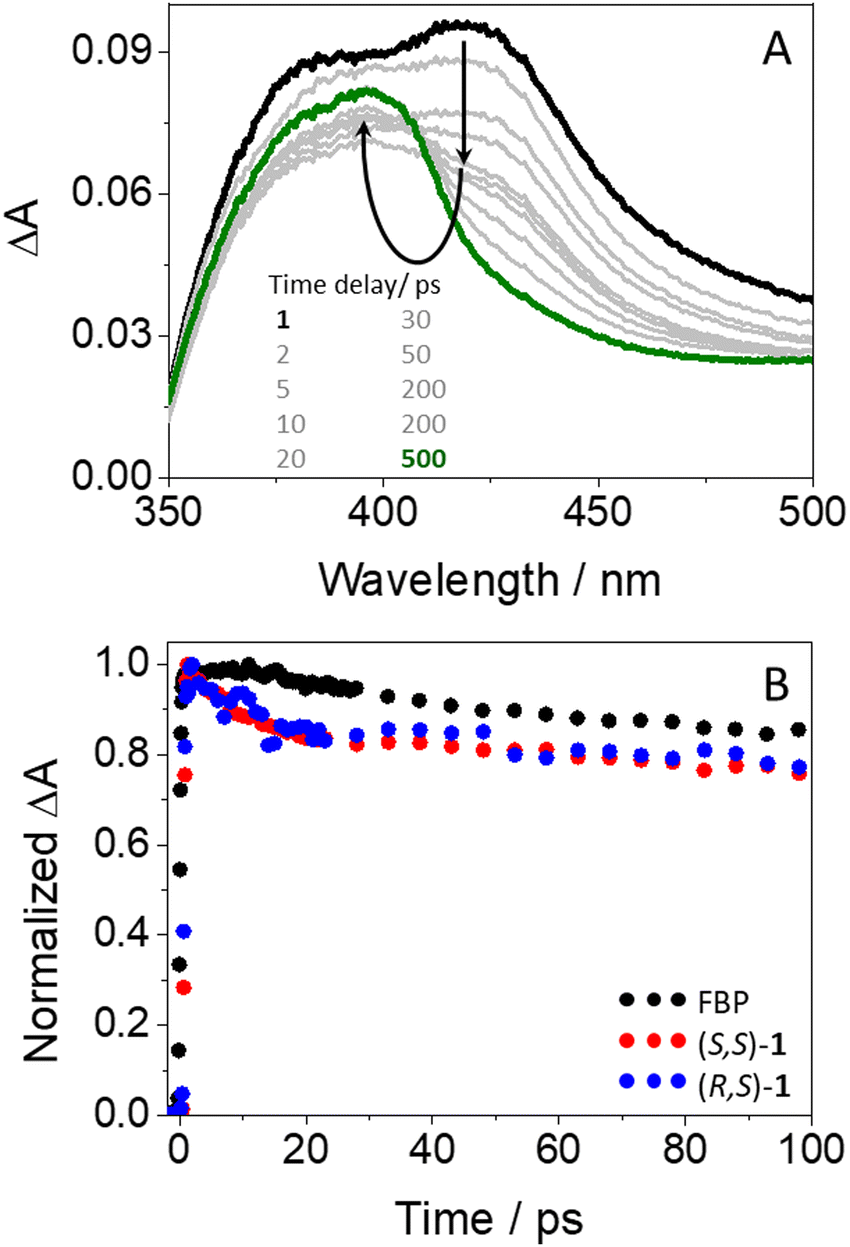 | ||
| Fig. 5 (A) Femtosecond transient absorption spectra for (S,S)-1 from 1 ps (black) to 0.5 ns (green). (B) Kinetic traces for (S)-FBP (black), (S,S)-1 (red) and (R,S)-1 (blue) at 425 nm. All measurements were performed upon excitation at 250 nm in acetonitrile. | ||
It is known that intramolecular photoprocesses related to those herein investigated are markedly influenced by the conformational arrangement of the systems.4,5 In order to better understand the stereodifferentiation observed for the dyads, and to get more insight into the relative orientation adopted by FBP and Trp in the more stable conformations, preliminary molecular modelling (PM3) was performed. The results showed that the two chromophores are closer to each other in (S,S)-1, which agrees with its higher fluorescence quenching compared with (R,S)-1, where both FBP and Trp are further apart (see Fig. S12 in ESI†).
To complete the study of excited state dynamics for longer-lived transient species, laser flash photolysis (LFP) experiments were performed with (S,S)-1 and (R,S)-1 after excitation at 266 nm in deaerated acetonitrile. The transient absorption spectra of both dyads were identical to that of isolated FBP (see Fig. S13 in ESI†), which was previously assigned to 3FBP*.41 However, the intensities were much lower for the dyads than for the parent drug, with estimated ϕT of about 0.07 and 0.11 for (S,S)-1 and (R,S)-1, respectively, which is not surprising due to the competing intramolecular quenching of their precursor 1FBP*. Concerning the triplet lifetimes (τT), of about 7.0 μs, they were also similar to that of FBP, which points to a lack of interaction between both chromophores in this excited state.
Concerning dyads (S,S)- and (R,S)-2, where FBP and Trp are separated by a long and flexible linear spacer, a parallel study was performed. Again, a strong and stereoselective fluorescence quenching was also observed (see Fig. 6A). Interestingly, (R,S)-2 exhibited a clearly lower emission quantum yield (ϕF = 0.07) than its (S,S)- counterpart (ϕF = 0.10), showing an apparent reverse behavior to (S,S)- and (R,S)-1. As in the case of 1, FBP and Trp contributed to the emission in both diastereomers of 2, since the fine structure from the drug between 300–315 nm was clearly observed, and the emission tail extended above 375 nm.
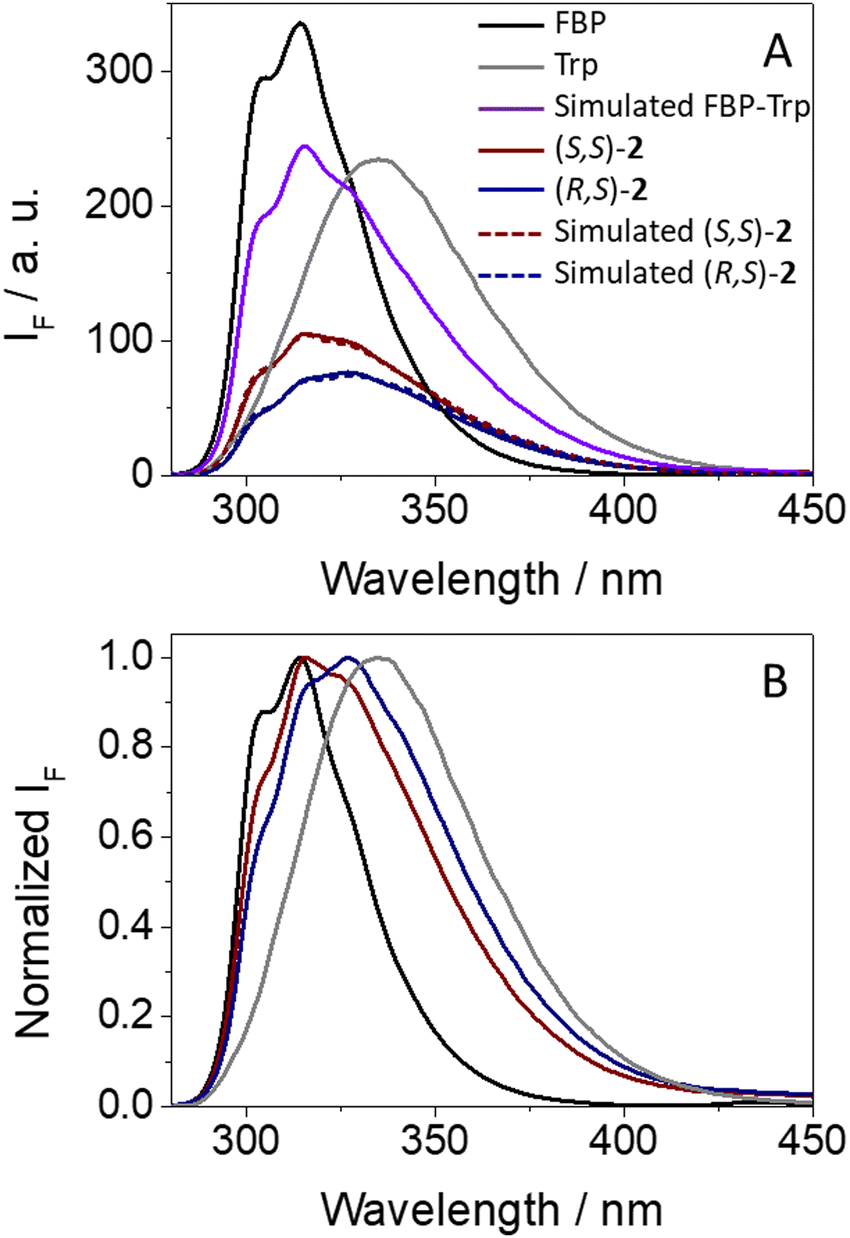 | ||
| Fig. 6 (A) Fluorescence spectra (λexc = 266 nm) for (S)-FBP (black), (S)-TrpMe (gray), (S,S)-2 (dark red) and (R,S)-2 (dark blue) in acetonitrile. The simulated spectrum that is obtained considering the percentage of photons absorbed by isolated FBP and TrpMe at 266 nm and assuming no interactions between them in their excited states is shown in violet. The simulated emissions for (S,S)-2 and (R,S)-2 considering a quenching process as explained in the text are shown in dashed dark red and dashed dark blue, respectively. (B) Normalized spectra at the emission maximum. | ||
The mathematical approach to estimate the quenching of either 1FBP* or 1Trp* in (S,S)-2 and (R,S)-2 is shown in eqn (6) and (7), respectively, which led to calculated curves (dashed lines in Fig. 6A) that matched with those obtained experimentally.
| AF((S,S)-2) = 0.24 × AF(FBP) + 0.22 × AF(Trp) | (6) |
| AF((R,S)-2) = 0.14 × AF(FBP) + 0.22 × AF(Trp) | (7) |
In this case, stereodifferentiation in the deactivation of 1FBP* was noticed, being higher for (R,S)-2 (77% quenching) than for its (S,S)- analog (60% quenching); by contrast, no differences were observed for 1Trp*, where 45% reduction of the emission intensity was determined for both diastereomers. Thus, it can be concluded that stereodifferentiation arises from 1FBP*. As it has been discussed above for dyads 1, irradiation of (S,S)-2 and (R,S)-2 at 250 nm induces a higher decrease of the emission of 1FBP* compared with the simulated one assuming no interactions between the two subunits (see Fig. S14 in ESI†), which is in line with the energy transfer process. Concerning the excited state dynamics, the fluorescence of (R,S)-2 decayed slightly faster than that of (S,S)-2 (see Fig. S15 in ESI†), displaying τF of ca. 1.1 and 1.2 ns, respectively. Interestingly, formation of exciplex-like species was negligible for dyads 2, since no emission was detected above 420 nm (Fig. 6B).
The photobehavior of (S,S)-2 and (R,S)-2 at the very early steps after excitation was also investigated by femtosecond transient absorption spectroscopy. Thus, excitation of the dyads at 250 nm resulted in the formation of the same transients (see Fig. S16 in ESI†) as those observed for FBP, but with different relaxation dynamics. The ultrafast processes (33 ps for (S,S)-2 and 12 ps (R,S)-2) were again in accordance with an energy transfer from 1FBP* to Trp. In this regard, the kinetics was faster for (R,S)-2 (see Fig. 7), in line with the steady-state observations. Again, the components of ca. 50 ps and 1.2 ns (see Table S1, ESI†) are assigned to ISC and deactivation of 1FBP* to the ground state.
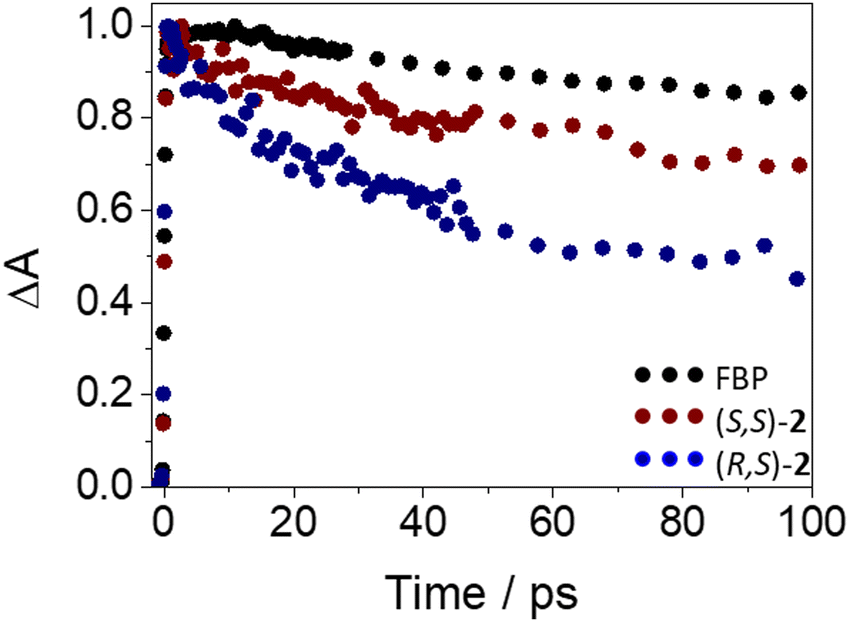 | ||
| Fig. 7 Femtosecond transient absorption decay traces at 425 nm for (S)-FBP (black), (S,S)-2 (dark red) and (R,S)-2 (dark blue) after excitation at 250 nm in acetonitrile. | ||
As mentioned above for dyads 1, the conformational arrangements adopted by FBP and Trp in (S,S)- and (R,S)-2 were also explored by means of simple molecular modelling (PM3). The two chromophores displayed a closer and more favorable orientation to interact with each other in (R,S)-2 (see Fig. S17 in ESI†), which totally agrees with the experimental results.
To complete the photophysical study of dyads 2, LFP measurements at λexc = 266 nm in deaerated acetonitrile revealed the formation of 3FBP* in both diastereomers (see Fig. S18 in ESI†). Interestingly, (R,S)-2 displayed lower intensity (estimated ϕT ∼ 0.14, while for (S,S)-2 it was ca. 0.27), which is not surprising since its fluorescence was quenched to a greater extent. The triplet lifetimes of dyads 2 (τT ∼ 7 μs) were coincident with that of isolated FBP, evidencing the lack of interaction between FBP and Trp from these excited states.
Finally, the photobehavior of flurbiprofen bound to HSA was investigated by means of femtosecond transient absorption spectroscopy. Previous reports discussing the ultrafast fluorescence dynamics of (S)- and (R)-FBP within HSA at the very early events after excitation have shown a stereoselective quenching of both 1FBP* and the single 1Trp* unit from the protein. The latter has been attributed to exciplex formation and/or electron transfer.18 However, although energy transfer from the drug to HSA was proposed to explain 1FBP* quenching, the ultrafast fluorescence technique presented some limitations for the study, due to the absorption of light at 266 nm by both FBP and HSA and to the strong overlap of their emission spectra. In this regard, femtosecond transient absorption spectroscopy could be a more appropriate tool to investigate this process.
Concerning the photobehavior in the protein-bound flurbiprofen, it is worth to note that HSA has multiple cavities that can host small organic moieties, and the most important ones to accommodate drugs are the so-called sites I, II and III.50,51 The only Trp residue of HSA is located in site I.52 In this context, previous studies have demonstrated that FBP mainly interacts to site II of HSA, but also to site I.53,54 This fact would be consistent with the possibility of energy transfer from 1FBP* to the protein, which would follow a Förster-type mechanism55 due to the large size of site I. In this regard, we have determined the R0 parameter from eqn (8) and (9), and the distance (r) for energy transfer through FRET mechanism from eqn (10), which was found to be ca. 19 Å. Hence, this mechanism might occur in the protein-bound flurbiprofen.
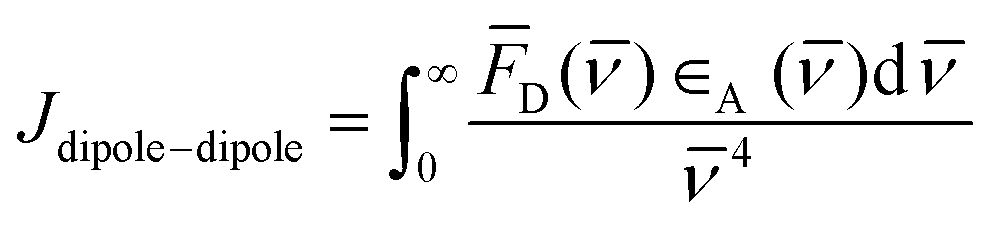 | (8) |
 | (9) |
 | (10) |
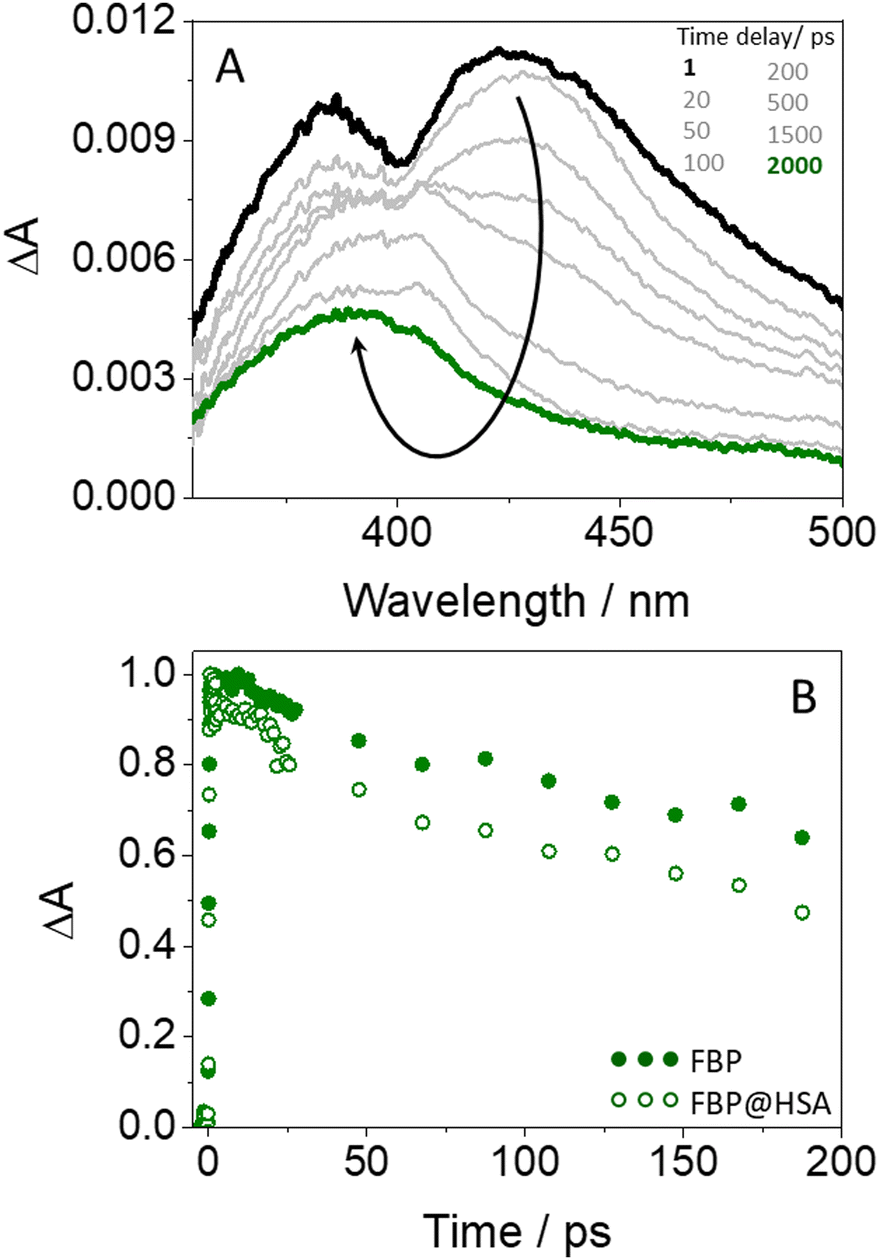 | ||
| Fig. 8 (A) Femtosecond transient absorption spectra from 1 ps (black) to 2 ns (green) for FBP@HSA. (B) Decay traces at 425 nm for FBP (solid green) and FBP@HSA (opened green). All measurements were performed at λexc = 250 nm in PBS. | ||
Conclusions
The photobehavior of model dyads composed of flurbiprofen (FBP) and tryptophan (Trp) separated by a short and rigid cyclic or a long and flexible linear spacer has been compared with that of the biologically relevant FBP@HSA complexes in order to get mechanistic insight into the photoinduced processes (e. g. energy and/or electron transfer) that may arise upon their exposure to light. To this end, fluorescence, both in the steady-state and time-resolved modes, in addition to ultrafast transient absorption spectroscopies have been used. Additionally, simple molecular modelling calculations in the dyads have explained why the observed photoinduced processes are clearly affected by the relative orientation of the two chromophores.In general, the photoreactivity is highly influenced by topological aspects such as the conformational arrangement and the distance between FBP and Trp. In this regard, a marked and stereoselective intramolecular quenching of the fluorescence of both units has been detected. In the case of dyads 1, this effect is stronger in the (S,S)- diastereomer, whereas the reverse is true for dyads 2, where the (R,S)- analog exhibits weaker emission. These results can be correlated with those of simple molecular modelling (PM3), where a closer arrangement is found for dyads (S,S)-1 and (R,S)-2, in line with the experimental results.
In general, the observed quenching is dynamic in nature and is associated with different processes depending on the investigated chromophore. For 1FBP*, energy transfer to Trp is proposed in view of the experimental results obtained from ultrafast transient absorption spectroscopy. On the other hand, electron transfer and/or exciplex formation occurs along deactivation of 1Trp*. The detected stereodifferentiation is markedly affected by topological factors. In this context, for dyads 1, where a short and rigid cyclic spacer is used to link FBP and Trp, stereodifferentiation occurs upon quenching of 1Trp*. Conversely, in the case of dyads 2, where both chromophores are separated by a linear and long flexible spacer, the stereoselectivity is detected during deactivation of 1FBP*. Finally, the same photoinduced processes detected for the model dyads are observed in the supramolecular FBP@HSA complexes. Hence, energy transfer from the excited drug to the protein is supported by femtosecond transient absorption spectroscopy. Overall, the obtained results prove the value of properly designed dyads as models to investigate relevant interactions between photoactive drugs and the amino acids located in the binding sites of proteins. This provides a useful tool to investigate the photoinduced processes that may occur upon exposure of complex biological systems to light, which could eventually lead to the occurrence of photobiological damage.
Author contributions
Research was conceived by all authors. Experiments were performed by L. T. and L. G.-G. with the aid of I. V. and M. C. J. The research was supervised by I. V. and M. A. M. All authors contributed to the writing of the manuscript and ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
Grant PID2020-115010RB-I00 funded by MCIN/AEI/10.13039/501100011033 and grant from Conselleria d’Innovació, Universitats, Ciència i Societat Digital (CIAICO/2021/061) are gratefully acknowledged.References
- R. Mang and J. Krutmann, Textbook of Contact Dermatitis, Springer Berlin Heidelberg, 2001, pp. 133–143 Search PubMed.
- B. Quintero and M. A. Miranda, Ars Pharm., 2000, 41, 27–46 CAS.
- V. Ramamurthy and K. S. Schanze, Organic Photochemistry and Photophysics, CRC Press/Taylor & Francis, Boca Raton, 2006 Search PubMed.
- V. Lhiaubet-Vallet and M. A. Miranda, Pure Appl. Chem., 2006, 78, 2277–2286 CrossRef CAS.
- I. Vayá, V. Lhiaubet-Vallet, M. C. Jimenez and M. A. Miranda, Chem. Soc. Rev., 2014, 43, 4102–4122 RSC.
- I. Vayá, I. Andreu, M. C. Jimenez and M. A. Miranda, Photochem. Photobiol. Sci., 2014, 13, 224–230 CrossRef PubMed.
- I. Vayá, T. Gustavsson, D. Markovitsi, M. A. Miranda and M. C. Jiménez, J. Photochem. Photobiol., A, 2016, 322, 95–101 CrossRef.
- I. Vayá, M. C. Jimenez and M. A. Miranda, J. Phys. Chem. B, 2007, 111, 9363–9371 CrossRef PubMed.
- I. Vayá, R. Perez-Ruiz, V. Lhiaubet-Vallet, M. C. Jimenez and M. A. Miranda, Chem. Phys. Lett., 2010, 486, 147–153 CrossRef.
- H. Ling, J. T. Luomab and D. Hilleman, Cardiol. Res., 2013, 4, 47–55 Search PubMed.
- I. Vayá, I. Andreu, V. T. Monje, M. C. Jimenez and M. A. Miranda, Chem. Res. Toxicol., 2016, 29, 40–46 Search PubMed.
- T. Peters, All About Albumin - Biochemistry, Genetics, and Medical Applications, Elsevier, Academic Press, San Diego, 1995, ch. 3, pp. 76–132 Search PubMed.
- P. L. Lomen, L. F. Turner, K. R. Lamborn, M. A. Winblad, R. L. Sack and E. L. Brinn, Am. J. Med., 1986, 80, 134–139 CrossRef CAS PubMed.
- J. Rovensky and D. Micekova, Drugs Exp. Clin. Res., 2000, 26, 19–24 CAS.
- G. D. Solomon and R. S. Kunkel, Cleveland Clin. J. Med., 1993, 60, 43–48 CrossRef CAS PubMed.
- J. Stamp, V. Rhind and I. Haslock, Br. J. Clin. Pract., 1989, 43, 24–26 CrossRef CAS PubMed.
- P. Tan, F. P. Flowers, O. E. Araujo and P. Doering, Drug Intell. Clin. Pharm., 1986, 20, 496–499 CAS.
- I. Vayá, P. Bonancia, M. C. Jimenez, D. Markovitsi, T. Gustavsson and M. A. Miranda, Phys. Chem. Chem. Phys., 2013, 15, 4727–4734 RSC.
- M. El-Kemary, M. Gil and A. Douhal, J. Med. Chem., 2007, 50, 2896–2902 CrossRef CAS PubMed.
- N. Seedher and S. Bhatia, J. Pharm. Biomed. Anal., 2005, 39, 257–262 CrossRef CAS PubMed.
- P. G. Takla, S. G. Schulman and J. H. Perrin, J. Pharm. Biomed. Anal., 1985, 3, 41–50 CrossRef CAS PubMed.
- D. Zhong, S. K. Pal, C. Wan and A. H. Zewail, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 11873–11878 CrossRef CAS PubMed.
- A. Buzády, J. Savolainen, J. Erostyak, P. Myllyperkio, B. Somogyi and J. Korppi-Tommola, J. Phys. Chem. B, 2003, 107, 1208–1214 CrossRef.
- I. Andreu, E. Lence, C. Gonzalez-Bello, C. Mayorga, M. C. Cuquerella, I. Vayá and M. A. Miranda, Front. Pharmacol., 2020, 11, 576495 CrossRef CAS PubMed.
- I. Vayá, I. Andreu, E. Lence, C. González-Bello, M. C. Cuquerella, M. Navarrete-Miguel, D. Roca-Sanjuan and M. A. Miranda, Chem. – Eur. J., 2020, 26, 15922–15930 CrossRef PubMed.
- L. Tamarit, M. El Ouardi, I. Andreu, I. Vayá and M. A. Miranda, Chem. Sci., 2021, 12, 12027–12035 RSC.
- L. Tamarit, M. El Ouardi, E. Lence, I. Andreu, C. González-Bello, I. Vayá and M. A. Miranda, Chem. Sci., 2022, 13, 9644–9654 RSC.
- M. D. Li, W. Li, J. Ma, T. Su, M. Liu, Y. Du and D. L. Phillips, J. Phys. Chem. A, 2011, 115, 14168–14174 CrossRef CAS PubMed.
- T. Su, J. Ma, N. Wong and D. L. Phillips, J. Phys. Chem. B, 2013, 117, 8347–8359 CrossRef CAS PubMed.
- M. Liu, M. D. Li, J. Huang, T. Li, H. Liu, X. Li and D. L. Phillips, Sci. Rep., 2016, 6, 21606 CrossRef PubMed.
- E. Bignon, M. Marazzi, V. Besancenot, H. Gattuso, G. Drouot, C. Morell, L. A. Eriksson, S. Grandemange, E. Dumont and A. Monari, Sci. Rep., 2017, 7, 8885 CrossRef PubMed.
- C. Ruckebusch, M. Sliwa, P. Pernot, A. de Juan and R. Tauler, J. Photochem. Photobiol., C, 2012, 13, 1–27 CrossRef CAS.
- V. Balevicius, Jr., T. Wei, D. Di Tommaso, D. Abramavicius, J. Hauer, T. Polivka and C. D. P. Duffy, Chem. Sci., 2019, 10, 4792–4804 RSC.
- N. L. Spector, W. Xia, H. Burris, 3rd, H. Hurwitz, E. C. Dees, A. Dowlati, B. O'Neil, B. Overmoyer, P. K. Marcom, K. L. Blackwell, D. A. Smith, K. M. Koch, A. Stead, S. Mangum, M. J. Ellis, L. Liu, A. K. Man, T. M. Bremer, J. Harris and S. Bacus, J. Clin. Oncol., 2005, 23, 2502–2512 CrossRef CAS PubMed.
- C. H. Yun, T. J. Boggon, Y. Li, M. S. Woo, H. Greulich, M. Meyerson and M. J. Eck, Cancer Cell, 2007, 11, 217–227 CrossRef CAS PubMed.
- N. U. Lin, L. A. Carey, M. C. Liu, J. Younger, S. E. Come, M. Ewend, G. J. Harris, E. Bullitt, A. D. Van den Abbeele, J. W. Henson, X. Li, R. Gelman, H. J. Burstein, E. Kasparian, D. G. Kirsch, A. Crawford, F. Hochberg and E. P. Winer, J. Clin. Oncol., 2008, 26, 1993–1999 CrossRef CAS PubMed.
- N. U. Lin, V. Dieras, D. Paul, D. Lossignol, C. Christodoulou, H. J. Stemmler, H. Roche, M. C. Liu, R. Greil, E. Ciruelos, S. Loibl, S. Gori, A. Wardley, D. Yardley, A. Brufsky, J. L. Blum, S. D. Rubin, B. Dharan, K. Steplewski, D. Zembryki, C. Oliva, D. Roychowdhury, P. Paoletti and E. P. Winer, Clin. Cancer Res., 2009, 15, 1452–1459 CrossRef CAS PubMed.
- A. Blasco-Brusola, I. Vayá and M. A. Miranda, Org. Biomol. Chem., 2020, 18, 9117–9123 RSC.
- A. Blasco-Brusola, I. Vayá and M. A. Miranda, J. Org. Chem., 2020, 85, 14068–14076 CrossRef CAS PubMed.
- A. Blasco-Brusola, M. Navarrete-Miguel, A. Giussani, D. Roca-Sanjuán, I. Vayá and M. A. Miranda, Phys. Chem. Chem. Phys., 2020, 22, 20037–20042 RSC.
- M. C. Jiménez, M. A. Miranda, R. Tormos and I. Vayá, Photochem. Photobiol. Sci., 2004, 3, 1038–1041 CrossRef PubMed.
- H. Lemmetyinen, N. Tkachenko, A. Efimov and M. Niemi, J. Porphyrins phthalocyanines, 2009, 13, 1090–1097 CrossRef CAS.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Plenum Press, New York, 2006 Search PubMed.
- M. Montalti, A. Credi, L. Prodi and M. T. Gandolfi, Handbook of Photochemistry, CRC Press, Taylor and Francis Group, Boca Raton, FL, 2006 Search PubMed.
- A. Z. Weller, Phys. Chem., 1982, 133, 93–98 CAS.
- D. L. Dexter, J. Chem. Phys., 1953, 21, 836–850 CrossRef CAS.
- S. S. Skourtis, C. Liu, P. Antoniou, A. M. Virshup and D. N. Beratan, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 8115–8120 CrossRef CAS PubMed.
- A. Saeki, T. Kozawa, Y. Ohnishi and S. Tagawa, J. Phys. Chem. A, 2007, 111, 1229–1235 CrossRef CAS PubMed.
- Y. P. Tsentalovich, O. A. Snytnikova and R. Z. Sagdeev, J. Photochem. Photobiol., A, 2004, 162, 371–379 CrossRef CAS.
- G. Sudlow, D. J. Birkett and D. N. Wade, Mol. Pharmacol., 1976, 12, 1052–1061 CAS.
- F. Zsila, Mol. Pharm., 2013, 10, 1668–1682 CrossRef CAS PubMed.
- X. M. He and D. C. Carter, Nature, 1992, 358, 209–215 CrossRef CAS PubMed.
- I. Vayá, C. J. Bueno, M. C. Jimenez and M. A. Miranda, ChemMedChem, 2006, 1, 1015–1020 CrossRef PubMed.
- S. Amezqueta, J. L. Beltran, A. M. Bolioli, L. Campos-Vicens, F. J. Luque and C. Rafols, Pharmaceuticals, 2021, 14, 214 CrossRef CAS PubMed.
- T. Förster, Discuss. Faraday Soc., 1959, 27, 7–17 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section and characterization of the different investigated intermediates and dyads by means of 1H- and 13C-NMR, in addition to spectroscopic results from UV, fluorescence, LFP and femtosecond transient absorption can be found in ESI. Besides, geometry optimized structures (PM3) for both dyads 1 and 2 are also shown. See DOI: https://doi.org/10.1039/d3cp01082a |
| This journal is © the Owner Societies 2023 |