
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
On the nature of inter-anion coinage bonds†
Jiayao
Li
a,
Qiuyan
Feng
a,
Changwei
Wang
*a and
Yirong
Mo
 *b
*b
aKey Laboratory for Macromolecular Science of Shaanxi Province, School of Chemistry & Chemical Engineering, Shaanxi Normal University, Xi’an 710119, P. R. China. E-mail: snnu.changweiwang@snnu.edu.cn
bDepartment of Nanoscience, Joint School of Nanoscience & Nanoengineering, University of North Carolina at Greensboro, Greensboro, NC 27401, USA. E-mail: y_mo3@uncg.edu
First published on 10th May 2023
Abstract
To explore the binding energy profiles and elucidate the bonding nature in counter-intuitive anion⋯anion coinage bonds (CiBs), thirty-one complexes were constructed, and the inter-anion CiBs were studied theoretically. The metastability was evidenced by the characteristic potential wells in six cases, demonstrating that anions [Au(CN)4]−, [Ag(CN)2]− and [AuO]− are appropriate building blocks for CiBs. The kinetic stability was further supported by ab initio molecular dynamics (AIMD) simulations and the analyses based on the local vibrational mode and quantum theory of atoms in molecules (QTAIM) methods. The anion⋯anion CiBs in the dimers of [AuCl4]− and [Au(CN)4]− previously observed in condensed phases were confirmed to be thoroughly repulsive under vacuum, but turned attractive in the crystal environment which was simulated using the solvation model based on density (SMD). However, the intrinsic strength of the inter-anion bonding is barely variated by the environment, as it is the combination of the inter-anion interaction and the environment effect that stabilizes the anion pairs. The block-localized wavefunction (BLW) method and its corresponding energy decomposition (BLW-ED) approach were further employed aiming at a chemically meaningful explanation for these counterintuitive phenomena. By inspecting the profiles of energy components, we identified the vital distinction between inter-anion CiBs and conventional non-covalent interactions lying in the electrostatic interaction, which variates nonmonotonically in the inter-anion complexes. The electrostatic interaction also dominates the depth of potential wells, which is commonly used to evaluate the kinetic stability, while Pauli exchange repulsion is the most repulsive factor preventing the formation of anion adducts. The importance of the Pauli exchange repulsion was further highlighted by comparing cases with and without metastability, in which the absence of a potential well is solely caused by the enhancement of the Pauli exchange repulsion.
1 Introduction
The spectrum of the noncovalent interactions has been remarkably expanded with newly recognized types, among which elements of both the p and d blocks on the periodic table can be directly involved as electrophilic sites.1–11 Particularly, coinage bonds (CiBs, or regium bonds),12,13 which refer to the close–shell interaction between the electron-deficient region on a coinage atom and the nucleophilic center of a Lewis base and have been determined and applied experimentally,14–17 attract attention for their unusual nature. Unconventional cases or findings challenge and eventually enrich our understanding of noncovalent interactions even for the classical hydrogen bond.18–20 A prominent example is the counterintuitive inter-anion hydrogen bond,18,21–26 which has already been utilized as a novel architectural linker in experimental studies.23,27–32 The inter-anion hydrogen bond challenges the classical electrostatic model and the force field description of hydrogen bonds, given the incredible Coulombic repulsion between anions.33,34 Similarly, inter-anion halogen bonds were also confirmed experimentally,20,35–37 after pioneering theoretical predictions.20 So far, the inter-anion/cation hydrogen bond, halogen bonds and many others have been well explored,18,23,24,27,30–32,35,38–67 forming a diverse family of counterintuitive bonding. The inter-anion CiB term was first coined by Daolio et al.,14 who studied the crystal structures containing tetrachloridoaurates and subsequently characterized the Au⋯Cl short contact between [AuCl4]− anions by employing the quantum theory of atom in molecule (QTAIM)68,69 and natural bond orbital (NBO) theories.70–75 Notably, coinage metal complexes exhibit different oxidation states for central metals and various geometries, and may involve undiscovered inter-anion CiBs. Elucidating the nature of chemical bonds involving transition metals is also challenging due to their specific electronic structures.76–84The history of CiBs may date back to the year 2000, when the Gerry lab studied the microwave spectra of Rg⋯M–X complexes, where Rg, M and X stand for rare gas, group 11 and halogen atoms respectively.85–90 Complexes formed between M–X and a series of Lewis bases have been determined in experimental studies ever since.91–95 Moreover, the Au⋯Cl bonding was spotted in the cytosine-derived gold(III) complex, according to the X-ray characterization and NBO analysis.96 Various theoretical studies were carried out aiming at elucidating the bonding nature of CiBs, and the electrostatic model based on the σ-hole concept was usually adopted.12,13,97–99 Before the remarkable work of Daolio et al.,14 other earlier experimental evidence for inter-anion CiBs can be found in the studies of the Leznoff group, who built coordination polymers using the [Au(CN)4]− units,100 and analyzed the Au⋯N interaction systematically in a subsequent work.101
Theoretical investigations of inter-anion CiBs can follow the example of inter-anion hydrogen bonds, which have been intensively studied. Inter-anion hydrogen bonds can be analyzed from the perspectives of energy, force constant and electron density. The energy indicator was first introduced by Mata et al., who explored the energy profile along the inter-anion distance and found that there is a local minimum followed by a transition state (TS) impending the anion adduct from dissociation.22 Hence, the energy difference between the local minimum and TS (well-depth) serves as an energy measurement for the metastability. Alternatively, the local bond stretching force constant,102–105 which is an “in situ” descriptor for the strength of a specified chemical bond,106–112 has been utilized to describe the inter-anion hydrogen bonds.113 Criteria for the inter-anion phenomena have also been developed based on the QTAIM approach,68,69 which has been broadly applied to from the interpretations of novel chemical bonding114 to the developments of QM-based force fields for large-scale molecular simulations.115,116 But the limitations of the QTAIM theory can be exemplified by the controversies over the H⋯H interaction in phenanthrene117,118 and a series of intramolecular interactions.119–121 Weinhold and Klein argued that the electrostatic interaction cannot be responsible for the inter-anion hydrogen bond, considering the Coulombic repulsion between net charges. Instead they proposed the charge transfer explanation and highlighted the governing role of orbital interactions with their NBO method.70–75 But this charge transfer model was queried by Frenking and Caramori,21 who stressed that it is unreliable to evaluate the electrostatic interaction based on a fallacious model where each entire anion is oversimplified to a point charge. There are also concerns that the NBO method tends to overestimate orbital interactions due to the non-optimization of bond orbitals,21,33,122–124 and suffers from the basis set superposition error (BSSE),125 though it has often been employed to probe the conventional and inter-anion CiBs.12–14,98 By contrary, the electrostatic explanation of the kinetic stability has been supported by energy decomposition analyses (EDAs) with the key role of the electrostatic interaction in well-depths confirmed.20,113,126 Notably, Alkorta et al. found that inter-anion interactions exhibit similar electron density distributions which are characterized by a series of chemical descriptors, to their conventional counterparts, which can be well understood with the classical electrostatic model.24,37,127 Similarly, a series of inter-cation non-covalent interactions have also been studied with their origins analyzed.128–131
Obviously, theoretical methods with the capacity of quantify inter-anion physical factors, especially, the electrostatic and charge transfer interactions, are crucial for exploring the nature of inter-anion CiBs and resolving controversies. Many EDA methods have been developed and applied so far.132–142 However, Foroutan-Nejad et al. pointed out that EDA approaches are path dependent.143,144 Hence, it is necessary to examine the roles of key physical factors in the chemical bonding from multiple perspectives. For instance, the electrostatic interaction, which is homologous in most EDA approaches,134,139,141,142,145–147 can be simulated using atomic multipole expansions as in force fields.148–151 For the electron transfer interaction, ab initio valence bond (VB) theory152,153 is preferable since it uses localized Lewis states to define a molecular wavefunction. As the simplest variant of VB theory, the block-localized wavefunction (BLW) method154–156 retains the characteristics of VB theory by using localized (fragmental) orbitals but simplifies the wavefunction with a single determinant for computational efficiency. Its energy decomposition (BLW-ED) approach157,158 has also been developed and applied to inter-anion hydrogen, halogen and chalcogen bonds.20,54,113
In this work, we intend to probe the nature of counterintuitive interactions in inter-anion CiBs theoretically. Representative coinage metal-containing anions with different geometries and oxidation states for central metals were selected14,15,159–162 and their interactions with a series of simple anions were explored, with the goal of identifying new members of the inter-anion CiBs. In addition, the inter-anion CiBs found in crystal structures14,100,101 were inspected in vacuum and condensed phase by employing the solvation model based on density (SMD).163 Subsequently, the metastability was further examined using the QTAIM method,68,69 the local vibrational mode theory,104,105,164–166 and the ab initio molecular dynamics (AIMD) simulations. At last, key factors for the kinetic stability were elucidated by employing the BLW-ED approach.157,158
2 Methods and computational details
2.1 BLW-ED approach
In the BLW-ED approach, the binding energy is defined as the overall variation in energy upon the formation of a complex from isolated and optimal monomers, and can be decomposed into several physically meaningful components, as shown in eqn (1). The deformation energy (ΔEdef) stands for the energy cost to distort monomer structures from their free and optimal states to the geometries in the complex. Bringing the deformed monomers from infinity to their positions in the complex without disturbing their respective electron densities leads to the frozen energy (ΔEF), as defined in eqn (2), where the initial block-localized state (ΨBLW0 in eqn (3)) is constructed using the wavefunction of distorted monomers (Φ0A and Φ0B), and  means the antisymmetric operator. The frozen energy can be further separated into the electrostatic energy and Pauli exchange repulsion at the Hartree–Fock (HF) level (eqn (2)).| ΔEb = ΔEdef + ΔEele + ΔEPauli + ΔEpol + ΔECT + ΔEec | (1) |
| ΔEF = E(ΨBLW0) − E(Φ0A) − E(Φ0B) = ΔEele + ΔEPauli | (2) |
| ΨBLW0 = Â(Φ0AΦ0B) | (3) |
The electron densities of monomers can be adjusted within each of them to respond to the electric field generated by others. This process corresponds to the self-consistent optimization of the electron-localized state (ΨBLW in eqn (4)), and the subsequent stability gained is defined as the polarization energy (ΔEpol in eqn (5)). The complex can be further stabilized by extending the movement of electrons to the entire complex, and this delocalization leads to the final HF wavefunction. The corresponding energy lowering is defined as the charge transfer energy, supplemented by the basis set superposition error (BSSE) (eqn (6)). Finally, the electron correlation contribution is defined as the difference in electron correlation energy between the complex and sum of deformed monomers (eqn (7)). In this work, the domain based local pair natural orbital coupled cluster method with single- and double-excitations (DLPNO-CCSD)167 was employed for the calculation of electron correlations. Moreover, the BBSE is also divided into the HF and electron correlation parts, and assigned to the charge transfer and electron correlation components correspondingly (eqn (6) and (7)). Notably, the electron correlation term can be further decomposed based on the local energy decomposition (LED) analysis168 within the DLPNO-CCSD(T) framework. However, the overall electron correlation is the least important contributor to the metastability among all energy terms as will be demonstrated, and therefore no further decomposition is conducted in this work.
| ΨBLW = Â(ΦAΦB) | (4) |
| ΔEpol = E(ΨBLW) − E(ΨBLW0) | (5) |
| ΔECT = E(ΨHF) − E(ΨBLW) + BSSEHF | (6) |
| ΔEec = EABcorr − EAcorr − EBcorr + BSSEcorr | (7) |
To study the binding energy in the crystal state, the SMD method is adopted to simulate the crystal environment. The total energy of the system (ESMD in eqn (8)) is composed of the internal energy of the focused complex (EI) and the energy of complex-surrounding interaction (V), which is dominated by the electrostatic forces. Therefore, the total binding energy in the crystal environment (ΔESMDb in eqn (9)) can be simply divided into the intrinsic component (ΔEIb), which stands for the difference in internal energy between the complex and optimal monomers, and the complex-surrounding component (ΔV), which corresponds to the variation in the energy of the complex-surrounding interaction from the isolated and optimized monomers to the formation of the complex.
| ΔESMD = EI + V | (8) |
| ΔESMDb = ΔEIb + ΔV | (9) |
2.2 Computational details
Thirty-one anion⋯anion complexes were constructed and are shown in Scheme 1. The def2-TZVP basis set was selected and the small-core relativistic pseudopotential (def2-TZVP-PP) was utilized for gold and silver.169,170 For complexes 1–6, the energy profiles along the inter-anion distance were evaluated by first performing constrained geometry optimizations, in which only the bond distance (R in Scheme 1) was fixed at a series of values with all the rest of the geometrical parameters optimized. A series of theoretical methods were tested, including B97-D3,171 PBE0-D3,172 M06-2X-D3,173 B3LYP-D3,174 CAM-B3LYP-D3,175 ωB97x-D2176 and SCS-MP2.177 Afterwards, single point calculations were carried out at each point by employing the DLPNO-CCSD method,167 with the resolution of the identity approximation178 and frozen core technique adopted (RI-DLPNO-CCSD) for computational efficiency. Complex 4 is a crucial case because [Au(CN)4]− and I− become thoroughly repulsive according to the RI-DLPNO-CCSD energy profile, while shallow potential wells (−3.5 to −5.2 kJ mol−1) were observed based on all other methods except for ωB97x-D2 and SCS-MP2 (Fig. S1v-a and b, ESI†). In general, well-depths derived from ωB97x-D2 and SCS-MP2 are nearly the same and both larger than the RI-DLPNO-CCSD results (Fig. S1, ESI†). To examine the reliability of our above computations, we performed single point calculations for complex 1 with the perturbative triples (T) correction incorporated (i.e., RI-DLPNO-CCSD(T)), and also re-optimized the geometries using enlarged basis sets. It turned out that increased theoretical levels barely influence the well-depths (within 0.4 kJ mol−1). Therefore, the ωB97x-D2 method was eventually selected for geometry optimizations and vibrational frequency calculations, and the RI-DLPNO-CCSD method was employed for the subsequent BLW-ED analysis of the binding energy profiles, aiming at a rigorous examination of the metastability.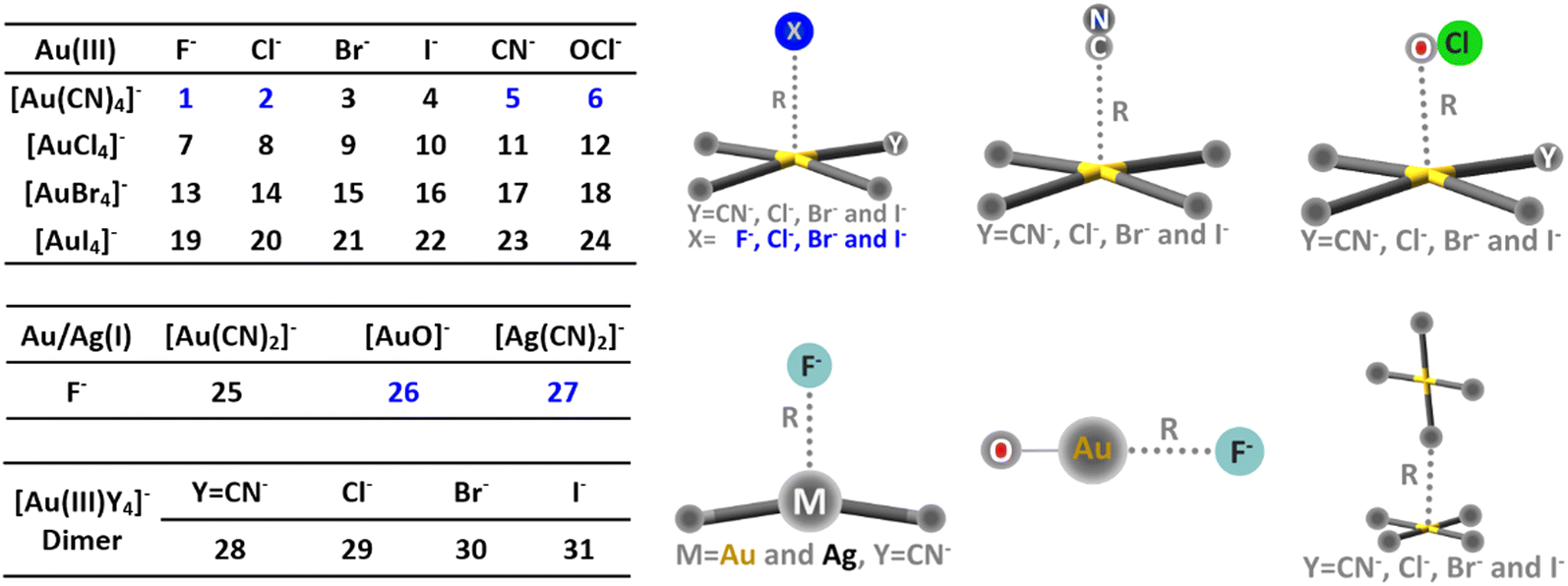 | ||
| Scheme 1 Complexes 1–31 studied in this work. | ||
AIMD simulations in vacuum at 298 K were carried out at the ωB97x-D2/def2-TZVP(-PP) theoretical level for the anion adducts which exhibit metastability in vacuum. Local minima were chosen as the initial structures and the initial atomic velocities were obtained based on the Boltzmann–Maxwell distribution randomly. The Berendsen thermostat was utilized and a step size of 1 fs was selected. Simulations with 5000 steps were accomplished for each case with the quantitative statics performed using results after the equilibration (i.e., 1500–5000 fs in each simulation).
Geometrical optimizations, vibrational frequency calculations, BLW-ED and GKS-EDA at the HF level were performed using the in-house version of GAMESS (US).179 Orca180,181 was used for the electron correlation energies and AIMD simulations. In addition, local bond stretching force constants of inter-anion CiBs were derived from the local vibrational mode theory.104,105,164–166 QTAIM analysis was performed using the Multiwfn package.182 QM-based atomic charges and multipoles were obtained by using the natural population analysis (NPA) and the GDMA183 program respectively. For these NPA and GDMA calculations, the ωB97x functional and the 6-311G basis set were selected except that for Au and Ag the Lanl2DZ pseudopotentials were adopted. Afterwards, the electrostatic energy was reevaluated along the inter-anion distance using the atomic multipole expansion scheme in the AMOEBA polarizable force field184–187 incorporated in the Tinker software.188
3 Results and discussions
Binding energy profiles for complex 1 ([Au(CN)4]−⋯F−) and 4 ([Au(CN)4]−⋯I−) are plotted in Fig. 1. The metastability of [Au(CN)4]−⋯F− was evidenced by the local minimum, which can trap the anion adduct. On the contrary, the binding energy between [Au(CN)4]− and I− monotonously gets repulsive as the inter-anion distance shrinks, suggesting a repulsive nature of the inter-anion interaction. All cases were similarly examined (Fig. S2, ESI†) and the characteristic potential wells were found in complexes 1, 2, 5, 6, 26 and 27 (colored in blue in Scheme 1). Among all tetracoordinated coinage metal complexes, only [Au(CN)4]− can form inter-anion CiBs. This is due to the fact that cyanide is the strongest electron-withdrawing group tested, and can considerably reduce the electron density of Au. Furthermore, a considerable barrier height (−42.5 kJ mol−1) was found in [Au(CN)4]−⋯F−, in which F− has the smallest radius among anions and therefore, the closest contact with the coinage center. Metastability was absent in [Au(CN)2]−⋯F− (complex 25), while observed in [Ag(CN)2]−⋯F− (complex 27), providing us comparative examples to clarify the roles of coinage metals to the metastability. Inter-anion CiB was also observed in [AuO]−⋯F− (complex 26), which exhibits a linear structure as the conventional CiBs illuminated previously in a theoretical study.11 Besides, substitution reactions were observed in complexes 7, 13, 19, 23 and 24. For the example of complex 7, one Cl− in [AuCl4]− is switched with F− after reaching the (Coulombic) barrier height, and the infinitely separated Cl− and [AuF(Cl)3]− anions are eventually observed (Fig. S2g, ESI†).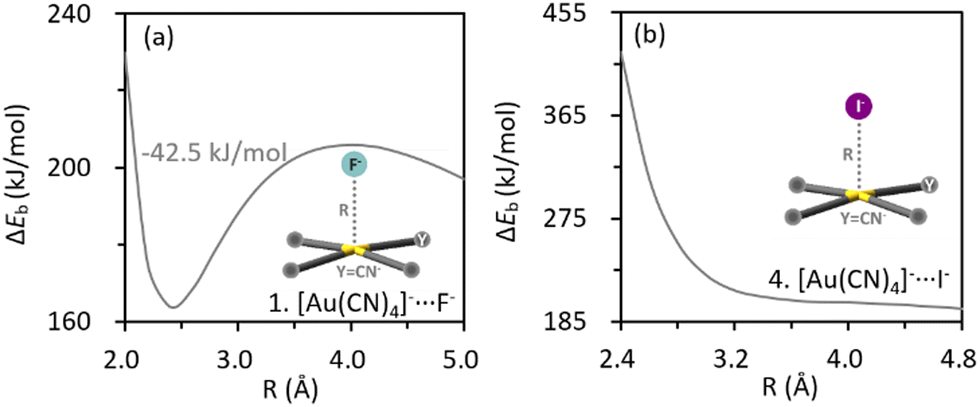 | ||
| Fig. 1 Profiles of binding energies along the inter-anion distances for complexes 1 (a) and 4 (b) computed at the RI-DLPNO-CCSD//ωB97x-D2/def2-TZVP theoretical level. | ||
Intriguingly, the dimer of [AuCl4]− turns more destabilizing as the inter-anion distance decreases, suggesting that the experimentally observed inter-anion interaction cannot hold in vacuum. This repulsive nature was also confirmed by re-evaluating the binding energy profile at higher theoretical levels in vacuum (Fig. S2a and c, ESI†). Obviously, the packing effect in the crystal structure plays an essential role in stabilizing the anion⋯anion pairs. The effect of the crystal environment on the binding energy profile was subsequently simulated using the SMD method, with a series of dielectric constants tested (Fig. 2). Indeed, the characteristic potential well appeared when the environmental effect was incorporated, and even negative binding energies were observed. Importantly, the intrinsic binding energy in the [AuCl4]− dimer is barely variated by the SMD method (Fig. 2b), while the solute–solvent term, which stands for the interaction between anion pair and the crystal environment in this work, turns more stabilizing drastically at short ranges (Fig. 2c) and gives rise to the local minimum. In other words, it is the interaction between the anion pair and crystal environment that stabilizes the system in condensed phases. Similarly, the key role of the surrounding to the inter-anion CiBs between [Au(CN)4]− anions was also confirmed (Fig. 2d-f). The intrinsic binding energies of aforementioned dimers were not further decomposed because of the absence of metastability. Besides, the absence of metastability in vacuum was observed in all AuY4− (Y = CN−, Cl−, Br− and I−) dimers (Fig. S2ab–ae, ESI†). In the following part, we performed theoretical analyses only for cases exhibiting inter-anion CiBs in vacuum.
 | ||
| Fig. 2 Profiles of total binding energy (a and d, ΔESMDb in kJ mol−1), intrinsic binding energy (b and e, ΔEIb in kJ mol−1) and the solute–solvent interaction term (c and f, ΔV in kJ mol−1) along the inter-anion distance for complexes [AuCl4]−⋯[AuCl4]− and [Au(CN)4]−⋯[Au(CN)4]−, obtained from single point calculations at the ωB97X-D2/def2-TZVP theoretical level with a series of dielectric constants selected. | ||
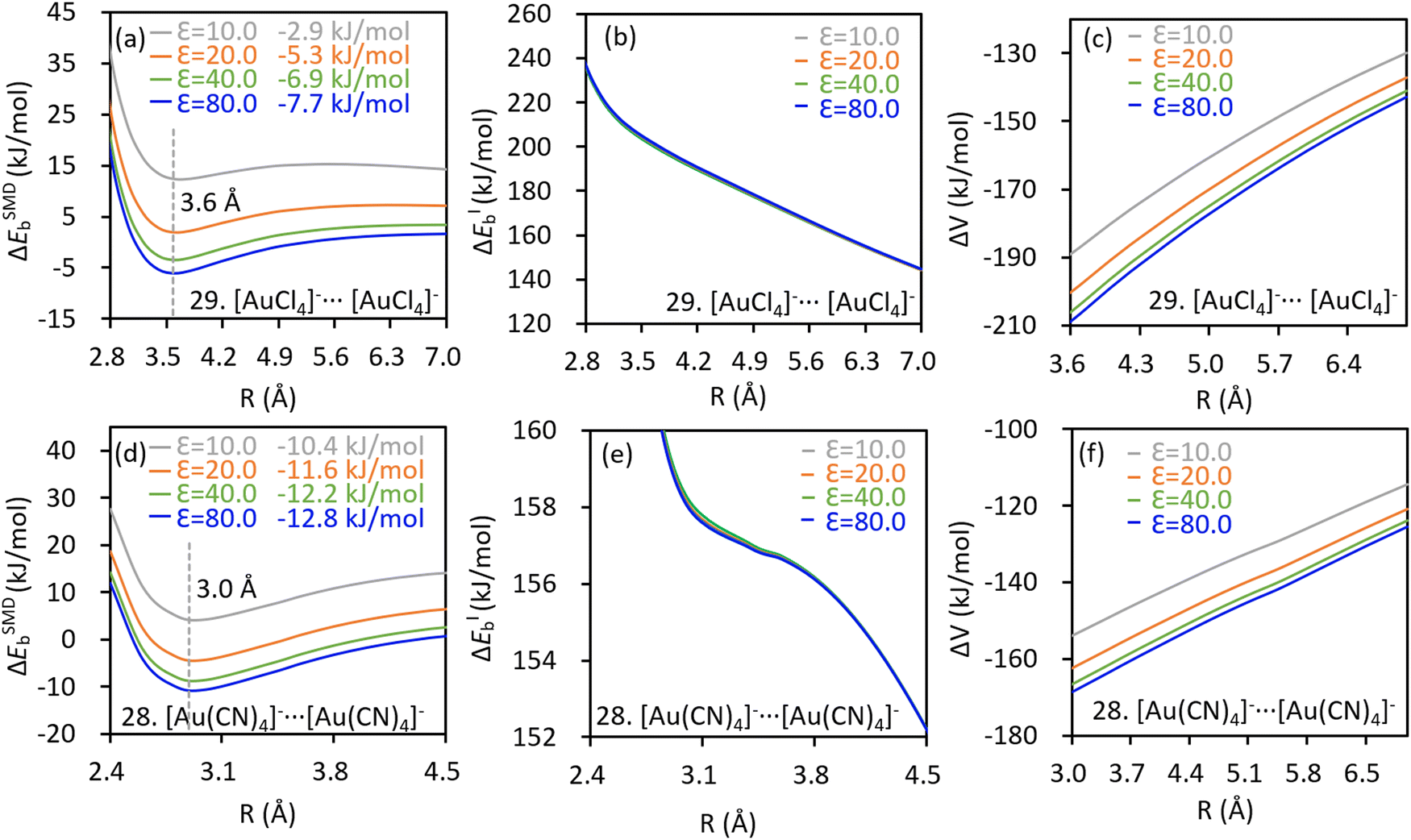
The kinetic stabilities of complex [Au(CN)4]−⋯F− were also evidenced by the AIMD simulations, in which dissociation of the anion adduct was not observed. As shown in Fig. 3a, the bond distance deviates from the average value (![[R with combining macron]](https://www.rsc.org/images/entities/i_char_0052_0304.gif) ) insignificantly. AIMD results of the other complexes exhibiting the inter-anion CiBs are shown in Fig. S3 (ESI†). Notably, the maximum/minimum deviations of the bond distance correlate with the localized bond force constants, which stand for the curvatures of binding energy profiles around the local minima, in an appropriate linear manner (Fig. 3b). In other words, a steeper curve leads to less significant deviations of bond lengths in the AIMD simulation. For example, complex 26 ([AuO]−⋯F−) exhibits the greatest bond force constant (0.982 mDyn Å−1) and therefore, the least deviation in bond length (Fig. S3e, ESI†). Besides, the average bond distance in the AIMD simulations is close to the optimal value (Table 1). The inter-anion CiBs were further supported by the QTAIM theory, because a bond critical point (BCP) with appropriate electron density and positive Laplace values were observed in each case with metastability, suggesting close–shell interactions.
) insignificantly. AIMD results of the other complexes exhibiting the inter-anion CiBs are shown in Fig. S3 (ESI†). Notably, the maximum/minimum deviations of the bond distance correlate with the localized bond force constants, which stand for the curvatures of binding energy profiles around the local minima, in an appropriate linear manner (Fig. 3b). In other words, a steeper curve leads to less significant deviations of bond lengths in the AIMD simulation. For example, complex 26 ([AuO]−⋯F−) exhibits the greatest bond force constant (0.982 mDyn Å−1) and therefore, the least deviation in bond length (Fig. S3e, ESI†). Besides, the average bond distance in the AIMD simulations is close to the optimal value (Table 1). The inter-anion CiBs were further supported by the QTAIM theory, because a bond critical point (BCP) with appropriate electron density and positive Laplace values were observed in each case with metastability, suggesting close–shell interactions.
 | ||
| Fig. 3 (a) Evolution of the Au⋯F− distance over time (after the 1500st fs) in the AIMD simulation of complex 1 and (b) correlation between the maximum/minimum deviations of the bond distances from the average values and localized bond force constants. | ||
, in Å) from the AIMD simulations, electron density (ρ, in a.u.) and the Laplace (∇2ρ, in a.u.) values at the BCPs of complexes with metastability
| No. | Complexes | R |
|
ρ | ∇2ρ |
|---|---|---|---|---|---|
| 1 | [Au(CN)4]−⋯F− | 2.360 | 2.360 | 0.052 | 0.226 |
| 2 | [Au(CN)4]−⋯Cl− | 2.980 | 3.024 | 0.026 | 0.075 |
| 5 | [Au(CN)4]−⋯CN− | 2.620 | 2.656 | 0.042 | 0.112 |
| 6 | [Au(CN)4]−⋯OCl− | 2.535 | 2.561 | 0.040 | 0.142 |
| 26 | [AuO]−⋯F− | 2.157 | 2.159 | 0.072 | 0.354 |
| 27 | [Ag(CN)2]−⋯F− | 2.284 | 2.274 | 0.050 | 0.254 |
Roles of individual energy components in the metastability can be revealed intuitively by examining their corresponding profiles along the inter-anion distances (Fig. 4). In each complex, the polarization, charge transfer and electron correlation interactions all become more attractive as anions approach each other, suggesting a preference for the local minimum over the transition states (TS), which has a stretched inter-anion distance. Conversely, both the Pauli exchange repulsion and deformation energy grow more destabilizing as the inter-anion distance shrinks, contributing negatively to the well depths. The repulsive electrostatic interaction increases as the length of inter-anion CiB shrinks at long ranges, but turns to decrease drastically after reaching the Coulombic repulsion barrier, indicating a positive contribution to the well depth. The decreasing trend of electrostatic interaction can be rationalized by the nucleus–electron attraction, which can overturn the electrostatic repulsion among electrons and among nuclei at short ranges. Actually, the nonmonotonic profile of electrostatic interaction was also observed in other inter-anion interactions,54,113 and can be regarded as the key distinction between inter-anion and conventional interactions, in which all energy terms variate monotonically along the bond length. The electrostatic energy profiles of complexes 1, 26 and 27 were reevaluated using the QM-based atomic multipoles, and the vital nonmonotonicity was reproduced (Fig. S4, ESI†). Hence, the electrostatic interaction in inter-anion CiBs can be simulated and explained based on the classical distributed atomic multipole expansion.
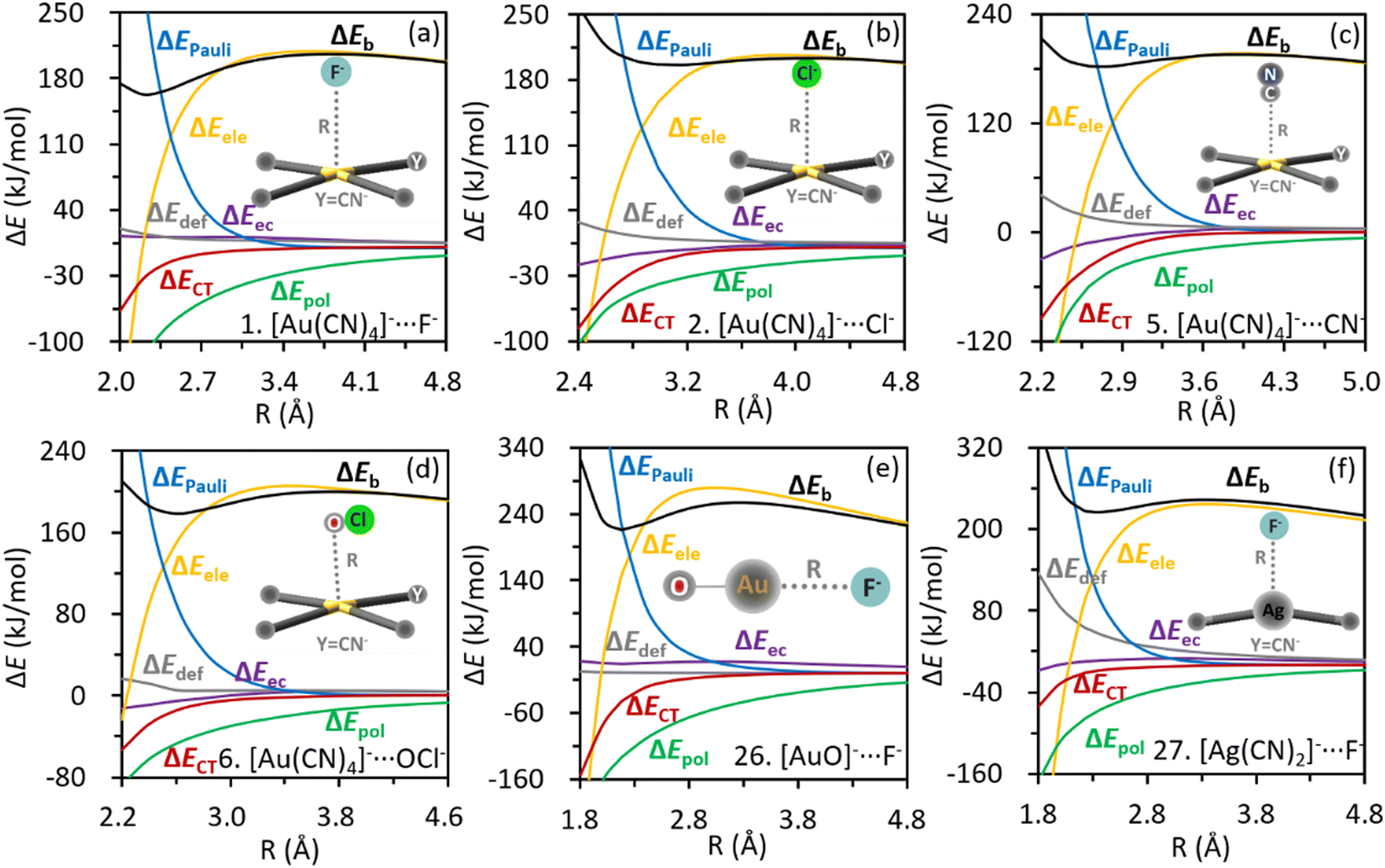 | ||
| Fig. 4 Variations of energy components along with the inter-anion distances in (a) [Au(CN)4]−⋯F−; (b) [Au(CN)4]−⋯Cl−; (c) [Au(CN)4]−⋯CN−; (d) [Au(CN)4]−⋯OCl−; (e) [AuO]−⋯F−; (f) [Ag(CN)2]−⋯F−. | ||
The origin of potential wells can be clarified by examining the differences in energy components between local minima and the corresponding TSs as listed in Table 2. Negative values mean the stabilities (lowering in energy) gained from a TS to a local minimum, following the convention in the BLW-ED scheme. All the well depths (ΔΔEb) are dominated by the electrostatic interaction, with the polarization interaction acting as the second most important factor. Both the charge transfer and electron correlation contribute positively to the kinetic stability, while Pauli exchange repulsion and deformation tend to shallow the potential wells. The governing role of electrostatic interactions is in line with its decreasing trend at short ranges as shown in Fig. 4. The positive role of polarization can be rationalized by the considerable polarizability of the coinage donor (Table S1, ESI†). All the aforementioned contributions of physical factors were also observed in our previous studies of inter-anion hydrogen, halogen and chalcogen interactions.20,54,113 The electrons are shifted from F− or OCl− to the metal complexes according to the natural population analysis (NPA in Table S2, ESI†), exhibiting a similar pattern as the conventional non-covalent interactions (from Lewis base to acid). Besides, we redrew the energy profiles by using the def2-TZVPD basis set and found that the metastability and the lengths of inter-anion CiBs (Table S3, ESI†) are barely variated.
| No. | Complexes | ΔΔEdef | ΔΔEele | ΔΔEPauli | ΔΔEpol | ΔΔECT | ΔΔEec | ΔΔEb |
|---|---|---|---|---|---|---|---|---|
| 1 | [Au(CN)4]−⋯F− | 5.0 | −101.7 | 135.9 | −69.1 | −16.8 | 4.2 | −42.5 |
| 2 | [Au(CN)4]−⋯Cl− | 2.8 | −20.4 | 41.1 | −18.0 | −6.9 | −5.6 | −7.1 |
| 5 | [Au(CN)4]−⋯CN− | 7.2 | −80.9 | 124.3 | −29.9 | −23.0 | −10.9 | −13.3 |
| 6 | [Au(CN)4]−⋯OCl− | 3.6 | −53.4 | 89.1 | −33.8 | −14.2 | −9.6 | −18.3 |
| 26 | [AuO]−⋯F− | 0.7 | −121.9 | 198.1 | −77.2 | −36.9 | −3.0 | −40.2 |
| 27 | [Ag(CN)2]−⋯F− | 26.3 | −77.0 | 82.1 | −40.6 | −6.6 | −2.0 | −17.7 |
The potential well vanishes when Ag(I) in [Ag(CN)2]−⋯F− (complex 27) is replaced by Au(I) (complex 25). We analyzed the role of the central metal by comparing individual energy terms in complexes 27 and 25 at a series of fixed inter-anion distances, ranging from the local minimum of [Ag(CN)2]−⋯F− to the TS. Fig. 5a illustrates the correlations of important energy components. The electrostatic interactions in both cases are pretty close within the interval of distances selected, according to the linear correlation with a slope of 1.0. Interestingly, the [Au(CN)2]−⋯F− even exhibits more stabilizing polarization and charge transfer interactions than [Ag(CN)2]−⋯F− (slopes greater than one). This is understandable because [Au(CN)2]− has a higher polarizability than [Ag(CN)2]− (Table S1, ESI†), and therefore, stronger polarization. Moreover, Au(I) has a larger size than Ag(I), leading to stronger inter-anion orbital overlap. Nevertheless, the enhanced orbital overlap also increases the destabilizing Pauli exchange repulsion, which eventually leads to the absence of metastability in [Au(CN)4]−⋯F−. We also compared the BLW-ED results of the [Au(CN)4]− dimer with the complex [Au(CN)4]−⋯F− (Fig. 5b). In general, from the TS to the local minimum of [Au(CN)4]−⋯F−, the variation of the overall stabilizing factors is −187.6 kJ mol−1, which is almost the same as the corresponding change in the [Au(CN)4]− dimer (−183.3 kJ mol−1). However, the enhancement in Pauli exchange repulsion is much greater in the [Au(CN)4]− dimer (240.5 vs. 135.9 kJ mol−1) and eventually erases the metastability. Liner correlations between energy components of these two complexes were also observed (Fig. 5b). Comparable electrostatic interactions were found, while stronger charge transfer interaction was discovered in the [Au(CN)4]− dimer. It is reasonable because F− is the weakest electron donor studied in this work. F− has more a concentrated electron density distribution than [Au(CN)4]−, and therefore imposes a stronger electric field to [Au(CN)4]−, leading to stronger polarization in [Au(CN)4]−⋯F−.
 | ||
| Fig. 5 Correlations of energy components calculated at a series of fixed inter-anion distances between complexes with and without kinetic stability: (a) [Au(CN)2]−⋯F−vs. [Ag(CN)2]−⋯F− and (b) [Au(CN)4]− dimer Vs [Au(CN)4]−⋯F−. | ||
4. Conclusions
Binding energy profiles of thirty-one anion pairs involving CiBs were explored along the inter-anion distances, employing state-of-the-art theoretical methods. It turns out that the anions [Au(CN)4]−, [Ag(CN)2]− and [AuO]− are all possible candidates for the formation of inter-anion CiBs in vacuum. The kinetic stability was further supported by the AIMD simulations, the QTAIM theory and the local vibrational analyses. Two general characters can be derived from the cases with metastability. One is the coordination of CN− on the coinage metal, and the other is the small size of the anion acting as the Lewis base. This is understandable because cyanide usually leads to a remarkable electron deficiency (relatively) of the metal, and the anions with small radius may approach the central metal closely.The inter-anion CiBs in [AuCl4]−⋯[AuCl4]− and [Au(CN)4]−⋯[Au(CN)4]− complexes identified in experimentally obtained crystal structures were proved to be absent in vacuum, but slightly attractive when the crystal environment is simulated with the SMD method. However, it should be emphasized that the intrinsic strength of the inter-anion interaction was barely variated by the environment, and the changes of the binding energy profiles originate from the solute–solvent term, which stands for the interaction between the anion pair and the crystal environment in this work.
Variations in individual energy components along the inter-anion distances were inspected and the distinction of inter-anion CiBs from conventional non-covalent interactions was found to be the electrostatic interaction, which variates nonmonotonically in anion⋯anion complexes. The distinctive nonmonotonicity of electrostatic profiles was captured by the classical atomic multipole expansion, offering us a concise model to rationalize and simulate the electrostatic interaction in these counterintuitive phenomena. The electrostatic interaction also governs the well-depths. In addition, the polarization, charge transfer and electron correlation all prefer the local minima over TSs, while the deformation energy, and especially the Pauli exchange repulsion become more destabilizing from TSs to local minima, playing negative roles in the well-depths. The significance of Pauli exchange repulsion in the metastability was highlighted by comparing the BLW-ED results of cases with and without metastability at a series of fixed inter-anion distances. In detail, from [Ag(CN)2]−⋯F− to [Au(CN)2]−⋯F−, the increasing size of the central metal strengthens the destabilizing Pauli exchange repulsion, which eliminates the potential well. Similarly, the Pauli exchange repulsion is remarkably enhanced when F− in [Au(CN)4]−⋯F− is replaced by [Au(CN)4]−, leading to the absence of metastability.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
CW acknowledges the support from the Natural Science Foundation of China (No. 22073060). This work was performed in part at the Joint School of Nanoscience and Nanoengineering, a member of the National Nanotechnology Coordinated Infrastructure (NNCI), which is supported by the National Science Foundation (Grant ECCS-2025462).Notes and references
- A. Daolio, A. Pizzi, M. Calabrese, G. Terraneo, S. Bordignon, A. Frontera and G. Resnati, Angew. Chem., Int. Ed., 2021, 60, 20723–20727 CrossRef CAS PubMed.
- A. Bauza, I. Alkorta, J. Elguero, T. J. Mooibroek and A. Frontera, Angew. Chem., Int. Ed., 2020, 59, 17482–17487 CrossRef CAS PubMed.
- A. Bauza, T. J. Mooibroek and A. Frontera, Angew. Chem., Int. Ed., 2013, 52, 12317–12321 CrossRef CAS PubMed.
- J. Fanfrlik, A. Prada, Z. Padelkova, A. Pecina, J. Machacek, M. Lepsik, J. Holub, A. Ruzicka, D. Hnyk and P. Hobza, Angew. Chem., Int. Ed., 2014, 53, 10139–10142 CrossRef CAS PubMed.
- S. Zahn, R. Frank, E. Hey-Hawkins and B. Kirchner, Chem. – Eur. J., 2011, 17, 6034–6038 CrossRef CAS PubMed.
- A. Daolio, A. Pizzi, G. Terraneo, A. Frontera and G. Resnati, ChemPhysChem, 2021, 22, 2281–2285 CrossRef CAS PubMed.
- I. Alkorta, J. Elguero and A. Frontera, Crystals, 2020, 10, 180 CrossRef CAS.
- S. Mirdya, A. Frontera and S. Chattopadhyay, CrystEngComm, 2019, 21, 6859–6868 RSC.
- L. Guan and Y. Mo, J. Phys. Chem. A, 2014, 118, 8911–8921 CrossRef CAS PubMed.
- A. C. Legon, Phys. Chem. Chem. Phys., 2010, 12, 7736–7747 RSC.
- A. C. Legon and N. R. Walker, Phys. Chem. Chem. Phys., 2018, 20, 19332–19338 RSC.
- A. Frontera and A. Bauza, Chem. – Eur. J., 2018, 24, 7228–7234 CrossRef CAS PubMed.
- R. Wang, Z. Wang, X. Yu and Q. Li, ChemPhysChem, 2020, 21, 2426–2431 CrossRef CAS PubMed.
- A. Daolio, A. Pizzi, G. Terraneo, M. Ursini, A. Frontera and G. Resnati, Angew. Chem., Int. Ed., 2021, 60, 14385–14389 CrossRef CAS PubMed.
- A. Pizzi, M. Calabrese, A. Daolio, M. Ursini, A. Frontera and G. Resnati, CrystEngComm, 2022, 24, 3846–3851 RSC.
- M. L. N. Pina, A. Frontera and A. Bauza, J. Phys. Chem. Lett., 2020, 11, 8259–8263 CrossRef CAS PubMed.
- J. Halldin Stenlid, A. J. Johansson and T. Brinck, Phys. Chem. Chem. Phys., 2018, 20, 2676–2692 RSC.
- F. Weinhold and R. A. Klein, Angew. Chem., Int. Ed., 2014, 53, 11214–11217 CrossRef CAS PubMed.
- X. Zhang, H. Dai, H. Yan, W. Zou and D. Cremer, J. Am. Chem. Soc., 2016, 138, 4334–4337 CrossRef CAS PubMed.
- C. Wang, Y. Fu, L. Zhang, D. Danovich, S. Shaik and Y. Mo, J. Comput. Chem., 2018, 39, 481–487 CrossRef CAS PubMed.
- G. Frenking and G. F. Caramori, Angew. Chem., Int. Ed., 2015, 54, 2596–2599 CrossRef CAS PubMed.
- I. Mata, I. Alkorta, E. Molins and E. Espinosa, ChemPhysChem, 2012, 13, 1421–1424 CrossRef CAS PubMed.
- M. O. Miranda, D. J. R. Duarte and I. Alkorta, ChemPhysChem, 2020, 21, 1052–1059 CrossRef CAS PubMed.
- L. M. Azofra, J. Elguero and I. Alkorta, J. Phys. Chem. A, 2020, 124, 2207–2214 CrossRef CAS PubMed.
- I. Mata, E. Molins, I. Alkorta and E. Espinosa, J. Phys. Chem. A, 2015, 119, 183–194 CrossRef CAS PubMed.
- I. Iribarren, M. M. Montero-Campillo, I. Alkorta, J. Elguero and D. Quinonero, Phys. Chem. Chem. Phys., 2019, 21, 5796–5802 RSC.
- E. M. Fatila, E. B. Twum, A. Sengupta, M. Pink, J. A. Karty, K. Raghavachari and A. H. Flood, Angew. Chem., Int. Ed., 2016, 55, 14057–14062 CrossRef CAS PubMed.
- J. S. McNally, X. P. Wang, C. Hoffmann and A. D. Wilson, Chem. Commun., 2017, 53, 10934–10937 RSC.
- E. M. Fatila, E. B. Twum, J. A. Karty and A. H. Flood, Chem. – Eur. J., 2017, 23, 10652–10662 CrossRef CAS PubMed.
- W. Zhao, A. H. Flood and N. G. White, Chem. Soc. Rev., 2020, 49, 7893–7906 RSC.
- N. G. White, CrystEngComm, 2019, 21, 4855–4858 RSC.
- T. S. C. MacDonald, B. L. Feringa, W. S. Price, S. J. Wezenberg and J. E. Beves, J. Am. Chem. Soc., 2020, 142, 20014–20020 CrossRef CAS PubMed.
- F. Weinhold, Angew. Chem., Int. Ed., 2003, 42, 4188–4194 CrossRef CAS.
- F. Weinhold, Molecules, 2022, 27, 377 CrossRef CAS PubMed.
- J. M. Holthoff, E. Engelage, R. Weiss and S. M. Huber, Angew. Chem., Int. Ed., 2020, 59, 11150–11157 CrossRef CAS PubMed.
- C. Loy, J. M. Holthoff, R. Weiss, S. M. Huber and S. V. Rosokha, Chem. Sci., 2021, 12, 8246–8251 RSC.
- D. Quinonero, I. Alkorta and J. Elguero, Phys. Chem. Chem. Phys., 2016, 18, 27939–27950 RSC.
- A. Strate, T. Niemann, D. Michalik and R. Ludwig, Angew. Chem., Int. Ed., 2017, 56, 496–500 CrossRef CAS PubMed.
- R. Barbas, R. Prohens, A. Bauza, A. Franconetti and A. Frontera, Chem. Commun., 2019, 55, 115–118 RSC.
- D. A. Cullen, M. G. Gardiner and N. G. White, Chem. Commun., 2019, 55, 12020–12023 RSC.
- R. Prohens, A. Portell, M. Font-Bardia, A. Bauza and A. Frontera, Chem. Commun., 2018, 54, 1841–1844 RSC.
- F. Zapata, L. Gonzalez, A. Bastida, D. Bautista and A. Caballero, Chem. Commun., 2020, 56, 7084–7087 RSC.
- G. Dos Passos Gomes, G. Xu, X. Zhu, L. M. Chamoreau, Y. Zhang, O. Bistri-Aslanoff, S. Roland, I. V. Alabugin and M. Sollogoub, Chem. – Eur. J., 2021, 27, 8127–8142 CrossRef CAS PubMed.
- J. M. Holthoff, R. Weiss, S. V. Rosokha and S. M. Huber, Chem. – Eur. J., 2021, 27, 16530–16542 CrossRef CAS PubMed.
- E. M. Fatila, M. Pink, E. B. Twum, J. A. Karty and A. H. Flood, Chem. Sci., 2018, 9, 2863–2872 RSC.
- A. Knorr, P. Stange, K. Fumino, F. Weinhold and R. Ludwig, ChemPhysChem, 2016, 17, 458–462 CrossRef CAS PubMed.
- T. Niemann, P. Stange, A. Strate and R. Ludwig, ChemPhysChem, 2018, 19, 1691–1695 CrossRef CAS PubMed.
- D. Quinonero, I. Alkorta and J. Elguero, ChemPhysChem, 2020, 21, 1597–1607 CrossRef CAS PubMed.
- R. Wysokinski, W. Zierkiewicz, M. Michalczyk and S. Scheiner, ChemPhysChem, 2020, 21, 1119–1125 CrossRef CAS PubMed.
- R. Wysokinski, W. Zierkiewicz, M. Michalczyk and S. Scheiner, ChemPhysChem, 2021, 22, 818–821 CrossRef CAS PubMed.
- W. Zierkiewicz, R. Wysokinski, M. Michalczyk and S. Scheiner, ChemPhysChem, 2020, 21, 870–877 CrossRef CAS PubMed.
- F. Weinhold, Inorg. Chem., 2018, 57, 2035–2044 CrossRef CAS PubMed.
- W. Zhao, J. Tropp, B. Qiao, M. Pink, J. D. Azoulay and A. H. Flood, J. Am. Chem. Soc., 2020, 142, 2579–2591 CrossRef CAS PubMed.
- D. Fan, L. Chen, C. Wang, S. Yin and Y. Mo, J. Chem. Phys., 2021, 155, 234302 CrossRef CAS PubMed.
- S. Scheiner, J. Chem. Phys., 2020, 153, 140901 CrossRef PubMed.
- S. Scheiner, R. Wysokinski, M. Michalczyk and W. Zierkiewicz, J. Phys. Chem. A, 2020, 124, 4998–5006 CrossRef CAS PubMed.
- J. Chen, K. Qian, K. Xiao, J. Luo, H. Li, T. Ma, U. Kortz, M. Tsige and T. Liu, Langmuir, 2020, 36, 10519–10527 CrossRef CAS PubMed.
- F. Khorrami and M. H. Kowsari, J. Phys. Chem. B, 2020, 124, 3770–3783 CrossRef CAS PubMed.
- A. Grabarz, M. Michalczyk, W. Zierkiewicz and S. Scheiner, Molecules, 2021, 26, 2116 CrossRef CAS PubMed.
- J. K. Philipp and R. Ludwig, Molecules, 2020, 25, 4972 CrossRef CAS PubMed.
- L. M. Azofra, J. Elguero and I. Alkorta, Phys. Chem. Chem. Phys., 2020, 22, 11348–11353 RSC.
- T. Niemann, P. Stange, A. Strate and R. Ludwig, Phys. Chem. Chem. Phys., 2019, 21, 8215–8220 RSC.
- R. Wysokinski, W. Zierkiewicz, M. Michalczyk and S. Scheiner, Phys. Chem. Chem. Phys., 2021, 23, 13853–13861 RSC.
- A. Gogoi, D. Dutta, A. K. Verma, H. Nath, A. Frontera, A. K. Guha and M. K. Bhattacharyya, Polyhedron, 2019, 168, 113–126 CrossRef CAS.
- A. Knorr and R. Ludwig, Sci. Rep., 2015, 5, 17505 CrossRef PubMed.
- S. Scheiner, J. Comput. Chem., 2022, 43, 1814–1824 CrossRef CAS PubMed.
- L. Andreo, R. M. Gomila, E. Priola, A. Giordana, S. Pantaleone, E. Diana, G. Mahmoudi and A. Frontera, Cryst. Growth Des., 2022, 22, 6539–6544 CrossRef CAS PubMed.
- I. Mata, E. Molins, I. Alkorta and E. Espinosa, J. Chem. Phys., 2009, 130, 044104 CrossRef PubMed.
- R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, Oxford, UK, 1990 Search PubMed.
- F. Weinhold and C. R. Landis, Chem. Educ. Res. Pract., 2001, 2, 91–104 RSC.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- F. Weinhold and C. R. Landis, Discovering Chemistry With Natural Bond Orbitals, Wiley, Hoboken, NJ, 2012 Search PubMed.
- F. Weinhold, C. R. Landis and E. D. Glendening, Int. Rev. Phys. Chem., 2016, 35, 399–440 Search PubMed.
- E. D. Glendening, C. R. Landis and F. Weinhold, J. Comput. Chem., 2019, 40, 2234–2241 CrossRef CAS PubMed.
- E. D. Glendening, C. R. Landis and F. Weinhold, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 2, 1–42 Search PubMed.
- G. Ohanessian and W. A. Goddard, Acc. Chem. Res., 1990, 23, 386–392 CrossRef CAS.
- M. Kaupp, Angew. Chem., Int. Ed., 2001, 40, 3534–3565 CrossRef CAS PubMed.
- S. Radenković, D. Danovich, S. Shaik, P. C. Hiberty and B. Braïda, Comput. Theor. Chem., 2017, 1116, 195–201 CrossRef.
- J. Joy, D. Danovich, M. Kaupp and S. Shaik, J. Am. Chem. Soc., 2020, 142, 12277–12287 CrossRef CAS PubMed.
- J. B. Mann, T. L. Meek, E. T. Knight, J. F. Capitani and L. C. Allen, J. Am. Chem. Soc., 2000, 122, 5132–5137 CrossRef CAS.
- D. G. Liakos and F. Neese, J. Chem. Theory Comput., 2011, 7, 1511–1523 CrossRef CAS PubMed.
- M. Kaupp, J. Comput. Chem., 2007, 28, 320–325 CrossRef CAS PubMed.
- J. M. Galbraith, A. Shurki and S. Shaik, J. Phys. Chem. A, 2000, 104, 1262–1270 CrossRef CAS.
- S. Dordevic, S. Radenkovic, S. Shaik and B. Braida, Molecules, 2022, 27, 490 CrossRef PubMed.
- C. J. Evans, A. Lesarri and M. C. L. Gerry, J. Am. Chem. Soc., 2000, 122, 6100–6105 CrossRef CAS.
- J. M. Michaud and M. C. L. Gerry, J. Am. Chem. Soc., 2006, 128, 7613–7621 CrossRef CAS PubMed.
- J. M. Thomas, N. R. Walker, S. A. Cooke and M. C. L. Gerry, J. Am. Chem. Soc., 2004, 126, 1235–1246 CrossRef CAS PubMed.
- C. J. Evans and M. C. L. Gerry, J. Chem. Phys., 2000, 112, 9363–9674 CrossRef CAS.
- C. J. Evans and M. C. L. Gerrya, J. Chem. Phys., 2000, 112, 1321–1329 CrossRef CAS.
- C. J. Evans, D. S. Rubinoff and M. C. L. Gerry, Phys. Chem. Chem. Phys., 2000, 2, 3943–3948 RSC.
- S. G. Francis, S. L. Matthews, O. Poleshchuk, N. R. Walker and A. C. Legon, Angew. Chem., Int. Ed., 2006, 45, 6341–6343 CrossRef CAS PubMed.
- S. J. Harris, A. C. Legon, N. R. Walker and D. E. Wheatley, Angew. Chem., Int. Ed., 2010, 49, 181–183 CrossRef CAS PubMed.
- C. Medcraft, E. Gougoula, D. M. Bittner, J. C. Mullaney, S. Blanco, D. P. Tew, N. R. Walker and A. C. Legon, J. Chem. Phys., 2017, 147, 234308 CrossRef PubMed.
- S. L. Stephens, W. Mizukami, D. P. Tew, N. R. Walker and A. C. Legon, J. Chem. Phys., 2012, 136, 064306 CrossRef PubMed.
- J. C. Mullaney, S. L. Stephens, D. P. Zaleski, M. J. Sprawling, D. P. Tew, N. R. Walker and A. C. Legon, J. Phys. Chem. A, 2015, 119, 9636–9643 CrossRef CAS PubMed.
- A. Terron, J. Buils, T. J. Mooibroek, M. Barcelo-Oliver, A. Garcia-Raso, J. J. Fiol and A. Frontera, Chem. Commun., 2020, 56, 3524–3527 RSC.
- J. H. Stenlid and T. Brinck, J. Am. Chem. Soc., 2017, 139, 11012–11015 CrossRef CAS PubMed.
- W. Zierkiewicz, M. Michalczyk and S. Scheiner, Phys. Chem. Chem. Phys., 2018, 20, 22498–22509 RSC.
- C. Trujillo, G. Sánchez-Sanz, J. Elguero and I. Alkorta, Struct. Chem., 2020, 31, 1909–1918 CrossRef CAS.
- C. J. Shorrock, H. Jong, R. J. Batchelor and D. B. Leznoff, Inorg. Chem., 2003, 42, 3917–3924 CrossRef CAS PubMed.
- A. R. Geisheimer, J. E. Wren, V. K. Michaelis, M. Kobayashi, K. Sakai, S. Kroeker and D. B. Leznoff, Inorg. Chem., 2011, 50, 1265–1274 CrossRef CAS PubMed.
- D. Cremer and E. Kraka, Dalton Trans., 2017, 46, 8323–8338 RSC.
- E. Kraka and D. Cremer, Int. J. Quantum Chem., 2019, 119, e25849 CrossRef.
- Y. Tao, W. Zou, S. Nanayakkara and E. Kraka, J. Chem. Theory Comput., 2022, 18, 1821–1837 CrossRef CAS PubMed.
- D. Cremer, A. Wu, A. Larsson and E. Kraka, J. Mol. Model., 2000, 6, 396–412 CrossRef CAS.
- Y. Tao, W. Zou and E. Kraka, Chem. Phys. Lett., 2017, 685, 251–258 CrossRef CAS.
- V. Oliveira, E. Kraka and D. Cremer, Inorg. Chem., 2017, 56, 488–502 CrossRef CAS PubMed.
- W. Zou, X. Zhang, H. Dai, H. Yan, D. Cremer and E. Kraka, J. Organomet. Chem., 2018, 865, 114–127 CrossRef CAS.
- R. Kalescky, E. Kraka and D. Cremer, J. Phys. Chem. A, 2013, 117, 8981–8995 CrossRef CAS PubMed.
- V. Oliveira, D. Cremer and E. Kraka, J. Phys. Chem. A, 2017, 121, 6845–6862 CrossRef CAS PubMed.
- D. Setiawan, E. Kraka and D. Cremer, J. Phys. Chem. A, 2015, 119, 1642–1656 CrossRef CAS PubMed.
- D. Sethio, V. Oliveira and E. Kraka, Molecules, 2018, 23, 2763 CrossRef PubMed.
- L. Chen, Q. Feng, C. Wang, S. Yin and Y. Mo, J. Phys. Chem. A, 2021, 125, 10428–10438 CrossRef CAS PubMed.
- S. Shaik, D. Danovich, W. Wu and P. C. Hiberty, Nat. Chem., 2009, 1, 443–449 CrossRef CAS PubMed.
- F. Jimenez-Gravalos and D. Suarez, J. Chem. Theory Comput., 2021, 17, 4981–4995 CrossRef CAS PubMed.
- T. Xu, W. Wang and S. Yin, J. Chem. Theory Comput., 2018, 14, 3728–3739 CrossRef CAS PubMed.
- S. Grimme, C. Muck-Lichtenfeld, G. Erker, G. Kehr, H. Wang, H. Beckers and H. Willner, Angew. Chem., Int. Ed., 2009, 48, 2592–2595 CrossRef CAS PubMed.
- Y. Mo, J. Phys. Chem. A, 2012, 116, 5240–5246 CrossRef CAS PubMed.
- E. Cerpa, A. Krapp, A. Vela and G. Merino, Chem. – Eur. J., 2008, 14, 10232–10234 CrossRef CAS PubMed.
- L. J. Farrugia, C. Evans and M. Tegel, J. Phys. Chem. A, 2006, 110, 7952–7961 CrossRef CAS PubMed.
- P. Cassam-Chenaï and D. Jayatilaka, Theor. Chem. Acc., 2001, 105, 213–218 Search PubMed.
- F. M. Bickelhaupt and E. J. Baerends, Angew. Chem., 2003, 115, 4315–4320 CrossRef.
- P. R. Schreiner, Angew. Chem., Int. Ed., 2002, 41, 3579–3582 CrossRef CAS PubMed.
- Y. Mo, W. Wu, L. Song, M. Lin, Q. Zhang and J. Gao, Angew. Chem., Int. Ed., 2004, 43, 1986–1990 CrossRef CAS PubMed.
- A. J. Stone, J. Phys. Chem. A, 2017, 121, 1531–1534 CrossRef CAS PubMed.
- P. R. Horn, Y. Mao and M. Head-Gordon, Phys. Chem. Chem. Phys., 2016, 18, 23067–23079 RSC.
- I. Alkorta, I. Mata, E. Molins and E. Espinosa, Chem. – Eur. J., 2016, 22, 9226–9234 CrossRef CAS PubMed.
- A. Mele, G. Romano, M. Giannone, E. Ragg, G. Fronza, G. Raos and V. Marcon, Angew. Chem., Int. Ed., 2006, 45, 1123–1126 CrossRef CAS PubMed.
- M. Holz and K. J. Patil, Ber. Bunsenges. Phys. Chem., 1991, 95, 107–113 CrossRef CAS.
- S. Grimme and J. P. Djukic, Inorg. Chem., 2011, 50, 2619–2628 CrossRef CAS PubMed.
- O. Shih, A. H. England, G. C. Dallinger, J. W. Smith, K. C. Duffey, R. C. Cohen, D. Prendergast and R. J. Saykally, J. Chem. Phys., 2013, 139, 035104 CrossRef PubMed.
- K. Morokuma, Acc. Chem. Res., 1977, 10, 294–300 CrossRef CAS.
- R. Z. Khaliullin, A. T. Bell and M. Head-Gordon, Chem. – Eur. J., 2009, 15, 851–855 CrossRef CAS PubMed.
- B. Jeziorski, R. Moszynski and K. Szalewicz, Chem. Rev., 1994, 94, 1887–1930 CrossRef CAS.
- K. Kitaura and K. Morokuma, Int. J. Quantum Chem., 1976, 10, 325–340 CrossRef CAS.
- R. Z. Khaliullin, A. T. Bell and M. Head-Gordon, J. Chem. Phys., 2008, 128, 184112 CrossRef PubMed.
- P. Su and H. Li, J. Chem. Phys., 2009, 131, 014102 CrossRef PubMed.
- P. Su, H. Liu and W. Wu, J. Chem. Phys., 2012, 137, 034111 CrossRef PubMed.
- W. Chen and M. S. Gordon, J. Phys. Chem., 1996, 100, 14316–14328 CrossRef CAS.
- R. Z. Khaliullin, E. A. Cobar, R. C. Lochan, A. T. Bell and M. Head-Gordon, J. Phys. Chem. A, 2007, 111, 8753–8765 CrossRef CAS PubMed.
- P. Su, Z. Jiang, Z. Chen and W. Wu, J. Phys. Chem. A, 2014, 118, 2531–2542 CrossRef CAS PubMed.
- P. Su, Z. Tang and W. Wu, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2020, 10, e1460 CAS.
- D. M. Andrada and C. Foroutan-Nejad, Phys. Chem. Chem. Phys., 2020, 22, 22459–22464 RSC.
- J. Poater, D. M. Andrada, M. Sola and C. Foroutan-Nejad, Phys. Chem. Chem. Phys., 2022, 24, 2344–2348 RSC.
- C. D. Sherrill, Acc. Chem. Res., 2013, 46, 1020–1028 CrossRef CAS PubMed.
- M. O. Sinnokrot and C. D. Sherrill, J. Am. Chem. Soc., 2004, 126, 7690–7697 CrossRef CAS PubMed.
- Y. Geng, T. Takatani, E. G. Hohenstein and C. D. Sherrill, J. Phys. Chem. A, 2010, 114, 3576–3582 CrossRef CAS PubMed.
- S. A. Bojarowski, P. Kumar, C. M. Wandtke, B. Dittrich and P. M. Dominiak, J. Chem. Theory Comput., 2018, 14, 6336–6345 CrossRef CAS PubMed.
- C. Liu, J. P. Piquemal and P. Ren, J. Chem. Theory Comput., 2019, 15, 4122–4139 CrossRef CAS PubMed.
- Q. Wang, J. A. Rackers, C. He, R. Qi, C. Narth, L. Lagardere, N. Gresh, J. W. Ponder, J. P. Piquemal and P. Ren, J. Chem. Theory Comput., 2015, 11, 2609–2618 CrossRef CAS PubMed.
- A. J. Stone, J. Phys. Chem. A, 2011, 115, 7017–7027 CrossRef CAS PubMed.
- W. Wu, P. Su, S. Shaik and P. C. Hiberty, Chem. Rev., 2011, 111, 7557–7593 CrossRef CAS PubMed.
- Z. Chen and W. Wu, J. Chem. Phys., 2020, 153, 090902 CrossRef CAS PubMed.
- Y. Mo and S. D. Peyerimhoff, J. Chem. Phys., 1998, 109, 1687–1697 CrossRef CAS.
- Y. Mo, J. Chem. Phys., 2003, 119, 1300–1306 CrossRef CAS.
- Y. Mo, L. Song and Y. Lin, J. Phys. Chem. A, 2007, 111, 8291–8301 CrossRef CAS PubMed.
- Y. Mo, J. Gao and S. D. Peyerimhoff, J. Chem. Phys., 2000, 112, 5530–5538 CrossRef CAS.
- Y. Mo, P. Baob and J. Gao, Phys. Chem. Chem. Phys., 2011, 13, 6760–6775 RSC.
- M. De Santis, S. Rampino, L. Storchi, L. Belpassi and F. Tarantelli, Inorg. Chem., 2019, 58, 11716–11729 CrossRef CAS PubMed.
- M. A. Rawashdeh-Omary, M. A. Omary and H. H. Patterson, J. Am. Chem. Soc., 2000, 122, 10371–10380 CrossRef CAS.
- R. Zhang, Y. Yu, T. C. Steimle and L. Cheng, J. Chem. Phys., 2017, 146, 064307 CrossRef PubMed.
- T. Ichino, A. J. Gianola, D. H. Andrews and W. C. Lineberger, J. Phys. Chem. A, 2004, 108, 11307–11313 CrossRef CAS.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- Y. Tao, W. Zou, D. Sethio, N. Verma, Y. Qiu, C. Tian, D. Cremer and E. Kraka, J. Chem. Theory Comput., 2019, 15, 1761–1776 CrossRef CAS PubMed.
- E. Kraka, W. Zou and Y. Tao, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2020, 10, e1480 CAS.
- W. Zou, Y. Tao, M. Freindorf, D. Cremer and E. Kraka, Chem. Phys. Lett., 2020, 748, 137337 CrossRef CAS.
- Y. Minenkov, E. Chermak and L. Cavallo, J. Chem. Theory Comput., 2015, 11, 4664–4676 CrossRef CAS PubMed.
- C. Geng, S. Ye and F. Neese, Angew. Chem., Int. Ed., 2010, 49, 5717–5720 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2007, 120, 215–241 Search PubMed.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- J. D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- S. Grimme, J. Chem. Phys., 2003, 118, 9095–9102 CrossRef CAS.
- T. Shen, Z. Zhu, I. Y. Zhang and M. Scheffler, J. Chem. Theory Comput., 2019, 15, 4721–4734 CrossRef CAS PubMed.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. Su, T. L. Windus, M. Dupuis and J. A. Montgomery Jr, J. Comput. Chem., 1993, 11, 1347–1363 CrossRef.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 2, 73–78 Search PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1606 Search PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- A. J. Stone, J. Chem. Theory Comput., 2005, 1, 1128–1132 CrossRef CAS PubMed.
- P. Ren and J. W. Ponder, J. Phys. Chem. B, 2003, 107, 5933–5947 CrossRef CAS.
- J. W. Ponder, C. Wu, P. Ren, V. S. Pande, J. D. Chodera, M. J. Schnieders, I. Haque, D. L. Mobley, D. S. Lambrecht, J. Robert, A. DiStasio, M. Head-Gordon, G. N. I. Clark, M. E. Johnson and T. Head-Gordon, J. Phys. Chem. B, 2010, 114, 2549–2564 CrossRef CAS PubMed.
- P. Ren, C. Wu and J. W. Ponder, J. Chem. Theory Comput., 2011, 7, 3143–3161 CrossRef CAS PubMed.
- Y. Shi, Z. Xia, J. Zhang, R. Best, C. Wu, J. W. Ponder and P. Ren, J. Chem. Theory Comput., 2013, 9, 4046–4063 CrossRef CAS PubMed.
- J. A. Rackers, Z. Wang, C. Lu, M. L. Laury, L. Lagardere, M. J. Schnieders, J. P. Piquemal, P. Ren and J. W. Ponder, J. Chem. Theory Comput., 2018, 14, 5273–5289 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp00978e |
| This journal is © the Owner Societies 2023 |