
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tetrel bonds involving a CF3 group participate in protein–drug recognition: a combined crystallographic and computational study†
María de las Nieves
Piña
a,
Akshay Kumar
Sahu
bc,
Antonio
Frontera
 a,
Himansu S.
Biswal
*bc and
Antonio
Bauzá
*a
a,
Himansu S.
Biswal
*bc and
Antonio
Bauzá
*a
aDepartment of Chemistry, Universitat de les Illes Balears, Crts de Valldemossa km 7.6, 07122 Palma de Mallorca, Baleares, Spain. E-mail: antonio.bauza@uib.es
bSchool of Chemical Sciences, National Institute of Science Education and Research (NISER), PO-Bhimpur-Padanpur, Via-Jatni, Khurda 752050, Bhubaneswar, India. E-mail: himansu@niser.ac.in
cHomi Bhabha National Institute, Training School Complex, Anushakti Nagar, Mumbai 400094, India
First published on 10th April 2023
Abstract
In this study, the ability of CF3 groups to bind to the electron-rich side chains and backbone groups of proteins has been investigated by combining a Protein Data Bank (PDB) survey and ab initio quantum mechanics calculations. More precisely, an inspection of the PDB involving organic ligands containing a CF3 group and electron-rich atoms (A = N, O and S) in the vicinity revealed 419 X-ray structures exhibiting CF3⋯A tetrel bonds (TtBs). In a posterior stage, those hits that exhibited the most relevant features in terms of directionality and intermolecular distance were selected for theoretical calculations at the RI-MP2/def2-TZVPD level of theory. Also, Hammett's regression plots of several TtB complexes involving meta- and para-substituted benzene derivatives were computed to shed light on the substituent effects. Moreover, the TtBs were characterized through several state-of-the-art computational techniques, such as the Quantum Theory of Atoms in Molecules (QTAIM) and Noncovalent Interactions plot (NCIplot) methodologies. We believe that the results gathered from our study will be useful for rational drug design and biological communities as well as for further expanding the role of this interaction to biomedical applications.
Introduction
During the last decade, noncovalent interactions (NCIs) have undergone a fast-growing revolution which has led them to become essential resources in the chemist's toolbox owing to their crucial role in several fields of modern chemistry, such as supramolecular chemistry,1 molecular recognition2 and crystal engineering.3 Despite the great roles that hydrogen bonding interactions (HBs) play in many chemical and biological systems,4–6 such as in enzyme catalysis and protein folding,7 other noncovalent interactions based on the p-block of elements (aerogen,8 halogen,9 chalcogen,10 pnictogen11 and tetrel bonds)12 have emerged during the last decade. These family of interactions are also known as “σ-hole interactions”, since they are based on positive electrostatic potential regions located on the extension of covalent X–Ae, X–Hal, X–Ch, X–Pn and X–Tr bonds, which are able to interact in a favourable manner with electron-rich species (i.e. a lone pair or an anion). Their study and recognition by the scientific community has led to their powerful and novel applications in the fields of rational drug design,13–15 molecular aggregation16–18 or even tuning self-assembly phenomena,19–21 among others.22In this context, tetrel bonds (TtBs), which involve a σ-hole located on an element from group IV and a Lewis base, were theoretically described by the groups of Frontera12 and Arunan in 2013.23 From these initial studies, several computational and experimental works have analysed the physical nature of the interaction as well as its impact on the fields of crystal engineering, materials science, supramolecular chemistry and enzyme chemistry.24–30 These studies typically involved the heavier tetrel atoms (Sn or Pb) since they are more polarizable than C and Si and thus, more prone to establish a stronger interaction with an electron-rich specie. However, while carbon is the lightest tetrel element, sp3 hybridized C atoms are also able to undergo quite stable TtB interactions when suitable spatial and chemical conditions are fulfilled. That normally implies the utilization of small-size substituents, which makes the σ-hole sterically accessible along with the inclusion of strong electron-withdrawing substituents (EWG), such as fluorine or cyano groups.31–36
In the PDB, more than 1200 ligands contain at least one CF3 group in their structure, which is typically incorporated to increase the polarity of the molecule or the hydrogen bonding (HB) acceptor ability through the three F atoms. This implies effective protein inhibitors for the treatment of cancer,37 dengue,38 tuberculosis,39,40 and ulcerative colitis,41 among other diseases. Interestingly, computational studies (e.g. docking, molecular dynamics as well as quantum mechanics calculations) have been demonstrated to be reliable tools for predicting protein–ligand relative binding affinities as well as for providing both physical and chemical background of the NCIs that are present in those systems.42–46 Therefore, they are also routinely used as complementary techniques to experiments and also for studying novel ways of interactions between molecules by means of the statistical analysis of the Protein Data Bank (PDB).47
In this context, our research group revealed in 2017 the existence of tetrel bonds in several biological systems.48 Building upon our previous work, in this study our goal was to (i) expand the current biological knowledge on the interaction by inspecting the entire PDB database (from this inspection several biological examples have been selected, and the biological implications of the TtB interaction have been discussed), (ii) analyse the physical nature and interplay between –CF3 tetrel bonds and other NCIs that also stabilize the protein–drug complex and (iii) study the correlation between the Hammett's σ constants for a series of TtB complexes involving meta- and para-substituted benzene derivatives and two electron-rich molecules (formaldehyde and dimethylether). We expect that the results derived from our study will be useful for those scientists devoted to the fields of supramolecular chemistry and pharmacology.
Methods
PDB inspection
Only protein structures with a resolution better than 3 Å were downloaded from the PDB.47 In total, 1661 PDB files containing at least one CF3 group were screened for the tetrel bonding interaction. The PDB files where TtB interactions were identified are gathered in a GitHub repository accessible through this link: https://github.com/tonibr9/TtB-CF3-PDB-survey.git.The following geometrical criteria were used during the search:
(1) The distance (C⋯A) between the CF3 carbon (C) and the Lewis base (A = N, O, and S) was maintained in between 2.5 Å and 4 Å. The electronegative atoms considered here belong to the amino acids and ligands. Interactions with the solvent molecules were not taken into account.
(2) The angle criteria used for the search was 120° ≤ ∠X–C⋯A ≤ 180° where X is any atom attached to the C of the CF3 group.
All the PDB analyses were carried out using an in-home written Python program. The Stride standalone program49 was used for the analysis of the protein secondary structures.
Quantum mechanics calculations
From the structures derived from the search, those showing the most promising features regarding the intermolecular TtB distance and directionality features were selected for calculations. Initially, the H atoms from the structure were optimized at the BP8650-D351/def2-SVP52 level of theory to obtain a reliable position prior to the evaluation of the interaction strength at the RI-MP253/def2-TZVPD52 level of theory by means of single point calculations. This level of theory has achieved success in accurately representing interaction energies involving both neutral and charged electron donors.54 The calculations were carried out using TURBOMOLE 7.0 software.55 The binding energy values (ΔE) were calculated as the energy difference between the complex and the isolated monomers following the supermolecule approximation (ΔEbinding = Ecomplex − EmonomerA − EmonomerB) and were corrected using the Boys and Bernardi counterpoise technique.56 The theoretical models used to compute the interaction energies on the selected biological examples were composed of the interacting amino acid capped at the two sides of the structure by methyl groups and the CF3-bearing molecule. On the other hand, complexes 11 to 30 were fully optimized at the RI-MP2/def2-TZVPD level of theory. Frequency calculations demonstrated that the geometries obtained represented true minima. The Cartesian coordinates of all theoretical models used in this study (both selected biological examples and complexes 11 to 30) are gathered in the ESI.†The MEP surfaces have been computed using the Gaussian-16 software.57 In particular, single point calculations at the MP2/def2-TZVP level of theory on the optimized geometries were performed and the .cube files obtained were processed using Gaussview 6.0 software (using a 0.001 isocontour value).58 Two MEP images were generated, one with the colour scale adjusted to the max and min of the molecule's electrostatic potential and another one (gathered inside a square) where a close value to the C σ-hole's electrostatic potential was used as an MEP max., in order to assist in the visualization of this electropositive region. The final editing of Fig. 2–4 was performed using Photoshop software. Bader's AIM theory59 has been used to analyse and describe the interactions discussed in this work using the AIMall calculation package.60 The RI-MP2/def2-TZVP level of theory was also used for the wavefunction analysis (also using Gaussian-16 software). The NCIplot61 isosurfaces acknowledge the presence of both attractive and repulsive interactions, as denoted by the sign of the second-density Hessian eigenvalue and characterized by the isosurface colour. The colour scheme is composed of a red–yellow–green–blue scale using red for repulsive (λcut+) and blue for attractive (λcut−) NCI interaction densities. Weak repulsive and weak attractive interactions are identified by yellow and green surfaces, respectively.
Results and discussion
PDB survey
We found a total number of 419 unique interactions that satisfied the mentioned geometrical criteria. The radial distribution of C⋯A distance in proteins is shown in Fig. 1a, where the angle (θ) made by A with respect to the C–X (∠X–C⋯A) bond is plotted against the distance of C to the A (![[R with combining circumflex]](https://www.rsc.org/images/entities/i_char_0052_0302.gif) C⋯A) atom, in which the sum of van der Waals radii of C and A atoms was taken as the normalization factor. The TtB distances range between 1 and 1.2 times C⋯A, disclosing that most of the structures retrieved from the PDB exhibit distances up to 20% longer than the sum of the vdW radii (ΣRvdW) (see Table S1 in the ESI† for the vdW radius values used). Also, while a great number of structures are gathered between interaction angles comprised in the region of 160°–180° (as expected for a σ-hole interaction), there is also an accumulation of hits in the region around 120°, which accounts for potential hydrogen bonding (HB) interactions with the negative fluorine atoms of the CF3 group.
C⋯A) atom, in which the sum of van der Waals radii of C and A atoms was taken as the normalization factor. The TtB distances range between 1 and 1.2 times C⋯A, disclosing that most of the structures retrieved from the PDB exhibit distances up to 20% longer than the sum of the vdW radii (ΣRvdW) (see Table S1 in the ESI† for the vdW radius values used). Also, while a great number of structures are gathered between interaction angles comprised in the region of 160°–180° (as expected for a σ-hole interaction), there is also an accumulation of hits in the region around 120°, which accounts for potential hydrogen bonding (HB) interactions with the negative fluorine atoms of the CF3 group.
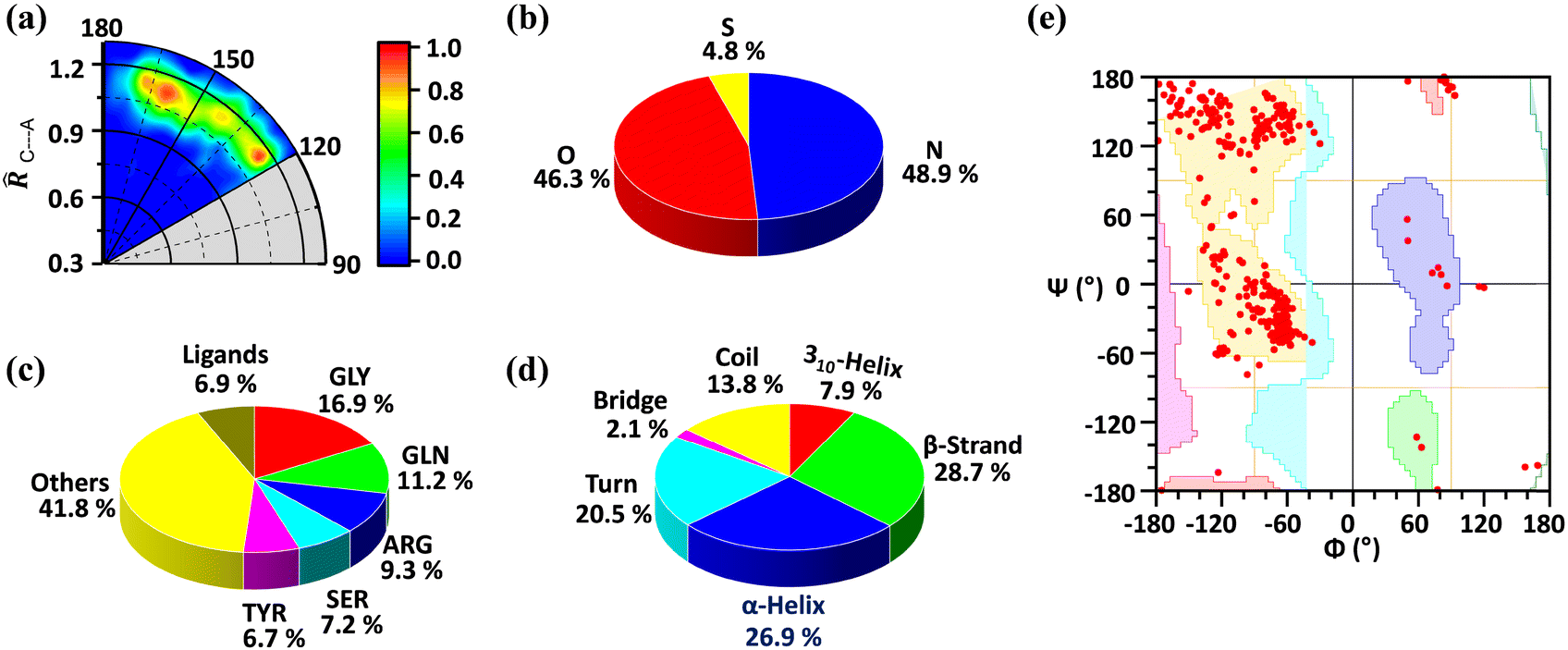 | ||
| Fig. 1 The radial distribution of C⋯A distance in proteins. The angle (θ) made by A with respect to the C–X (∠X–C⋯A) bond is plotted against the distance of C to the A (C⋯A) atom, in which the sum of van der Waals radii of C and A atoms was taken as the normalization factor (a). Pie chart for electron-rich partners involved in the carbon bonding (b). Pie chart for the amino-acid residues involved in carbon bonding (c). Pie chart of the secondary structure of the interacting amino-acid residues distribution (d). Ramachandran plot of combined N/O/S atoms belonging to the amino acid residue (e). | ||
A careful statistical analysis suggests that the C⋯O and C⋯N interactions are the prominent ones. The C⋯O interaction contributes 46.3% of the total interactions while the C⋯N interaction contributes 48.9%. Even though being less in number sulphur atoms also participate in carbon bonding (Fig. 1b). We further analysed the residues involved in this interaction and found that glycine (GLY, 16.9%) is the major contributor, followed by glutamine (GLN, 11.2%), arginine (ARG, 9.3%), serine (SER, 7.2%), and tyrosine (TYR, 6.7%). As shown in Fig. 1c rest of the amino acids contribute 41.8% and the ligand contribution is 6.9%. From these results, we can say that all the amino acids are involved in this interaction but due to its small size, glycine is preferred over other amino acids. The secondary structure analysis is shown in Fig. 1d. It suggests that this interaction is present over all the secondary structures by slightly preferring the β-strand with 28.7% and α-helix (26.9%). This is further confirmed by the Ramachandran plot as shown in Fig. 1e. The distribution of red points over all the allowed region (highlighted with different colours) shows that these interactions are present in all the secondary structure regions.
Selected examples
We have selected three examples from the PDB search to give a general view of the impact of TtBs involving protein–ligand chemistry. For each selected example, the physical nature of the TtB has been analyzed using state-of-the-art QM calculations at the RI-MP2/def2-TZVPD level of theory. This involves (i) the study of the strength and directionality of the interaction, including the interplay between the TtB and other noncovalent interactions, (ii) the calculation of the electrostatic potential value over the C atom from CF3 to demonstrate the presence of a σ-hole and (iii) the use of the QTAIM and NCIplot methodologies to demonstrate the presence and extension in real space of the TtBs studied herein.The first biological example corresponds to the structure of HER2, a protein member of the epidermal growth factor receptor (EGFR) family (PDBID 3POZ).62 This protein plays a crucial role in the regulation of cell proliferation, differentiation, and migration processes.63 In fact, several tumor families are associated with abnormal signal transduction through activated receptor tyrosine kinases belonging to the human EGFR family.64 In this context, several antibody-based drugs consisting of small molecule tyrosine kinase inhibitors that compete with ATP binding have received regulatory approval for the treatment of cancer.65,66
The study from Aertgeerts and collaborators62 reports the crystal structure of two protein–ligand complexes of EGFR with TAK-285 and SYR127063 inhibitors, which are dual HER2/EGFR inhibitors with a pyrrolo[3,2-d]pyrimidine-based backbone.67 More in particular, the TAK-285 inhibitor (03P1023 in Fig. 2a) presents a CF3 group attached to an aromatic moiety on its structure. Interestingly, in the 3POZ structure (protein–TAK 285 complex) the position of the trifluoromethylphenyl group was shifted 150° compared to that in the protein–SYR127063 complex structure, which had deep implications in binding affinity, as stated by the original authors. Two key protein residues are crucial to understanding the differences in binding mode and affinity observed by the authors, which are SER783 (present in HER2) and CYS775 (present in EGFR). In the case of the EGFR protein, the TAK-285 inhibitor can adopt two types of conformations (hence being a dual inhibitor), due to a larger active site in EGFR (provoked by a different spatial arrangement of the residues). In addition, the O atom from a carbonyl group belonging to CYS775 (Fig. 2a) is involved in a TtB interaction with the –CF3 group from the inhibitor molecule (dO⋯C = 3.679 Å and ∢O–C–C = 162.4°), which helps to stabilize a particular conformation of the TAK-285 molecule in the active site. In addition, the ∢F–C–O angles were comprised between 55 and 85°, with the C atom exhibiting the typical sp3 pyramidalization, as it can be noted in Fig. 2.
 | ||
| Fig. 2 Partial view of the X-ray structure 3POZ with an indication of the energetics and directionality of the TtB interaction (ΔETtB1) between 03P1023 and CYS775. In addition, the contribution of HalB and NH–π interactions (involving LEU788 and ASP855 residues) to the strength of the TtB is also indicated (ΔETtB2) (a). MEP surface of the theoretical model of ligand 03P1023 with an indication of the electrostatic potential on the C's σ-hole (0.001 a.u.) (b). Distribution of bond critical points (colored in red) and bond paths for the CYS775⋯03P1023 TtB complex. Ancillary HB interactions are highlighted in blue. NCIPlot colour range −0.002 a.u. ≤ (signλ2)ρ ≤ −0.002 a.u. (c). | ||
 | ||
| Fig. 3 Partial view of the X-ray structure 4J8M with an indication of the energetics and directionality of the TtB interaction (ΔETtB1) between CJ5501 and GLY145. In addition, the contribution of a double HB interaction (involving the ASP274 residue) to the strength of the TtB is also indicated (ΔETtB2) (a). MEP surface of the theoretical model of ligand CJ5501 with an indication of the electrostatic potential on the C's σ-hole (0.001 a.u.) (b). Distribution of bond critical points (colored in red) and bond paths for the GLY145⋯CJ5501 TtB complex. NCIPlot colour range −0.002 a.u. ≤ (signλ2)ρ ≤ −0.002 a.u. (c). | ||
 | ||
| Fig. 4 Partial view of the X-ray structure 7OT2 with an indication of the energetics and directionality of the TtB interaction (ΔETtB1) between 1FI1602 and GLN1237. In addition, the contribution of π–π stacking interaction (involving the PHE1167 residue) to the strength of the TtB is also indicated (ΔETtB2) (a). MEP surface of the theoretical model of ligand 1FI1602 with an indication of the electrostatic potential on the C's σ-hole (0.001 a.u.) (b). Distribution of bond critical points (colored in red) and bond paths for the GLY145⋯CJ5501 TtB complex. Ancillary HB interactions are highlighted in blue. NCIPlot colour range −0.002 a.u. ≤ (signλ2)ρ ≤ −0.002 a.u. (c). | ||
Using a computational model, we were able to estimate the strength of the interaction, resulting in −5.8 kJ mol−1 (ΔETtB1 in Fig. 2a). In addition, a halogen bond (HalB) and NH–π interactions are also established between (i) a Cl atom and the aromatic system of the trifluoromethylphenyl moiety and (ii) an O atom from a leucine residue and an NH group from an aspartate residue, respectively. Interestingly, when both interactions are considered in our theoretical model and the energetics of the TtB interaction is evaluated, we observed a reinforcement of around 7 kJ mol−1, resulting in −12.6 kJ mol−1 (ΔETtB2 in Fig. 2a). We computed also the MEP surface of the trifluoromethylphenyl group present in the TAK-285 inhibitor structure (Fig. 2b), observing a σ-hole over the C atom with a slightly positive electrostatic potential value (+1.9 kJ mol−1). Finally, we were interested in analyzing the interaction from several points of view: (i) first, we computed the QTAIM analysis of the theoretical model used (Fig. 2c), which did not show a bond critical point (bcp) connecting the O and C atoms. Instead, a bcp was found connecting CH groups from the protein backbone to a negative F atom from the inhibitor molecule, therefore characterizing an ancillary HB. In this representation, we have also included the NCIplot graph, which showed a green isosurface between the carbonyl O atom from the CYS775 residue and the CF3 group, thus evidencing the presence of a weak noncovalent force (TtB).
The second selected example involves the study from Gustafson and collaborators (PDBID 4J8M),68 in which they designed and synthesized a class of inhibitors able to disrupt the native conformation of Aurora A. This protein plays a key regulatory role in the proteolytic degradation of MYC proteins, which are involved in a range of cancers, including neuroblastoma and medulloblastoma.69,70 The inhibitor CJ5501 is composed of an aminopyrazole-pyrimidine ATP-mimetic backbone and a 3-trifluoromethyl biphenyl urea moiety (Fig. 3a). The authors determined that a double HB interaction established between the urea moiety of the inhibitor and the catalytic ASP274 residue was an important source of inactivation of Aurora A. In addition, the accommodation of the trifluoromethyl group inside the active site led to a displacement of two β strands, which disrupted the active conformation of Aurora A (apo).
Interestingly, the bulky –CF3 group is stabilized through the establishment of a TtB that involves an O atom from the carbonyl group of GLY145 belonging to one of these two β strands, with an O⋯C distance of 3.577 Å and an ∢O–C–C of 167.3° (Fig. 2a). In this case, the ∢F–C–O lied between 60 and 84°, similarly to the previous example. The computed interaction energy for this TtB interaction is −8.4 kJ mol−1 (ΔETtB1). We have also computed the strength of this TtB in the presence of the double HB between the inhibitor and the catalytic residue ASP274 as well as a CH–π interaction involving a CH group from PRO282 and the aromatic system of the inhibitor, resulting in a reinforcement of the interaction of around 3 kJ mol−1 (ΔETtB2 = −11.5 kJ mol−1). This was rationalized by computing the electrostatic potential map of the inhibitor molecule (Fig. 2b), which revealed the presence of a σ-hole over the C atom from the CF3 group, exhibiting an MEP value of +5.2 kJ mol−1. Finally, we computed the QTAIM and NCIplot analysis on the same graph (Fig. 2c). In this system, we did not find any intermolecular bcp that connected both counterparts; however, we found a green isosurface between the carbonyl's O atom and the C atom from the trifluoromethylphenyl moiety that allowed the identification of the TtB interaction.
The last biological example encompasses the work from Pang and collaborators (PDBID 7OT2, Fig. 4),71 which involves the inhibition of human cytosolic prolyl-tRNA synthetase (HcProRS). The protein family of aminoacyl-tRNA synthetases (aaRSs) is responsible for the ligation of an amino acid to its cognate tRNA in an ATP-dependent manner,72 leading to the formation of charge tRNAs which are subsequently used for protein synthesis at the ribosome. Many human diseases are associated with aaRS dysfunction, such as the overexpression and the enhancement of aaRS catalytic activity in some cancers73 and the emergence of clinically relevant aaRS mutants in genetic diseases.74–78
In this context, the human ProRS (HcProRS) activity is exhibited at the C-terminal region of this protein and inhibition of this enzyme has been recognized as a promising approach for the treatment of HcProRS-related diseases. In their study, the authors combined computational (docking and molecular dynamics simulations) and experimental (thermal shift assays and X-ray diffraction studies) efforts to design novel drug candidates for new applications such as therapeutics in HcProRS-related diseases. One of the structural modifications that the authors introduced was the substitution around the benzene moiety of the inhibitor. More in detail, the authors used both EDG (e.g. CH3, OCH3) and EWG (e.g. Cl, CF3) groups, observing that the inclusion of the latter led to a 7-fold increase in binding potency.
The X-ray structure 7OT2 (Fig. 4a) shows the main noncovalent interactions that stabilize the inhibitor 1F11602 inside the active site. As noted, a TtB interaction is formed between an O atom from the protein's backbone (belonging to GLN1237) and the CF3 group from the inhibitor, acting as an overlooked stabilization resource (dO⋯C = 3.802 Å and ∢O–C–C = 159.3°). In this example, the ∢F–C–O was between 50 and 88°, in line with the previously discussed examples, with the C atom of the –CF3 moiety being disposed in an sp3 pyramidal fashion. The computed interaction energy value is −3.1 kJ mol−1 (ΔETtB1). This result is in line with the MEP surface shown in Fig. 4b, which shows a positive potential on the carbon's σ-hole (+15.7 kJ mol−1). However, another important interaction that is established in the enzyme's active site is a π–π stacking between a benzene moiety from a phenylalanine residue (PHE1167) and the pyrazine ring of the inhibitor structure, with an intermolecular distance of 3.541 Å. When the TtB interaction strength was evaluated considering the π–π stacking, it resulted in −2.5 kJ mol−1 (ΔETtB2 in Fig. 4a). Therefore, the interplay between both types of interactions led to a weakening of 0.6 kJ mol−1 of the TtB.
In a similar fashion to the other two selected examples (see above), the QTAIM and NCIplot analyses were computed for the TtB complex (Fig. 4c). They revealed the presence of two HBs denoted by the two bcps and bond paths connecting the CH groups from the protein backbone and the F atoms from the CF3 substituent, which also contributed to the binding. Also, the NCIplot analysis showed a green isosurface between the C and O atoms, which indicated the presence of a weak interaction (TtB).
Energetic study
Since most of the TtB examples found in the PDB search involved a –CF3 group attached to an aromatic ring and a lone pair donor molecule consisting of either an sp2 O atom from a carbonyl group or an sp3 O atom from a protein residue, we computed the binding energies of a series of meta- and para-substituted benzene derivatives with either formaldehyde or dimethylether as electron donor molecules (see Fig. 5, 6 and Table 1) in order to shed light on substituent effects. | ||
| Fig. 5 Compounds 1 to 10 and complexes 11 to 30 used in this study. | ||
 | ||
| Fig. 6 Molecular Electrostatic Potential (MEP) surfaces of compounds 1 to 10. Energy values gathered at specific points of the surface are given in kJ mol−1 (0.001 a.u.). See the ESI† for the rest of the MEP surfaces. | ||
| Complex | ΔEBSSE | d | σ |
|---|---|---|---|
| 11 | −0.3 | 3.436 | −0.07 |
| 12 | −0.8 | 3.350 | 0.12 |
| 13 | −0.7 | 3.383 | 0 |
| 14 | −2.6 | 3.318 | 0.56 |
| 15 | −2.7 | 3.372 | 0.71 |
| 16 | −3.1 | 3.373 | −0.07 |
| 17 | −3.5 | 3.286 | 0.12 |
| 18 | −3.3 | 3.256 | 0 |
| 19 | −5.2 | 3.366 | 0.56 |
| 20 | −5.0 | 3.244 | 0.71 |
| 21 | −0.5 | 3.422 | −0.17 |
| 22 | −0.7 | 3.421 | −0.37 |
| 23 | −1.0 | 3.364 | 0 |
| 24 | −2.8 | 3.329 | 0.66 |
| 25 | −2.8 | 3.345 | 0.78 |
| 26 | −3.1 | 3.254 | −0.17 |
| 27 | −3.1 | 3.503 | −0.37 |
| 28 | −3.5 | 3.248 | 0 |
| 29 | −5.3 | 3.298 | 0.66 |
| 30 | −5.2 | 3.235 | 0.78 |
As noted, in all the cases attractive binding energy values were obtained, ranging between −0.3 (complex 11) and −5.2 kJ mol−1 (complex 30). In particular, those complexes involving formaldehyde as an electron donor molecule (11 to 20) showed a weaker TtB interaction strength compared to their respective dimethylether analogs (21 to 30), due to the high basicity of the sp3 hybridized O atom. Besides, while the ∢C–C–O was close to 180°, the ∢F–C–O was around 70° in all the cases, being far from the 90° observed in transition state structures involving SN2 mechanisms.
In general, the same behavior was observed no matter the type of aromatic substitution (meta- or para-) and electron donor molecule considered, that is, complexes involving strong EWG substituents on the aromatic ring (CN and NO2) achieved larger TtB interaction energies (see for instance complexes 14, 15, 19, 20, 24, 25, 29 and 30) than those complexes involving electron donor groups (EDG) such as CH3 and OH substituents (complexes 11, 12, 16, 17, 21, 22, 26 and 27). These results can be rationalized with the visualization of the Molecular Electrostatic Potential (MEP) surfaces of compounds 1 to 10. In Fig. 6 only six representative compounds are shown, which are compounds 2 and 7 involving an EDG (OH), compounds 3 and 8 involving H and compounds 5 and 10 implicating an EWG (NO2). In all cases, a positive electrostatic potential surface was obtained over the C atom (magnified into the square parts of the figure) located along the vector of the C–CF3 covalent bond, also known as a “σ-hole”. Depending on the position (meta- or para-) of the aromatic substituent, a different MEP value was obtained over the C σ-hole. For instance, in the case of compounds 2 and 7 (OH), the former achieved a more positive σ-hole MEP value (+5.7 kJ mol−1), due to a more pronounced induction effect from the electronegative O atom. On the other hand, the opposite behaviour was obtained when comparing compounds 5 and 10, where the former showed a less positive σ-hole MEP value. In compound 10, one of the possible resonance forms of the nitrobenzene molecule leaves a positive formal charge over the para-C atom, which in turn attracts the electron density from the C–CF3 bond, leading to a more positive electrostatic potential value. Finally, in the case of compounds 3 and 8, the latter showed a slightly more positive MEP value, likely due to the distribution of the four F atoms attached to the benzene ring, which are closer to the CF3 group in compound 8 (for additional details regarding the rest of the compounds, see Fig. S1 in the ESI†).
In addition, in Fig. 7 we represented the relationship between the BSSE corrected interaction energies (ΔEBSSE in kJ mol−1) from complexes 11 to 30 and the electrostatic potential values over the C's σ-hole for compounds 1 to 10 (ESP σ-hole in kJ mol−1). As noted, two series were plotted involving the two electron donor molecules used (formaldehyde in blue and dimethylether in orange). Interestingly, in both series we obtained a very good agreement between the strength of the interaction and the σ-hole ESP potentials (R = 0.993 and 0.994, respectively).
 | ||
| Fig. 7 Regression plot of the electrostatic potential value (ESP in kJ mol−1) calculated at the σ-hole of the carbon atom vs. the BSSE corrected binding energy values (ΔEBSSE, in kJ mol−1) for complexes 11 to 30. | ||
This indicates that the value of the potential at the C's σ-hole is a good predictor of the TtB interaction strength. Also, the fact that using both meta- and para-substituted benzene derivatives in the same series yields such a good R value points out that resonance effects are not an important factor in the stabilization of the TtB complexes, while on the hand induction effects are a prominent player.
In Fig. 8, several Hammett's plots are represented, either considering meta- or para-substituted complexes for each electron donor molecule (Fig. 8a and b) or combining both types of substitution in a single plot (Fig. 8c). As noticed by Hammett's plot involving one type of substitution, we obtained very good correlations for both series of TtB complexes (R values 0.998, 0.973 and 0.981). Therefore, Hammett's σ-constants can also be used as a reliable predictor of the TtB interaction strength. In addition, while mixing the two types of substitutions (Fig. 8c) the behavior observed was similar to that shown in Fig. 7 (using the σ-hole ESP). Hence, the fact that both types of substitutions can be combined in the same representation points to induction effects as the main contributor to the interaction strength. These results agree well with those obtained from the energetic study, which showed similar interaction energies while comparing the meta- and para-substituted complexes that belong to the same substituent (for instance complexes 11 and 21, 16 and 26 or 20 and 30). Hence, the position of the substituent para or meta is not relevant, as it has been also obtained in the case of the halogen bonding interaction.79
 | ||
| Fig. 8 (a and b) Hammett's plots (ΔEBSSEvs. σ) of formaldehyde (in blue) and dimethylether (in orange)⋯meta-C7F7X complexes (left) and ⋯para-C7F7X complexes (right). (c) Hammett's plots (ΔEBSSEvs. σ) of both formaldehyde⋯meta- and para-C7F7X complexes (in blue) and dimethylether ··meta- and para-C7F7X complexes (in orange). | ||
Conclusions
The results gathered in this manuscript remark on the ability of –CF3 groups to behave as efficient and directional tetrel bond donors in protein–drug systems. A PDB search was conducted, resulting in 419 unique interactions. Interestingly, almost every amino acid residue is involved in tetrel bonding, suggesting that it might be an underlooked binding force in biological systems. Computations at the RI-MP2/def2-TZVPD level of theory shed light on the energetics of the interaction, as well as the plausible interplay between tetrel bonds and other NCIs present in the active site of proteins (e.g. halogen bonding, π–π stacking or hydrogen bonding interactions). The NCIplot analysis via the RDG isosurfaces revealed the TtBs in real space disclosing their existence and attractive nature in the PDB structures studied herein. Also, Hammett's regression plots for a series of tetrel bond complexes involving meta- and para-substituted benzene derivatives shed light on substituent effects, which pointed out a minor role of resonance in induction. We believe that the results gathered from our study will be useful for rational drug design and biological communities as well as for further expanding the role of this interaction to biomedical applications.Author contributions
Most of the computational studies were conducted by M. N. P., A. K. S. and A. F. H. S. B. carried out the PDB search while A. B. wrote the article and directed the study.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the MICIU/AEI of Spain (project PID2020-115637GB-I00 FEDER funds). We thank the CTI (UIB) for computational facilities. H. S. B. thanks the Department of Atomic Energy (DAE) India for financial support.References
- H. J. Schneider, Binding Mechanisms in Supramolecular Complexes, Angew. Chem., Int. Ed., 2009, 48, 3924–3977 CrossRef CAS PubMed.
- C. A. Hunter and J. K. M. Sanders, The Nature of π–π Interactions, J. Am. Chem. Soc., 1990, 112, 5525–5534 CrossRef CAS.
- W. J. Vickaryous, R. Herges and D. W. Jonhson, Arsenic–π Interactions Stabilize a Self-Assembled As2L3 Supramolecular Complex, Angew. Chem., Int. Ed., 2004, 43, 5831–5833 CrossRef CAS PubMed.
- S. J. Grabowski, What Is the Covalency of Hydrogen Bonding?, Chem. Rev., 2011, 111, 2597–2625 CrossRef CAS PubMed.
- P. Murrayrust and W. D. S. Motherwell, Computer Retrieval and Analysis of Molecular Geometry. 4. Intermolecular Interactions, J. Am. Chem. Soc., 1979, 101, 4374–4376 CrossRef CAS.
- A. Chand, D. K. Sahoo, A. Rana, S. Jena and H. S. Biswal, The Prodigious Hydrogen Bonds with Sulfur and Selenium in Molecular Assemblies, Structural Biology, and Functional Materials, Acc. Chem. Res., 2020, 53, 1580–1592 CrossRef CAS PubMed.
- Y. Bai, T. R. Sosnick, L. Mayne and S. W. Englander, Protein Folding Intermediates: Native-State Hydrogen Exchange, Science, 1995, 269, 192–197 CrossRef CAS PubMed.
- A. Bauzá and A. Frontera, Aerogen Bonding Interaction: A New Supramolecular Force?, Angew. Chem., Int. Ed., 2015, 54, 7340–7343 CrossRef PubMed.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, The Halogen Bond, Chem. Rev., 2016, 116, 2478–2601 CrossRef CAS PubMed.
- W. Wang, B. Ji and Y. Zhang, Assembling Structure of Single-Walled Carbon Nanotube Thin Bundles, J. Phys. Chem. A, 2009, 113, 8132–8135 CrossRef CAS PubMed.
- S. Scheiner, The Pnicogen Bond: Its Relation to Hydrogen, Halogen, and Other Noncovalent Bonds, Acc. Chem. Res., 2013, 46, 280–288 CrossRef CAS PubMed.
- A. Bauzá, T. J. Mooibroek and A. Frontera, Tetrel-Bonding Interaction: Rediscovered Supramolecular Force?, Angew. Chem., Int. Ed., 2013, 52, 12317–12321 CrossRef PubMed.
- Z. Xu, Z. Liu, T. Chen, T. Chen, Z. Wang, G. Tian, J. Shi, X. Wang, Y. Lu, X. Yan, G. Wang, H. Jiang, K. Chen, S. Wang, Y. Xu, J. Shen and W. Zhu, Utilization of Halogen Bond in Lead Optimization: A Case Study of Rational Design of Potent Phosphodiesterase Type 5 (PDE5) Inhibitors, J. Med. Chem., 2011, 54, 5607–5611 CrossRef CAS PubMed.
- A. Bauzá, D. Quiñonero, P. M. Deyà and A. Frontera, Pnicogen–π Complexes: Theoretical Study and Biological Implications, Phys. Chem. Chem. Phys., 2012, 14, 14061–14066 RSC.
- E. Margiotta, S. C. C. Van Der Lubbe, L. De Azevedo Santos, G. Paragi, S. Moro, F. M. Bickelhaupt and C. Fonseca Guerra, Halogen Bonds in Ligand–Protein Systems: Molecular Orbital Theory for Drug Design, J. Chem. Inf. Model., 2020, 60, 1317–1328 CrossRef CAS PubMed.
- P. Guo, A. A. Farahat, A. Paul, A. Kumar, D. W. Boykin and W. D. Wilson, Extending the σ-Hole Motif for Sequence-Specific Recognition of the DNA Minor Groove, Biochemistry, 2020, 59, 1756–1768 CrossRef CAS PubMed.
- J. Y. C. Lim and P. D. Beer, Sigma-hole Interactions in Anion Recognition, Chem, 2018, 4, 731–783 CAS.
- A. Bauzá and A. Frontera, σ/π-Hole noble gas bonding interactions: Insights from Theory and Experiment, Coord. Chem. Rev., 2020, 404, 213112 CrossRef.
- A. Bauzá, S. K. Seth and A. Frontera, Tetrel bonding Interactions at Work: Impact on Tin and Lead Coordination Compounds, Coord. Chem. Rev., 2019, 384, 107–125 CrossRef.
- B. V. Pandiyan, P. Deepa and P. Kolandaivel, Studies on the σ–hole Bonds (Halogen, Chalcogen, Pnicogen and Carbon bonds) Based on the Orientation of Crystal Structure, Mol. Phys., 2016, 114, 3629–3642 CrossRef CAS.
- R. Zeng, Z. Gong and Q. Yan, Chalcogen-Bonding Supramolecular Polymers, J. Org. Chem., 2020, 85, 8397–8404 CrossRef CAS PubMed.
- S. Jena, J. Dutta, K. D. Tulsiyan, A. K. Sahu, S. S. Choudhury and H. S. Biswal, Noncovalent interactions in proteins and nucleic acids: beyond hydrogen bonding and π-stacking, Chem. Soc. Rev., 2022, 51, 4261–4286 RSC.
- D. Mani and E. Arunan, The X–C⋯Y (X = O/F, Y = O/S/F/Cl/Br/N/P) ‘carbon bond’ and hydrophobic interactions, Phys. Chem. Chem. Phys., 2013, 15, 14377–14383 RSC.
- D. Sethio, V. Oliveira and E. Kraka, Quantitative Assessment of Tetrel Bonding Utilizing Vibrational Spectroscopy, Molecules, 2018, 23, 2763 CrossRef PubMed.
- V. L. Heywood, T. P. J. Alford, J. J. Roeleveld, S. J. L. Deprez, A. Verhoofstad, J. I. van der Vlugt, S. R. Domingos, M. Schnell, A. P. Davis and T. J. Mooibroek, Observations of tetrel bonding between sp3-carbon and THF, Chem. Sci., 2020, 11, 5289–5293 RSC.
- Y. Zhang, W. Wang and Y.-B. Wang, Comput. Theor. Chem., 2019, 1147, 8–12 CrossRef CAS.
- M. S. Taylor, Anion recognition based on halogen, chalcogen, pnictogen and tetrel bonding, Coord. Chem. Rev., 2020, 413, 213720 CrossRef.
- A. Bauzá, S. K. Seth and A. Frontera, Tetrel bonding interactions at work: Impact on tin and lead coordination compounds, Coord. Chem. Rev., 2019, 384, 107–125 CrossRef.
- V. R. Mundlapati, D. K. Sahoo, S. Bhaumik, S. Jena, A. Chandrakar and H. S. Biswal, Noncovalent Carbon-Bonding Interactions in Proteins, Angew. Chem., Int. Ed., 2018, 57, 16496 CrossRef CAS PubMed.
- J. Dutta, D. K. Sahoo, S. Jena, K. D. Tulsiyan and H. S. Biswal, Non-covalent interactions with inverted carbon: a carbo-hydrogen bond or a new type of hydrogen bond?, Phys. Chem. Chem. Phys., 2020, 22, 8988–8997 RSC.
- J. J. Roeleveld, J. L. Deprez, A. Verhoofstad, A. Frontera, J. I. van der Vlugt and T. J. Mooibroek, Engineering Crystals Using sp3-C Centred Tetrel Bonding Interactions, Chem. – Eur. J., 2020, 26, 10126–10132 CrossRef CAS PubMed.
- A. Frontera, Tetrel Bonding Interactions Involving Carbon at Work: Recent Advances in Crystal Engineering and Catalysis, C, 2020, 6, 60 CAS.
- A. Bauzá, T. J. Mooibroek and A. Frontera, Non-covalent sp3 carbon bonding with ArCF3 is analogous to CH–π interactions, Chem. Commun., 2014, 50, 12626–12629 RSC.
- A. Bauzá, T. J. Mooibroek and A. Frontera, Small cycloalkane (CN)2C-C(CN)2 structures are highly directional non-covalent carbon-bond donors, Chemistry, 2014, 20, 10245–10248 CrossRef PubMed.
- A. Bauzá, T. J. Mooibroek and A. Frontera, Tetrel Bonding Interactions, Chem. Rec., 2016, 16, 473–487 CrossRef PubMed.
- S. J. Grabowski, Tetrel bond–σ-hole bond as a preliminary stage of the SN2 reaction, Phys. Chem. Chem. Phys., 2014, 16, 1824–1834 RSC.
- Y. Y. Chu, H. J. Cheng, Z. H. Tian, J. C. Zhao, G. Li, Y. Y. Chu, C. J. Sun and W. B. Li, Rational drug design of indazole-based diarylurea derivatives as anticancer agents, Chem. Biol. Drug Des., 2017, 90, 609–617 CrossRef CAS PubMed.
- A. C. Gibbs, R. Steele, G. Liu, B. A. Tounge and G. T. Montelione, Inhibitor bound dengue NS2B-NS3pro reveals multiple dynamic binding modes, Biochemistry, 2018, 57, 1591–1602 CrossRef CAS PubMed.
- J. Piton, A. Vocat, C. S. Lupien, O. Foo, V. Riabova, V. Makarov and S. T. Cole, Structure-based drug design and characterization of sulfonyl-piperazine benzothiazinone inhibitors of DprE1 from Mycobacterium tuberculosis, Antimicrob. Agents Chemother., 2018, 62, e00681 CrossRef CAS PubMed.
- S. S. Kar and C. A. Thomas, Strategically Placed Trifluoromethyl Substituent in the Realm of Antitubercular Drug Design, Curr. Drug Delivery, 2019, 14, 114–123 CAS.
- H. He, M. Xie, H. Zhang, H. Zhang, H. Zhu, Y. Fang, Z. Shen, R. Wang, Z. Zhao and L. Zhu, Design, synthesis, and biological evaluation of potent and selective S1PR1 agonists for the treatment of Ulcerative Colitis, Chin. J. Chem., 2022, 40, 2625–2632 CrossRef CAS.
- T. Ginex, E. Herrero, E. Gibert and F. J. Luque, On the Use of Quantum Mechanical Solvation Continuum Models in Drug Design: IEF/PCM-MST Hydrophobic Descriptors in 3D-QSAR Analysis of AMPA Inhibitors in Theoretical and Quantum Chemistry at the Dawn of the 21st Century, Taylor and Francis Group, UK, 1st edn, 2018 Search PubMed.
- J. Shawon, A. M. Khan, M. M. Rahman, M. M. Hoque, M. A. K. Khan, M. G. Sarwar and M. A. Halim, Molecular recognition of azelaic acid and related molecules with DNA polymerase I investigated by molecular modeling calculations, Interdiscip. Sci., 2018, 10, 525–537 CrossRef CAS PubMed.
- Y. Sun, H. Tang, X. Wang, F. Feng, T. Fan, D. Zhao, B. Xiong, H. Xie and T. Liu, Identification of 1 H-pyrazolo [3, 4-b] pyridine derivatives as novel and potent TBK1 inhibitors: design, synthesis, biological evaluation, and molecular docking study, J. Enzyme Inhib. Med. Chem., 2022, 37, 1411–1425 CrossRef CAS PubMed.
- S. Banerjee, S. Yadav, S. Banerjee, S. O. Fakayode, J. Parvathareddy, W. Reichard, S. Surendranathan, F. Mahmud, R. Whatcott and J. Thammathong, Drug repurposing to identify nilotinib as a potential SARS-CoV-2 main protease inhibitor: insights from a computational and in vitro study, J. Chem. Inf. Model., 2021, 61, 5469–5483 CrossRef CAS PubMed.
- S. Keretsu, S. P. Bhujbal and S. J. Cho, Docking and 3D-QSAR studies of hydrazone and triazole derivatives for selective inhibition of GRK2 over ROCK2, Lett. Drug Des. Discovery, 2020, 17, 618–632 CrossRef CAS.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, The Protein Data Bank, Nucleic Acids Res., 2000, 28, 235–242 CrossRef CAS PubMed.
- X. García-Llinás, A. Bauzá, S. K. Seth and A. Frontera, Importance of R–CF3⋯O Tetrel Bonding Interactions in Biological Systems, J. Phys. Chem. A, 2017, 121, 5371–5376 CrossRef PubMed.
- M. Heinig and D. Frishman, STRIDE: a Web server for secondary structure assignment from known atomic coordinates of proteins, Nucleic Acids Res., 2004, 32, W500 CrossRef CAS PubMed.
- A. D. Becke, Density-functional exchange-energy approximation with correct asymptotic behavior, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098–3100 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. A. Krieg, consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Weigend and M. Häser, RI-MP2: first derivatives and global consistency, Theor. Chem. Acc., 1997, 97, 331–340 Search PubMed.
- A. Bauzá, I. Alkorta, A. Frontera and J. Elguero, On the Reliability of Pure and Hybrid DFT Methods for the Evaluation of Halogen, Chalcogen, and Pnicogen Bonds Involving Anionic and Neutral Electron Donors, J. Chem. Theory Comput., 2013, 9, 5201–5210 CrossRef PubMed.
- R. Ahlrichs, M. Bar, M. Haser, H. Horn and C. Kolmel, Electronic Structure Calculations on Workstation Computers – the Program System turbomole, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- S. B. Boys and F. Bernardy, The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- R. Dennington, T. A. Keith and J. M. Millam, GaussView, Version 6.0, Semichem Inc., Shawnee Mission, KS, 2016 Search PubMed.
- R. F. W. Bader, A quantum theory of molecular structure and its applications, Chem. Rev., 1991, 91, 893–928 CrossRef CAS.
- T. A. Keith, TK Gristmill Software, AIMAll (Version 13.05.06), Overland Park KS, USA, 2013 Search PubMed.
- J. Contreras-García, E. R. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. N. Beratan and W. Yang, NCIPLOT: A Program for Plotting Noncovalent Interaction Regions, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef PubMed.
- K. Aertgeerts, R. Skene, J. Yano, B. C. Sang, H. Zou, G. Snell, A. Jennings, K. Iwamoto, N. Habuka, A. Hirokawa, T. Ishikawa, T. Tanaka, H. Miki, Y. Ohta and S. Sogabe, Structural Analysis of the Mechanism of Inhibition and Allosteric Activation of the Kinase Domain of HER2 Protein, J. Biol. Chem., 2011, 286, 18756–18765 CrossRef CAS PubMed.
- J. Schlessinger, Common and distinct elements in cellular signaling via EGF and FGF receptors, Science, 2004, 306, 1506–1507 CrossRef CAS PubMed.
- Y. Yarden and M. X. Sliwkowski, Untangling the ErbB signalling network, Nat. Rev. Mol. Cell Biol., 2001, 2, 127–137 CrossRef CAS PubMed.
- T. Wada, J. N. Myers, Y. Kokai, V. I. Brown, J. Hamuro, C. M. LeVea and M. I. Greene, Anti-receptor antibodies reverse the phenotype of cells transformed by two interacting proto-oncogene encoded receptor proteins, Oncogene, 1990, 5, 489–495 CAS.
- D. Ye, J. Mendelsohn and Z. Fan, Augmentation of a humanized Anti-HER2 mAb 4D5 induced growth inhibition by a human-mouse chimeric anti-EGF receptor mAb C225, Oncogene, 1999, 18, 731–738 CrossRef CAS PubMed.
- T. Ishikawa, T. Taniguchi, H. Banno and M. Seto, WO. Pat., 118588, 2005 Search PubMed.
- W. C. Gustafson, J. G. Meyerowitz, E. A. Nekritz, J. Chen, C. Benes, E. Charron, E. F. Simonds, R. Seeger, K. K. Matthay, N. T. Hertz, M. Eilers, K. M. Shokat and W. A. Weiss, Drugging MYCN through an Allosteric Transition in Aurora Kinase A, Cell, 2014, 26, 414–427 CAS.
- C. Y. Lin, J. Lovén, P. B. Rahl, R. M. Paranal, C. B. Burge, J. E. Bradner, T. I. Lee and R. A. Young, Transcriptional amplification in tumor cells with elevated c-Myc, Cell, 2012, 151, 56–67 CrossRef CAS PubMed.
- Z. Nie, G. Hu, G. Wei, K. Cui, A. Yamane, W. Resch, R. Wang, D. R. Green, L. Tessarollo, R. Casellas, K. Zhao and D. Levens, c-Myc Is a Universal Amplifier of Expressed Genes in Lymphocytes and Embryonic Stem Cells, Cell, 2012, 151, 68–79 CrossRef CAS PubMed.
- L. Pang, S. D. Weeks, M. Juhás, S. V. Strelkov, J. Zitko and A. Van Aerschot, Towards Novel 3-Aminopyrazinamide-Based Prolyl-tRNA Synthetase Inhibitors: In Silico Modelling, Thermal Shift Assay and Structural Studies, Int. J. Mol. Sci., 2021, 22, 7793 CrossRef CAS PubMed.
- M. Ibba and D. Soll, Aminoacyl-TRNA Synthesis, Annu. Rev. Biochem., 2000, 69, 617–650 CrossRef CAS PubMed.
- S. Kim, S. You and D. Hwang, Aminoacyl-TRNA Synthetases and Tumorigenesis: More than Housekeeping, Nat. Rev. Cancer, 2011, 11, 708–718 CrossRef CAS PubMed.
- P. Yao and P. L. Fox, Aminoacyl-TRNA Synthetases in Medicine and Disease, EMBO Mol. Med., 2013, 5, 332–343 CrossRef CAS PubMed.
- M. Datt and A. Sharma, Evolutionary and Structural Annotation of Disease-Associated Mutations in Human Aminoacyl-TRNA Synthetases, BMC Genomics, 2014, 15, 1063 CrossRef PubMed.
- R. Meyer-Schuman and A. Antonellis, Emerging Mechanisms of Aminoacyl-TRNA Synthetase Mutations in Recessive and Dominant Human Disease, Hum. Mol. Genet., 2017, 26, R114–R127 CrossRef CAS PubMed.
- S.-H. Kim, S. Bae and M. Song, Recent Development of Aminoacyl-TRNA Synthetase Inhibitors for Human Diseases: A Future Perspective, Biomolecules, 2020, 10, 1625 CrossRef CAS PubMed.
- C. S. Francklyn and P. Mullen, Progress and Challenges in Aminoacyl-TRNA Synthetase-Based Therapeutics, J. Biol. Chem., 2019, 294, 5365–5385 CrossRef CAS PubMed.
- A. Bauzá, D. Quiñonero, A. Frontera and P. M. Deyà, Substituent effects in halogen bonding complexes between aromatic donors and acceptors: a comprehensive ab initio study, Phys. Chem. Chem. Phys., 2011, 13, 20371–20379 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1, Cartesian coordinates of the PDB models and of complexes 11 to 30. See DOI: https://doi.org/10.1039/d3cp00839h |
| This journal is © the Owner Societies 2023 |