
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLooking for chiral recognition in photoinduced bimolecular electron transfer using ultrafast spectroscopy†
Pragya
Verma
 ,
Christoph
Nançoz
,
Johann
Bosson
,
Géraldine M.
Labrador
,
Jérôme
Lacour
and
Eric
Vauthey
*
,
Christoph
Nançoz
,
Johann
Bosson
,
Géraldine M.
Labrador
,
Jérôme
Lacour
and
Eric
Vauthey
*
Department of Physical Chemistry, University of Geneva, 30 Quai Ernest-Ansermet, CH-1211 Geneva 4, Switzerland. E-mail: eric.vauthey@unige.ch
First published on 21st March 2023
Abstract
Occurrence of chiral recognition in bimolecular photoinduced electron transfer (ET) is difficult to identify because of the predominant role of diffusion. To circumvent this problem, we apply a combination of ultrafast time-resolved fluorescence and transient electronic absorption to look for stereoselectivity in the initial, static stage of ET quenching, where diffusion is not relevant. The fluorophore and electron acceptor is a cationic hexahelicene, whereas the quencher has either stereocentered (tryptophan) or axial (binaphthol) chirality. We found that, in all cases, the quenching dynamics are the same, within the limit of error, for different diastereomeric pairs in polar and medium-polar solvents. The same absence of chiral effect is observed for the recombination of the radical pair, which results from the quenching. Molecular dynamics simulations suggest that the distribution of inter-reactant distance is independent of the chirality of the acceptor and the donor. Close contact resulting in large electronic coupling is predicted to be possible with all diastereomeric pairs. In this case, ET is an adiabatic process, whose dynamics do no longer depend on the coupling, but are rather controlled by high-frequency intramolecular modes.
1 Introduction
Although chirality is known to be a key factor in many areas of chemistry,1–4 its role in charge-transfer processes has not been much studied so far. Electron transmission through chiral molecules was shown to depend on the spin state of the electron.5,6 This chiral-induced spin selectivity (CISS) was mostly found with thermal electron transfer (ET). Photoinduced CISS was reported with quantum dots attached to a metallic surface via a chiral spacer,7,8 but there is still no fully unambiguous observation of this effect with a photoinduced intramolecular ET in a donor–bridge–acceptor molecular system.9By contrast, several investigations reported on a significant effect of chirality on the photoinduced intermolecular ET between chiral electron acceptor (A) and donor (D) connected via an achiral bridge.10–13 For example, Miranda and coworkers investigated diastereomeric dyads consisting of an hydroxybiphenyl drug linked to a tryptophan donor and found variations in the fluorescence lifetime as large as a factor of four depending on the diastereomer.11 This effect as well as that observed with other diastereomeric dyads were attributed to the linker that limits the conformational space available to the reactants. This leads to marked differences in the electronic coupling and thus in the ET quenching dynamics.14 Evidence of stereodifferentiation in the charge recombination process was also obtained from 1H-CIDPN studies of similar dyads.12,13 This sensitivity of intramolecular ET to chirality can be exploited to achieve stereocontrolled photoredox catalysis, as demonstrated with chiral transition metal complexes.15–18 Besides ET, other intramolecular photoinduced processes such as hydrogen abstraction and excitation energy transfer were also found to exhibit some stereodifferentiation.19,20
Occurrence of chiral recognition in intermolecular photoinduced ET is more ambiguous. Stern–Volmer analysis of the fluorescence quenching of binaphthyl by a chiral aniline pointed to a stereodifferentiation of 1.9 in cyclohexane, decreasing to 1.5 in dichloroethane and vanishing in a highly polar solvent.21 Subsequent studies with other AD pairs found weak stereoselectivity, ≤1.15, in non-polar solvents, and very small (<1.05) if any in polar solvents.22–24 This dependence on the solvent polarity was attributed to the formation of an exciplex in weakly and non-polar solvents and to remote ET in polar media.
Involvement of a ground state complex was also found to favour stereoselectivity in intermolecular ET. For example, a chiral effect of 1.3 was reported for the ET quenching of Zn-myoglobin in the triplet state by a chiral methyl-viologen in an aqueous solution.25 It was found to be twice as large for the subsequent charge recombination to the ground state. This significant chiral recognition in a polar environment was explained by the adsorption of the quencher on the protein surface, leading to different electronic coupling for the two quencher enantiomers. Similarly, significant stereoselectivity was observed for the formation of chiral AD complexes.26,27 However, to the best of our knowledge, the charge-transfer dynamics of such complexes upon photoexcitation has not been investigated so far. Chiral recognition involving complex formation through H-bond interactions is also exploited for fluorescence sensing of chiral analytes. In this case, binding to the analyte suppresses intramolecular ET in the photoexcited sensor, which becomes highly fluorescent.28–30
In the absence of a ground-state complex, observation of chiral recognition in bimolecular ET is complicated by the presence of the diffusion step, which is not sensitive to chirality, at least in an achiral environment. Because of this, the ET quenching should not be in the diffusion-controlled regime. This problem can be circumvented by considering that, directly after excitation, quenching is mostly static and occurs first in reactant pairs at distances and with mutual orientations enabling ET without significant diffusion.31–34 Previous studies of chirality effects in photoinduced bimolecular ET were performed using stationary fluorescence quenching or ns and slower time-resolved spectroscopy and were, thus, not sensitive to this static stage of the quenching.
Herein, we present our investigation of the occurrence of chiral recognition in photoinduced ET in polar environments using ultrafast spectroscopy, concentrating on the early stage of quenching. We tested several cationic hexahelicenes as electron acceptors and chromophores as well as potential chiral donors, with either stereocentered or axial chirality (Fig. S1, ESI†). Detailed studies were then performed with AD pairs exhibiting efficient quenching without follow-up reactions (Fig. 1). Our results reveal that the ET quenching dynamics is indeed non-exponential and takes place on timescales ranging from less than a ps to a few ns. However, these dynamics are the same for different diastereomeric AD pairs, for all the systems investigated. Furthermore, our measurements point to identical recombination dynamics of the ET quenching product. Molecular dynamics simulations were also carried out to better understand this absence of chiral recognition, even in the static stage of quenching. They suggest that encounters resulting in a large coupling occur for all diastereomeric reactant pairs.
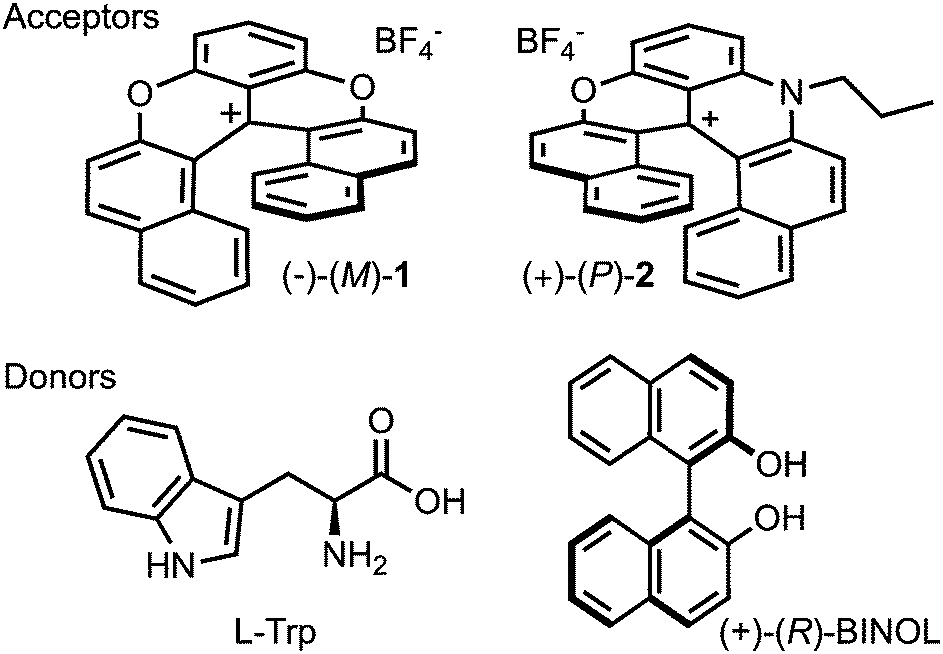 | ||
| Fig. 1 Chiral electron acceptors and donors, with only one enantiomer shown. | ||
2 Experimental
2.1 Samples
The cationic dioxa 1 and azaoxa 2 [6]helicenes were synthesised with BF4− as the counterion according to ref. 35. Their enantiomers were isolated through chiral stationary phase HPLC resolution.36 The electron donors, D- and L-tryptophan (D-Trp, 99% Acros Organics, L-Trp, 99% Fluka), (+)- and (−)1,1′-bi-2-naphthol (+)-(−)-BINOL, (99%, Alfa Aesar), L-proline (Pro, 99%, Sigma-Aldrich) and as well as the solvents, water (deionised, Sigma-Aldrich), acetonitrile (ACN, for spectroscopy, Acros Organics), ethanol (EtOH, Acroseal, Acros Organics), dichloromethane (DCM, Rotidry Sept, Carl Roth), and tetrahydrofuran (THF, for spectroscopy, Sigma-Aldrich) were of the highest purity commercially available and were used as received. N,N-Dimethyl-1-phenylethylamine (DMPEA, Sigma-Aldrich) was distilled before use.2.2 Spectroscopy
The stationary electronic absorption spectra and the circular dichroism spectra were recorded using a Cary 50 and a JASCO J-815 spectrometer. Fluorescence spectra were recorded using a Horiba Fluoromax 4 fluorometer and were corrected using a set of secondary emission standards.37Fluorescence dynamics on the nanosecond time scale was measured using a time-correlated single photon counting (TCSPC) setup described in detail previously.38 Excitation was performed at 469 nm using 60 ps pulses at 10 MHz produced by a laser diode (Picoquant, LDH-PC-470). The full width at half maximum (FWHM) of the instrument response function (IRF) was around 200 ps. Faster dynamics was investigated by fluorescence up-conversion using the setup described in ref. 39. Excitation was performed at 450 nm using 100 fs pulses produced by frequency doubling the output of a Ti:Sapphire oscillator (Spectra-Physics, Mai Tai). The pump irradiance on the sample was around 5 μJ cm−2, and the FWHM of the IRF was ca. 200 fs. The sample solutions were located in a 0.4 mm rotating cell and had an absorbance of about 0.1 at the excitation wavelength.
Electronic transient absorption (TA) spectra were recorded with the setup described in ref. 40 and 41. In brief, 35 fs pulses centred at 800 nm at a repetition rate of 5 kHz were produced using an amplified Ti:Sapphire system (Solstice Ace, Spectra-Physics). A fraction of this output fed a TOPAS-Prime combined with a NirUVis module (Light Conversion) producing tunable pump pulses with an irradiance of 0.15–0.75 mJ cm−2 at the sample position. Probing was achieved at magic angle using white-light pulses generated in a 3 mm CaF2 plate. The sample pathlength was 1 mm and the IRF had an FWHM varying between 80 and 250 fs depending on the probe wavelength.
2.3 Molecular dynamics simulations
Molecular dynamics (MD) simulations were carried out using GROMACS 2021.2.42 The optimised structure of both enantiomers of 1, Trp and BINOL, was determined from quantum-chemical calculations at the DFT level (B3LYP/6-31G+d)43 using Gaussian 16.44 The same topology file was used for the two enantiomers of a given molecule. They were generated using the Antechamber Python parser interface (ACPYPE)45 with the OPLS-AA force field.46 The force field parameters for the dihedral angle of BINOL were determined from DFT calculations. The atomic charges were determined from CHELPG fits of the electrostatic potential obtained from the quantum-chemical calculations.47 The TIP3P model was used for water,48 whereas for the other solvents, the GAFF-ESP-2018 force field was used.49 A periodic cubic box (5 × 5 × 5 nm3) was used for the simulations, which were performed at constant pressure (1 atm) and temperature (295 K) with 2 fs steps for 50 ns. Further details on the simulation parameters can be found in the ESI† (Section S6).3 Results
3.1 Stationary spectroscopy: selection of the AD pairs
As chiral chromophores and potential electron acceptors, we selected the cationic dioxa and azaoxa [6]helicenes (1–2, Fig. 1), whose electronic absorption, emission and CD spectra are illustrated in Fig. 2 and Fig. S2 (ESI†). As possible electron donors, we chose the amino acids tryptophan (Trp) and proline (Pro), as well as a chiral aromatic amine, N,N-dimethyl-1-phenylethylamine (DMPEA), and 1,1′-bi-2-naphthol (BINOL). The ET driving force, −ΔGET, estimated from the Weller equation,50 for the different AD pairs is listed in Table S1 (ESI†). Occurrence of quenching was determined using stationary fluorescence and/or TCSPC measurements using the chromophores in the racemic form.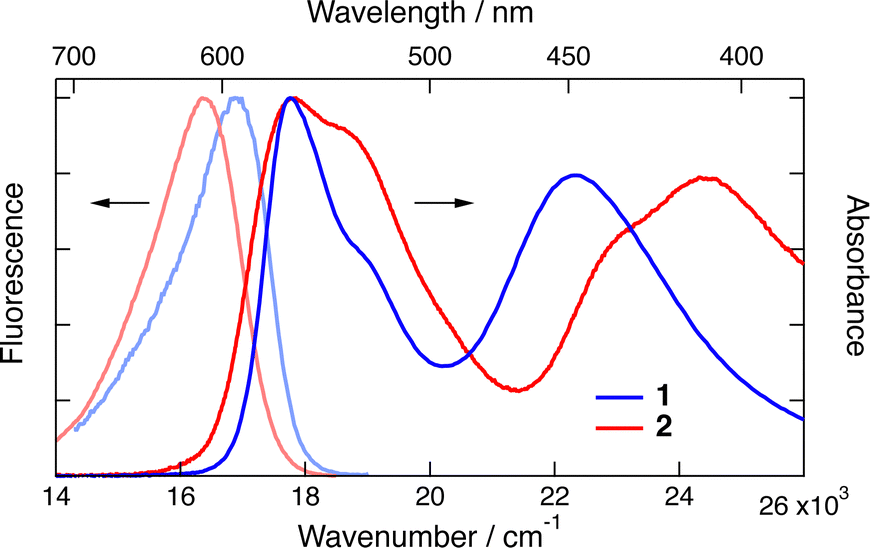 | ||
| Fig. 2 Stationary electronic absorption and emission spectra measured with dioxa 1 and azaoxa 2 [6]helicenes in acetonitrile. | ||
Trp was found to quench both 1 and 2 in water/ACN mixtures. No quenching could be observed with Pro in water, despite a favourable driving force estimated from the Weller equation for 1. This discrepancy could arise from the uncertainty on the oxidation potential of Pro as well as the possible difference of potential for 1 in water compared to ACN.51 On the other hand, DMPEA quenched efficiently 1 and 2 in ACN, but rapid photodegradation was found after TCSPC experiments. Finally, BINOL was also found to quench the fluorescence of 1 and 2 in ACN. In the medium polar DCM, however, efficient quenching was only observed with 1. No new emission band, that could be assigned to an exciplex, was observed. Based on these results, the helicenes 1 and 2 as well as Trp and BINOL were selected for detailed measurements.
3.2 Time-resolved fluorescence measurements of the quenching dynamics
Fig. 3 depicts Stern–Volmer (SV) plots obtained from stationary and ns-resolved measurements of the fluorescence of (rac)-1 and (−)-(M)-1 in the presence of L-Trp in a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 (v/v) water/ACN mixture. This solvent mixture was chosen to favour sufficient solubility of both the helicenes and Trp. The SV plots obtained from the fluorescence lifetimes are linear, and their slopes, 17.0 ± 0.2 and 17.1 ± 0.2 M−1 for (rac)-1 and (−)-1, are identical within the limit of error. Given the fluorescence lifetime of 2.3 ns in the absence of a quencher (Fig. S3, ESI†), these values correspond to a quenching rate constant of 7.4 × 109 M−1 s−1. This is close to the diffusion limit, which can be estimated to be 9 × 109 M−1 s−1 considering the viscosity of this solvent mixture.52 By contrast, the SV plots obtained from stationary measurements are not linear. At the highest Trp concentration, the decrease of fluorescence intensity is more than twice as large as the decrease of lifetime. This difference points to the presence of fast decay components, which are not resolved with the TCSPC setup and can be attributed to the static and transient stages of the quenching.32 In this case again, no significant difference between (rac)-1 and (−)-1 can be detected.
:50 (v/v) water/ACN mixture. This solvent mixture was chosen to favour sufficient solubility of both the helicenes and Trp. The SV plots obtained from the fluorescence lifetimes are linear, and their slopes, 17.0 ± 0.2 and 17.1 ± 0.2 M−1 for (rac)-1 and (−)-1, are identical within the limit of error. Given the fluorescence lifetime of 2.3 ns in the absence of a quencher (Fig. S3, ESI†), these values correspond to a quenching rate constant of 7.4 × 109 M−1 s−1. This is close to the diffusion limit, which can be estimated to be 9 × 109 M−1 s−1 considering the viscosity of this solvent mixture.52 By contrast, the SV plots obtained from stationary measurements are not linear. At the highest Trp concentration, the decrease of fluorescence intensity is more than twice as large as the decrease of lifetime. This difference points to the presence of fast decay components, which are not resolved with the TCSPC setup and can be attributed to the static and transient stages of the quenching.32 In this case again, no significant difference between (rac)-1 and (−)-1 can be detected.
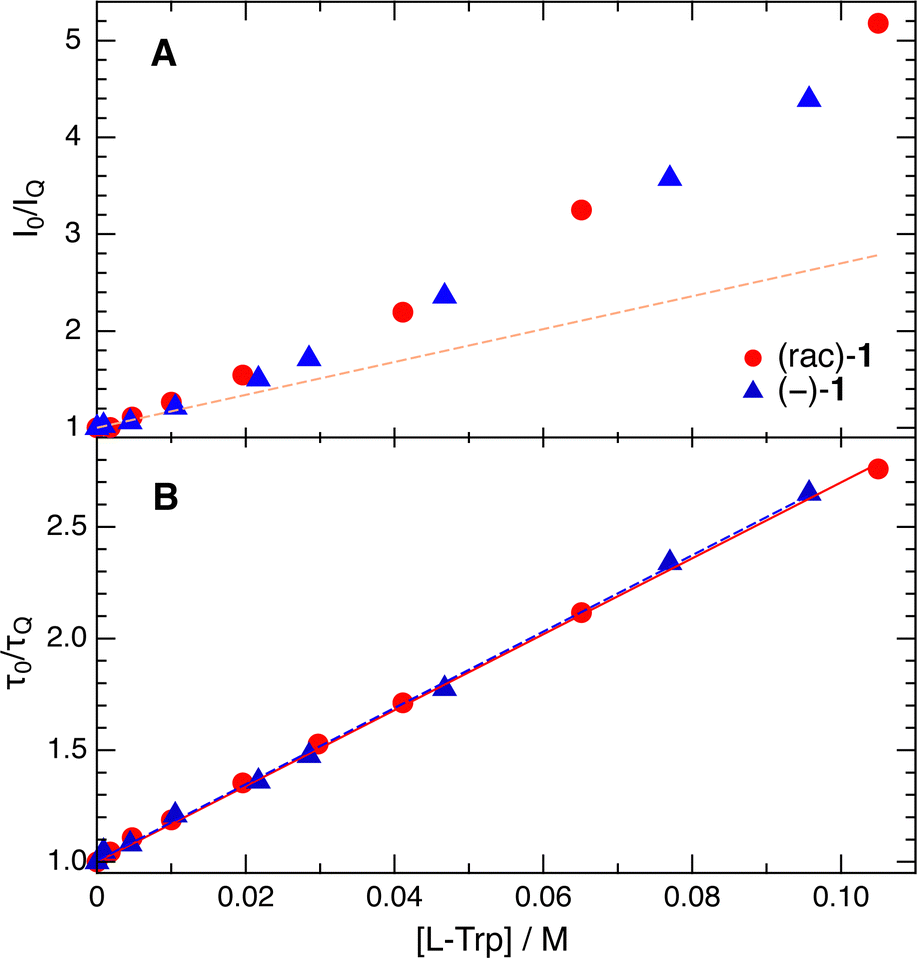 | ||
| Fig. 3 Stern–Volmer plots obtained from stationary (A) and time-resolved (B) measurements of the fluorescence quenching of (rac)-1 and (−)-1 by L-Trp in a 50:50 (v/v) water/ACN mixture. The dashed line in panel (A) is the best linear fit of the data with (rac)-1 shown in panel (B). | ||
The fluorescence dynamics at 610 nm of the two enantiomers of 1 in water/ACN are, as expected, identical within the limit of error, the data with (+)-(P)-1 being illustrated in Fig. 4A. They exhibit a partial decay within the first 5 ps and a slower one on the ns timescale, in agreement with the 2.3 ns lifetime of 1 in this solvent mixture. This initial decay can be attributed to vibrational and/or solvent relaxation.53Fig. 4A also depicts time profiles measured in the same conditions with the two enantiomers of 1 and 50 mM L-Trp. Their decay is significantly accelerated compared to those without Trp. To better evidence the effect of Trp, the time-dependent fluorescence intensity with Trp, IQ(t), was divided by that without quencher, I0(t), to obtain the pure quenching dynamics, R(t) = IQ(t)/I0(t). As illustrated in Fig. 4B, the pure quenching dynamics are the same within the limit of error for both enantiomers, pointing to an absence of significant stereoselectivity. These results confirm the presence of short fluorescence decay components that are too fast to be resolved by TCSPC.
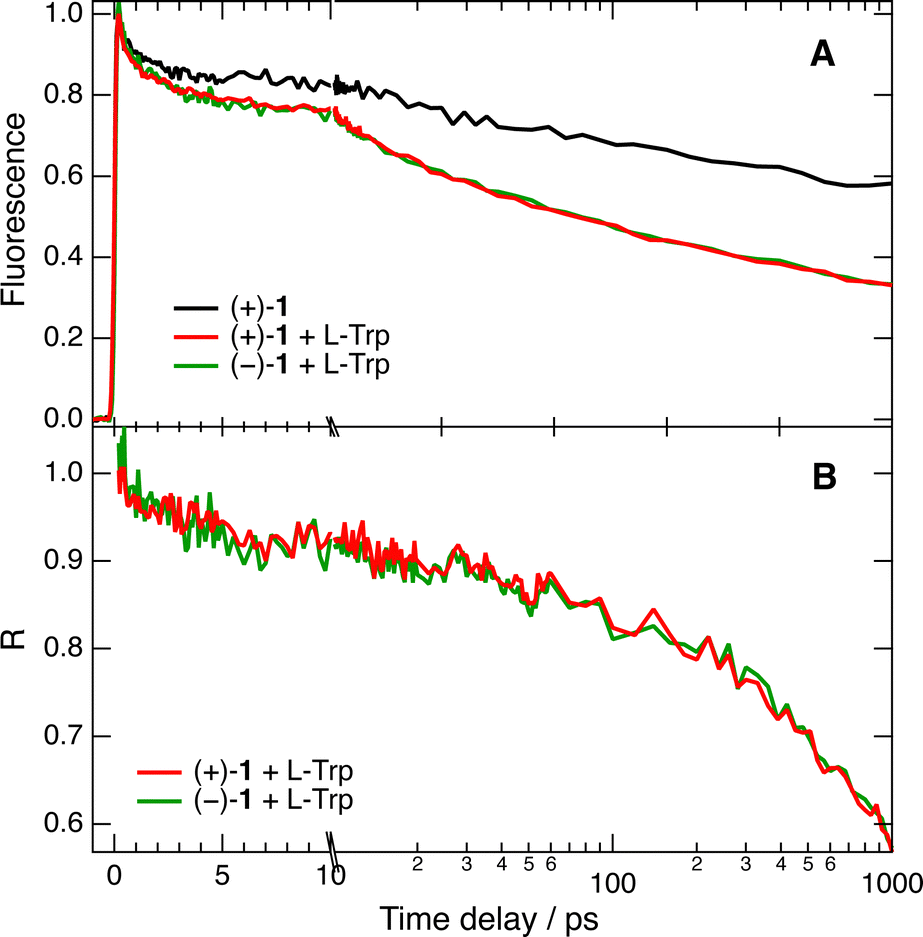 | ||
| Fig. 4 (A) Time evolution of the fluorescence intensity at 610 nm upon 450 nm excitation of (+)-1 alone and of the two enantiomers of 1 with 0.05 M L-Trp in 50:50 (v/v) water/ACN. (B) Pure quenching dynamics, R(t), obtained from the data in panel (A). The time axis is linear up to 10 ps and logarithmic afterwards. | ||
These dynamics are strongly non-exponential and their quantitative analysis requires the use of proper theoretical approaches, such as the differential encounter theory (DET),32–34 that take into account the time dependence of the reactant-pairs distribution, and hence the time dependence of the quenching rate. However, to be insightful, these models require precise knowledge of a large number of parameters, such as the driving force, the reorganisation energy or the diffusion constants, that are not readily available for the AD pairs and solvent mixtures investigated here. However, chiral recognition in bimolecular photoinduced ET can be identified without applying such models. Instead, a multiexponential analysis of the R(t) profiles was performed in order to have a measure of the quenching timescales and of the amplitude of its static stage. The sum of not less than three exponential functions was required to properly reproduce these dynamics, with a time constant close to the ∼200 fs IRF, another around 20 ps and a third larger than 2 ns. The slow component, with a relative amplitude of about 0.85, reflects the dynamic quenching, for which no chiral effect is expected. The two faster components can be associated with the static and transient stages of the quenching. However, because of their relatively small amplitude, the presence of a weak stereoselectivity cannot be totally ruled out.
Despite the limited solubility of the helicenes, fluorescence up-conversion measurements could also be carried out in pure water. As illustrated in Fig. 5A with the quenching of (−)-1, the amplitude of the fast decay components of R(t) is significantly larger than in water/ACN. Despite this, no significant difference between L- and D-Trp could be observed (Fig. 5A). Multiexponential analysis reveals that about 40% of the amplitude of R(t) decays within the IRF. Subsequently, about 13% of the amplitude decays in 5 ± 1 ps and, finally, the diffusion-controlled regime takes place on the ns timescale.
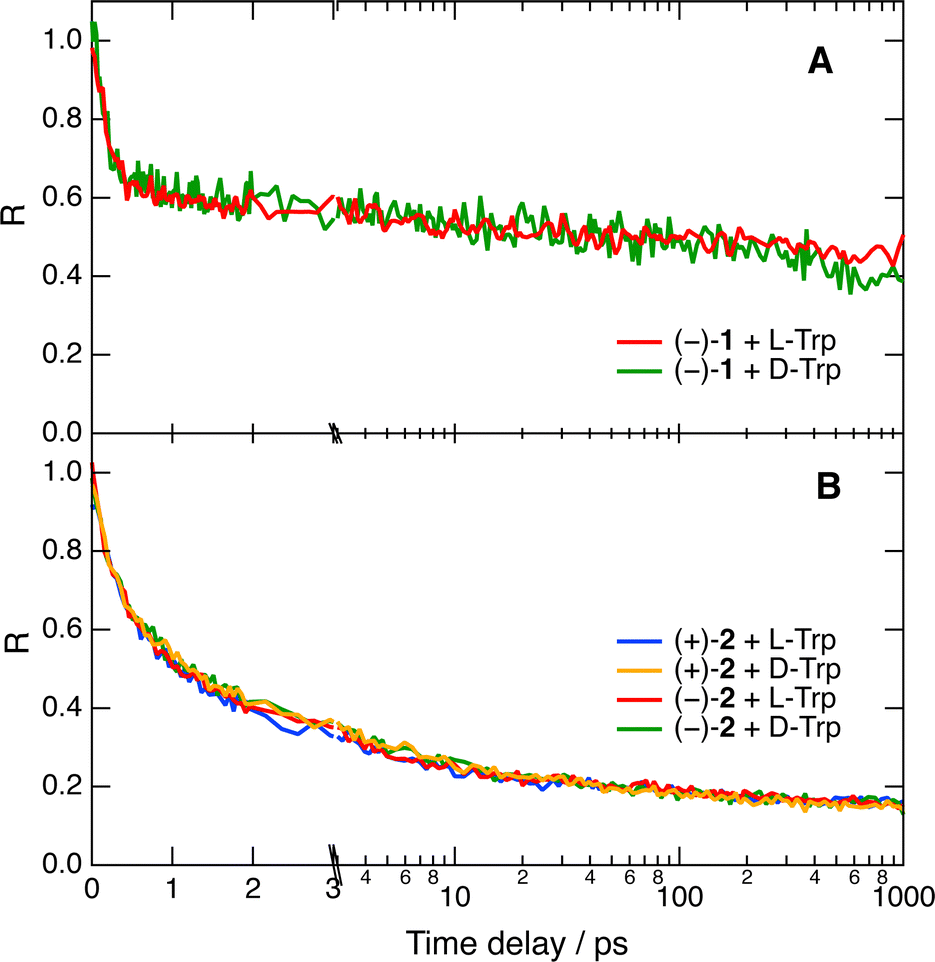 | ||
| Fig. 5 Pure quenching dynamics of the fluorescence of 1 (A) and 2 (B) by 0.05 M Trp in water after 450 nm excitation. The time axis is linear up to 3 ps and logarithmic afterwards. | ||
The limited IRF of the experiment does not allow the detection of stereoselectivity in the fastest quenching component of 1 by Trp. To try slowing down the dynamics, we repeated the measurement with azaoxa helicene 2, which is a weaker acceptor than 1 (Table S1, ESI†). Fig. 5B reveals that with this fluorophore, about 75% of the quenching occurs before the stationary diffusion-assisted regime becomes operative. This is probably due to the reduced solubility of 2 in water that favours association with Trp through dispersion interactions and hydrophobic effects. In agreement with a smaller ET driving force, the fastest quenching component is slower than with 1 and is associated with a 750 fs time constant for both diastereomeric AD pairs, well above the IRF. A slower, 7–8 ps component with a smaller relative amplitude is also present, additionally to the ns diffusion-controlled component. In this case again, no significant stereoselectivity can be detected.
This absence of chiral recognition observed with these cationic [6]helicenes and Trp in water and a water/ACN mixture is consistent with previous studies where no stereoselectivity was detected in a polar environment.21–24 As discussed below, one could also argue that the stereogenic centre of Trp is far from the aromatic ring and does not contribute much to the electronic coupling with the helicene acceptors. Therefore, trying to increase the probability to observe a chiral effect, we changed the quencher to BINOL, which possesses axial chirality and a better-defined C2-symmetry conformational environment, and we decreased the solvent polarity by using DCM and THF. Fig. 6 shows time profiles of the fluorescence intensity measured with the two enantiomers of 1 alone and with 0.15 M of the either enantiomer of BINOL in DCM, whereas the pure quenching dynamics are presented in Fig. S4 (ESI†). It should first be noted that the fluorescence lifetime of 1 in DCM of 10.8 ns is markedly larger than that of 2.3 ns in water/ACN (Fig. S3, ESI†). Similar solvent dependence was also observed with the parent cationic diaza [4]helicene54 and was attributed to hydrogen-bond induced non-radiative decay.55,56 However, given the long wavelength emission of these dyes, quenching by energy transfer to water could also contribute to this lifetime shortening.57
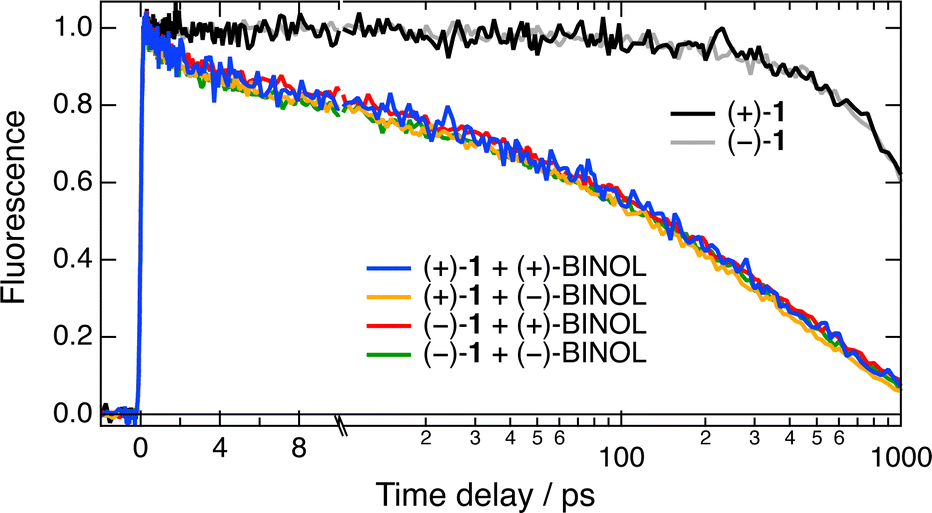 | ||
| Fig. 6 Time evolution of the fluorescence intensity at 610 nm upon 450 nm excitation of the two enantiomers of 1 alone and with 0.15 M of either enantiomer of BINOL in DCM. The time axis is linear up to 10 ps and logarithmic afterward. | ||
Coming back to the ET quenching, Fig. 6 points to an absence of significant stereoselectivity with this AD pair as well, despite the axial chirality of the quencher and the lower solvent polarity. About 40% of the intensity decays with 3 and ≈50 ps time constants, well within the time resolution of the experiment. The signal-to-noise ratio of the data in THF was lower because of the inferior solubility of 1. As illustrated in Fig. S5 (ESI†), the R(t) profiles measured with the four combinations of A and D enantiomers are the same within the limit of error and are similar to those in DCM.
3.3 Transient absorption measurements of the recombination dynamics
ET quenching of the cationic [6]helicenes by Trp and BINOL should result in the formation of the neutral [6]helicene radicals and the radical cation of the quenchers. After their formation, these radical pairs can either undergo geminate recombination or diffuse apart to free radicals, which then recombine on the microsecond timescale.50 The latter process is diffusion controlled and is, thus, not expected to depend on the chirality of the radicals. By contrast, geminate recombination does not involve significant diffusion and might thus exhibit stereoselectivity. To test this, we performed transient absorption (TA) measurements with 1 and both Trp and BINOL in various environments. Because of the limited solubility of 1 in water, TA measurements with Trp were done in 90:10 (v/v) water/ACN and 60:40 (v/v) water/ethanol mixtures. The second mixture was selected to try slowing down the charge-transfer dynamics, because of the lower polarity and slower relaxation dynamics of EtOH.58
The TA spectra recorded upon 470 or 530 nm excitation of 1 alone in both solvent mixtures are dominated by a negative band in the 700–510 nm region, which can be attributed to the S1 → S0 stimulated emission and the bleach of the S1 ← S0 absorption (Fig. 7B). Another negative band centered at 450 nm due to the bleach of the S2 ← S0 absorption and two positive features that can be assigned to Sn ← S1 excited-state absorption (ESA) are also visible. These spectra exhibit small initial dynamics, which can be attributed to vibrational/solvent relaxation, before decaying on the ns timescale, in agreement with the fluorescence lifetime. In the presence of 50 mM Trp, all these features lose more than 50% of their intensity within the first few ps, before decaying on the ns timescale (Fig. 7A). To better appreciate the spectral changes, global analysis was performed assuming a series of successive exponential steps (Fig. 7B). The resulting evolution-associated difference spectra (EADS) cannot be necessarily assigned to a given species/state but allow for a visualisation of the spectral changes and their timescale.59,60 As shown in Fig. S6 (ESI†), the EADS and time constants obtained from the analysis of the TA data measured with (−)-1 and 50 mM Trp are the same within the limit of error for both D- and L-Trp enantiomers, pointing to an absence of chiral effect on the observed dynamics in both solvent mixtures. Comparison of EADS A and B reveals significant intensity decay in the stimulated-emission region that can be interpreted as an ultrafast quenching component of the excited state. In the 440–350 nm ESA region, differences compared to the spectra without quencher can also be observed. The red side of this band is more intense in the presence of Trp. This difference is no longer present in EADS C and D, where this band is similar to that measured without quencher.
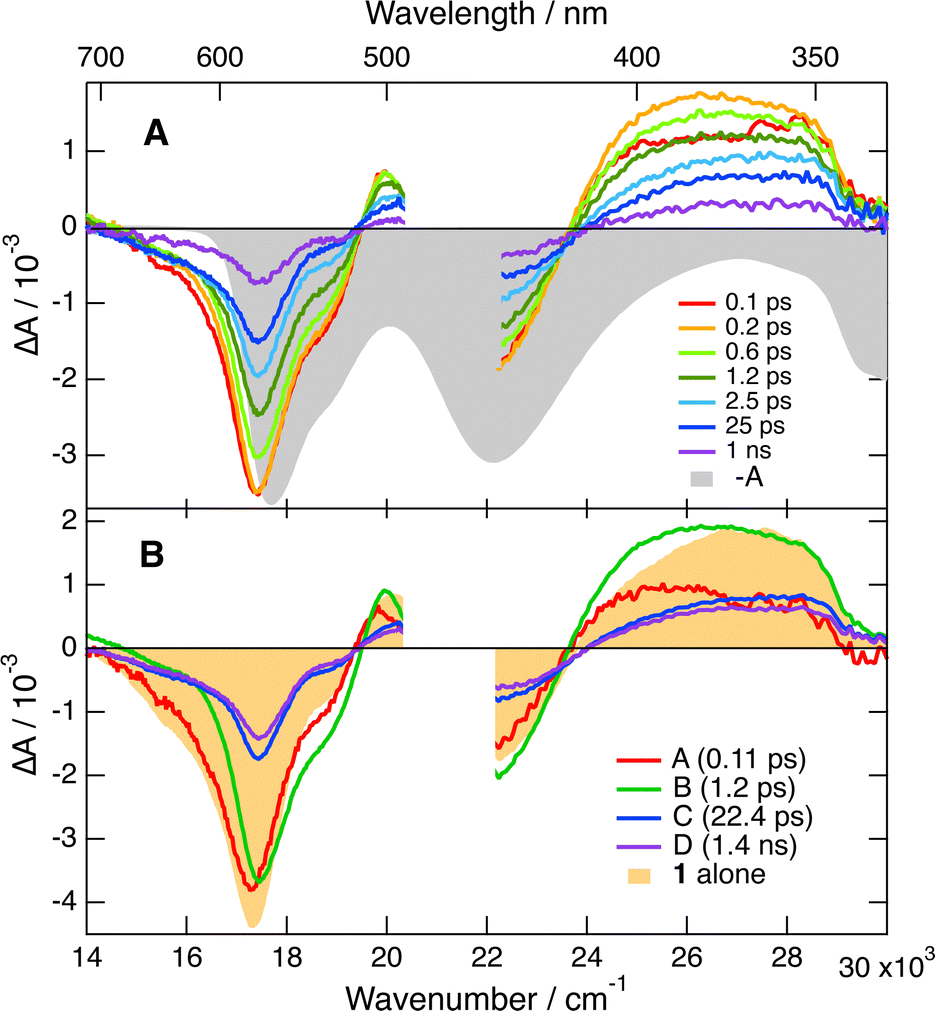 | ||
| Fig. 7 (A) Transient absorption spectra recorded upon 470 nm excitation of (−)-1 with 50 mM L-Trp in 60:40 (v/v) water/ethanol and negative stationary absorption spectrum. (B) Evolution-associated difference spectra (EADS) and time constants obtained from a global analysis of the same data assuming four successive exponential steps (A → B → C → D →). Because of the larger IRF at a shorter wavelength, the shape of EADS A below 420 nm should be considered with caution. A transient absorption spectrum recorded with (−)-1 alone is also shown for comparison. | ||
This spectral evolution with Trp can be interpreted as follows: the ultrafast, ∼100 fs, decay component in the stimulated-emission region (EADS A → B), is due to static quenching and produces the neutral [6]helicene radical as well as Trp radical cation, Trp˙+. The enhanced absorption around 400 nm is attributed to the dioxa helicene radical because it is also observed with BINOL, as discussed below. Moreover, Trp˙+ is known to absorb in the 650–500 nm range, in the same region as the S1 ← S0 absorption of 1.61 However, its small absorption coefficient relative to 1, i.e. 2700 vs. 16000 M−1 cm−1,51,61 makes its presence difficult to detect here. Consequently, the concurrent decays of the 440–350 nm ESA band and of the 550 nm bleach, visible in the transition from EADS B to C, mostly reflect the geminate recombination of the radical pair and the recovery of the ground-state population of 1. According to the global analysis (Fig. S6, ESI†), this process occurs on the sub-ps timescale (∼0.8 ps) in water/ACN, and is, as expected, significantly slower in water/EtOH (∼1.2 ps). Like the ET quenching, the recombination dynamics cannot be described using a simple exponential model.62 Its analysis requires accounting for the intrinsically non-Markovian nature of this process. Indeed, the recombination dynamics depend on the radical pair distribution, which is itself determined by the preceding quenching process. All these aspects are accounted for in the integral encounter theory (IET) and equivalent models.34,63,64 However, in this case again, to be insightful their application requires knowledge of a large number of parameters which are not readily available for the AD pairs and solvent mixtures used here. However, the analysis performed here, although not appropriate for obtaining a quantitative description of the quenching and recombination dynamics, gives access to the timescales on which these processes take place and, more importantly, allows for the detection of a possible stereoselectivity. Whereas the spectral evolution visible by going from EADS A to C is mostly due to the static quenching and the recombination of the ensuing radical pairs, the subsequent C → D → steps mainly reflect the slower dynamic quenching. The absence of any feature of the radical pair product in EADS C and D is consistent with the recombination being much faster than the quenching itself. Once generated upon dynamic quenching, the radical pairs most probably recombine efficiently without major diffusive motion.
Consequently, these TA results not only confirm the absence of significant stereoselectivity in the quenching dynamics of 1 by Trp but also reveal that chirality does not influence the recombination dynamics as well. Given the high signal-to-noise ratio of the TA data, a stereodifferentiation in the overall dynamics larger than 1.05 can be excluded.
TA measurements were also carried out with BINOL as the electron donor in both ACN and DCM. As illustrated in Fig. S7 (ESI†), the transient spectra in ACN are essentially the same as those measured without a quencher except for the dynamics and do not present any feature of the quenching product. This implies that recombination occurs faster than the photoinduced ET, even for those radical pairs generated during the fastest stages of quenching. This suggests that these pairs recombine on a sub-ps timescale. This is consistent with the recombination timescales found above with Trp in the water/ACN mixture. The EADS and time constants obtained from global analysis with (+)- and (−)-BINOL coincide almost perfectly, confirming the absence of a chiral effect for the quenching dynamics. As recombination is much faster than quenching and cannot be resolved here, some stereoselectivity in recombination can not be totally ruled out.
The recombination dynamics is markedly slowed down when going to the medium polar DCM but do not depend on the chirality of the reactants (Fig. S8, ESI†). As shown in Fig. 8A, C and Fig. S8 (ESI†), the shape of the 440–350 nm ESA band changes with time, with the maximum shifting to about 400 nm, indicative of the presence of the neutral radical of 1. Unfortunately, the absorption spectrum of this radical does not differ sufficiently from that of the S1 state of 1 to allow disentangling of the quenching and recombination dynamics. The observation of quenching product features indicates that recombination is no longer much faster than the early stage of quenching, as also found with Trp. This is confirmed by comparing the time evolution of the TA intensity in the stimulated-emission and the ground-state-bleach regions (Fig. 8B). The decay of the stimulated emission during the first 100 ps is significantly faster than the ground-state recovery. Consequently, the fact that the TA dynamics measured with two 1/BINOL diastereomeric pairs in DCM is exactly the same (Fig. S8, ESI†) points to an absence of significant chiral recognition for both the photoinduced ET and the subsequent recombination.
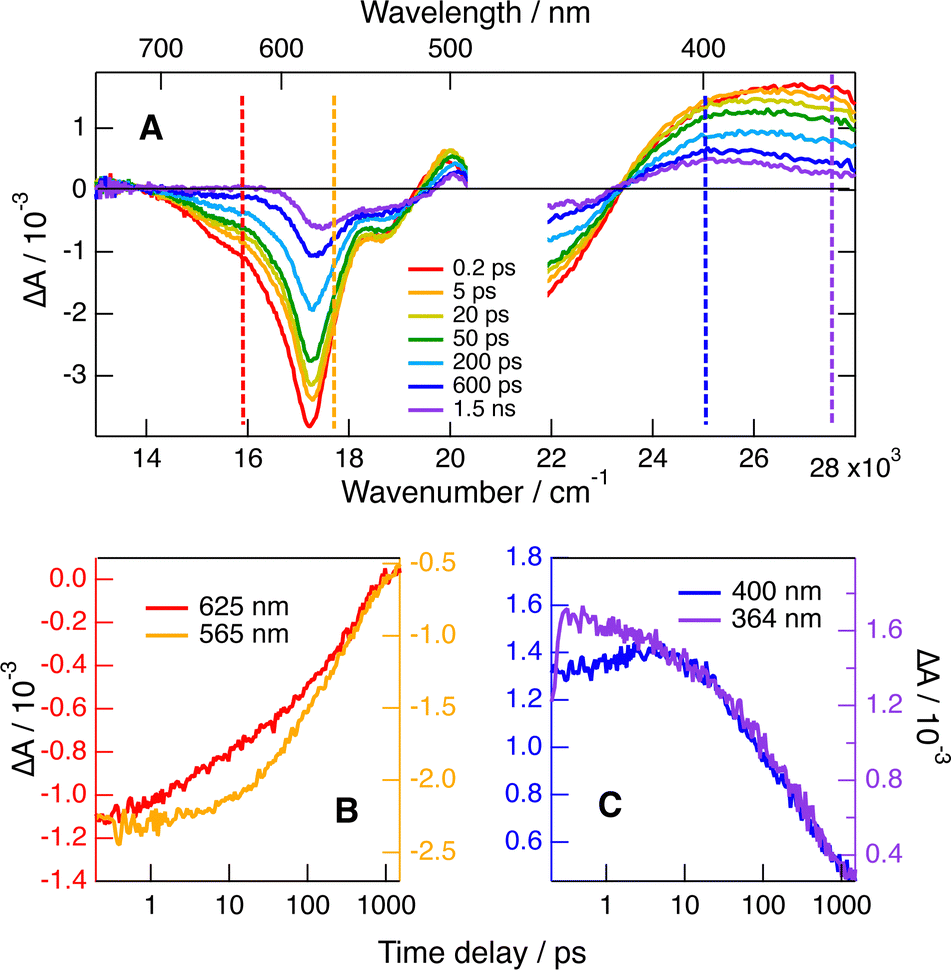 | ||
| Fig. 8 (A) Transient absorption spectra recorded upon 470 nm excitation of (+)-1 with 150 mM (−)-BINOL in DCM. (B) Time evolution of the transient absorption in the stimulated emission and ground-state bleach regions and (C) in the band below 420 nm (dashed lines in panel A). | ||
4 Discussion and MD simulations
All these results point to an absence of significant stereoselectivity in bimolecular photoinduced ET and in the subsequent recombination in high and medium polarity solvents. The previous studies were mainly measuring the dynamic stage of quenching, which is strongly influenced by the diffusion of the reactants. In these cases, stereoselectivity in the intrinsic ET step can be hidden, at least partially, by the chirality-independent diffusion step. Here, we could resolve the initial, static, stage of quenching where diffusion does not play a significant role.According to Marcus ET theory,65 the only parameter that is affected by the chirality of the reactants is the electronic coupling, V, which depends on their distance and mutual orientation. The absence of significant stereoselectivity found here with all AD pairs points to a similar electronic coupling for diastereomeric reactant pairs. MD simulations were performed to gain a better insight into how chirality affects the mutual orientation and distance between 1 and the quenchers. The results with (−)-1 and both Trp enantiomers in pure water reveal that, as soon as the two reactants collide, they remain in close contact over the whole 50 ns simulations. Histograms of the minimum distance between any pair of atoms of the two molecules, rmin, as well as the centre-of-mass distance, rcom, are presented in Fig. 9A and Fig. S9 (ESI†) together with plots of the radial distribution function, g(r). All of them indicate that the distribution of distances is very similar for both Trp enantiomers. This result implies that the 1/Trp pairs experience very similar interaction energy independently of their chirality.
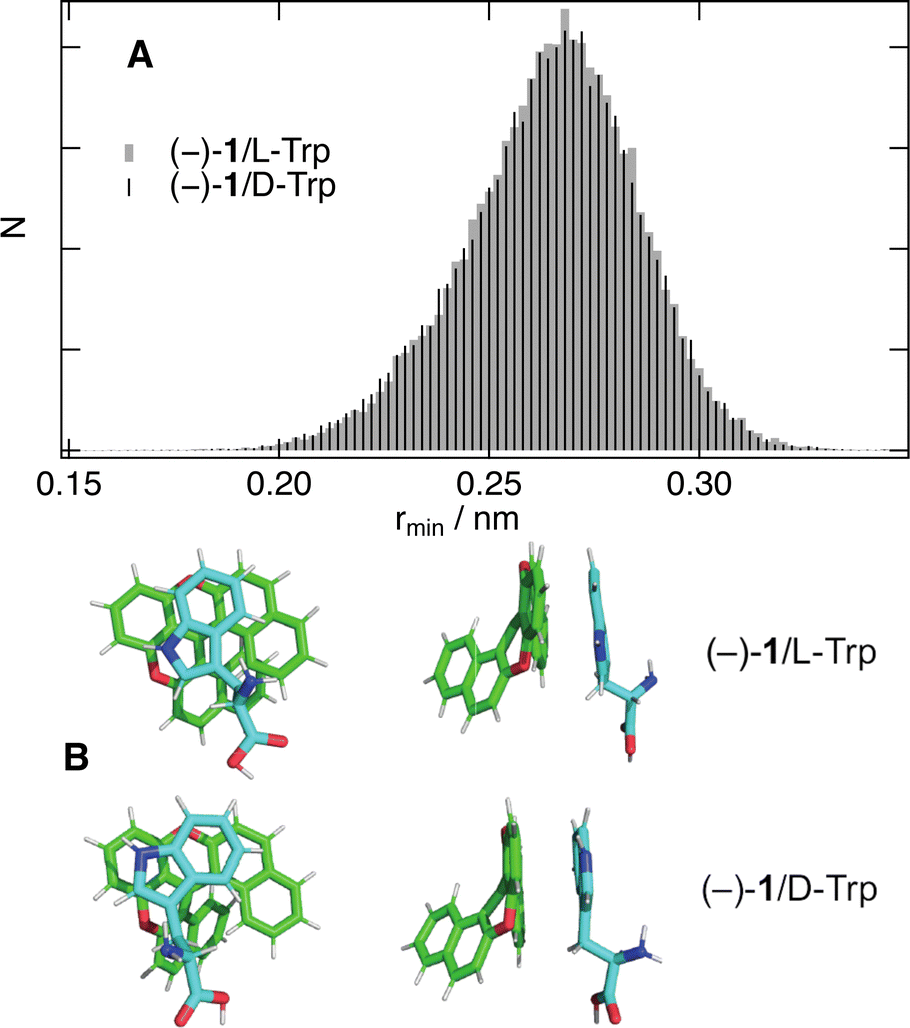 | ||
| Fig. 9 (A) Histograms of the minimum AD distance obtained from 50 ns MD trajectories of (−)-1 and Trp in water. (B) Snapshots from MD simulations of the two diastereomeric pairs with a centre of mass distance of 0.5 nm (top (left) and side views (right)). | ||
Fig. 9B depicts snapshots of two diastereomeric pairs with rcom = 0.5 nm. In both cases, the indol rings of Trp adopt a π-stack configuration with the central rings of the helicene. The electronic coupling for the photoinduced ET depends on the overlap of the HOMOs of both A and D. As illustrated in Fig. S10 (ESI†), the HOMO of 1 is delocalised over the whole molecule. The situation differs for Trp, whose HOMO is almost entirely localised on the aromatic systems. Given the small size of the indol rings relatively to 1, large coupling for the ET can be realised with a large distribution of mutual orientations. As the stereogenic centre of Trp is relatively far from the aromatic system and has negligible HOMO amplitude, it does not contribute significantly to the electronic coupling. These results fully supported the absence of stereoselectivity found experimentally.
The situation for the recombination of the radical-pair product is not expected to differ much. As 1 is a closed-shell cation, ET with a neutral donor produces its neutral radical and the radical cation of the donor. Consequently, the Coulombic interactions between the reaction partners should not change significantly upon ET. Moreover, given the ultrashort timescale on which recombination takes place, this process should occur at essentially the same geometry as that for the initial ET step. However, contrary to the photoinduced ET, the electronic coupling for the recombination depends on the overlap of the acceptor LUMO with the donor HOMO. As shown in Fig. S10 (ESI†), the LUMO of 1 is also delocalized over the whole molecule. Consequently, large electronic coupling for recombination can be achieved independently of the chirality of the reactants.
MD simulations were also carried out with BINOL in ACN and THF, the least polar solvent used in this study. Simulations in ACN with a single BINOL molecule in the box pointed to relatively rare close contacts with 1 (Fig. S11, ESI†). Simulations were then carried out with 11 BINOL molecules in the ACN box, corresponding to a quencher concentration of 0.15 M. The radial distribution functions determined from 50 ns simulations of diastereomeric AD pairs are qualitatively similar, with a major peak at a centre-of-mass distance of 0.8 nm and a smaller one close to 0.5 nm (Fig. S12, ESI†). Thus, chirality is not predicted to have a significant effect on the distribution of AD distances, hence on the interaction energy. As the molecular orbitals involved in both the photoinduced ET and the recombination steps are delocalised over the whole aromatic systems of both A and D (Fig. S10, ESI†), chirality should likewise not have a significant influence on the electronic coupling.
Contrary to ACN, simulations with a single quencher molecule in THF show that, once the reactants are in contact, they remain close together during the rest of the trajectory. Fig. 10A and Fig. S13 (ESI†) depict the histograms of rmin and rcom for two diastereomeric AD pairs. The differences between these histograms are of the same order of magnitude as those observed between histograms constructed from two different 50 ns simulations with the same diastereomer. Fig. 10B shows snapshots extracted from the simulations with a center-of-mass distance of 0.55 nm. They point to π stacking between one naphthol group of BINOL and the helicene. Such a mutual orientation can be realised independently of the chirality of the reactants. In this case again, the simulations support the absence of stereoselectivity observed experimentally.
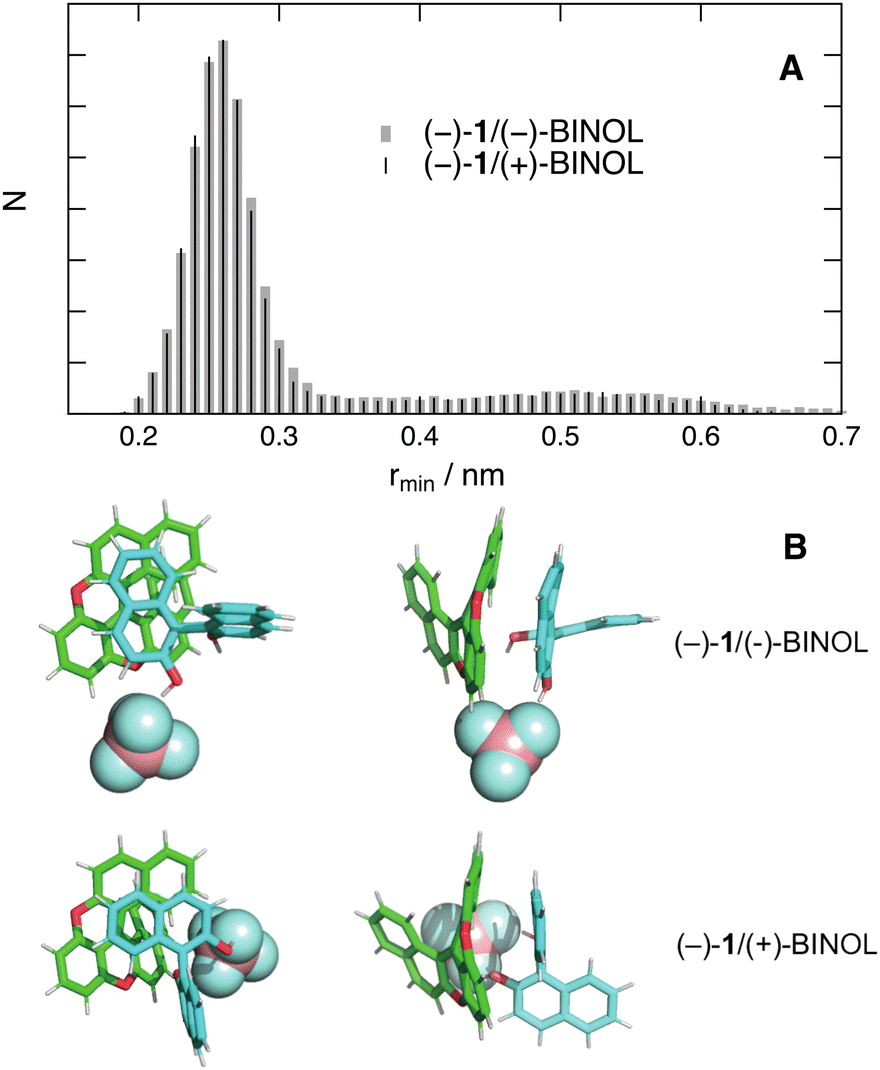 | ||
| Fig. 10 (A) Histograms of the minimum inter-reactant distance obtained from 50 ns MD trajectories of (−)-1 and one BINOL molecule in THF. (B) Snapshots from MD simulations of two diastereomeric pairs with a centre of a mass distance of 0.55 nm (top (left) and side views (right)). | ||
The dramatic solvent dependence of the distribution of AD distances can be attributed to the BF4− counter-ion. According to simulations in ACN in the absence of BINOL, the cationic helicene and BF4− are not often in contact, as expected from the favourable solvation energy in polar media and the efficient dielectric screening of the Coulombic interaction. Simulations in the medium polar THF reveal that BF4− has a much higher probability to be found in contact with the cation (Fig. S14, ESI†). In the presence of quencher, BF4− tends to bind to BINOL via H-bond interactions. This can be seen in Fig. 10B, where the anion is bound to one or both hydroxyl groups of BINOL. This binding and the stronger Coulombic interaction in THF favour close contact between the cationic helicene and BINOL. In ACN, this binding is also operative, although apparently less efficient. In any case, close contact between the BINOL⊂BF4− complex and the cationic helicene is disfavoured in ACN because it results in a smaller solvation energy.
The absence of stereoselectivity in polar solvents, ACN and water, is consistent with previous studies based on stationary quenching experiments.21–24 However, the occurrence of long-distance ET, which was proposed to explain this result, cannot be invoked here, because static quenching in highly coupled reactant pairs could be unambiguously identified. According to a recent investigation based on MD simulations and quantum-chemical calculations,66 the electronic coupling for bimolecular photoinduced ET between aromatic reactants is typically of the order of 0.1 to 0.3 eV up to a minimum AD distance of 0.3 nm. The same order of magnitude can be expected for the AD pairs investigated here. With such a large electronic coupling, ET can no longer be considered as a non-adiabatic process, where the ET probability depends on the square modulus of the coupling.65 As initially suggested by Tachiya and coworkers67,68 and supported by more recent investigations,66,69,70 ET in such highly-coupled reactant pairs is an adiabatic process, and, thus, cannot be discussed in terms of Marcus ET theory. In these cases, the reactant pair should rather be viewed as a ‘supermolecule’. Photoinduced ET is equivalent to an internal conversion from a locally-excited state to a charge-transfer state and recombination to an internal conversion to the ground state. Here, the ET dynamics is mostly controlled by the reorganisation of intramolecular modes and depend only weakly on the solvent.
5 Conclusions
By using a combination of ultrafast time-resolved fluorescence and transient electronic absorption spectroscopy, we could investigate the effect of chirality on the initial stage of the ET quenching dynamics and on the ensuing recombination, where the influence of diffusion is minimal. Our results with a helical acceptor fluorophore and quenchers with either stereocentered or axial chirality did not evidence any stereoselectivity, neither for the photoinduced ET step nor for the recombination step. Contrary to many bimolecular reactions like, e.g., proton transfer, ET does not require a very specific orientation to occur. This is particularly the case for aromatics, where significant electronic coupling is possible in a broad range of orientations. Our MD simulations suggest that close contact between the reactants, hence high coupling, can be easily realised independently of the chirality of the reactants. Even if present, small stereoselectivity in the electronic coupling, which could not be identified in the MD simulations, is not expected to result in significant chiral recognition, because ET in such highly coupled reactants is an adiabatic process, whose dynamics is mostly controlled by high-frequency modes and no longer depends on the coupling itself.One can anticipate that the AD pairs investigated here would exhibit significant chiral recognition if they were connected via a linker, which would inhibit to close a distance, keeping ET in the non-adiabatic regime, and would narrow the distribution of mutual orientations.
The results obtained here should be quite general. Stereoselectivity in bimolecular ET might be relevant with specific reactant pairs, such as a pair of helical molecules of similar size, or non-aromatic donors and acceptors with the reactive sites localised on the stereogenic centres.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Swiss National Science Foundation (grants 200020-184607, 200020-184843 and 200020-207539) and the University of Geneva for financial support. All MD simulations were performed using the High Performance Computing facilities of the University of Geneva.Notes and references
- R. Bentley, Chirality in Biology, ed. R. A. Meyers, 2006 Search PubMed.
- J. Gal, Helv. Chim. Acta, 2013, 96, 1617–1657 CrossRef CAS.
- Q. Sallembien, L. Bouteiller, J. Crassous and M. Raynal, Chem. Soc. Rev., 2022, 51, 3436–3476 RSC.
- Y.-D. Yang, B.-B. Yang and L. Li, Chirality, 2022, 34, 1355–1370 CrossRef CAS PubMed.
- R. Naaman, Y. Paltiel and D. H. Waldeck, Acc. Chem. Res., 2020, 53, 2659–2667 CrossRef CAS PubMed.
- Z. Shang, T. Liu, Q. Yang, S. Cui, K. Xu, Y. Zhang, J. Deng, T. Zhai and X. Wang, Small, 2022, 18, 2203015 CrossRef CAS PubMed.
- J. M. Abendroth, D. M. Stemer, B. P. Bloom, P. Roy, R. Naaman, D. H. Waldeck, P. S. Weiss and P. C. Mondal, ACS Nano, 2019, 13, 4928–4946 CrossRef CAS PubMed.
- T. P. Fay and D. T. Limmer, Nano Lett., 2021, 21, 6696–6702 CrossRef CAS PubMed.
- A. Privitera, E. Macaluso, A. Chiesa, A. Gabbani, D. Faccio, D. Giuri, M. Briganti, N. Giaconi, F. Santanni, N. Jarmouni, L. Poggini, M. Mannini, M. Chiesa, C. Tomasini, F. Pineider, E. Salvadori, S. Carretta and R. Sessoli, Chem. Sci., 2022, 13, 12208–12218 RSC.
- S. Abad, U. Pischel and M. A. Miranda, Photochem. Photobiol. Sci., 2005, 4, 69–74 CrossRef CAS PubMed.
- P. Bonancía, I. Vayá, D. Markovitsi, T. Gustavsson, M. C. Jiménez and M. A. Miranda, Org. Biomol. Chem., 2013, 11, 1958–1963 RSC.
- E. A. Khramtsova, D. V. Sosnovsky, A. A. Ageeva, E. Nuin, M. L. Marin, P. A. Purtov, S. S. Borisevich, S. L. Khursan, H. D. Roth, M. A. Miranda, V. F. Plyusnin and T. V. Leshina, Phys. Chem. Chem. Phys., 2016, 18, 12733–12741 RSC.
- A. A. Ageeva, A. B. Doktorov, O. Y. Selyutina, I. M. Magin, M. G. Ilyina, S. S. Borisevich, R. Y. Rubtsov, S. L. Khursan, A. A. Stepanov, S. F. Vasilevsky, N. E. Polyakov and T. V. Leshina, Int. J. Mol. Sci., 2022, 23, 38 CrossRef CAS PubMed.
- A. J. Stasyuk, O. A. Stasyuk, S. Filippone, N. Martin, M. Sola and A. A. Voityuk, Chem. – Eur. J., 2018, 24, 13020–13025 CrossRef CAS PubMed.
- L.-A. Chen, W. Xu, B. Huang, J. Ma, L. Wang, J. Xi, K. Harms, L. Gong and E. Meggers, J. Am. Chem. Soc., 2013, 135, 10598–10601 CrossRef CAS PubMed.
- H. Huo, X. Shen, C. Wang, L. Zhang, P. Röse, L.-A. Chen, K. Harms, M. Marsch, G. Hilt and E. Meggers, Nature, 2014, 515, 100–103 CrossRef CAS PubMed.
- J. Ma, X. Zhang, X. Huang, S. Luo and E. Meggers, Nat. Protoc., 2018, 13, 605–632 CrossRef CAS PubMed.
- A. G. Amador and T. P. Yoon, Angew. Chem., Int. Ed., 2020, 55, 2304–2306 CrossRef PubMed.
- U. Pischel, S. Abad, L. R. Domingo, F. Boscá and M. A. Miranda, Angew. Chem., Int. Ed., 2003, 42, 2531–2534 CrossRef CAS PubMed.
- M. C. Jiménez, U. Pischel and M. A. Miranda, J. Photochem. Photobiol., C, 2007, 8, 128–142 CrossRef.
- M. Irie, T. Yorozu and K. Hayashi, J. Am. Chem. Soc., 1978, 100, 2236–2237 CrossRef CAS.
- H. Rau and F. Totter, J. Photochem. Photobiol., A, 1992, 63, 337–347 CrossRef CAS.
- M. Woerner, G. Greiner and H. Rau, J. Phys. Chem., 1995, 99, 14161 CrossRef CAS.
- T. Nishiyama, K. Mizuno, Y. Otsuji and H. Inoue, Chem. Lett., 1994, 2227–2228 CrossRef CAS.
- H. Takashima, M. Tanaka, Y. Hasegawa and K. Tsukahara, J. Biol. Inorg. Chem., 2003, 8, 499–506 CrossRef CAS PubMed.
- T. Suzuki, K. Ichioka, H. Higuchi, H. Kawai, K. Fujiwara, M. Ohkita, T. Tsuji and Y. Takahashi, J. Org. Chem., 2005, 70, 5592–5598 CrossRef CAS PubMed.
- Y. Imai, K. Kamon, T. Kinuta, N. Tajima, T. Sato, R. Kuroda and Y. Matsubara, Tetrahedron, 2007, 63, 11928–11932 CrossRef CAS.
- J. Zhao, T. M. Fyles and T. D. James, Angew. Chem., Int. Ed., 2004, 43, 3461–3464 CrossRef CAS PubMed.
- Z.-B. Li, J. Lin, M. Sabat, M. Hyacinth and L. Pu, J. Org. Chem., 2007, 72, 4905–4916 CrossRef CAS PubMed.
- Y. Wu, H. Guo, X. Zhang, T. D. James and J. Zhao, Chem. – Eur. J., 2011, 17, 7632 CrossRef CAS PubMed.
- S. Murata and M. Tachiya, J. Phys. Chem., 1996, 100, 4064–4070 CrossRef CAS.
- A. I. Burshtein, Adv. Chem. Phys., 2004, 129, 105–418 CrossRef CAS.
- A. Rosspeintner and E. Vauthey, Phys. Chem. Chem. Phys., 2014, 16, 25741–25754 RSC.
- G. Angulo and A. Rosspeintner, J. Chem. Phys., 2020, 153, 040902 CrossRef CAS PubMed.
- F. Torricelli, J. Bosson, C. Besnard, M. Chekini, T. Bürgi and J. Lacour, Angew. Chem., Int. Ed., 2013, 52, 1796–1800 CrossRef CAS PubMed.
- G. M. Labrador, J. Bosson, Z. S. Breitbach, Y. Lim, E. R. Francotte, R. Sabia, C. Villani, D. W. Armstrong and J. Lacour, Chirality, 2016, 28, 282–289 CrossRef CAS PubMed.
- J. A. Gardecki and M. Maroncelli, Appl. Spectrosc., 1998, 52, 1179–1189 CrossRef CAS.
- A. Fürstenberg and E. Vauthey, Photochem. Photobiol. Sci., 2005, 4, 260–267 CrossRef PubMed.
- G. Duvanel, J. Grilj, H. Chaumeil, P. Jacques and E. Vauthey, Photochem. Photobiol. Sci., 2010, 9, 908–915 CrossRef CAS PubMed.
- A. Aster, G. Licari, F. Zinna, E. Brun, T. Kumpulainen, E. Tajkhorshid, J. Lacour and E. Vauthey, Chem. Sci., 2019, 10, 10629–10639 RSC.
- J. S. Beckwith, A. Aster and E. Vauthey, Phys. Chem. Chem. Phys., 2022, 24, 568–577 RSC.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Rev. B.01, 2016 Search PubMed.
- A. W. Sousa da Silva and W. F. Vranken, BMC Res. Notes, 2012, 5, 367 CrossRef PubMed.
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, J. Am. Chem. Soc., 1996, 118, 11225–11236 CrossRef CAS.
- L. E. Chirlian and M. M. Francl, J. Comput. Chem., 1987, 8, 894–905 CrossRef CAS.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- D. van der Spoel, M. M. Ghahremanpour and J. A. Lemkul, J. Phys. Chem. A, 2018, 122, 8982–8988 CrossRef CAS PubMed.
- A. Weller, Pure Appl. Chem., 1982, 54, 1885–1888 CrossRef CAS.
- J. Bosson, G. M. Labrador, S. Pascal, F.-A. Miannay, O. Yushchenko, H. Li, L. Bouffier, N. Sojic, R. C. Tovar, G. Muller, D. Jacquemin, A. D. Laurent, B. Le Guennic, E. Vauthey and J. Lacour, Chem. – Eur. J., 2016, 22, 18394–18403 CrossRef CAS PubMed.
- H. Wode and W. Seidel, Ber. Bunsenges. Phys. Chem., 1994, 98, 927–934 CrossRef CAS.
- A. Rosspeintner, B. Lang and E. Vauthey, Annu. Rev. Phys. Chem., 2013, 64, 247–271 CrossRef CAS PubMed.
- O. Kel, P. Sherin, N. Mehanna, B. Laleu, J. Lacour and E. Vauthey, Photochem. Photobiol. Sci., 2012, 11, 623–631 CrossRef CAS PubMed.
- H. Shimada, A. Nakamura, T. Yoshihara and S. Tobita, Photochem. Photobiol. Sci., 2005, 4, 367–375 CrossRef CAS PubMed.
- B. Dereka and E. Vauthey, Chem. Sci., 2017, 8, 5057–5066 RSC.
- J. Maillard, K. Klehs, C. Rumble, E. Vauthey, M. Heilemann and A. Fürstenberg, Chem. Sci., 2021, 12, 1352–1362 RSC.
- M. L. Horng, J. A. Gardecki, A. Papazyan and M. Maroncelli, J. Phys. Chem., 1995, 99, 17311–17337 CrossRef CAS.
- I. H. M. van Stokkum, D. S. Larsen and R. van Grondelle, Biochim. Biophys. Acta, Bioenerg., 2004, 1657, 82–104 CrossRef CAS PubMed.
- J. S. Beckwith, C. A. Rumble and E. Vauthey, Int. Rev. Phys. Chem., 2020, 39, 135–216 Search PubMed.
- I. A. Shkrob, J. Sauer, C. Myran, A. D. Liu, R. A. Crowell and A. D. Trifunac, J. Phys. Chem. A, 1998, 102, 4976–4989 CrossRef CAS.
- G. Angulo, A. Rosspeintner, B. Lang and E. Vauthey, Phys. Chem. Chem. Phys., 2018, 20, 25531–25546 RSC.
- A. Burshtein, Adv. Chem. Phys., 2000, 114, 419–587 Search PubMed.
- A. Goun, K. Glusac and M. D. Fayer, J. Chem. Phys., 2006, 124, 084504 CrossRef PubMed.
- R. A. Marcus and N. Sutin, Biochim. Biophys. Acta, 1985, 811, 265–322 CrossRef CAS.
- C. A. Rumble and E. Vauthey, J. Phys. Chem. B, 2021, 125, 10527–10537 CrossRef CAS PubMed.
- S. Iwai, S. Murata, R. Katoh, M. Tachiya, K. Kikuchi and Y. Takahashi, J. Chem. Phys., 2000, 112, 7111–7117 CrossRef CAS.
- S. Murata and M. Tachiya, J. Phys. Chem. A, 2007, 111, 9240–9248 CrossRef CAS PubMed.
- C. Nançoz, C. Rumble, A. Rosspeintner and E. Vauthey, J. Chem. Phys., 2020, 152, 244501 CrossRef PubMed.
- E. Vauthey, J. Phys. Chem. B, 2022, 126, 778–788 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: ET driving forces, additional stationary spectra, fluorescence up-conversion and transient absorption data, additional results from MD simulations. See DOI: https://doi.org/10.1039/d3cp00760j. All data can be downloaded from: https://doi.org/10.26037/yareta:pn7f26ulenhoxlfvtlsf7n3u4q |
| This journal is © the Owner Societies 2023 |