
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInfrared spectroscopy of [H2O–Xn]+ (n = 1–3, X = N2, CO2, CO, and N2O) radical cation clusters: competition between hydrogen bond and hemibond formation of the water radical cation†
Mizuhiro
Kominato
and
Asuka
Fujii
 *
*
Department of Chemistry, Graduate School of Science, Tohoku University, Sendai 980-8578, Japan. E-mail: asuka.fujii.c5@tohoku.ac.jp
First published on 15th May 2023
Abstract
The water radical cation H2O+ is an important intermediate in radiation chemistry and radiobiology, and its role in radical reactions has recently attracted much attention. However, knowledge of intermolecular interactions of H2O+ remains very limited due to its high reactivity. We focus on structures of [H2O–X]+, formed by H2O+ with a counter molecule X, as a model for intermediates in reactions of H2O+. Such structural information provides the basis for understanding reaction processes of H2O+. Two structural motifs for [H2O–X]+ have been known: hydrogen bond and hemibond, which are expected to have very different reactivities from each other. Due to the high acidity of H2O+, the H-bonded form is mostly considered to be preferred. However, it has recently been reported that the hemibonded form is preferred in some cases. We perform infrared photodissociation spectroscopy and quantum chemical calculations on [H2O–Xn]+ (n = 1–3, X = N2, CO2, CO, and N2O) to determine their structural motifs. The competition between the hydrogen bond and hemibond formation is systematically examined based on the firm structure information. The competition is interpreted in terms of the proton affinity (PA) and the ionization potential (IP) of X. The rough ranges of PA and IP for the priority of the hemibond motif are determined. The impact of other factors on the competition is also discussed.
1. Introduction
Water ionization and subsequent processes are central issues in the aqueous environmental chemistry and biology of various radiation-related processes.1 Water (in aqueous solutions) ionized upon the interaction with ionizing radiation initially forms the water radical cation H2O+, which is immediately followed by the formation of the active chemical species, the hydrated electron e− and OH radical. The OH radical is well known as an important intermediate of radiation-induced damage in hydrated environments due to its high reactivity. In recent years, however, the role of H2O+ itself as well as the OH radical has attracted much attention in radical chemistry of water induced by ionizing radiation.2–13 Direct reactions with H2O+ can be important when water molecules are ionized in close proximity to other molecules, e.g., at the interface between water and biomolecules. Otherwise, H2O+ should undergo ultrafast proton transfer with surrounding water molecules to form the OH radical, as described above. Indeed, in the highly concentrated solutions,2–9 where water molecules are in contact with other molecules, and in microdroplets,10–13 it has been indicated that H2O+ plays a key role in chemical reactions. Despite such importance, H2O+ is still challenging to study, especially for direct measurements in the liquid phase, because of its high reactivity. Although there have been some reports,14–20 knowledge of H2O+ remains scarce. Gas-phase cluster studies are a powerful approach to such ultrashort-lived radical cation species, as their reactions are well controlled under isolated conditions. In the present study, we focus on structures of [H2O–X]+ formed by H2O+ and a counter molecule X as a model for reaction intermediates of H2O+. Such structural information is expected to provide the basis for understanding reaction processes of H2O+.Structures of the [H2O–X]+ (X = He, Ne, Ar, Kr, CO2, N2O, and H2O) radical cation clusters have been studied by infrared (IR) spectroscopy in the gas phase.21–31 Two structural motifs are confirmed: hydrogen bond (H-bond) and hemibond. The H-bonded type has a structure in which X is H-bonded to one of the OH groups of H2O+ (HOH⋯X). On the other hand, the hemibonded structure (H2O⋯X) has a non-classical (two center-three electron, 2c–3e) covalent bond formed by overlapping lone pair orbitals of H2O+ and X. The hemibond has attracted strong attention in radiation chemistry, mainly of sulfur-containing systems.32–47 Recently, it has been reported that OH radicals form hemibonds in the aqueous phase.48,49 However, roles of hemibonds of H2O+ in aqueous systems have been scarcely evidenced so far. The formation of these two possible motifs, H-bond and hemibond, should compete with each other, and the preferential motif is expected to depend on X.
The X = H2O case, i.e., the (H2O)n+ cluster, has been actively studied as the simplest model for ionization of water.21–23,50–89 Some early theoretical calculations showed that the hemibond formation is preferred. However, many recent high-level theoretical computations agree well that the hemibonded form is less stable than the H-bonded form. In fact, only the H-bonded form has been observed by IR spectroscopic experiments of (H2O)n+.21–23 Therefore, it can be concluded that the H-bond formation is favored for X = H2O.
As for other molecules X, structural studies have been performed on rare gas atoms and triatomic molecules. For X = He, Ne, and Ar, it was concluded that the H-bond formation is preferred.24–28 Inokuchi et al. also concluded that the H-bond formation is preferred over the hemibond formation for X = CO2.29 Thus, H2O+ seems to favor H-bond formation because of its high acidity. Recently, however, it was found that the hemibond formation is favored over the H-bond formation in the cases of X = N2O and Kr.30,31 Here, a question arises; what factors govern the competition between H-bond and hemibond formation.
Plausible factors that determine the competition between H-bond and hemibond formation are the proton affinity (PA) and the ionization potential (IP) of X. When the PA of X is high, the H-bond strength would increase, resulting in the preference for H-bond formation, while when the IP of X is close to H2O, the strong charge resonance interaction would lead to hemibond formation.34–36 In fact, the IPs of N2O and Kr are close to H2O, which would be a favorable condition for the hemibond formation. Then, we have an alternative question; what are the ranges of IP and PA of X for the preferential hemibond formation in [H2O–X]+. This range may provide a guide to judge the realization of hemibond formation of H2O+ in the condensed phase.
The structural motifs of [H2O–X]+ have been studied individually for each molecule X so far. However, this competition between H-bond and hemibond formation has not been fully systematically examined and is still poorly understood. In this study, we perform IR predissociation spectroscopy and quantum chemical calculations of [H2O–Xn]+ (n = 1–3, X = N2, CO2, CO, and N2O) to determine their structural motif. Here, as a counterpart molecule X, we choose molecules that have IPs relatively close to H2O and are expected to have high hemibond strength comparable to H-bond strength. We should also note that the acidy of water is highly enhanced with ionization, and its H-bond is expected to be much stronger than typical H-bonds among neutral molecules, even though these X molecules are ordinarily categorized into very weak proton acceptors. Based on the results obtained in this study and those of the previous studies, the correlation between the competing H-bond/hemibond formation and the PA/IP values is investigated to determine the conditions for the preference of hemibond formation of H2O+.
For IR spectroscopy of cluster ions in the gas phase, we ordinately employ the infrared predissociation (IRPD) spectroscopic technique,90,91 in which predissociation fragments upon vibrational excitation of parent cluster ions are detected as a measure of IR absorption. This is because the concentration of cluster ions is too low to detect their IR absorption directly. Here, we note that for n = 1 of [H2O–Xn]+, an IR spectrum of the most stable isomer may not be obtained by IRPD spectroscopy because its dissociation energy is estimated to be much higher than the IR photon energy. Moreover, considering dissociation energies, it is expected that higher energy (less stable) isomers are rather observed. Therefore, in the present study, the Ar-“tagging” technique91–94 is employed for n = 1 to observe low-energy isomers. In this technique, an Ar atom is attached to the cluster of interest. The Ar atom is easily released from the cluster with the IR vibrational excitation, and this is detected as a measure of the IR absorption in the scheme of the dissociation spectroscopy. We can safely suppose that the attachment of Ar is inert enough not to perturb the structural motif of the “bare” cluster (the smaller interaction energy of Ar than the X molecules will be shown later). Moreover, the Ar-tagged cluster becomes cold because only clusters of low internal (vibrational) energy can hold the Ar atom until the interaction region of the spectrometer (irradiation of the IR light). The structural motifs for X = CO2 and N2O have already been reported.29,30 However, in these previous studies, there remains some ambiguity in the structure determination of n = 1. For X = CO2, a high-energy isomer could be observed for n = 1, as mentioned above. For N2O, the IR spectrum was not measured for n = 1, and the structural motif was determined based on theoretical computations and the analogy to the n ≥ 2 clusters. The present measurements of the Ar-tagged clusters of n = 1 provide firm and direct experimental evidence for the most stable structural motif of [H2O–X]+.
2. Experimental and computational methods
Details of the experimental apparatus have been described elsewhere.93 In brief, [H2O–Xn]+ (n = 1–3, X = N2, CO2, CO, and N2O) and their Ar-tagged clusters (n = 1 only) were generated by electron ionization in a supersonic expansion. A gaseous mixture of water, X, and Ar or He carrier (∼8 MPa) was expanded through a high-pressure pulsed valve (Even–Lavie valve95) into a vacuum chamber. An electron beam accelerated to 200 V from an electron gun (Omegatron Corporation) was used for electron ionization. The produced ions were introduced into a tandem quadrupole mass spectrometer. The cluster ions of interest were mass-selected at the first stage of the mass spectrometer. The mass resolution was set higher than Δm/z ≤ 1, and the corresponding protonated cluster, H3O+–Xn, was carefully removed. The mass-selected ions were then introduced into the octopole ion guide and were irradiated by IR light. The IR light was the output of an IR optical parametric oscillator/amplifier (LaserVision) pumped by the fundamental output of a Nd:YAG laser (Spectra Physics GCR 230). Fragment ions produced by vibrational predissociation were mass-selected in the second stage of the mass spectrometer and were detected by a channel electron multiplier. An IR spectrum was measured by monitoring the photofragment intensity as a function of the IR frequency. The loss channel of a single X molecule was monitored for the bare clusters, and the loss channel of Ar for the Ar-tagged clusters.Quantum chemical computations of [H2O–Xn]+ were performed using the Gaussian 16 program package.96 Energy-optimized structure search and harmonic vibrational simulations were performed at the MP2/aug-cc-pVTZ level for X = N2, CO2, and CO. Only for X = N2O, the B3LYP/aug-cc-pVTZ level was employed. This is because, for X = N2O, the SCF calculations for the MP2 calculation did not converge. The calculated frequencies were scaled by a factor of 0.955 for the MP2 calculations and 0.958 for the B3LYP calculations. Only for the n = 1 clusters, we also performed second order vibrational perturbation theory (VPT2) calculations for anharmonic analyses of minor combination and overtone bands.
3. Results and discussion
3.1. [H2O–(N2)n]+ radical cation clusters
Fig. 1 shows the observed and simulated IR spectra of [H2O–N2]+–Ar. As expected, the H-bonded and hemibonded isomers were obtained for the calculated stable structures. Two labels H and h represent the H-bond and hemibond in the isomers, respectively. For [H2O–X]+–Ar, the first label corresponds to the bonding type formed by H2O+ and X, and the second label to that of Ar (e.g., in [H2O–N2]+–Ar, isomer h-H has H2O+ forming a hemibond with N2 and a H-bond with Ar).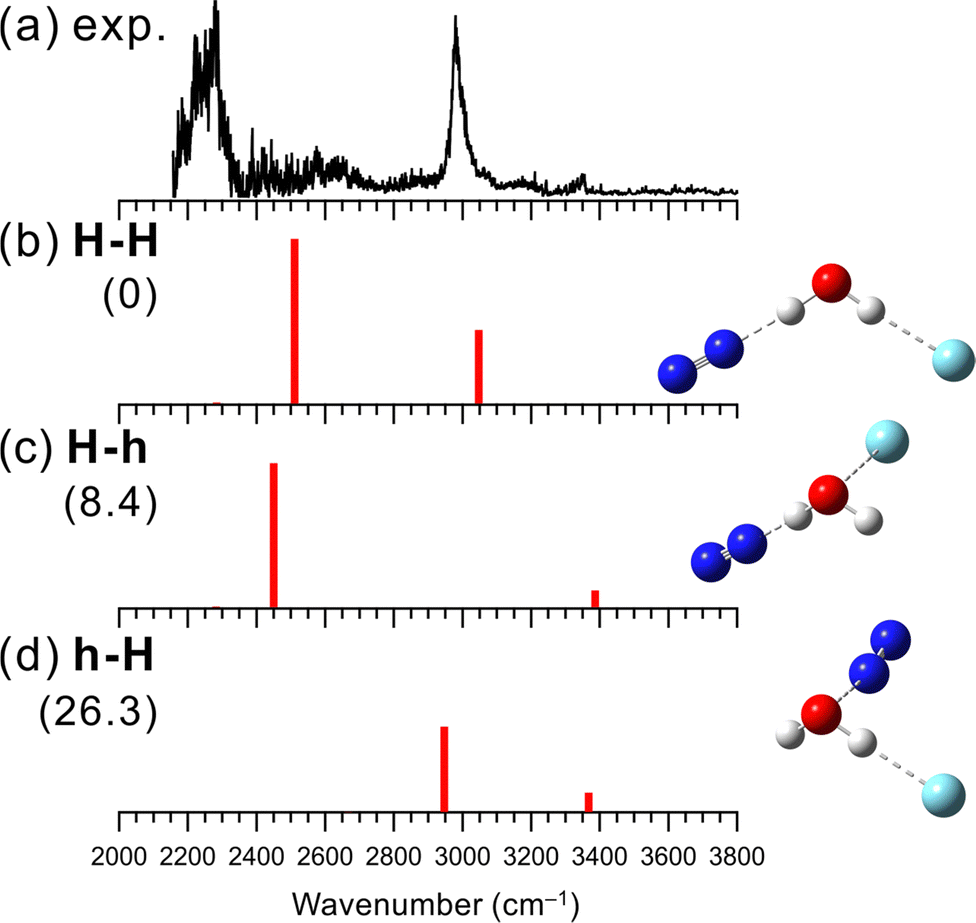 | ||
| Fig. 1 Comparison of (a) observed IRPD spectrum of [H2O–N2]+–Ar and (b–d) simulated spectra of the stable isomers calculated at MP2/aug-cc-pVTZ. The simulated spectra were scaled by a factor of 0.955. The intensities of the simulated spectra of the isomers are plotted in the same scale. The schematic structures of the isomers are also shown. Numbers in parentheses are ZPE-corrected energies (in kJ mol−1). | ||
In the observed spectrum, strong bands were seen at 2990 and around 2250 cm−1, which are attributed to the Ar-bound and N2-bound OH stretching vibrations of H2O+, respectively. This indicates that in [H2O–N2]+–Ar, both the OH groups of the H2O+ moiety are H-bonded to the N2 and Ar, respectively. Moreover, there are no remarkable bands in the 3300–3500 cm−1 region, where free OH stretching bands of H2O+ are expected. These spectral features mean that an isomer without free OH should be the major carrier of the spectrum. Therefore, it is concluded that H-bond formation is preferred over hemibond formation for X = N2. Some minor bands are observed at ∼2600, 3160 and 3350 cm−1. The former two bands could be a combination bands of the N2 (or Ar)-bound OH stretching and the intermolecular stretching vibration between H2O+ and H-bonded N2 (or Ar), respectively. The latter could be attributed to contributions from other isomers (see below).
Three stable isomers are found in the theoretical calculations of [H2O–N2]+–Ar at the MP2/aug-cc-pVTZ level. Their schematic structures, relative energies at the zero-point energy (ZPE) level, and harmonic vibrational spectra are shown in Fig. 1(b)–(d). The most stable structure of [H2O–N2]+–Ar is isomer H-H, in which both N2 and Ar are H-bonded to the OH groups of H2O+. Isomer H-h has a structure in which a water molecule is H-bonded to N2, and the oxygen atom of water and Ar form a hemibond, while h-H is the reversed one: N2 forms a hemibond, and Ar forms a H-bond. These computational results are consistent with the experimental result. Isomer H-H, in which both OH groups are H-bonded, is calculated to be the most stable, and its simulated spectrum well reproduces the major features of the observed spectrum. In addition, as shown in Fig. S1 in the ESI,† the VPT2 anharmonic vibrational calculations for isomer H-H reproduce the two minor bands, the combination bands at ∼2600 and 3160 cm−1. Therefore, isomer H-H is assigned to the major carrier of the observed spectrum. This means that N2 favors the H-bond formation with H2O+ over the hemibond formation. This is also consistent with the MP2/aug-cc-pVTZ level computations of the “bare” [H2O–N2]+ cluster shown Fig. S2 in ESI;† the H-bonded (H) isomer is calculated to be more stable (−32.1 kJ mol−1) than the hemibonded (h) isomer.
Here, we discuss the contributions of the other isomers in the observed IR spectrum. The weak band observed at 3350 cm−1 can be attributed to the free OH stretching vibrational band of higher energy isomers H-h or h-H, of which the population would be much less than isomer H-H. It is difficult at the present stage to clearly identify the spectral carrier of this band. The calculated relative energies show that isomer H-h, where Ar forms a hemibond, is favored. However, the contribution of isomer h-H cannot be ruled out since we have obtained an IR spectrum attributed to the N2-hemibonded (h) form in the measurement of the “bare” [H2O–N2]+ cluster, in which higher energy isomers tend to appear because predissociation following IR excitation can occur only with the assistance of internal energy (see Fig. S2 in ESI† for details).
Similarly, IRPD spectroscopy and quantum chemical calculations were performed for n = 2 and 3 of [H2O–(N2)n]+. The results are summarized in Fig. S3 and S4 in ESI,† respectively. For n = 2, a strong band attributed to the N2-bound OH stretching vibrations appears, while no obvious bands corresponding to free OH stretching vibrations are seen, as in the case of [H2O–N2]+–Ar. In addition, isomer H-H is the most stable one in the theoretical calculations. Therefore, both the N2 molecules are preferentially bonded to the OH groups of H2O+ for n = 2. This also indicates again that the H-bond formation is favored for X = N2 over the hemibond formation. For n = 3, an IR spectrum similar to that of n = 2 was obtained. Based on the experimental and calculational results, the spectrum was attributed to the structure in which two N2 molecules are H-bonded to the OH groups and the third one is hemibonded to the O atom of H2O+.
3.2. [H2O–(CO2)n]+ radical cation clusters
The IR photodissociation spectra of bare [H2O–(CO2)n]+ (n = 1–7) have been reported by Inokuchi et al., and they have concluded that the H-bond formation is favored over the hemibond formation.29 In n = 1, however, because of its large dissociation energy, the dissociation efficiency decreases as the excitation energy decreases. As a result, a remarkable disagreement of the vibrational band intensities between the observed and calculated spectra was seen, which leaves the ambiguity of the spectral and structural assignment. In the present study, we measured an IR spectrum of n = 1 using Ar-tagging for clearer assignments.Fig. 2 shows the IR spectrum obtained in the present experiment of [H2O–CO2]+–Ar and the calculated vibrational spectra of the stable isomers. In the observed spectrum, a strong band is seen at 3070 cm−1, which is attributed to the Ar-bound OH stretching vibration. The bands at 2390, 3560, and 3740 cm−1 are attributed to the antisymmetric CO stretching vibration (v3) and the v1 + v3 and 2v2 + v3 combination bands of CO2, respectively, as proposed in the previous studies.29,97 Here, v1 and v2 represent the symmetric stretching and bending vibrations of CO2, respectively. The other weak peaks around 3230 and 3370 cm−1 are attributed to a combination band between Ar-bound OH stretching and intermolecular stretching vibrations and to contributions of higher energy isomers, respectively, as in the case of [H2O–N2]+–Ar shown in the previous subsection. It is noted that no remarkable free OH band is seen in the observed spectrum of [H2O–CO2]+–Ar. Therefore, we also conclude that the H-bond formation is preferred over the hemibond formation for X = CO2, as Inokuchi et al. have shown. The band of the CO2-bound OH stretching vibration is not clearly seen in the observed spectrum; however, the rising of the signal below 2300 cm−1 would correspond to the tail of the CO2-bound OH stretching band. CO2 has a larger proton affinity than N2 (541 and 494 kJ mol−1, respectively98). Therefore, the CO2-bound OH stretching vibration of [H2O–CO2]+–Ar is expected to show a lower frequency than that of the N2-bound OH stretching vibration (∼2250 cm−1) of [H2O–N2]+–Ar, and the band would be shifted to out of the measured frequency range. Inokuchi et al. have attributed a weak and broad band appearing at ∼2600 cm−1 to CO2-bound OH, which was not obtained in the present measurement. Therefore, the present Ar-tagged measurement provides the firm structural and spectral assignments of [H2O–CO2]+.
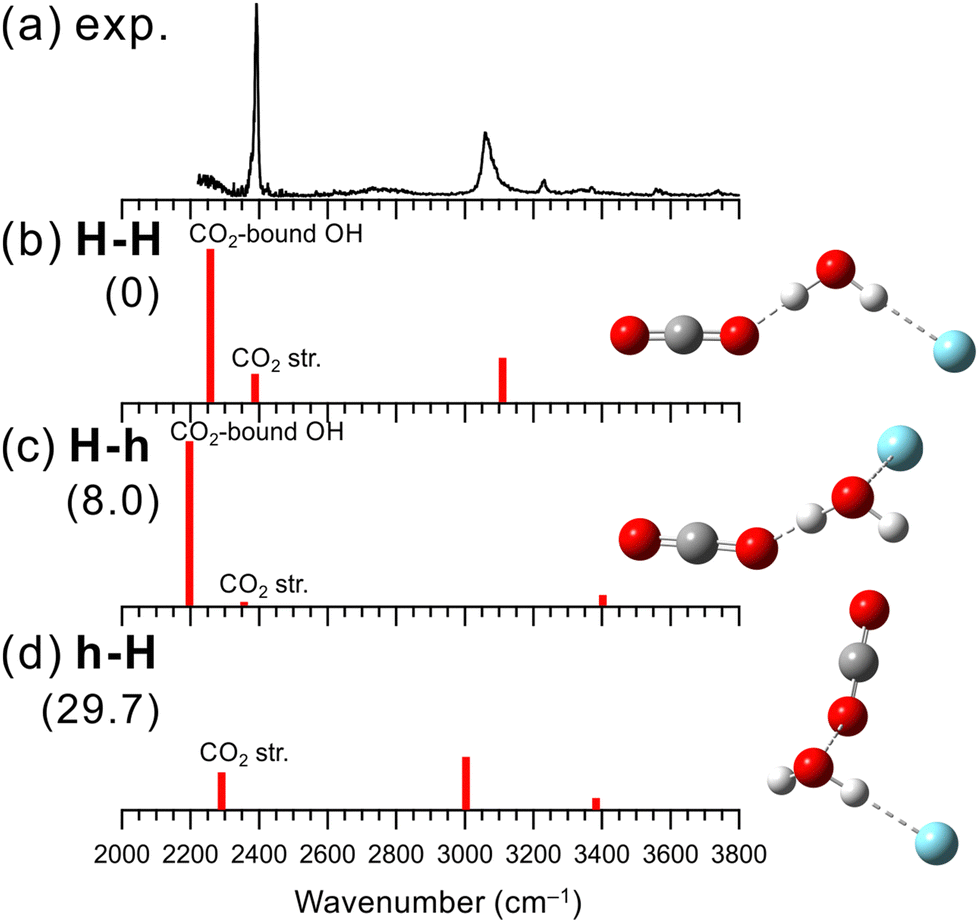 | ||
| Fig. 2 Comparison of (a) observed IRPD spectrum of [H2O–CO2]+–Ar and (b–d) simulated spectra of the stable isomers calculated at MP2/aug-cc-pVTZ. The simulated spectra were scaled by a factor of 0.955. The intensities of the simulated spectra of the isomers are plotted in the same scale. The schematic structures of the isomers are also shown. Numbers in parentheses are ZPE-corrected energies (in kJ mol−1). | ||
The structures, energies, and simulated spectra of three isomers of [H2O–CO2]+–Ar obtained by the MP2 calculations are shown in Fig. 2(b)–(d). The obtained stable structures have the same structural motifs as for X = N2. The most stable structure is isomer H-H. For CO2, forming a H-bond with H2O+ is 29.7 kJ mol−1 more stable than forming a hemibond (isomer h-H). Considering the missing of the largely red-shifted CO2-bound OH stretching vibration, all the major features of the observed spectrum are consistent well with the calculated spectrum of isomer H-H. The anharmonic calculation results shown in Fig. S5 (ESI†) also demonstrate that the most stable structure, isomer H-H, well reproduces the features of the observed spectrum.
Finally, we note that the weak band near 3370 cm−1 in the observed IR spectrum of [H2O–CO2]+–Ar can be attributed to the minor population of the Ar-hemibonded isomer, H-h, or the CO2-hemibonded isomer, h-H. Isomer H-h is energetically more stable than h-H, while the spectrum of bare [H2O–CO2]+ measured in the present study suggested the hemibonded (h) isomer formation with CO2 (see Fig. S6 in ESI† for details), as in the case of [H2O–N2]+. Therefore, it is difficult to identify which isomer is the spectral carrier of this minor band.
We also re-measured the IR spectra of n = 2 and 3 and performed their theoretical computations. The results are summarized in Fig. S7 and S8 in ESI,† and they provide the same conclusion (the superiority of the H-bond formation over the hemibond formation between H2O+ and CO2) as reported in the previous study by Inokuchi et al.29
3.3. [H2O–(CO)n]+ radical cation clusters
Fig. 3 shows the observed IR spectrum of [H2O–CO]+–Ar and the simulated vibrational spectra of the stable isomers. The peaks at 3180 and 3300 cm−1 in the observed spectrum can be attributed to the Ar-bound OH stretching vibration and its combination band with the intermolecular stretching vibration, respectively. In addition, the shoulder around 3070 cm−1 could be a bending overtone of H2O+. Unlike the cases of X = N2 and CO2 mentioned above, a strong band is seen at 3470 cm−1, which should be attributed to a free OH stretching vibration. The presence of the free OH stretching vibrational band indicates a preference for a different structural motif from the H-bonded form for X = CO.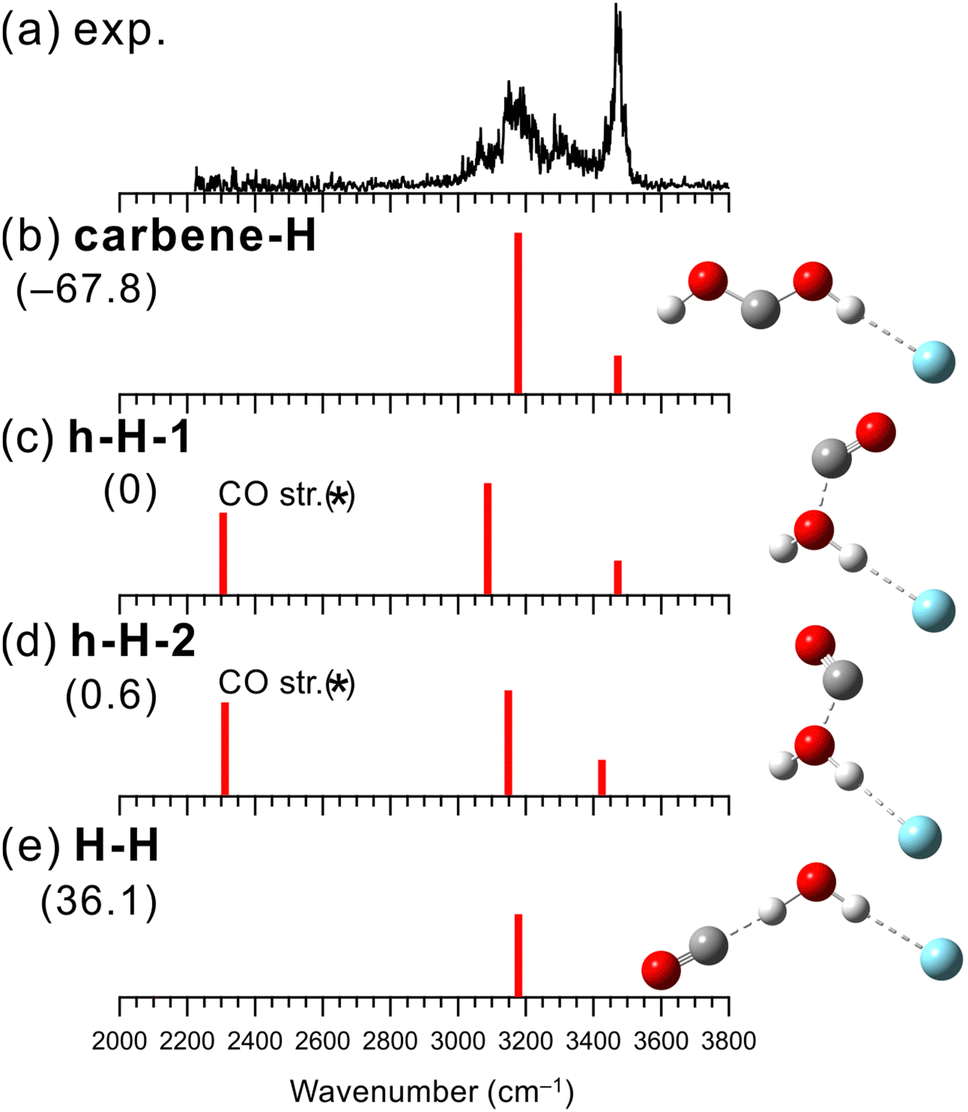 | ||
| Fig. 3 Comparison of (a) observed IRPD spectrum of [H2O–CO]+–Ar and (b–e) simulated spectra of the stable isomers calculated at MP2/aug-cc-pVTZ. The simulated spectra were scaled by a factor of 0.955. The intensities of the simulated spectra of the isomers are plotted in the same scale. The schematic structures of the isomers are also shown. Numbers in parentheses are ZPE-corrected energies (in kJ mol−1). In calculated spectra (c) and (d), the frequency of the CO stretching vibration (asterisked band) could be overestimated with the MP2 calculation (see text for details.). | ||
The structures, energies, and spectra of the four stable isomers of [H2O–CO]+–Ar obtained by the MP2 calculations are shown in Fig. 3(b)–(e). The most stable structural motif is the carbene-type isomer (carbene-H), which is formed by the rearrangement of the covalent bonds between the two molecules. The hemibonded isomers (h-H-1 and h-H-2) are more stable than the H-bonded isomer (H-H) by 36 kJ mol−1 while 68 kJ mol−1 more unstable than the carbene-type isomer. It would be straightforward to assume that the carbene-type isomer is dominantly generated. However, the previous computational studies showed that the isomerization barrier between the hemibonded and carbene-type isomers is calculated to be quite high (∼170 kJ mol−1),99,100 and the isomerization of all the produced clusters seems unlikely to occur.
When we compare the observed and simulated spectra in Fig. 3, we note that in the calculated spectra of h-H-1 and h-H-2, a band corresponding to the CO stretching vibration is predicted at ∼2300 cm−1, though the experimental spectrum shows no band in this region. However, the stretching vibration of bare neutral CO is 2147 cm−1,101 suggesting that the MP2 calculation overestimates the CO stretching frequency. Indeed, the harmonic vibrational analysis at the MP2/aug-cc-pVTZ level of CO and CO+ calculated their frequencies to be 2015 and 2745 cm−1, respectively, while the experimental values were 2147 and 2188 cm−1, respectively.101 For CO+, the extremely large overestimation occurs. The CO site is partially charged in the hemibonded isomer, and this would lead to an overestimation of the frequency of the CO site. The CO stretching vibration of the hemibonded isomers is expected out of the measured spectral range. Therefore, the main features of the observed spectra are in good agreement with both the calculated spectra of the carbene-type and hemibonded isomers. However, the shoulder band around 3070 cm−1, which is not predicted in both the harmonic vibrational simulations, could be attributed to a bending overtone of H2O+. Since isomer carbene-H has no H2O subunit, this could be a marker band for the presence of the hemibonded isomer. The results of the anharmonic vibrational calculations are shown in Fig. S9 in ESI.† For isomer carbene-H, only the Ar-bound OH stretching band is predicted in the 3000–3200 cm−1 region, while for the hemibonded isomers, a water bending overtone is calculated in this region in addition to the Ar-bound OH stretching band (the Fermi resonance occurs). Therefore, the calculated anharmonic spectra of the hemibonded isomers (especially h-H-2) well reproduce the observed spectrum with its shoulder band around 3070 cm−1. To summarize for [H2O–CO]+–Ar, the contribution of the most stable structure, the carbene-type isomer, to the observed spectrum is highly plausible. However, because the isomerization barrier to the carbene form is quite high, some contribution of the hemibonded isomers could occur, as evidenced by the shoulder band around 3070 cm−1. Therefore, it is concluded that the coexistence of the carbene-type and hemibonded isomers are plausible. Finally, we emphasize that the experimental spectral features clearly exclude the dominance of the H-bonded isomer (h-H). In addition, the H-bonded isomer is calculated to be much less stable than the hemibonded isomers. In other words, both the observed spectral features and the calculated energies indicate that hemibond formation is preferred over H-bond formation for X = CO.
Experimental measurements and theoretical calculations were also performed for n = 2–4. The results are summarized in Fig. S10–S12 in ESI.† For n ≧ 2, it has been calculated that CO acts as a catalyst and significantly reduces the isomerization barrier between the hemibonded and carbene-type isomers (∼0.8 kJ mol−1).100 Therefore, it is likely that the carbene-type isomer is predominantly formed for n ≥ 2 since the carbene form is energetically much more stable than the hemibonded form.
3.4. [H2O–(N2O)n]+ radical cation clusters
Structures of [H2O–(N2O)n]+ have been investigated by collision induced dissociation (CID), photodissociation, and IR spectroscopic experiments.30,102–105 Matsushima et al. have measured the IR predissociation spectra of [H2O–(N2O)n]+ at n = 2–7 and have performed their theoretical computations.30 They have shown that N2O favors the hemibond formation with H2O+ over the H-bond formation. However, the experimental spectrum of n = 1 was not obtained because of its large dissociation energy, and the structure of [H2O–N2O]+ was inferred by the theoretical computations and analogy with the larger clusters (n ≥ 2). Therefore, in the present study, we measured an IR spectrum of [H2O–N2O]+ by using Ar-tagging to determine the binding motif unequivocally.Fig. 4 shows the observed IR spectrum of [H2O–N2O]+–Ar and the simulated vibrational spectra of its stable isomers. The observed spectrum has similar spectral features to those of [H2O–CO]+–Ar. The peaks at 3240, 3390, and 3460 cm−1 in the observed spectrum can be assigned to an Ar-bound OH stretching band, its combination band with intermolecular stretching, and a free OH stretching band, respectively. In addition, the very weak peak at 3090 cm−1 could be a bending overtone of H2O+. The presence of the free OH stretching band indicates that the hemibond formation is preferred over the H-bond formation for X = N2O.
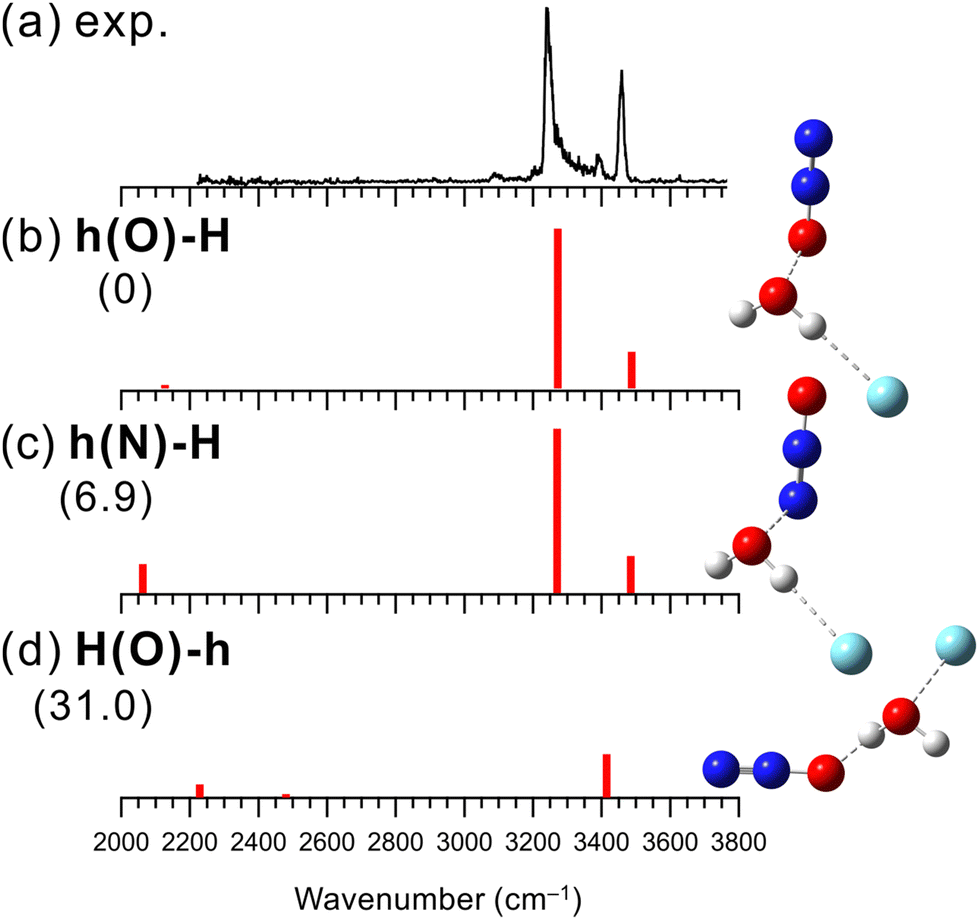 | ||
| Fig. 4 Comparison of (a) observed IRPD spectrum of [H2O–N2O]+–Ar and (b–d) simulated spectra of the stable isomers calculated at B3LYP/aug-cc-pVTZ. The simulated spectra were scaled by a factor of 0.958. The intensities of the simulated spectra of the isomers are plotted in the same scale. The schematic structures of the isomers are also shown. Numbers in parentheses are ZPE-corrected energies (in kJ mol−1). | ||
Fig. 4(b)–(d) show the three lowest energy stable isomer structures, relative energies, and vibrational spectra of [H2O–N2O]+–Ar. Here, the B3LYP/aug-cc-pVTZ level of theory was used for the calculations. This is because the SCF calculations for the MP2 calculations did not converge in this system, and the alternative choice of B3LYP/aug-cc-pVTZ follows the theory employed in the previous study by Matsushima et al.30 The calculated relative energies show that hemibonded isomer h(O)-H is more stable than H-bonded isomer H(O)-h by 31 kJ mol−1, which supports the present experimental result. Also, this preference for the hemibond over the H-bond is consistent with the previous theoretical results.30,106 Since isomers h(O)-H and h(N)-H exhibit the calculated IR spectra similar to each other, it is difficult to distinguish the contribution of each isomer to the observed spectrum. However, h(O)-H is more stable than h(N)-H by 6.9 kJ mol−1 and could be predominantly present. As shown in Fig. S13 in ESI,† the anharmonic vibrational calculations for the hemibonded isomers reproduce the observed spectrum including the minor bands, combination band between the Ar-bound OH stretching and the intermolecular stretching vibration and bending overtone of H2O+. We can conclude that X = N2O favors the hemibond formation in [H2O–N2O]+–Ar. This conclusion is consistent with the previous study of bare [H2O–(N2O)n]+.30,106 However, the present study provided firm experimental evidence of the hemibond formation in [H2O–N2O]+.
We also measured the IR spectra of the bare clusters of n = 1–3 and performed their theoretical computations. The results are summarized in Fig. S14–S16 in ESI.† In the present study, we could observe the spectrum of bare n = 1. However, its fragment signal intensity was very weak, and this suggests that we might observe very hot components, in which internal energy assists photodissociation. For n = 2 and 3, we have essentially the same results and conclusions as those in the previous study by Matsushima et al.30
3.5. Correlation between the competing H-bond/hemibond formation and the PA/IP
The results obtained in this study (Sections 3.1–3.4) and the previous studies21–31 reveal the preferred binding motif of [H2O–X]+ for the various X molecules (X = He, Ne, Ar, Kr, N2, CO2, CO, N2O, and H2O). Fig. 5 shows the PA-IP plot of the various X molecules,98,107 color-coded with the preferred binding motifs (hemibond or H-bond) of [H2O–X]+. In this plot, points of X favoring the H-bond formation are colored in blue, and those favoring the hemibond formation are in red. This plot shows the ranges of PA and IP of X for the preference of the hemibond formation with H2O+. The preference for hemibond formation is limited only to X of which IP is in the narrow range within ∼1.5 eV from that of H2O (12.6 eV). This trend is consistent with the previous theoretical studies34–36 that the hemibond strength decreases exponentially with increases of ΔIP, the difference in the ionization potentials of the two units. When X is H2O ([H2O–H2O]+), ΔIP is 0, and therefore, the strongest hemibond formation is expected. However, it has been known that the H-bond formation is actually favored in [H2O–H2O]+. This means that the PA of water (691 kJ mol−1) is high enough to form the H-bond stronger than the hemibond. Therefore, based on the plot in Fig. 5, we conclude that the hemibond formation in [H2O–X]+ can be superior to the H-bond formation only when the IP of X is within ∼1.5 eV from that of water (12.6 eV) and the PA is lower than that of water (691 kJ mol−1).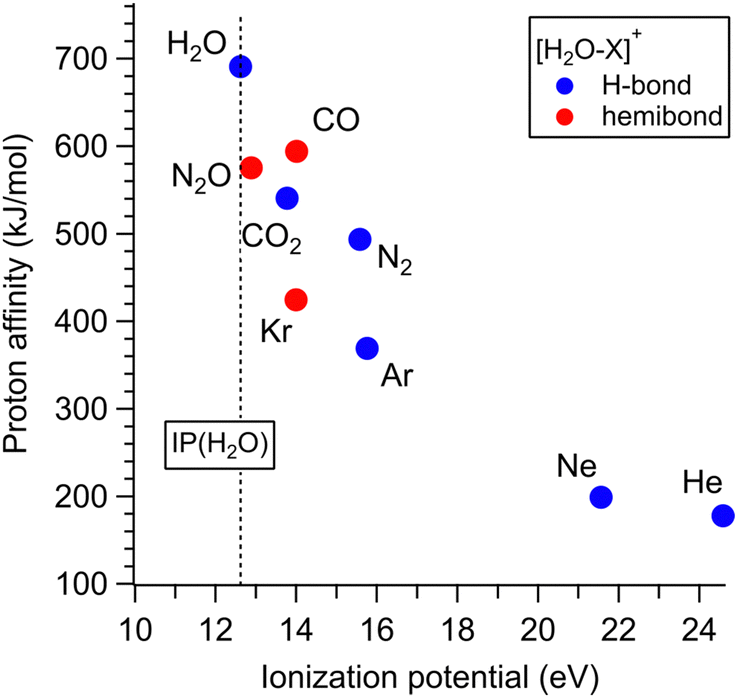 | ||
| Fig. 5 PA-IP plot of the various X molecules color-coded with the preferred binding motifs of [H2O–X]+. Points of X favoring the H-bond formation are colored in blue and those favoring the hemibond formation are in red. This plot is based on the preferential structure determinations in the present and previous studies.21–31 The dotted line shows the IP of H2O. | ||
However, the PA-IP plot does not fully explain the competition between the hemibond and H-bond formation. The point of X = CO2 seems exceptional; though one would expect the preference of the hemibond formation for CO2 because its IP is closer to H2O than CO and its PA is also lower than CO, [H2O–CO2]+ actually favors the H-bond formation. Therefore, further consideration of the competition is required.
To investigate this correlation between the competing bond formations and PA/IP in more detail, the correlation between the H-bond strength and PA of X and that between the hemibond strength and IP was evaluated using binding energy D0, respectively. In the following, all computations were performed at the CCSD/aug-cc-pVTZ level. This is because we frequently had failure of MP2 calculations for the homodimer cations, which are requested in the later discussion on hemibonds. Note that no stable hemibonded structure was obtained for X = Ne and He (only a hemibond-like structure was obtained; however, the charge is almost localized on H2O+, suggesting that its hemibond nature is weak.), and the SCF calculation did not converge for X = N2O. The calculation results are shown in Fig. 6. Fig. 6 (top) shows the correlation between the binding energy of the H-bonded isomer and the PA value of X. The binding energy increases monotonically with increasing PA. This indicates that, as expected, the H-bond strength becomes higher as the PA of X increases. Fig. 6 (bottom) shows the correlation between the binding energy of the hemibonded isomer and the IP value of X. The binding energy tends to increase as the IP approaches that of H2O. However, the point of X = CO2 seems exceptional; the binding energy of X = CO2 is lower than those of X = Kr and CO, even though the IP of CO2 is lower than those of Kr and CO and closer to that of H2O. Therefore, the hemibond strength cannot be simply correlated only to IP (or ΔIP).
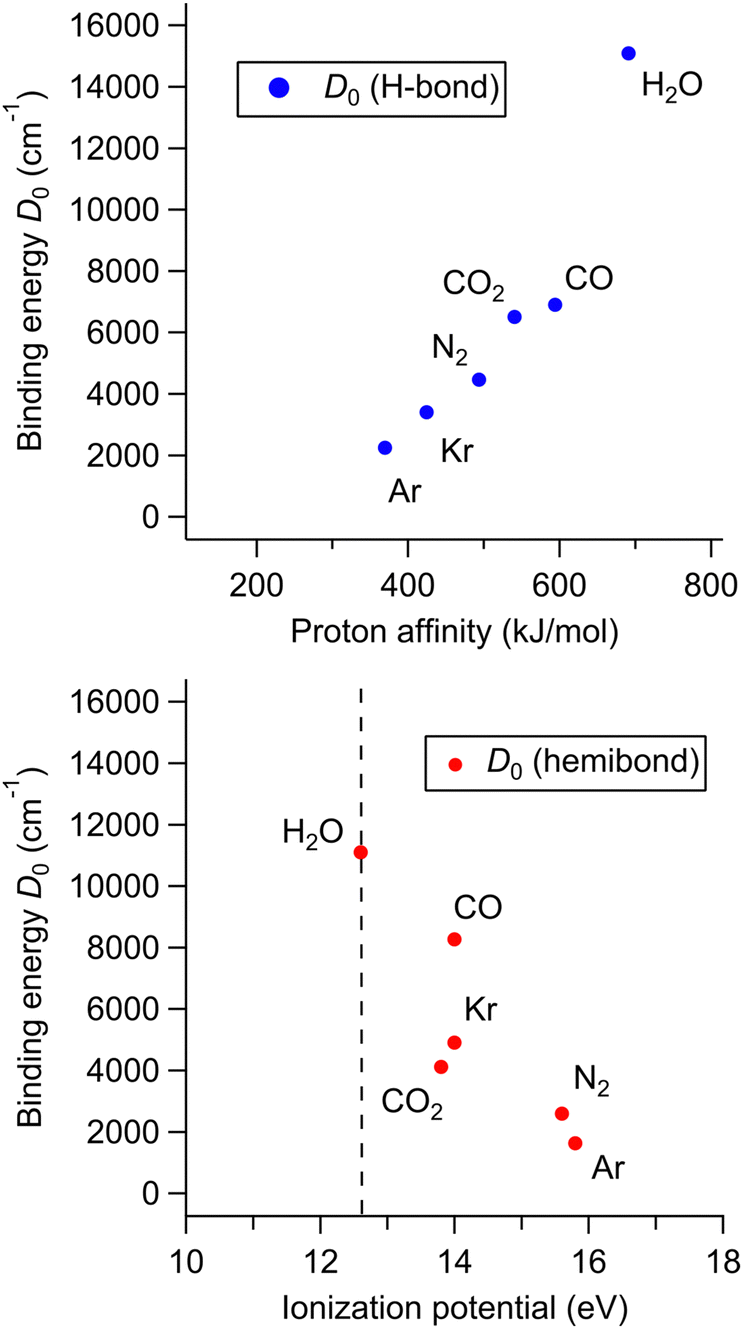 | ||
| Fig. 6 (top) Correlation between D0 of the H-bonded isomers of [H2O–X]+ and the PA of X. (bottom) Correlation between D0 of the hemibonded isomers and the IP of X. In both the plots, the D0 values were calculated at CCSD/aug-cc-pVTZ. | ||
According to the previous theoretical studies,34–36 the binding energy DAB of a hemibond is given by:
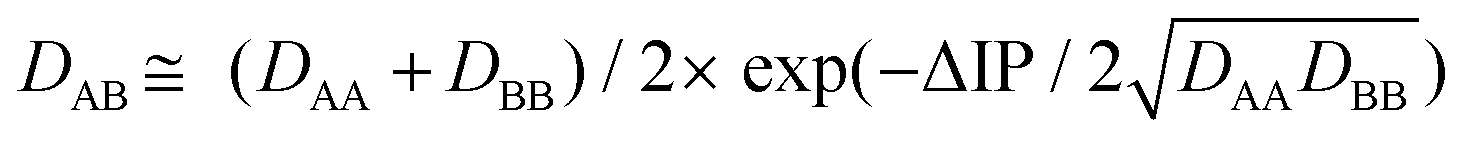 | (1) |
| X | IP (eV) | D AA (cm−1) | D AB (cm−1) | D 0 (cm−1) |
|---|---|---|---|---|
| Ar | 15.8 | 9703 | 3071 | 1626 |
| N2 | 15.6 | 9725 | 3300 | 2589 |
| Kr | 14.0 | 9769 | 6118 | 4904 |
| CO | 14.0 | 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 942 942 |
9308 | 8264 |
| CO2 | 13.8 | 4198 | 3873 | 4116 |
| H2O | 12.6 | 11100 |
11100 |
11100 |
4. Conclusions
Infrared photodissociation spectroscopy and quantum chemical calculations of [H2O–Xn]+ (n = 1–3, X = N2, CO2, CO, and N2O) were performed to systematically investigate the competition between the hydrogen (H-) bond and hemibond formation of H2O+ and its correlation with the proton affinity (PA) and ionization potential (IP) values of counter molecule X in the intermolecular bond formation. We found that the H-bond formation is dominant for X = N2 and CO2, while the hemibond formation is preferred for X = CO and N2O. The correlation of the preferred binding motif (hemibond or H-bond) with the PA/IP values was examined on the basis of the most stable isomer structures of [H2O–X]+ obtained in this study and the previous studies. It was concluded that this competition was basically explained by the magnitudes of the PA and IP, which essentially reflect the magnitudes of the H-bond and hemibond, respectively. We roughly estimated the ranges of the PA and IP of X for the preferential hemibond formation with H2O+; the hemibond formation in [H2O–X]+ can overcome the competitive H-bond formation only when the IP of X is within 1.5 eV from that of water and the PA is lower than that of water. Therefore, when water is ionized in close proximity to molecules whose IP is within 1.5 eV from that of water, not only H-bond but also hemibond formation with H2O+ can be considered. As has been pointed out theoretically, however, the magnitude of the hemibond between H2O+ and X would also depend on some other properties of X, such as orbital overlap. Thus, some exceptional H-bond preference can occur even in the PA/IP ranges for the hemibond preference. The present results are hoped to advance understanding of the detailed processes of ionizing radiation reactions in aqueous solutions.Author contributions
M.K.: investigation, calculations, data analysis, and writing. A.F.: conceptualization, supervision, funding acquisition, and writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study is supported by a Grant-in-Aid for Scientific Research (Project No. 21H04671) from JSPS. M. K. was supported by JST SPRING, Grant Number JPMJSP2114. A part of the computation was performed at the Research Center for Computational Science, Okazaki, Japan.References
- B. C. Garrett, D. A. Dixon, D. M. Camaioni, D. M. Chipman, M. A. Johnson, C. D. Jonah, G. A. Kimmel, J. H. Miller, T. N. Rescigno, P. J. Rossky, S. S. Xantheas, S. D. Colson, A. H. Laufer, D. Ray, P. F. Barbara, D. M. Bartels, K. H. Becker, K. H. Bowen Jr, S. E. Bradforth, I. Carmichael, J. V. Coe, L. R. Corrales, J. P. Cowin, M. Dupuis, K. B. Eisenthal, J. A. Franz, M. S. Gutowski, K. D. Jordan, B. D. Kay, J. A. Laverne, S. V. Lymar, T. E. Madey, C. W. McCurdy, D. Meisel, S. Mukamel, A. R. Nilsson, T. M. Orlando, N. G. Petrik, S. M. Pimblott, J. R. Rustad, G. K. Schenter, S. J. Singer, A. Tokmakoff, L.-S. Wang, C. Wettig and T. S. Zwier, Chem. Rev., 2005, 105, 355–390 CrossRef CAS PubMed.
- J. Ma, F. Wang and M. Mostafavi, Molecules, 2018, 23, 244 CrossRef PubMed.
- J. Ma, S. A. Denisov, A. Adhikary and M. Mostafavi, Int. J. Mol. Sci., 2019, 20, 4963 CrossRef CAS PubMed.
- D. Mi and K. Chingin, Molecules, 2020, 25, 3490 CrossRef CAS PubMed.
- J. Ma, U. Schmidhammer, P. Pernot and M. Mostafavi, J. Phys. Chem. Lett., 2014, 5, 258–261 CrossRef CAS PubMed.
- J. Ma, U. Schmidhammer and M. Mostafavi, J. Phys. Chem. A, 2014, 118, 4030–4037 CrossRef CAS PubMed.
- J. Ma, U. Schmidhammer and M. Mostafavi, J. Phys. Chem. B, 2015, 119, 7180–7185 CrossRef CAS PubMed.
- F. Wang, U. Schmidhammer, A. de La Lande and M. Mostafavi, Phys. Chem. Chem. Phys., 2017, 19, 2894–2899 RSC.
- R. Musat, S. A. Denisov, J.-L. Marignier and M. Mostafavi, J. Phys. Chem. B, 2018, 122, 2121–2129 CrossRef CAS PubMed.
- M. Wang, X.-F. Gao, R. Su, P. He, Y.-Y. Cheng, K. Li, D. Mi, X. Zhang, X. Zhang, H. Chen and G. R. Cooks, CCS Chem., 2022, 4, 1224–1231 CrossRef CAS.
- L. Qiu, M. D. Psimos and R. G. Cooks, J. Am. Soc. Mass Spectrom., 2022, 33, 1362–1367 CrossRef CAS PubMed.
- L. Qiu, N. M. Morato, K.-H. Huang and R. G. Cooks, Front. Chem., 2022, 10, 903774 CrossRef CAS PubMed.
- L. Qiu and R. G. Cooks, Angew. Chem., Int. Ed., 2022, 61, e202210765 CAS.
- Y. Gauduel, S. Pommeret, A. Migus and A. Antonetti, Chem. Phys., 1990, 149, 1–10 CrossRef CAS.
- O. Marsalek, C. G. Elles, P. A. Pieniazek, E. Pluhařová, J. VandeVondele, S. E. Bradforth and P. Jungwirth, J. Chem. Phys., 2011, 135, 224510 CrossRef PubMed.
- S. Thürmer, M. Ončák, N. Ottosson, R. Seidel, U. Hergenhahn, S. E. Bradforth, P. Slavíček and B. Winter, Nat. Chem., 2013, 5, 590–596 CrossRef PubMed.
- J. Li, Z. Nie, Y. Y. Zheng, S. Dong and Z.-H. Loh, J. Phys. Chem. Lett., 2013, 4, 3698–3703 CrossRef CAS.
- V. Svoboda, R. Michiels, A. C. LaForge, J. Med, F. Stienkemeier, P. Slavíček and H. J. Wörner, Sci. Adv., 2020, 6, eaaz0385 CrossRef CAS PubMed.
- Z.-H. Loh, G. Doumy, C. Arnold, L. Kjellsson, S. H. Southworth, A. Al Haddad, Y. Kumagai, M.-F. Tu, P. J. Ho, A. M. March, R. D. Schaller, M. S. Bin Mohd Yusof, T. Debnath, M. Simon, R. Welsch, L. Inhester, K. Khalili, K. Nanda, A. I. Krylov, S. Moeller, G. Coslovich, J. Koralek, M. P. Minitti, W. F. Schlotter, J.-E. Rubensson, R. Santra and L. Young, Science, 2020, 367, 179–182 CrossRef CAS PubMed.
- M.-F. Lin, N. Singh, S. Liang, M. Mo, J. P. F. Nunes, K. Ledbetter, J. Yang, M. Kozina, S. Weathersby, X. Shen, A. A. Cordones, T. J. A. Wolf, C. D. Pemmaraju, M. Ihme and X. J. Wang, Science, 2021, 374, 92–95 CrossRef CAS PubMed.
- G. H. Gardenier, M. A. Johnson and A. B. McCoy, J. Phys. Chem. A, 2009, 113, 4772–4779 CrossRef CAS PubMed.
- K. Mizuse, J.-L. Kuo and A. Fujii, Chem. Sci., 2011, 2, 868–876 RSC.
- K. Mizuse and A. Fujii, J. Phys. Chem. A, 2013, 117, 929–938 CrossRef CAS PubMed.
- D. Roth, O. Dopfer and J. P. Maier, Phys. Chem. Chem. Phys., 2001, 3, 2400–2410 RSC.
- O. Dopfer, D. Roth and J. P. Maier, J. Chem. Phys., 2001, 114, 7081–7093 CrossRef CAS.
- O. Dopfer, J. Phys. Chem. A, 2000, 104, 11693–11701 CrossRef CAS.
- O. Dopfer, D. Roth and J. P. Maier, J. Phys. Chem. A, 2000, 104, 11702–11713 CrossRef CAS.
- J. P. Wagner, D. C. McDonald II and M. A. Duncan, J. Chem. Phys., 2017, 147, 104302 CrossRef PubMed.
- Y. Inokuchi, Y. Kobayashi, A. Muraoka, T. Nagata and T. Ebata, J. Chem. Phys., 2009, 130, 154304 CrossRef PubMed.
- R. Matsushima, T. Ebata and Y. Inokuchi, J. Phys. Chem. A, 2010, 114, 11037–11042 CrossRef CAS PubMed.
- J.-M. Liu, T. Nishigori, T. Maeyama, Q.-R. Huang, M. Katada, J.-L. Kuo and A. Fujii, J. Phys. Chem. Lett., 2021, 12, 7997–8002 CrossRef CAS PubMed.
- K. D. Asmus, Acc. Chem. Res., 1979, 12, 436–442 CrossRef CAS.
- M. Goebl, M. Bonifacic and K. D. Asmus, J. Am. Chem. Soc., 1984, 106, 5984–5988 CrossRef CAS.
- T. Clark, J. Am. Chem. Soc., 1988, 110, 1672–1678 CrossRef CAS.
- P. C. Hiberty, S. Humbel and P. Archirel, J. Phys. Chem., 1994, 98, 11697–11704 CrossRef CAS.
- T. Clark, ChemPhysChem, 2017, 18, 2766–2771 CrossRef CAS PubMed.
- C. Schöneich, D. Pogocki, G. L. Hug and K. Bobrowski, J. Am. Chem. Soc., 2003, 125, 13700–13713 CrossRef PubMed.
- A. Adhikary, A. Kumar, B. J. Palmer, A. D. Todd and M. D. Sevilla, J. Am. Chem. Soc., 2013, 135, 12827–12838 CrossRef CAS PubMed.
- C. H. Hendon, D. R. Carbery and A. Walsh, Chem. Sci., 2014, 5, 1390–1395 RSC.
- J. Jie, Y. Xia, C.-H. Huang, H. Zhao, C. Yang, K. Liu, D. Song, B.-Z. Zhu and H. Su, Nucleic Acids Res., 2019, 47, 11514–11526 CAS.
- D. Wang and A. Fujii, Chem. Sci., 2017, 8, 2667–2670 RSC.
- M. Xie, Z. Shen, D. Wang, A. Fujii and Y.-P. Lee, J. Phys. Chem. Lett., 2018, 9, 3725–3730 CrossRef CAS PubMed.
- D. Wang, K. Hattori and A. Fujii, Chem. Sci., 2019, 10, 7260–7268 RSC.
- K. Hattori, D. Wang and A. Fujii, Phys. Chem. Chem. Phys., 2019, 21, 16064–16074 RSC.
- M. Xie, H.-R. Tsai, A. Fujii and Y.-P. Lee, Phys. Chem. Chem. Phys., 2019, 21, 16055–16063 RSC.
- X. Sun, M. Xie, W. Qiu, C. Wei, X. Chen and Y. Hu, Phys. Chem. Chem. Phys., 2022, 24, 19354–19361 RSC.
- T. Kato and A. Fujii, J. Phys. Chem. A, 2023, 127, 742–750 CrossRef CAS PubMed.
- B. Rana and J. M. Herbert, Phys. Chem. Chem. Phys., 2020, 22, 27829–27844 RSC.
- B. Rana and J. M. Herbert, J. Phys. Chem. Lett., 2021, 12, 8053–8060 CrossRef CAS PubMed.
- H. Shinohara, N. Nishi and N. Washida, J. Chem. Phys., 1986, 84, 5561–5567 CrossRef CAS.
- P. M. W. Gill and L. Radom, J. Am. Chem. Soc., 1988, 110, 4931–4941 CrossRef CAS.
- R. T. Jongma, Y. Huang, S. Shi and A. M. Wodtke, J. Phys. Chem. A, 1998, 102, 8847–8854 CrossRef CAS.
- M. Sodupe, J. Bertran, L. Rodríguez-Santiago and E. J. Baerends, J. Phys. Chem. A, 1999, 103, 166–170 CrossRef CAS.
- L. Angel and A. J. Stace, Chem. Phys. Lett., 2001, 345, 277–281 CrossRef CAS.
- H. Tachikawa, J. Phys. Chem. A, 2002, 106, 6915–6921 CrossRef CAS.
- S. Yamaguchi, S. Kudoh, Y. Kawai, Y. Okada, T. Orii and K. Takeuchi, Chem. Phys. Lett., 2003, 377, 37–42 CrossRef CAS.
- H. Tachikawa, J. Phys. Chem. A, 2004, 108, 7853–7862 CrossRef CAS.
- Y. V. Novakovskaya, Russ. J. Phys. Chem. A, 2007, 81, 216–224 CrossRef CAS.
- Y. V. Novakovskaya, Russ. J. Phys. Chem. A, 2007, 81, 225–234 CrossRef CAS.
- A. Kumar, M. Kołaski, H. M. Lee and K. S. Kim, J. Phys. Chem. A, 2008, 112, 5502–5508 CrossRef CAS PubMed.
- P. A. Pieniazek, J. VandeVondele, P. Jungwirth, A. I. Krylov and S. E. Bradforth, J. Phys. Chem. A, 2008, 112, 6159–6170 CrossRef CAS PubMed.
- Q. Cheng, F. A. Evangelista, A. C. Simmonett, Y. Yamaguchi and H. F. Schaefer III, J. Phys. Chem. A, 2009, 113, 13779–13789 CrossRef CAS PubMed.
- E. Kamarchik, O. Kostko, J. M. Bowman, M. Ahmed and A. I. Krylov, J. Chem. Phys., 2010, 132, 194311 CrossRef PubMed.
- E. Livshits, R. S. Granot and R. Baer, J. Phys. Chem. A, 2011, 115, 5735–5744 CrossRef CAS PubMed.
- P.-R. Pan, Y.-S. Lin, M.-K. Tsai, J.-L. Kuo and J.-D. Chai, Phys. Chem. Chem. Phys., 2012, 14, 10705–10712 RSC.
- M.-K. Tsai, J.-L. Kuo and J.-M. Lu, Phys. Chem. Chem. Phys., 2012, 14, 13402–13408 RSC.
- H. Tachikawa and T. Takada, Chem. Phys., 2013, 415, 76–83 CrossRef CAS.
- H. Do and N. A. Besley, J. Phys. Chem. A, 2013, 117, 5385–5391 CrossRef CAS PubMed.
- O. Svoboda, D. Hollas, M. Ončák and P. Slavíček, Phys. Chem. Chem. Phys., 2013, 15, 11531–11542 RSC.
- H. Do and N. A. Besley, Phys. Chem. Chem. Phys., 2013, 15, 16214–16219 RSC.
- Z.-L. Lv, K. Xu, Y. Cheng, X.-R. Chen and L.-C. Cai, J. Chem. Phys., 2014, 141, 054309 CrossRef PubMed.
- T. Stein, C. A. Jiménez-Hoyos and G. E. Scuseria, J. Phys. Chem. A, 2014, 118, 7261–7266 CrossRef CAS PubMed.
- E.-P. Lu, P.-R. Pan, Y.-C. Li, M.-K. Tsai and J.-L. Kuo, Phys. Chem. Chem. Phys., 2014, 16, 18888–18895 RSC.
- H. Tachikawa and T. Takada, RSC Adv., 2014, 5, 6945–6953 RSC.
- J. D. Herr, J. Talbot and R. P. Steele, J. Phys. Chem. A, 2015, 119, 752–766 CrossRef CAS PubMed.
- Z.-L. Lv, Y. Cheng, X.-R. Chen and L.-C. Cai, Chem. Phys., 2015, 452, 25–30 CrossRef CAS.
- P.-R. Pan, E.-P. Lu, J.-L. Kuo and M.-K. Tsai, J. Chin. Chem. Soc., 2016, 63, 488–498 CrossRef CAS.
- H. Tachikawa and T. Takada, Comput. Theor. Chem., 2016, 1089, 13–20 CrossRef CAS.
- J. J. Talbot, X. Cheng, J. D. Herr and R. P. Steele, J. Am. Chem. Soc., 2016, 138, 11936–11945 CrossRef CAS PubMed.
- L. Liu, C.-E. Hu, M. Tang, X.-R. Chen and L.-C. Cai, J. Chem. Phys., 2016, 145, 154307 CrossRef PubMed.
- M. Tang, C.-E. Hu, Z.-L. Lv, X.-R. Chen and L.-C. Cai, J. Phys. Chem. A, 2016, 120, 9489–9499 CrossRef CAS PubMed.
- D. M. Chipman, J. Phys. Chem. A, 2016, 120, 9618–9624 CrossRef CAS PubMed.
- G.-J. Li, C.-E. Hu, M. Tang, X.-R. Chen and L.-C. Cai, Comput. Theor. Chem., 2017, 1099, 123–132 CrossRef CAS.
- L.-T. Shi, M. Tang, X.-R. Chen, C.-E. Hu and Y. Cheng, Comput. Theor. Chem., 2017, 1120, 102–111 CrossRef CAS.
- J. Chalabala, F. Uhlig and P. Slavíček, J. Phys. Chem. A, 2018, 122, 3227–3237 CrossRef CAS PubMed.
- W.-Q. Chen, M. Fu, H.-Y. Wang, Z.-Y. Zeng and B.-R. Yu, Struct. Chem., 2018, 29, 1273–1285 CrossRef CAS.
- W. Zhao-Qi, W. Hai-Yan, Z.-Y. Zeng and C. Yan, Struct. Chem., 2019, 30, 151–165 CrossRef.
- Y.-M. Wen, S.-K. Zhang, C.-E. Hu and Y. Cheng, Theor. Chem. Acc., 2019, 138, 83 Search PubMed.
- A. Ünal and U. Bozkaya, Int. J. Quantum Chem., 2020, 120, e26100 CrossRef.
- L. I. Yeh, M. Okumura, J. D. Myers, J. M. Price and Y. T. Lee, J. Chem. Phys., 1989, 91, 7319–7330 CrossRef CAS.
- D. J. Goebbert, T. Wende, R. Bergmann, G. Meijer and K. R. Asmis, J. Phys. Chem. A, 2009, 113, 5874–5880 CrossRef CAS PubMed.
- M. Okumura, L. I. Yeh, J. D. Myers and Y. T. Lee, J. Chem. Phys., 1986, 85, 2328–2329 CrossRef CAS.
- K. Mizuse and A. Fujii, Phys. Chem. Chem. Phys., 2011, 13, 7129–7135 RSC.
- T. Shimamori, J.-L. Kuo and A. Fujii, J. Phys. Chem. A, 2016, 120, 9203–9208 CrossRef CAS PubMed.
- U. Even, J. Jortner, D. Noy, N. Lavie and C. Cossart-Magos, J. Chem. Phys., 2000, 112, 8068–8071 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Rev. C.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- Y. Inokuchi, A. Muraoka, T. Nagata and T. Ebata, J. Chem. Phys., 2008, 129, 044308 CrossRef PubMed.
- E. P. L. Hunter and S. G. Lias, J. Phys. Chem. Ref. Data, 1998, 27, 413–656 CrossRef CAS.
- E. Uggerud, W. Koch and H. Schwarz, Int. J. Mass Spectrom. Ion Processes, 1986, 73, 187–196 CrossRef CAS.
- C. Y. Wong, P. J. A. Ruttink, P. C. Burgers and J. K. Terlouw, Chem. Phys. Lett., 2004, 390, 176–180 CrossRef CAS.
- K. P. Huber and G. Herzberg, Molecular Spectra and Molecular Structure IV. Constants of Diatomic Molecules, Van Nostrand Reinhold Co, New York, 1979 Search PubMed.
- S. T. Graul, H.-S. Kim and M. T. Bowers, Int. J. Mass Spectrom. Ion Processes, 1992, 117, 507–536 CrossRef CAS.
- M. J. Bastian, R. A. Dressler, D. J. Levandier, E. Murad, F. Muntean and P. B. Armentrout, J. Chem. Phys., 1997, 106, 9570–9579 CrossRef CAS.
- S. Williams, Y.-H. Chiu, D. J. Levandier and R. A. Dressler, J. Chem. Phys., 1998, 109, 7450–7461 CrossRef CAS.
- S. Williams, Y.-H. Chiu, D. J. Levandier and R. A. Dressler, J. Chem. Phys., 1998, 108, 9383–9389 CrossRef CAS.
- J. E. Stevens, M. C. Holthausen and K. Morokuma, J. Chem. Phys., 1999, 111, 7766–7773 CrossRef CAS.
- In CRC handbook of chemistry and physics, ed. D. R. Lide, CRC Press, Boca Raton, FL, 90th edn, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Comparison of the observed and simulated spectra of [H2O–Xn]+ (n =1–3, X = N2, CO2, CO, and N2O). See DOI: https://doi.org/10.1039/d3cp00753g |
| This journal is © the Owner Societies 2023 |