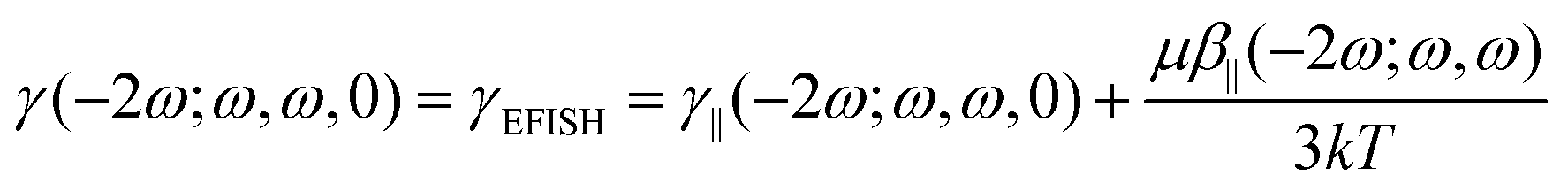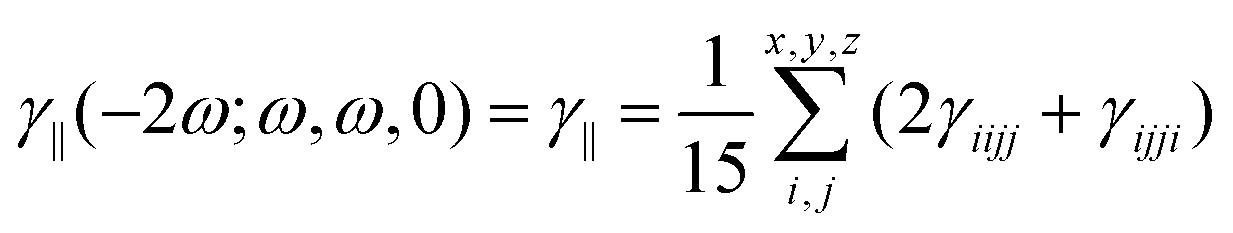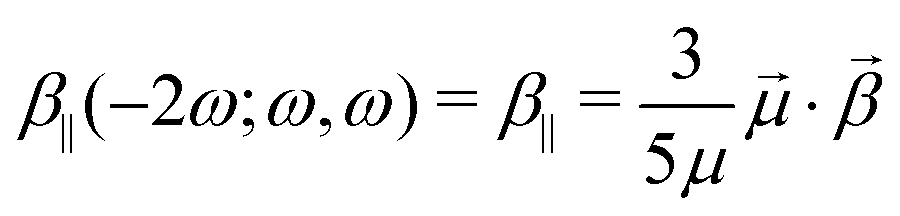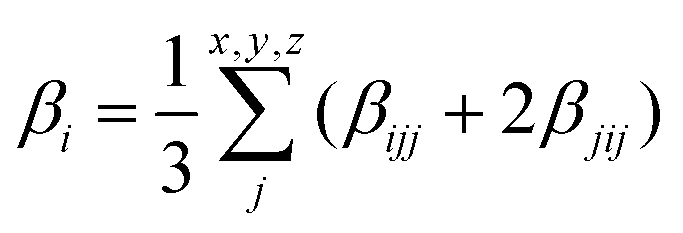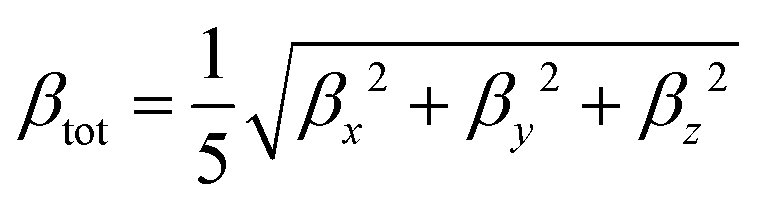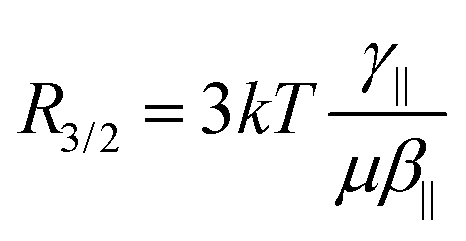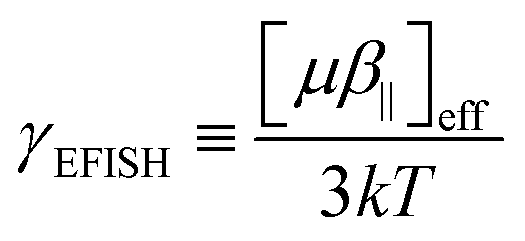Electric-field induced second harmonic generation responses of push–pull polyenic dyes: experimental and theoretical characterizations†
Received
16th February 2023
, Accepted 7th May 2023
First published on 8th May 2023
Abstract
The second-order nonlinear optical properties of four series of amphiphilic cationic chromophores involving different push–pull extremities and increasingly large polyenic bridges have been investigated both experimentally, by means of electric field induced second harmonic (EFISH) generation, and theoretically, using a computational approach combining classical molecular dynamics (MD) and quantum chemical (QM) calculations. This theoretical methodology allows to describe the effects of structural fluctuations on the EFISH properties of the complexes formed by the dye and its iodine counterion, and provides a rationale to EFISH measurements. The good agreement between experimental and theoretical results proves that this MD + QM scheme constitutes a useful tool for a rational, computer-aided, design of SHG dyes.
1. Introduction
The design of organic dyes displaying high second-order nonlinear optical (NLO) properties is an important issue in many (bio)technological fields for probing asymmetric media such as artificial interfaces or cell membranes.1 In particular, the exogenous labeling of lipid bilayers by amphiphilic potential-sensitive dyes displaying large second harmonic generation (SHG) responses is at the heart of high resolution imaging microscopy techniques.2–9 SHG probes are usually designed by functionalizing the two extremities of a π-conjugated linker with electron-donating and electron-withdrawing substituents, which provide the asymmetry required for quadratic NLO phenomena. The elaboration of SHG chromophores based on this dipolar architecture,10–13 including responsive systems such as NLO switches,14–16 has been the object of intense research in the last 30 years. In this context, two of us reported the synthesis and characterization of the SHG responses of amphiphilic chromophores incorporating a pyridinium acceptor and a dibutylaminophenyl donor as terminal groups, connected by increasingly large polyenic bridges (series A, Fig. 1).17 The presence of the hydrophobic butyl chains on the donor group, together with the positively-charged hydrophilic acceptor moiety, confers an amphiphilic character to these push–pull dyes that facilitates their interaction with a lipidic membrane. In addition to their use as SHG probes, pyridinium-based derivatives have been widely used in the last decades for producing materials with large quadratic NLO responses, owing to their structural diversity and ability to form different non-centrosymmetric crystal packings when associated with different anions.18,19
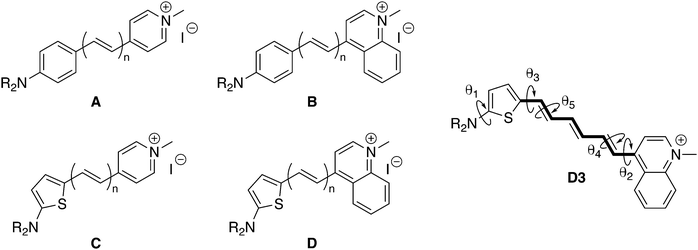 |
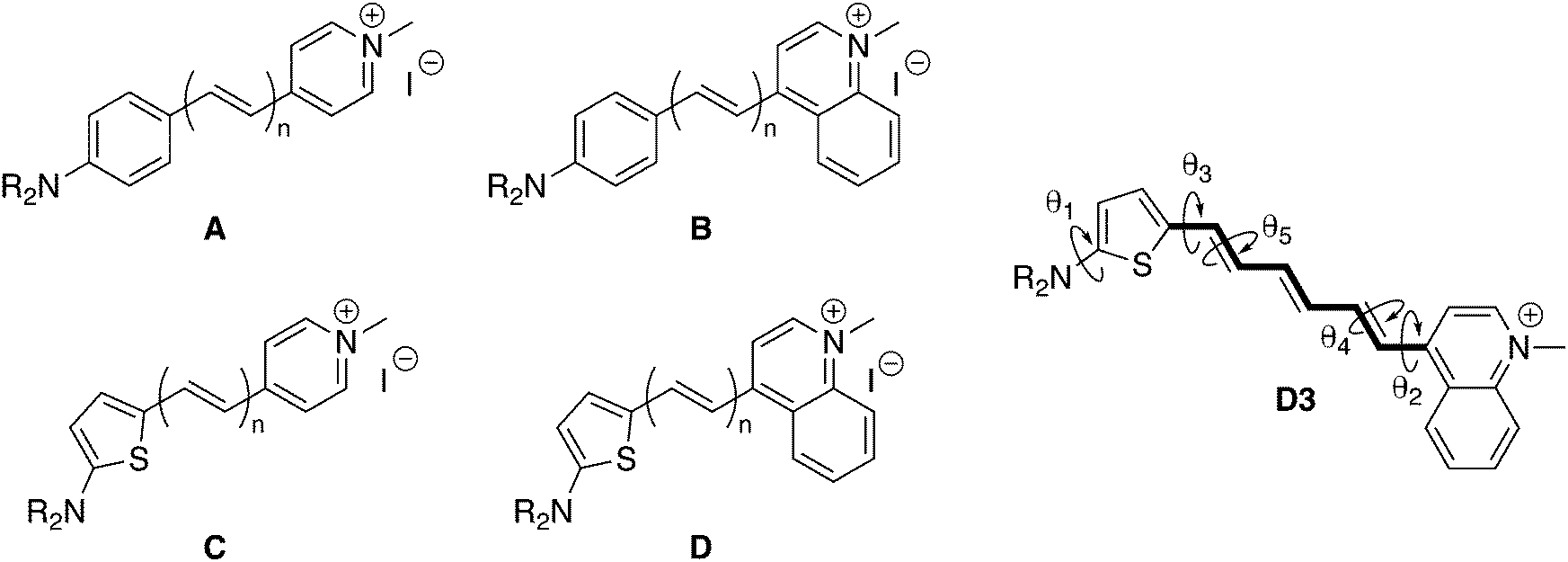
| | Fig. 1 Structure of the push–pull polyenic dyes A–D investigated in this study (n = 1–5), together with their iodide counterion. R = nBu (series A and B), R = nHex (series C and D except C1: R = nBu). On the right: structure of D3 with the π-conjugated segment (in bold) used to calculate the bond length alternation (BLA) and the torsional angles θi discussed in the text. See the ESI† for a more detailed definition of the geometrical parameters. | |
In continuation of our efforts in designing new SHG chromophores, we report in this contribution three new families of derivatives (Fig. 1), for which enhanced optical nonlinearities are expected compared to the original series. In series B and D the pyridinium acceptor is replaced by a quinolinium, whose increased electron-withdrawing strength has been shown to enhance the second-order NLO properties.8,20 In series C and D, the original phenyl is replaced by a thienyl linker, as it was also shown that substituting 1,4-phenylene by 2,5-thienylene π-linkers led to increased NLO responses.21 The SHG responses of these dyes are probed by means of electric-field induced second harmonic (EFISH) generation. The use of the EFISH technique was made possible for these cationic chromophores by using a solvent of low relative permittivity and a short duration (1 μs) of the applied pulsed electric field, which prevents the ion pairs from dissociating into the positively-charged dye and its iodide counterion. The role of such ion pairs interactions in the second-order NLO responses of pyridinium-based salts was investigated in several experimental22 and theoretical works.23,24 EFISH measurements on other methylated azaheterocyclic cations interacting with iodide counter anions have also been reported.25,26
In this study, experimental characterizations are complemented by theoretical chemistry calculations, to provide a fundamental understanding of the origin of the NLO responses in these complexes. The computational approach is based on a methodology we developed in previous works, and combines classical molecular dynamics (MD) simulations and quantum chemical (QM) calculations using time-dependent density functional theory (TD-DFT). As recently reviewed,27 this MD + QM scheme was formerly employed to investigate ion pairs in solution,23,24,28 but also more complex systems such as organic nanoparticles,29 self-assembled monolayers,30,31 and stained lipid bilayers.32–34 Here, MD simulations carried out on the reference D3/iodine complex bring key information on the average position of the iodide anion with respect to the organic chromophore, as well as on the effect of dynamical structural fluctuations on the NLO properties. Then, systematic TD-DFT calculations performed on the whole set of systems provide a rationale to experimental data and allow to establish precise relationships between the structure of the dye and the magnitude of the second- and third-order contributions to the EFISH intensity.
2. Experimental and computational details
2.1 Synthesis
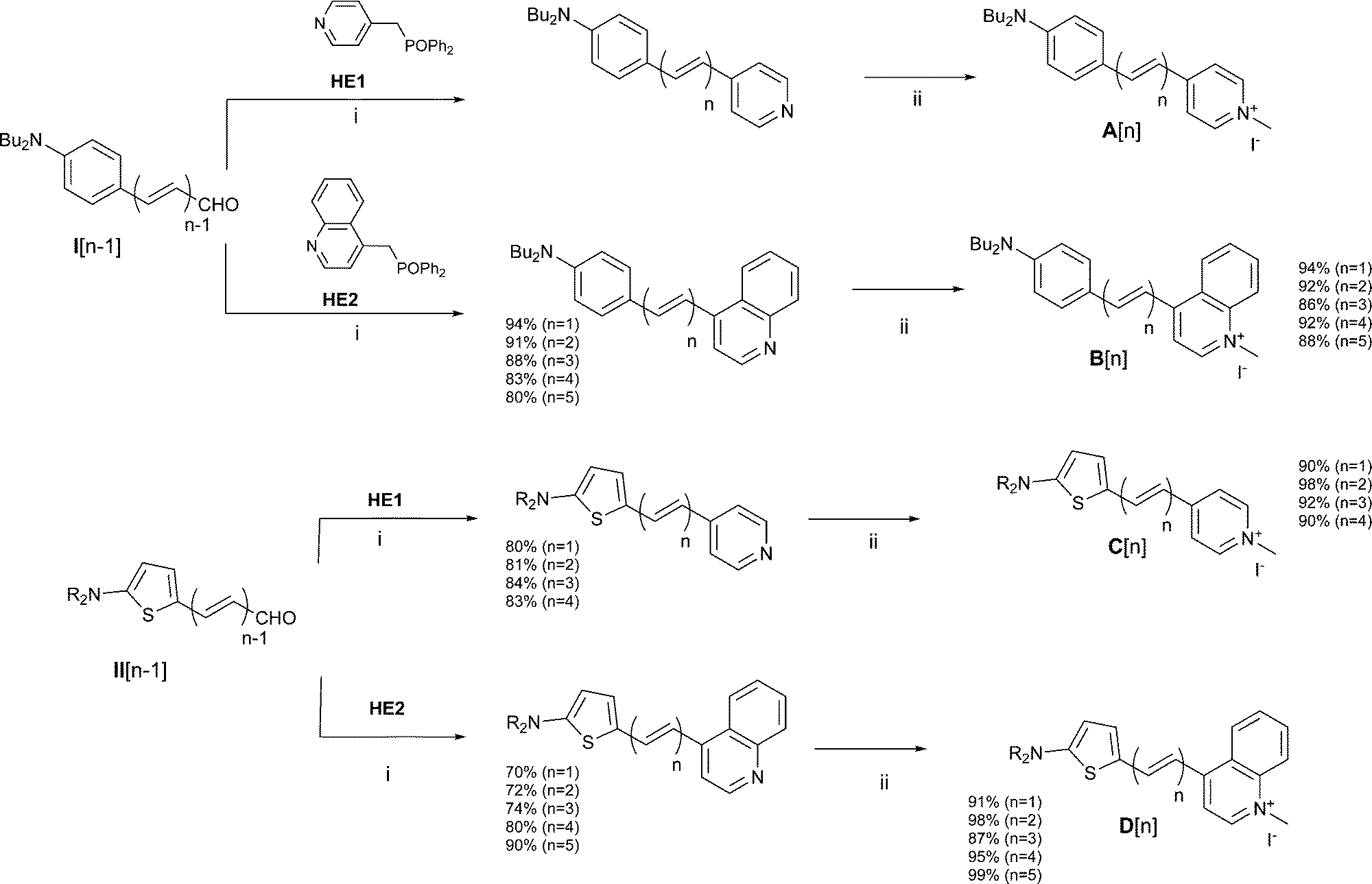
Push–pull dyes of series A have been reported in ref. 17. Compounds of series B were prepared from polyenal I using a similar synthetic scheme based on a Wittig Horner reaction with reagent HE28 followed by alkylation in neat methyl iodide (Fig. 2). Compounds of series C and D were obtained using similar experimental protocols starting from polyenals II12 in place of polyenals I (Fig. 2). More details on synthesis are provided in the ESI.†
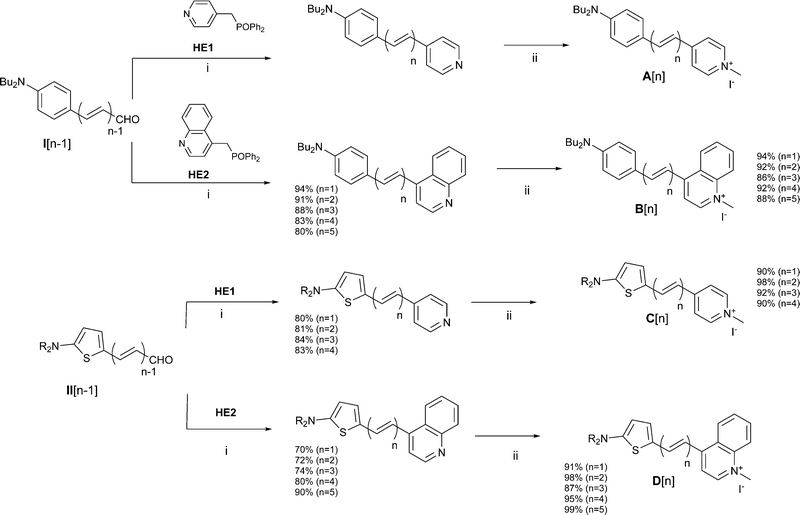 |
| | Fig. 2 Synthesis of compounds A–D. C1: R = nBu; C2–C5 and D1–D5: R = nHex. Reaction conditions: (i) NaH/THF, r.t., (ii) MeI, r.t. | |
2.2 EFISH measurements
Investigation of the molecular nonlinearities was carried out by performing EFISH experiments35,36 using a Q-switched Nd3+:YAG laser emitting pulse trains at 1.06 μm and pumping a Hydrogen Raman cell, resulting in the emission of a coherent Stokes radiation at 1.91 μm. EFISH experiments were performed by using, for each molecule, solutions of increasing concentration in chloroform. Measurements were calibrated relative to the pure solvent. The experimental accuracy ranges between 5 and 10%.
In the EFISH setup, the probed SHG response results from a third-order process described by γ(−2ω; ω, ω, 0). This term is formally expressed as the sum of a pure third-order contribution and a second-order one:
| | 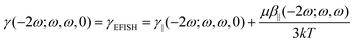 | (1) |
where
μ is the norm of the ground state dipole moment,
k is the Boltzmann constant and 3
kT = 2.833 × 10
−3 a.u. at room temperature (
T = 298.15 K). The
γ‖ (− 2
ω;
ω,
ω, 0) contribution corresponds to the isotropic invariant of the
γ tensor:
| | 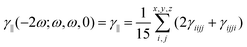 | (2) |
The second term in
eqn (1) implies the projection of the vectorial representation of the
β tensor on the dipole moment:
| | 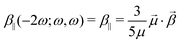 | (3) |
where the
β vector components read:
| | 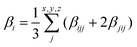 | (4) |
and
| | 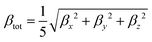 | (5) |
The relative amplitudes of the second- and third-order contributions in
eqn (1) can be analyzed using the
R3/2 ratio:
| | 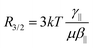 | (6) |
In principle, the use of the EFISH technique is precluded in the case of ionic species, because the
dc electric field necessary to break centrosymmetry induces the migration of ions. However, operating in a solvent of low polarity favors the formation of electrically neutral dye/iodine ion pairs, which enables the characterization of their NLO responses by means of EFISH. Apart from our first study on series
A,
17 it was demonstrated for various positively charged organic derivatives with a range of counteranions.
22,25,26,28 Moreover, using short pulses for the poling electric fields precludes the migration of ions towards electrodes, then avoiding the presence of an ionic current in the solution. In this work, all experiments were carried out in chloroform, with dielectric constants
ε0 = 4.711 in the static limit and
ε∞ = 2.091 at infinite frequency. As mentioned above, an incident laser wavelength of 1907 nm was used, in order to minimize the frequency dispersion effects on
β values due to absorption of the second harmonic light. Moreover, as evidenced from
eqn (1), measurements should be performed at different temperatures to evaluate the second- and third-order contributions and their relative amplitude in the total EFISH response. Here, experimental results were analyzed assuming that the
γ‖ contribution is negligible with respect to the
μβ‖/3
kT term, so that
R3/2 ∼ 0. Therefore, EFISH responses are analyzed as effective second-order responses:
| | 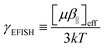 | (7) |
The validity of this assumption is discussed hereafter in light of the computational results.
2.3 Computational methodology
The computational methodology consisted of two steps. Using D3 as reference system, we first addressed the dynamical behavior of the complex composed by the dye and its counterion, as well as the impact of structural fluctuations on the EFISH response. This first step was achieved by adopting the same framework as used in previous studies:23,24,28 (i) the structures of chloroform-solvated ion pairs were generated using classical molecular dynamics (MD) simulations employing a system-specific force field (see next section), and (ii) non-correlated structural snapshots were eventually extracted at regular time intervals of the trajectories to calculate their EFISH responses by means of time-dependent density functional theory (TD-DFT). This MD + TD-DFT approach allowed us to assess the relative magnitude of the γ‖ and μβ‖/3kT contributions (R3/2, eqn (6)), as well as to identify the average position of the iodine with respect to the cationic chromophore. In a second step, we extended the calculations to the whole set of molecules by optimizing the structure of the dye/iodine complexes at the DFT level. The average position of the iodine with respect to the chromophore derived from MD simulations on D3 was used to prepare the initial dye/iodine structures for geometry optimization. On the basis of the relaxed structures, the EFISH responses of all complexes were calculated using TD-DFT and compared to experimental results.
2.3.1 Force field parameterization.
The general AMBER force field (FF) was partially modified to finely reproduce the equilibrium geometry and torsional degrees of freedom of D3, which is a prerequisite to obtain a reliable description of the NLO responses. In particular, we have modified the FF bond lengths to accurately match (with a mean absolute error of 0.002 Å) those calculated at the M06-2X/6-311G(d) level, in which solvent effects (chloroform) were accounted for by using the integral equation formalism of the polarizable continuum model (IEF-PCM).37 Electrostatic potential-fitted (ESP) atomic charges of the chromophore, iodide anion and chloroform have been obtained at the same level of theory. In addition, relaxed potential energy scans were performed at the M06-2X/6-311G(d) level in gas phase for five relevant dihedral angles (θ1–θ5, see Fig. 1) and fitted following the procedure described in ref. 38. Reparameterization of the bonds and torsion potentials was done iteratively until convergence. All the details of force field parameterization are reported in the ESI.†
2.3.2 Molecular dynamics simulations.
Starting from a low density cubic box of size 120 Å containing the dye/iodide ion pair and 1600 chloroform molecules, the system was equilibrated for 10 ns in the NpT ensemble (p = 1 atm and T = 298.15 K). In order to better span the conformational degrees of freedom of the dye, two different initial conformations corresponding to different values of the θ3 dihedral were considered. Production runs were then carried out in the NVT ensemble for 20 ns with timestep of 1 fs, and performed by rescaling the temperature to 298.15 K every 100 steps. 200 structural snapshots were extracted from each MD trajectory, providing a total set of 400 structures for calculating the NLO properties. The probability distributions of the values of relevant geometrical parameters (bond length alternation, torsional angles, anion–cation distances, see ESI†) for the 400 geometrical snapshots were found to coincide with the distributions obtained by using the 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 structures extracted every 1000 timesteps of the simulation, confirming that the set of selected geometries is representative of the dynamics of the system. All MD simulations have been performed using the NAMD software.39
000 structures extracted every 1000 timesteps of the simulation, confirming that the set of selected geometries is representative of the dynamics of the system. All MD simulations have been performed using the NAMD software.39
2.3.3 Calculation of the EFISH responses.
For all compounds, the molecular geometries of the dye/iodide complexes were optimized at IEF-PCM:M06-2X/6-311G(d) level in chloroform (considering nBu chains in all compounds). The optimized structures were confirmed to be real minima of the potential energy surface on the basis of their harmonic vibrational frequencies, which were found real for all normal modes. Calculations of hyperpolarizabilities have been performed at the TD-DFT level employing the M06-2X exchange–correlation (XC) functional.40 First hyperpolarizabilities were computed analytically using the standard TD-DFT method,41,42 while calculations of the second hyperpolarizabilities were performed using the first-order numerical derivatives of the analytical first hyperpolarizabilities. The suitability of M06-2X for computing the second-order NLO properties of push–pull π-conjugated dyes was demonstrated in previous computational works.43,44 To further assess its adequacy for the compounds investigated here, static first hyperpolarizabilities of cationic dyes of series D were also computed using the second-order Møller–Plesset perturbation theory (MP2), in which the energy derivatives are calculated using a numerical finite field (FF) procedure and refined by using a Romberg scheme. As reported in the ESI† (Fig. S15), a very good correlation was found between the two data sets. Preliminary calculations on series D were also performed to choose the most appropriate basis set. Three basis sets of decreasing flexibility were tested for computing the static first hyperpolarizability of the chromophores, namely aug-cc-pVTZ, aug-cc-pVDZ and 6-311+G(d). As reported in Fig. S16 (ESI†), both the 6-311+G(d) and aug-cc-pVDZ basis sets provide static β values very similar to those obtained using the larger aug-cc-pVTZ basis. Therefore, the computationally-cheaper 6-311+G(d) basis was chosen to describe the chromophores. To model the iodide anion, we employed the aug-cc-pVDZ basis set and the associated pseudopotential, although the basis set used for the counterion does not have a significant impact on the first hyperpolarizability of the complexes (Table S5, ESI†). All DFT calculations were performed using Gaussian 16.45 Graphical representations of the molecules were realized with the Chemcraft package.46
2.3.4 Calculation of conjugation and aromaticity measures.
The atomic partition employed to compute the aromaticity indices and the bond-order alternation (BOA) is the quantum theory of atoms in molecules (QTAIM).47 The QTAIM atomic overlap matrices were obtained from the AIMAll software,48 and the aromaticity indices were calculated with the in-house ESI-3D program.49–51 This program provides AV1245,52 AVmin,53 BLA, BOA,54 FLU,50 HOMA,55Iring,56 and MCI57 values. The definitions and expressions of all the indices are provided in ESI.† Note that we have included AV1245 and AVmin in our study due to the limitations of Iring and MCI in measuring large conjugated circuits.52 The former indices were specifically designed to replicate the values of MCI in larger conjugated chains.
3. Results and discussion
3.1 Dynamics of the EFISH response of the D3/iodide complex
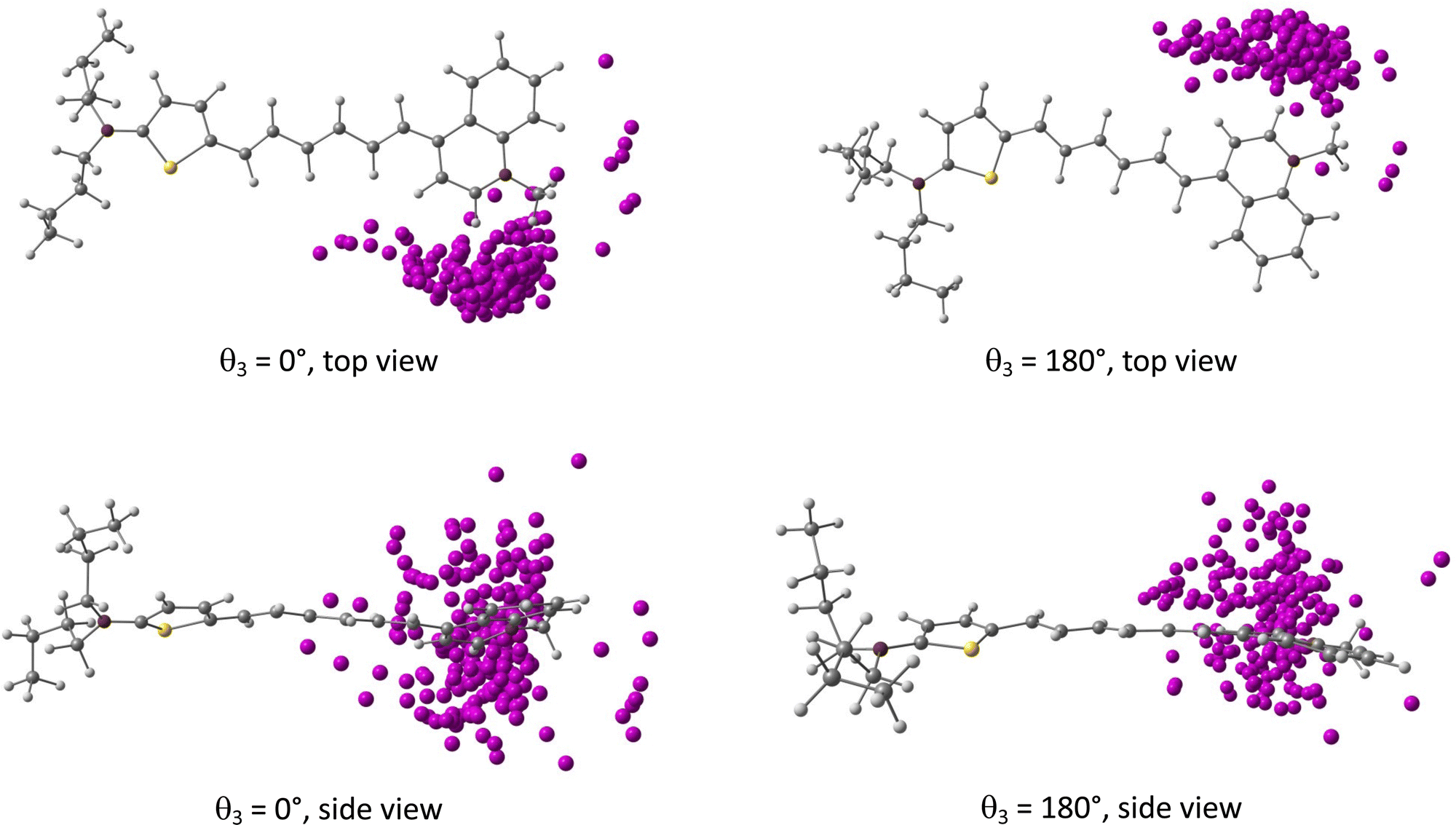
The probability distributions issued from MD samplings reveal large fluctuations in the bond length alternation (BLA, see Fig. 1) along the conjugated linker, with BLA = (−0.045 ± 0.023) Å when considering the 400 geometries used for NLO calculations. MD simulations also show that the position of the iodide with respect to the chromophore is highly dynamical, the counterion nevertheless remaining in close proximity to the dye with an average distance from the quinolinium nitrogen dNI = (4.57 ± 0.47) Å. Moreover, the iodide anion oscillates around an average position located within the mean plane of the quinolinium moiety, with values of the dihedral angle between the terminal phenyl and the anion θPhI = 175.1° ± 45.9° (Fig. S9, ESI†). Fig. 3 illustrates how the iodide anion fluctuates around the dye for the 400 selected snapshots extracted from the MD simulations.
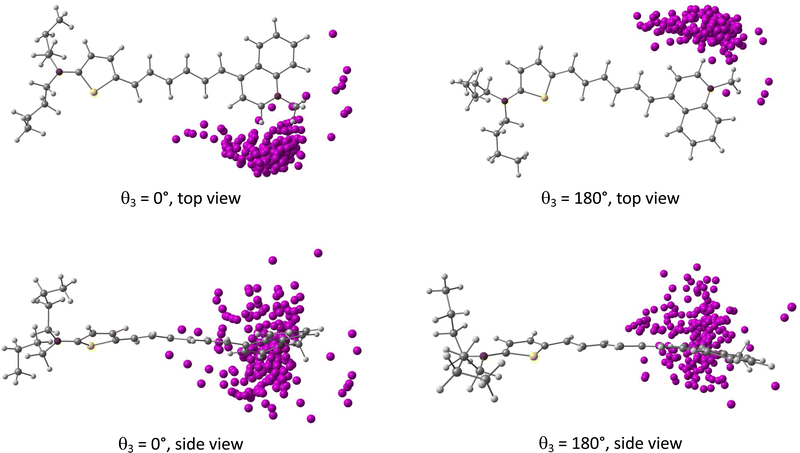 |
| | Fig. 3 Distribution of the iodide counterion positions with respect to the D3 chromophore (fixed in its initial position) for the two different initial conformations of the dihedral θ3. | |
The time evolution of the various terms involved in the EFISH signal of the D3/iodide complex (namely γEFISHG, γ‖, μ, β‖ and μβ, see eqn (1)–(4)), as well as of the angle θ between the ![[small mu, Greek, vector]](https://www.rsc.org/images/entities/i_char_e0e9.gif) and
and ![[small beta, Greek, vector]](https://www.rsc.org/images/entities/i_char_e115.gif) vectors, are collected in Fig. S10. Their average values and standard deviations are reported in Table 1. The results show that both the second- and third-order contributions of the EFISH response display high sensitivity to structural fluctuations, with standard deviations reaching about 50% of their average values. Moreover, the EFISH response is strongly dominated by the second-order term μβ‖/3kT, which is two orders of magnitude larger than γ‖. This confirms the validity of the experimental hypothesis, assuming that R3/2 ∼ 0 and the interpretation of the EFISH signal as an effective second-order response (eqn (7)). As reported in Table 1, the [μβ‖]eff value predicted by MD + TD-DFT calculations (3.5 × 106 a.u.) is overestimated compared to experimental measurements (1.5 × 106 a.u.), but the order of magnitude of the NLO response is well reproduced.
vectors, are collected in Fig. S10. Their average values and standard deviations are reported in Table 1. The results show that both the second- and third-order contributions of the EFISH response display high sensitivity to structural fluctuations, with standard deviations reaching about 50% of their average values. Moreover, the EFISH response is strongly dominated by the second-order term μβ‖/3kT, which is two orders of magnitude larger than γ‖. This confirms the validity of the experimental hypothesis, assuming that R3/2 ∼ 0 and the interpretation of the EFISH signal as an effective second-order response (eqn (7)). As reported in Table 1, the [μβ‖]eff value predicted by MD + TD-DFT calculations (3.5 × 106 a.u.) is overestimated compared to experimental measurements (1.5 × 106 a.u.), but the order of magnitude of the NLO response is well reproduced.
Table 1 Average values and standard deviations (σ) of the EFISH responses of the D3/iodide complex, computed at the IEF-PCM:M06-2X/6-311+G(d) level in chloroform, using the 400 snapshots extracted from the MD trajectories. The NLO properties and the norm of the dipole moment are given in atomic units, the angle θ is given in degrees
| Property |
Average |
σ
|
|
γ
EFISH × 10−7 |
124.1 |
55.4 |
|
μβ
‖/3kT × 10−7 |
121.6 |
55.5 |
|
γ
‖ × 10−7 |
2.6 |
1.3 |
|
R
3/2
|
0.02 |
0.07 |
|
μβ
‖ × 10−6 |
3.4 |
1.6 |
|
β
‖ × 10−4 |
16.8 |
6.1 |
|
μ
|
12 |
2 |
|
θ
|
48 |
15 |
| [μβ‖]eff × 10−6 |
3.5 |
1.6 |
| [μβ‖]eff × 10−6 (exp.) |
1.5 |
— |
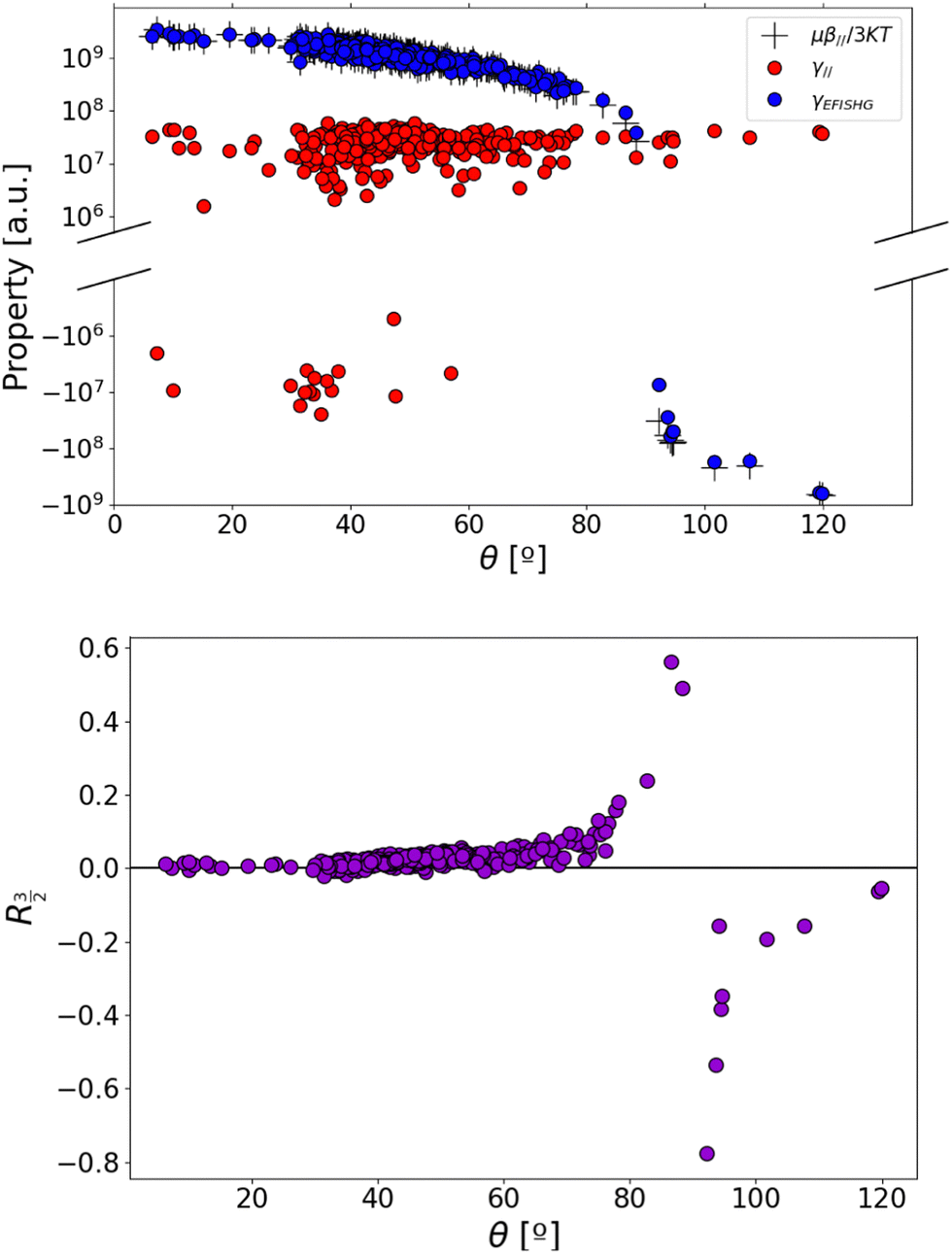
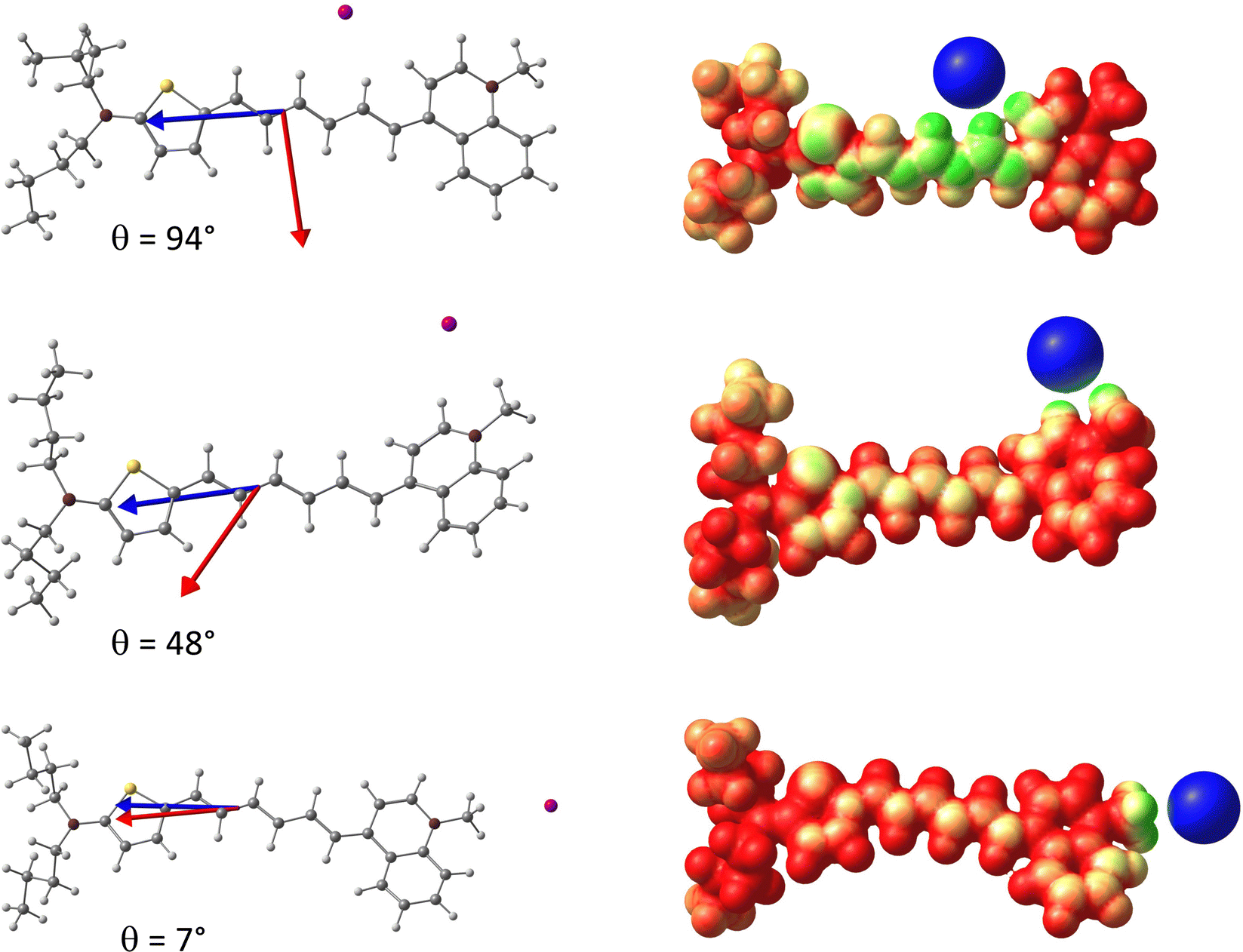
To gain further insight into the relationship between the EFISH response and the dynamical structure of the dye/iodide complex, Fig. 4a reports the distribution of the second- and third-order contributions to the EFISH response with respect to the angle θ between the and vectors. This plot shows that γ‖ displays relatively weak variations with respect to θ, despite a small set of structures gives rise to negative γ‖ values, as discussed later on. Fig. S13e (ESI†) further shows that the variations of the γ‖ values are not correlated to those of the θ angle. On the contrary, the second-order contribution μβ‖/3kT strongly depends on θ, since β‖ involves the scalar product between the and vectors (eqn (3)). Therefore, μβ‖/3kT progressively decreases as the θ value increases, and cancels out for θ = 90° before changing sign. In the θ ∼ 90° region, the EFISH response is thus dominated by the third-order term γ‖, which translates into a divergence of the R3/2 ratio (Fig. 4b). As illustrated in Fig. 5, the value of the θ angle is entirely driven by the position of the iodine anion with respect to the chromophore. When I− is located close to the polyenic bridge, the and vectors are quasi-perpendicular (θ ∼ 90°), giving rise to a vanishing μβ‖/3kT contribution, while when I− is found close to the electron-withdrawing extremity of the dye (θ ∼ 0°), the μβ‖/3kT contribution is maximized. The average position of the anion deduced from the MD trajectories corresponds to an intermediate value of θ = 48°, a situation in which γ‖ is negligible with respect to μβ‖/3kT (R3/2 = 0.02). Note that, in addition to β‖, the norm of the total dipole moment is also strongly correlated with the θ angle, and thus with the position of the iodide anion. The evolution of β‖ and μ with respect to θ is illustrated in Fig. S13 (ESI†). Consistent with the dipole variations, we can also notice in Fig. 5 that the position of the counterion induces significant variation in the electrostatic potential within the molecule. For θ ∼ 0°, the charge transfer along the long molecular axis is enhanced, reinforcing the asymmetry of the electron density, while for θ ∼ 90° the potential displays a more symmetrical shape. However, as illustrated in Fig. S13 (ESI†), the total first hyperpolarizability (βtot) does not exhibit any clear correlation with θ, indicating that the changes in the electronic density of the chromophore induced by the iodide position has no significant impact on the second-order NLO response itself, but only on its projection onto the dipole moment direction. This is further illustrated in Fig. S12 (ESI†), which shows that βtot values calculated in the presence of the iodide and those calculated for the same geometries of the chromophore after removing the anion evolve similarly.
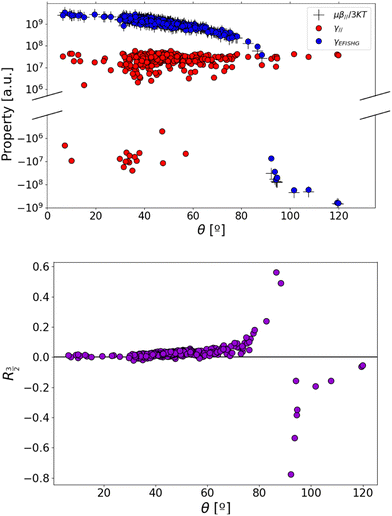 |
| | Fig. 4 (top) Distribution of the total EFISH response of the D3/iodide complex, and of its second- and third-order contributions (μβ‖/3kT and γ‖); (bottom) distribution of the R3/2 ratio as a function of the angle θ. The y-axis in the top plot is in logarithmic scale. | |
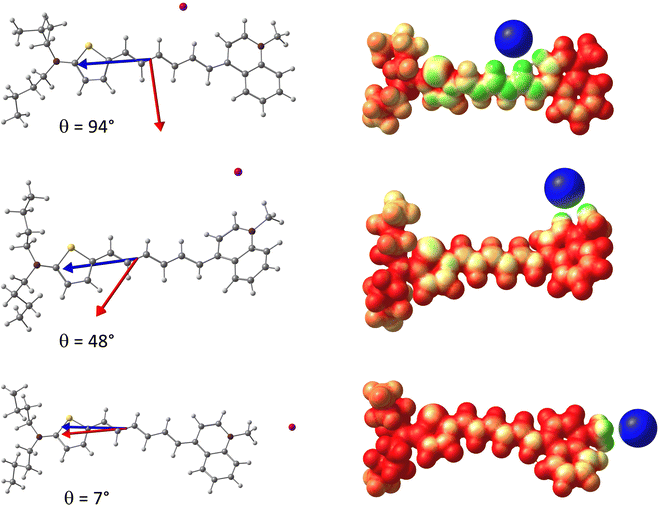 |
| | Fig. 5 Left: Structure of the D3/iodine complex for three different positions of the anion, with the value of the θ angle between the (normalized) (red) and (blue) vectors; Right: Molecular electrostatic potential (MEP) for the three configurations. | |
It is also instructive to address the variations of the NLO properties with respect to the BLA along the conjugated segment of the chromophore. Fig. S11 (ESI†) first reveals that the BLA values calculated along the MD trajectories are spread over a broad range, from negative to positive values. In addition, the BLA globally increases as the θ angle decreases, which indicates that the position of the iodide anion somehow influences the conjugation along the polyenic linker, although the two quantities do not show a clear, direct correlation. Recent calculations also demonstrated a similar influence of the position of the anion on the BLA of the organic cation in cyanine crystals.58
As illustrated in Fig. S14 (ESI†), γ‖ evidences a global reverse correlation with BLA, negative γ‖ values being associated with highly conjugated structures having BLA values close to zero. A similar relationship between γ‖ and BLA values was recently observed for the phenol blue, another typical cyanine dye.59 Interestingly, the variations of the isotropic linear polarizability (αiso) of D3 are also clearly correlated with BLA, while μ and βtot are not. This evidences that odd-order optical quantities are mainly impacted by the fluctuations in the degree of conjugation within the polyenic bridge, while even-order ones mainly depend on the fluctuations in the iodide position.
3.2 EFISH response of the dye/iodide complexes
In a second step, the structures of all the dye/iodide complexes represented in Fig. 1 were optimized at the IEF-PCM:M06-2X/6-311G(d) level, with the initial position of the anion set to its average position according to the MD samplings. The BLA along the polyenic linker of the dye, the value of the θ2 and θ3 torsional angles, as well as the dNI and θPhI values associated with the position of the iodide anion are reported in Table 2 for all complexes. As indicated by the latter parameters, the optimization process did not change significantly the position of the iodine (see Fig. S17–S20 (ESI†) a scheme of the optimized geometries). In series A and B, the absolute BLA values slightly decrease with increasing n, while they are significantly smaller and regularly increase in series C and D, indicating that the conjugation along the polyenic bridge is mostly driven by the nature of the donor moiety. Note that, for very extended linkers (n ≫ 5), the BLA is expected to converge towards similar values independently of the nature of the donor and acceptor moieties, as the conjugated segment resembles in this case an unsubstituted polyene. Furthermore, the values of θ2 show that molecules incorporating a quinolinium acceptor (series B and D) display larger deviation from planarity than their pyridinium analogs.
Table 2 Bond length alternation (BLA, Å), torsional angles θ2 and θ3 (degrees), as well as dNI (Å) and θPhI (degrees) values associated to the position of the iodide anion in all dye/iodide complexes
| Molecule |
BLA |
θ
2
|
θ
3
|
d
NI
|
θ
PhI
|
|
A1
|
−0.096 |
−177.6 |
−177.6 |
4.49 |
−177.6 |
|
A2
|
−0.088 |
−179.5 |
−178.0 |
4.49 |
174.2 |
|
A3
|
−0.086 |
179.2 |
177.3 |
4.49 |
−177.2 |
|
A4
|
−0.087 |
−178.4 |
179.3 |
4.49 |
−179.3 |
|
A5
|
−0.087 |
178.9 |
178.1 |
4.47 |
172.8 |
|
B1
|
−0.095 |
156.9 |
174.0 |
4.50 |
−175.2 |
|
B2
|
−0.086 |
−160.2 |
−178.9 |
4.51 |
−179.3 |
|
B3
|
−0.086 |
−159.7 |
−177.6 |
4.50 |
177.0 |
|
B4
|
−0.085 |
−161.6 |
178.5 |
4.51 |
−177.9 |
|
B5
|
−0.087 |
−159.4 |
179.4 |
4.50 |
−177.0 |
|
C1
|
−0.066 |
−179.8 |
−179.3 |
4.50 |
−178.7 |
|
C2
|
−0.069 |
−179.6 |
−179.6 |
4.49 |
−178.1 |
|
C3
|
−0.074 |
177.6 |
179.8 |
4.48 |
−173.0 |
|
C4
|
−0.077 |
179.8 |
−179.5 |
4.49 |
177.9 |
|
C5
|
−0.080 |
−179.3 |
−179.3 |
4.49 |
177.5 |
|
D1
|
−0.055 |
−172.1 |
−178.3 |
4.53 |
−179.1 |
|
D2
|
−0.061 |
−167.7 |
−179.2 |
4.52 |
−179.9 |
|
D3
|
−0.067 |
167.2 |
−179.1 |
4.52 |
178.3 |
|
D4
|
−0.074 |
163.0 |
−180.0 |
4.51 |
−177.9 |
|
D5
|
−0.077 |
163.8 |
−179.1 |
4.51 |
176.8 |
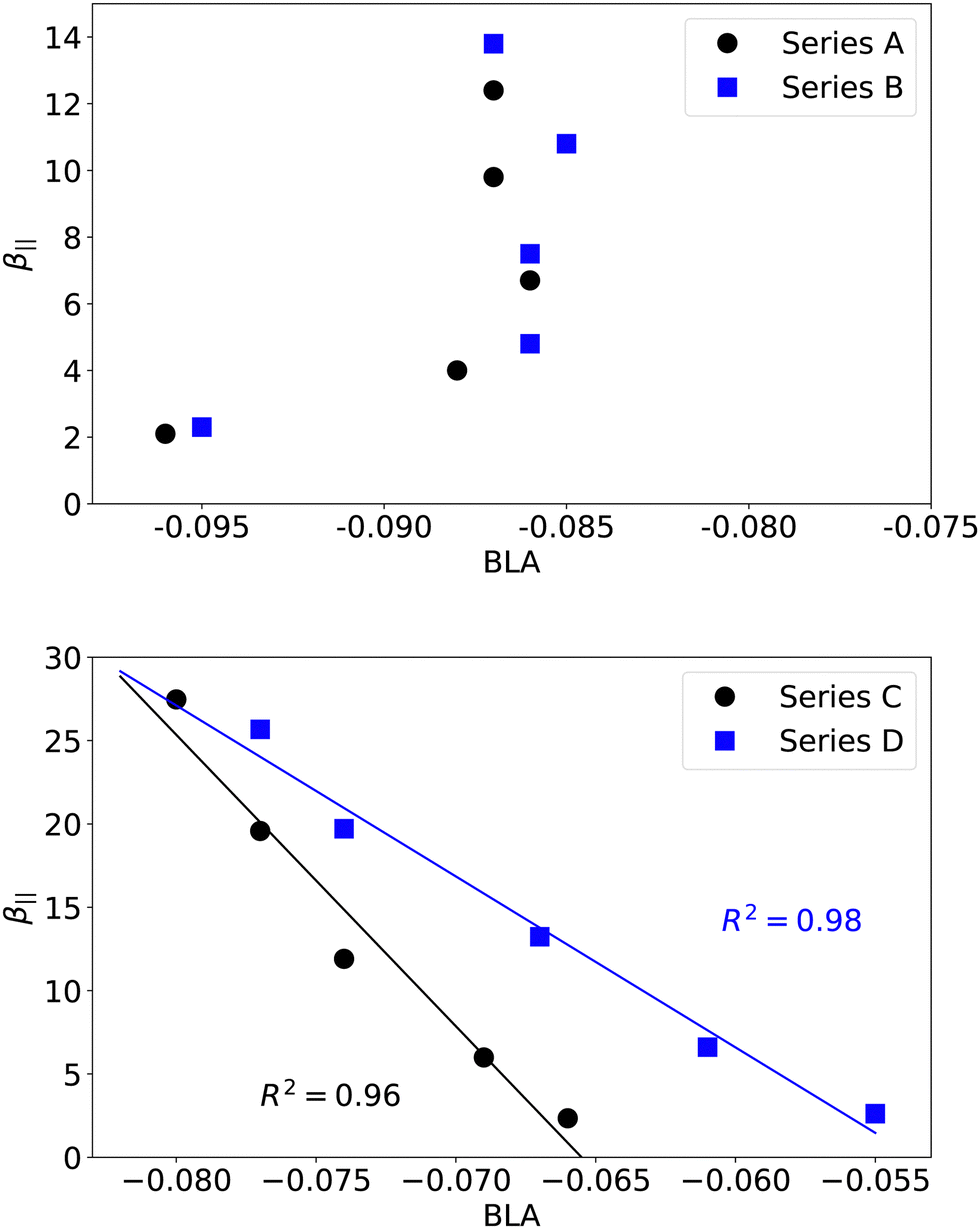
The computed NLO responses collected in Table 3 show that, whatever the nature of the dye, the third-order contribution to the total EFISH response remains small with respect to the second-order contribution, with R3/2 ratios not exceeding 8%. This result is consistent with our previous report on stilbazolium–anion complexes.23 Nevertheless, the R3/2 ratio monotonically increases in the four series when elongating the polyenic linker, indicating that the γ‖ contribution should be considered in the case of highly extended systems. Series C and D incorporating a dibutyl-aminothienyl donor display larger second-order β‖ responses that inversely correlate with the BLA values (Fig. 6, bottom). Consistently, series A and B, which display small variations of BLA with n, show a lower enhancement of β‖ when increasing the size of the bridge. However, there is no correlation between BLA and β‖ values in the last two families of compounds (Fig. 6, top). Thus, the BLA along the conjugated polyenic bridge, as defined in Fig. 1, is a relevant structural indicator only for derivatives incorporating a thienyl group, but not for those incorporating a phenyl.
Table 3 Components of the dynamic (λ = 1907 nm) EFISH response of the dye/iodide complexes calculated at the IEF-PCM:M06-2X/6-311+G(d) level: dipole moment norm (μ, a.u.), parallel first hyperpolarizability (β‖, 104 a.u.), angle between and (θ, degrees), μβ‖ (105 a.u.), μβ‖/3kT (108 a.u.), parallel second hyperpolarizability (γ‖, 106 a.u.), total EFISH response (γEFISH, 108 a.u.), effective EFISH response ([μβ‖]eff, 105 a.u.), and R3/2(%) ratio
| Molecule |
μ
|
β
‖
|
θ
|
μβ
‖
|
μβ
‖/3kT |
γ
‖
|
γ
EFISH
|
[μβ‖]eff |
R
3/2
|
|
A1
|
10.5 |
2.1 |
45 |
3.7 |
1.3 |
2.4 |
1.3 |
3.8 |
1.8 |
|
A2
|
10.5 |
4.0 |
47 |
6.9 |
2.4 |
6.5 |
2.5 |
7.1 |
2.7 |
|
A3
|
10.6 |
6.7 |
47 |
11.9 |
4.2 |
15.2 |
4.3 |
12.3 |
3.6 |
|
A4
|
10.7 |
9.8 |
48 |
17.4 |
6.2 |
28.5 |
6.4 |
18.2 |
4.6 |
|
A5
|
10.5 |
12.4 |
51 |
21.7 |
7.7 |
46.2 |
8.1 |
23.0 |
6.0 |
|
B1
|
10.8 |
2.3 |
53 |
4.2 |
1.5 |
3.7 |
1.5 |
4.3 |
2.5 |
|
B2
|
10.7 |
4.8 |
53 |
8.5 |
3.0 |
10.3 |
3.1 |
8.8 |
3.4 |
|
B3
|
10.7 |
7.5 |
54 |
13.4 |
4.7 |
22.5 |
5.0 |
14.1 |
4.7 |
|
B4
|
10.6 |
10.8 |
56 |
19.1 |
6.7 |
42.2 |
7.1 |
20.3 |
6.3 |
|
B5
|
10.6 |
13.8 |
56 |
24.3 |
8.6 |
66.0 |
9.2 |
26.1 |
7.7 |
|
C1
|
11.5 |
2.3 |
37 |
4.5 |
1.6 |
1.3 |
1.6 |
4.5 |
0.8 |
|
C2
|
12.2 |
6.0 |
30 |
12.2 |
4.3 |
5.5 |
4.4 |
12.4 |
1.3 |
|
C3
|
12.6 |
11.9 |
27 |
25.0 |
8.8 |
17.4 |
9.0 |
25.5 |
2.0 |
|
C4
|
12.8 |
19.6 |
24 |
41.9 |
14.8 |
40.6 |
15.2 |
43.0 |
2.7 |
|
C5
|
13.0 |
27.5 |
22 |
59.3 |
20.9 |
74.2 |
21.7 |
61.4 |
3.5 |
|
D1
|
12.1 |
2.6 |
44 |
5.3 |
1.9 |
1.3 |
1.9 |
5.3 |
0.7 |
|
D2
|
12.0 |
6.6 |
47 |
13.2 |
4.6 |
7.6 |
4.7 |
13.4 |
1.6 |
|
D3
|
12.0 |
13.2 |
48 |
26.4 |
9.3 |
28.1 |
9.6 |
27.2 |
3.0 |
|
D4
|
11.8 |
19.7 |
51 |
38.7 |
13.7 |
68.4 |
14.3 |
40.6 |
5.0 |
|
D5
|
11.5 |
25.7 |
53 |
49.4 |
17.4 |
124.6 |
18.7 |
52.9 |
7.1 |
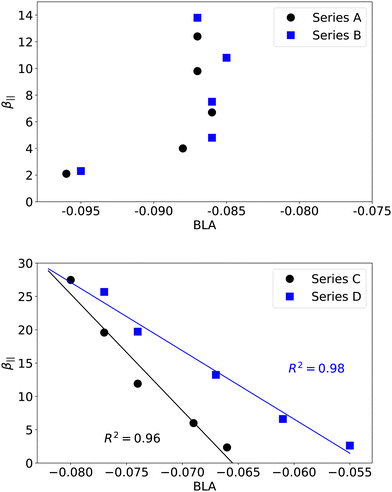 |
| | Fig. 6 Evolution of β‖ (104 a.u.) as a function of the BLA (Å) computed along the conjugated bridge (see Fig. 1) in series A and B (top) and series C and D (bottom). | |
It is also interesting to note that the values of the dipole moment do not vary significantly with n in any of the molecular series. This is related to the fact that the dipole moment is predominantly determined by the mutual position of the iodide anion and positively charged nitrogen atom, which, as mentioned above, is similar for all systems (see Fig. S17–S20, ESI†) As a consequence, the magnitude of the second-order contribution of the EFISH response, μβ‖/3kT, is essentially driven by the value of β‖ and that of the θ angle between the and vectors. While β‖ smoothly increases with n in the four series, θ increases in the A, B and D families, thus dampening the increase of the μβ‖/3kT term. On the contrary, the θ angle decreases in the C series, making the and vectors increasingly parallel as the linker lengthens (see Fig. S19, ESI†), and thus contributing to the enhancement of the second-order response.
3.3 Further study of structure–properties relationships
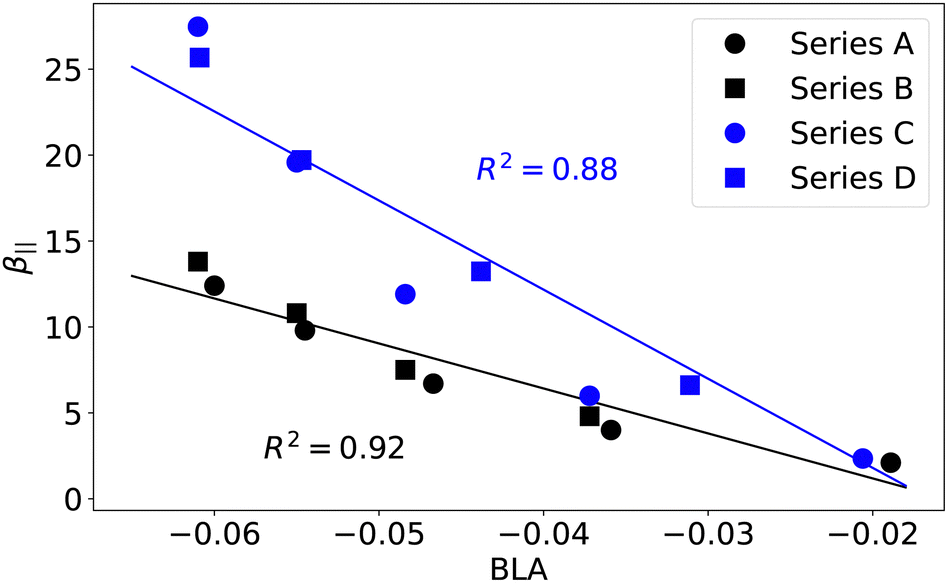
In order to gain deeper insights into structure–property relationships in the four molecular families, we investigated more direct measures of electron conjugation, such as AVmin,52,53 the bond-order alternation (BOA),54 and FLU.50 We computed these descriptors along the polyenic bridge highlighted in Fig. 1, but also along the more extended fragment that includes the thienyl or phenyl ring (see Tables S6–S8, ESI†). The pyridinium and quinolinium acceptor units were excluded from this second conjugated circuit since results in Fig. 6 clearly show that the different behavior observed in series A–B compared to series C–D is related to the nature of the donor moiety rather than the acceptor. The different evolutions of the molecular families A–B and C–D can be rationalized by the different nature of the thienyl and phenyl rings compared to their isolated thiophene and benzene counterparts. According to the MCI and Iring aromaticity indicators,60 benzene is more aromatic than thiophene. Hence, the phenyl ring is expected to be more resilient to the loss of aromaticity. Calculations indicate that the phenyl ring in the compounds of series A–B is about 60–63% less aromatic than benzene. In contrast, the thienyl ring in series C–D is 45–50% less aromatic than thiophene. This difference results in a larger contribution of the phenyl ring to the BLA. The latter contribution is only included if we consider the conjugated segment going from the amine nitrogen to the first carbon of the pyridinium (series A and B) or quinolinium (series C and D), as shown in Fig. 7. Since the BLA of the polyenic bridge defined in Fig. 1 did not show a good correlation with β‖ and γ‖ for the series A–B, we are deemed to conclude that the aromaticity of the phenyl ring plays an important role in the magnitude of the NLO responses. A similar conclusion can be reached using other indicators of electron conjugation, such as BOA, FLU, and AVmin. Correlations of β‖ and γ‖ with these indices for the series A–B and C–D are reported in Fig. S25–S27 (ESI†).
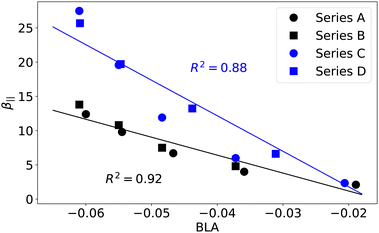 |
| | Fig. 7 Evolution of β‖ (104 a.u.) as a function of the BLA (Å) computed along the conjugated path going from the amine nitrogen to the first carbon of the pyridinium (series A and B) or quinolinium (series C and D). See ESI† for details. | |
3.4 Comparison to experiment
The experimental EFISH data are collected in Table 4, together with the maximum absorption wavelengths. The static EFISH responses extrapolated using the two-state approximation for β are also reported. Consistently with the red shift of the main absorption band, the EFISH response increases in each series with the size of the polyenic bridge. As expected, removing frequency dispersion effects by extrapolating responses to the infinite-wavelength limit attenuates the increase of μβ‖ with chain length. EFISH measurements also globally confirm the anticipated enhancement of the quadratic NLO response upon replacement of the pyridinium by a quinolinium acceptor (A → B and C → D), and upon replacement of the 1,4-phenylene by a 2,5-thienylene π-linker (A → C and B → D). A few exceptions to this general trend are nevertheless observed when considering μβ‖(2ω) values in Table 4: B2 < A2, D1 < B1, and C1 < A1. As a matter of fact, the difference in the μβ‖(2ω) values of C1 and A1 lies within the experimental error, and the same is true when comparing C1 and A1. The difference is more significant for D1 and B1, for which the expected increase of the NLO response due to the use of a thienyl linker is not observed.
Table 4 Experimental EFISH responses (μβ‖(2ω) ≡ [μβ‖ (−2ω; ω, ω)]eff) measured at λ = 1907 nm, maximum absorption wavelength (λmax, nm), two-state frequency dispersion factors F(ω),a and static EFISH responses extrapolated as μβ‖(0) = μβ‖(2ω)/F(ω). All NLO data are given in 10−48 esu (1 a.u. of β = 8.6392 × 10−33 esu)
| Molecule |
μβ
‖(0)c |
μβ
‖ (2ω)c |
λ
max
|
F(ω) |
|
1/F(ω) = (1 − λmax2/λ2) (1 − 4λmax2/λ2).
From ref. 17.
Experimental errors correspond to 10% of the measured value.
|
|
A1
|
549 ± 55 |
841 ± 84 |
517 |
1.53 |
|
A2
|
1338 ± 134 |
2230 ± 223 |
558 |
1.67 |
|
A3
|
1537 ± 154 |
2700 ± 270 |
581 |
1.76 |
|
A4
|
1887 ± 189 |
3380 ± 338 |
589 |
1.79 |
|
A5
|
2014 ± 201 |
3690 ± 369 |
598 |
1.83 |
|
B1
|
783 ± 78 |
1380 ± 138 |
582 |
1.76 |
|
B2
|
1098 ± 110 |
2156 ± 216 |
624 |
1.96 |
|
B3
|
1566 ± 157 |
3250 ± 325 |
643 |
2.08 |
|
B4
|
4050 ± 405 |
8750 ± 875 |
656 |
2.16 |
|
B5
|
4284 ± 428 |
9435 ± 944 |
662 |
2.20 |
|
C1
|
523 ± 52 |
891 ± 89 |
568 |
1.70 |
|
C2
|
1332 ± 133 |
2669 ± 267 |
631 |
2.00 |
|
C3
|
2946 ± 295 |
6366 ± 637 |
656 |
2.16 |
|
C4
|
4954 ± 495 |
10946 ± 1095 |
663 |
2.21 |
|
D1
|
445 ± 45 |
900 ± 90 |
634 |
2.02 |
|
D2
|
1793 ± 179 |
4770 ± 477 |
714 |
2.66 |
|
D3
|
4083 ± 408 |
13600 ± 1360 |
763 |
3.33 |
|
D4
|
5710 ± 571 |
18170 ± 1817 |
754 |
3.18 |
|
D5
|
7281 ± 728 |
22500 ± 2250 |
748 |
3.09 |
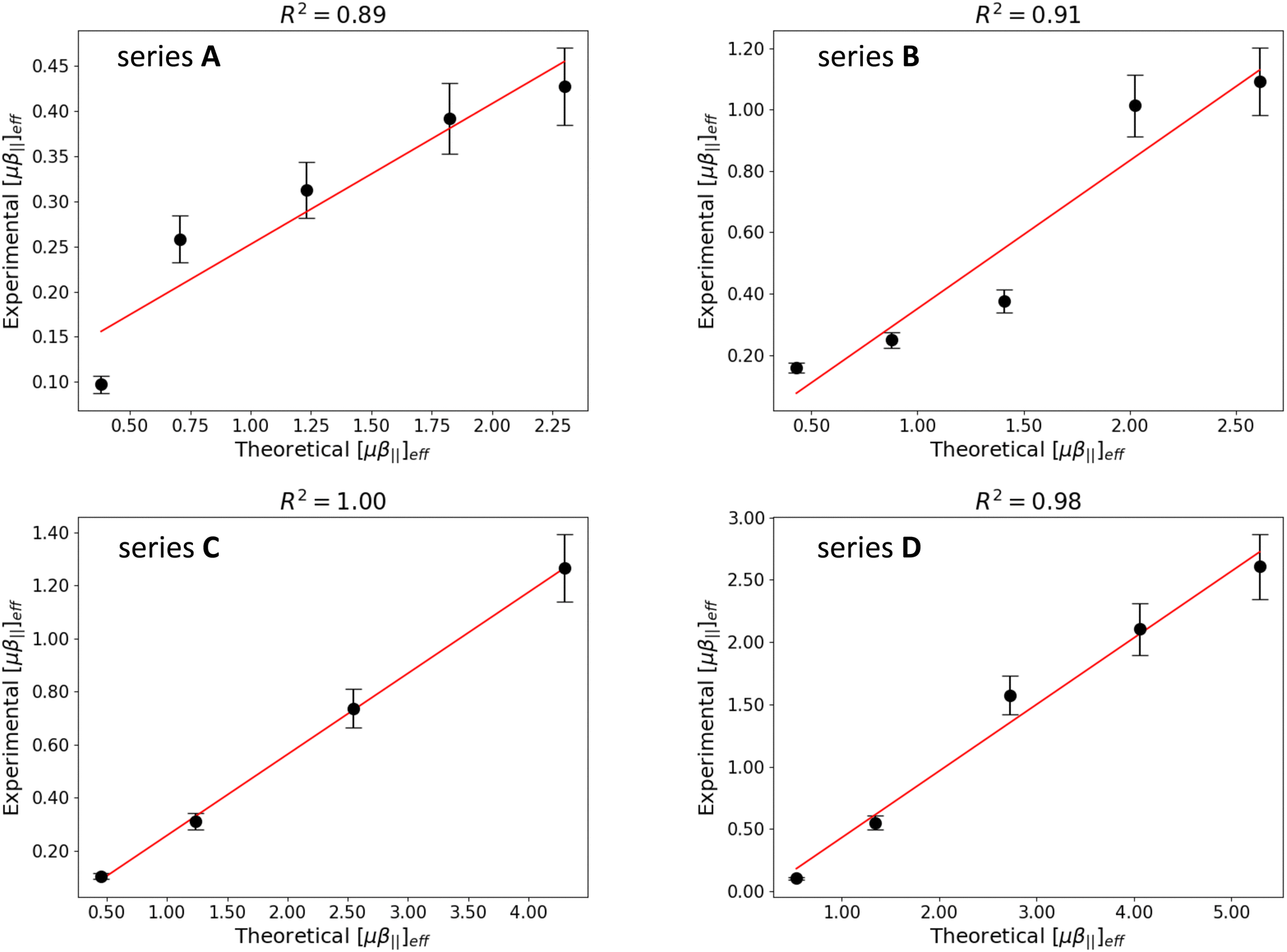
Although the computed [μβ‖]eff values overestimate the experimental ones by a factor 2–4, Fig. 8 reveals a very good agreement between theory and experiment. All series display Pearson correlation coefficients larger than 0.89 between calculated and measured [μβ‖]eff values, with R2 values very close to 1.0 for series C and D. However, the relative ordering of the [μβ‖]eff values in the different series is only partially reproduced: while DFT calculations predict that series A and B display the smallest NLO responses in agreement with experimental results, the relative magnitude of the EFISH signal is inverted for the longer derivatives in series C and D. The enhancement of the [μβ‖]eff values with the elongation of the polyenic chain is also underestimated in the two latter series. These remaining discrepancies between calculations and measurements may be ascribed to the use of the continuum solvation model, which do not describe explicitly the interactions between the ion pair and the chloroform solvent. They might also originate from local field effects, which make the field felt by the dye/iodide complex in the solution different from the external electrical field applied to the medium and used in the calculations.61
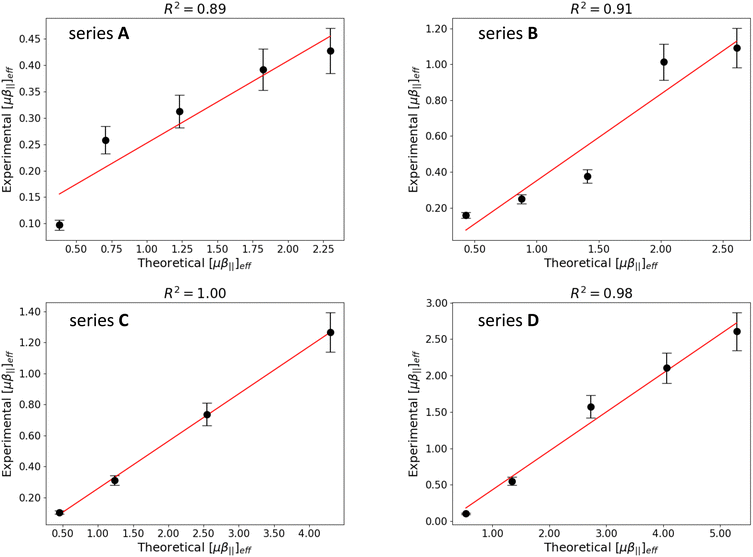 |
| | Fig. 8 Correlation between experimental and computational μβ‖(2ω) values (in 106 a.u.) for the four series of molecules. Errors bars on experimental data correspond to 10% of the measured value. | |
4. Conclusions
In this work, the NLO responses of four series of amphiphilic cationic chromophores have been investigated by means of EFISH experiments and quantum chemical calculations. EFISH measurements were made possible owing to the electric neutrality of the pairs formed, in chloroform, by the cationic dye and the iodine anion. The computational approach combining MD simulations and time-dependent DFT calculations performed on a representative dye/iodide pair allowed us to describe the geometrical fluctuations of the complex, and to highlight their impact on the NLO responses. These calculations confirmed that the iodide anion remains in the proximity of the dye all along the simulations, with an average position close to the charged heterocyclic acceptor. The position of the iodide relative to the dye was also shown to be at the origin of the relative magnitude of the second- and third-order contributions of the EFISH signal. In the investigated system, the third-order component was found negligible compared to the second-order one, supporting the experimental assumptions. Overall, the good agreement between experimental and theoretical characterizations demonstrates the reliability of the computational protocol, which provides a tractable strategy towards systematic and accurate in silico characterization of novel molecular probes for SHG imaging.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank J. Daniel for experimental support. C. N. acknowledges the University of Bordeaux Initiative of Excellence (IDEX) for his PhD grant. C. T. is supported by Donostia International Physics Center (DIPC) and Gipuzkoa Council Joint Program “Women and Science”. This work was supported by the Transnational Common Laboratory QuantumChemPhys (Theoretical Chemistry and Physics at the Quantum Scale, grant number ANR-10-IDEX-03-02) established between the Université de Bordeaux (UB), Euskal Herriko Unibertsitatea (UPV/EHU) and DIPC. It was also funded by the DIPC (DIPC_INV_003132) and the Basque Government/Eusko Jaurlaritza (GV/EJ) (grant number IT1584-22). Calculations were performed on the computing facilities provided by the DIPC, and the Mésocentre de Calcul Intensif Aquitain (MCIA) of the University of Bordeaux and of the Université de Pau et des Pays de lAdour. Technical and human support provided by IZO-SGI, SGIker (UPV/EHU, MICINN, GV/EJ, ERDF and ESF), and MCIA is also gratefully acknowledged.
Notes and references
- L. R. Dalton, P. A. Sullivan and D. H. Bale, Chem. Rev., 2010, 110, 25–55 CrossRef CAS PubMed.
- P. J. Campagnola, M. D. Wei and L. M. Loew, Biophys. J., 1999, 77, 3341–3349 CrossRef CAS PubMed.
- L. Moreaux, O. Sandre, S. Charpak, M. Blanchard-Desce and J. Mertz, Biophys. J., 2001, 80, 1568–1574 CrossRef CAS PubMed.
- D. A. Dombeck, M. Blanchard-Desce and W. W. Webb, J. Neurosci., 2004, 24, 999–1003 CrossRef CAS PubMed.
- J. E. Reeve, H. L. Anderson and K. Clays, Phys. Chem. Chem. Phys., 2010, 12, 13484–13498 RSC.
- T. Mizuguchi and M. Nuriya, Biophys. Rev., 2020, 12, 1321–1329 CrossRef CAS.
- V. Parodi, E. Jacchetti, R. Osellame, G. Cerullo, D. Polli and M. T. Raimondi, Front. Bioeng. Biotechnol., 2020, 8, 585363 CrossRef PubMed.
- G. Prévot, T. Bsaibess, J. Daniel, C. Genevois, G. Clermont, I. Sasaki, S. Marais, F. Couillaud, S. Crauste-Manciet and M. Blanchard-Desce, Nanoscale Adv., 2020, 2, 1590–1602 RSC.
- Y. de Coene, S. Jooken, O. Deschaume, V. Van Steenbergen, P. Vanden Berghe, C. Van den Haute, V. Baekelandt, G. Callewaert, S. Van Cleuvenbergen, T. Verbiest, C. Bartic and K. Clays, Small, 2022, 18, 2200205 CrossRef CAS.
- S. R. Marder, L.-T. Cheng, B. G. Tiemann, A. C. Friedli, M. Blanchard-Desce, J. W. Perry and J. Skindhøj, Science, 1994, 263, 511–514 CrossRef CAS.
- G. Bourhill, J.-L. Bredas, L.-T. Cheng, S. R. Marder, F. Meyers, J. W. Perry and B. G. Tiemann, J. Am. Chem. Soc., 1994, 116, 2619–2620 CrossRef CAS.
- V. Parthasarathy, R. Pandey, M. Stolte, S. Ghosh, F. Castet, F. Würthner, P. K. Das and M. Blanchard-Desce, Chem. – Eur. J., 2015, 21, 14211–14217 CrossRef CAS PubMed.
- V. Parthasarathy, R. Pandey, P. K. Das, F. Castet and M. Blanchard-Desce, Chem. Phys. Chem., 2018, 19, 187–197 CrossRef CAS PubMed.
- B. J. Coe, Chem. – Eur. J., 1999, 5, 2464–2471 CrossRef CAS.
- F. Castet, V. Rodriguez, J.-L. Pozzo, L. Ducasse, A. Plaquet and B. Champagne, Acc. Chem. Res., 2013, 46, 2656–2665 CrossRef CAS PubMed.
- P. Beaujean, F. Bondu, A. Plaquet, J. Garcia-Amorós, J. Cusido, F. M. Raymo, F. Castet, V. Rodriguez and B. Champagne, J. Am. Chem. Soc., 2016, 138, 5052–5062 CrossRef CAS PubMed.
- V. Alain, M. Blanchard-Desce, I. Ledoux-Rak and J. Zyss, Chem. Commun., 2000, 353–354 RSC.
- M. Jazbinsek, L. Mutter and P. Gunter, IEEE J. Sel. Top. Quantum Electron., 2008, 14, 1298–1311 CAS.
- S. Kannan, A. Sekar and K. Sivaperuman, J. Mater. Chem. C, 2020, 8, 16668–16690 RSC.
- J.-H. Jeong, J.-S. Kim, J. Campo, S.-H. Lee, W.-Y. Jeon, W. Wenseleers, M. Jazbinsek, H. Yun and O.-P. Kwon, Dyes Pigm., 2015, 113, 8–17 CrossRef CAS.
- F. Bureš, RSC Adv., 2014, 4, 58826–58851 RSC.
- F. Tessore, E. Cariati, F. Cariati, D. Roberto, R. Ugo, P. Mussini, C. Zuccaccia and A. Macchioni, Chem. Phys. Chem., 2010, 11, 495–507 CrossRef CAS.
- T. N. Ramos, S. Canuto and B. Champagne, J. Chem. Inf. Model., 2020, 60, 4817–4826 CrossRef CAS PubMed.
- T. N. Ramos, F. Castet and B. Champagne, J. Phys. Chem. B, 2021, 125, 3386–3397 CrossRef CAS.
- S. Achelle, S. Kahlal, A. Barsella, J.-Y. Saillard, X. Che, J. Vallet, F. Bureš, B. Caro and F. R. le Guen, Dyes Pigm., 2015, 113, 562–570 CrossRef CAS.
- R. J. Durand, S. Gauthier, S. Achelle, T. Groizard, S. Kahlal, J.-Y. Saillard, A. Barsella, N. Le Poul and F. R. Le Guen, Dalton Trans., 2018, 47, 3965–3975 RSC.
- F. Castet, C. Tonnelé, L. Muccioli and B. Champagne, Acc. Chem. Res., 2022, 55, 3716–3726 CrossRef CAS.
- K. Pielak, C. Tonnelé, L. Sanguinet, E. Cariati, S. Righetto, L. Muccioli, F. Castet and B. Champagne, J. Phys. Chem. C, 2018, 122, 26160–26168 CrossRef CAS.
- L. Lescos, P. Beaujean, C. Tonnelé, P. Aurel, M. Blanchard-Desce, V. Rodriguez, M. de Wergifosse, B. Champagne, L. Muccioli and F. Castet, Phys. Chem. Chem. Phys., 2021, 23, 23643–23654 RSC.
- C. Tonnelé, K. Pielak, J. Deviers, L. Muccioli, B. Champagne and F. Castet, Phys. Chem. Chem. Phys., 2018, 20, 21590–21597 RSC.
- C. Tonnelé, B. Champagne, L. Muccioli and F. Castet, Chem. Mater., 2019, 31, 6759–6769 CrossRef.
- C. Bouquiaux, C. Tonnelé, F. Castet and B. Champagne, J. Phys. Chem. B, 2020, 124, 2101–2109 CrossRef CAS PubMed.
- C. Bouquiaux, F. Castet and B. Champagne, J. Phys. Chem. B, 2021, 125, 10195–10212 CrossRef CAS PubMed.
- C. Bouquiaux, F. Castet and B. Champagne, J. Phys. Chem. B, 2023, 127, 528–541 CrossRef CAS PubMed.
- B. F. Levine and C. G. Bethea, J. Chem. Phys., 1975, 63, 2666–2682 CrossRef CAS.
- I. Ledoux and J. Zyss, Chem. Phys., 1982, 73, 203–213 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS.
- A. Pizzirusso, M. Savini, L. Muccioli and C. Zannoni, J. Mater. Chem., 2011, 21, 125–133 RSC.
- J. C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kalé and K. Schulten, J. Comput. Chem., 2005, 26, 1781–1802 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- S. J. A. van Gisbergen, J. G. Snijders and E. J. Baerends, J. Chem. Phys., 1998, 109, 10644–10656 CrossRef CAS.
- T. Helgaker, S. Coriani, P. Jørgensen, K. Kristensen, J. Olsen and K. Ruud, Chem. Rev., 2012, 112, 543–631 CrossRef CAS.
- L. E. Johnson, L. R. Dalton and B. H. Robinson, Acc. Chem. Res., 2014, 47, 3258–3265 CrossRef CAS PubMed.
- L. Lescos, S. Sitkiewicz, P. Beaujean, M. Blanchard-Desce, B. R. Champagne, E. Matito and F. Castet, Phys. Chem. Chem. Phys., 2020, 22, 16579–16594 RSC.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Revision C.01, Gaussian Inc., Wallingford CT, 2016 Search PubMed.
- Chemcraft-Graphical Software for Visualization of Quantum Chemistry Computations, https://www.chemcraftprog.com.
-
R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, Oxford, 1990 Search PubMed.
-
T. A. Keith, AIMAll (Version 14.11.23), TK Gristmill Software, Overland Park KS, USA, 2014 (http://aim.tkgristmill.com).
-
E. Matito, Electron Sharing Indices Program for Molecular Space Partitioning, 2006–2023, Institute of Computational Chemistry and Catalysis (Girona Univ.) and Donostia International Physics Center (DIPC).
- E. Matito, M. Duran and M. Solà, J. Chem. Phys., 2005, 122, 014109 CrossRef.
- E. Matito, M. Solà, P. Salvador and M. Duran, Faraday Discuss., 2007, 135, 325–345 RSC.
- E. Matito, Phys. Chem. Chem. Phys., 2016, 18, 11839–11846 RSC.
- C. Garca-Fernández, E. Sierda, M. Abadia, B. E. C. Bugenhagen, M. H. Prosenc, R. Wiesendanger, M. Bazarnik, J. E. Ortega, J. Brede, E. Matito and A. Arnau, J. Phys. Chem. C, 2017, 121, 27118–27125 CrossRef.
- I. Casademont-Reig, E. Ramos-Cordoba, M. Torrent-Sucarrat and E. Matito, Molecules, 2020, 25, 711 CrossRef CAS.
- J. Kruszewski and T. M. Krygowski, Tetrahedron Lett., 1972, 13, 3839–3842 CrossRef.
- M. Giambiagi, M. S. de Giambiagi, C. D. dos Santos Silva and A. P. de Figueiredo, Phys. Chem. Chem. Phys., 2000, 2, 3381–3392 RSC.
- P. Bultinck, R. Ponec and S. Van Damme, J. Phys. Org. Chem., 2005, 18, 706–718 CrossRef CAS.
- M. Eskandari, J. C. Roldao, J. Cerezo, B. Milián-Medina and J. Gierschner, J. Am. Chem. Soc., 2020, 142, 2835–2843 CrossRef CAS PubMed.
- I. Brandão, T. L. Fonseca, L. R. Franco, H. C. Georg and M. A. Castro, Chem. Phys. Lett., 2022, 796, 139549 CrossRef.
- F. Feixas, E. Matito, J. Poater and M. Solà, J. Comput. Chem., 2008, 29, 1543–1554 CrossRef CAS PubMed.
- R. Wortmann and D. M. Bishop, J. Chem. Phys., 1998, 108, 1001–1007 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details on the force field parameterization for the D3 dye, results from MD simulations, assessment of the DFT approximation for computing NLO properties, and correlation between hyperpolarizabilities and aromaticity indices. See DOI: https://doi.org/10.1039/d3cp00750b |
|
| This journal is © the Owner Societies 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Raphaël
Vangheluwe
a,
Isabelle
Ledoux-Rak
c,
Benoît
Champagne
ab,
Raphaël
Vangheluwe
a,
Isabelle
Ledoux-Rak
c,
Benoît
Champagne