
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Time-resolved detection of light-induced conformational changes of heliorhodopsin†
Yusuke
Nakasone
 a,
Yuma
Kawasaki
b,
Masae
Konno
bc,
Keiichi
Inoue
b and
Masahide
Terazima
*a
a,
Yuma
Kawasaki
b,
Masae
Konno
bc,
Keiichi
Inoue
b and
Masahide
Terazima
*a
aDepartment of Chemistry, Graduate School of Science, Kyoto University, Kyoto, Japan. E-mail: mterazima@kuchem.kyoto-u.ac.jp
bThe Institute for Solid State Physics, The University of Tokyo, Kashiwa, Chiba, Japan
cPRESTO, Japan Science and Technology Agency, Kawaguchi, Saitama, Japan
First published on 19th April 2023
Abstract
Heliorhodopsins (HeRs) are a new category of rhodopsins. They exist as a dimer and exhibit a characteristic inverted topology. HeRs bind all-trans-retinal as a chromophore in the dark, and its isomerization to the 13-cis form by light illumination leads to a photocyclic reaction involving several photo-intermediates: K, L, M, and O. In this study, the kinetics of conformational changes of HeR from Thermoplasmatales archaeon SG8-52-1 (TaHeR) were studied by the transient grating (TG) and circular dichroism (CD) methods. The TG method reveals that the diffusion coefficient (D) does not change until the O formation suggesting no significant conformation change at the surface of the protein during the early steps of the reaction. Subsequently, D decreases upon the O formation. Although two time constants (202 μs and 2.6 ms) are observed for the conversion from the M to O by the absorption detection, D decreases only at the first step (202 μs). Light-induced unfolding of helical structure is detected by the CD method. To examine the contribution of a characteristic helix in the intracellular loop 1 (ICL1 helix), Tyr93 on the ICL1 helix was replaced by Gly (Y93G), and the reaction of this mutant was also investigated. It was found that this replacement partially suppresses the D-change, although the CD-change is almost the same as that of the wild type. These results are interpreted in terms of different sensitivities of TG and CD methods, that is, D is sensitive to the structure of the solvent-exposed surface and selectively observes the conformational change in the ICL1 region. It is suggested that the structure of hydrophilic residues in the ICL1 helix is changed during this process.
Introduction
Rhodopsin is a ubiquitous family of photoreceptive membrane proteins. Two distinctive types of rhodopsins, microbial (type 1) and animal (type 2) rhodopsins, are known.1–3 Both microbial and animal rhodopsins consist of seven transmembrane helical architecture (TM1–7), in which a retinylidene Schiff base (retinal chromophore) is covalently bound to a lysine residue in TM7. Microbial rhodopsins exhibit a wide variety of functions, light-driven ion pumps, light-gated ion channels, light-dependent enzymes, and so on,4,5 whereas most animal rhodopsins activate heterotrimeric G proteins in a light-dependent manner as a sub-group of the G protein-coupled receptor (GPCR) superfamily.6In 2018, the third class of rhodopsin, heliorhodopsin (HeR), was discovered by the functional metagenome analysis.7 Although the biological function of most HeRs is not known except for a few members,8–10 they exhibit a characteristic inverted orientation relative to the canonical microbial and animal rhodopsins with the N- and C-termini facing the intracellular and extracellular sides, respectively.7,11 HeR 48C12 and HeR from Thermoplasmatales archaeon SG8-52-1 (TaHeR) were revealed to form dimers by X-ray crystallography in which protomers are bridged to each other by their long extracellular loops between TM1 and 2 (Fig. 1(a)).12–14
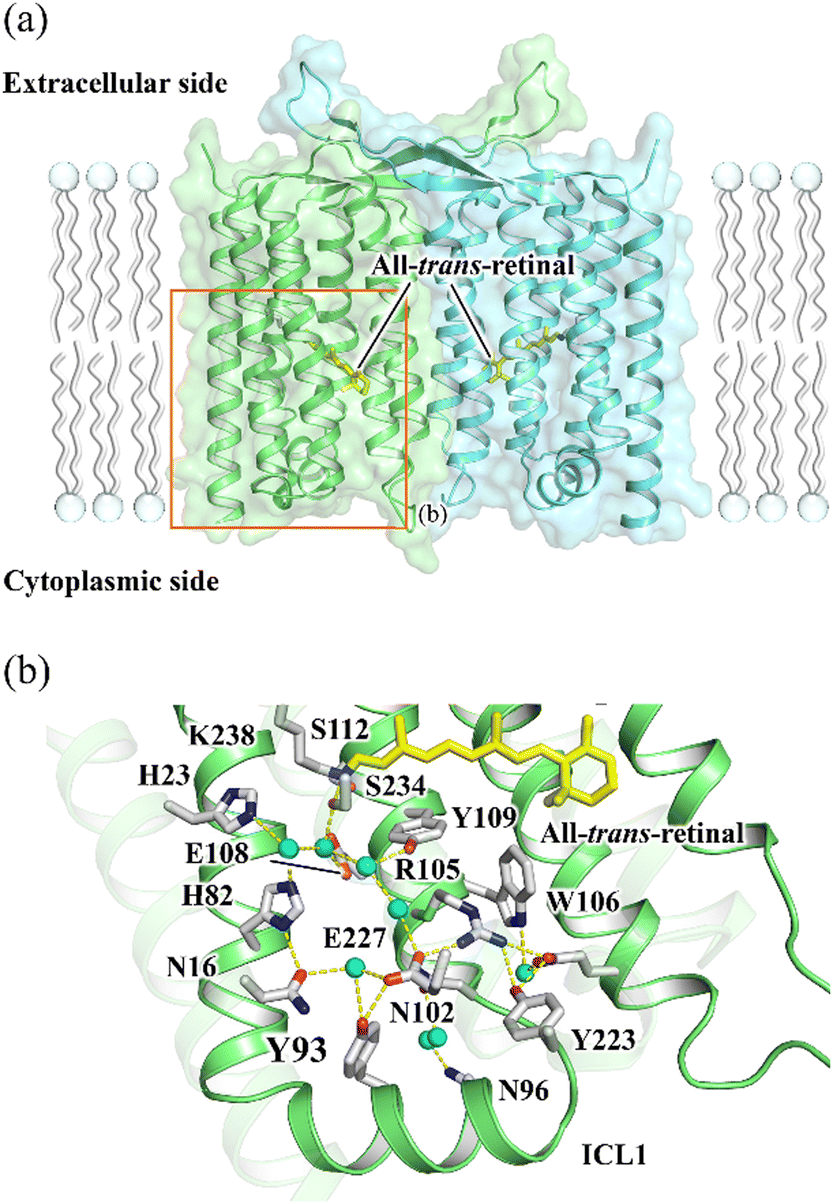 | ||
| Fig. 1 (a) The X-ray crystallographic structure of TaHeR dimer (PDB ID: 6IS6).11 The area shown in b is indicated by an orange rectangle. (b) The extracellular hydrogen bonding network of TaHeR connecting the retinal Schiff base (RSB) region and ICL1. | ||
HeRs bind an all-trans-retinal chromophore in the dark, and it isomerized to the 13-cis form upon light illumination leading to a photocyclic reaction involving several photo-intermediates: K, L, M, and O. The K and O intermediates exhibit red-shifted visible absorption spectra compared to the dark state, while the absorption spectrum of the M intermediate, in which the Schiff-base linkage of the retinal chromophore is deprotonated, is highly blue-shifted. The turnover rate of HeRs is relatively longer (0.6–11 s) compared with most abundant H+-pumping type 1 rhodopsins. Because the presence of the long-lived photo-intermediate without ion transport is a common feature of sensory and enzyme rhodopsins,15–18 HeR is thought to be involved in unidentified signal transduction or cellular metabolic process.7,12,18 In addition, the putative function of HeR transporting amphiphilic molecules was suggested based on the absence of HeRs in diderm (Gram-negative) bacteria.19 A study using the HPLC analysis and the Fourier transform infrared (FTIR) spectroscopy revealed a large change in the backbone of HeR in the last O intermediate having 13-cis-retinal chromophore.7 Although this conformational change may represent the formation of functionally active state, the reaction kinetics of the change have not been elucidated.
The transient grating (TG) method measures a diffraction of a probe beam (TG signal) by a periodic refractive index change in a protein solution associated with the photoreaction in the time domain.20–22 The decay rate of the TG signal due to chemical species reflects the translational diffusion coefficient (D) of a protein, and time-dependent D can be determined by changing the grating wavenumber (q). If there is a structural change affecting the interaction between the protein and solvent, D of the product can be different from that of the reactant and even can alter over time during the photoreaction. Here, to reveal the dynamics of the formation of the active state of HeR, we studied the global conformational change of TaHeR by monitoring D-change dynamics in the time domain using the TG method. In particular, the contribution of a characteristic helix for HeR (ICL1 helix in Fig. 1(b)) was investigated using a mutant of Y93G.
Experimental
Cloning of TaHeR wild type (WT) and Y93G
The gene encoding TaHeR with codons optimized for Escherichia coli expression was synthesized (Genscript, NJ, USA) and cloned into NdeI–XhoI site of pET21a (+) vector (Novagen, Merck KGaA, Germany). The plasmid was transformed into E. coli C43 (DE3) strain (Lucigen, WI, USA). For mutagenesis to construct TaHeR Y93G, the QuikChange site-directed mutagenesis method (Agilent Technologies, CA, USA) was used according to a standard protocol. The sequences of the primers used in mutagenesis are listed in Table S1 (ESI†).Purification of TaHeR WT and Y93G
E. coli cells harboring the TaHeR WT or Y93G-cloned plasmids were cultured in 2× YT medium containing 50 μg mL−1 ampicillin. The expression of C-terminal 6× His-tagged proteins was induced by 0.1 mM isopropyl-β-D-thiogalactopyranoside (IPTG) in the presence of 10 μM all-trans-retinal (Toronto Research Chemicals, Canada) for 4.5 h at 37 °C. The harvested cells were sonicated (Ultrasonic Homogeniser VP-300N; TAITEC, Japan) for disruption in a buffer containing 50 mM Tris–HCl (pH 8.0) and 5 mM MgCl2. The membrane fraction was collected by ultracentrifugation (CP80NX; Eppendorf Himac Technologies, Japan) at 142![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g for 1 h. The proteins were solubilized in a buffer containing 50 mM MES–NaOH (pH 6.5), 300 mM NaCl, 5 mM imidazole, 5 mM MgCl2, and 1% n-dodecyl-β-D-maltopyranoside (DDM). The solubilized proteins were separated from the insoluble fractions by ultracentrifugation at 142000 × g for 1 h. The proteins were purified using a Co-NTA affinity column (HiTrap TALON® crude; Cytiva, MA, USA). The resin was washed with buffer containing 50 mM MES–NaOH (pH 6.5), 300 mM NaCl, 50 mM imidazole, 5 mM MgCl2, and 0.1% DDM. The proteins were eluted in a buffer containing 50 mM MES–NaOH (pH 6.0), 300 mM NaCl, 500 mM imidazole, 5 mM MgCl2, and 0.1% DDM. The eluted proteins were dialyzed in a buffer containing 20 mM Tris–HCl (pH 8.0), 100 mM NaCl, 0.05% DDM to remove imidazole.
000 × g for 1 h. The proteins were solubilized in a buffer containing 50 mM MES–NaOH (pH 6.5), 300 mM NaCl, 5 mM imidazole, 5 mM MgCl2, and 1% n-dodecyl-β-D-maltopyranoside (DDM). The solubilized proteins were separated from the insoluble fractions by ultracentrifugation at 142000 × g for 1 h. The proteins were purified using a Co-NTA affinity column (HiTrap TALON® crude; Cytiva, MA, USA). The resin was washed with buffer containing 50 mM MES–NaOH (pH 6.5), 300 mM NaCl, 50 mM imidazole, 5 mM MgCl2, and 0.1% DDM. The proteins were eluted in a buffer containing 50 mM MES–NaOH (pH 6.0), 300 mM NaCl, 500 mM imidazole, 5 mM MgCl2, and 0.1% DDM. The eluted proteins were dialyzed in a buffer containing 20 mM Tris–HCl (pH 8.0), 100 mM NaCl, 0.05% DDM to remove imidazole.
Transient absorption measurement
The detail of transient absorption measurement was previously reported.20TaHeR WT and Y93G were solubilized in 20 mM Tris–HCl (pH 8.0), 100 mM NaCl, 0.05% DDM. The absorption of the protein solution was adjusted to O.D.λmax = 0.5 (protein concentration = 0.25 mg mL−1). The sample was illuminated with a beam of second harmonics of a nanosecond-pulsed Nd:YAG laser (λ = 532 nm, 5.7 mJ cm−2, 0.06–0.1 Hz) (INDI40, Spectra-Physics, CA, USA). Light from a Xe arc lamp (L9289-01, Hamamatsu Photonics, Japan) monochromated by a monochrometer (S-10, SOMA OPTICS, Japan) was used for probe light. The time evolution of the transient absorption change was monitored by a photomultiplier tube (R10699, Hamamatsu Photonics, Japan) equipped with a notch filter (532 nm, bandwidth = 17 nm) (Semrock, NY, USA) to remove scattered excitation light. To increase the signal-to-noise (S/N) ratio, 3–20 signals were averaged. The signals were global-fitted with a multi-exponential function to determine the lifetimes of photo-intermediates.Transient grating measurement
The experimental setup for the TG measurement was similar to that reported previously.20–22 A laser pulse from the second harmonic of an Nd:YAG laser (GCR-170-10, Spectra-Physics, CA, USA) was used as an excitation beam. A diode laser (785 nm, L785P090, Thorlabs, NJ, USA) was used as the probe beam. The TG signal was detected by a photomultiplier tube (R1477, Hamamatsu Photonics, Japan) and recorded with a digital oscilloscope (DSOS054A, Agilent technologies, CA, USA). The q-value was determined from the decay rate of the thermal grating signal of a calorimetric reference sample, bromocresol purple in aqueous solution. More than 50 signals were averaged to improve the S/N ratio. The repetition rate of the excitation pulse was set to be 0.02 Hz to avoid photoexcitation of the photo-product. All measurements were carried out at 23 °C.CD spectroscopy
Circular dichroism (CD) spectra were recorded with a spectropolarimeter (J-720WS, JASCO, Japan) with flowing N2 gas. The background signal from the spectrum of the buffer solution was subtracted from all other measurements. An optical path length of a sample cell was 1.0 cm and the protein concentration was 0.5 μM. It took about 30 s for recording one CD spectrum. To improve the signal-to-noise ratio (S/N), 20 spectra were averaged. To measure the CD spectrum of the light-adapted state, blue light from a LED (480 nm) was illuminated during the CD measurement. Since the detector used for the CD measurement was sensitive to light only in far UV region, blue light illumination did not disturb the measurement. The CD measurements were performed at 4 °C to slow down the thermal recovery process for efficient accumulation of the light-adapted state. In the case of Y93G mutant, the CD spectrum of the light-adapted state could not be recorded due to light-induced denaturation. However, probing thermal recovery curve after blue light irradiation of a short period (6 s) by a CD signal was possible at one probe wavelength. The sample solution was exchanged to fresh one to minimize possible photodamage. The thermal recovery measurements were performed at 23 °C and over the time-profiles of 10 signals were averaged to improve the S/N ratio.Results
Transient absorption measurement of TaHeR
To determine the kinetics of the photocycle of TaHeR in DDM by absorption method, transient absorption signals were measured upon pulsed photoexcitation (Fig. S1(a), ESI†). We observed four photo intermediates, K, L, M, and O, which are similar to those observed in lipids, by probing at 409, 549, and 605 nm.11 All traces are reproduced by the global fitting with a five-exponential function leading the photocycle model (Fig. S1(a), right, ESI†). Although the recovery of the initial state from the O intermediate was slightly slower than that of the protein in lipids (23.6 ± 0.8 s vs. 11.1 ± 0.1 s8), the overall photocycle of TaHeR WT in DDM is the same as that in lipids.TG measurement of TaHeR
The TG signal of TaHeR (6 μM) was measured at q2 = 1.1 × 1011 m−2 and shown in Fig. 2(a). The signal rose quickly within the response time of our system and showed a rise-decay signal in the time range of 10 μs to 500 μs. Subsequently, the signal exhibited a rise-decay profile after once reaching the baseline. The initial rise and the subsequent decay components (<1 ms of Fig. 2a) are expressed by a tri-exponential function with rate constants of k1, k2 (k1 > k2), and Dthq2,| ITG(t) = α{δn1exp(−k1t) + δn2exp(−k2t) + δnthexp(−Dthq2t) + δnspe(t)}2 | (1) |
 | ||
| Fig. 2 (a) The TG signal of TaHeR (6 μM) obtained at q2 = 1.1 × 1011 m−2. The black broken line is the best-fitted curve based on eqn (1) and (2). (b) Granting wavenumber dependence on the molecular diffusion signal of TaHeR. q2 are 180, 85, 23, 4.4, 1.1, 0.29 × 1011 m−2 from left to right. The black broken lines are the best-fitted curves based on eqn (3). | ||
Since the time ranges of the rise-decay signals observed on a slower time scale (1 ms–1 s) are dependent on q2 (Fig. 2(b)), the signal is attributed to the molecular diffusion (diffusion signal). As described previously,20–22 if D is a constant within the observation time range, the diffusion signal should be expressed by the following bi-exponential function:
| δnspe(t) = δnPexp(−DPq2t) − δnRexp(−DRq2t) | (2) |
In general, D depends on the molecular size. The above observed D is close to those of proteins such as lactoperoxidase (6.0 × 10−11 m2 s−1, 93 kDa)23 and aldolase (4.3 × 10−11 m2 s−1, 158 kDa).24TaHeR is reported to exist as a dimer, and its molecular mass (60 kDa as a dimer) is smaller than those proteins. However, since TaHeR is solubilized by DDM, the bound DDM must be included in the molecular size. According to a previous study using several types of chromatography, it has been calculated that about 100–200 surfactants are bound around one rhodopsin molecule.25 Using these numbers with the molecular mass of DDM (510 Da), the molecular mass of the protein–detergent complex is calculated to be around 160–260 kDa. Indeed, the molecular mass of the protein-detergent complex has been previously estimated to be 193 kDa by size exclusion chromatography combined with multi-angle laser light scattering (SEC-MALLS) for TaHeR,11 which is in the range of the above estimation. Hence, D from the diffusion signal is reasonable for the molecular size, suggesting that the reactant is the dimer.
When the concentration of TaHeR is increased from 6 μM to 500 μM, the shape of the diffusion signal is significantly altered (Fig. S2(a), ESI†). This concentration dependence suggests that TaHeR forms larger oligomers at high concentrations, and the oligomers also undergo a reaction which is different from that of the dimer. We speculate that the concentration of TaHeR in living cells would be lower than 10 μM. Hence, the higher oligomers might be non-physiological forms. Since the signal does not depend on the concentration below 12 μM, the contribution of the higher oligomers should be negligible at <12 μM. We do not discuss further the reaction observed at high concentrations and we performed all TG experiments at 6 μM.
The diffusion signals normalized by the signal intensity due to the absorption changes of the L and M states were sensitively dependent on q2 (Fig. 2(b)). The peak intensity was weak in a fast time range (large q2) and became stronger in the slower time range. This q2 dependence indicates that D of the photoexcited protein gradually changed in the observed time range. The time development became negligible after 10 ms, indicating that the D-change was almost completed within 10 ms. According to the transient absorption measurement, there are two steps in the time range of the diffusion signal appears (100 μs–1 s). Hence, the time development of the diffusion signal was analyzed based on the following reaction scheme:
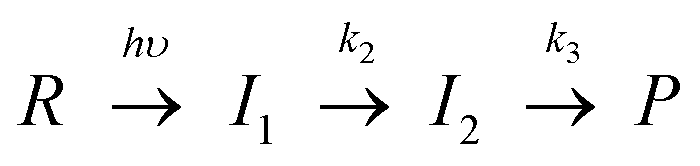
| δnspe(t) = −δnRexp(−DRq2t) + [δnI1 − δnI2{k2/(k2 − k3)} + δnP{k2k3/(k2 − k3)}{1/(DI1 − DP)q2 + k2}]exp{(−DI1q2 + k2)t} + [δnI2{k2/(k2 − k3)} − δnP{k2k3/(k2 − k3)}{1/(DI2 − DP)q2 + k3}]exp{(−DI2q2 + k3)t} + δnP{k2k3/(k2 − k3)}[{1/(DI2 − DP)q2 + k3} − {1/(DI1 − DP)q2 + k2}]exp(−DPq2t) | (3) |
| K/L → M (μs) | M → M′/O (μs) | M′/O → O′ (ms) | Thermal recovery (s) | Diffusion coefficient (10−11 m2 s−1) | Amplitude of CD change (103 deg cm2 dmol−1) | ||||
|---|---|---|---|---|---|---|---|---|---|
| Reactant | I 1 | I 2 | Product | ||||||
| WT | 7.5 ± 0.1 | 202 ± 1 | 2.6 ± 0.1 | 23.6 ± 0.2 | 5.4 ± 0.2 | 5.4 ± 0.3 | 4.5 ± 0.3 | 4.5 ± 0.2 | 8.5 ± 0.6 |
| Y93G | 6.2 ± 0.3 | 167 ± 4 | 3.0 ± 0.3 | 10.7 ± 0.1 | 5.2 ± 0.2 | 5.2 ± 0.3 | 4.7 ± 0.3 | 4.6 ± 0.3 | 8.3 ± 0.8 |
Since TaHeR exists as a dimer, the photoexcitation of one or two protomers in the dimer may induce different conformational changes. This possibility was investigated by changing the excitation light intensity. If two protomers can be excited by strong pulsed light and the conformation change is different from that upon one protomer excitation, the diffusion signal should be light intensity dependent. However, the observed time-profile of the TG signal was almost light-intensity independent (SI-3, ESI†). This result might be explained by assuming that the conformation change does not depend on the number of photoexcited protomers in the dimer. However, we found that this is not the case, because the relative diffusion signal intensity against the signal intensity before the diffusion signal, which represents amount of the reaction intermediates, are almost light intensity independent (Fig. S3(c), ESI†). This light intensity independence indicates that we cannot photoconvert two protomers even at the strongest light intensity we used. Possible reasons of the negligible photoconversion of two protomers in the dimer is discussed in SI-3 (ESI†).
Nevertheless, D change was observed for the dimer having one protomer in the light state, indicating that a conformational change of the protein moiety occurs when one protomer in the dimer enters the photocycle. We consider that this situation, photoconversion of one protomer, is important in physiological environments, because the continuous sun-light photons will be absorbed by the long-lived O intermediate, and the 13-cis-retinal in the O intermediate of some rhodopsins is known to be converted back to the all-trans form by absorbing visible light.27 This behaviour was also confirmed for TaHeR; i.e., the O decay is accelerated by increasing the probe light intensity (SI-4, Fig. S4, ESI†). Hence, the activated state is not fully accumulated even under steady-state light illumination conditions in nature. Therefore, dimers containing one dark- and one light-adapted protomers are likely to be functional under physiological conditions.
To investigate the structural change further, we measured the CD spectra in the dark- and light-adapted states (Fig. 3(a)). Decrease in the CD intensity was observed upon light irradiation. The difference spectrum is also shown in Fig. 3(a). Although the difference is rather small, the difference spectrum seems to resemble a typical spectrum of the helix.28 This observation suggests unfolding of α-helices upon photoexcitation. Fig. 3(b) shows the time course of the CD intensity at 222 nm during the thermal recovery process. A refolding process was observed as an increase in the negative CD intensity, and it was reproduced by a single exponential function. The rate of the recovery of the CD intensity (23 s) was the same as that of absorption change in the visible region (Fig. S1, ESI†), representing that recovery of the helical structure was synchronized with the recovery of the absorption change of the retinal chromophore. This confirms that the observed CD change is related to the photocycle of TaHeR.
 | ||
| Fig. 3 (a) CD spectra of TaHeR (0.5 μM) at the dark- (blue) and light-adapted (red) states. The light-minus-dark difference spectrum and its magnified (×10) spectrum are shown in a black solid and black broken lines, respectively. (b) A thermal recovery of CD intensity monitored at 222 nm. The black broken line is the best-fitted curve based on a single exponential function. | ||
Photoreaction of Y93G mutant of TaHeR
The decrease in D by the photoreaction is a characteristic feature. Previous studies have suggested that one molecular origin of this decrease is the unfolding of α-helices, which has been demonstrated in poly-L-glutamic acid and several light-sensor proteins such as phototropin.22,29 The friction for the translational diffusion increases by the increase in the intermolecular interaction between the protein and water. On the basis of this mechanism, we consider that the structural changes in the transmembrane regions may not cause the observed D-changes, because they are highly hydrophobic and covered by detergent (DDM). Therefore, we consider that the unfolding reaction in the helical region on the hydrophilic surface is a cause of the D-change. According to the crystal structure,11 a characteristic helix exists in the solvent-exposed region (ICL1 helix in Fig. 1(b)). This helix does not exist in other rhodopsins and is unique to HeRs.11,12 Furthermore, the light-induced conformational change on the cytoplasmic side is relevant for the function of rhodopsins in many cases.9,10,30 Hence, we speculate that this helix is involved in the photoreaction of HeR. For examining conformation change in this region, we prepared a mutant TaHeR Y93G and investigated the reaction, because Y93 locates in the ICL1 helix to be a part of a hydrogen bonding network that extending from the retinal (Fig. 1(a)), and it could be a key residue in signal transduction from the chromophore to the ICL1 helix.The absorption spectra in the dark and light states are very similar to those of WT (Fig. S1(b), inset, ESI†), and the rates of M and O formations are close to those of WT. However, the transient absorption signal is slightly different (Fig. S1(b), ESI†); in particular, the amplitude of the absorption change due to the formation of the M intermediate is smaller than that of WT. The recovery rate of the initial state (10.7 ± 0.1 s) was approximately 2-fold faster compared to WT, indicating Y93 regulates the turnover rate of the photocycle of TaHeR. Even after the full-decay of the O intermediate, a weak bleach signal remained at 549 nm, suggesting a small proportion of the excited proteins, which was estimated to be ∼4% from its intensity relative to the full-bleach signal, was denatured.
The TG signal of TaHeR Y93G (6 μM) was measured at q2 = 1.1 × 1011 m−2 (Fig. 4(a)). Similar to the case of WT, the concentration-dependent analysis confirmed that Y93G exists as a dimer at 6 μM (Fig. S2(b), ESI†). The TG signal of WT measured under the identical experimental conditions (excitation pulse energy, protein concentration, experimental setup) is also shown in Fig. 4(a). The time-profile of the TG signal of Y93G is similar to that of WT and is reproduced well by the same analytical equations (eqn (1) and (2)). The TG signal on a fast time scale (10 μs–500 μs) reflects changes in absorption due to the decays of the L and M intermediates. The rate constants are in good agreement with those obtained for transient absorption signal (Fig. S1(b), ESI†), and their amplitudes are smaller than those of WT, which also corresponds well with the transient absorption measurements. The signal observed at a slower time scale (10 ms–1 s) is attributed to be a molecular diffusion signal, since the time scale varies with q2 (Fig. S5, ESI†). Interestingly, although the signal intensity before the diffusion signal (1–10 ms in Fig. 4(a)), which represents the difference in absorption spectrum between the ground state and the O intermediate, is almost identical to that of WT, the diffusion signal intensity was weaker than that of WT. This difference suggests that the structural change observed as the D-change is partially suppressed by the mutation. Global analysis of the diffusion signals using the same analytical model (eqn (3)) reproduced the signal well, and the determined parameters are summarized in Table 1. Similar to WT, the D change mainly occurs at the k2 step (167 μs), but DP is slightly larger in the mutant, representing that the D-change is decreased by the mutation. Quantitatively, the change in the friction (Δf) was calculated by Δf = kBT(1/DP − 1/DR), where kB is the Boltzmann constant and T is the temperature, and the enhanced frictions were determined to be 1.5 × 10−11 kg s−1 for WT and 1.0 × 10−11 kg s−1 for Y93G. The smaller friction change suggests that the conformational change is decreased in the Y93G mutant. Hence, we consider that Y93 is involved in the conformational change and that movement in the ICL1 region contributes to the D change.
 | ||
| Fig. 4 (a) The TG signal of Y93G mutant (green) and WT (red) of TaHeR obtained at 6 μM and at q2 = 1.1 × 1011 m−2. The black broken lines are the best-fitted curves based on eqn (1) and (2). (b) CD spectra of Y93G (green) and WT (red) of TaHeR (0.5 μM) at the dark state. (c) A thermal recovery of CD intensity of Y93G monitored at 222 nm. The black broken line is the best-fitted curve based on a single exponential function. | ||
Fig. 4(b) shows the CD spectra of WT and Y93G mutant in the dark. Fig. 4(c) shows the thermal recovery of CD intensity at 222 nm. The recovery was observed as the case of WT and the rate constant of the CD change (11 s) was again the same as that of the recovery of the absorption spectrum (Fig. S1(b), ESI†). Interestingly, the amplitude of the CD intensity change was almost identical to that of WT. Since the CD spectrum of Y93G in the light-adapted state could not be measured due to light induced degradation, we could not conclude if the light induced CD spectrum change of Y93G is the same as that of WT. However, if the spectrum is the same, this observation indicates that the light-induced unfolding of the helical structure is not affected by the Y93G mutation. This point is discussed below.
Discussion
In this study, we observed two features on the conformation changes: D-change and CD-change. The CD measurements showed a decrease in the helical structure upon light irradiation, and the intensity does not change by the Y93G mutation, whereas the D-change is sensitive to the mutation. This observation may indicate that the conformation changes detected by D and CD are different. Since Y93 locates on the ICL1 helix, the conformation change detected by D-change should represent the change in the ICL1 helix. However, since this helix is short (ca. 5% among the total helices of TaHeR), this change might be difficult to be detected by CD. The secondary structure change detected by CD should come from the conformation changes in other parts of this protein. On the other hand, D is dependent on the nature of surface-exposed residues, e.g., hydrophobicity. Therefore, if the mutation alters surface properties such as a change in the number of hydrophilic residues exposed to the solvent, D-change is observed. Since the ICL1 helix (L89YYRYVQNLKN99) indeed contains both hydrophilic and hydrophobic residues, and the binding site of ICL1 helix also contains hydrophilic residues such as N16 and N102, the solvent exposed residues might be changed by the mutation as well as the light illumination, which is observed as changes in D. However, this movement could be CD spectrally silent.The TG method revealed the decrease in D at the formation of the O intermediate. During the conversion from M to O, two time constants (202 μs and 2.6 ms) are observed and the D change occurs at the first step (202 μs). The two-step reaction between specific intermediates is commonly observed for other rhodopsins.11,31 In the M intermediate, the pKa of the retinal Schiff base (RSB) (pKa,RSB) is smaller than that of the proton accepting group (PAG) (pKa,PAG), so that H+ exists at PAG.7,11 During the M to O process, pKa's change to pKa,RSB > pKa,PAG to exhibit the spectral red shift. This pKa shift is caused by a reorganization of hydrogen-bonding network on the hydrophilic intracellular side. We consider that this reorganization induces structural change around ICL1 to change D. At the second step, the difference between pKa,RSB and pKa,PAG becomes larger by changes in the hydrogen bonding network inside the protein, but this change does not affect the conformation of ICL1 so that D does not change.
In this study, we analysed the reaction from M to O based on sequential reaction scheme and the TG signals were reproduced well. However, even using a parallel reaction scheme suggesting structural heterogeneity, a similar result of the analysis was obtained. Hence, we could not distinguish which scheme is appropriate. Nonetheless, it is interesting to note that, although the absorption spectrum change is the same for the first and second steps, the difference in the conformation change is apparent as the D-change.
It should be noted that Y93G mutation accelerates the thermal recovery, suggesting that Y93 contributes to the stabilization of the active state. Since Y93 is involved in the hydrogen bonding network from the retinal, the replacement may perturb this network and destabilize the active form. The long-lived active state is important for efficient signalling by TaHeR.7,14 Since Y93 is highly (∼80%) conserved in HeR family, it may contribute to extend the lifetime of the active state. Furthermore, the partial irreversible bleaching of Y93G upon light illumination indicates, Y93 also critical to prevent photo degradation.
Previously, photoreactions of octopus rhodopsin and a few sensory rhodopsins have been studied by the TG method.30,32–34 In the case of octopus rhodopsin, a pronounced D change was observed (DR = 6.1 × 10−11 m2 s−1, DP = 2.9 × 10−11 m2 s−1), which was assigned to the opening movement of the transmembrane helices.32,33 In the case of sensory rhodopsins, no detectable change in D was induced by photoexcitation. In the presence of their downstream transducer proteins, however, significant D changes were observed, and they were attributed to conformational changes at the cytoplasmic extension site (so called ![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) istidine kinases,
istidine kinases, ![[A with combining low line]](https://www.rsc.org/images/entities/char_0041_0332.gif) denylyl cyclases,
denylyl cyclases, ![[M with combining low line]](https://www.rsc.org/images/entities/char_004d_0332.gif) ethylaccepting chemotaxis proteins, and
ethylaccepting chemotaxis proteins, and ![[P with combining low line]](https://www.rsc.org/images/entities/char_0050_0332.gif) hosphatases (HAMP) domain) of the Halobacterial transducer protein II (HtrII) (DR = 6.3 × 10−11 m2 s−1, DP = 2.9 × 10−11 m2 s−1)30 or to changes in intermolecular interaction (dissociation) with the soluble transducer (DR = 2.5 × 10−11 m2 s−1, DP = 7.5 × 10−11 m2 s−1).34 Compared to these D changes, the amount of D change for TaHeR was small, suggesting that it is activated by a localized and minor conformational change on the intracellular side where the signal transduction occur.7 Also, previous structural analysis has reported a crystal structure of HeR 48C12 at acidic pH mimicking the M intermediate with an acetate ion incorporated inside the molecule, which is suggested to be relevant for the putative enzymatic function of HeR for redox reaction.12 If TaHeR has the ability to incorporate ions in a light-dependent manner, the conformational change in the ICL1 region may be responsible for the uptake of substrate ions from the cytoplasmic side, since the binding site of acetate (inner cavity) is on the cytoplasmic side and the ICL1 helix locates on the presumed pathway connecting the inner cavity and cytoplasmic side of the protein.
hosphatases (HAMP) domain) of the Halobacterial transducer protein II (HtrII) (DR = 6.3 × 10−11 m2 s−1, DP = 2.9 × 10−11 m2 s−1)30 or to changes in intermolecular interaction (dissociation) with the soluble transducer (DR = 2.5 × 10−11 m2 s−1, DP = 7.5 × 10−11 m2 s−1).34 Compared to these D changes, the amount of D change for TaHeR was small, suggesting that it is activated by a localized and minor conformational change on the intracellular side where the signal transduction occur.7 Also, previous structural analysis has reported a crystal structure of HeR 48C12 at acidic pH mimicking the M intermediate with an acetate ion incorporated inside the molecule, which is suggested to be relevant for the putative enzymatic function of HeR for redox reaction.12 If TaHeR has the ability to incorporate ions in a light-dependent manner, the conformational change in the ICL1 region may be responsible for the uptake of substrate ions from the cytoplasmic side, since the binding site of acetate (inner cavity) is on the cytoplasmic side and the ICL1 helix locates on the presumed pathway connecting the inner cavity and cytoplasmic side of the protein.
Conclusions
In this study, the photoreaction of TaHeR was investigated mainly by TG and CD methods. The TG method revealed that the D does not change until the O formation in spite of large spectral shifts. Hence, the spectral shift is originated by rather localized structural changes around the chromophore. D decreases during the formation of the O intermediate and this reaction occurs even when one protomer in the dimer reacts. During the conversion from M to O, two time constants are observed without spectral shape change, but the D-change occurs at the first step. Since D is highly sensitive to conformational changes in the solvent-exposed region, we suggest that the first step involves the reorganization of hydrogen-bonding network on the hydrophilic intracellular side, whereas the second step is minor change inside the protein. The CD measurement showed that the helical structure is partially unfolded in the light state. Interestingly, however, the mutational effects on the D change and the CD change are different: in Y93G, D change is significantly suppressed, but CD change is not. This difference implies that the sensitivities of these methods are different; the TG method is more sensitive to the reactions on the hydrophilic surfaces, which may be important for the function of HeRs. For the O formation, the absorption changes by pKa shift due to two step reorganization processes of hydrogen-bonding network around the chromophore. The observed D-change dynamics indicates that only the first step of the reorganization induces structural change around ICL1 to change D. We also found that Y93 contributes to the stabilization of the active state.Author contributions
Conceptualization, Y. N., K. I., and M. T.; project administration, Y. N., K. I., and M. T.; methodology, Y. N., Y. K., M. K., and K. I.; resources, Y. K., M. K., and K. I.; investigation, Y. N., Y. K., M. K., and K. I.; formal analysis, Y. N., Y. K., and K. I.; supervision, Y. N., K. I., and M. T.; validation, Y. N., K. I., and M. T.; visualization, Y. N., and K. I.; writing – original draft, Y. N., Y. K., M. K., and K. I.; Writing – review & editing, M. T.; funding acquisition, Y. N., K. I., and M. T.;Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Grants-in-Aid for Scientific Research from JSPS (Grant Numbers: JP20H04708 to Y. N., JP21H01875, JP20K21383 to K. I., 19H01863, 21H01885, 21K19218 to M. T.), MEXT KAKENHI, Grant-in-Aid for Transformative Research Areas (B) “Low-energy manipulation” (Grant Number: JP20H05758 to K. I.).References
-
J. L. Spudich and K.-H. Jung, in Handbook of Photosensory Receptors, ed. W. R. Briggs and J. L. Spudich, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2005, pp. 1–23 Search PubMed
.
- O. P. Ernst, D. T. Lodowski, M. Elstner, P. Hegemann, L. S. Brown and H. Kandori, Chem. Rev., 2014, 114, 126–163 CrossRef CAS PubMed
- T. Nagata and K. Inoue, J. Cell Sci., 2021, 134, jcs258989 CrossRef CAS PubMed
- E. G. Govorunova, O. A. Sineshchekov, H. Li and J. L. Spudich, Annu. Rev. Biochem., 2017, 86, 845–872 CrossRef CAS PubMed
- A. Rozenberg, K. Inoue, H. Kandori and O. Béjà, Annu. Rev. Microbiol., 2021, 75, 427–447 CrossRef PubMed
- Y. Shichida and T. Morizumi, Photochem. Photobiol., 2007, 83, 70–75 CAS
- A. Pushkarev, K. Inoue, S. Larom, J. Flores-Uribe, M. Singh, M. Konno, S. Tomida, S. Ito, R. Nakamura, S. P. Tsunoda, A. Philosof, I. Sharon, N. Yutin, E. V. Koonin, H. Kandori and O. Béjà, Nature, 2018, 558, 595–599 CrossRef CAS PubMed
- S. Hososhima, R. Mizutori, R. Abe-Yoshizumi, A. Rozenberg, S. Shigemura, A. Pushkarev, M. Konno, K. Katayama, K. Inoue, S. P. Tsunoda, O. Beja and H. Kandori, eLife, 2022, 11, e78416 CrossRef PubMed
- S. Cho, M. Song, K. Chuon, J. Shim, S. Meas and K. Jung, PLoS Biol., 2022, 20, E3001817 CrossRef CAS PubMed
- J. Shim, S. Cho, S. Kim, K. Chuon, S. Meas, A. Choi and K. Jung, Microbiol. Spectrum, 2022, e02215 Search PubMed
- W. Shihoya, K. Inoue, M. Singh, M. Konno, S. Hososhima, K. Yamashita, K. Ikeda, A. Higuchi, T. Izume, S. Okazaki, M. Hashimoto, R. Mizutori, S. Tomida, Y. Yamauchi, R. Abe-Yoshizumi, K. Katayama, S. P. Tsunoda, M. Shibata, Y. Furutani, A. Pushkarev, O. Béjà, T. Uchihashi, H. Kandori and O. Nureki, Nature, 2019, 574, 132–136 CrossRef CAS PubMed
- K. Kovalev, D. Volkov, R. Astashkin, A. Alekseev, I. Gushchin, J. M. Haro-Moreno, I. Chizhov, S. Siletsky, M. Mamedov, A. Rogachev, T. Balandin, V. Borshchevskiy, A. Popov, G. Bourenkov, E. Bamberg, F. Rodriguez-Valera, G. Büldt and V. Gordeliy, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 4131–4141 CrossRef CAS PubMed
- Y. Lu, X. E. Zhou, X. Gao, N. Wang, R. Xia, Z. Xu, Y. Leng, Y. Shi, G. Wang, K. Melcher, H. E. Xu and Y. He, Cell Res., 2020, 30, 88–90 CrossRef CAS PubMed
- K. Inoue, T. Tsukamoto and Y. Sudo, Biochim. Biophys. Acta, 2013, 1837, 562–577 CrossRef PubMed
- M. Luck, T. Mathes, S. Bruun, R. Fudim, R. Hagedorn, T. M. Tran Nguyen, S. Kateriya, J. T. Kennis, P. Hildebrandt and P. Hegemann, J. Biol. Chem., 2012, 287, 40083–40090 CrossRef CAS PubMed
- K. Yoshida, S. P. Tsunoda, L. S. Brown and H. Kandori, J. Biol. Chem., 2017, 292, 7531–7541 CrossRef CAS PubMed
- U. Scheib, M. Broser, O. M. Constantin, S. Yang, S. Gao, S. Mukherjee, K. Stehfest, G. Nagel, C. E. Gee and P. Hegemann, Nat. Commun., 2018, 9, 2046 CrossRef PubMed
- P. A. Bulzu, V. S. Kavagutti, M.-C. Chiriac, C. D. Vavourakis, K. Inoue, H. Kandori, A.-S. Andrei, R. Ghai and S. J. Hallam, mSphere, 2021, 6, e00661 CrossRef CAS PubMed
- J. Flores-Uribe, G. Hevroni, R. Ghai, A. Pushkarev, K. Inoue, H. Kandori and O. Béjà, Environ. Microbiol. Rep., 2019, 11, 419–424 CrossRef CAS PubMed
- M. Terazima, Acc. Chem. Res., 2021, 54, 2238–2248 CrossRef CAS PubMed
- Y. Nakasone and M. Terazima, Front. Genet., 2021, 12, 691010 CrossRef CAS PubMed
- M. Terazima, Phys. Chem. Chem. Phys., 2006, 8, 545–557 RSC
- A. Carlström, Acta Chem. Scand., 1969, 23, 185–202 CrossRef PubMed
- P. Illien, X. Zhao, K. K. Dey, P. J. Butler, A. Sen and R. Golestanian, Nano Lett., 2017, 17, 4415–4420 CrossRef CAS PubMed
- J. V. Møller and M. le Maire, J. Biol. Chem., 1993, 268, 18659–18672 CrossRef
- K. Tanaka, Y. Nakasone, K. Okajima, M. Ikeuchi, S. Tokutomi and M. Terazima, J. Mol. Biol., 2011, 409, 773–785 CrossRef CAS PubMed
- K. Ludmann, C. Ganea and G. Váró, J. Photochem. Photobiol., B, 1999, 49, 23–28 CrossRef CAS
- S. Brahms and J. Brahms, J. Mol. Biol., 1980, 138, 149–178 CrossRef CAS PubMed
- K. Inoue, N. Baden and M. Terazima, J. Phys. Chem. B, 2005, 109, 22623–22628 CrossRef CAS PubMed
- K. Inoue, J. Sasaki, J. L. Spudich and M. Terazima, J. Mol. Biol., 2008, 376, 963–970 CrossRef CAS PubMed
- I. Chizhov, G. Schmies, R. Seidel, J. R. Sydor, B. Lüttenberg and M. Engelhard, Biophys. J., 1998, 75, 999–1009 CrossRef CAS PubMed
- K. Inoue, M. Tsuda and M. Terazima, Biophys. J., 2007, 92, 3643–3651 CrossRef CAS PubMed
- Y. Nishioku, M. Nakagawa, M. Tsuda and M. Terazima, Biophys. J., 2002, 83, 1136–1146 CrossRef CAS PubMed
- M. Kondoh, K. Inoue, J. Sasaki, J. L. Spudich and M. Terazima, J. Am. Chem. Soc., 2011, 133, 13406–13412 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp00711a |
| This journal is © the Owner Societies 2023 |