
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The path towards type V deep eutectic solvents: inductive effects and steric hindrance in the system tert-butanol + perfluoro tert-butanol†
Inês C. M.
Vaz
 ad,
Ana I. M. C.
Lobo Ferreira
a,
Gonçalo M. C.
Silva
bc,
Pedro
Morgado
b,
Dinis O.
Abranches
c,
Margarida
Bastos
a,
Luís M. N. B. F.
Santos
a,
Eduardo J. M.
Filipe
b and
João A. P.
Coutinho
*c
ad,
Ana I. M. C.
Lobo Ferreira
a,
Gonçalo M. C.
Silva
bc,
Pedro
Morgado
b,
Dinis O.
Abranches
c,
Margarida
Bastos
a,
Luís M. N. B. F.
Santos
a,
Eduardo J. M.
Filipe
b and
João A. P.
Coutinho
*c
aCIQUP, Institute of Molecular Sciences (IMS) – Departamento de Química e Bioquímica, Faculdade de Ciências da Universidade do Porto, Rua do Campo Alegre, P-4169-007 Porto, Portugal
bCQE, Centro de Química Estrutural, Instituto Superior Técnico, Universidade de Lisboa, Lisboa, Portugal
cCICECO – Aveiro Institute of Materials, Department of Chemistry, University of Aveiro, Aveiro, Portugal. E-mail: jcoutinho@ua.pt
dLaboratoire de Chimie de l’ENS Lyon, CNRS and Université de Lyon, 46 allée d’Italie, 69364 Lyon, France
First published on 28th March 2023
Abstract
The solid–liquid phase behaviour of two tertiary alcohols, perfluoro-tert-butanol and tert-butanol, was studied here using experimental (ITC, DSC and density measurements) and theoretical (MD simulations) approaches. The phase diagram of the binary mixture reveals highly negative deviations from ideality at low concentrations, as well as the formation of co-crystals and is characterized by two eutectic points, a congruent melting point and a peritectic reaction corresponding to TBH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :TBF stoichiometries of 2:1 and 1:1 respectively. Excess molar enthalpies and volumes were calculated, showing negative and positive deviations from ideality, respectively. The effect of acidity, stereochemical hindrance and phobic effects and how they affect intermolecular interactions in these binary mixtures is discussed, with the aim of designing and fine-tuning type V deep eutectic solvents. The results showed that the fluorination of tertiary alcohols can be used for the tuning of the mixing properties and solid–liquid phase diagrams.
:TBF stoichiometries of 2:1 and 1:1 respectively. Excess molar enthalpies and volumes were calculated, showing negative and positive deviations from ideality, respectively. The effect of acidity, stereochemical hindrance and phobic effects and how they affect intermolecular interactions in these binary mixtures is discussed, with the aim of designing and fine-tuning type V deep eutectic solvents. The results showed that the fluorination of tertiary alcohols can be used for the tuning of the mixing properties and solid–liquid phase diagrams.
1. Introduction
Deep eutectic solvents (DESs) are liquid systems obtained by physically mixing solid compounds.1–3 Because DESs are mixtures, their physicochemical properties can be easily tailored by carefully selecting their precursors and varying mole ratios. Together with the sustainable character of common DES constituents and the absence of synthesis steps in their preparation, this led to extensive research on the use of DESs as green solvents.3,4 However, most DESs proposed in the past contain at least one ionic precursor (types I–IV), limiting their application scope. Recently, though, a new type of DES based solely on non-ionic compounds has been proposed (type V DES), opening the road to creating a whole range of non-ionic systems that better mimic the properties of conventional organic solvents.5,6The liquid phase of DESs arises from the establishment of an eutectic-type solid–liquid equilibrium (SLE) between the solid precursors, leading to a melting temperature depression of the mixture and, consequently, a liquid phase, at a given operational temperature. Because this depression is heightened by negative deviations from thermodynamic ideality,2,6 the design of DESs entails selecting precursors that display strong cross intermolecular interactions, usually hydrogen bond donors (HBDs) and hydrogen bond acceptors (HBAs).2 Please note the use of the term “cross interactions”: negative deviations are favoured by strong HBD–HBA interactions and weak HBD–HBD or HBA–HBA association. For type V DESs where electrostatic interactions are not present, this is commonly achieved by selecting HBDs with weak HBA capability (also known as asymmetric HBDs) or HBAs without any HBD sites (also known as lone HBAs) as precursors.6 For example, the prototypical type V DES is the thymol–menthol system. Despite the structural similarity between thymol and menthol, the aromatic ring in thymol acts as an electron withdrawing agent to its hydroxyl group, leading to a stronger HBD but weaker HBA site.5 This results in a weaker thymol–thymol hydrogen bond (or, in other words, less thymol–thymol association) and a stronger thymol–menthol hydrogen bond, leading to a DES that is liquid at room temperature in a wide composition range.
Tertiary alcohols are a particularly interesting class of compounds in this area. By changing the substituents around the tertiary carbon, it is possible to drastically influence the molecule's electronic distribution. In fact, akin to the aromaticity in the case of thymol explained above, fluorination can be used to increase the acidity of the alcohol hydroxyl group, with tert-butanol (TBH) and perfluoro tert-butanol (TBF) being excellent examples of this behaviour. Previous studies7–23 have shown the structure of TBH to be formed by coexisting monomers, dimers, and cyclic tetramers and hexamers. The population of these cyclic oligomers represents more than 65% of the total species in the liquid phase. However, the structure of TBF is markedly different.24,25 Its liquid phase is essentially constituted by monomers (70–80%), with a minor fraction of dimers and almost no trimers. This weak tendency for self-association is due to the electron-withdrawing effect of the –CF3 groups26 that confers a strong acidic character on TBF that is reflected on its pKa = 5.4,27 which is significantly lower than TBH, which has a pKa close to 19.28 Additionally, given the bulky size of –CF3 groups, steric hindrance also affects its hydrogen bonding capabilities, preventing aggregation beyond the dimer. All these properties of TBF suggest that it could be an excellent candidate to act as an asymmetric HBD in the formulation of type V DESs.
The behaviour of TBF as an asymmetric HBD is further supported by previous studies of the liquid phase of mixtures of TBF–TBH (note the resemblance between TBF–TBH and thymol–menthol).29,30 Using a variety of techniques including vibrational spectroscopy (Raman and infrared) and X-ray diffraction, supported by DFT and MD simulations, it was possible to show that TBF acts as a structure breaker of TBH alcohol networks and that the 1:1 TBF–TBH hetero-dimer is the dominant species in the mole fraction concentration range of 0.3–0.7. For mixtures richer in TBF the characteristic monomers of the pure TBF liquid phase become dominant, while for mixtures richer in TBH oligomers start to appear with some relevance of the 2:1 trimer TBH–TBF–TBH. Once again, these characteristics of the TBF–TBH system are identical to those observed for the thymol–menthol system.5
Obtaining a rigorous understanding of the behaviour of the TBF–TBH system connects not only to the formulation of DESs, but also to how the simultaneous presence of hydrogenated and perfluorinated chains affects the organization of a fluid. It is well known that hydrogenated and perfluorinated chains are mutually phobic, despite interacting through similar dispersion forces.31 Their mixtures display positive deviations from Raoult's law, liquid–liquid immiscibility, large positive excess enthalpies and volumes, and nanosegregation,31–33 all clear signs of weak unlike intermolecular forces. Anomalies in the transport,34 surface,35,36 and conformational37 properties have also been reported. Liquids in which the two types of chains coexist are thus prone to organization38 and self-assembly.39 In recent work we have shown that mixtures of hydrogenated and perfluorinated alcohols display clear evidence of coexisting polar, hydrogenated and perfluorinated domains.40–42 A network of hydrogen bonds is formed, zigzagging throughout the liquid, while the hydrogenated and perfluorinated segments occupy the remaining space while trying to avoid each other. The TBH–TBF mixture studied in this work is a further example of this type of mixture. In this case, the presence of tertiary groups on both alcohols eliminates the influence of flexibility vs. stiffness and gauche conformers on the organization of the liquid, which was shown to be important in mixtures involving linear alcohols.
In order to probe the organization of the liquid mixture and its impact on the liquid phase non-ideality and the solid–liquid equilibrium of the system, the excess enthalpies and excess volumes of TBH–TBF mixtures were measured across the entire composition range. These results were interpreted in terms of intermolecular interactions with the aid of molecular dynamics simulations, while gaining molecular level information on the organization and energetics of the tertiary alcohol binary system. This combination of excess properties, molecular dynamics, and hydrogen bonding analysis has been extensively and successfully used in the field of DESs to probe their liquid structures.43–45
2. Experimental and computational details
The alcohols used in this study, pure (99.5%, anhydrous) tert-butanol and pure (99%) perfluoro-tert-butanol, were obtained from Thermo Scientific and Alfa Aesar, respectively. To ensure the dryness of the alcohol, activated molecular sieves of pore size 0.3 nm with a humidity indicator, obtained from Metrohm AG, were used.2.1. Excess enthalpies
A high sensitivity heat conduction isothermal titration microcalorimeter from Thermometric AB/TA (now WatersTM), together with a prototype water bath from Lund University, Sweden, was used to measure the partial excess molar enthalpies of mixtures of TBF in TBH, at atmospheric pressure and at 298.15 ± 0.01 K.46 A 71/2 digit Agilent nanovoltmeter (model 34420A) is connected to the calorimetric channel and interfaced with the computer, where data acquisition and the syringe pump control are performed through a modified version of the program LABTERMO.46,47 The system was electrically calibrated by release of controlled amounts of heat through a ‘homemade’ insertion heater with accurately known resistance, as well as chemically by use of the test reaction of dilution of a 10% propan-1-ol solution in water.48A Hastelloy® made titration cell of 1 mL volume equipped with a gold propeller (Lund University) was used for the ITC experiments, together with a stainless-steel solid cell as the reference cell. Previous to the titration run the titration cell was warmed up to 310 K to prevent the crystallization of the tert-butanol. The sample cell was charged with solutions of different molar fraction of the two alcohols and the perfluoro-tert-butanol was titrated in aliquots of 10–40 μL through a modified gas-tight Hamilton syringe.
The system was allowed to equilibrate between injections, re-establishing the baseline. After each injection, a peak corresponding to the heat generated or absorbed by the mixing process is recorded. The signal obtained was corrected using Tian's equation, and the corresponding peaks integrated numerically.49 The area of each peak is proportional to the heat involved in the mixing experiment and is thus used to calculate the partial excess molar enthalpy of TBF, ![[H with combining macron]](https://www.rsc.org/images/entities/i_char_0048_0304.gif) 2E. The experimental data were fitted using the derivative of the Redlich–Kister equation and from the obtained Ai parameters the excess molar enthalpy of mixing (ΔmixH) was calculated. To reproduce closely the composition dependence of the partial excess molar enthalpy while avoiding introduction of unphysical behaviour into the fitting equation, five (n = 4) fitting parameters were used in eqn (1)–(4)
2E. The experimental data were fitted using the derivative of the Redlich–Kister equation and from the obtained Ai parameters the excess molar enthalpy of mixing (ΔmixH) was calculated. To reproduce closely the composition dependence of the partial excess molar enthalpy while avoiding introduction of unphysical behaviour into the fitting equation, five (n = 4) fitting parameters were used in eqn (1)–(4)
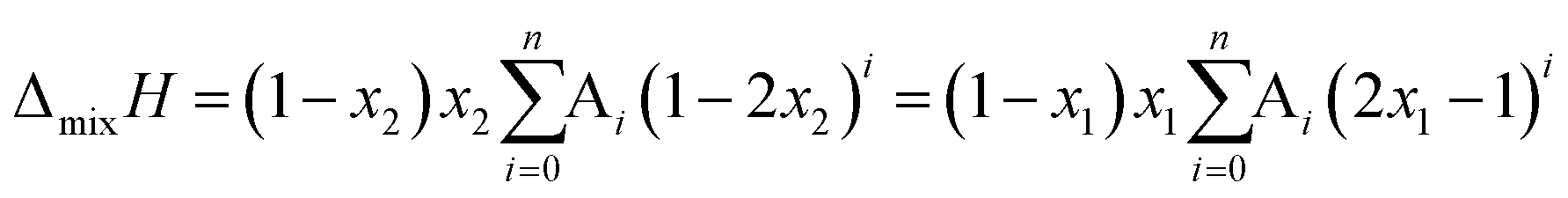 | (1) |
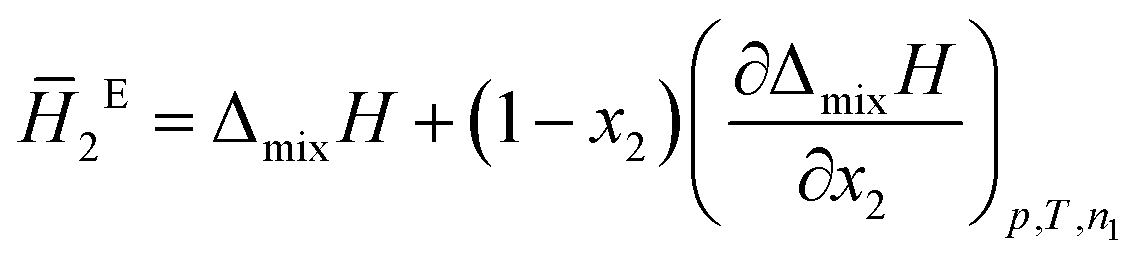 | (2) |
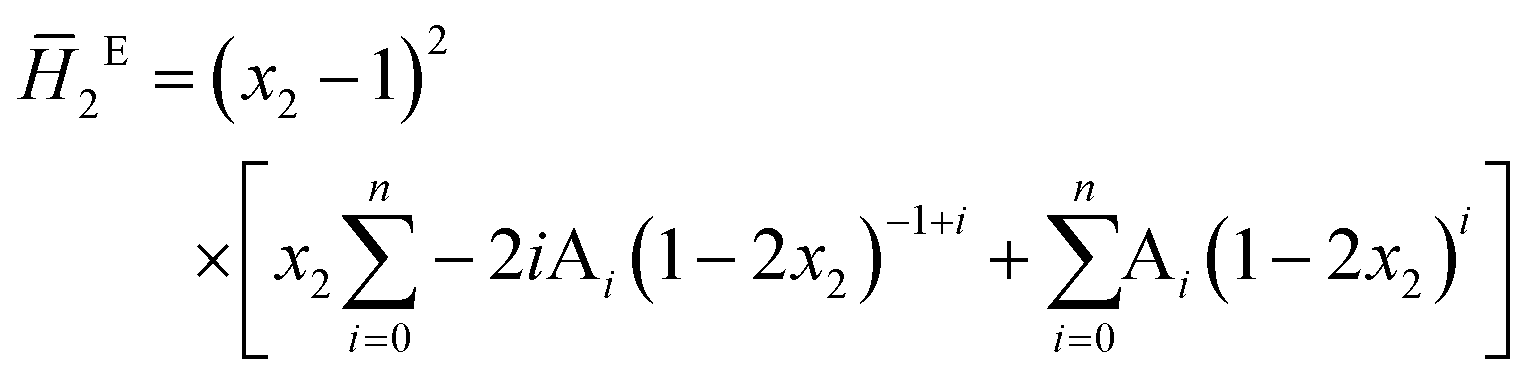 | (3) |
| ΔmixH (T, p, x) = x11E (T, p, n2) + x22E (T, p, n1) | (4) |
2.2. Excess volumes
The density of TBF–TBH liquid mixtures was measured at 298.15 K, using an Anton Paar DMA 5000 vibrating tube densimeter. The densimeter was previously calibrated with atmospheric air and freshly boiled Millipore water at several temperatures, and the maximum deviation from literature values was found to be less than 0.00002 g cm−3. Between each measurement, the cell was thoroughly cleaned with isopropanol and dried with air and the density of air checked. Samples of the binary mixtures were prepared gravimetrically in 4 mL screw-cap glass vials, using a Mettler AE 240 analytical balance. The composition uncertainty was estimated as better than ± 0.0005 mole fraction units, leading to uncertainties in the individual values of the excess volumes under 0.05 cm3 mol−1.2.3. Differential scanning calorimetry
Thermal behaviour and temperatures of phase transition and fusion for TBF–TBH mixtures were evaluated in a power compensation differential scanning calorimeter, PerkinElmer model Pyris Diamond DSC, using hermetically sealed aluminium crucibles, samples of about 20 mg, and a constant flow of nitrogen (50 mL min−1). The temperature and heat flux scales of the power compensation DSC were calibrated by measuring the temperature and the enthalpy of fusion of some reference materials namely, benzoic acid, 4-methoxybenzoic acid, triphenylene, naphthalene, anthracene, 1,3,5-triphenylbenzene, diphenylacetic acid, perylene, o-terphenyl, and 9,10-diphenylanthracene, at different scanning rates (2, 5 and 10 K min−1).50–53The TBH–TBF mixtures at different compositions were prepared gravimetrically in a glove box under dry nitrogen in batches of 1 g and kept in sealed glass bottles under nitrogen, to avoid contamination and sample evaporation. The batches of each mixture were stored for at least 24 hours (mixture homogenization) prior to the DSC crucibles preparation under dry N2(g). All measurements were performed under a constant flow of nitrogen (50 mL min−1), using varying heating rates, and 50 μL sealed aluminium crucibles and samples were used. The mass of each experiment was selected according to the needs of the studied process as described previously.54 The composition uncertainty was estimated to be better than ±0.01 mole fraction. Each sample was cooled until 173 K and maintained at that temperature to ensure complete crystallization. A final scan at 5 K min−1 was performed to determine the temperatures of the solid–solid and solid–liquid phase transitions.
2.4. Molecular dynamics simulations
Atomistic molecular dynamics simulations of the studied TBF–TBH mixtures were performed using the GROMACS 5.0.7 simulation package.55 The tertiary alcohols were modelled with the OPLS-AA force field,56 except for the partial charges which were obtained from DFT calculations. Full details of the used models were described in a previous publication.25The unlike dispersive parameters were calculated using geometric combining rules, as prescribed by the OPLS-AA force field, with the exception of the interactions between hydrogen and fluorine atoms, where factors of 1.035 and 0.8 were applied to the crossed size (σH−F) and energy (εH−F) parameters, respectively. These deviations from the combining rules account for the peculiar volumetric and enthalpic effects that occur when perfluorinated and hydrogenated compounds are mixed, and were determined in a previous work for the n-butanol + 1H,1H-perfluorobutanol system.40
Cubic simulation boxes with periodic boundary conditions, containing 300 molecules were generated using the GROMACS “insert-molecules” program and pre-equilibrated in NpT using a Berendsen thermostat and barostat until the density reached an approximately constant value. The systems were then run in the NpT ensemble, using the leapfrog integrator with a 2 fs time step, for at least 35 ns, of which the first 5 ns were discarded as equilibration time and the remainder used to compute the averaged properties. Temperature and pressure were held constant during the production run, using a Nosé–Hoover thermostat57,58 and a Parrinello–Rahman barostat59 with coupling times of 0.5 and 10 ps, respectively. The LINCS algorithm was applied to constrain all bonds containing hydrogen atoms to the respective equilibrium length.60 A cut-off distance of 1.4 nm was used to calculate both dispersive and electrostatic interactions. Beyond the cut-off distance, standard analytical corrections to the energy and pressure dispersive terms were applied and the particle-mesh Ewald method was used to account for long-range electrostatic interactions.61,62
Physical properties obtained from molecular simulation models are well known to depend significantly on the electrostatic characteristics, especially in the case of highly polar and hydrogen bonding compounds such as the alcohols studied in this work. On the other hand, charge distributions obtained by ab initio calculations vary significantly with the details of the calculation, namely the level of theory, basis sets and algorithm used to attribute the point charges from the full electronic density distribution. In this work, a sensitivity analysis was performed on the partial charges of the hydroxyl groups, to evaluate its effect on the shape and magnitude of the calculated excess enthalpies and volumes. Several combinations of the charge distributions of both compounds were tested, and those that yielded the most relevant results are detailed in Table 1.
| TBF atom | Model 1 | Model 2 | TBH atom | Model 1 | Model 2 | Model 3 |
|---|---|---|---|---|---|---|
| C | −0.141 | −0.171 | C | 0.778 | 0.808 | 0.908 |
![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) F3 F3 |
0.579 | 0.579 | H3 |
−0.352 | −0.352 | −0.352 |
| F | −0.171 | −0.171 | ![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) C C |
0.071 | 0.071 | 0.071 |
![[O with combining low line]](https://www.rsc.org/images/entities/char_004f_0332.gif) H H |
−0.479 | −0.464 | H |
−0.746 | −0.761 | −0.866 |
| O |
0.422 | 0.437 | O |
0.385 | 0.370 | 0.375 |
In this table, the molecular models with the original point charges previously determined25 are designated as “Model 1”. In “Model 2”, the difference between the partial charges of the O–H groups of each alcohol was increased relatively to one another, while keeping the O–H charge separation in each pure compound constant. This change increases the more probable cross-association (the H-bond between the oxygen of TBF and the hydrogen of TBH). As will be shown below, this combination of charges reproduces the experimental HE curve but not the asymmetry of the VE curve. For “Model 3”, the difference between the partial charges within the O–H group of TBH was increased, and this model was used together with the original (Model 1) of TBF. This combination increases both the self-association of TBH and the more probable cross-association in the mixture and is able to reproduce the highly asymmetric shape of the VE curve.
3. Results and discussion
3.1. Excess properties
The study of the binary system TBF + TBH should be informative as to how a significant difference in acidity between two compounds is reflected in the non-ideality of their liquid mixture. As such, to probe the non-ideality of this system, excess enthalpies were measured by isothermal titration calorimetry (ITC). The experimental partial excess molar enthalpies2E (TBH being defined as the component (1) and TBF as the component (2)) were fitted using the derivative of the Redlich–Kister equation, with five (n = 4) fitting parameters. Table 2 lists the obtained Redlich–Kister coefficients Ai (n = 4), with additional information being provided in the ESI.† Moreover, the Redlich–Kister equation was used to estimate the partial excess molar enthalpy of TBH, 1E and the excess molar enthalpies of mixing, as described in the ESI.† The excess molar enthalpies and partial excess molar enthalpies at 298.15 K are shown in Fig. 1.
| A i | H/kJ mol−1 | V/cm3 mol−1 |
|---|---|---|
| A 0 | −25.8873 | 1.4249 |
| A 1 | 3.0927 | 3.7453 |
| A 2 | 8.9913 | −1.2840 |
| A 3 | −3.8657 | −4.6380 |
| A 4 | −3.8094 | — |
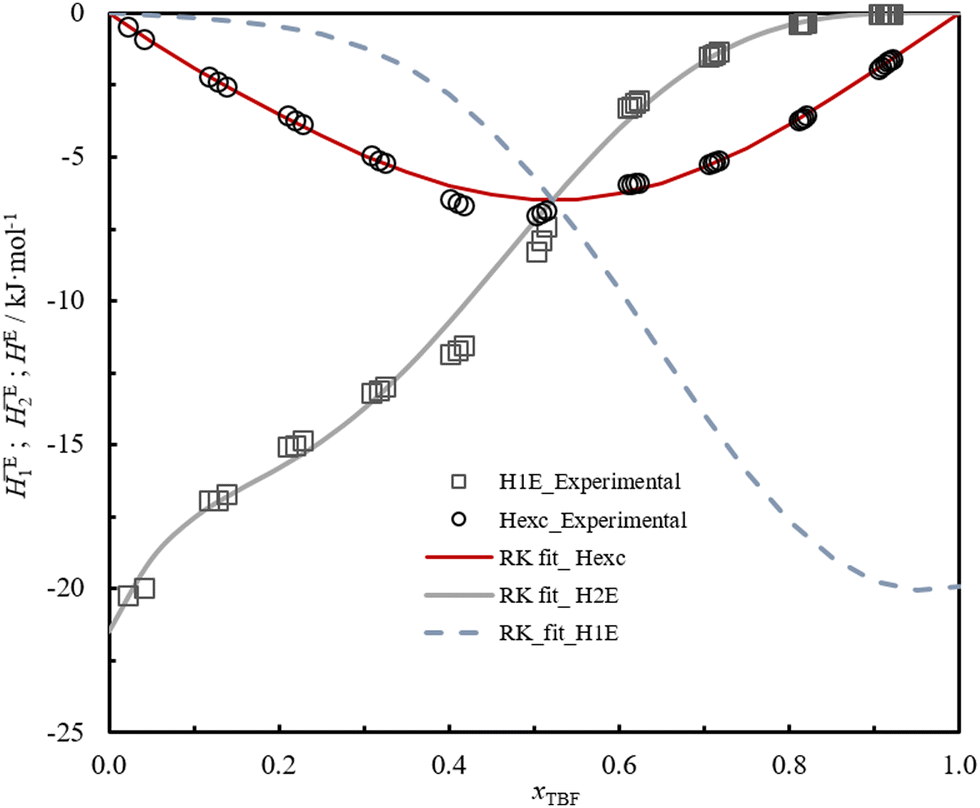 | ||
| Fig. 1 Experimental and fitted excess and partial excess molar enthalpies (1E, 2E) of the binary system TBH(x1) +TBF(x2) at T = 298.15 K. Gray squares – experimental partial excess molar enthalpies 2E; gray line – partial excess enthalpy 2E, derived from the Redlich–Kister equation; black circles – derived experimental excess molar enthalpies E; red line – excess enthalpy derived from the Redlich–Kister equation; dashed blue line – partial excess enthalpy, 1E, derived from the Redlich–Kister equation. | ||
The very large negative deviations seen in Fig. 1, around −7 kJ mol−1, confirm that, upon mixing, there is a considerable release of energy conferred by the preferred cross-association interaction. The perfluorinated compound TBF, which in its pure liquid phase is mostly found as isolated monomers, is able to form a very strong hydrogen bond in the presence of TBH.30 Due to the lower ability of TBF to associate in the pure form, which can be attributed to the steric hindrance and inductive effects created by the –CF3 substituents, the magnitude of the excess molar enthalpy is comparable to the energy of a single hydrogen bond. As such, the very negative HE values in this mixture derive essentially from the “excess” of hydrogen bonds formed, in comparison with the average of the pure compounds. The enthalpies of solution at infinite dilution of TBF in TBH and TBH in TBF, derived from the extrapolation of TBF partial excess molar enthalpies 2E and 1E to infinite dilution, were found to be −(20.7 ± 0.1) kJ mol−1 and −(19 ± 2) kJ mol−1 respectively, which is related to the strong hydrogen bond interaction between TBF and TBH.
To further understand this behaviour, Fig. 2 presents the experimental excess molar enthalpies along with the values calculated from the MD simulations.
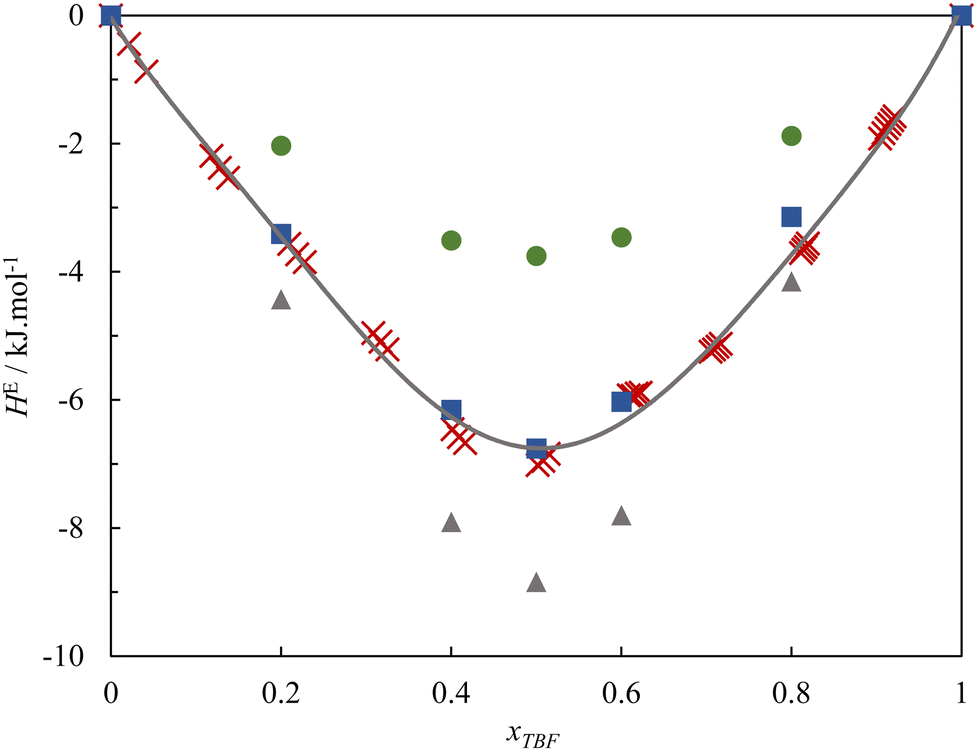 | ||
| Fig. 2 Excess molar enthalpies of the binary system TBF + TBH at T = 298.15 K. Red crosses – experimental data derived from ITC; solid curve – second order Redlich–Kister equation fitted to the experimental points; circles – simulations with the original (Model 1) force field; squares – simulations with Model 2 for both TBF and TBH. Triangles – simulations with the Model 3 of TBH and Model 1 of TBF. | ||
The MD simulations, using the original charge distributions, can reproduce the sign, shape and order of magnitude of the HE curve, predicting a value that is around 50% of the experimental for the equimolar composition. This agreement can be considered remarkable, as the MD excess enthalpies are very sensitive to small variations in the partial charges of the hydroxyl groups. A sensitivity analysis was performed, and it was found that very small corrections of 0.015 to the charges of the two OH groups (Model 2), to the effect of promoting the preferred cross-association, increase the (absolute) calculated excess enthalpy enough to quantitatively reproduce the experimental values. It should be noted that these corrections are fully within the uncertainty of the ab initio scheme used to attribute the point charges.
The origin of the very negative values of HE in this system was further interpreted by analysing the MD trajectories to calculate the average distribution of hydrogen bonds in the studied mixtures, which has been used in the past to understand the liquid structure of DESs. The results are shown in Fig. 3, where the distribution of the four possible types of H-bonds and their sum is represented as a function of composition. For this calculation, any two molecules were counted as hydrogen bonded if the intermolecular O–H distance was under 0.27 nm, and 100% corresponds to a total limit number of two hydrogen bonds per molecule. The results shown were calculated from the simulations with the original charges (Model 1) for both compounds, and the alternative models tested present very similar results. As can be seen, in the mixtures the total number of hydrogen bonds formed is always larger than the average of the pure compounds and, moreover, there is always a clear preference for the formation of the most energetically favourable cross hydrogen bond between the oxygen of TBH and the hydrogen of TBF.
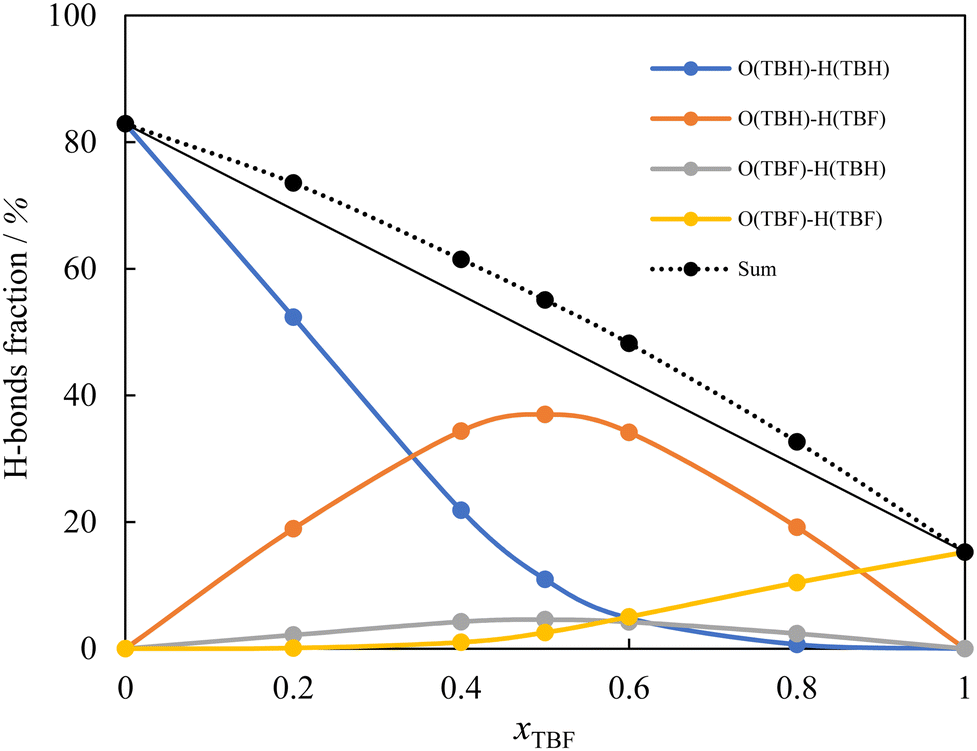 | ||
| Fig. 3 Hydrogen-bond distribution in the (TBH + TBF) mixture as a function of composition, obtained from the MD trajectories for the original (Model 1) point charge models. Lines are visual guides. | ||
From the results reported thus far, the enthalpic behaviour of the TBF–TBH system is clearly dominated by the changes in hydrogen bonding behaviour from the pure compounds to the mixtures. On the contrary, the well-known phobicity between hydrogenated and fluorinated chains, which normally results in positive excess enthalpies for mixtures of alkanes + perfluoroalkanes and hydrogenated alcohols + fluorinated alcohols,40,63,64 plays a secondary role in the case of these tertiary alcohols and, instead, large negative deviations are observed due to the increased association in the mixture.
The volumetric behaviour of the TBH + TBF system was also studied at 298.15 K, and the respective excess volumes are presented as Table S6 (ESI†) and in Fig. 4, along with the results obtained using MD simulations. The experimental data have been fitted to a four parameter (n = 3) Redlich–Kister equation (analogous to eqn (1)), and the obtained coefficients are reported in Table 2. As can be seen, the excess volume curve is very asymmetrical, with positive values on the TBH-rich side which peak at a value of 0.5 cm3 mol−1 at the 1(TBF): 2(TBH) proportion and then decrease, becoming very close to zero for mixtures at the 1(TBF):1(TBH) proportion. This unusual VE shape should be linked both with the variety of associating behaviours observed along the composition range8,26,27 and with the volume expansion that is typically observed when perfluorinated and hydrogenated chains are mixed.65,66 At low TBF compositions, the volumetric behaviour follows the characteristic expansion of hydrogenated + perfluorinated systems, even though clearly attenuated by the enthalpically favourable mixing process. On the opposite side of the diagram, for TBF-rich mixtures, this characteristic expansion is countered, possibly because the hydrogen-bonded aggregates (especially the very stable heterodimer) are able to accommodate more efficiently.
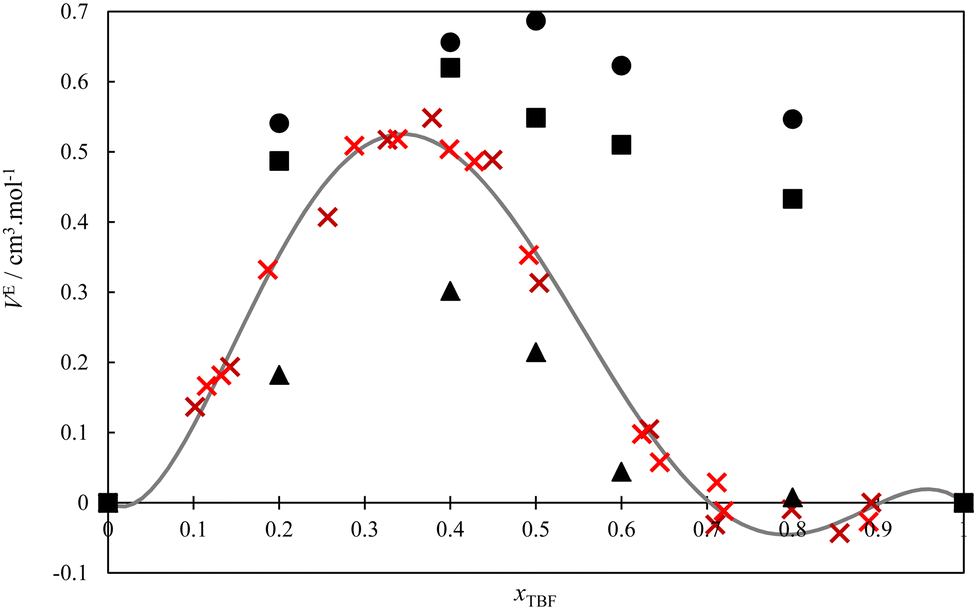 | ||
| Fig. 4 Excess molar volumes of the binary system TBF + TBH at T = 298.15 K. Crosses – experimental data; dashed curve –Redlich–Kister equation fitted to the experimental points; circles – simulations with the original (Model 1) force field; squares – simulations with Model 2 for TBH and TBF. Triangles – Simulations with the Model 3 of TBH and Model 1 of TBF. | ||
The MD simulation results are also shown in Fig. 4. As can be seen, the simulations with Models 1 and 2 predict positive excess volume curves of the correct order of magnitude but fail to reproduce the peculiar shape of the experimental curve, predicting VE values that are essentially symmetric with composition (as is observed in mixtures of linear alcohols). Thus, a sensitivity analysis was again performed on the charge distribution of the OH group of the pure alcohols, to evaluate its influence in the shape of the simulated VE curve. It was found that, if the difference between O and H partial charges of TBH is slightly increased (keeping the original model for TBF), the simulated VE curve reproduces the experimental behaviour, albeit with smaller absolute values. These changes correspond to increasing the self-association of TBH and the intensity of the O(TBH)–H(TBF) cross-association. This combination of Model 3 of TBH and Model 1 of TBF slightly overpredicts the experimental excess enthalpy values, as seen in Fig. 2. In conclusion, these results show that the peculiar properties of the TBH + TBF mixture result from a very subtle balance between the hydrogen-bonding characteristics of the pure compounds, combined with the unfavorable dispersive interactions between their hydrogenated and perfluorinated moieties.
3.2. Solid–liquid equilibrium
Thermal behaviour and temperatures of phase transition phase and fusion of tert-butanol and perfluoro-tert-butanol were evaluated on a DSC, PERKIN ELMER model Pyris Diamond DSC, using hermetically sealed aluminium crucibles. The obtained results (average of five independent experiments) are listed in Table 3. The experimental results indicate an identical enthalpy and entropy of isotropization for both compounds (TBH and TBF). However, the perfluorinated tert-butanol sample shows a pre-melting solid–solid transition (SS) at 246.2 K with a strong enthalpic/entropic effect.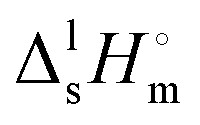 (Tfus), and entropy,
(Tfus), and entropy, 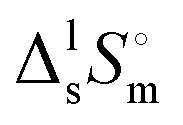 (Tfus), of fusion for TBH and TBF
(Tfus), of fusion for TBH and TBF
| T ss (K) | T fus (K) |
|
|
|
|
|
|---|---|---|---|---|---|---|
The expanded uncertainties of the experimental results, including the calibration uncertainty, were assigned based on independent experiments as 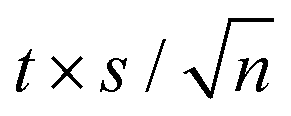 , where t is obtained from Student's t-distribution (0.95 level of confidence), s is the standard deviation and n is the number of independent experiments.a Sum of the enthalpic contributions (SS + isotropization).b Sum of the entropic contributions (SS + isotropization). , where t is obtained from Student's t-distribution (0.95 level of confidence), s is the standard deviation and n is the number of independent experiments.a Sum of the enthalpic contributions (SS + isotropization).b Sum of the entropic contributions (SS + isotropization). |
||||||
| TBH | 298.7 ± 0.3 | 5.6 ± 0.5 | 18.6 ± 1.7 | |||
| TBF | 246.2 ± 0.5 | 251.7 ± 0.5 | 3.0 ± 1.1 | 5.2 ± 0.9 (8.2 ± 1.4)a | 12.2 ± 4.5 | 20.6 ± 3.6 (32.8 ± 5.6)b |
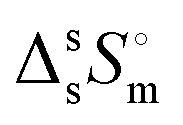
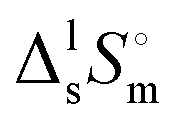
The obtained SLE phase diagram of the binary mixture is depicted in Fig. 5. It is characterized by the formation of two eutectic points, a congruent melting point and a peritectic reaction. The complexity of the obtained phase diagram is generated by the formation of two co-crystals. The congruent melting point and peritectic compositions, corresponding to TBH:TBF stoichiometries of 2:1 and 1:1 respectively, result from the formation of two co-crystals designated as α and β in Fig. 5.
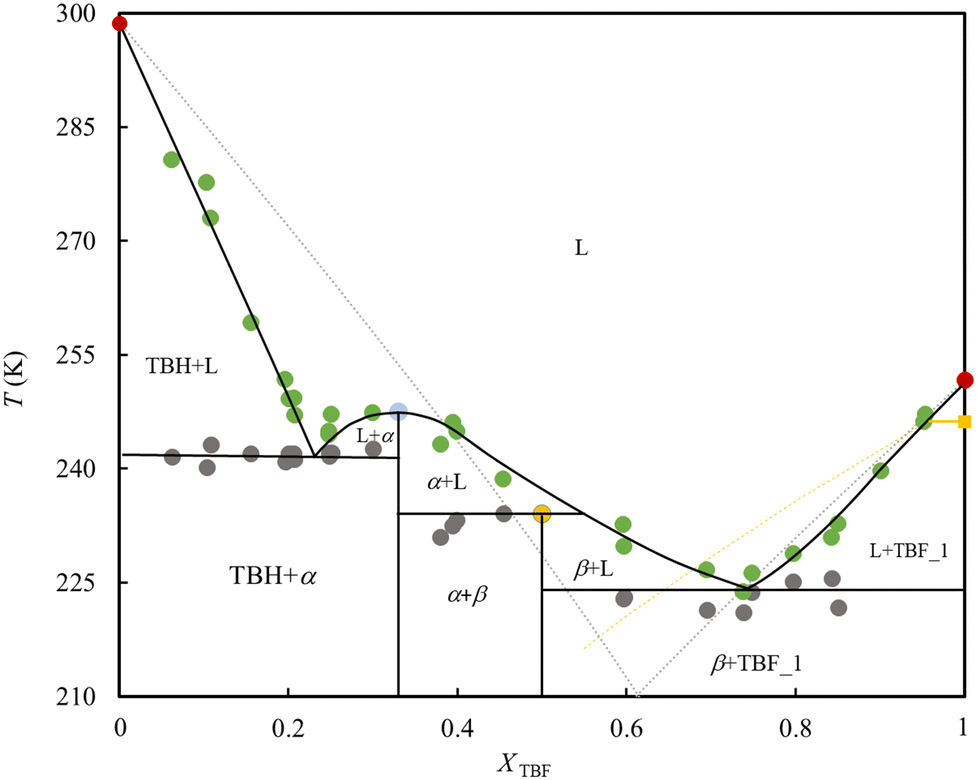 | ||
| Fig. 5 Experimental solid–liquid equilibrium phase diagram at the atmospheric pressure for TBF + TBH. Green and grey circle symbols represent the data along the liquidus and solidus curves, respectively. Red circles represent the isotropization temperature of the pure samples, yellow square is the solid–solid transition of the TBF. The solid lines are a guide for the eye following the experimental data, and dotted curves represent the ideal behaviour (grey disregarding the solid–solid transition of TBF, yellow including it). The α and β correspond to the co-crystals of 1:2 and 1:1 stoichiometry, respectively, with their melting temperatures highlighted (blue and yellow circles, respectively). L represents the liquid phase. | ||
The asymmetry in melting temperatures of two seemingly similar compounds is related to differentiation of the hydrogen bonding capabilities in the pure form. Due to their low cohesive interaction in the liquid phase and high conformational rigidity, fluorinated compounds usually have lower boiling points and higher melting points than the hydrogenated counterparts. However, TBF is an exception to the typical behaviour because the lower ability of TBF to establish hydrogen bonds (strong hindrance) translates into weaker intermolecular interactions in its pure liquid phase, when compared to TBH. The observed lower melting point of TBF and weaker intermolecular interactions in the liquid is supported by the differentiation of the overall enthalpic (TBH: (5.6 ± 0.5) kJ· mol−1 | TBF: (8.2 ± 1.4) kJ mol−1) and entropic (TBH: (20.6 ± 3.6) J K−1 mol−1 | TBF: (32.8 ± 5.6) J K−1 mol−1) contributions of the pre-melting solid–solid transition (ss) and the isotropization of the TBF when compared with the TBH.
Another important aspect of this phase diagram is the formation of two co-crystals driven by the favourable interactions between two compounds and their favourable ability (size and/shape) to pack in a co-crystal form. It is not surprising that the stoichiometries of solids α and β are the most common aggregation states observed in the liquid mixtures; the hetero-dimer TBH–TBF and the trimer TBH–TBF–TBH.30 The formation of stoichiometric compounds could be mitigated by decreasing the extent of fluorination and further decreasing the melting temperature of eutectic mixtures.
The most relevant feature of the SLE diagram is the considerable negative deviation from the ideality at compositions in-between the pure TBH and the eutectic points. This is particularly striking given that hydrocarbons and fluorocarbons mixtures are known to interact weakly as compared to their pure state, generally resulting in positive deviations to ideality and often leading to a liquid–liquid phase separation. However, here this effect is heavily displaced by the strong hydrogen bonding cross-interactions, particularly the one between the proton of TBF and the oxygen of TBH. In fact, the cross-associative interactions are so strong that the two compounds react to form stoichiometric co-crystals. Thus, this clearly supports the idea that fluorinated compounds act as asymmetric hydrogen bond donors, with their liquid mixtures behaving as type V DESs, displaying severe negative deviations from ideality.
4. Conclusions
In this work, we covered the potential of fluorine atoms to increase the acidity of hydroxyl groups in an attempt to understand and address the design of novel type V deep eutectic solvents. Besides the determination of the SLE diagram for the TBH + TBF system, energetic and volumetric characterizations of this system were also made, and the interpretation was aided by the use of MD simulations. The results show significant negative deviations in the solid–liquid transitions and excess molar enthalpies, as well as positive deviations in the excess molar volumes. The strong cross-association between the two tertiary alcohols is responsible for the non-ideality of these mixtures. Despite the importance of the hetero-molecular interactions in the tendency of the two compounds of a binary mixture to display very low eutectic points, this aspect only reveals a part of the picture. The governing interactions of the pure constituents must not be overlooked, as well as their potential to neatly pack or form stable co-crystals. Moreover, the results here reported showed that the degree of fluorination in tertiary alcohols can be an interesting strategy for the tuning of the melting points, mixing properties, and control of co-crystallization.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partly developed within the scope of the project CICECO-Aveiro Institute of Materials (UIDB/50011/2020, UIDP/50011/2020 and LA/P/0006/2020), CIQUP, Centro de Investigação em Química da Universidade do Porto (UIDB/00081/2020); IMS, Institute of Molecular Sciences (LA/P/0056/2020), and Centro de Química Estrutural, Institute of Molecular Sciences, (UIDB/00100/2020/LA/P/0056/2020), financed by national funds through the FCT/MEC (PIDDAC). AIMCLF is financed by national funds through the FCT-I.P., in the framework of the execution of the program contract provided in paragraphs 4, 5, and 6 of art. 23 of Law no. 57/2016 of 29 August, as amended by Law no. 57/2017 of 19 July.References
- D. O. Abranches and J. A. P. Coutinho, Everything You Wanted to Know about Deep Eutectic Solvents but Were Afraid to Be Told, Annu. Rev. Chem. Biomol. Eng., 2023, 14(1) DOI:10.1146/annurev-chembioeng-101121-085323.
- M. A. R. Martins, S. P. Pinho and J. A. P. Coutinho, Insights into the Nature of Eutectic and Deep Eutectic Mixtures, J. Solution Chem., 2019, 48(7), 962–982, DOI:10.1007/s10953-018-0793-1.
- E. L. Smith, A. P. Abbott and K. S. Ryder, Deep Eutectic Solvents (DESs) and Their Applications, Chem. Rev., 2014, 114(21), 11060–11082, DOI:10.1021/cr300162p.
- F. M. Perna, P. Vitale and V. Capriati, Deep Eutectic Solvents and Their Applications as Green Solvents, Curr. Opin. Green Sustainable Chem., 2020, 21, 27–33, DOI:10.1016/j.cogsc.2019.09.004.
- D. O. Abranches, M. A. R. Martins, L. P. Silva, N. Schaeffer, S. P. Pinho and J. A. P. Coutinho, Phenolic Hydrogen Bond Donors in the Formation of Non-Ionic Deep Eutectic Solvents: The Quest for Type V DES, Chem. Commun., 2019, 55(69), 10253–10256, 10.1039/C9CC04846D.
- D. O. Abranches and J. A. P. Coutinho, Type V Deep Eutectic Solvents: Design and Applications, Curr. Opin. Green Sustainable Chem., 2022, 35, 100612, DOI:10.1016/j.cogsc.2022.100612.
- C. R. Yonker, S. L. Wallen, B. J. Palmer and B. C. Garrett, Effects of Pressure and Temperature on the Dynamics of Liquid Tert-Butyl Alcohol, J. Phys. Chem. A, 1997, 101(50), 9564–9570, DOI:10.1021/jp972154f.
- M. Saunders and J. B. Hyne, Trimeric Association of t-Butanol by NMR, J. Chem. Phys., 1958, 29(1), 253–254, DOI:10.1063/1.1744453.
- P. P. Nath, S. Sarkar, P. S. R. Krishna and R. N. Joarder, Intermolecular Structure of Liquid D- Tert-Butanol by Neutron-Diffraction Data, Appl. Phys. A: Mater. Sci. Process., 2002, 74, s348–s351, DOI:10.1007/s003390201546.
- P. P. Nath, S. Sarkar, P. S. R. Krishna and R. N. Joarder, Molecular Conformation and Structural Correlations of Liquid D-Tert-Butanol at Room Temperature by Neutron Diffraction, Z. Naturforsch., A: Phys. Sci., 2001, 56(12), 825–831, DOI:10.1515/zna-2001-1205.
- B. D. T. Bowron, J. L. Finney and A. K. Soper, The Structure of Pure Tertiary Butanol, Mol. Phys., 1998, 93(4), 531–543, DOI:10.1080/002689798168871.
- D. Zimmermann, T. Häber, H. Schaal and M. A. Suhm, Hydrogen Bonded Rings, Chains and Lassos: The Case of t-Butyl Alcohol Clusters, Mol. Phys., 2001, 99(5), 413–425, DOI:10.1080/00268970010017009.
- S. Sarkar, P. P. Nath and R. N. Joarder, Orientation Correlation versus Cluster Correlation in Molecular Liquids-Signature through Diffraction Data, Phys. Lett. A, 2000, 275(1–2), 138–141, DOI:10.1016/S0375-9601(00)00560-0.
- A. H. Narten and S. I. Sandler, X-Ray Diffraction Study of Liquid Tertiary Butyl Alcohol at 26 °C, J. Chem. Phys., 1979, 71(5), 2069, DOI:10.1063/1.438576.
- A. R. Abdel Hamid, R. Lefort, Y. Lechaux, A. Moréac, A. Ghoufi, C. Alba-Simionesco and D. Morineau, Solvation Effects on Self-Association and Segregation Processes in Tert-Butanol–Aprotic Solvent Binary Mixtures, J. Phys. Chem. B, 2013, 117(35), 10221–10230, DOI:10.1021/jp402380f.
- P. Sassi, F. Palombo, R. S. Cataliotti, M. Paolantoni and A. Morresi, Distributions of H-Bonding Aggregates in Tert-Butyl Alcohol: The Pure Liquid and Its Alkane Mixtures, J. Phys. Chem. A, 2007, 111(27), 6020–6027, DOI:10.1021/jp071609q.
- F. Palombo, M. Paolantoni, P. Sassi, A. Morresi and R. S. Cataliotti, Spectroscopic Studies of the “Free” OH Stretching Bands in Liquid Alcohols, J. Mol. Liq., 2006, 125(2–3), 139–146, DOI:10.1016/j.molliq.2005.11.006.
- J. M. Andanson, Etude de La Liaison Hydrogène Par Spectroscopie Vibrationnelle et Simulation de Dynamique Moléculaire d’alcools En Condition Supercritique, PhD thesis, Université Bordeaux I, 2005 Search PubMed.
- J.-M. Andanson, J.-C. Soetens, T. Tassaing and M. Besnard, Hydrogen Bonding in Supercritical Tert-Butanol Assessed by Vibrational Spectroscopies and Molecular-Dynamics Simulations, J. Chem. Phys., 2005, 122(17), 174512, DOI:10.1063/1.1886730.
- P. K. Kipkemboi, P. C. Kiprono and J. J. Sanga, Vibrational Spectra of T-Butyl Alcohol, t-Butylamine and t-Butyl Alcohol + t-Butylamine Binary Liquid Mixtures, Bull. Chem. Soc. Ethiop., 2003, 17(2), 211–218, DOI:10.4314/bcse.v17i2.61689.
- G. Larsen and Z. K. Ismail, Monomeric Tert-Butanol in Benzene and Cyclohexane Solutions: A Fourier-Transform Infrared Spectroscopy Study, J. Solution Chem., 1998, 27(10), 901–909, DOI:10.1023/A:1022659111851.
- J. Korppi-Tommola, Tert-Butyl Alcohol—Matrix i.r. Spectra and Vibrational Assignment, Spectrochim. Acta, Part A, 1978, 34(11), 1077–1085, DOI:10.1016/0584-8539(78)80130-5.
- J. Korppi-Tommola, Association of Tert-Butyl Alcohol: A Matrix Infrared Study, J. Mol. Struct., 1977, 40(1), 13–23, DOI:10.1016/0022-2860(77)80002-1.
- J. Murto, A. Kivinen, J. Korppi-Tommola, R. Viitala, J. Hyömäki and C.-G. Swahn, Fluoroalcohols. Part 18. Infrared and Raman Spectra of the Perfluorinated t-Butyl Alcohols (CF3)3COH and (CF3)3COD, Acta Chem. Scand., 1973, 27, 107–120, DOI:10.3891/acta.chem.scand.27-0107.
- M. Isabel Cabaço, M. Besnard, C. Cruz, P. Morgado, G. M. C. Silva, E. J. M. Filipe, J. A. P. Coutinho and Y. Danten, The Structure of Liquid Perfluoro Tert-Butanol Using Infrared, Raman and X-Ray Scattering Analyzed by Quantum DFT Calculations and Molecular Dynamics, Chem. Phys. Lett., 2021, 779, 138844, DOI:10.1016/j.cplett.2021.138844.
- D. Vuluga, J. Legros, B. Crousse, A. M. Z. Slawin, C. Laurence, P. Nicolet and D. Bonnet-Delpon, Influence of the Structure of Polyfluorinated Alcohols on Brønsted Acidity/Hydrogen-Bond Donor Ability and Consequences on the Promoter Effect, J. Org. Chem., 2011, 76(4), 1126–1133, DOI:10.1021/jo1023816.
- I. L. Knunyants and B. L. Dyatkin, Perfluoro-Tertiary Butyl Alcohol and the Dissociation Constants of Trifluoromethylcarbinols, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1964, 13(5), 863–865, DOI:10.1007/BF00846509.
- E. Serjeant and B. Dempsey, Ionisation Constants of Organic Acids in Aqueous Solution, In IUPAC chemical data series, Pergamon Press, Oxford, 1979, p. 989 Search PubMed.
- M. Isabel Cabaço, M. Besnard, P. Morgado, E. J. M. Filipe, J. A. P. Coutinho and Y. Danten, Gaseous Hetero Dimers of Perfluoro Tert-Butyl Alcohol with Hydrogenated Alcohols by Infrared Spectroscopy and Quantum DFT Calculations, Chem. Phys., 2021, 544, 111110, DOI:10.1016/j.chemphys.2021.111110.
- M. I. Cabaço, M. Besnard, C. Cruz, P. Morgado, G. M. C. Silva, E. J. M. Filipe, J. A. P. Coutinho and Y. Danten, Breaking the Structure of Liquid Hydrogenated Alcohols Using Perfluorinated Tert-Butanol: A Multitechnique Approach (Infrared, Raman, and X-Ray Scattering) Analyzed by DFT and Molecular Dynamics Calculations, J. Phys. Chem. B, 2022, 126(9), 1992–2004, DOI:10.1021/acs.jpcb.1c10776.
- J. C. S. Costa, C. F. R. A. C. Lima, A. Mendes and L. M. N. B. F. Santos, Fluorination Effect on the Thermodynamic Properties of Long-Chain Hydrocarbons and Alcohols, J. Chem. Thermodyn., 2016, 102, 378–385, DOI:10.1016/j.jct.2016.07.037.
- S. Brode and I. R. McDonald, Excess Thermodynamic Properties of Liquid Mixtures of Methane and Perfluoromethane, Mol. Phys., 1988, 65(4), 1007–1012, DOI:10.1080/00268978800101561.
- J. H. Simons and R. D. Dunlap, The Properties of n-Pentforane and Its Mixtures with n-Pentane, J. Chem. Phys., 1950, 18(3), 335–346, DOI:10.1063/1.1747628.
- P. Morgado, J. Black, J. Ben Lewis, C. R. Iacovella, C. McCabe, L. F. G. Martins and E. J. M. Filipe, Viscosity of Liquid Systems Involving Hydrogenated and Fluorinated Substances: Liquid Mixtures of (Hexane + perfluorohexane), Fluid Phase Equilib., 2013, 358, 161–165, DOI:10.1016/j.fluid.2013.07.060.
- T. Handa and P. Mukerjee, Surface Tensions of Nonideal Mixtures of Fluorocarbons and Hydrocarbons and Their Interfacial Tensions against Water, J. Phys. Chem., 1981, 85(25), 3916–3920, DOI:10.1021/j150625a042.
- P. Morgado, J. Gaspar and E. J. M. Filipe, Liquid–Liquid Interfaces: Water–Perfluoroalkanes and Water–Perfluoroalkylalkanes, Experimental Interfacial Tensions and Molecular Simulation, J. Mol. Liq., 2020, 312, 113385, DOI:10.1016/j.molliq.2020.113385.
- P. Morgado, A. R. Garcia, L. F. G. Martins, L. M. Ilharco and E. J. M. Filipe, Alkane Coiling in Perfluoroalkane Solutions: A New Primitive Solvophobic Effect, Langmuir, 2017, 33(42), 11429–11435, DOI:10.1021/acs.langmuir.7b02516.
- P. Morgado, J. Barras and E. J. M. Filipe, From Nano-Seggregation to Mesophases: Probing the Liquid Structure of Perfluoroalkylalkanes with 129 Xe NMR Spectroscopy, Phys. Chem. Chem. Phys., 2020, 22(26), 14736–14747, 10.1039/D0CP02123G.
- G. M. C. Silva, P. Morgado, P. Lourenço, M. Goldmann and E. J. M. Filipe, Spontaneous Self-Assembly and Structure of Perfluoroalkylalkane Surfactant Hemimicelles by Molecular Dynamics Simulations, Proc. Natl. Acad. Sci. U. S. A., 2019, 116(30), 14868–14873, DOI:10.1073/pnas.1906782116.
- P. Morgado, A. R. Garcia, L. M. Ilharco, J. Marcos, M. Anastácio, L. F. G. Martins and E. J. M. Filipe, Liquid Mixtures Involving Hydrogenated and Fluorinated Alcohols: Thermodynamics, Spectroscopy, and Simulation, J. Phys. Chem. B, 2016, 120(38), 10091–10105, DOI:10.1021/acs.jpcb.6b04297.
- P. Duarte, M. Silva, D. Rodrigues, P. Morgado, L. F. G. Martins and E. J. M. Filipe, Liquid Mixtures Involving Hydrogenated and Fluorinated Chains: (P, ρ, T, x) Surface of (Ethanol + 2,2,2-Trifluoroethanol), Experimental and Simulation, J. Phys. Chem. B, 2013, 117(33), 9709–9717, DOI:10.1021/jp3105387.
- G. M. C. Silva, P. Morgado and E. J. M. Filipe, Towards Aqueous – Fluorous – Hydrogenous Emulsions: Phase Equilibria and Liquid Structure of (Water + 1H,1H-Perfluorobutanol + 1-Butanol) Ternary Mixture, Fluid Phase Equilib., 2020, 522, 112737, DOI:10.1016/j.fluid.2020.112737.
- E. Cea-Klapp, J. M. Garrido and H. Quinteros-Lama, Insights into the Orientation and Hydrogen Bond Influence on Thermophysical and Transport Properties in Choline-Based Deep Eutectic Solvents and Methanol, J. Mol. Liq., 2022, 345, 117019, DOI:10.1016/j.molliq.2021.117019.
- V. P. Cotroneo-Figueroa, N. F. Gajardo-Parra, P. López-Porfiri, Á. Leiva, M. Gonzalez-Miquel, J. M. Garrido and R. I. Canales, Hydrogen Bond Donor and Alcohol Chain Length Effect on the Physicochemical Properties of Choline Chloride Based Deep Eutectic Solvents Mixed with Alcohols, J. Mol. Liq., 2022, 345, 116986, DOI:10.1016/j.molliq.2021.116986.
- J. Baz, C. Held, J. Pleiss and N. Hansen, Thermophysical Properties of Glyceline–Water Mixtures Investigated by Molecular Modelling, Phys. Chem. Chem. Phys., 2019, 21(12), 6467–6476, 10.1039/C9CP00036D.
- G. Bai, L. M. N. B. F. Santos, M. Nichifor, A. Lopes and M. Bastos, Thermodynamics of the Interaction between a Hydrophobically Modified Polyelectrolyte and Sodium Dodecyl Sulfate in Aqueous Solution, J. Phys. Chem. B, 2004, 108(1), 405–413, DOI:10.1021/jp036377j.
- L. M. N. B. F. Santos, M. T. Silva, B. Schröder and L. Gomes, Labtermo: Methodologies for the Calculation of the Corrected Temperature Rise in Isoperibol Calorimetry, J. Therm. Anal. Calorim., 2007, 89(1), 175–180, DOI:10.1007/s10973-006-7509-2.
- R. Adão, G. Bai, W. Loh and M. Bastos, Chemical Calibration of Isothermal Titration Calorimeters: An Evaluation of the Dilution of Propan-1-Ol into Water as a Test Reaction Using Different Calorimeters, Concentrations, and Temperatures, J. Chem. Thermodyn., 2012, 52, 57–63, DOI:10.1016/j.jct.2011.12.018.
- I. Wadsö, Trends in Isothermal Microcalorimetry, Chem. Soc. Rev., 1997, 26(2), 79–86, 10.1039/CS9972600079.
- R. Sabbah, A. Xu-wu, J. S. Chickos, M. L. P. Leitão, M. V. Roux and L. A. Torres, Reference Materials for Calorimetry and Differential Thermal Analysis, Thermochim. Acta, 1999, 331(2), 93–204, DOI:10.1016/S0040-6031(99)00009-X.
- G. Blanquart and H. Pitsch, Thermochemical Properties of Polycyclic Aromatic Hydrocarbons (PAH) from G3MP2B3 Calculations, J. Phys. Chem. A, 2007, 111(28), 6510–6520, DOI:10.1021/jp068579w.
- G. Della Gatta, M. J. Richardson, S. M. Sarge and S. Stølen, Standards, Calibration, and Guidelines in Microcalorimetry. Part 2. Calibration Standards for Differential Scanning Calorimetry* (IUPAC Technical Report), Pure Appl. Chem., 2006, 78(7), 1455–1476, DOI:10.1351/pac200678071455.
- E. S. Domalski and E. D. Hearing, Heat Capacities and Entropies of Organic Compounds in the Condensed Phase. Volume III, J. Phys. Chem. Ref. Data, 1996, 25(1), 1–525, DOI:10.1063/1.555985.
- A. I. M. C. Lobo Ferreira, S. M. Vilas-Boas, R. M. A. Silva, M. A. R. Martins, D. O. Abranches, P. C. R. Soares-Santos, F. A. Almeida Paz, O. Ferreira, S. P. Pinho, L. M. N. B. F. Santos and J. A. P. Coutinho, Extensive Characterization of Choline Chloride and Its Solid–Liquid Equilibrium with Water, Phys. Chem. Chem. Phys., 2022, 24(24), 14886–14897, 10.1039/D2CP00377E.
- D. Van Der Spoel, E. Lindahl, B. Hess, G. Groenhof, A. E. Mark and H. J. C. Berendsen, GROMACS: Fast, Flexible, and Free, J. Comput. Chem., 2005, 26(16), 1701–1718, DOI:10.1002/jcc.20291.
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids, J. Am. Chem. Soc., 1996, 118(45), 11225–11236, DOI:10.1021/ja9621760.
- S. Nosé, A Molecular Dynamics Method for Simulations in the Canonical Ensemble, Mol. Phys., 1984, 52(2), 255–268, DOI:10.1080/00268978400101201.
- W. G. Hoover, Canonical Dynamics: Equilibrium Phase-Space Distributions, Phys. Rev. A, 1985, 31(3), 1695–1697, DOI:10.1103/PhysRevA.31.1695.
- M. Parrinello and A. Rahman, Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method, J. Appl. Phys., 1981, 52(12), 7182–7190, DOI:10.1063/1.328693.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, Lincs: A Linear Constraint Solver for Molecular Simulations, J. Comput. Chem., 1997, 18(12), 1463–1472, DOI:10.1002/(SICI)1096-987X(199709)18:12<1463::AID-JCC4>3.0.CO;2-H.
- T. Darden, D. York and L. Pedersen, Particle Mesh Ewald: An N log(N) Method for Ewald Sums in Large Systems, J. Chem. Phys., 1993, 98(12), 10089–10092, DOI:10.1063/1.464397.
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, A Smooth Particle Mesh Ewald Method, J. Chem. Phys., 1995, 103(19), 8577–8593, DOI:10.1063/1.470117.
- P. Morgado, J. Barras, A. Galindo, G. Jackson and E. J. M. Filipe, Modeling the Fluid-Phase Equilibria of Semifluorinated Alkanes and Mixtures of (n -Alkanes + n -Perfluoroalkanes) with the SAFT-γ Mie Group-Contribution Approach, J. Chem. Eng. Data, 2020, 65(12), 5909–5919, DOI:10.1021/acs.jced.0c00785.
- T. Minamihonoki, H. Ogawa, H. Nomura and S. Murakami, Thermodynamic Properties of Binary Mixtures of 2,2,2-Trifluoroethanol with Water or Alkanols at T = 298.15K, Thermochim. Acta, 2007, 459(1–2), 80–86, DOI:10.1016/j.tca.2007.04.012.
- W. Mahler, D. Guillon and A. Skoulios, Smectic Liquid Crystal from (Perfluorodecyl)Decane, Mol. Cryst. Liq. Cryst., Lett., 1985, 2(3–4), 111–119, DOI:10.1080/01406566.1985.10767000.
- P. Morgado, C. McCabe and E. J. M. Filipe, Modelling the Phase Behaviour and Excess Properties of Alkane + Perfluoroalkane Binary Mixtures with the SAFT–VR Approach, Fluid Phase Equilib., 2005, 228–229, 389–393, DOI:10.1016/j.fluid.2004.08.002.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp00701d |
| This journal is © the Owner Societies 2023 |