
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComputational investigations of stable multiple-cage-occupancy He clathrate-like hydrostructures†
Raquel
Yanes-Rodríguez
 ab and
Rita
Prosmiti
*a
ab and
Rita
Prosmiti
*a
aInstitute of Fundamental Physics (IFF-CSIC), CSIC, Serrano 123, 28006 Madrid, Spain. E-mail: rita@iff.csic.es; Tel: +34 915616800
bDoctoral Programme in Theoretical Chemistry and Computational Modelling, Doctoral School, Universidad Autónoma de Madrid, Spain
First published on 17th May 2023
Abstract
One of the several possibilities offered by the interesting clathrate hydrates is the opportunity to encapsulate several atoms or molecules, in such a way that more efficient storage materials could be explored or new molecules that otherwise do not exist could be created. These types of applications are receiving growing attention from technologists and chemists, given the future positive implications that they entail. In this context, we investigated the multiple cage occupancy of helium clathrate hydrates, to establish stable novel hydrate structures or ones similar to those predicted previously by experimental and theoretical studies. To this purpose, we analyzed the feasibility of including an increased number of He atoms inside the small (D) and large (H) cages of the sII structure through first-principles properly assessed density functional approaches. On the one hand, we have computed energetic and structural properties, in which we examined the guest–host and guest–guest interactions in both individual and two-adjacent clathrate-like sII cages by means of binding and evaporation energies. On the other hand, we have carried out a thermodynamical analysis on the stability of such He-containing hydrostructures in terms of changes in enthalpy, ΔH, Gibbs free energy, ΔG, and entropy, ΔS, during their formation process at various temperature and pressure values. In this way, we have been able to make a comparison with experiments, reaffirming the ability of computational DFT approaches to describe such weak guest–host interactions. In principle, the most stable structure involves the encapsulation of one and four He atoms inside the D and H sII cages, respectively; however, more He atoms could be entrapped under lower temperature and/or higher pressure thermodynamic conditions. We foresee such accurate computational quantum chemistry approaches contributing to the current emerging machine-learning model development.
Computational tools are in constant development, allowing one to obtain deep knowledge of physico-chemical properties beyond what can be directly measured experimentally, and providing valuable information on new molecules and materials that can be supplemented by experimentation.1 Density functional theory (DFT) arose as the method of choice to study the electronic structure of chemical species, especially for large molecular systems, such as clusters and solids. This has enabled the availability of multiple datasets for an incredibly huge number of molecules, which nowadays, with the advancements in machine learning techniques, elicits a revolution in the field.2,3 The combination of computational methods and data-driven techniques results in reduction of time and cost, and multiple studies have already been reported in assorted areas, such as molecular design4–7 and force-field development.8,9
One of the systems that is in the spotlight for these novel technologies is the clathrate hydrates. These inclusion compounds are predicted to have promising applications,10–15 such as in storage materials or energy sources, and are considered as future “green tools” to reduce the greenhouse gas emissions from industrial processes,16 such as chemical and power plants.17,18 Therefore, further knowledge on these complex structures in terms of formation, stabilization, dissociation, phase behaviors or thermodynamic properties19 would facilitate the necessary progress to achieve a feasible industrial implementation.13,20,21
Within the group of such inclusion compounds, helium clathrate hydrates are one of the less-explored systems.22–29 Although their structure was predicted a long time ago,22,26 it was not until 2018 that the first helium clathrate hydrate was experimentally formed.29 This synthesis was possible thanks to a previous discovery30 in which it was confirmed that guest Ne atoms can be removed from clathrate hydrates, giving rise to new stable or metastable structures that can remain empty (the case of ice XVI) or be filled by another guest atom (the case of He@sII). Precisely, the guest atom not only determines the type of structure that is formed, but also the occupancy of the water cages present in the structure. For example, He hydrate forms are based on filled ice II, while Ar has not been found in any filled ice structure yet. Also, certain small molecules, such as noble gases, H2 or N2,31–33 allow multiple cage occupancy, whereas other larger ones, such as CH4, only permit a single molecule per water cage.34–38 The large cages usually have around full occupancy, but it is not mandatory for the stabilization of the crystals. In some cases there may even be empty cages if the occupancy of the guests is sufficient to stabilize the whole hydrate lattice.39–41
Despite the fact that a priori helium clathrate hydrates may not seem as interesting as those of methane or carbon dioxide (given their environmental impact), the importance of studying this noble gas compound lies in the fact that He atoms are one of the most promising guest species for the synthesis of new water structures.29,30,42–47 The small size and low reactivity of this noble gas atom facilitate its removal by pumping from the structure where it is contained, with the consequent discovery of new icy-water structures. In this context arise the ultra-low-density ices,48–54 structures belonging to the less-examined pressure regions of the phase diagram, which are predicted to be the most stable solid phases of water. Other interesting compounds that have attracted attention in both theoretical and experimental studies are the superionic states of helium–water systems at high pressures.55 Such new phases combine liquid-like and solid-like behavior, and are characterized by a fixed sublattice of oxygen atoms, while the hydrogen (and/or helium) atoms diffuse through it almost freely. Understanding the mechanism of diffusion and formation conditions in these states is of great relevance in developing battery materials and new models in planetary science.55,56
Therefore, given the great interest aroused by the aforementioned helium clathrate hydrates, in this work we share new insights into these inclusion compounds as a part of our line of research.57–59 In previous studies, we have benchmarked the performance of different DFT functionals through a validation protocol in comparison with well-converged wavefunction (WF)-based reference data, and we have identified the best-performing functionals in describing such weak guest–host interactions, together with the appropriate dispersion correction schemes. Now we intend to go one step further by analyzing the effect of multiple cage occupancy in the sII clathrate hydrate in terms of structural, energetic and thermochemical properties. Having in mind that experimentalists have predicted that in the He@sII crystal structure the small cage is occupied by only one He atom, and the large cage by four He atoms,29 our present study aims to find out if such a prediction on multiple cage filling holds for the individual and two-adjacent sII clathrate-like cages from first-principles DFT computations. In fact, our current research focus has been to establish the connection with nanoscale science, as cluster models could serve as building blocks for solid-state materials.60 In this sense, finite-size cluster systems may evolve smoothly toward bulk properties, allowing extrapolation from few-body to many-body behavior, while sometimes significant changes in the nanoscopic properties occur, enabling different applications. To this purpose, we have examined how many helium atoms could in principle be encapsulated inside the isolated cages (energetically favored); we have then tested if there is any effect on the interaction of the closest-neighbor cages, and next, we have analyzed the stability of the cage systems trapping an increased number of He atoms by means of thermochemistry calculations under a range of temperature (T) and pressure (P) conditions comparable to those reported by experiments.
1. Computational details
1.1 Target hydrate structures
In Fig. 1, we display all clathrate-like cluster structures under consideration in this work, which consist of the individual small 512 or D and large 51264 or H cages, formed by 20 water molecules with a diameter of 7.4 Å, and 28 water molecules with a diameter of 9.0 Å, respectively, as well as two adjacent cages contained in the sII hydrate crystal. Taking into account the connections established in the sII lattice (see Fig. 1), three such cage systems will be considered: the small–small (DD) system composed of 35 water molecules, and the small–large (DH) system formed by 43 water molecules, both sharing a pentagonal face between the cages, and the large–large (HH) system containing 50 water molecules, with the H cage sharing, an hexagonal face.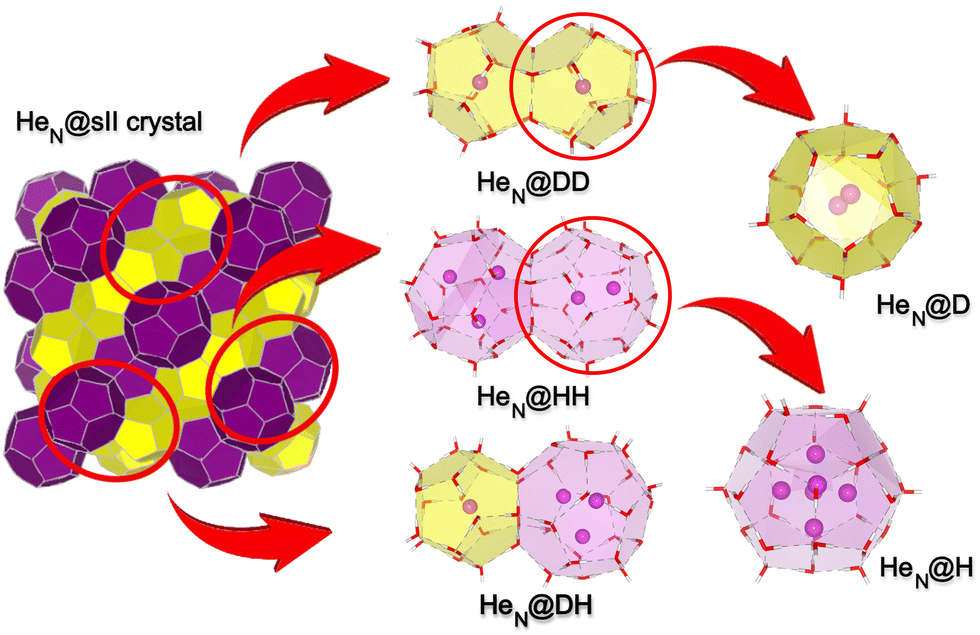 | ||
| Fig. 1 He@sII crystal structure and clathrate-like hydrostructures. | ||
All cage system configurations were extracted, using the DENEB software package,61 from the three-dimensional (3D) crystalline frameworks of the sII clathrate hydrates, as have been determined in ref. 62. The positions of the water oxygen atoms were experimentally determined by X-ray diffraction analysis of clathrate hydrate structures, while the positions of the water hydrogen atoms were ordered in such a way that they satisfy the ice rules, having the lowest potential energy orientation for the protons, and a net zero dipole moment. It has been found62 that different proton distributions show a fairly narrow potential energy spread, and thus a rather small perturbative effect on the interaction energies of the guests in the cage systems is expected. However, such water rearrangements have been found to affect the nuclear quantum translational–rotational dynamics of guest molecules.63,64 In turn, taking into account that in crystal clathrates each water molecule participates in four hydrogen bonds, which probably restricts its orientation, it could be considered that guest–host effects should be described better in the case of the rigid cages. Hence, at least initially, the guest–cage can be treated as an isolated system to a very good approximation, providing an excellent starting point for gradual inclusion of the weak interactions with the extended environment, such as guest–guest in the two-adjacent cages or guest-lattice effects in periodic crystals later on. Thus, each of these empty (rigid) sII cage structures is gradually filled with an increasing number (N) of He atoms (see Fig. 1), and their structures (HeN@D/H, HeN@DD/DH/HH) and energies have been then computed, as described below.
1.2 Electronic structure calculations
Geometry optimization and single point calculations were carried out for the empty and filled cage systems using both WF-based methods, such as CCSD(T), domain-based local pair natural orbital coupled-cluster (DLPNO-CCSD(T)) and its explicitly correlated (-F12) counterpart,65,66 and DFT approaches, such as the GGA PW86PBE67,68 and the hybrid PBE069 methods. The choice of these DFT functionals in describing the interactions is based on their performance against WF-based methods, as reported in previous studies among various GGA, meta-GGA and hybrid functionals for similar clathrate-like cage and crystal structures.57,58,70,71 Thus, the DFT computations were carried out with the Gaussian72 and QE73–75 software, while the CCSD(T) and DLPNO-CCSD(T)/DLPNO-CCSD(T)-F12 calculations were performed with the MOLPRO76–78 and ORCA79,80 packages of codes, respectively. Since dispersion forces have been found to significantly affect the underlying interactions in these systems,57,58 all DFT calculations have taken into account such contributions through the D481–83 and XDM84,85 dispersion correction schemes, as implemented in the DFTD486 and POSTG87,88 or QE codes, respectively.In the DLPNO-CCSD(T) calculations, the augmented correlation consistent basis sets, AVXZ, were used, in addition to the equivalent correlation fitting AVXZ/C basis set for the resolution of the identity (RI). The tight threshold for both self-consistent field (SCF) and pair natural orbital (PNO) settings was specified to achieve better converged wave-functions and to reduce numerical noise, respectively. In the case of DLPNO-CCSD(T)-F12 computations, correlation consistent basis sets, VXZ, were employed, together with the equivalent correlation fitting, AVXZ/C, and a complementary auxiliary basis set, VXZ-F12/CABS. In the rest of the WF-based and density-based methods, the AVXZ basis sets were used. In particular, geometry optimization calculations of N isolated He atoms were carried out using CCSD(T)/AVXZ (with X = 6 for N = 2–4 He atoms, X = 5 for N = 5–6, X = Q for N = 7–8, and X = T for 9 He atoms) and for both isolated HeN and those encapsulated inside the sII (D, H, DD, HH, DH) cage hydrostructures, the PW86PBE/AVTZ level of theory was used. The basis set superposition error (BSSE) through the counterpoise correction (CP)89 was applied to the energies in the CCSD(T), DLPNO-CCSD(T) and DLPNO-CCSD(T)-F12 results, while the weighted average complete basis set (CBS) extrapolation scheme of the CP-corrected and uncorrected energies developed by Lee et al.90 was used in the case of the CCSD(T) calculations to obtain well-converged CCSD(T)/CBS interaction energies.
In order to analyze the stability (energetically favored) of the different cage systems under study, two quantities are considered: the binding energies,  , and evaporation energies,
, and evaporation energies,  , where
, where  corresponds to the total energy of the optimized HeN@cage/s system formed by the rigid cage/s encapsulating HeN atoms, Ecage/s to the total energy of the clathrate cage/s,
corresponds to the total energy of the optimized HeN@cage/s system formed by the rigid cage/s encapsulating HeN atoms, Ecage/s to the total energy of the clathrate cage/s,  to the total energy of the optimized isolated HeN atoms, and EHe to the total energy of a single He atom.
to the total energy of the optimized isolated HeN atoms, and EHe to the total energy of a single He atom.
Moreover, we also examined the thermodynamic stability by analysing the enthalpy (ΔH), Gibbs free energy (ΔG) and entropy (ΔS) variations, under different T–P conditions, involved in the encapsulation of one or several (N) He atoms inside the cages of the sII structure, both individual (D and H) and connected to their neighbor cage (DD, HH and DH). Consequently, these properties can give us an idea about the thermodynamic feasibility and the spontaneous nature of the clathrate formation process. The thermochemical quantities have been determined through standard statistical thermodynamics techniques for finite-size systems,91 as implemented in the Gaussian package,72,92 by carrying out DFT calculations. Therefore, the geometry optimizations of the HeN atoms inside the rigid cage systems are followed by frequency calculations at PBE0-D4/AVTZ level of theory. The ΔH, ΔG and ΔS are calculated for the HeN@cage/s clathrate formation reactions as ΔH/G/S = H/G/SHeN@cage/s − H/G/Scage/s − H/G/SHeN, where H = E + ZPE + ε0, G = H − T × S and Stot = Se+ Sv + Sr + St, with E being the total electronic energies, ZPE the zero-point vibrational energy corrections, ε the internal thermal corrections to the energy (given by electronic, vibrational, rotational and translational contributions) and Se, Sv, Sr and St the electronic, vibrational, rotational and translational contributions to the entropy.
2 Results and discussion
2.1 Benchmarking the simplest He–He and He–H2O interactions
In our previous studies57,58 a variety of DFT functionals were assessed for different He clathrate hydrate structures, by extensive and systematic comparisons against well-converged WF-based reference data, concluding that the PW86PBE-XDM/D4 and PBE0-D4 functionals are the best-performing. As we are interested in the multiple cage occupancy of such systems, we decided to check the performance of these DFT-D functionals on the simplest He–He and He–H2O interactions involved in the present case too.Thus, in Fig. 2 we plot the potential curves for the He2 (left panel) and He–H2O (right panel) systems as a function of the He–He and He–O distances (see inset plots), respectively, as obtained from the CCSD(T), DLPNO-CCSD(T)/F12, PW86PBE/XDM/D4 and PBE0/XDM/D4 calculations in this work, as well as from the semiempirical and ab initio model potentials available in the literature.57,93–97 Starting with the PW86PBE functional, one can see that dispersion correction schemes are needed to correctly represent both interactions. The D4 scheme substantially improves the description of the minima, giving results close to those of DLPNO-CCSD(T)-F12/AVQZ (within 0.1 cm−1) in the He–He case, while for the He–H2O interaction the PW86PBE-D4 energies are lying between the DLPNO-CCSD(T)/AVQZ (above by ≈1.4 cm−1) and DLPNO-CCSD(T)-F12/AVQZ (below by ≈0.6 cm−1) values. As regards the PW86PBE-XDM energies, there is a surprising level of agreement with the accurate He–He model potential of ref. 96, and a difference of 0.8 cm−1 with respect to the CCSD(T)/CBS56 results. Moreover, we have also examined the efficiency of the PW86PBE-XDM/D4 functional in the He3 case, as one can see in Fig. S1 in the ESI,† verifying once again that PW86PBE-XDM provides CCSD(T)-like results. Now, in the case of He–H2O, the PW86PBE-XDM energy values are found to be between the CCSD(T)/AV5Z and DLPNO-CCSD(T)/AVQZ results, shifted by ≈1.5 cm−1 from each of them. As for the PBE0 and PBE0-D4/XDM functionals, we found that they overestimate binding, although in the He–H2O case the pure PBE0 functional provides energy values that almost overlap with those from the DLPNO-CCSD(T)/AVQZ calculations. Such behavior has been also observed previously in the clathrate-like cages;58 thus, together with PW86PBE-XDM/D4, we will also take into consideration the PBE0-D4/XDM functional.
 | ||
| Fig. 2 Interaction energy curves for the He–He and He–H2O systems obtained from the indicated WF-based and DFT-based methods, as well as with various potential models. [1] CCSD(T)/CBS[56] for He2 and CCSD(T)/AV5Z for He–H2O. | ||
2.2 Multiple-cage-occupancy effects: structures and energetics
The first helium clathrate hydrate, He@sII, was synthesized in 2018,29 and its structure has been analyzed by neutron diffraction experiments. Previous reported data from thermodynamic modeling26 and ab initio molecular dynamics98 have indicated that the large H cages of the sII structure could encapsulate up to 4 He atoms in tetrahedrally degenerate positions, while the small D cages could be occupied by one or two He atoms. Similar findings have been also reported for the multiple occupancy in H2@sII,99–102 where the large cages could entrap up to 4 H2 molecules, although the double and triple occupancy appeared to be the most stable, while the small cages remained with only one guest hydrogen molecule. Although the H atom is larger than the He one, we can observe comparable tendencies in terms of multi-occupancy of the cages and in general, it seems that the most stable configuration is the one that minimizes the occupancy of the small cages and maximizes that of the large cages. Therefore, we first analyze here the multiple helium occupancy in the D and H cages of the sII structure on the basis of DFT calculations on binding and evaporation energies.We start by performing geometry optimization calculations for both isolated He atoms (at CCSD(T)/AV(T/Q/5/6)Z and PW86PBE/AV(T/Q)Z levels of theory), and those encapsulated inside the rigid D and H sII cages (at the PW86PBE/AVTZ level of theory). For this purpose, we introduced up to 4 and 10 He atoms inside the D and H cavities, respectively. The resulting configurations can be seen in the left panel of Fig. 3, with the corresponding energies listed in Tables S1–S3 (see ESI†). One can distinguish ordered positions in the HeN complexes with up to 6 He atoms, such as triangles, pyramids or even rhombuses, while for species formed from 7 to 9 He atoms it starts to become difficult to recognize orderly shapes. We have also optimized the He10@H system; however, the results show that one He is ejected from the cage (see Fig. S2 in the ESI†). In the upper right panel of Fig. 3, the average He–He bond lengths of isolated HeN and encapsulated HeN@D/H atoms are represented from the PW86PBE calculations. As is shown, the PW86PBE functional does not adequately estimate the He–He bond lengths, so we simply concentrate our attention on the observed trends. As expected, the He–He bond length decreases as we move from the free to trapped He atoms in the H and D cages, and also as the number of caged He atoms is increasing. Such modifications in the bond distances for noble-gas molecules encapsulated in fullerenes have already been reported in the literature, resulting in a change in the noble gas reactivity too.103
 | ||
| Fig. 3 Left panel: Optimized structures for the isolated HeN atoms and encapsulated HeN@D/H at CCSD(T) and PW86PBE levels of theory, respectively. Right upper panel: Average He–He bond-lengths (in Å) from the geometry optimization PW86PBE/AVTZ calculations. Right lower panel: Evaporation energies (in cm−1) from the PW86PBE/XDM/D4 calculations. | ||
In the lower right panel of Fig. 3, evaporation energies calculated at the PW86PBE/AVTZ level, including D4 and XDM dispersion corrections, are plotted as a function of the number of trapped He atoms. This quantity gives us an idea of the energy gain when an extra He atom is added in the cage/s. One can see that the pure PW86PBE functional predicts unbound cage systems, and therefore, we will only discuss the DFT-D results. As we can see, the energy shoots up when more than one He atom is encapsulated in the D cage, although in principle the inclusion of two He atoms is energetically favored too. Regarding the H cage, the energy goes slightly down until it is filled with four He atoms, which is the most stable configuration in this case, and then it begins to rise, remaining favorable up to seven trapped He atoms. In order to further analyze the energetic stability of these cage systems, the binding energies calculated at PW86PBE-D4/XDM and PBE0-D4/XDM levels of theory are shown in Fig. 4. Once we had checked that there was no significant difference between the AVTZ and AVQZ results (see Fig. S3 in the ESI†), we decided to analyze the AVTZ curves, considering the PW86PBE/AVTZ optimized geometries. It should be noted that whilst the evaporation energies refer to the energy gain with respect to the addition of an extra He atom, the binding energies correspond to the total energy gain of the cage system in contrast with its isolated fragments (water cages and He-atoms) in their equilibrium.
 | ||
| Fig. 4 Binding energies for the HeN@D/H systems, with N up to 4 atoms inside the D cage and N up to 9 atoms in the H cage, from the PW86PBE-XDM/D4 and PBE0-XDM/D4 calculations. | ||
Starting with the D cage, both the PW86PBE-D4/XDM and PBE0-D4/XDM functionals predict the same behavior as that found for the evaporation energies, although PBE0-D4 estimates that He2@D is slightly more favored than He@D in terms of energy, with the inclusion of 3 or more He atoms being totally unfeasible. As for the H cage, a different trend is observed. The PW86PBE-D4/XDM functional predicts that the most favorable configurations are those entrapping 4 to 6 He atoms, and in principle up to 7 He atoms can be encapsulated, while the PBE0-D4/XDM results show that the inclusion of 6 He atoms implies the most stable configurations, and that up to 9 (or even a few more) He atoms could get trapped.
So far, we have analyzed the guest–host interactions in individual clathrate-like cages, and in turn we intend to explore the inter-cage effects by means of two-adjacent DD, HH and DH cage systems. As before, the computations for double clathrate-like cages have been carried out for empty and single or multiple He cage filling. On the basis of the individual cage results, we considered examining a total of 18 different configurational combinations, consisting of up to two He atoms inside the D cages and 4 He atoms trapped in the H cages, such as He(1/0)@DD, He(1/1)@DD, He(2/0)@DD, He(1/2)@DD, He(2/2)@DD, He(0/1)@DH, …, He(4/4)@HH, with the (n/m) index indicating the number of He atoms in each (D/H) cage of the DD, DH and HH systems. In Fig. 5 the binding energies obtained from the PW86PBE-XDM and PW86PBE-D4 calculations are presented, and both results are in accord.
The first aspect that stands out is the surprisingly strong binding for the He(1/0)@DD and He(2/2)@DD cage systems. Such a trend has already been observed for the individual cages, where the inclusion of one He atom inside the D cage involved a fairly large gain in energy, being even more favorable than introducing more He atoms into the H cage. For the DD cages, the most stable configuration is predicted to be the one with one singly occupied cage (1/0), although the one with maximum (2/2) occupancy in both D cages is also close in energy. In turn, for the HH cage system, once again the most stable configuration is the one with the maximum number of He's inside both cages, He(4/4)@HH. In fact, the maximum occupancy of 4 He's in at least one H cage is always prioritized. As regards the DH cage system, the most energetically favored configuration is found to be the He(1/4)@DH one, in agreement with the experiment, while He(2/4)@DH is also very close in energy, just within 100 cm−1 above it. Moreover, as just mentioned, the presence of one He inside the D cage (1/0) is preferred over the (0/1) in the H cage. In general, it holds again that the maximum occupancy prevails in the H cages. Indeed, the (0/4) configuration is one of the most energetically favored, which may support some investigations that defend H cages needing to be completely filled or nearly so, while some D cages could even remain empty if the stabilization provided by the rest of the cages in the clathrate hydrate is sufficient.39–41 Finally, for the PW86PBE-D4 results (see Fig. 5), the same tendency is observed for the DD and HH cage systems. Nonetheless, some differences are encountered in the DH cages, such as the second most energetically favored configuration being (0/4) rather than the (2/4) one, and the least stable one being (2/0) instead of (0/1).
 | ||
| Fig. 5 Binding energies (in cm−1) of two-adjacent DD, DH, and HH HeN@sII cage systems computed at PW86PBE-XDM and PW86PBE-D4 levels of theory, assuming the indicated single and multiple He cage occupancy. | ||
Taking into account all this information, we can infer that the most energetically efficient filling processes correspond to just one singly occupied D cage, He(1/0)@DD, at an energy of −1813.4 cm−1, or doubly occupied cages He(2/2)@DD, at −1661.4 cm−1, followed by tetrahedrally occupied cages He(4/4)@HH, at −1068.7 cm−1, and by the singly occupied D in combination with tetrahedrally occupied H in He(1/4)@DH systems, at an energy of −643.5 cm−1. The fact that the He(1/0)@DD cage system is the most stable configuration is astonishing, although consistent with the results obtained for the individual D sII cages. Among the 18 configurations analyzed here, we found that the presence or absence of He atoms in the neighbor cage has a clear impact on the energy, and altogether, the H cages seek to be overloaded, whereas the D cages prefer minimal He occupancy.
2.3 Multiple cage occupancy effects: thermochemical properties
The determination of thermochemical properties is crucial to ascertain the viability of a particular reaction and to know if it entails an energy absorption or release. In this context, the calculation of enthalpy (ΔH), Gibbs free energy (ΔG) and entropy (ΔS) variations is a valuable tool to understand the stability of a system and its spontaneous evolution towards an equilibrium state. Further, when experimental data are available, it can serve as a point of comparison to verify the reliability of theoretical results, and moreover could be useful for the development of corrections in theoretical predictive methodologies. In the case of the helium clathrate hydrates, only the He@sII structure has been observed in recent laboratory experiments,29 and its synthesis has been reported in the range of 80–120 K and 50–150 MPa. Having this in mind, our next step consists of evaluating ΔH, ΔG and ΔS for the HeN + D/H → HeN@cage formation reactions by means of DFT-D geometry optimizations and frequency calculations under a wide variety of temperature (T) and pressure (P) conditions, ranging from 50–298 K and 1–1500 atm, respectively.As in a previous work58 the ΔH and ΔG variations have been examined for the single He occupation of the individual D and H cages of the sII structure, we here focused on studying the multiple He cage occupancies. First, we have analyzed the He2@D and He4@H cases by studying the influence of more He guests on the thermodynamic stability of these systems. The ΔH as a function of temperature is displayed in Fig. 6 in comparison with the corresponding single He occupancy cases. The range of experimental conditions is highlighted in the plot, and as one can see, the effect resulting from the inclusion of more than one He atom in the D cage is not visible at these temperatures. Translated into numbers, at 50–80 K the difference between enthalpies is approximately 1%, at 100 K it is 3% and at 120 K it is around 64%. In spite of the fact that this difference is greater as the temperature increases, in any case ΔH is negative, which means that the encapsulation of one or two He atoms inside the D cage is an exothermic process with a consequent release of energy. Nevertheless, at temperatures higher than 298 K the process is expected to become endothermic, especially in the He2@D case. Continuing with the H cage, in contrast to the D case, the entrapment of one or four He's has a noticeable impact on ΔH values. Although in both systems the process is exothermic, the trend observed as the temperature increases is totally opposite. While in He@D the higher the temperature, the lower the ΔH, in He4@H the tendency is contrary to this, as observed in the D cage. Furthermore, the influence of the temperature on the ΔH variation is more prominent in the H cages than in the D cages.
 | ||
| Fig. 6 ΔH (in kcal mol−1) as a function of temperature (in K) for the multiply occupied He2@D (squares) and He4@H (downwards triangle) sII clathrate-like cages in comparison with the data from ref. 58 for the singly occupied He@D (circles) and He@H (upwards triangle) systems. The dashed red boxes indicate the reported experimental conditions. | ||
In turn, Fig. 7 shows the corresponding results of the ΔG energies as a function of temperature and pressure. The formation of the single-occupied He@D/H cages has been reported to be spontaneous in the range of experimental conditions.58 Nonetheless, the incorporation of more He atoms inside these hydrostructures may entail a modification of the spontaneity of the reaction. Starting with He2@D, we see that ΔG is positive in all the T and P values considered, with the exception at 50 K between 500–1500 atm, which signifies that the encapsulation of two He's inside the D cage is a spontaneous process, and therefore, the reaction benefits by itself the formation of the products under this given set of conditions. In the case of He4@H, ΔG also takes positive values under most of the conditions studied. Surprisingly, however, at 80–100 K and 500–1500 atm, which are within the experimental regime, ΔG is negative and therefore, the reaction is thermodynamically favored. These outcomes are very satisfactory, since they fully correspond to those observed in experiments.29 In principle, it seems that the inclusion of one He inside the D cage and four He's inside the H one corresponds to spontaneous processes in the range of experimental conditions, while the encapsulation of two He's in the D case is also spontaneous only at lower temperatures.
 | ||
| Fig. 7 ΔG (in kcal mol−1) as a function of temperature (in K) at various pressure values (in atm) for the multiply occupied He2@D (squares) and He4@H (downwards triangle) sII clathrate-like cages in comparison with the data from ref. 58 (see dashed lines in the inset plots) for the singly occupied He@D (circles) and He@H (upwards triangle) systems. The dashed black boxes indicate the experimental conditions. | ||
Finally, in order to complete the thermodynamic stability analysis of the individual D and H cages, we have also examined the change in entropy, ΔS, as shown in Fig. 8. We see that in both singly, He@D/H (except at high pressures), and multiply occupied, He2@D and He4@H, cages, ΔS has negative values, whatever the temperature and pressure values are. This indicates that the disorder of the reactants has decreased or, in other words, the encapsulated He atoms inside the individual D/H cages are more ordered than in their isolated configurations. This makes sense, given that the He's inside the clathrate-like cages become closer, and have less randomness as a result of the volume decrease. Moreover, taking into account that ΔG = ΔH − TΔS and that ΔH and ΔS are negative, this means that the spontaneity of the reaction will depend on the temperature value, as in fact we have verified in the analysis of ΔG. Finally, we should note two peculiarities. On the one hand, different slopes are observed in the curves: while in the He@D, He2@D and He4@H cases ΔS increases with the temperature, in the He@H case the tendency is the opposite. This behavior was already observed for ΔH. In any case, the change suffered as a consequence of the temperature is not significant (10−4 order of magnitude between temperature values), ΔS being more affected by the pressure (10−3 order of magnitude between pressure values), although also to a very low extent. On the other hand, the more He atoms are encapsulated, the more negative ΔS is, as it implies a higher ordering with respect to the initial isolated structures.
 | ||
| Fig. 8 ΔS (in kcal mol−1 K−1) as a function of temperature (in K) at various pressure values (in atm) for He@D (circles), He2@D (squares), He@H (upwards triangles) and He4@H (downwards triangles) cage systems. The dashed black boxes indicate the experimental conditions. | ||
In the same vein, we next examined the evolution of ΔH, ΔG and ΔS in the two-adjacent sII cage systems. Given the size of the double-cage systems and the increase in computational resources, we decided to analyze the stability of the experimentally observed He(1/4)@DH structure, through the He + He4 + DH → He(1/4)@DH formation reaction, considering only the range of experimental T–P conditions, between 80–120 K for temperatures and 500–1500 atm for pressures. The obtained results are shown in Fig. 9, and we comment below the observed changes with respect to the individual D/H cages and their possible extrapolation from these smaller entities.
 | ||
| Fig. 9 ΔH, ΔG and ΔS as a function of T (in K) and P (in atm) values comparable to the experiment for the He(1/4)@DH cage system. | ||
In general, we observe similar trends to those in the individual D and H sII cages. The ΔH values are also negative (exothermic reaction), presenting the same behavior as in He@H, that is, the higher the temperature the more negative ΔH is. ΔG only corresponds to a spontaneous process for T = 80 K and T = 500–1500 atm (similar to He4@H), meaning that this reaction is favored at lower T and higher P values. Finally, ΔS is positive throughout the studied range, indicating that the encapsulated structure is becoming more disordered than the reactant fragments, which are indeed thermodynamically more stable. So, if we recapitulate, we see that the quantity that is most affected by the effects of neighbor-cage occupation is ΔG, showing that low temperatures and high pressures conditions are necessary to favor the corresponding formation reactions. Indeed, this fact matches with the standard conditions under which such clathrate hydrates are formed.
3 Summary and conclusions
We have evaluated the multiple cage occupancy effects on the stability of the HeN@sII clathrate-like cage systems through first-principles DFT-D approaches and under a variety of P–T conditions, motivated by previously reported experimental and theoretical studies. Once we had ensured the reliability of the PW86PBE-XDM and PBE0-D4 functionals on the simplest He–He and He–H2O interactions, we performed geometry optimizations for both isolated gas-phase HeN complexes and their encapsulated analogs inside the individual (D/H) and two-adjacent (DD/DH/HH) sII clathrate-like cage systems, determining evaporation and binding energies as the number of trapped He atoms increases. Our results predict that both He@D and He2@D structures are energetically favored, with the inclusion of just one He inside the D cage being the most stable configuration, while for the H cage, the most energetically favored configurations are those including 4–6 He's, although up to 7–8 He's could be encapsulated. Next, by analyzing the inter-cage effects of first-neighbors, we have concluded that for the DD systems the most energetically efficient filling processes are that with just one singly occupied cage, He(1/0)@DD, and the one with doubly occupied cages, He(2/2)@DD, while in HH cages the most favored one is the doubly tetrahedrally occupied He(4/4)@HH, and in DH it is the one with singly and tetrahedrally occupied D and H cages, He(1/4)@DH. Furthermore, we have been able to verify that the presence/absence of He atoms in the neighbor sII cage clearly influences the energy and in general, the H cages seek multiple He occupancy, while on the contrary, D cages prefer minimal He filling.Finally, we have examined the stability of the multi-occupied individual D and H, as well as the double DH sII clathrate-like cages, by means of enthalpy, ΔH, Gibbs free energy, ΔG, and entropy, ΔS, variations. To this purpose, a wide range of temperature (50–298 K) and moderate pressure (1–1500 atm) values were selected, including the regime of experimental conditions. Our results reveal that while the formation reactions of He@D and He@H are exothermic and spontaneous in the range of experimental conditions, those associated with the formation of He2@D, He4@H and He(1/4)@DH are exothermic and mostly non-spontaneous. In order to make them thermodynamically favored, lower temperature and/or higher pressure values are needed. In all individual cage cases studied (except He@D at high pressures) the encapsulated He atoms inside the sII cages are more orderly structured than their isolated counterparts.
In summary, we have verified that the conclusions extracted from the study of the individual cages, as well as from the two-adjacent cage systems, are very similar and are in accord with the experimental observations of a multiple cage filling of He atoms (up to 4) in the H cages of the sII clathrate hydrates and single He occupancy of the D cages. Moreover, we have verified that some D sII cages may even stay empty if the stabilization provided by the overloaded H cages is enough, while the inclusion of two He atoms in D cages is possible by adjusting the thermodynamical conditions.
On the one hand, such findings are in line with available experimental and theoretical predictions, indicating that the analysis of the fundamental entities (such as the building block clathrate-like cages) may be representative enough to get a first idea of the stability of such inclusion compounds under specific thermodynamic conditions, although several methodological limitations, such as the pressure control on finite size systems,104–106 should be further examined. On the other hand, current investigations involve exploration of the multiple cage occupancy effects by means of DFT-D computations in periodic crystals, in order to check if the predictions observed here on stability and He clustering inside the clathrate-like cages are also fulfilled in He@sII crystal structures. Additionally, other clathrate hydrates and ice polymorphs for which there is experimental evidence or a theoretical prediction can be investigated, such as He@III, He@Ih or He@C0. Both fundamental and technological progress is essential to be able to exploit the multiple auspicious applications that clathrate hydrates offer in the near future, which would be beneficial not only for the battle against global warming and other environmental concerns, but also for the discovery of materials with unique and advantageous properties, such as new ice phases, and development of novel models of relevance in planetary science.
4. Data availability
The data that support the findings of this study are available within the article and its ESI,† as well as from the authors upon reasonable request.Author contributions
The authors confirm contributions to the paper as follows: R. Y.-R. (data curation, formal analysis, investigation, methodology, validation, writing – original draft preparation) and R. P. (funding acquisition, project administration, conceptualization, formal analysis, investigation, methodology, validation, writing – review and editing).Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the “Centro de Cálculo del IFF-CSIC and SGAI-CSIC” and CESGA-Supercomputing Centre for allocation of computer time. This work has been supported by MICINN grant no. PID2020-114654GB-I00, and COST Actions CA18212(MD-GAS) and CA21101(COSY).Notes and references
- Nat. Commun., 2020, 11, 4811 Search PubMed.
- A. Tkatchenko, Nature, 2020, 11, 4125 CAS.
- J. A. Keith, V. Vassilev-Galindo, B. Cheng, S. Chmiela, M. Gastegger, K.-R. Müller and A. Tkatchenko, Chem. Rev., 2021, 121, 9816–9872 CrossRef CAS PubMed.
- V. Singh, S. Patra, N. A. Murugan, D.-C. Toncu and A. Tiwari, Mater. Adv., 2022, 3, 4069–4087 RSC.
- J. Westermayr, M. Gastegger, K. T. Schütt and R. J. Maurer, J. Chem. Phys., 2021, 154, 230903 CrossRef CAS PubMed.
- E. A. Engel, CrystEngComm, 2021, 23, 252–263 RSC.
- M. Staszak, K. Staszak, K. Wieszczycka, A. Bajek, K. Roszkowski and B. Tylkowski, WIREs Comput. Mol. Sci., 2022, 12, e1568 CrossRef.
- O. T. Unke, S. Chmiela, H. E. Sauceda, M. Gastegger, I. Poltavsky, K. T. Schütt, A. Tkatchenko and K.-R. Müller, Chem. Rev., 2021, 121, 10142–10186 CrossRef CAS PubMed.
- O. T. Unke, S. Chmiela, M. Gastegger, K. T. Schütt, H. E. Sauceda and K.-R. Müller, Nat. Commun., 2021, 12, 7273 CrossRef CAS PubMed.
- C. A. Koh and E. D. Sloan, AIChE J., 2007, 53, 1636–1643 CrossRef CAS.
- X. Wang, M. Dennis and L. Hou, Renew. Sust. Energ. Rev., 2014, 36, 34–51 CrossRef CAS.
- A. Davoodabadi, A. Mahmoudi and H. Ghasemi, iScience, 2021, 24, 101907 CrossRef CAS PubMed.
- P. Sahu, J. Water Process. Eng., 2021, 41, 102058 CrossRef.
- Y. Kuang, L. Zhang and Y. Zheng, Energy, 2022, 252, 124082 CrossRef CAS.
- J. A. Ripmeester and S. Alavi, Clathrate Hydrates: Molecular Science and Characterization, Wiley-VCH GmbH, 2022 Search PubMed.
- A. Li, J. Wang and B. Bao, Greenh. Gases: Sci. Technol., 2019, 9, 175–193 CrossRef CAS.
- Y.-H. Ahn, D. Lim, J. Min, J. Kim, B. Lee, J. W. Lee and K. Shin, Chem. Eng. J., 2019, 359, 1629–1634 CrossRef CAS.
- N. Thakre and A. K. Jana, Renew. Sust. Energ. Rev., 2021, 135, 110150 CrossRef CAS.
- H. Tanaka, T. Yagasaki and M. Matsumoto, AIChE J., 2020, 67, e17009 Search PubMed.
- M. Khurana, Z. Yin and P. Linga, ACS Sustainable Chem. Eng., 2017, 5, 11176–11203 CrossRef CAS.
- C. I. Ratcliffe, Energy Fuels, 2022, 36, 10412–10429 CrossRef CAS.
- D. Londono, J. L. Finney and W. F. Kuhs, J. Chem. Phys., 1992, 97, 547–552 CrossRef CAS.
- K. A. Udachin, J. Lipkowski and M. Tkacz, Supramol. Chem., 1994, 3, 181–183 CrossRef CAS.
- Y. Dyadin, E. Larionov, E. Aladko, A. Y. Manakov, F. V. Zhurko, T. V. Mikina, V. Y. Komarov and E. V. Grachev, J. Struct. Chem., 1999, 40, 790–795 CrossRef CAS.
- C. Lobban, J. L. Finney and W. F. Kuhs, J. Chem. Phys., 2002, 117, 3928–3934 CrossRef CAS.
- V. Belosludov, O. Subbotin, Y. Bozhko, R. Belosludov, H. Mizuseki, Y. Kawazoe and V. Fomin, Proceedings of the 7th International Conference on Gas Hydrates (ICGH 2011), Edinburgh, Scotland, United Kingdom, 2011, pp. 1–9.
- A. V. Ildyakov, A. Y. Manakov, E. Y. Aladko, V. I. Kosyakov and V. A. Shestakov, J. Phys. Chem. B, 2013, 117, 7756–7762 CrossRef CAS PubMed.
- G. Malenkov, J. Struct. Chem., 2017, 58, 159–166 CrossRef CAS.
- W. F. Kuhs, T. C. Hansen and A. Falenty, J. Phys. Chem. Lett., 2018, 9, 3194–3198 CrossRef CAS PubMed.
- A. Falenty, T. Hansen and W. Kuhs, Nature, 2014, 516, 231–233 CrossRef CAS PubMed.
- E. D. Sloan Jr, Nature, 2003, 426, 353–363 CrossRef PubMed.
- J. S. Loveday and R. J. Nelmes, Phys. Chem. Chem. Phys., 2008, 10, 937–950 RSC.
- A. Omran, N. Nesterenko and V. Valtchev, Int. J. Hydrog., 2022, 47, 8419–8433 CrossRef CAS.
- Y.-A. Chen, L.-K. Chu, C.-K. Chu, R. Ohmura and L.-J. Chen, Sci. Rep., 2019, 9, 12345 CrossRef PubMed.
- A. Cabrera-Ramírez, D. J. Arismendi-Arrieta, A. Valdés and R. Prosmiti, Chem. Phys. Chem., 2021, 22, 359–369 CrossRef PubMed.
- D. T. Wilson, B. C. Barnes, D. T. Wu and A. K. Sum, Fluid Phase Equilib., 2016, 413, 229–234 CrossRef CAS.
- P. E. Brumby, D. Yuhara, D. T. Wu, A. K. Sum and K. Yasuoka, Fluid Phase Equilib., 2016, 413, 242–248 CrossRef CAS.
- A. Hassanpouryouzband, E. Joonaki, M. V. Farahani, S. Takeya, C. Ruppel, J. Yang, N. J. English, J. M. Schicks, K. Edlmann, H. Mehrabian, Z. M. Aman and B. Tohidi, Chem. Soc. Rev., 2020, 49, 5225–5309 RSC.
- S. Takeya, K. A. Udachin, I. L. Moudrakovski, R. Susilo and J. A. Ripmeester, J. Am. Chem. Soc., 2010, 132, 524–531 CrossRef CAS PubMed.
- C. Yu, L. Chen and B. Sun, Chin. J. Chem. Eng., 2019, 27, 2189–2206 CrossRef CAS.
- W. Cai, X. Huang and H. Lu, Energies, 2022, 15, 485 CrossRef CAS.
- H. Liu, Y. Yao and D. D. Klug, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 014102 CrossRef.
- L. del Rosso, M. Celli and L. Ulivi, Nat. Commun., 2016, 7, 133894 Search PubMed.
- M. Catti, L. del Rosso, L. Ulivi, M. Celli, F. Grazzi and T. C. Hansen, Phys. Chem. Chem. Phys., 2019, 21, 14671–14677 RSC.
- C. G. Salzmann, Chem. Phys., 2019, 150, 060901 Search PubMed.
- T. Hansen, Nat. Commun., 2021, 12, 3161 CrossRef CAS PubMed.
- Y. Liu, Y. Pu and X. C. Zeng, Nanoscale, 2023, 15, 92–100 RSC.
- Y. Huang, C. Zhu, L. Wang, J. Zhao and X. C. Zeng, Chem. Phys. Lett., 2017, 671, 186–191 CrossRef CAS.
- Y. Huang, C. Zhu, L. Wang, X. Cao, Y. Su, X. Jiang, S. Meng, J. Zhao and X. C. Zeng, Sci. Adv., 2016, 2, e1501010 CrossRef PubMed.
- T. Matsui, M. Hirata, T. Yagasaki, M. Matsumoto and H. Tanaka, Chem. Phys., 2017, 147, 091101 Search PubMed.
- Y. Liu, Y. Huang, C. Zhu, H. Li, J. Zhao, L. Wang, L. Ojamáe, J. S. Francisco and X. C. Zeng, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 12684–12691 CrossRef CAS PubMed.
- Y. Liu, Y. Huang, C. Zhu, H. Li, J. Zhao, L. Wang, L. Ojamáe, J. S. Francisco and X. C. Zeng, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 12684–12691 CrossRef CAS PubMed.
- Y. Liu, W. Zhu, J. Jiang, C. Zhu, C. Liu, B. Slater, L. Ojamáe, J. S. Francisco and X. C. Zeng, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2104442118 CrossRef CAS PubMed.
- Q. Lu, J. Ren and J. Li, Phys. Lett. A, 2021, 401, 127330 CrossRef CAS.
- C. Liu, H. Gao, Y. Wang, R. J. Needs, C. J. Pickard, J. Sun, H.-T. Wang and D. Xing, Nat. Phys., 2019, 15, 1065–1070 Search PubMed.
- C. Liu, H. Gao, A. Hermann, Y. Wang, M. Miao, C. J. Pickard, R. J. Needs, H.-T. Wang, D. Xing and J. Sun, Phys. Rev. X, 2020, 10, 021007 Search PubMed.
- R. Yanes-Rodríguez, D. J. Arismendi-Arrieta and R. Prosmiti, J. Chem. Inf. Model., 2020, 60, 3043–3056 CrossRef PubMed.
- R. Yanes-Rodríguez and R. Prosmiti, Phys. Chem. Chem. Phys., 2022, 24, 1475–1485 RSC.
- R. Yanes-Rodríguez, A. Cabrera-Ramírez and R. Prosmiti, Phys. Chem. Chem. Phys., 2022, 24, 13119–13129 RSC.
- A. Pinkard, A. Champsaur and X. Roy, Acc. Chem. Res., 2018, 51, 919–929 CrossRef CAS PubMed.
- Atelgraphics, DENEB 1.30 beta: the Nanotechnology Software, 2020, https://www.atelgraphics.com.
- F. Takeuchi, M. Hiratsuka, R. Ohmura, S. Alavi, A. K. Sum and K. Yasuoka, J. Chem. Phys., 2013, 138, 124504 CrossRef PubMed.
- A. Powers, O. Marsalek, M. Xu, L. Ulivi, D. Colognesi, M. E. Tuckerman and Z. Bacić, J. Phys. Chem. Lett., 2016, 7, 308–313 CrossRef CAS PubMed.
- D. M. Benoit, D. Lauvergnat and Y. Scribano, Faraday Discuss., 2018, 212, 533–546 RSC.
- C. Riplinger and F. Neese, J. Chem. Phys., 2013, 138, 034106 CrossRef PubMed.
- C. Riplinger, B. Sandhoefer, A. Hansen and F. Neese, J. Chem. Phys., 2013, 139, 134101 CrossRef PubMed.
- J. P. Perdew and W. Yue, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8800 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- A. Cabrera-Ramírez, D. J. Arismendi-Arrieta, A. Valdés and R. Prosmiti, Chem. Phys. Chem., 2020, 21, 2618–2628 CrossRef PubMed.
- A. Cabrera-Ramírez, R. Yanes-Rodríguez and R. Prosmiti, J. Chem. Phys., 2021, 154, 044301 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Revision C1, Gaussian Inc., Wallingford CT, 2016 Search PubMed.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. dal Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- P. Giannozzi, O. Andreussi, T. Brumme, O. Bunau, M. Buongiorno Nardelli, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, M. Cococcioni, N. Colonna, I. Carnimeo, A. Dal Corso, S. de Gironcoli, P. Delugas, R. A. DiStasio, A. Ferretti, A. Floris, G. Fratesi, G. Fugallo, R. Gebauer, U. Gerstmann, F. Giustino, T. Gorni, J. Jia, M. Kawamura, H.-Y. Ko, A. Kokalj, E. Küçükbenli, M. Lazzeri, M. Marsili, N. Marzari, F. Mauri, N. L. Nguyen, H.-V. Nguyen, A. Otero-de-la Roza, L. Paulatto, S. Poncé, D. Rocca, R. Sabatini, B. Santra, M. Schlipf, A. P. Seitsonen, A. Smogunov, I. Timrov, T. Thonhauser, P. Umari, N. Vast, X. Wu and S. Baroni, J. Phys.: Condens. Matter, 2017, 29, 465901 CrossRef CAS PubMed.
- P. Giannozzi, O. Baseggio, P. Bonfá, D. Brunato, R. Car, I. Carnimeo, C. Cavazzoni, S. de Gironcoli, P. Delugas, F. Ferrari Ruffino, A. Ferretti, N. Marzari, I. Timrov, A. Urru and S. Baroni, J. Chem. Phys., 2020, 152, 154105 CrossRef CAS PubMed.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby and M. Schütz, WIREs Comput. Mol. Sci., 2012, 2, 242–253 CrossRef CAS.
- H.-J. Werner, P. J. Knowles, F. R. Manby, J. A. Black, K. Doll, A. Heβelmann, D. Kats, A. Köhn, T. Korona, D. A. Kreplin, Q. Ma, T. F. Miller, A. Mitrushchenkov, K. A. Peterson, I. Polyak, G. Rauhut and M. Sibaev, J. Chem. Phys., 2020, 152, 144107 Search PubMed.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby, M. Schütz, P. Celani, W. Györffy, D. Kats, T. Korona, R. Lindh, A. Mitrushenkov, G. Rauhut, K. R. Shamasundar, T. B. Adler, R. D. Amos, S. J. Bennie, A. Bernhardsson, A. Berning, D. L. Cooper, M. J. O. Deegan, A. J. Dobbyn, F. Eckert, E. Goll, C. Hampel, A. Hesselmann, G. Hetzer, T. Hrenar, G. Jansen, C. Köppl, S. J. R. Lee, Y. Liu, A. W. Lloyd, Q. Ma, R. A. Mata, A. J. May, S. J. McNicholas, W. Meyer, T. F. M. III, M. E. Mura, A. Nicklass, D. P. O'Neill, P. Palmieri, D. Peng, K. Pflüger, R. Pitzer, M. Reiher, T. Shiozaki, H. Stoll, A. J. Stone, R. Tarroni, T. Thorsteinsson, M. Wang and M. Welborn, MOLPRO, a package of ab initio programs, https://www.molpro.net.
- F. Neese, WIREs Comput. Mol. Sci., 2012, 2, 73–78 CrossRef CAS.
- F. Neese, WIREs Comput. Mol. Sci., 2018, 8, e1327 Search PubMed.
- E. Caldeweyher, C. Bannwarth and S. Grimme, J. Chem. Phys., 2017, 147, 034112 CrossRef PubMed.
- E. Caldeweyher, S. Ehlert, A. Hansen, H. Neugebauer, S. Spicher, C. Bannwarth and S. Grimme, J. Chem. Phys., 2019, 150, 154122 CrossRef PubMed.
- E. Caldeweyher, J.-M. Mewes, S. Ehlert and S. Grimme, Phys. Chem. Chem. Phys., 2020, 22, 8499–8512 RSC.
- E. R. Johnson and A. D. Becke, J. Chem. Phys., 2005, 123, 024101 CrossRef PubMed.
- A. D. Becke and E. R. Johnson, J. Chem. Phys., 2005, 122, 154104 CrossRef PubMed.
- U. Bonn, D4-A Generally Applicable Atomic-ChargeDependent London Dispersion Correction, https://www.chemie.uni-bonn.de/pctc/mulliken-center/software/dftd4.
- A. Otero-de-la Roza and E. R. Johnson, J. Chem. Phys., 2013, 138, 204109 CrossRef CAS PubMed.
- F. O. Kannemann and A. D. Becke, J. Chem. Theory Comput., 2010, 6, 1081–1088 CrossRef CAS.
- S. Boys and F. Bernardi, Mol. Phys., 1970, 19, 553–566 CrossRef CAS.
- E. C. Lee, D. Kim, P. Jurecka, P. Tarakeshwar, P. Hobza and K. S. Kim, J. Phys. Chem. A, 2007, 111, 3446–3457 CrossRef CAS PubMed.
- D. McQuarrie and J. Simon, Molecular Thermodynamics, University Science Books, 1999 Search PubMed.
- J. Ochterski, Thermochemistry in Gausssian, https://gaussian.com/thermo/.
- H. J. C. Berendsen, J. R. Grigera and T. P. Straatsma, J. Chem. Phys., 1987, 91, 6269–6271 CrossRef CAS.
- J. L. F. Abascal, E. Sanz, R. García Fernández and C. Vega, J. Chem. Phys., 2005, 122, 234511 CrossRef CAS PubMed.
- R. V. Belosludov, Y. Y. Bozhko, O. S. Subbotin, V. R. Belosludov, H. Mizuseki, Y. Kawazoe and V. M. Fomin, J. Phys. Chem. C, 2014, 118, 2587–2593 CrossRef CAS.
- R. A. Aziz and M. J. Slaman, J. Chem. Phys., 1991, 94, 8047–8053 CrossRef CAS.
- J. Makarewicz, J. Chem. Phys., 2008, 129, 184310 CrossRef PubMed.
- S. Mondal and P. K. Chattaraj, Phys. Chem. Chem. Phys., 2014, 16, 17943–17954 RSC.
- D.-Y. Koh, H. Kang, J. Jeon, Y.-H. Ahn, Y. Park, H. Kim and H. Lee, J. Phys. Chem. C, 2014, 118, 3324–3330 CrossRef CAS.
- C. J. Burnham, Z. Futera and N. J. English, Phys. Chem. Chem. Phys., 2017, 19, 717–728 RSC.
- A. Rasoolzadeh and A. Shariati, Fluid Phase Equilib., 2019, 494, 8–20 CrossRef CAS.
- Y. Krishnan, M. R. Ghaani, A. Desmedt and N. J. English, Appl. Sci., 2021, 11, 282 CrossRef CAS.
- M. Li, X. He, B. Wang, D. Zhao, C. Rong, P. K. Chattaraj and S. Liu, Front. Chem., 2020, 8, 566 CrossRef CAS PubMed.
- A. Vítek, D. J. Arismendi-Arrieta, M. Sarmanová, R. Kalus and R. Prosmiti, J. Phys. Chem. A, 2020, 124, 4036–4047 CrossRef PubMed.
- A. Vítek, D. J. Arismendi-Arrieta, R. Rodríguez-Cantano, R. Prosmiti, P. Villarreal, R. Kalus and G. Delgado-Barrio, Phys. Chem. Chem. Phys., 2015, 17, 8792–8801 RSC.
- D. J. Arismendi-Arrieta, A. Vítek and R. Prosmiti, J. Phys. Chem. C, 2016, 120, 26093–26102 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3cp00603d |
| This journal is © the Owner Societies 2023 |